

The title compound, a charge-neutral bis{5-(3,4-dimethoxyphenyl)-1,2,4-triazol-3-ato)-6-(pyrazol-1-yl)pyridine}iron(II) dimethanol dichloroform solvate, is a high-spin complex with a distorted pseudooctahedral coordination environment of the metal ion. Due to the tapered shape and polar nature, the molecules stack in one-dimensional columns that are bound by weak hydrogen bonds into layers, which, in turn, are arranged in a three-dimensional network without interlayer interactions below van der Waals radii.
Keywords: crystal structure, iron(II) complexes, neutral complexes
Abstract
The unit cell of the title compound, [Fe(C18H15N6O2)2]·2CH3OH·2CHCl3, consists of a charge-neutral complex molecule, two methanol and two chloroform molecules. In the complex, the two tridentate 2-(5-(3,4-dimethoxyphenyl)-1,2,4-triazol-3-yl)-6-(pyrazol-1-yl)pyridine ligands coordinate to the central FeII ion through the N atoms of the pyrazole, pyridine and triazole groups, forming a pseudo-octahedral coordination sphere. Neighbouring tapered molecules are linked through weak C—H(pz)⋯π(ph) interactions into one-dimensional chains, which are joined into two-dimensional layers through weak C—H⋯N/C/O interactions. Furthermore, the layers stack in a three-dimensional network linked by weak interlayer C—H⋯π interactions of the methoxy and phenyl groups. The intermolecular contacts were quantified using Hirshfeld surface analysis and two-dimensional fingerprint plots, revealing the relative contributions of the contacts to the crystal packing to be H⋯H 32.0%, H⋯C/C⋯H 26.3%, H⋯N/N⋯H 13.8%, and H⋯O/O⋯H 7.5%. The average Fe—N bond distance is 2.185 Å, indicating the high-spin state of the FeII ion. Energy framework analysis at the HF/3–21 G theory level was performed to quantify the interaction energies in the crystal structure.
1. Chemical context
A broad class of coordination compounds exhibiting spin-state switching between low- (total spin S = 0) and high-spin states (total spin S = 2) is represented by FeII complexes based on tridentate bisazolepyridine ligands (Halcrow, 2014 ▸; Suryadevara et al., 2022 ▸; Halcrow et al., 2019 ▸). In the case of asymmetric ligand design, where one of the azole groups carries a hydrogen on a nitrogen heteroatom and acts as a Brønsted acid, deprotonation can produce neutral complexes that can be either high spin (Schäfer et al., 2013 ▸) or low spin (Shiga et al., 2019 ▸) or exhibit temperature-induced transition between the spin states of the central atom (Seredyuk et al., 2014 ▸; Grunwald et al., 2023 ▸) depending on the ligand field strength. The periphery of the molecule, i.e. ligand substituents, also plays an important role in the behaviour, determining the way that molecules are packed in the crystal and their interactions with each other, and therefore further influencing the spin state adopted by the central atom. For example, the dynamic rearrangement of the methoxy group between bent and extended configurations can lead to a highly hysteretic spin transition via a supramolecular blocking mechanism (Seredyuk et al., 2022 ▸).

Having interest in spin-transition 3d-metal complexes formed by polydentate ligands (Bartual-Murgui et al., 2017 ▸; Bonhommeau et al., 2012 ▸; Valverde-Muñoz et al., 2020 ▸), we report here a new [FeII L 2] complex based on the asymmetric deprotonable ligand with two substituents on the phenyl group, L = 2-(5-(3,4-dimethoxyphenyl)-1,2,4-triazol-3-yl)-6- (pyrazol-1-yl)pyridine.
2. Structural commentary
The complex has a tapered structure with divergent phenyl groups. The ligand molecules are almost planar, including the methoxy substituents, which are also in the plane of the phenyl group. The independent methanol molecule forms O—H⋯N hydrogen bonds with the triazole (trz) rings of the ligand molecule (Fig. 1 ▸, Table 1 ▸). The chloroform molecules form double weak C—H⋯O bonds with the methoxy groups of the ligand. The central FeII ion of the complex has a distorted octahedral N6 coordination environment formed by the nitrogen donor atoms of two tridentate ligands (Fig. 1 ▸). The average bond length, <Fe—N> = 2.185 Å, is typical for high-spin complexes with an N6 coordination environment (Gütlich & Goodwin, 2004 ▸). The average trigonal distortion parameters Σ = Σ1 12(|90 − φ i|), where φ i is the angle N—Fe—N′ (Drew et al., 1995 ▸), and Θ = Σ1 24(|60 − θ i|), where θ i is the angle generated by superposition of two opposite faces of an octahedron (Chang et al., 1990 ▸) are 148.6 and 474.2°, respectively. The values reveal a deviation of the coordination environment from an ideal octahedron (where Σ = Θ = 0), which is, however, in the expected range for bisazolepyridine and similar ligands (see below). The calculated continuous shape measure (CShM) value relative to the ideal Oh symmetry is 5.391 (Kershaw Cook et al., 2015 ▸). The volume of the [FeN6] coordination polyhedron is 12.796 Å3.
Figure 1.

The molecular structure of a half of the title compound with displacement ellipsoids drawn at the 50% probability level. The strong O—H⋯N and weak C—H⋯O/N/C hydrogen bonds are shown with the nearest neighbours. Symmetry codes: (i) 1 − x, 1 + y,
 − z; (ii) −
− z; (ii) −
 + x,
+ y,
− z; (iii)
− x, −
+ y, z; (iv)
+ x,
+ y,
− z.
