

Visual Abstract

Keywords: CKD, metabolism
Abstract
Significance Statement
Hypoxia drives kidney damage and progression of CKD. Although erythrocytes respond rapidly to hypoxia, their role and the specific molecules sensing and responding to hypoxia in CKD remain unclear. In this study, we demonstrated in a mouse model that erythrocyte ENT1-AMPD3 is a master energy regulator of the intracellular purinergic hypoxic compensatory response that promotes rapid energy supply from extracellular adenosine, eAMPK-dependent metabolic reprogramming, and O2 delivery, which combat renal hypoxia and progression of CKD. ENT1-AMPD3-AMPK-BPGM comprise a group of circulating erythroid-specific biomarkers, providing early diagnostic and novel therapeutic targets for CKD.
Background
Hypoxia drives kidney damage and progression of CKD. Although erythrocytes respond rapidly to hypoxia, their role and the specific molecules sensing and responding to hypoxia in CKD remain unclear.
Methods
Mice with an erythrocyte-specific deficiency in equilibrative nucleoside transporter 1 (eEnt1−/−) and a global deficiency in AMP deaminase 3 (Ampd3−/−) were generated to define their function in two independent CKD models, including angiotensin II (Ang II) infusion and unilateral ureteral obstruction (UUO). Unbiased metabolomics, isotopic adenosine flux, and various biochemical and cell culture analyses coupled with genetic studies were performed. Translational studies in patients with CKD and cultured human erythrocytes examined the role of ENT1 and AMPD3 in erythrocyte function and metabolism.
Results
eEnt1−/− mice display severe renal hypoxia, kidney damage, and fibrosis in both CKD models. The loss of eENT1-mediated adenosine uptake reduces intracellular AMP and thus abolishes the activation of AMPKα and bisphosphoglycerate mutase (BPGM). This results in reduced 2,3-bisphosphoglycerate and glutathione, leading to overwhelming oxidative stress in eEnt1−/− mice. Excess reactive oxygen species (ROS) activates AMPD3, resulting in metabolic reprogramming and reduced O2 delivery, leading to severe renal hypoxia in eEnt1−/− mice. By contrast, genetic ablation of AMPD3 preserves the erythrocyte adenine nucleotide pool, inducing AMPK-BPGM activation, O2 delivery, and antioxidative stress capacity, which protect against Ang II-induced renal hypoxia, damage, and CKD progression. Translational studies recapitulated the findings in mice.
Conclusion
eENT1-AMPD3, two highly enriched erythrocyte purinergic components that sense hypoxia, promote eAMPK-BPGM–dependent metabolic reprogramming, O2 delivery, energy supply, and antioxidative stress capacity, which mitigates renal hypoxia and CKD progression.
Introduction
CKD is a worldwide health problem with high morbidity and mortality and a global estimated prevalence of 13.4%.1 When CKD progresses to ESKD, no effective therapy is available except renal dialysis or organ transplantation, making ESKD one of the most expensive diseases to treat per-patient basis.2,3 CKD is caused by multiple chronic conditions, including hypertension, diabetes, and GN. In clinics, CKD is mainly assessed by eGFR, proteinuria levels, and invasive renal biopsy or noninvasive but expensive renal molecular imaging, such as positron emission tomography (PET), single-photon emission computed tomography (SPECT), and magnetic resonance imaging (MRI).4 When these measurements are abnormal, it often indicates that renal damage has already occurred. Although substantial efforts have been made in basic and clinical research, our current understanding of the pathogenesis of CKD is extremely limited due to lack of early diagnosis and early interventions for CKD management. Therefore, we desperately need to explore the pathogenesis of CKD from a new perspective with the hope of identifying early diagnostic biomarkers and innovative therapies.
Kidneys are one of the most sensitive organs to hypoxia. A large number of studies indicate that CKD is driven by renal tissue hypoxia.5,6 It is commonly accepted that chronic renal hypoxia promotes persistently elevated hypoxia-inducible factor-1α (HIF-1α), a hypervasoactive renin–angiotensin–aldosterone system, hemodynamic disturbances, impaired metabolism, excessive inflammatory response, cellular fate transition, and renal fibrosis, eventually resulting in the progression of CKD to end-stage renal failure.7–9 Most studies have focused on dysfunction of HIF-1α in CKD. Although the renal tissue response to hypoxia has been extensively examined, the function and regulatory mechanisms for erythrocytes to sense and respond to hypoxia in CKD are rarely studied.
As the only cells that carry and deliver oxygen (O2), erythrocytes play a vital role in maintaining the normal energy metabolism, function, and survival of every cell and organ within our body. Uniquely, each hemoglobin (Hb) tetramer in erythrocytes carries four molecules of O2. However, under normoxic conditions, Hb does not release all four O2 molecules. Thus, mechanisms for decreasing Hb-O2 binding affinity to promote more O2 release are critical to counteract tissue hypoxia. Notably, 2,3-bisphosphoglycerate (2,3-BPG) generated by erythroid-specific glycolytic Rapoport–Luebering Shunt (RLS) is a negative allosteric modulator of Hb-O2 affinity.10–12 More than 5 decades ago, studies revealed that 2,3-BPG and O2 delivery capacity were elevated in humans exposed to high-altitude hypoxia.13 Subsequently, elevated erythrocyte 2,3-BPG levels were discovered in multiple diseases associated with hypoxia, including CKD.14–16 The molecular basis of elevated 2,3-BPG production-mediated O2 delivery from erythrocytes and its specific roles to counteract hypoxic tissue damage and CKD progression have only recently been explored. For example, recent studies showed that elevated adenosine signaling through erythrocyte adenosine A2B receptors (ADORA2B) followed by the activation of AMPK-dependent bisphosphoglycerate mutase (BPGM) is a critical erythrocyte adaptive metabolic response underlying 2,3-BPG induction and enhanced O2 delivery in counteracting high-altitude hypoxia and renal hypoxia and progression of CKD.17,18 Moreover, in mature erythrocytes, equilibrative nucleoside transporter 1 (ENT1) is the major transporter for rapid extracellular adenosine uptake and was recently identified as a critical regulator for “hypoxic adaptation memory”19 and also contributes to normal fetal growth by counteracting placental hypoxia.20 Like eENT1, AMP deaminase 3 (AMPD3) is highly enriched in erythrocytes and plays an important role to maintain erythrocyte intracellular purine nucleoside/nucleotide homeostasis.21 However, to the best of our knowledge, the effect and molecular bases of ENT1 and AMPD3 on the erythrocyte hypoxic response and the progression of CKD remain undetermined. Thus, here, we used mouse genetic, two independent CKD models and human translational studies coupled with high throughput metabolomics and isotopically labeled adenosine flux analyses to precisely determine the function, molecular and metabolic bases, and human significance of erythrocyte Ent1 and AMPD3 in renal damage and the progression of CKD.
Methods
Human Subjects
Patients with CKD admitted to the Department of Nephrology between January 2018 and March 2018 were identified by nephrologists of Xiangya Hospital, Central South University at Changsha, Hunan, China. Inclusion criteria were the presence of CKD stages 4–5, defined as an eGFR of <30 ml/min per 1.73 m2. Exclusion criteria were the initiation of maintenance dialysis or kidney transplantation. eGFR of the human subjects was calculated based on the Modification of Diet in Renal Disease (MDRD) equation: eGFR=186×serum creatinine−1.154×age−0.203×0.742 (if female). Twenty-two subjects were initially eligible for inclusion. Age-matched and sex-matched healthy individuals, free of kidney disease from the Health Physical Examination Center, were included as controls. Demographic and clinical information of human subjects are listed in Table 1 and Supplemental Table 1. Blood samples were collected by venous puncture and then centrifuged at 2000×g for 5 minutes at room temperature followed by 70% percoll purification. For long-term storage, samples were stored at −80°C. Written informed consent was obtained from all human subjects, and the research protocol was approved by the Central South University Committee for the Protection of Human Subjects (No. 201512551, 2019030128).
Table 1.
Clinical information of human subjects
| Variable | Control (n=18) | CKD (n=18) |
|---|---|---|
| Age (yr) | 47.00±11.79 | 48.83±9.82 |
| Sex (Female/Male) | 9/9 | 8/10 |
| Hematuria (n) | 0 | 10 |
| Proteinuria (g/24 h) | — | 2.55±1.66 |
| SBP (mm Hg) | 112.33±7.33 | 149.94±7.67a |
| DBP (mm Hg) | 77.27±5.16 | 102.16±8.46a |
| RBC (1012/L) | 4.95±0.32a | 3.06±0.95a |
| Hb (g/L) | 144.83±12.34 | 89.11±28.04a |
| HCT (%) | 42.88±3.04 | 27.37±8.47a |
| TP (g/L) | 70.83±4.24 | 58.79±8.51a |
| ALB (g/L) | 45.44±1.82 | 30.47±7.47a |
| GLO (g/L) | 29.52±3.59 | 28.31±3.81 |
| BUN (mmol/L) | 4.72±1.28 | 18.33±11.99a |
| Scr (μmol/L) | 70.75±11.10 | 548.85±227.66a |
| eGFR | 97.18±9.38 | 12.30±9.27a |
| Etiology | ||
| Chronic GN (n) | — | 8 |
| Diabetes (n) | — | 6 |
| Hypertension (n) | — | 4 |
| Medications | ||
| Antihypertensive drugs (n) | — | 14 |
| Antianemia drugs (n) | — | 9 |
Data were expressed as mean±SD. P values of age, RBC, Hb, HCT, TP, ALB, BUN, Scr, and eGFR were assessed using two-tailed unpaired t test with Welch correlation; P value of SBP, DBP, and GLO were assessed using two-tailed unpaired t test. SBP, systolic BP; DBP, diastolic BP; RBC, red blood cell; Hb, hemoglobin; HCT, hematocrit; TP, total serum protein; ALB, serum albumin; GLO, serum globulin; Scr, serum creatinine.
a P < 0.05.
