

In the crystal of the title compound, C—H⋯O hydrogen bonds link the molecules, enclosing
 (10) and
(16) ring motifs, into layers almost parallel to the bc plane. The layers are further connected by π–π stacking interactions.
(10) and
(16) ring motifs, into layers almost parallel to the bc plane. The layers are further connected by π–π stacking interactions.
Keywords: crystal structure, π-stacking, C—H⋯O hydrogen bonds, dihydroquinoline
Abstract
In the title molecule, C14H11NO3, the dihydroquinoline core deviates slightly from planarity, indicated by the dihedral angle of 1.07 (3)° between the two six-membered rings. In the crystal, layers of molecules almost parallel to the bc plane are formed by C—H⋯O hydrogen bonds. These are joined by π–π stacking interactions. A Hirshfeld surface analysis revealed that the most important contributions to the crystal packing are from H⋯H (36.0%), H⋯C/C⋯H (28.9%) and H⋯O/O⋯H (23.5%) interactions. The evaluation of the electrostatic, dispersion and total energy frameworks indicates that the stabilization is dominated by the dispersion energy contribution. Moreover, the molecular structure optimized by density functional theory (DFT) at the B3LYP/6-311G(d,p) level is compared with the experimentally determined molecular structure in the solid state. The HOMO–LUMO behaviour was elucidated to determine the energy gap.
1. Chemical context
Quinoline derivatives form a class of heterocyclic compounds that have received much attention due to their biological and pharmacological activities (Filali Baba et al., 2019 ▸; Hayani et al., 2021 ▸). They are used in the pharmaceutical industry because of their antimicrobial (Katoh et al., 2004 ▸; Abdel-Wahab et al., 2012 ▸), anti-inflammatory (Leatham et al., 1983 ▸), antihypertensive (Muruganantham et al., 2004 ▸), antibiotic (Mahamoud et al., 2006 ▸), anti-HIV (Wilson et al., 1992 ▸; Strekowski et al., 1991 ▸) and corrosion inhibitive activities (Filali Baba et al., 2016a ▸,b ▸). They are also considered as an important scaffold for the development of new pharmaceutically active agents (Filali Baba et al., 2020 ▸; Bouzian et al., 2018 ▸).
In continuation of our research work devoted to the study of O-alkylation and N-alkylation reactions involving quinoline derivatives, we report herein the synthesis and the molecular and crystal structures of methyl 2-oxo-1-(prop-2-ynyl)-1,2-dihydroquinoline-4-carboxylate, obtained by an alkylation reaction of methyl 2-oxo-1,2-dihydroquinoline-4-carboxylate using an excess of propargyl bromide as an alkylating reagent in phase transfer catalysis (PTC). Moreover, a Hirshfeld surface analysis and interaction energy and energy framework calculations were performed. The molecular structure optimized by density functional theory (DFT) at the B3LYP/6-311G(d,p) level is compared with the experimentally determined molecular structure in the solid state.
2. Structural commentary
The dihydroquinoline core of the title molecule (Fig. 1 ▸) deviates slightly from planarity, as indicated by the dihedral angle of 1.07 (3)° between the mean planes of the A (C1–C5/N1) and B (C4–C9) rings. Atoms O1, O2, O3, C10, C13 and C14 are −0.1294 (11), 0.1907 (12), −0.2708 (15), 0.0177 (14), −0.0267 (13) and 0.0953 (23) Å from the least-squares plane of the A ring. The O2—C13 [1.3123 (17) Å] and O3—C13 [1.1955 (16) Å] distances in the ester group indicate localized single and double bonds, rather than delocalized bonding arrangements. The O2—C13—O3 bond angle [122.55 (12)°] seems to be slightly increased with respect to that present in a free acid (122.2°; Sim et al., 1955 ▸). The O2—C13—O3 bond angle may be compared with the corresponding value of 124.27 (17)° in diaquabis(2-bromobenzoato-κO)bis(nicotinamide-κN 1)zinc(II) (Hökelek et al., 2009 ▸).
Figure 1.

The molecular structure of the title compound with the atom-labeling scheme and displacement ellipsoids drawn at the 50% probability level.
3. Supramolecular features
In the crystal, C—H⋯O hydrogen bonds (Table 1 ▸) link the molecules, enclosing
(10) and
(16) ring motifs, into layers almost parallel to the bc plane (Fig. 2 ▸). These layers are further connected by π–π stacking interactions between the A and B(x − 1, y, z) rings [centroid-to-centroid distance = 3.5629 (7) Å, α = 1.13° and slippage = 1.221 Å] to form a triperiodic network.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C9—H9⋯O3 | 0.969 (17) | 2.210 (15) | 2.8807 (19) | 125.3 (12) |
| C10—H10A⋯O1 | 0.922 (16) | 2.277 (17) | 2.6961 (17) | 107.1 (12) |
| C14—H14B⋯O1i | 0.95 (2) | 2.56 (2) | 3.433 (2) | 153.8 (18) |
Symmetry code: (i)
 .
.
Figure 2.

A partial packing diagram, viewed down the a axis, with C—H⋯O hydrogen bonds shown as dashed lines.