+ x,
+ y,
− z; (iii)
− x, −
+ y, z; (iv)
+ x,
+ y,
− z.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C2—H2⋯C14i | 0.95 | 2.85 | 3.676 (5) | 146 |
| C2—H2⋯C15i | 0.95 | 2.64 | 3.585 (5) | 171 |
| C7—H7⋯C1ii | 0.95 | 2.81 | 3.705 (5) | 158 |
| C1—H1⋯N6iii | 0.95 | 2.34 | 3.267 (5) | 166 |
| C12—H12⋯C9iv | 0.95 | 2.85 | 3.541 (5) | 130 |
| C20—H20⋯O1 | 1.00 | 2.28 | 3.115 (6) | 141 |
| C20—H20⋯O2 | 1.00 | 2.39 | 3.179 (6) | 135 |
| O3—H3A⋯N5 | 0.84 | 1.94 | 2.775 (4) | 177 |
| C3—H3⋯O3v | 0.95 | 2.33 | 3.238 (5) | 161 |
| C5—H5⋯O3v | 0.95 | 2.47 | 3.401 (6) | 167 |
Symmetry codes: (i)
 ; (ii)
; (ii)
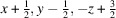 ; (iii)
; (iii)
 ; (iv)
; (iv)
 ; (v)
; (v)
 .
.
3. Supramolecular features
Due to the tapered structure, neighbouring complex molecules fit into each other and interact through a weak C—H(pz)⋯π(ph) intermolecular contact between the pyrazole (pz) and phenyl (ph) groups respectively [the C2⋯Cg(ph) distance is 3.574 (5) Å]. The one-dimensional supramolecular chains formed extend along the b-axis direction with the stacking periodicity equal to 10.281 (3) Å (= cell parameter b) (Fig. 2 ▸). Through weak intermolecular C—H(pz, py)⋯ N/C(pz, trz) interactions in the range 3.115–3.705 (5) Å (Table 1 ▸), neighbouring chains are joined into corrugated two-dimensional layers in the ab plane (Fig. S1a,b in the supporting information). The layers stack without any interlayer interactions below the van der Waals radii (Fig. S1b in the supporting information). The voids between the layers are occupied by solvent molecules, which also participate in the bonding within separate layers. The methanol molecule forms a strong O—H⋯N hydrogen bond with the deprotonated triazole group, and a chloroform molecule located between two methoxy groups of the phenyl substituent forms a five-membered cyclic motif with two C—H⋯O bonds (see Fig. 1 ▸). A complete list of the considered intermolecular interactions is given in Table 1 ▸.
Figure 2.

One-dimensional supramolecular chain formed by stacking molecules of the title compound. Red dashed lines correspond to contacts between the pyrazole and phenyl groups of neighbouring molecules below the sum of van der Waals radii.
4. Hirshfeld surface and 2D fingerprint plots
Hirshfeld surface analysis was performed and the associated two-dimensional fingerprint plots were generated using CrystalExplorer (Spackman et al., 2021 ▸), with a standard resolution of the three-dimensional d norm surfaces plotted over a fixed colour scale of −0.6492 (red) to 1.3918 (blue) a.u. (Fig. 3 ▸ a). The pale-red spots symbolize short contacts and negative d norm values on the surface corresponding to the interactions described above. The overall two-dimensional fingerprint plot is illustrated in Fig. 4 ▸. The two-dimensional fingerprint plots, with their relative contributions to the Hirshfeld surface, are shown for the H⋯H, H⋯C/C⋯H, H⋯N/N⋯H and H⋯O/O⋯H contacts together with the . At 32.0%, the largest contribution to the overall crystal packing is from H⋯H interactions, which are located in the middle region of the fingerprint plot. H⋯C/C⋯H contacts contribute 26.3% to the Hirshfeld surface and result in a pair of characteristic wings. The H⋯N/N⋯H contacts, represented by a pair of sharp spikes in the fingerprint plot, make a 13.8% contribution to the Hirshfeld surface. Finally, H⋯O/O⋯H contacts, which account for a 7.5% contribution, are mostly distributed in the middle part of the plot. The electrostatic potential energy calculated using the HF/3-21G basis set localizes the negative charge on the trz-ph moieties of the complex molecule, while the pz-py moieties are relatively positively charged (Fig. 3 ▸ b). The polar nature of the molecule justifies the stacking in columns.
Figure 3.

(a) A projection of d norm mapped on the Hirshfeld surfaces, showing the intermolecular interactions within the molecule. Red/blue and white areas represent regions where contacts are shorter/larger than the sum and close to the sum of the van der Waals radii, respectively. (b) Electrostatic potential for the title compound derived from a HF/3–21 G wavefunction mapped on the Hirshfeld surface in the range −0.1658 (red) to 0.1235 a.u. (blue).
Figure 4.

The overall two-dimensional fingerprint plot and those decomposed into specified interactions.
5. Energy framework analysis
The energy framework (Spackman et al., 2021 ▸), calculated using the wave function at the HF/3-21G theory level, including the electrostatic potential forces (E ele), the dispersion forces (E dis) and the total energy diagrams (E tot), is shown in Fig. S2 in the supporting information. The cylindrical radii, adjusted to the same scale factor of 100, are proportional to the relative strength of the corresponding energies. The major contribution is due to the dispersion forces (E dis), reflecting dominating interactions in the crystal of the neutral asymmetric molecules. The topology of the energy framework resembles the topology of the interactions within and between the layers described above. The calculated value Etot for the intrachain interaction is −57.2 kJ mol−1 and for interchain interactions are down to −114.6 kJ mol−1. The interlayer interaction energies are close to zero. The colour-coded interaction mappings within a radius of 5.0 Å of a central reference molecule for the title compound together with full details of the various contributions to the total energy (E ele, E pol, E dis, E rep) are shown in the table in Figure S2.