Animals
All animal protocols were in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Welfare Committee of the University of Texas Health Science Center at Houston (AWC-19-0112) and the Ethics Committee of Xiangya Hospital, Central South University, Changhan, Hunan Province, China. All experimental animals were housed under controlled conditions with the temperature of 22°C and humidity around 50%. The lights are programmed to start “sunrise” at 7 am and “sunset” at 7 pm. Food and water were available ad libitum for animals. Erythrocyte-specific deletion of Ent1 was generated by crossing mice homozygous for floxed Ent1 allele (from Dr. Holger Eltzschig's laboratory, University of Colorado) with EpoR-Cre+ mice.22 Ampd3-/- mice were obtained from Dr. Cheng Chi Lee's laboratory, The University of Texas Health Science Center at Houston.23 C57BL/6 mice were purchased from Jackson Laboratory. In total, 8- to 12- week age-matched and sex-matched C57BL/6 and mutant mice were used for experiments. C57BL/6 (WT) mice were used as controls for the Ampd3−/− mice; EpoR-Cre+ mice were used as controls for EpoR-Cre+ mice to eliminate the possibility of Cre recombinase expression contributing the following phenotype.
Angiotensin II Infusion
ALZET Osmotic Pumps (model 2002, Palo Alto, CA) were filled with angiotensin II (Ang II) (Sigma-Aldrich A9525, MI) or saline as per manufacturer's guidelines. Mice were anesthetized with isoflurane (2%) and placed on a heating pad at 37°C. Osmotic pumps were implanted subcutaneously by an incision in the nape of the mouse neck. Ang II was delivered at a fixed rate of 500 ng/kg per minute for 14 days, and saline was infused in control mice.
Unilateral Ureteral Obstruction
Unilateral ureteral obstruction (UUO) was induced as described previously.9 In brief, mice were anesthetized and placed on a heating pad at 37°C. The left ureter was ligated with 6-0 silk through a midline incision. Sham-operated mice were also performed with the same operation except ureter ligation and used as controls.
Mouse Blood Pressure Measurements
Mouse systolic BP was measured using the noninvasive BP system for mice (CODA High Throughput System with 8 Activated Channels, Kent Scientific, Torrington, CT) before and after osmotic pump implantation as previously described.24,25 In brief, mice were placed on warming platform to keep tail temperature between 32°C and 35°C, and BP was measured in the tail of the mouse using volume pressure recording sensor technology. Training was conducted once a day for 3 days before minipump implantation. For every set of BP measurements, the first five cycles were regarded as acclimation and 20 cycles were recorded for the BP measurement. The average value of 20 cycles was used as the analysis of BP. BP was monitored before pump implantation considered as baseline day 0 and measured on days 3, 7, and 14 after minipump implantation.
Mouse Sample Collection
Metabolic cages (Nalgene) were used for 24-hour urine collection. Mice were anesthetized with 2.5% avertin, and whole blood was withdrawn by heart puncture. Blood samples were centrifuged at 2000 g for 5 minutes at 4°C. Plasma was collected in the upper layer, and mature erythrocytes were purified by 70% percoll (Sigma-Aldrich). Transcardiac perfusion with 20 ml ice–cold phosphate-buffered saline (PBS) was immediately performed after blood withdrawn. Kidney tissue was harvested and fixed in 10% buffered formalin phosphate or stored in −80°C for further experiments. All samples were stored in −80°C for further analysis.
Renal Function Measurement
Plasma urea and creatinine were determined using a urea assay kit (Sigma-Aldrich, MAK006) and a colorimetric assay kit (Abcam, ab65340), respectively. Proteinuria was indicated by urinary albumin-to-creatinine ratio, which was determined by a commercial kit (Exocell 1011 and 1012).
Metabolomic Profiling
Erythrocytes, plasma, and urine were lysed with lysis solution (methanol:acetonitrile:water 5:3:2 v/v/v) at 1:10, 1:25, and 1:25 dilutions, respectively. Kidneys were suspended with lysis buffer to a concentration of 15 mg/ml and were homogenized with small glass beads for 5 minutes by the Next Advance Bullet Blender. Suspensions were then vortexed continuously for 30 minutes at 4°C and then centrifuged at 18,213 g for 10 minutes at 4°C. Supernatants were injected into the Ultra-High-Pressure Liquid Chromatography–Mass Spectrometry (UHPLC-MS) using a Vanquish UHPLC coupled to a Q Exactive MS (Thermo Fisher, Bremen, Germany). Samples were analyzed using a 5-minute gradient as previously described.26–29 In brief, metabolites were separated on a Kinetex C18 column (150×2.1 mm, 1.7 um, Phenomenex, 00F-4475-AN) by the following chromatography conditions: flow rate 0.45 ml/min, column temperature 45°C, and sample compartment temperature 7°C. Positive solvent gradient was as follows: 0–0.5 minute 5% B, 0.5–1.1 minute 5%–95% B, 1.1–2.75 minutes hold at 95% B, 2.75–3 minutes 95%–5% B, and 3–5 minutes hold at 5% B (A: 0.1% formic acid in water; B: 0.1% formic acid in acetonitrile). Negative solvent gradient was as follows: 0–0.5 minute 0% B, 0.5–1.1 minute 0%–100% B, 1.1–2.75 minutes hold at 100% B, 2.75–3 minutes 100%–0% B, and 3–5 minutes hold at 0% B (A: 5% acetonitrile/95% water/1 mM ammonium acetate; B: 95% acetonitrile/5% water/1 mM ammonium acetate). Samples were randomized and run in positive and negative ion modes independently. The mass spectrometer was operated in full MS mode at resolution of 70,000, scan range 65–900 m/z, maximum injection time 200 ms, microscans 2, automatic gain control 3×106 ions, source voltage 4.0 kV (for both positive and negative ion modes), capillary temperature 320°C, and sheath gas 45, auxiliary gas 15, and sweep gas 0 (all nitrogen). For 2,3-BPG quantification, samples were prepared and run on the UHPLC-MS in negative mode. 2,3-BPG was quantified based on the isotopic-labeled 2,3-BPG standard (Toronto Research Chemicals, D491993).
Raw data files were converted to mzXML format using RawConverter (Scripps Research Institute) and analyzed via Maven (Princeton University, Princeton, NJ). Quantification is based on integrated peak areas of extracted ion chromatograms at the MS1 level. Instrument stability and quality control were assessed through replicate injections of a technical mixture every ten runs as described.
The metabolomic data were normalized based on website MetaboAnalyst (https://www.metaboanalyst.ca/MetaboAnalyst/). Sample normalization was conducted by the sum.
Adenosine Isotopic-Labeled Flux
Erythrocytes were isolated from blood with heparin as an anticoagulant for adenosine flux experiment. Packed erythrocytes were purified via Percoll density purification (Sigma-Aldrich) as previously described.30 Packed erythrocytes from WT and Ent1−/− mice were washed 3 times with F-10 Nutrient Mix (Invitrogen) and suspended to 4% hematocrit. Erythrocytes (1 ml) were added to each well of a 12-well plate and cultured with 5 μM of 13C10, 15N5-labeled adenosine (Sigma-Aldrich) either under normoxia or hypoxia (1% O2) for 10 minutes, 1, 3, and 6 hours. Erythrocytes and supernatants were collected and lysed as mentioned above and analyzed via Vanquish UHPLC coupled to a Q Exactive MS (Thermo Fisher, Bremen, Germany). Metabolite assignments and isotopologue distributions were performed through the Maven (Princeton, NJ).31
Erythrocyte 2,3-BPG and P50 Measurement
The erythrocyte 2,3-BPG level was measured as previously described.18 In brief, 20 μl erythrocyte pellet added into 100 μl 0.6 M cold perchloric acid, vortexed, and centrifuged at 10,000 g for 10 minutes at 4°C. In total, 80 μl supernatant was transferred to a new tube and neutralized with 10 μl 2.5 M K2CO3 and then centrifuged at 10,000 g for 5 minutes at 4°C; 10 μl supernatant was used to quantify 2,3-BPG via commercial kit (Roche 10148334001, Roche, Nutley, NJ); and 20 μl of whole blood was used for P50 measurement by mixing with 3 ml Hemox Buffer (TCS Scientific Corporation, PA), 5 μl antifoaming reagent (TCS Scientific Corporation, PA), and 5 μl 22% BSA in PBS. The Hemox Analyzer (TCS Scientific Corporation, PA) was applied for the measurement of oxygen equilibrium curve at 37°C.