4. Hirshfeld surface analysis
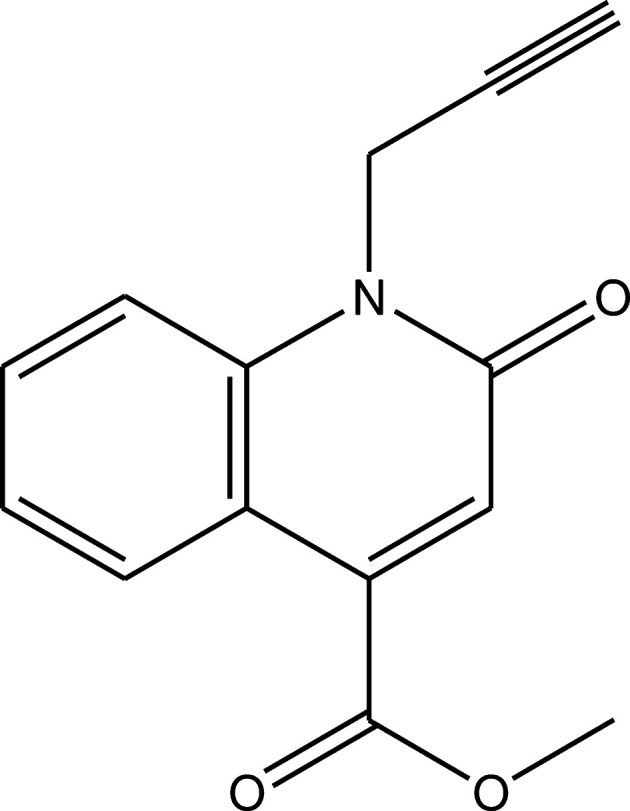
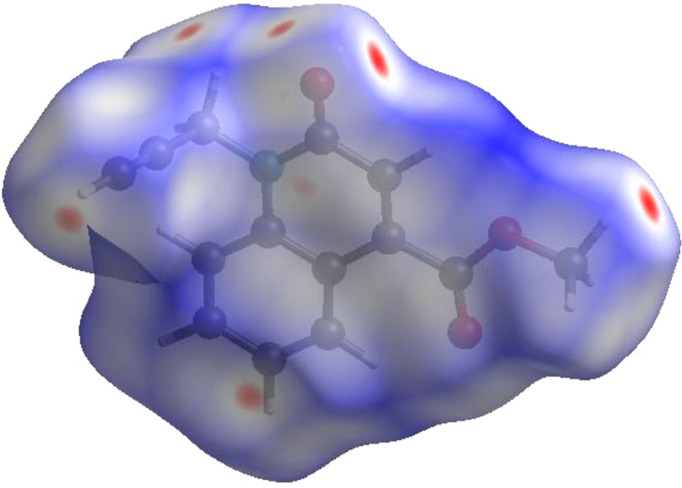
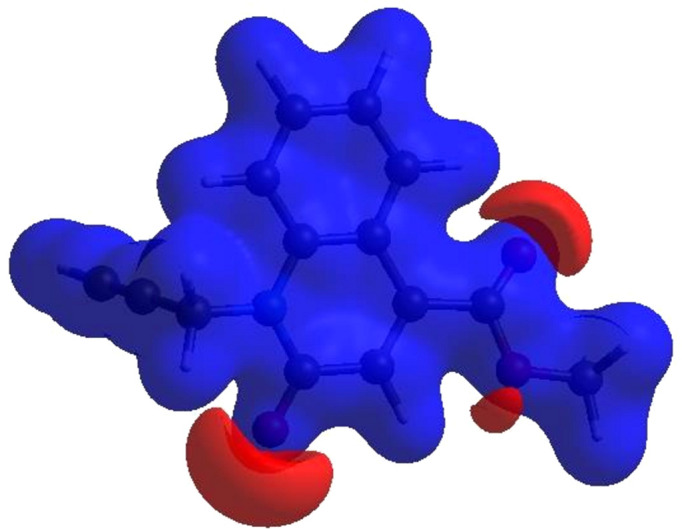
In order to visualize the intermolecular interactions in the crystal of the title compound, a Hirshfeld surface (HS) analysis (Hirshfeld, 1977 ▸) was carried out by using CrystalExplorer (Spackman et al., 2021 ▸). In the HS plotted over d norm (Fig. 3 ▸), the white surface indicates contacts with distances equal to the sum of the van der Waals radii, and the red and blue colours indicate distances shorter (in close contact) or longer (distinct contact) than the sum of the van der Waals radii (Venkatesan et al., 2016 ▸). The bright-red spots indicate their roles as respective donors and/or acceptors; they also appear as blue and red regions corresponding to positive and negative potentials on the HS mapped over electrostatic potential (Spackman et al., 2008 ▸; Jayatilaka et al., 2005 ▸), as shown in Fig. 4 ▸. The blue regions indicate positive electrostatic potential (hydrogen-bond donors), while the red regions indicate negative electrostatic potential (hydrogen-bond acceptors). The shape-index of the HS is a tool to visualize the π–π stacking interactions by the presence of adjacent red and blue triangles (Fig. 5 ▸). The overall two-dimensional fingerprint plot [Fig. 6 ▸(a)] and those delineated into H⋯H, H⋯C/C⋯H, H⋯O/O⋯H, C⋯C, C⋯O/O⋯C, H⋯N/N⋯H, C⋯N/N⋯C and N⋯O/O⋯N contacts (McKinnon et al., 2007 ▸) are illustrated in Figs. 6 ▸(b)–(i), respectively, together with their relative contributions to the Hirshfeld surface. The most important interaction is H⋯H, contributing 36.0% to the overall crystal packing, which is reflected in Fig. 6 ▸(b) as widely scattered points of high density due to the large hydrogen content of the molecule with the tip at d e = d i = 1.22 Å. In the absence of C—H⋯π interactions, the pair of characteristic wings resulting in the fingerprint plot delineated into H⋯C/C⋯H contacts [Fig. 6 ▸(c)] have a 28.9% contribution to the HS, with the tips at d e + d i = 2.68 Å. The pair of the scattered points of spikes resulting in the fingerprint plot delineated into H⋯O/O⋯H contacts [Fig. 6 ▸(d)], with a 23.5% contribution to the HS, has an almost symmetric distribution of points, with the tips at d e + d i = 2.44 Å. The C⋯C contacts [Fig. 6 ▸(e)] appear as an arrow-shaped distribution of points and have a contribution of 7.0% to the HS with the tip at d e = d i = 1.69 Å. The tiny spikes of C⋯O/O⋯C contacts [Fig. 6 ▸(f)], with a 2.5% contribution to the HS, are visible at d e + d i = 3.58 Å. Finally, the H⋯N/N⋯H [Fig. 6 ▸(g)], C⋯N/N⋯C [Fig. 6 ▸(h)] and N⋯O/O⋯N [Fig. 6 ▸(i)] contacts contribute 1.4, 0.4 and 0.4%, respectively, to the HS.