6. Database survey
A search of the Cambridge Structural Database (CSD, Version 5.42, last update February 2021; Groom et al., 2016 ▸) reveals several similar neutral FeII complexes with a deprotonable azole group, for example, derivatives of a pyrazolepyridinetetrazole, IGERIX and LUTGEO (Gentili et al., 2015 ▸; Senthil Kumar et al., 2015 ▸) and pyrazole-pyridine-benzimidazole XODCEB (Shiga et al., 2019 ▸). In addition, there are related complexes based on phenathrolinetetrazole, such as QIDJET (Zhang et al., 2007 ▸), phenanthroline-benzimidazole, DOMQUT (Seredyuk et al., 2014 ▸), dipyridylpyrrol, NIRLOT (Grunwald et al., 2023 ▸). The Fe—N distances of these complexes in the low-spin state are 1.933–1.959 Å, while in the high-spin state they are in the range 2.179–2.185 Å. The values of the trigonal distortion and CShM(Oh ) change correspondingly, and in the low-spin state they are systematically lower than in the high-spin state. Table 2 ▸ collates the structural parameters of the complexes and of the title compound.
Table 2. Computed distortion indices (Å, °) for the title compound and for similar complexes reported in the literature.
| CSD Code | Spin state | <Fe—N> | Σ | Θ | CShM(Oh ) |
|---|---|---|---|---|---|
| Title compound | High-spin | 2.185 | 148.6 | 474.2 | 5.39 |
| IGERIX | High spin | 2.179 | 149.7 | 553.2 | 6.06 |
| IGERIX01 | Low spin | 1.986 | 105.6 | 350.6 | 2.85 |
| LUTGEO | Low spin | 1.933 | 85.0 | 309.6 | 2.10 |
| XODCEB | Low spin | 1.950 | 87.4 | 276.6 | 1.93 |
| DOMQIH | Low spin | 1.962 | 83.8 | 280.7 | 2.02 |
| QIDJET01 | Low spin | 1.970 | 90.3 | 341.3 | 2.47 |
| QIDJET | High spin | 2.184 | 145.5 | 553.3 | 5.88 |
| DOMQUT | Low spin | 1.991 | 88.5 | 320.0 | 2.48 |
| DOMQUT02 | High spin | 2.183 | 139.6 | 486.9 | 5.31 |
| NIRLOT | Low spin | 1.939 | 77.3 | 255.6 | 1.68 |
7. Synthesis and crystallization
The synthesis of the title compound is identical to that reported recently for a similar complex (Seredyuk et al., 2022 ▸). It was produced by using a layering technique in a standard test tube. The layering sequence was as follows: the bottom layer contained a solution of [Fe(L 2)](BF4)2 prepared by dissolving L = 2-[5-(3,4-dimethoxyphenyl)-1,2,4-triazol-3-yl]-6-(pyrazol-1-yl)pyridine (88 mg, 0.252 mmol) and Fe(BF4)2·6H2O (43 mg, 0.126 mmol) in boiling acetone, to which chloroform (5 ml) was then added. The middle layer was a methanol–chloroform mixture (1:10, 10 ml), which was covered by a layer of methanol (10 ml), to which 100 ml of NEt3 were added dropwise. The tube was sealed, and black–orange single crystals appeared after 3–4 weeks (yield ca 60%). Elemental analysis calculated for C40H40Cl6FeN12O6: C, 45.61; H, 3.83; N, 15.96. Found: C, 45.52; H, 3.77; N, 15.77.
8. Refinement
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. H atoms were refined as riding [C—H = 0.95–0.98 Å with U iso(H) = 1.2–1.5U eq(C)]. The O-bound H atom was refined with U iso(H) = 1.5U eq(O).
Table 3. Experimental details.
| Crystal data | |
| Chemical formula | [Fe(C18H15N6O2)2]·2CH4O·2CHCl3 |
| M r | 1053.39 |
| Crystal system, space group | Orthorhombic, P b c n |
| Temperature (K) | 180 |
| a, b, c (Å) | 12.7195 (9), 10.281 (3), 36.735 (3) |
| V (Å3) | 4804.0 (13) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.71 |
| Crystal size (mm) | 0.25 × 0.2 × 0.03 |
| Data collection | |
| Diffractometer | Xcalibur, Eos |
| Absorption correction | Multi-scan (CrysAlis PRO; Rigaku OD, 2022 ▸) |
| T min, T max | 0.995, 1.000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 18857, 5510, 2962 |
| R int | 0.092 |
| (sin θ/λ)max (Å−1) | 0.688 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.079, 0.145, 1.03 |
| No. of reflections | 5510 |
| No. of parameters | 298 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.39, −0.44 |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989023008423/tx2075sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989023008423/tx2075Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989023008423/tx2075Isup3.cdx
Packing drawings and energy framework analysis data and drawings. DOI: 10.1107/S2056989023008423/tx2075sup5.doc
CCDC reference: 2297496
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
Author contributions are as follows: Conceptualization, KZ and MS; methodology, KZ; formal analysis, IOF; synthesis, SOM; single-crystal measurements, SS; writing (original draft), MS; writing (review and editing of the manuscript), TYS, MS; visualization and calculations, KZ, VMA; funding acquisition, MS, IOF, VMA.