BPGM Activity Measurement
A total of 10 μl erythrocytes were lysed in Tris-HCl (pH=7.4) buffer containing 0.5% Triton X-100 and protease inhibitors (Roche, Cat# 04906837001); 25 ug erythrocyte lysates were incubated in 100 μl prepared reaction mixture (100 mM Triethanolamine pH 7.6, 1 mM MgSO4, 4 mM adenosine triphosphate (ATP), 3 mM 3-phosphoglycerate, 10 units phosphoglycerate kinase) for 30 minutes at room temperature. The reaction was stopped by adding 5 µl of 11.63M PCA and subsequently centrifuged at 10,000 g for 5 minutes at 4°C. In total, 8 μl supernatant was mixed with 10 μl of 2.5 M K2CO3, vortexed, and centrifuged at 10,000 g for 5 minutes at 4°C; 10 μl supernatant was used for 2,3-BPG quantification by using a commercial kit as previously described.18
AMPD3 Activity Measurement
AMPD3 activity was measured according to previous reports with slight modification.32,33 Purified erythrocytes were collected in a buffer containing 150 mM KCL, 20 mM Tris-HCL, 1 mM EDTA, and 1 mM dithiothreitol; vortexed; and centrifuged at 3700 rpm for 30 minutes at 4°C. Protein concentration of the supernatant was determined by a BCA Protein Assay Kit (Thermo Scientific). The reaction mixture consisted of 25 mM sodium citrate pH 6.0, 50 mM potassium chloride, and 20 mM AMP. The enzyme reaction was initiated by the addition of 100 μl supernatant with 100 μl reaction mixture, and the mixture was incubated for 20 minutes at 37°C. AMPD3 activity was determined using a commercial assay kit (Wako Pure Chemical Industries, Osaka, Japan) and normalized to protein concentration.
Total AMPKα and p-AMPKα Measurement
Purified erythrocytes protein from human and mouse were collected as previously described.34 Total AMPKα and p-AMPKα were measured using commercially available ELISA kits (Cell Signaling #7959C and #7961C, Danvers) and normalized to protein concentration.
RBC Reactive Oxygen Species (ROS) Detection
The RBC ROS level was measured using a ROS Assay Kit (BL714A, Biosharp) according to the manufacturer's protocol. In brief, 1 μl RBC pellet was incubated with ROS fluorescence probe (H2DCFDA, 2 μM) for 45 minutes at 37°C protected from light. Samples were then washed 3 times with PBS and resuspended with 200 μl PBS. ROS levels were detected by fluorescence intensity, which was measured by a microplate reader (Synergy HTX Multi-Mode Reader, BioTek).
Tissue Hypoxia Detection
The tissue hypoxia level was detected using a Hypoxyprobe kit (Hypoxyprobe Omni Kit, Hypoxyprobe, Inc.) as previously described.24,25 In brief, mice were intraperitoneally injected with pimonidazole (50 mg/kg body weight) 30 minutes before anesthetization. Transcardiac perfusion was performed before kidneys were harvested. Tissues were fixed with 10% buffered formalin and embedded in paraffin for section of 4 μm. Slides were deparaffinized through serial baths in xylene and rehydrated in a graded series of alcohol and distilled water. Antigen retrieval was performed by boiling slides in sodium citrate buffer (pH 6.0) at 95°C for 30 minutes. After blocking with 3% BSA-PBS for 1 hour at room temperature, the slides were then incubated with rabbit antipimonidazole antibody (PAb2627AP, 1:100 in 0.5% BSA) in a humidified chamber at 4°C overnight. After the primary antibody incubation, Alexa Fluor 594 conjugated goat anti-rabbit secondary antibody (A-11037, 1:1000 in 3% BSA-PBS, Thermo Fisher) was applied. Negative control was incubated with secondary antibody alone. Slides were mounted with ProLong Gold Antifade Mountant with DAPI (P36935, Invitrogen). Quantification of the fluorescent signal intensity was determined using Image J software. The average fluorescence densities of randomized 8–10 areas per blinded sample were determined.
Histology
Mouse kidney tissue was fixed in 10% buffered formalin and sent to the Histology Laboratory (Department of Pathology and Laboratory Medicine, University of Texas Health Science Center at Houston) and Biossci Biotechnology Co. Ltd. (Hubei, China) for sectioning at 4 μm and hematoxylin and eosin stain or Masson trichrome staining. The slides were examined under light microscopy and assessed for renal injury and fibrosis. The renal injury assessment was determined by quantifying three characteristic components (glomeruli, tubules, and tubulointerstitial lesions) as previous described.7 The final score for each sample was obtained by the average of scores observed in ten randomized microscopic fields per blinded slide. Tubular injury was defined as tubular dilation, brush border loss, cast deposition, and necrosis. Ten high-power fields (magnification, ×200) were chosen randomly. The tubular injury score was estimated by the scale of 0–4 as follows: 0, no tubular injury; 1, <25% injury; 2, 25%–50% injury; 3, 51%–75% injury; and 4, >75% injury. The percentage of blue-stained area in ten randomized microscopic fields per blinded sample was quantified using Image J in the Masson trichrome-stained slides to evaluate renal fibrosis, and the perivascular areas were excluded. All assessments were performed by two investigators blinded to experimental conditions.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total kidney RNA was extracted via TRIzol (Invitrogen) according to the manufacturer's protocol. RNA was reverse transcribed into cDNA via the Reverse Transcription Kit (205313, Qiagen), and qRT-PCR was performed by using a SYBR Green PCR Kit (204056, Qiagen) on LightCycler 480 Instrument II (Roche). Primers were purchased from IDT (Integrated DNA Technologies, Inc.), and sequences were as follows: mouse Tgf-β1, forward 5′-CCCCACTGATACGCCTGAGT-3′, reverse 5′-AGCCCTGTATTCCGTCTCCTT-3′; Acta2, forward 5′-GACTACTGCCGAGCGTGAGA-3′, reverse 5′-CAGGGAGGAAGAGGAGGCG-3′; Col1a1, forward 5′-ATCCGGTAA CAAGGGTGAGC-3′, reverse 5′-GAACCAGGGCTGCCTCTAAG-3′; Fn1, forward 5′-GCCCTTACAGTTCCAAGTTCC-3′, reverse 5′-ATCTGTAGGCTGGTTCAGGC-3′; 18S, forward 5′-GCAATTATTCCCCATGAACG-3′, reverse 5′-GGCCTCACTAAACCATCCAA-3′; RENIN1, forward 5′-CTCTCTGGGCACTCTTGTTGC-3′, reverse 5′-GGGAGGTAAGATTGGTCAAGGA-3′; ACE, forward 5′-AGGTTGGGCTACTCCAGGAC-3′, reverse 5′-GGTGAGTTGTTGTCTGGCTTC-3′; Agtr1a, forward 5′-AACAGCTTGGTGGTGATCGTC-3′, reverse 5′-CATAGCGGTATAGACAGCCCA-3′; Agtr1b, forward 5′-TGGCTTGGCTAGTTTGCCG-3′, reverse 5′-ACCCAGTCCAATGGGGAGT-3′; Agtr2, forward 5′-AACTGGCACCAATGAGTCCG-3′, reverse 5′-CCAAAAGGAGTAAGTCAGCCAAG-3′; IL-6, forward 5′-ACCAAGAGATAAGCTGGAGTCAC-3′, reverse 5′-TAACGCACTAGGTTTGCCGA-3′; IL-1β, forward 5′-CTGGTGTGTGACGTTCCCAT-3′, reverse 5′-TCGTTGCTTGGTTCTCCTTGT-3′; Tnf-α, forward 5′-CACCACGCTCTTCTGTCTACT-3′, reverse 5′-AACTGATGAGAGGGAGGCCAT-3′. The ΔΔCt method was used for analysis.
Mouse and Human Erythrocyte Purification and in vitro Culture
Mouse or human blood collected with heparin was centrifuged at 2,000 g for 5 minutes at room temperature. Erythrocytes were purified by 70% percoll as mentioned above. Purified erythrocytes were washed 3 times with culture media (F-10 Nutrient Mix, Invitrogen) and resuspended to 4% hematocrit. In total, 1 ml of RBCs was added to each well of a 12-well plate and treated with different concentrations of drugs (Angiotensin II 10 nM, ab120183, Abcam; H2O2 3%, 88597, Millipore; DCF 10 μM, 2033, TOCRIS Bioscience; Dip 10 μM, D9766, Sigma-Aldrich) with different time as indicated in the results.
In vitro Hypoxic Challenge
Hypoxic challenge was conducted by a hypoxic workstation (Defendor HW 1000, CII) with constant oxygen concentration and temperature (37°C) for 10, 30, and 60 minutes. Oxygen concentration was set at 1% for hypoxic challenge.
Western Blot Analyses
Western blot analyses were performed as described previously.18,24 In brief, erythrocytes were lysed in cold water at the ratio of 1:10 in the presence of 1X protease inhibitor cocktail (Roche, Cat# 04906837001), and then, 10X PBS was added to balance. Protein concentration was measured using a BCA Protein Assay Kit (Thermo Scientific). Samples were loaded to 10% SDS-PAGE gel for Western blot. Membranes were incubated with primary antibody ENT1 (Proteintech, #11337-1-AP, 1:1000) or β-actin (Proteintech, #66009-1-Ig, 1:3000) and then incubated with secondary antibodies.
Flow Cytometry Analyses
Flow cytometry analyses were conducted as described previously.35 In brief, bone marrow cells were isolated and suspended in flow buffer (HBSS/0.5% BSA) at the concentration of 106/80 μl and stained with BV510-CD16/CD32 (Biolegend 101333, 0.2 μg/106 cells), PE-Cy7-IL-7Ra (BD 560733, 0.2 μg/106 cells), PE-c-Kit (Biolegend 101333, 0.2 μg/106 cells), AF647-CD34 (BD 560230, 0.1 μg/106 cells), BV605-Scal1 (BD 563288, 0.2 μg/106 cells), and lineage-eFluor 450 (BD 561301, 20 μl/106 cells) 30 minutes on ice in the dark. Cells were washed twice with 3 ml flow buffer. Finally, cells were suspended in 100 ul flow buffer and stained with viability marker 7-AAD (Biolegend 420403, 5 μl/106 cells) on ice for 10 minutes in the dark. After staining, cells were suspended in 400 μl flow buffer and analyzed within 1 hour using NovoCyte Quanteon. Unstained cells were used as negative control.