Figure 3.
View of the three-dimensional Hirshfeld surface of the title compound, plotted over d norm in the range from −0.1226 to 1.1991 a.u.
Figure 4.
View of the three-dimensional Hirshfeld surface of the title compound plotted over electrostatic potential energy in the range from −0.0500 to 0.0500 a.u., using the STO-3G basis set at the Hartree–Fock level of theory.
Figure 5.
The Hirshfeld surface of the title compound plotted over shape-index.
Figure 6.

The full two-dimensional fingerprint plots for the title compound, showing (a) all interactions, (b) H⋯H, (c) H⋯C/C⋯H, (d) H⋯O/O⋯H, (e) C⋯C, (f) C⋯O/O⋯C, (g) H⋯N/N⋯H, (h) C⋯N/N⋯C and (i) N⋯O/O⋯N interactions. The d i and d e values are the closest internal and external distances (in Å) from given points on the Hirshfeld surface contacts.
The Hirshfeld surface representations with the function d norm plotted onto the surface are shown for the H⋯H and H⋯C/C⋯H interactions in Figs. 7 ▸(a)–(c), respectively.
Figure 7.

The Hirshfeld surface representations with the function d norm plotted onto the surface for (a) H⋯H, (b) H⋯C/C⋯H and (c) H⋯O/O⋯H interactions.
The Hirshfeld surface analysis confirms the importance of H-atom contacts in establishing the packing. The large number of H⋯H, H⋯C/C⋯H and H⋯O/O⋯H interactions suggest that van der Waals interactions play the major role in the crystal packing (Hathwar et al., 2015 ▸).
5. Interaction energy calculations and energy frameworks
Using CrystalExplorer (Spackman et al., 2021 ▸), the intermolecular interaction energies were calculated at the CEB3LYP/631G(d,p) energy level, where a cluster of molecules is generated by applying crystallographic symmetry operations with respect to a selected central molecule within a radius of 3.8 Å by default (Turner et al., 2014 ▸). The total intermolecular energy (E tot) is the sum of electrostatic (E ele), polarization (E pol), dispersion (E dis) and exchange–repulsion (E rep) energies (Turner et al., 2015 ▸), with scale factors of 1.057, 0.740, 0.871 and 0.618, respectively (Mackenzie et al., 2017 ▸). Energy frameworks combine the calculation of intermolecular interaction energies with a graphical representation of their magnitude (Turner et al., 2015 ▸). Energies between molecular pairs are represented as cylinders joining the centroids of pairs of molecules with the cylinder radius proportional to the relative strength of the corresponding interaction energy. Energy frameworks were constructed for E ele (red cylinders), E dis (green cylinders) and E tot (blue cylinders), and are shown in Figs. 8 ▸(a)–(c). The evaluation of the electrostatic, dispersion and total energy frameworks indicates that in the title compound the stabilization is dominated by the dispersion energy contribution.
Figure 8.

The energy frameworks, viewed down the c axis, for a cluster of molecules of the title compound, showing the (a) electrostatic energy, (b) dispersion energy and (c) total energy diagrams, where the b axis is vertical and the c axis is horizontal. The cylindrical radius is proportional to the relative strength of the corresponding energies and was adjusted to the same scale factor of 80 with a cut-off value of 5 kJ mol−1 within 2 × 2 × 2 unit cells.
6. DFT calculations
The optimized structure of the title compound in the gas phase was computed on the basis of density functional theory (DFT) using the standard B3LYP functional and the 6311G(d,p) basis set (Becke, 1993 ▸), as implemented in GAUSSIAN09 (Frisch et al., 2009 ▸). Comparisons of calculated bond lengths and angles with those of the experimental study are compiled in Table 2 ▸. The frontier orbitals were also investigated, and the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) orbitals are depicted in Fig. 9 ▸. It can be seen that the electron density of the HOMO is mostly distributed within the quinoline moiety, while that of the LUMO is mostly distributed over the carboxylate group.
Table 2. Comparison (X-ray and DFT) of selected bond lengths and angles (Å, °).
| Bonds/angles | X-ray | B3LYP/6-311G(d,p) |
|---|---|---|
| O1—C1 | 1.2288 (14) | 1.2231 |
| N1—C5 | 1.3993 (14) | 1.3955 |
| N1—C10 | 1.4742 (14) | 1.4727 |
| N1—C1 | 1.3774 (16) | 1.4035 |
| O2—C13 | 1.3123 (17) | 1.3460 |
| O2—C14 | 1.4491 (17) | 1.4399 |
| O3—C13 | 1.1955 (16) | 1.2081 |
| C5—C4 | 1.4159 (16) | 1.4234 |
| C5—C6 | 1.4011 (17) | 1.4062 |
| C4—C3 | 1.4516 (16) | 1.4539 |
| C4—C9 | 1.4064 (16) | 1.4096 |
| C5—N1—C10 | 120.18 (10) | 120.925 |
| C1—N1—C5 | 123.16 (9) | 123.436 |
| C1—N1—C10 | 116.52 (10) | 115.623 |
| C13—O2—C14 | 116.39 (12) | 115.680 |
| N1—C5—C4 | 120.23 (10) | 120.142 |
| N1—C5—C6 | 119.97 (10) | 120.504 |
| C6—C5—C4 | 119.80 (10) | 119.355 |
| C5—C4—C3 | 117.38 (10) | 117.701 |
| C9—C4—C5 | 118.06 (11) | 118.477 |
Figure 9.