supplementary crystallographic information
Crystal data
| [Fe(C18H15N6O2)2]·2CH4O·2CHCl3 | Dx = 1.456 Mg m−3 |
| Mr = 1053.39 | Mo Kα radiation, λ = 0.71073 Å |
| Orthorhombic, Pbcn | Cell parameters from 2694 reflections |
| a = 12.7195 (9) Å | θ = 2.0–22.1° |
| b = 10.281 (3) Å | µ = 0.71 mm−1 |
| c = 36.735 (3) Å | T = 180 K |
| V = 4804.0 (13) Å3 | Prism, clear light yellow |
| Z = 4 | 0.25 × 0.2 × 0.03 mm |
| F(000) = 2160 |
Data collection
| Xcalibur, Eos diffractometer | 5510 independent reflections |
| Radiation source: fine-focus sealed X-ray tube, Enhance (Mo) X-ray Source | 2962 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.092 |
| Detector resolution: 16.1593 pixels mm-1 | θmax = 29.3°, θmin = 2.0° |
| ω scans | h = −17→14 |
| Absorption correction: multi-scan (CrysAlisPro; Rigaku OD, 2022) | k = −11→13 |
| Tmin = 0.995, Tmax = 1.000 | l = −30→45 |
| 18857 measured reflections |
Refinement
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.079 | H-atom parameters constrained |
| wR(F2) = 0.145 | w = 1/[σ2(Fo2) + (0.0352P)2 + 1.3579P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.03 | (Δ/σ)max < 0.001 |
| 5510 reflections | Δρmax = 0.39 e Å−3 |
| 298 parameters | Δρmin = −0.44 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Fe1 | 0.500000 | 0.76448 (9) | 0.750000 | 0.0222 (2) | |
| Cl1 | 0.66889 (10) | 0.30702 (16) | 0.48491 (4) | 0.0718 (5) | |
| Cl3 | 0.45098 (10) | 0.2575 (2) | 0.46990 (4) | 0.0951 (7) | |
| Cl2 | 0.60243 (13) | 0.05279 (18) | 0.46193 (4) | 0.0862 (6) | |
| N3 | 0.6647 (2) | 0.7695 (3) | 0.76381 (8) | 0.0212 (8) | |
| O1 | 0.6486 (2) | 0.1131 (3) | 0.56399 (8) | 0.0363 (8) | |
| N4 | 0.5728 (2) | 0.6285 (3) | 0.71371 (8) | 0.0221 (8) | |
| N2 | 0.6252 (2) | 0.9304 (3) | 0.80493 (9) | 0.0236 (8) | |
| N5 | 0.5447 (2) | 0.5444 (3) | 0.68666 (8) | 0.0223 (8) | |
| O2 | 0.4939 (2) | 0.2752 (3) | 0.56650 (8) | 0.0427 (9) | |
| N1 | 0.5214 (2) | 0.9131 (3) | 0.79464 (9) | 0.0246 (8) | |
| O3 | 0.3669 (2) | 0.6194 (4) | 0.64823 (9) | 0.0525 (10) | |
| H3A | 0.421091 | 0.594475 | 0.659283 | 0.079* | |
| N6 | 0.7165 (2) | 0.5103 (3) | 0.69940 (8) | 0.0221 (8) | |
| C9 | 0.6755 (3) | 0.6051 (4) | 0.72010 (10) | 0.0216 (10) | |
| C8 | 0.7306 (3) | 0.6878 (4) | 0.74638 (10) | 0.0208 (9) | |
| C14 | 0.6502 (3) | 0.1976 (4) | 0.59249 (11) | 0.0272 (10) | |
| C4 | 0.7027 (3) | 0.8539 (4) | 0.78750 (10) | 0.0222 (10) | |
| C16 | 0.5594 (3) | 0.3751 (4) | 0.62214 (11) | 0.0261 (10) | |
| H16 | 0.501808 | 0.433833 | 0.623005 | 0.031* | |
| C3 | 0.6331 (3) | 1.0150 (4) | 0.83276 (12) | 0.0348 (12) | |
| H3 | 0.696474 | 1.042065 | 0.844178 | 0.042* | |
| C12 | 0.7199 (3) | 0.2910 (4) | 0.64749 (11) | 0.0296 (11) | |
| H12 | 0.773273 | 0.291996 | 0.665593 | 0.036* | |
| C7 | 0.8379 (3) | 0.6927 (4) | 0.75259 (11) | 0.0295 (11) | |
| H7 | 0.884266 | 0.634916 | 0.740344 | 0.035* | |
| C10 | 0.6324 (3) | 0.4768 (4) | 0.67851 (10) | 0.0222 (10) | |
| C5 | 0.8097 (3) | 0.8673 (4) | 0.79519 (11) | 0.0300 (11) | |
| H5 | 0.835528 | 0.930144 | 0.811906 | 0.036* | |
| C1 | 0.4682 (3) | 0.9884 (4) | 0.81718 (11) | 0.0281 (11) | |
| H1 | 0.393795 | 0.996704 | 0.816911 | 0.034* | |
| C13 | 0.7257 (3) | 0.2015 (4) | 0.61954 (11) | 0.0304 (11) | |
| H13 | 0.782566 | 0.141665 | 0.618878 | 0.036* | |
| C11 | 0.6377 (3) | 0.3795 (4) | 0.64968 (10) | 0.0246 (10) | |
| C2 | 0.5343 (3) | 1.0548 (5) | 0.84167 (13) | 0.0399 (13) | |