Erythrocyte Life Span Measurement
Erythrocytes were labeled in vivo by using EZ-Link NHS-Biotin (Thermo Fisher Scientific, #20217), and the life span of circulating erythrocytes was measured as described previously.36 In brief, NHS-Biotin was injected by retro-orbital plexus of mice at a dose of 50 mg/kg. Blood samples were collected at days 1, 4, 7, 14, 21, and 28 after NHS-Biotin injection. The percentage of biotin-labeled erythrocytes was calculated by the fraction of Ter119-positive cells that were also labeled with streptavidin by flow cytometry.
Statistical Analyses
All data were expressed as mean±SEM. Statistical analyses were performed via GraphPad Prism 8 (GraphPad Software, San Diego, CA). D'Agostino Pearson omnibus and Shapiro–Wilk tests were applied to evaluate normality distribution. Equality of variance was determined using F test. Two-tailed unpaired t test, one-way ANOVA, and Holm–Sidak post hoc correction were conducted for normal distribution data, and Welch correlation was conducted for heterogeneity of variance. For abnormal distribution data, unpaired two-tailed Mann–Whitney U test or Kruskal–Wallis with Dunn post hoc test was conducted. Two-way ANOVA was conducted to evaluate the mouse BP. For the UHPLC-MS data, the two-tailed unpaired t test was conducted between two groups.
Results
eENT1 Combats Renal Hypoxia, Damage, and Progression of CKD
Given the fact that ENT1 is the major transporter expressed in both human and murine erythrocytes,19 we conducted genetic studies to determine its function in CKD. In brief, we generated erythrocyte-specific knockout mice by mating mice with floxed Ent1 alleles (Ent1f/f) with mice expressing the recombinase Cre only in the erythroid lineage (EpoR-Cre+)22 (Supplemental Figure 1). The Ent1f/f/EpoR-Cre+ mice (eEnt1−/−) were viable, fertile, and phenotypically indistinguishable from control littermates through adulthood.
Next, we chose a well-accepted mouse model of CKD on the basis of subcutaneous minipump infusion of Ang II (500 ng/kg per minute) for 14 days, while using saline as control8,17,25 (Figure 1A). Consistent with previous studies,7,17,24,25,37 systolic BP (SBP) increased continuously for 2 weeks in control mice with uninterrupted minipump infusion of Ang II. Genetic ablation of eENT1 had no effect on the basal level of SBP or the level of Ang II-induced hypertension (Figure 1B). However, the lack of eENT1 led to severe kidney damage, fibrosis, inflammation, and renal hypoxia compared to control mice with Ang II infusion. Specifically, proteinuria (urinary albumin-to-creatinine ratio), plasma creatinine and urea, renal injury score on the basis of HE staining and renal fibrosis quantified by Masson trichrome staining, hypoxyprobe staining, and hypoxia-inducible factor 1α (HIF-1α) in kidney sections were significantly elevated in Ang II-infused eEnt1−/− mice compared with the controls (Figure 1, C and D and Supplemental Figure 2). Moreover, the fibrotic gene expression (transforming growth factor β1, Tgf-β1; alpha smooth muscle actin 2, Acta2; collagen type I α1, Col1α1; fibronectin, Fn1) and inflammation-related gene expression (IL-6, IL-1β, Tnf-a) revealed that the eEnt1−/− mice with Ang II-infusion underwent severe fibrosis and inflammation compared with control mice (Figure 1E). By contrast, the eENT1 ablation did not affect the mRNA levels of local renal renin–angiotensin system (RAS)–related genes in either controls or eEnt1−/− mice with or without Ang II infusion (Supplemental Figure 3).
Figure 1.

eENT1 deficiency aggravates renal hypoxia, kidney damage, and progression to fibrosis in Ang II-infused CKD model. (A) Schematic representation of experimental design. Ang II or saline was infused subcutaneously by osmotic minipump into EpoR-Cre+ or eEnt1−/− mice (8–12 week) for 14 days. (B) Systolic BP was measured on days 0, 3, 7, and 14 by the tail-cuff method. Data are expressed as mean±SEM. N=4–6 in each group. P values were assessed by repeated measures two-way ANOVA. *P < 0.05, EpoR-Cre+ +Ang II/Saline, #P < 0.05, eEnt1−/− +Ang II/Saline. (C) Proteinuria measured by the 24-hour urine albumin-to-creatinine ratio was quantified by a commercially available kit. Data are expressed as mean±SEM; plasma urea and creatinine were quantified using a commercially available kit. Data are expressed as mean±SEM. N=4–6 in each group. *P < 0.05. (D) Representative hematoxylin and eosin (H&E)–stained, trichrome-stained, or hypoxyprobe-stained kidney sections after 14 days of Ang II infusion are shown. Renal injury score for H&E staining and fibrosis score for trichrome staining from ten randomly selected fields of each mouse from each group. Hypoxyprobe signal intensity quantified by ten randomly selected fields of each mouse from each group. Scale bar=50 μm. Data are expressed as mean±SEM. *P < 0.05. (E) Transforming growth factor β1(Tgf-β1), alpha smooth muscle actin 2 (Acta2), collagen type I α1 (Col1a1), and fibronectin (Fn1) and (F) IL-6, IL-1β, and Tnf-α mRNA levels in the mouse kidney. Data are expressed as mean±SEM. N=3–6 for each group. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test *P < 0.05.
Renal anemia is frequently developed in patients with CKD and is largely attributed to the decreased production of erythropoietin which is mainly produced by the kidney. Early studies showed that eENT1 is involved in erythroid commitment38 and stress erythropoiesis in response to anemia.35 Thus, we closely monitored erythrocyte parameters to determine whether the severe hypoxic microenvironment induced by Ang II infusion is associated with anemia in Ang II-infused eEnt1−/− mice. Consistent with earlier studies,35 we observed increased mean cell volume of erythrocytes in eEnt1−/− mice with slightly, but not significantly, decreased erythrocyte numbers at a basal level (Supplemental Figure 4, A and F). Moreover, Ang II infusion led to slightly decreased total erythrocyte numbers and total hemoglobin concentrations in eENT1−/− mice compared with control mice (Supplemental Figure 4, A and F) (but still within the normal range).39 Supporting these findings, no significant differences in the total or percentage of circulating reticulocytes were observed among all four groups (Supplemental Figure 4, G–H). Furthermore, we conducted flow cytometry and biotin-labeling experiments to determine whether the eENT1 ablation affects the erythroid differentiation in the bone marrow and erythrocyte life span, respectively. No differences were observed in the number of megakaryocytic-erythroid progenitor (MEP) cells and the erythrocyte life span (Supplemental Figure 5). Thus, these results led us to conclude that the severe hypoxia and CKD phenotype seen in the Ang II-infused eEnt1−/− mice are not associated with anemia but likely due to mature erythrocyte functional and metabolic impairment.
eENT1 Mitigates Severe Hypoxia-Induced Renal Oxidative Stress and Damage by Promoting Erythrocyte BPGM Activation and 2,3-BPG Production
To gain a comprehensive understanding of how eENT1 affects erythrocyte function and combats CKD progression, we performed a high throughput untargeted metabolomics profiling of erythrocytes, plasma, kidney, and urine from control and eEnt1−/− mice with or without Ang II infusion (Figure 2A). All metabolites detected are shown in Supplemental Spreadsheet Dataset 1. First, Ang II induced similar elevated levels of lactate in kidneys and plasma of controls and eEnt1−/− mice compared with saline-injected groups, indicating increased glycolysis is a renal metabolic compensatory response to counteract Ang II-induced renal hypoxia (Figure 2B). The lack of eENT1 does not affect renal hypoxia-mediated adaptive glycolysis (Supplemental Figure 6A). Tricarboxylic acid (TCA) cycle, and pentose phosphate pathway (PPP) intermediates showed similar levels among the four groups, implicating that the TCA cycle and PPP are not affected by the lack of eENT1 in response to Ang II (Supplemental Figure 6, B–C). Renal glutathione (GSH) decreased insignificantly in Ang II-infused eENT1−/− mice compared with Ang II-infused controls, and the ratio of glutathione disulfide (GSSG) to GSH was similar across the four groups (Supplemental Figure 7, A–C). Notably, Ang II infusion led to significantly decreased renal ATP production in eENT1−/− mice but not in control mice (Supplemental Figure 7D), implicating renal energy insufficiency in mutant mice with CKD. Overall amino acids (AAs) were increased in kidneys, plasma, and urine of Ang II-infused eENT1−/− mice compared with Ang II-infused controls, whereas no differences in AA levels were seen in the basal levels between controls and eENT1−/− mice (Supplemental Figure 8 and Supplemental Spreadsheet Dataset 1). These results indicate that severe hypoxia leads to increased renal damage, less renal tubular AAs reabsorption, and thereby more urinary loss in Ang II-infused eENT1−/− mice. As such, eENT1 is critical to protect against renal hypoxia, insufficient energy supply, dysfunctional renal reabsorption of AAs, urinary AAs loss, and progression of CKD.