The energy band gap of the title compound.
Other chemistry descriptors (chemical hardness η, softness S, electronegativity χ and electrophilicity ω) derived from the conceptual DFT calculations are given in Table 3 ▸. The HOMO and LUMO are localized in the plane extending from the methyl 2-oxo-1-(prop-2-ynyl)-1,2-dihydroquinoline-4-carboxylate ring. The energy band gap [ΔE = E LUMO − E HOMO] (Fig. 9 ▸) of the molecule is about −4.0 eV, with individual frontier molecular orbital energies, E HOMO and E LUMO, of −6.35 and −2.35 eV, respectively.
Table 3. Calculated energies for compound (I).
| Total energy, TE (eV) | −22331.1678 |
| E HOMO (eV) | –6,35 |
| E LUMO (eV) | –2.35 |
| Gap, ΔE (eV) | –4.0 |
| Dipole moment, μ (Debye) | 2.1062 |
| Ionization potential, I (eV) | 6.35 |
| Electron affinity, A | 2.35 |
| Electronegativity, χ | 4.35 |
| Hardness, η | 2 |
| Electrophilicity index, ω | 4.73 |
| Softness, σ | 0.5 |
| Fraction of electron transferred, ΔN | 0.66 |
7. Database survey
A search of the Cambridge Structural Database (CSD, updated 20 March 2023; Groom et al., 2016 ▸) using fragment (II) (Fig. 10 ▸) returned 20 hits, 16 of which contained an ester group attached to C7 (the rest contained an alkyl group at this position) and, only two of them, with refcodes ROKCIG (Filali Baba et al., 2019 ▸) and REYREV (Filali Baba et al., 2017 ▸), contain halogen atoms attached to aromatic rings. The former is more closely related to the title molecule due to the presence of an ethyl group on the nitrogen and ester substituents. Unlike the title molecule, that of ROKCIG forms an inverted dimer via C—H⋯O hydrogen bonds (instead of ribbons), with layer-by-layer connections approximately parallel to (10
 ), but it has no C—H⋯Cl hydrogen bonds or π–π stacking interactions. The halogen-free analogue of ROKCIG (ROKCOM; Filali Baba et al., 2019 ▸) uses C—H⋯O hydrogen bonds to form molecular bands along the c axis, which are connected by weak π–π interactions.
), but it has no C—H⋯Cl hydrogen bonds or π–π stacking interactions. The halogen-free analogue of ROKCIG (ROKCOM; Filali Baba et al., 2019 ▸) uses C—H⋯O hydrogen bonds to form molecular bands along the c axis, which are connected by weak π–π interactions.
Figure 10.
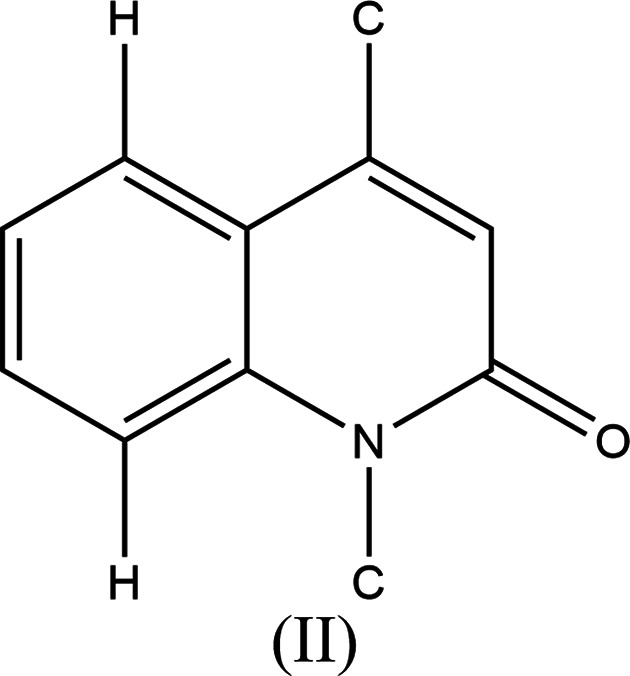
The molecular moiety used for the database search.
8. Refinement
Crystal, data collection and refinement details are presented in Table 4 ▸. H atoms were included as riding contributions in idealized positions with isotropic displacement parameters tied to those of the attached atoms. Two reflections obscured by the beamstop were omitted from the final refinement.