| H2 | 0.514382 | 1.114171 | 0.860259 | 0.048* | |
| C15 | 0.5664 (3) | 0.2858 (4) | 0.59409 (11) | 0.0266 (10) | |
| C6 | 0.8757 (3) | 0.7825 (5) | 0.77677 (12) | 0.0361 (12) | |
| H6 | 0.949305 | 0.786965 | 0.781044 | 0.043* | |
| C20 | 0.5675 (3) | 0.1923 (5) | 0.48712 (13) | 0.0490 (14) | |
| H20 | 0.556297 | 0.166973 | 0.513117 | 0.059* | |
| C17 | 0.7283 (3) | 0.0157 (5) | 0.56289 (13) | 0.0488 (14) | |
| H17A | 0.720138 | −0.036527 | 0.540761 | 0.073* | |
| H17B | 0.721896 | −0.040564 | 0.584307 | 0.073* | |
| H17C | 0.797583 | 0.057357 | 0.562860 | 0.073* | |
| C19 | 0.3952 (4) | 0.7063 (6) | 0.62079 (14) | 0.0644 (17) | |
| H19A | 0.433569 | 0.659748 | 0.601683 | 0.097* | |
| H19B | 0.440099 | 0.774643 | 0.630986 | 0.097* | |
| H19C | 0.331711 | 0.745638 | 0.610407 | 0.097* | |
| C18 | 0.4062 (4) | 0.3632 (6) | 0.56791 (15) | 0.085 (2) | |
| H18A | 0.357693 | 0.343789 | 0.547915 | 0.127* | |
| H18B | 0.431588 | 0.452834 | 0.565572 | 0.127* | |
| H18C | 0.369588 | 0.353055 | 0.591206 | 0.127* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Fe1 | 0.0131 (4) | 0.0258 (5) | 0.0278 (4) | 0.000 | −0.0003 (3) | 0.000 |
| Cl1 | 0.0667 (9) | 0.0745 (13) | 0.0741 (11) | −0.0242 (8) | −0.0098 (7) | −0.0051 (10) |
| Cl3 | 0.0540 (9) | 0.1473 (19) | 0.0841 (12) | 0.0051 (10) | −0.0120 (7) | 0.0380 (14) |
| Cl2 | 0.1297 (13) | 0.0655 (12) | 0.0634 (11) | −0.0138 (11) | 0.0108 (9) | −0.0116 (11) |
| N3 | 0.0149 (15) | 0.023 (2) | 0.0261 (18) | 0.0011 (15) | −0.0012 (13) | −0.0012 (18) |
| O1 | 0.0454 (17) | 0.029 (2) | 0.0346 (18) | 0.0121 (16) | −0.0062 (14) | −0.0080 (17) |
| N4 | 0.0150 (15) | 0.026 (2) | 0.0250 (19) | −0.0001 (15) | 0.0003 (13) | −0.0049 (18) |
| N2 | 0.0133 (15) | 0.023 (2) | 0.034 (2) | 0.0000 (14) | 0.0004 (14) | −0.0089 (19) |
| N5 | 0.0204 (16) | 0.024 (2) | 0.0228 (18) | −0.0025 (15) | −0.0025 (13) | −0.0074 (18) |
| O2 | 0.0406 (17) | 0.044 (2) | 0.0439 (18) | 0.0137 (16) | −0.0179 (14) | −0.0162 (18) |
| N1 | 0.0110 (15) | 0.027 (2) | 0.036 (2) | −0.0004 (14) | −0.0007 (13) | −0.0012 (19) |
| O3 | 0.0282 (15) | 0.074 (3) | 0.056 (2) | −0.0040 (18) | −0.0061 (15) | 0.027 (2) |
| N6 | 0.0194 (16) | 0.024 (2) | 0.0232 (19) | −0.0008 (15) | −0.0015 (13) | 0.0019 (17) |
| C9 | 0.0167 (19) | 0.026 (3) | 0.022 (2) | −0.0009 (18) | −0.0007 (15) | 0.002 (2) |
| C8 | 0.0186 (19) | 0.019 (2) | 0.024 (2) | 0.0022 (17) | 0.0017 (16) | 0.000 (2) |
| C14 | 0.035 (2) | 0.022 (3) | 0.025 (2) | 0.004 (2) | 0.0003 (18) | −0.001 (2) |
| C4 | 0.0177 (19) | 0.023 (3) | 0.026 (2) | −0.0007 (18) | 0.0036 (16) | −0.003 (2) |
| C16 | 0.022 (2) | 0.020 (3) | 0.036 (3) | 0.0009 (18) | −0.0020 (17) | −0.004 (2) |
| C3 | 0.024 (2) | 0.034 (3) | 0.046 (3) | −0.004 (2) | −0.0011 (19) | −0.019 (3) |
| C12 | 0.031 (2) | 0.028 (3) | 0.030 (2) | 0.003 (2) | −0.0083 (18) | −0.001 (2) |
| C7 | 0.019 (2) | 0.034 (3) | 0.035 (2) | 0.0060 (18) | 0.0008 (17) | −0.010 (3) |
| C10 | 0.0180 (19) | 0.023 (3) | 0.025 (2) | −0.0001 (18) | 0.0001 (16) | −0.002 (2) |
| C5 | 0.0190 (19) | 0.034 (3) | 0.037 (3) | −0.006 (2) | −0.0029 (18) | −0.008 (2) |
| C1 | 0.0181 (19) | 0.026 (3) | 0.040 (3) | 0.0038 (18) | 0.0061 (18) | 0.000 (2) |
| C13 | 0.032 (2) | 0.026 (3) | 0.033 (3) | 0.008 (2) | −0.0017 (18) | 0.000 (2) |
| C11 | 0.024 (2) | 0.024 (3) | 0.025 (2) | −0.0024 (19) | 0.0018 (17) | 0.000 (2) |
| C2 | 0.032 (2) | 0.035 (3) | 0.053 (3) | 0.006 (2) | 0.009 (2) | −0.020 (3) |