Figure 2.
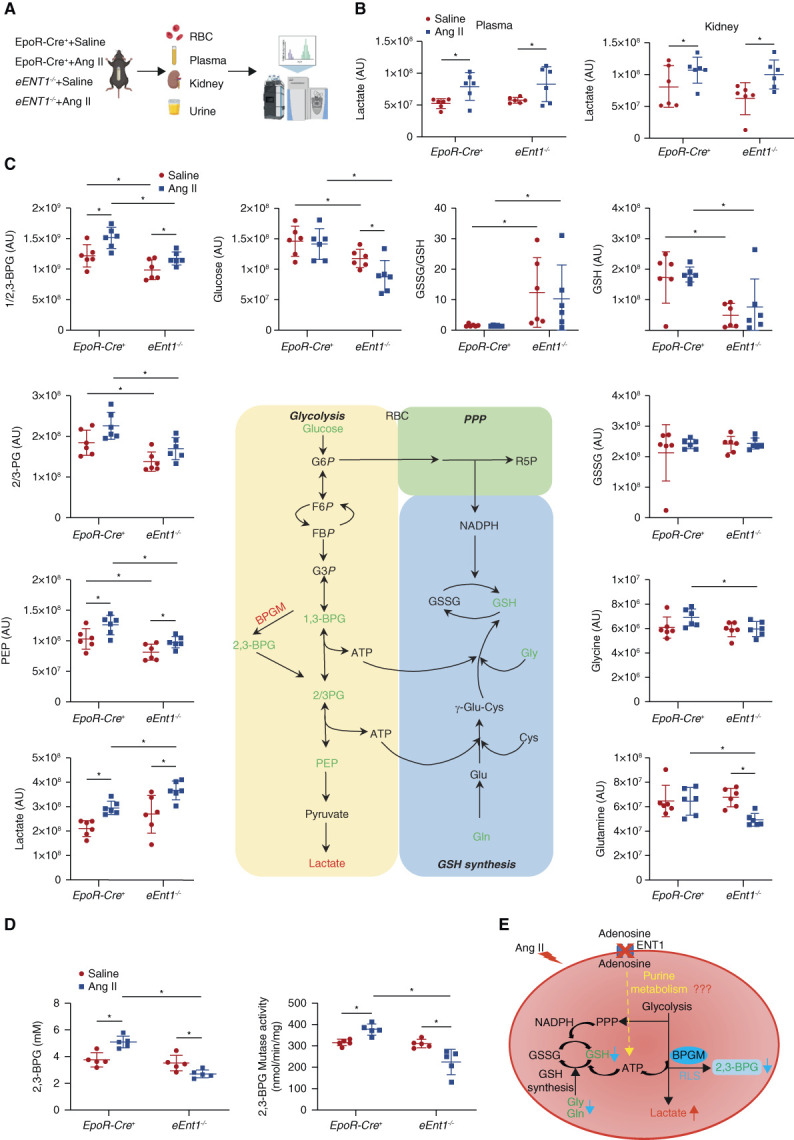
eENT1 mitigates severe hypoxia-induced renal oxidative stress and damage by enhancing erythrocyte BPGM activation and 2,3-BPG production. (A) Experimental design for mouse sample collection and metabolomics study. (B) Plasma and kidney lactate levels in metabolomics screening. Data are expressed as mean±SEM. N=6 for each group. P values were assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (C) Erythrocyte intermediates of glycolysis (glucose, 2,3-bisphosphoglycerate, 2/3-phosphoglycerate, phosphoenolpyruvate, and lactate), PPP, and glutathione synthesis (glycine, glutamine, GSH, and GSSG) were revealed by untargeted metabolomics. Data are expressed as mean±SEM. N=6 for each group. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. *P < 0.05. (D) Erythrocyte 2,3-BPG levels and BPGM activity were measured using commercial kit. Data were expressed as mean±SEM. N=5 mice in each group. P values were assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. *P < 0.05. (E) Proposed mechanism: loss of adenosine uptake in eEnt1−/− mice impairs purine metabolism, leading to reduced BPGM activity and 2,3-BPG production. Figure 2 can be viewed in color online at www.jasn.org.
By contrast, glutamine and glycine (key metabolites for GSH synthesis) concentrations in eENT1−/− mouse erythrocytes were significantly reduced (Figure 2C). As such, eEnt1−/− mice with or without Ang II infusion had significantly lower levels of GSH and a higher ratio of GSSG/GSH than the controls (Figure 2C). However, no significant alteration of PPP intermediates was seen between controls and mutant mice with or without Ang II infusion. Thus, we conclude that the lack of eENT1 leads to increasing oxidative stress within erythrocytes largely due to decreased glutamine and glycine availability for GSH production but independent of PPP (Figure 2C). Intriguingly, erythroid glycolytic Rapaport–Leubering Shunt (RLS) intermediates, including 2,3-bisphosphoglycerate and 2/3-phospho-D-glycerate, were remarkedly induced in control mice with Ang II infusion. Basal levels were reduced in the eEnt1−/− mice, but their levels were also increased in Ang II-infused eEnt1−/− mice. By contrast, lactate as the end metabolite of glycolysis was increased in erythrocytes of eEnt1−/− mice compared with controls and further increased with Ang II infusion (Figure 2C). These results suggest that glucose metabolism is increased in mice lacking eENT1 in the CKD model. However, the levels of 2,3-BPG (determined using an accurate quantification assay), the major product of glycolytic RLS, were significantly reduced in Ang II-infused eEnt1−/− mouse erythrocytes compared with controls with Ang II infusion (Figure 2D). Moreover, the activity of BPGM, the key enzyme for 2,3-BPG production, was reduced in response to Ang II in the eEnt1−/− mice (Figure 2D). Collectively, we demonstrated that eENT1-mediated adenosine uptake is essential for GSH homeostasis and BPGM activation, inducing glucose metabolism toward RLS to increase 2,3-BPG production and thus combatting renal hypoxia, oxidative stress, renal damage, dysfunction, and progression of CKD. Thus, our unexpected discovery of reduction of erythrocyte 2,3-BPG and GSH levels, without significant changes in GSSG levels in Ang II-infused eEnt1−/− mice (Figure 2, C and D), leads us to hypothesize that eENT1 is critical to control erythrocyte purine metabolism and downstream signaling cascades to promote the production ATP and 2,3-BPG, two key metabolites for GSH synthesis and O2 delivery in response to Ang II, respectively (Figure 2E).
eENT1 Orchestrates Intracellular Adenosine Metabolism to Induce BPGM Activation by Inhibiting AMPD3 and Activating AMPK in CKD
Supporting the above hypothesis, besides erythroid RLS and GSH metabolic pathways, our metabolic profiling revealed that erythrocyte intracellular purine metabolism is one of the most affected pathways when erythrocyte ENT1 was ablated in CKD (Figure 3A). Specifically, our metabolomic profiling revealed that AMP was decreased while ATP changed negligibly, leading to the reduced ratio of AMP to ATP in eEnt1−/− mice with Ang II infusion compared with Ang II-infused control mice (Figure 3, B–D). Realizing recent studies showed that AMPK, as a master energy sensor, activates BPGM, resulting in increased 2,3-BPG production,18 we hypothesized that the low levels of AMP underly decreased BPGM activity and 2,3-BPG production by reducing AMPK activity in Ang II-infused eEnt1−/− mice. AMPK activity was enhanced in Ang II-infused controls, whereas reduced in eEnt1−/− mice with Ang II infusion (Figure 3E). However, plasma adenosine did not significantly change among all four groups, ruling out the possibility that eENT1-induced AMPK-BPGM activation and in turn 2,3-BPG induction is dependent on erythrocyte ADORA2B receptor activation (Figure 3F).17,18
Figure 3.