Table 4. Experimental details.
| Crystal data | |
| Chemical formula | C14H11NO3 |
| M r | 241.24 |
| Crystal system, space group | Triclinic, P

|
| Temperature (K) | 296 |
| a, b, c (Å) | 4.7033 (2), 11.1113 (6), 11.3876 (5) |
| α, β, γ (°) | 81.759 (2), 83.356 (2), 85.564 (2) |
| V (Å3) | 583.89 (5) |
| Z | 2 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.10 |
| Crystal size (mm) | 0.24 × 0.14 × 0.11 |
| Data collection | |
| Diffractometer | Bruker DUO PHOTON III |
| Absorption correction | Multi-scan (SADABS; Krause et al., 2015 ▸) |
| T min, T max | 0.708, 0.746 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 45341, 3559, 2489 |
| R int | 0.045 |
| (sin θ/λ)max (Å−1) | 0.714 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.047, 0.148, 1.11 |
| No. of reflections | 3559 |
| No. of parameters | 207 |
| H-atom treatment | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.38, −0.28 |
9. Synthesis and crystallization
To a solution of methyl 2-oxo-1,2-dihydroquinoline-4-carboxylate (4.47 mmol) in 10 ml of dimethylformamide (DMF) were added propargyl bromide (9.83 mmol), K2CO3 (22.36 mmol) and tetra-n-butylammonium bromide (TBAB; 0.5 mmol). The reaction mixture was stirred at room temperature in DMF for 6 h. After removal of the formed salts, the solvent was evaporated under reduced pressure and the residue obtained was dissolved in dichloromethane. The organic phase was dried over Na2SO4 and then concentrated in vacuo. A pure compound was obtained after recrystallization from dichloromethane/hexane (2:3 v/v).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989023007557/wm5688sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989023007557/wm5688Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989023007557/wm5688Isup3.cdx
Supporting information file. DOI: 10.1107/S2056989023007557/wm5688Isup4.cdx
CCDC reference: 2291603
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
TH is grateful to Hacettepe University Scientific Research Project Unit.
supplementary crystallographic information
Crystal data
| C14H11NO3 | Z = 2 |
| Mr = 241.24 | F(000) = 252 |
| Triclinic, P1 | Dx = 1.372 Mg m−3 |
| a = 4.7033 (2) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 11.1113 (6) Å | Cell parameters from 9957 reflections |
| c = 11.3876 (5) Å | θ = 2.4–30.2° |
| α = 81.759 (2)° | µ = 0.10 mm−1 |
| β = 83.356 (2)° | T = 296 K |
| γ = 85.564 (2)° | Block, colourless |
| V = 583.89 (5) Å3 | 0.24 × 0.14 × 0.11 mm |
Data collection
| Bruker DUO PHOTON III diffractometer | Rint = 0.045 |
| Radiation source: microfocus sealed X-ray tube | θmax = 30.5°, θmin = 1.8° |
| φ and ω scans | h = −6→6 |
| Absorption correction: multi-scan (SADABS; Krause et al., 2015) | k = −15→15 |
| Tmin = 0.708, Tmax = 0.746 | l = −16→16 |
| 45341 measured reflections | 3 standard reflections every 1000 reflections |
| 3559 independent reflections | intensity decay: 1% |
| 2489 reflections with I > 2σ(I) |
Refinement
| Refinement on F2 | Primary atom site location: dual |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.047 | Hydrogen site location: difference Fourier map |
| wR(F2) = 0.148 | All H-atom parameters refined |
| S = 1.11 | w = 1/[σ2(Fo2) + (0.0712P)2 + 0.064P] where P = (Fo2 + 2Fc2)/3 |
| 3559 reflections | (Δ/σ)max < 0.001 |
| 207 parameters | Δρmax = 0.38 e Å−3 |
| 0 restraints | Δρmin = −0.28 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.3589 (2) | 0.16022 (9) | 0.53718 (9) | 0.0571 (3) | |
| N1 | 0.6716 (2) | 0.14916 (9) | 0.67741 (9) | 0.0381 (2) | |
| O2 | 0.2219 (3) | 0.56421 (9) | 0.64710 (10) | 0.0655 (3) | |
| O3 | 0.3962 (3) | 0.56270 (10) | 0.81846 (12) | 0.0812 (4) | |
| C5 | 0.7779 (2) | 0.19819 (10) | 0.76892 (10) | 0.0359 (2) | |