| C15 | 0.026 (2) | 0.023 (3) | 0.031 (2) | −0.0042 (19) | −0.0047 (17) | −0.002 (2) |
| C6 | 0.0129 (19) | 0.051 (4) | 0.045 (3) | 0.000 (2) | −0.0015 (18) | −0.011 (3) |
| C20 | 0.052 (3) | 0.054 (4) | 0.041 (3) | −0.008 (3) | −0.005 (2) | −0.002 (3) |
| C17 | 0.060 (3) | 0.036 (3) | 0.051 (3) | 0.014 (3) | −0.003 (2) | −0.017 (3) |
| C19 | 0.096 (4) | 0.051 (4) | 0.047 (3) | 0.007 (3) | 0.001 (3) | 0.013 (4) |
| C18 | 0.061 (3) | 0.100 (6) | 0.094 (5) | 0.049 (4) | −0.051 (3) | −0.051 (5) |
Geometric parameters (Å, º)
| Fe1—N3 | 2.156 (3) | C4—C5 | 1.398 (5) |
| Fe1—N3i | 2.156 (3) | C16—H16 | 0.9500 |
| Fe1—N4i | 2.142 (3) | C16—C11 | 1.420 (5) |
| Fe1—N4 | 2.142 (3) | C16—C15 | 1.383 (5) |
| Fe1—N1 | 2.258 (3) | C3—H3 | 0.9500 |
| Fe1—N1i | 2.258 (3) | C3—C2 | 1.363 (5) |
| Cl1—C20 | 1.749 (5) | C12—H12 | 0.9500 |
| Cl3—C20 | 1.745 (5) | C12—C13 | 1.380 (5) |
| Cl2—C20 | 1.764 (5) | C12—C11 | 1.389 (5) |
| N3—C8 | 1.348 (4) | C7—H7 | 0.9500 |
| N3—C4 | 1.320 (5) | C7—C6 | 1.369 (5) |
| O1—C14 | 1.361 (5) | C10—C11 | 1.458 (5) |
| O1—C17 | 1.425 (5) | C5—H5 | 0.9500 |
| N4—N5 | 1.365 (4) | C5—C6 | 1.386 (5) |
| N4—C9 | 1.349 (4) | C1—H1 | 0.9500 |
| N2—N1 | 1.385 (4) | C1—C2 | 1.408 (6) |
| N2—C4 | 1.414 (4) | C13—H13 | 0.9500 |
| N2—C3 | 1.346 (5) | C2—H2 | 0.9500 |
| N5—C10 | 1.347 (4) | C6—H6 | 0.9500 |
| O2—C15 | 1.374 (4) | C20—H20 | 1.0000 |
| O2—C18 | 1.437 (5) | C17—H17A | 0.9800 |
| N1—C1 | 1.320 (5) | C17—H17B | 0.9800 |
| O3—H3A | 0.8400 | C17—H17C | 0.9800 |
| O3—C19 | 1.394 (5) | C19—H19A | 0.9800 |
| N6—C9 | 1.342 (5) | C19—H19B | 0.9800 |
| N6—C10 | 1.362 (4) | C19—H19C | 0.9800 |
| C9—C8 | 1.465 (5) | C18—H18A | 0.9800 |
| C8—C7 | 1.385 (5) | C18—H18B | 0.9800 |
| C14—C13 | 1.382 (5) | C18—H18C | 0.9800 |
| C14—C15 | 1.401 (5) | ||
| N3i—Fe1—N3 | 177.28 (19) | C11—C12—H12 | 119.3 |
| N3—Fe1—N1 | 72.28 (11) | C8—C7—H7 | 120.7 |
| N3i—Fe1—N1 | 105.79 (11) | C6—C7—C8 | 118.5 (4) |
| N3i—Fe1—N1i | 72.29 (11) | C6—C7—H7 | 120.7 |
| N3—Fe1—N1i | 105.79 (11) | N5—C10—N6 | 113.2 (4) |
| N4i—Fe1—N3 | 106.79 (11) | N5—C10—C11 | 123.6 (3) |
| N4—Fe1—N3i | 106.79 (11) | N6—C10—C11 | 123.1 (3) |
| N4i—Fe1—N3i | 75.05 (12) | C4—C5—H5 | 122.3 |
| N4—Fe1—N3 | 75.05 (12) | C6—C5—C4 | 115.4 (4) |
| N4—Fe1—N4i | 98.53 (18) | C6—C5—H5 | 122.3 |
| N4i—Fe1—N1i | 147.30 (10) | N1—C1—H1 | 123.9 |
| N4i—Fe1—N1 | 92.40 (12) | N1—C1—C2 | 112.3 (3) |
| N4—Fe1—N1 | 147.30 (10) | C2—C1—H1 | 123.9 |
| N4—Fe1—N1i | 92.40 (12) | C14—C13—H13 | 119.4 |
| N1—Fe1—N1i | 94.81 (17) | C12—C13—C14 | 121.2 (4) |
| C8—N3—Fe1 | 118.5 (3) | C12—C13—H13 | 119.4 |
| C4—N3—Fe1 | 121.7 (2) | C16—C11—C10 | 120.5 (4) |
| C4—N3—C8 | 119.7 (3) | C12—C11—C16 | 117.8 (4) |
| C14—O1—C17 | 117.3 (3) | C12—C11—C10 | 121.7 (3) |
| N5—N4—Fe1 | 138.9 (2) | C3—C2—C1 | 104.6 (4) |
| C9—N4—Fe1 | 115.3 (3) | C3—C2—H2 | 127.7 |
| C9—N4—N5 | 105.5 (3) | C1—C2—H2 | 127.7 |
| N1—N2—C4 | 118.0 (3) | O2—C15—C14 | 115.3 (4) |
| C3—N2—N1 | 111.2 (3) | O2—C15—C16 | 124.0 (4) |
| C3—N2—C4 | 130.7 (3) | C16—C15—C14 | 120.6 (4) |
| C10—N5—N4 | 105.8 (3) | C7—C6—C5 | 121.9 (3) |
| C15—O2—C18 | 116.3 (3) | C7—C6—H6 | 119.1 |
| N2—N1—Fe1 | 113.6 (2) | C5—C6—H6 | 119.1 |
| C1—N1—Fe1 | 142.2 (2) | Cl1—C20—Cl2 | 109.8 (2) |
| C1—N1—N2 | 104.0 (3) | Cl1—C20—H20 | 109.0 |
| C19—O3—H3A | 109.5 | Cl3—C20—Cl1 | 110.5 (3) |
| C9—N6—C10 | 101.4 (3) | Cl3—C20—Cl2 | 109.6 (3) |
| N4—C9—C8 | 118.3 (3) | Cl3—C20—H20 | 109.0 |