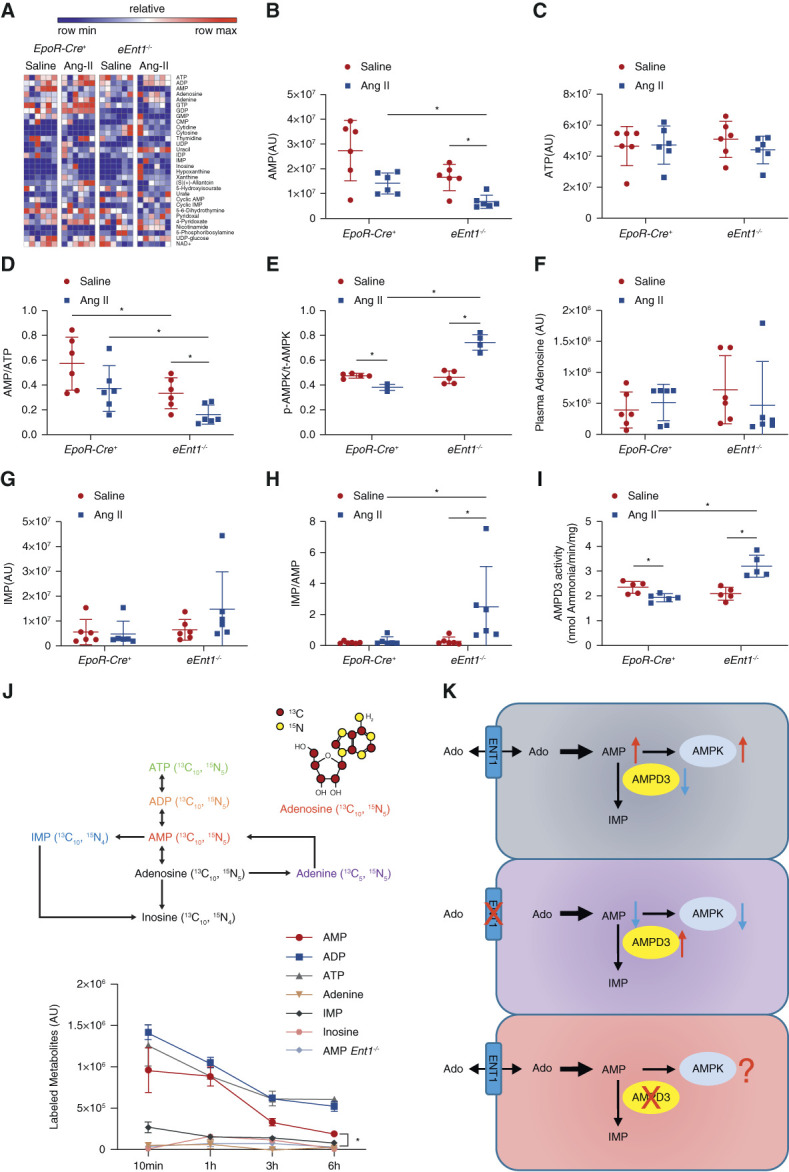
eENT1 orchestrates intracellular adenosine metabolism to promote BPGM activation by inhibiting AMPD3 and activating AMPK in CKD. (A) Heatmap of erythrocyte nucleotides and other metabolites measured by untargeted metabolomics. (B–D) AMP and ATP levels and the ratio of AMP to ATP. P values were assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. No significant differences were observed. (E) The ratio of erythrocyte phosphorylated AMPKα to total AMPKα measured using Elisa kits. Data are expressed as mean±SEM. N=5 mice in each group. *P < 0.05. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (F) Plasma adenosine levels. (G) Erythrocyte IMP levels, (H) the ratio of IMP to AMP, and (I) AMP deaminase activity in erythrocytes. Data are expressed as mean±SEM. N=5 or 6 mice per group. *P < 0.05. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (J) Isotopically labeled adenosine flux experiments were conducted in cultured WT and eENT1-deficient mouse erythrocytes to determine how eENT1-facilitated uptake of adenosine was metabolized intracellularly. Data are expressed as mean±SEM. (K) Proposed hypothesis: eENT1 is critical to promote AMPK-mediated BPGM activation and functional regulation by promoting the uptake of extracellular adenosine, the inhibition AMPD3, and the accumulation of AMP inside of erythrocytes.
To our surprise, in contrast to decreased AMP and AMP/ATP, our metabolomics data further revealed enhanced inosine monophosphate (IMP) levels and an increased ratio of IMP to AMP in Ang II-infused eEnt1−/− mice, while their levels were not altered at basal levels (Figure 3, G and H). These findings immediately suggest that AMPD3, as an intracellular component of purine nucleotide metabolism, is involved in eENT1-mediated accumulation of AMP and activation of AMPK in response to CKD. Indeed, Ang II infusion decreased eAMPD3 activities in control mice but increased AMPD3 activity in eEnt1−/− mice (Figure 3I). These findings support an intriguing possibility that inhibiting eAMPD3-mediated deamination of AMP to IMP and in turn allowing the accumulation of AMP is a critical intracellular purinergic regulatory mechanism for AMPK activation in erythrocytes during CKD.
To precisely follow the fate of ENT1-mediated adenosine uptake and its intracellular metabolism in erythrocytes, we conducted adenosine flux experiments by incubating erythrocytes acquired from wild type (WT) and global ENT1-deficient mice with isotopically 13C1015N5-labeled adenosine and tracing the intracellular adenosine metabolism after its uptake by ENT1 (Figure 3J). First, labeled adenosine could not be detected in the supernatant of cultured WT erythrocytes, but only in Ent1−/− mouse erythrocytes, indicating that ENT1 is critical for extracellular adenosine uptake in erythrocytes (Supplemental Figure 9). However, no labeled adenosine was detected inside the erythrocytes of either WT or Ent1−/− mice (Supplemental Table 1). Instead, high amounts of labeled AMP were detected in WT mouse erythrocytes, whereas labeled AMP was barely detected in Ent1−/− mouse erythrocytes (Figure 3J), indicating that eENT1-mediated uptake of adenosine is largely converted to AMP. Labeled AMP, ADP, and ATP were readily detected at 10 minutes and gradually declined over time, whereas adenine, inosine, and IMP were present at very low levels that showed little change over time. Thus, flux analyses provided us the deep understanding of intracellular adenosine metabolism after it is transported into the erythrocytes by ENT1. First, adenosine uptake into erythrocytes by ENT1 is quickly converted to AMP and further phosphorylated to adenosine diphosphate (ADP) and ATP, and very little is metabolized toward adenine or IMP. As such, our findings support the hypothesis that the inhibition of AMPD3 is critical for erythrocyte eENT1-mediated AMPKα activation by reducing the deamination of AMP to IMP and in turn allowing accumulation of AMP in CKD (Figure 3K).
Genetic Ablation of Murine AMPD3 Attenuates Renal Hypoxia, Kidney Damage, and CKD Progression
Next, we took advantage of Ampd3−/− mice to explore whether the loss of eAMPD3 plays any role in AMPK-BPGM activation in erythrocytes and in turn has a protective role in CKD. The Ampd3−/− mice were fertile and morphologically indistinguishable from WT mice. Then, we conducted the same Ang II-induced CKD model (Figure 4A). Consistent with in vivo results in EpoR-Cre+ mice, AMPD3 activity decreased in WT mice with Ang II infusion (Figure 4B). In contrast to eEnt1−/− mice, the Ampd3−/− mice with Ang II infusion displayed a protective phenotype. In brief, SBP increased as minipump continuously infusing Ang II into Ampd3−/− mice but slightly lower than WT mice (Figure 4C). Ampd3−/− mice attenuated Ang II infusion-induced kidney damage, fibrosis, and hypoxia compared with control mice. Proteinuria, plasma creatinine and urea, renal injury score, renal fibrosis determined by Masson trichrome staining, and the hypoxyprobe staining in kidney sections were significantly decreased in Ang II-infused Ampd3−/− mice compared with the controls (Figure 4, D and E). Finally, fibrotic and inflammatory gene expression indicated that the Ampd3−/− mice with Ang II-infusion experienced less renal fibrosis and inflammation than the control mice (Figure 4, F and G). Moreover, no significant differences were observed in complete blood count and RAS-related gene expression in the kidneys between WT and Ampd3−/− mice with or without Ang II infusion (Supplemental Figures 10 and 11), ruling out the possibilities that the protective role of ablation of AMPD3 in CKD is due to the differences of erythrocyte production or local renal vasoconstriction. Thus, genetic ablation of AMPD3 attenuates Ang II-induced renal hypoxia, tissue damage, fibrosis, inflammation, and progression of CKD.
Figure 4.

AMPD3 deficiency attenuates renal hypoxia, kidney damage, and CKD progression. (A) Schematic representation of experimental design. Ang II or saline was subcutaneously infused by osmotic minipump into wild type (WT) or Ampd3−/− mice (8–12 week) for 14 days. (B) Erythrocyte AMP deaminase activity. Data are expressed as mean±SEM. N=5 mice in each group. *P < 0.05. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (C) Systolic BP was measured on days 0, 3, 7, and 14 using the tail-cuff method. Data are expressed as mean±SEM. N=5 in each group. *P < 0.05, WT +Ang II/Saline, #P < 0.05, Ampd3−/− +Ang II/Saline. P values were assessed by repeated measures two-way ANOVA. (D) Proteinuria measured by the 24-hour urine albumin-to-creatinine ratio was quantified using a commercially available kit; plasma urea and creatinine were quantified by a commercially available kit. Data are expressed as mean±SEM. n=5 in each group. *P < 0.05. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (E) Representative hematoxylin and eosin (H&E)–stained, trichrome-stained, or hypoxyprobe-stained kidney sections are shown. Renal injury score from H&E staining, fibrosis score from trichrome staining, and hypoxyprobe signal intensity quantified by ten randomly selected fields of each mouse from each group. Data are expressed as mean±SEM. Scale bar=50 μm. (F) Transforming growth factor β1(Tgf-β1), alpha smooth muscle actin 2 (Acta2), collagen type I α1 (Col1a1), and fibronectin (Fn1) and (G) IL-6, IL-1β, and Tnf-α mRNA levels in the mouse kidney. Data are expressed as mean±SEM. N=5 for each group. *P < 0.05. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test.