| C4 | 0.6875 (2) | 0.31800 (10) | 0.79156 (10) | 0.0357 (2) | |
| C3 | 0.4848 (3) | 0.38599 (10) | 0.71625 (10) | 0.0372 (3) | |
| C10 | 0.7636 (3) | 0.02397 (11) | 0.65470 (13) | 0.0441 (3) | |
| C13 | 0.3695 (3) | 0.51328 (11) | 0.73414 (12) | 0.0434 (3) | |
| C11 | 0.6261 (3) | −0.06721 (11) | 0.74351 (13) | 0.0463 (3) | |
| C6 | 0.9724 (3) | 0.12821 (12) | 0.83921 (12) | 0.0439 (3) | |
| C1 | 0.4663 (3) | 0.20952 (11) | 0.60989 (11) | 0.0411 (3) | |
| C2 | 0.3857 (3) | 0.33419 (11) | 0.63055 (11) | 0.0422 (3) | |
| C9 | 0.8005 (3) | 0.36311 (12) | 0.88452 (12) | 0.0438 (3) | |
| C8 | 0.9935 (3) | 0.29352 (14) | 0.95173 (12) | 0.0500 (3) | |
| C7 | 1.0789 (3) | 0.17610 (14) | 0.92919 (13) | 0.0497 (3) | |
| C12 | 0.5140 (4) | −0.14126 (14) | 0.81495 (17) | 0.0605 (4) | |
| C14 | 0.0863 (5) | 0.68407 (15) | 0.6591 (2) | 0.0686 (5) | |
| H10A | 0.720 (4) | 0.0174 (14) | 0.5791 (15) | 0.053 (4)* | |
| H9 | 0.736 (3) | 0.4439 (15) | 0.9029 (14) | 0.054 (4)* | |
| H10B | 0.974 (3) | 0.0129 (13) | 0.6502 (12) | 0.044 (4)* | |
| H6 | 1.030 (3) | 0.0475 (15) | 0.8245 (14) | 0.052 (4)* | |
| H2 | 0.247 (4) | 0.3772 (15) | 0.5795 (15) | 0.056 (4)* | |
| H8 | 1.067 (4) | 0.3254 (16) | 1.0161 (17) | 0.070 (5)* | |
| H7 | 1.214 (4) | 0.1274 (17) | 0.9743 (16) | 0.066 (5)* | |
| H14A | 0.232 (6) | 0.741 (2) | 0.657 (2) | 0.111 (8)* | |
| H14B | −0.011 (5) | 0.7061 (19) | 0.590 (2) | 0.083 (6)* | |
| H14C | −0.036 (6) | 0.684 (2) | 0.729 (2) | 0.100 (8)* | |
| H12 | 0.435 (5) | −0.201 (2) | 0.874 (2) | 0.091 (7)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0699 (7) | 0.0505 (6) | 0.0589 (6) | 0.0021 (5) | −0.0237 (5) | −0.0248 (5) |
| N1 | 0.0416 (5) | 0.0328 (5) | 0.0416 (5) | 0.0006 (4) | −0.0036 (4) | −0.0131 (4) |
| O2 | 0.0899 (8) | 0.0451 (6) | 0.0658 (7) | 0.0234 (5) | −0.0303 (6) | −0.0185 (5) |
| O3 | 0.1245 (11) | 0.0485 (6) | 0.0815 (8) | 0.0261 (6) | −0.0469 (8) | −0.0339 (6) |
| C5 | 0.0346 (5) | 0.0359 (6) | 0.0379 (6) | −0.0031 (4) | −0.0010 (4) | −0.0092 (4) |
| C4 | 0.0357 (5) | 0.0355 (6) | 0.0367 (5) | −0.0027 (4) | −0.0009 (4) | −0.0097 (4) |
| C3 | 0.0401 (6) | 0.0323 (5) | 0.0395 (6) | −0.0015 (4) | −0.0015 (4) | −0.0083 (4) |
| C10 | 0.0468 (7) | 0.0369 (6) | 0.0508 (7) | 0.0033 (5) | −0.0026 (5) | −0.0189 (5) |
| C13 | 0.0488 (7) | 0.0337 (6) | 0.0488 (7) | −0.0004 (5) | −0.0053 (5) | −0.0097 (5) |
| C11 | 0.0466 (7) | 0.0352 (6) | 0.0607 (8) | 0.0035 (5) | −0.0097 (6) | −0.0185 (6) |
| C6 | 0.0423 (6) | 0.0402 (6) | 0.0491 (7) | 0.0027 (5) | −0.0061 (5) | −0.0075 (5) |
| C1 | 0.0461 (6) | 0.0387 (6) | 0.0407 (6) | −0.0015 (5) | −0.0056 (5) | −0.0125 (5) |
| C2 | 0.0485 (7) | 0.0368 (6) | 0.0425 (6) | 0.0022 (5) | −0.0101 (5) | −0.0085 (5) |
| C9 | 0.0460 (6) | 0.0440 (7) | 0.0447 (6) | −0.0034 (5) | −0.0051 (5) | −0.0161 (5) |
| C8 | 0.0501 (7) | 0.0602 (8) | 0.0438 (7) | −0.0047 (6) | −0.0105 (6) | −0.0154 (6) |
| C7 | 0.0454 (7) | 0.0572 (8) | 0.0472 (7) | 0.0010 (6) | −0.0119 (6) | −0.0055 (6) |
| C12 | 0.0642 (9) | 0.0447 (8) | 0.0737 (10) | −0.0063 (7) | −0.0086 (8) | −0.0093 (7) |
| C14 | 0.0861 (13) | 0.0412 (8) | 0.0810 (12) | 0.0209 (8) | −0.0280 (11) | −0.0148 (8) |
Geometric parameters (Å, º)
| O1—C1 | 1.2288 (14) | C10—H10B | 0.983 (15) |
| N1—C5 | 1.3993 (14) | C11—C12 | 1.180 (2) |
| N1—C10 | 1.4742 (14) | C6—C7 | 1.3785 (19) |
| N1—C1 | 1.3774 (16) | C6—H6 | 0.949 (17) |
| O2—C13 | 1.3123 (17) | C1—C2 | 1.4524 (17) |
| O2—C14 | 1.4491 (17) | C2—H2 | 0.977 (18) |
| O3—C13 | 1.1955 (16) | C9—C8 | 1.3750 (19) |
| C5—C4 | 1.4159 (16) | C9—H9 | 0.969 (17) |
| C5—C6 | 1.4011 (17) | C8—C7 | 1.386 (2) |
| C4—C3 | 1.4516 (16) | C8—H8 | 0.966 (19) |
| C4—C9 | 1.4064 (16) | C7—H7 | 0.950 (19) |