| N6—C9—N4 | 114.1 (3) | Cl2—C20—H20 | 109.0 |
| N6—C9—C8 | 127.5 (3) | O1—C17—H17A | 109.5 |
| N3—C8—C9 | 112.1 (3) | O1—C17—H17B | 109.5 |
| N3—C8—C7 | 120.8 (4) | O1—C17—H17C | 109.5 |
| C7—C8—C9 | 127.0 (4) | H17A—C17—H17B | 109.5 |
| O1—C14—C13 | 125.6 (4) | H17A—C17—H17C | 109.5 |
| O1—C14—C15 | 115.7 (3) | H17B—C17—H17C | 109.5 |
| C13—C14—C15 | 118.7 (4) | O3—C19—H19A | 109.5 |
| N3—C4—N2 | 114.2 (3) | O3—C19—H19B | 109.5 |
| N3—C4—C5 | 123.6 (4) | O3—C19—H19C | 109.5 |
| C5—C4—N2 | 122.2 (4) | H19A—C19—H19B | 109.5 |
| C11—C16—H16 | 119.8 | H19A—C19—H19C | 109.5 |
| C15—C16—H16 | 119.8 | H19B—C19—H19C | 109.5 |
| C15—C16—C11 | 120.5 (4) | O2—C18—H18A | 109.5 |
| N2—C3—H3 | 126.0 | O2—C18—H18B | 109.5 |
| N2—C3—C2 | 107.9 (4) | O2—C18—H18C | 109.5 |
| C2—C3—H3 | 126.0 | H18A—C18—H18B | 109.5 |
| C13—C12—H12 | 119.3 | H18A—C18—H18C | 109.5 |
| C13—C12—C11 | 121.3 (4) | H18B—C18—H18C | 109.5 |
| Fe1—N3—C8—C9 | −0.6 (4) | C9—N6—C10—N5 | −1.6 (4) |
| Fe1—N3—C8—C7 | 175.9 (3) | C9—N6—C10—C11 | 177.0 (4) |
| Fe1—N3—C4—N2 | 5.5 (5) | C9—C8—C7—C6 | 175.8 (4) |
| Fe1—N3—C4—C5 | −174.9 (3) | C8—N3—C4—N2 | −177.7 (3) |
| Fe1—N4—N5—C10 | −172.7 (3) | C8—N3—C4—C5 | 1.9 (6) |
| Fe1—N4—C9—N6 | 173.7 (3) | C8—C7—C6—C5 | 0.5 (7) |
| Fe1—N4—C9—C8 | −9.8 (4) | C4—N3—C8—C9 | −177.6 (3) |
| Fe1—N1—C1—C2 | 175.1 (3) | C4—N3—C8—C7 | −1.0 (6) |
| N3—C8—C7—C6 | −0.2 (6) | C4—N2—N1—Fe1 | −1.3 (4) |
| N3—C4—C5—C6 | −1.6 (6) | C4—N2—N1—C1 | 175.1 (3) |
| O1—C14—C13—C12 | −179.6 (4) | C4—N2—C3—C2 | −174.5 (4) |
| O1—C14—C15—O2 | −0.2 (5) | C4—C5—C6—C7 | 0.3 (7) |
| O1—C14—C15—C16 | 179.2 (4) | C3—N2—N1—Fe1 | −176.9 (3) |
| N4—N5—C10—N6 | 1.2 (4) | C3—N2—N1—C1 | −0.5 (4) |
| N4—N5—C10—C11 | −177.4 (3) | C3—N2—C4—N3 | 172.2 (4) |
| N4—C9—C8—N3 | 6.9 (5) | C3—N2—C4—C5 | −7.4 (7) |
| N4—C9—C8—C7 | −169.4 (4) | C10—N6—C9—N4 | 1.4 (4) |
| N2—N1—C1—C2 | 0.4 (5) | C10—N6—C9—C8 | −174.7 (4) |
| N2—C4—C5—C6 | 178.0 (4) | C13—C14—C15—O2 | −179.3 (4) |
| N2—C3—C2—C1 | −0.2 (5) | C13—C14—C15—C16 | 0.1 (6) |
| N5—N4—C9—N6 | −0.8 (4) | C13—C12—C11—C16 | 0.0 (6) |
| N5—N4—C9—C8 | 175.8 (3) | C13—C12—C11—C10 | −178.2 (4) |
| N5—C10—C11—C16 | 17.2 (6) | C11—C16—C15—O2 | 179.9 (4) |
| N5—C10—C11—C12 | −164.6 (4) | C11—C16—C15—C14 | 0.5 (6) |
| N1—N2—C4—N3 | −2.5 (5) | C11—C12—C13—C14 | 0.6 (6) |
| N1—N2—C4—C5 | 177.9 (4) | C15—C14—C13—C12 | −0.6 (6) |
| N1—N2—C3—C2 | 0.4 (5) | C15—C16—C11—C12 | −0.6 (6) |
| N1—C1—C2—C3 | −0.2 (5) | C15—C16—C11—C10 | 177.7 (4) |
| N6—C9—C8—N3 | −177.1 (4) | C17—O1—C14—C13 | 3.5 (6) |
| N6—C9—C8—C7 | 6.6 (7) | C17—O1—C14—C15 | −175.5 (4) |
| N6—C10—C11—C16 | −161.3 (4) | C18—O2—C15—C14 | 179.0 (4) |
| N6—C10—C11—C12 | 16.9 (6) | C18—O2—C15—C16 | −0.4 (6) |
| C9—N4—N5—C10 | −0.3 (4) |
Symmetry code: (i) −x+1, y, −z+3/2.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2···C14ii | 0.95 | 2.85 | 3.676 (5) | 146 |
| C2—H2···C15ii | 0.95 | 2.64 | 3.585 (5) | 171 |
| C7—H7···C1iii | 0.95 | 2.81 | 3.705 (5) | 158 |
| C1—H1···N6iv | 0.95 | 2.34 | 3.267 (5) | 166 |
| C12—H12···C9v | 0.95 | 2.85 | 3.541 (5) | 130 |
| C20—H20···O1 | 1.00 | 2.28 | 3.115 (6) | 141 |
| C20—H20···O2 | 1.00 | 2.39 | 3.179 (6) | 135 |
| O3—H3A···N5 | 0.84 | 1.94 | 2.775 (4) | 177 |
| C3—H3···O3vi | 0.95 | 2.33 | 3.238 (5) | 161 |
| C5—H5···O3vi | 0.95 | 2.47 | 3.401 (6) | 167 |
Symmetry codes: (ii) −x+1, y+1, −z+3/2; (iii) x+1/2, y−1/2, −z+3/2; (iv) x−1/2, y+1/2, −z+3/2; (v) −x+3/2, y−1/2, z; (vi) x+1/2, y+1/2, −z+3/2.