AMPD3 Ablation Promotes Erythrocyte Adaptive Metabolic Reprogramming to Counteract CKD by Promoting Energy Supply, O2 Delivery, and Anti-ROS Capacity
To gain a better understanding of how ablation of AMPD3 affects erythrocyte metabolism and in turn combats CKD progression, we conducted a high throughput untargeted metabolomic profiling of the erythrocytes, plasma, and kidney from control and Ampd3−/− mice with or without Ang II infusion. All metabolites detected are shown in Supplemental Spreadsheet Dataset 2. Genetic ablation of AMPD3 significantly induced multiple intermediates of glycolysis, the PPP, and glutathione homeostasis in erythrocytes and plasma at the basal levels and in response to Ang II compared with WT mice. These include 1/2,3-BPG, D-glucono-1-5-lactone 6-phosphate, γ-L-glutamyl-L-cysteine, and GSH/GSSG (Figure 5, A–E). Remarkably, metabolomic screening showed that erythrocyte nucleotides changed dramatically, and especially AMP, ADP and ATP levels were significantly increased in Ampd3−/− mice at the baseline, but not further elevated by Ang II infusion (Figure 5F). Because ATP can bind to Hb to trigger O2 delivery,40 P50 was significantly induced in Ampd3−/− mice without Ang II infusion (Figure 5G). Interestingly, erythroid glycolytic RLS was not elevated at the basal levels, whereas Ang II infusion could induce AMPKα-BPGM activity and thus 2,3-BPG production in Ampd3−/− mice compared with control mice (Figure 5, H–J). Thus, genetic ablation of AMPD3 is beneficial to combat Ang II-induced CKD by promoting energy supply and O2 delivery via triple mechanisms: (1) increasing adenine nucleotide pool as energy supply, (2) enhancing AMPKα-BPGM activity and 2,3-BPG production, and (3) lowering oxidative stress.
Figure 5.

AMPD3 deficiency promotes erythrocyte adaptive metabolic reprogramming to counteract CKD by enhancing energy supply, O2 delivery, and anti-ROS capacity. (A) Heatmap of erythrocytes, plasma and kidney glycolysis, PPP, glutathione homeostasis, and TCA cycle measured by untargeted metabolomics. (B) 2,3-BPG and (C) 6-phosphogluconolactone and (D) γ-L-glutamyl-L-cysteine levels in metabolomics screening. (E) The ratio of GSSG to GSH. Data are expressed as mean±SEM. N=5 for each group. *P < 0.05. P values were assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (F) Purine metabolism (ATP, ADP, AMP, IMP, adenosine, Inosine, adenine) in erythrocytes determined by untargeted metabolomics. Data are expressed as mean±SEM. N=5 for each group. *P < 0.05. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (G) Erythrocyte oxygen release capacity (P50) was measured using a Hemox analyzer. Data are expressed as mean±SEM. N=5 mice in each group. *P < 0.05. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (H) Erythrocyte AMPKα activity was measured by quantification of the optical density (OD) value of phospho-AMPKα (Thr172) using the ELISA kit. Data are expressed as mean±SEM. N=5 mice in each group. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (I) Erythrocyte 2,3-BPG and (J) BPGM activity were measured using a commercial kit. Data were expressed as mean±SEM. N=5 mice in each group. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test.
Importance of Erythrocyte ENT1 and AMPD3 in an Independent CKD Model Challenged by Unilateral Ureteral Obstruction (UUO)
To further determine the broad role of eENT1 and AMPD3 in CKD, we challenged both eENT1 and AMPD3-deficient mice with UUO. Consistently, severe renal hypoxia, tubular injury, inflammation, and fibrosis as well as fibrotic and inflammatory gene expression were further induced in eEnt1−/− mice compared with the controls with UUO challenge (Figure 6, A–C). Similar to Ang II infusion, eENT1 genetic ablation attenuated AMPD3 inhibition, AMPK activation, and 2,3-BPG elevation as well as increased oxygen delivery compared with the controls with UUO manipulation (Figure 6, D–F). In contrast to eEnt1−/− mice, less renal hypoxia, tubular injury, renal fibrosis, and inflammation were observed in Ampd3−/− mice with UUO challenge compared with controls (Figure 6, G–I). Furthermore, the ablation of Ampd3 led to decreased erythrocyte AMPD3 activity and increased p-AMPK/t-AMPK and 2,3-BPG production (Figure 6, J–L). Altogether, we demonstrated that eENT1 and AMPD3 as the erythrocyte hypoxia sensors play an importance role in two independent CKD models, Ang II infusion or UUO challenge.
Figure 6.

Validation of ENT1-AMPD3 signaling pathway in UUO model. (A) Representative hematoxylin and eosin (H&E)–stained, trichrome-stained, or hypoxyprobe-stained kidney sections after 10 days of UUO are shown. Tubular injury score for H&E staining and fibrosis score for trichrome staining from ten randomly selected fields of each mouse from each group. Hypoxyprobe signal intensity quantified by ten randomly selected fields of each mouse from each group. Scale bar=50 μm. Data are expressed as mean±SEM. *P < 0.05. (B) Transforming growth factor beta1 (Tgf-β1), alpha smooth muscle actin 2 (Acta2), collagen type I α1 (Col1a1), and fibronectin (Fn1) and (C) IL-6, IL-1β, and Tnf-α mRNA levels in the mouse kidney. Data are expressed as mean±SEM. N=4 for each group. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test *P < 0.05. (D) AMPD3 activity, (E) the ratio of erythrocyte phosphorylated AMPKα to total AMPKα, and (F) erythrocyte 2,3-BPG was measured using a commercial kit. Data were expressed as mean±SEM. N=4 mice in each group. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (G) Representative hematoxylin and eosin (H&E)–stained, trichrome-stained, or hypoxyprobe-stained kidney sections after 10 days of UUO are shown. Tubular injury score for H&E staining and fibrosis score for trichrome staining from ten randomly selected fields of each mouse from each group. Hypoxyprobe signal intensity quantified by ten randomly selected fields of each mouse from each group. Scale bar=50 μm. Data are expressed as mean±SEM. *P < 0.05. (H) Transforming growth factor beta 1 (Tgf-β1), α smooth muscle actin 2 (Acta2), collagen type I α1 (Col1a1), and fibronectin (Fn1) and (I) IL-6, IL-1β, and Tnf-α mRNA levels in the mouse kidney. Data are expressed as mean±SEM. N=4 for each group. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test *P < 0.05. (J) AMPD3 activity, (K) the ratio of erythrocyte phosphorylated AMPKα to total AMPKα, and (L) erythrocyte 2,3-BPG was measured using a commercial kit. Data were expressed as mean±SEM. N=4 mice in each group. P value was assessed using the ordinary one-way ANOVA with the Holm–Sidak post hoc multiple comparisons test.
Decreased Oxidative Stress Inhibits Erythrocyte AMPD3 Activity under Hypoxia in a Positive Forward Manner
Given that AMPD3 inhibition-mediated activation of AMPK-BPGM and 2,3-BPG and O2 delivery is a critical erythrocyte compensatory response to counteract CKD progression in two independent CKD models, we sought to probe molecular mechanisms controlling erythrocyte AMPD3 activity in CKD. Because of the complexity of an intact animal, we conducted a series of in vitro studies in cultured primary WT mouse erythrocytes to initially determine whether Ang II directly regulates AMPD3 activity. We found that Ang II treatment failed to regulate AMPD3 activity in cultured primary mouse erythrocytes (Supplemental Figure 12), ruling out the possibility of Ang II being involved in the direct regulation of AMPD3 activity in erythrocytes.
Next, we wondered whether erythrocyte purine metabolism is altered in response to hypoxia and in turn regulates AMPD3 and AMPKα activity. To test this possibility, we performed adenosine flux experiments to trace intracellular adenosine metabolism under hypoxia (Figure 7A). Compared with normoxia, hypoxia induced AMP and decreased ATP with no significant change in ADP in cultured WT mouse erythrocytes (Figure 7, B–D), indicating that AMPKα may be activated under hypoxia. Supporting this, the ratio of AMP to ATP increased under hypoxia (Figure 7E). Importantly, the ratio of IMP to AMP decreased with hypoxia treatment compared with normoxia (Figure 7F), suggesting that hypoxia inhibits AMPD3 and in turn activates AMPKα by promoting AMP levels (Figure 7G).
Figure 7.

Hypoxia directedly inhibits AMPD3 activity by reducing oxidative stress in cultured primary murine erythrocytes. (A) Isotopically labeled adenosine was used to trace intracellular adenosine metabolism in cultured WT erythrocytes under normoxia and hypoxia. (B) Labeled AMP, (C) ADP, and (D) ATP were detected by UHPLC-MS. (E) The ratio of labeled AMP to ATP. (F) The ratio of labeled IMP to AMP. Data are expressed as mean±SEM. N=3 mice in each group. *P < 0.05. P value was assessed using the repeated measures two-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (G) Experimental design for in vitro study. (H) ROS level, (I) AMPD3 activity, and (J) AMPKα activity in WT mouse primary erythrocytes cultured under normoxia and hypoxia (1% oxygen) at 10, 30, and 60 minutes. Data are expressed as mean±SEM. N=3 mice in each group. *P < 0.05. P value was assessed using the repeated measures two-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (K) ROS level, (L) AMPD3 activity, and (M) Phosphorylated AMPK1α in cultured mouse erythrocytes with or without H2O2 treatment under normoxia at 10, 30, and 60 minutes. Data are expressed as mean±SEM. N=3 mice in each group. *P < 0.05. P value was assessed using the repeated measures two-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. Figure 7 can be viewed in color online at www.jasn.org.