| C3—C13 | 1.5081 (16) | C12—H12 | 0.94 (2) |
| C3—C2 | 1.3449 (17) | C14—H14A | 0.97 (3) |
| C10—C11 | 1.457 (2) | C14—H14B | 0.95 (2) |
| C10—H10A | 0.922 (16) | C14—H14C | 0.93 (3) |
| C5—N1—C10 | 120.18 (10) | C7—C6—C5 | 120.28 (12) |
| C1—N1—C5 | 123.16 (9) | C7—C6—H6 | 120.2 (9) |
| C1—N1—C10 | 116.52 (10) | O1—C1—N1 | 121.71 (11) |
| C13—O2—C14 | 116.39 (12) | O1—C1—C2 | 122.56 (12) |
| N1—C5—C4 | 120.23 (10) | N1—C1—C2 | 115.73 (10) |
| N1—C5—C6 | 119.97 (10) | C3—C2—C1 | 123.09 (11) |
| C6—C5—C4 | 119.80 (10) | C3—C2—H2 | 121.9 (10) |
| C5—C4—C3 | 117.38 (10) | C1—C2—H2 | 114.9 (10) |
| C9—C4—C5 | 118.06 (11) | C4—C9—H9 | 119.0 (10) |
| C9—C4—C3 | 124.56 (11) | C8—C9—C4 | 121.30 (12) |
| C4—C3—C13 | 121.83 (10) | C8—C9—H9 | 119.6 (10) |
| C2—C3—C4 | 120.15 (10) | C9—C8—C7 | 120.08 (12) |
| C2—C3—C13 | 117.97 (11) | C9—C8—H8 | 120.2 (11) |
| N1—C10—H10A | 106.8 (10) | C7—C8—H8 | 119.7 (11) |
| N1—C10—H10B | 109.5 (8) | C6—C7—C8 | 120.47 (13) |
| C11—C10—N1 | 112.13 (10) | C6—C7—H7 | 118.6 (11) |
| C11—C10—H10A | 111.3 (10) | C8—C7—H7 | 120.9 (11) |
| C11—C10—H10B | 111.7 (8) | C11—C12—H12 | 176.4 (14) |
| H10A—C10—H10B | 105.1 (13) | O2—C14—H14A | 109.5 (16) |
| O2—C13—C3 | 111.85 (10) | O2—C14—H14B | 105.1 (13) |
| O3—C13—O2 | 122.55 (12) | O2—C14—H14C | 111.5 (16) |
| O3—C13—C3 | 125.54 (12) | H14A—C14—H14B | 107.6 (19) |
| C12—C11—C10 | 179.65 (16) | H14A—C14—H14C | 110 (2) |
| C5—C6—H6 | 119.5 (9) | H14B—C14—H14C | 113 (2) |
| O1—C1—C2—C3 | 174.62 (13) | C10—N1—C5—C4 | −179.38 (10) |
| N1—C5—C4—C3 | −0.33 (16) | C10—N1—C5—C6 | 0.02 (17) |
| N1—C5—C4—C9 | 179.99 (10) | C10—N1—C1—O1 | 2.22 (18) |
| N1—C5—C6—C7 | 179.74 (11) | C10—N1—C1—C2 | −177.92 (10) |
| N1—C1—C2—C3 | −5.23 (19) | C13—C3—C2—C1 | −175.93 (11) |
| C5—N1—C10—C11 | 75.59 (14) | C6—C5—C4—C3 | −179.73 (10) |
| C5—N1—C1—O1 | −173.59 (11) | C6—C5—C4—C9 | 0.59 (17) |
| C5—N1—C1—C2 | 6.27 (18) | C1—N1—C5—C4 | −3.71 (17) |
| C5—C4—C3—C13 | 178.69 (10) | C1—N1—C5—C6 | 175.68 (11) |
| C5—C4—C3—C2 | 1.31 (17) | C1—N1—C10—C11 | −100.36 (13) |
| C5—C4—C9—C8 | 0.10 (19) | C2—C3—C13—O2 | −11.40 (17) |
| C5—C6—C7—C8 | 0.4 (2) | C2—C3—C13—O3 | 165.96 (15) |
| C4—C5—C6—C7 | −0.86 (19) | C9—C4—C3—C13 | −1.66 (18) |
| C4—C3—C13—O2 | 171.16 (11) | C9—C4—C3—C2 | −179.04 (12) |
| C4—C3—C13—O3 | −11.5 (2) | C9—C8—C7—C6 | 0.3 (2) |
| C4—C3—C2—C1 | 1.54 (19) | C14—O2—C13—O3 | −1.1 (2) |
| C4—C9—C8—C7 | −0.5 (2) | C14—O2—C13—C3 | 176.33 (15) |
| C3—C4—C9—C8 | −179.56 (12) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C9—H9···O3 | 0.969 (17) | 2.210 (15) | 2.8807 (19) | 125.3 (12) |
| C10—H10A···O1 | 0.922 (16) | 2.277 (17) | 2.6961 (17) | 107.1 (12) |
| C14—H14B···O1i | 0.95 (2) | 2.56 (2) | 3.433 (2) | 153.8 (18) |
Symmetry code: (i) −x, −y+1, −z+1.
Funding Statement
Funding for this research was provided by: Hacettepe University Scientific Research Project Unit (grant No. 013 D04 602 004 to T. Hökelek).
References
- Abdel-Wahab, B. F., Khidre, R. E., Farahat, A. A. & El-Ahl, A. S. (2012). Arkivoc, pp. 211–276.
- Baba, Y. F., Gökce, H., Rodi, Y. K., Hayani, S., Chahdi, F. O., Boukir, A., Jasinski, J. P., Kaur, M., Hökelek, T., Sebbar, N. K. & Essassi, E. M. (2020). J. Mol. Struct. 1217, 128461.
- Becke, A. D. (1993). J. Chem. Phys. 98, 5648–5652.
- Bouzian, Y., Hlimi, F., Sebbar, N. K., El Hafi, M., Hni, B., Essassi, E. M. & Mague, J. T. (2018). IUCrData, 3, x181438.
- Bruker (2019). APEX3 and SAINT, Bruker AXS, Inc., Madison, Wisconsin, USA.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Filali Baba, Y., Elmsellem, H., Kandri Rodi, Y., Steli, H., Ouazzani Chahdi, F., Ouzidan, Y., Sebbar, N. K. & Essassi, E. M. (2016b). Environ. Sci. 7, 2424–2434.