Funding Statement
Funding for this research was provided by a grant from the Ministry of Education and Science of Ukraine for perspective development of the scientific direction ‘Mathematical sciences and natural sciences’ at Taras Shevchenko National University of Kyiv and by the Ministry of Education and Science of Ukraine (grant Nos. 22BF037-03, 22BF037-04).
References
- Bartual-Murgui, C., Piñeiro-López, L., Valverde-Muñoz, F. J., Muñoz, M. C., Seredyuk, M. & Real, J. A. (2017). Inorg. Chem. 56, 13535–13546. [DOI] [PubMed]
- Bonhommeau, S., Lacroix, P. G., Talaga, D., Bousseksou, A., Seredyuk, M., Fritsky, I. O. & Rodriguez, V. (2012). J. Phys. Chem. C, 116, 11251–11255.
- Chang, H. R., McCusker, J. K., Toftlund, H., Wilson, S. R., Trautwein, A. X., Winkler, H. & Hendrickson, D. N. (1990). J. Am. Chem. Soc. 112, 6814–6827.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Drew, M. G. B., Harding, C. J., McKee, V., Morgan, G. G. & Nelson, J. (1995). J. Chem. Soc. Chem. Commun. pp. 1035–1038.
- Gentili, D., Demitri, N., Schäfer, B., Liscio, F., Bergenti, I., Ruani, G., Ruben, M. & Cavallini, M. (2015). J. Mater. Chem. C. 3, 7836–7844.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Grunwald, J., Torres, J., Buchholz, A., Näther, C., Kämmerer, L., Gruber, M., Rohlf, S., Thakur, S., Wende, H., Plass, W., Kuch, W. & Tuczek, F. (2023). Chem. Sci. 14, 7361–7380. [DOI] [PMC free article] [PubMed]
- Gütlich, P. & Goodwin, H. A. (2004). Top. Curr. Chem. 233, 1–47.
- Halcrow, M. A. (2014). New J. Chem. 38, 1868–1882.
- Halcrow, M. A., Capel Berdiell, I., Pask, C. M. & Kulmaczewski, R. (2019). Inorg. Chem. 58, 9811–9821. [DOI] [PubMed]
- Kershaw Cook, L. J., Mohammed, R., Sherborne, G., Roberts, T. D., Alvarez, S. & Halcrow, M. A. (2015). Coord. Chem. Rev. 289–290, 2–12.
- Rigaku OD (2022). CrysAlis PRO. Rigaku Oxford Diffraction, Yarnton, England.
- Schäfer, B., Rajnák, C., Šalitroš, I., Fuhr, O., Klar, D., Schmitz-Antoniak, C., Weschke, E., Wende, H. & Ruben, M. (2013). Chem. Commun. 49, 10986–10988. [DOI] [PubMed]
- Senthil Kumar, K., Šalitroš, I., Heinrich, B., Fuhr, O. & Ruben, M. (2015). J. Mater. Chem. C. 3, 11635–11644.
- Seredyuk, M., Znovjyak, K., Valverde-Muñoz, F. J., da Silva, I., Muñoz, M. C., Moroz, Y. S. & Real, J. A. (2022). J. Am. Chem. Soc. 144, 14297–14309. [DOI] [PMC free article] [PubMed]
- Seredyuk, M., Znovjyak, K. O., Kusz, J., Nowak, M., Muñoz, M. C. & Real, J. A. (2014). Dalton Trans. 43, 16387–16394. [DOI] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Shiga, T., Saiki, R., Akiyama, L., Kumai, R., Natke, D., Renz, F., Cameron, J. M., Newton, G. N. & Oshio, H. (2019). Angew. Chem. Int. Ed. 58, 5658–5662. [DOI] [PubMed]
- Spackman, P. R., Turner, M. J., McKinnon, J. J., Wolff, S. K., Grimwood, D. J., Jayatilaka, D. & Spackman, M. A. (2021). J. Appl. Cryst. 54, 1006–1011. [DOI] [PMC free article] [PubMed]
- Suryadevara, N., Mizuno, A., Spieker, L., Salamon, S., Sleziona, S., Maas, A., Pollmann, E., Heinrich, B., Schleberger, M., Wende, H., Kuppusamy, S. K. & Ruben, M. (2022). Chem. A Eur. J. 28, e202103853. [DOI] [PMC free article] [PubMed]
- Valverde-Muñoz, F., Seredyuk, M., Muñoz, M. C., Molnár, G., Bibik, Y. S. & Real, J. A. (2020). Angew. Chem. Int. Ed. 59, 18632–18638. [DOI] [PubMed]
- Zhang, W., Zhao, F., Liu, T., Yuan, M., Wang, Z. M. & Gao, S. (2007). Inorg. Chem. 46, 2541–2555. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989023008423/tx2075sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989023008423/tx2075Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989023008423/tx2075Isup3.cdx
Packing drawings and energy framework analysis data and drawings. DOI: 10.1107/S2056989023008423/tx2075sup5.doc
CCDC reference: 2297496
Additional supporting information: crystallographic information; 3D view; checkCIF report