- Filali Baba, Y., Elmsellem, H., Kandri Rodi, Y., Steli, H., Ouazzani Chahdi, F., Ouzidan, Y., Sebbar, N. K., Essassi, E. M., El-Hajjaji, F. & Hammouti, B. (2016a). Pharm. Lett. 8, 128–137.
- Filali Baba, Y., Kandri Rodi, Y., Ouzidan, Y., Mague, J. T., Ouazzani Chahdi, F. & Essassi, E. M. (2017). IUCrData, 2, x171038.
- Filali Baba, Y., Sert, Y., Kandri Rodi, Y., Hayani, S., Mague, J. T., Prim, D., Marrot, J., Ouazzani Chahdi, F., Sebbar, N. K. & Essassi, E. M. (2019). J. Mol. Struct. 1188, 255–268.
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J. A. Jr, Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, O., Foresman, J. B., Ortiz, J. V., Cioslowski, J. & Fox, D. J. (2009). GAUSSIAN09. Gaussian Inc., Wallingford, CT, USA. https://gaussian.com/.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Hathwar, V. R., Sist, M., Jørgensen, M. R. V., Mamakhel, A. H., Wang, X., Hoffmann, C. M., Sugimoto, K., Overgaard, J. & Iversen, B. B. (2015). IUCrJ, 2, 563–574. [DOI] [PMC free article] [PubMed]
- Hayani, S., Sert, Y., Baba, Y. F., Benhiba, F., Chahdi, F. O., Laraqui, F. Z., Mague, J. T., El Ibrahimi, B., Sebbar, N. K., Rodi, Y. K. & Essassi, E. M. (2021). J. Mol. Struct. 1227, 129520.
- Hirshfeld, H. L. (1977). Theor. Chim. Acta, 44, 129–138.
- Hökelek, T., Dal, H., Tercan, B., Özbek, F. E. & Necefoğlu, H. (2009). Acta Cryst. E65, m607–m608. [DOI] [PMC free article] [PubMed]
- Jayatilaka, D., Grimwood, D. J., Lee, A., Lemay, A., Russel, A. J., Taylor, C., Wolff, S. K., Cassam-Chenai, P. & Whitton, A. (2005). TONTO – A System for Computational Chemistry. Available at: http://hirshfeldsurface.net/.
- Katoh, M., Matsune, R., Nagase, H. & Honda, T. (2004). Tetrahedron Lett. 45, 6221–6223.
- Krause, L., Herbst-Irmer, R., Sheldrick, G. M. & Stalke, D. (2015). J. Appl. Cryst. 48, 3–10. [DOI] [PMC free article] [PubMed]
- Leatham, P. A., Bird, H. A., Wright, V., Seymour, D. & Gordon, A. (1983). J. Rheumatol. Inflamm. 6, 209–211. [PubMed]
- Mackenzie, C. F., Spackman, P. R., Jayatilaka, D. & Spackman, M. A. (2017). IUCrJ, 4, 575–587. [DOI] [PMC free article] [PubMed]
- Mahamoud, A., Chevalier, J., Davin-Regli, A., Barbe, J. & Pages, J. (2006). Curr. Drug Targets, 7, 843–847. [DOI] [PubMed]
- McKinnon, J. J., Jayatilaka, D. & Spackman, M. A. (2007). Chem. Commun. pp. 3814–3816. [DOI] [PubMed]
- Muruganantham, N., Sivakumar, R., Anbalagan, N., Gunasekaran, V. & Leonard, J. T. (2004). Biol. Pharm. Bull. 27, 1683–1687. [DOI] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Sim, G. A., Robertson, J. M. & Goodwin, T. H. (1955). Acta Cryst. 8, 157–164.
- Spackman, M. A., McKinnon, J. J. & Jayatilaka, D. (2008). CrystEngComm, 10, 377–388.
- Spackman, P. R., Turner, M. J., McKinnon, J. J., Wolff, S. K., Grimwood, D. J., Jayatilaka, D. & Spackman, M. A. (2021). J. Appl. Cryst. 54, 1006–1011. [DOI] [PMC free article] [PubMed]
- Strekowski, L., Mokrosz, J. L., Honkan, V. A., Czarny, A., Cegla, M. T., Wydra, R. L., Patterson, S. E. & Schinazi, R. F. (1991). J. Med. Chem. 34, 1739–1746. [DOI] [PubMed]
- Turner, M. J., Grabowsky, S., Jayatilaka, D. & Spackman, M. A. (2014). J. Phys. Chem. Lett. 5, 4249–4255. [DOI] [PubMed]
- Turner, M. J., Thomas, S. P., Shi, M. W., Jayatilaka, D. & Spackman, M. A. (2015). Chem. Commun. 51, 3735–3738. [DOI] [PubMed]
- Venkatesan, P., Thamotharan, S., Ilangovan, A., Liang, H. & Sundius, T. (2016). Spectrochim. Acta A Mol. Biomol. Spectrosc. 153, 625–636. [DOI] [PubMed]
- Wilson, W. D., Zhao, M., Patterson, S. E., Wydra, R. L., Janda, L., Strekowski, L. & Schinazi, R. F. (1992). J. Med. Chem. 2, 102–110.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989023007557/wm5688sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989023007557/wm5688Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989023007557/wm5688Isup3.cdx
Supporting information file. DOI: 10.1107/S2056989023007557/wm5688Isup4.cdx
CCDC reference: 2291603
Additional supporting information: crystallographic information; 3D view; checkCIF report