Early studies demonstrated that 2,3-BPG plays an important role in the repression of the activity of AMPD3,21,41 while ROS is known to activate AMPD3.42 Supporting this notion, in WT mice, we observed elevated 2,3-BPG and O2 delivery along with the inhibition of AMPD3 activity, whereas eENT1 ablation significantly reduced 2,3-BPG and in turn increased ROS along with AMPD3 activity in CKD models. Given our in vivo findings and realizing that hypoxia is a common feature in CKD, that erythrocytes are quite sensitive to hypoxia by promoting more O2 release and in turn lowering its own ROS and that ROS is known to induce AMPD3 activity, we hypothesized that eAMPD3 activity is likely regulated by hypoxia, Hb-O2 binding affinity, and ROS levels (Figure 7G). To test this possibility, we cultured primary WT mouse erythrocytes under normoxia or hypoxia for different times. We found that hypoxia decreased ROS levels, as well as decreased AMPD3 activity and in turn induced AMPK activity, compared with normoxia in WT mouse erythrocytes up to 1 hour (Figure 7, H–J). Next, to determine whether oxidative stress directly activates AMPD3 activity and in turn inhibits AMPK activity, we treated WT mouse erythrocytes with ROS activator hydrogen peroxide (H2O2) under normoxia. We found that H2O2 treatment directly induced ROS levels and AMPD3 activity under normoxia and in turn decreased AMPK activity under normoxia in WT mice (Figure 7, K–M, Supplemental Figure 13, A–C). Altogether, we provided in vitro evidence that hypoxia-mediated rapid O2 delivery and subsequent reduction of ROS underlie the inhibition of erythrocyte AMPD3 in CKD. As such, the inhibition of eAMPD3 functions as a key intracellular purinergic hypoxia sensor, leading to the accumulation of AMP, AMPK-BPGM activation, enhanced O2 delivery, and further reduction of ROS to counteract CKD in a positive feedforward manner.
Human Translational Studies in Patients with CKD
To address the potential clinical relevance of our preclinical findings from animal studies, we performed human translational studies in patients with CKD (Figure 8A). Specifically, we analyzed the levels of 2,3-BPG, BPGM activity, AMPK activity, AMPD3 activity, and P50 in patients with CKD (n=18) compared with normal individuals (n=18) (demographic and clinical information for CKD and control subjects are summarized in Table 1 and Supplemental Table 2). Recapitulating mouse findings, the production of 2,3-BPG, BPGM activity, p-AMPK level, and P50 were increased, whereas AMPD3 activity was decreased in patients with CKD compared with normal controls (Figure 8, B–F). No noticeable differences between females and males were observed.
Figure 8.

Translational studies in patients with CKD. (A) Experimental design: in vivo validation of mouse findings in patients with CKD and in vitro confirmation of hypoxia-mediated reduction of ROS underlying inhibition of AMPD3 and thus AMPKα activation in cultured human erythrocytes. (B) 2,3-BPG, (C) BPGM activity, (D) Phospho-AMPKα, (E) AMPD3 activity, and (F) P50 in erythrocytes isolated from control individuals and patients with CKD. Data are expressed as mean±SEM. Black dots represent females; red dots represent males. N=18 in each group. *P < 0.05. P value was assessed using the repeated measures two-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (G) ROS level, (H) AMPD3 activity, and (I) Phospho-AMPKα in isolated human erythrocytes under normoxia and hypoxia (1% oxygen) at 10, 30, and 60 minutes. Data are expressed as mean±SEM. N=3 in each group. *P < 0.05. P value was assessed by repeated measures two-way ANOVA with Holm–Sidak post hoc multiple comparisons test. (J) ROS level, (K) AMPD3 activity and (L) Phospho-AMPKα in cultured primary human erythrocytes with or without H2O2 treatment under normoxia at 10, 30, and 60 minutes. Data are expressed as mean±SEM. N=3 mice in each group. *P < 0.05. P value was assessed using the repeated measures two-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. Figure 8 can be viewed in color online at www.jasn.org.
Moreover, we conducted in vitro experiments with erythrocytes isolated from normal human blood to validate the direct regulation of hypoxia-enhanced O2 delivery and subsequent downregulation of ROS in AMPD3 and AMPK activity. Consistent with the mouse results, we found that hypoxia decreased ROS levels and also AMPD3 activity, thus in turn induced p-AMPK (Figure 8, G–I). To determine whether ROS directly induces AMPD3 activity and inhibits AMPK activity, we conducted the same treatment as mouse in vitro experiments. Consistent with mouse findings, H2O2 directly increased ROS levels and AMPD3 activity and decreased p-AMPK (Figure 8, J–L, Supplemental Figure 13, D–E).
Molecular Links of Human eENT1-Mediated Adenosine Uptake with eAMPD3-AMPK Cascade and 2,3-BPG Production
To precisely delineate the molecular links of human eENT1-mediated adenosine uptake with the eAMPD3-eAMPK cascade and 2,3-BPG production, we performed in vitro experiments with erythrocytes isolated from normal human blood with dipyridamole (Dip, a specific ENT inhibitor) and deoxycoformycin (DCF, an AMP deaminase inhibitor) alone or in combination with the presence of adenosine (Figure 9A). As expected and consistent to early studies,43,44 DCF successfully inhibited AMPD3 activity from 10 minutes to 6 hours (Figure 9B). Notably, the inhibition of adenosine uptake by ENT1 (the predominant ENT in erythrocytes) with Dip treatment gradually activated AMPD3 activity and significantly induced its activity at 6 hours comparing with the controls (Figure 9B). Therefore, we chose 6 hours for cotreatment with DCF and Dip to probe the effect of inhibited AMPD3 in Dip-mediated suppressed ENT1-adenosine uptake-AMPK axis and 2,3-BPG production. Similar as murine genetic ablation of eENT1, we found that interfering eENT1-mediated adenosine uptake by Dip treatment alone induced AMPD3 activity, reduced AMPK activity, and increased 2,3-BPG production, while DCF treatment alone inhibited AMPD3 activity, activated AMPK activity, and increased 2,3-BPG production. Furthermore, cotreatment with DIP and DCF directly rescued suppressed eENT1-dependent adenosine uptake-mediated erythrocyte AMPK inhibition and restored 2,3-BPG production (Figure 9, C–E). Altogether, we provide the in vitro human molecular link that eENT1-mediated adenosine uptake coordinated with AMPD3 inhibition is essential for erythrocyte AMPK activation and 2,3-BPG production.
Figure 9.

Molecular links of human eENT1-mediated adenosine uptake with eAMPD3-AMPK cascade and 2,3-BPG production. (A) Experimental design for in vitro experiment. (B) AMPD3 activity in isolated human erythrocytes treated with DCF or Dip in the presence of adenosine at 0, 10 minutes, 1, 3, and 6 hours. Data are expressed as mean±SEM. N=3 in each group. *P < 0.05. P value was assessed using the repeated measures two-way ANOVA with the Holm–Sidak post hoc multiple comparisons test. (C) AMPD3 activity, (D) Phospho-AMPKα and (E) 2,3-BPG in isolated human erythrocytes treated with DCF or/and Dip in the presence or absence of adenosine at 6 hours. (F) Working Model: ENT1-AMPD3 signaling network is a critical “erythrocyte hypoxia sensor” to preferentially promote the intracellular purinergic signaling cascade underlying AMPK-BPGM-mediated elevation of 2,3-BPG and enhanced O2 delivery and anti-ROS capacity to combat renal hypoxia, damage, and progression of CKD. Figure 9 can be viewed in color online at www.jasn.org.
Discussion
Erythrocytes are the most abundant cells and only cell type responsible for delivering O2 to peripheral tissues in our body. Although both ENT1 and AMPD3 are highly enriched in erythrocytes, their role and underlying mechanisms in CKD have not been previously recognized. Here, we conducted mouse genetic studies and human translational studies and revealed that eENT1 and AMPD3 functioning as two critical “erythrocyte hypoxic adenosine sensors” coordinately mitigate renal hypoxia and CKD progression by controlling erythrocyte intracellular purine metabolism, promoting AMPK-BPGM–mediated metabolic reprogramming, and inducing 2,3-BPG production, O2 delivery, and antioxidative stress capacity. Overall, our studies have added a significant new chapter to a poorly understood beneficial role of intracellular purinergic metabolic regulation modulated by eENT1 and eAMPD3 in erythroid physiology and identified multiple unique erythroid hypoxic sensors to counteract renal hypoxia and progression of CKD (Figure 9F).
Extracellular adenosine is well known to be induced by hypoxia, which is largely generated from ATP by two ecto-nucleotidases, triphosphate diphosphohydrolase (CD39) and monophosphohydrolase (CD73).45 Substantial studies have revealed that extracellular adenosine regulates numerous physiological and pathological functions by engaging four G-protein–coupled receptors.46–48 Extracellular adenosine is tightly regulated49 and can be rapidly transported into cells by equilibrative nucleoside transporters (ENTs). Notably, recent studies showed that erythrocytes are enriched with eENT1, which plays an important role in transporting extracellular adenosine from blood and regulating hypoxic adaption to high altitude. Moreover, CD73 coupled with eENT1-dependent “erythrocyte hypoxic memory” has been revealed for fast acclimatization on reascent.19 More recent studies have revealed that maternal eENT1-dependent adenosine uptake is critical for normal fetal growth by promoting AMPK-BPGM activity and 2,3-BPG production and O2 delivery to counteract placental hypoxia.20 However, whether eENT1 is involved in CKD remains unknown. Moreover, how eENT1 transported extracellular adenosine is metabolized intracellularly in erythrocytes and whether eENT1 regulates erythrocyte metabolism and function in CKD remain as puzzles. Here, we conducted both mouse genetic studies and human translation studies to solve these puzzles. Functionally and metabolically, we demonstrated that eENT1 mitigates severe renal hypoxia, insufficient energy supply, and progression of CKD by inducing erythrocyte AMPK-BPGM activation, 2,3-BPG production, and O2 delivery. Like adenosine, AMPK is induced under the conditions of limited O2 availability to act as an intracellular energy master regulator to offset energy depletion.50 However, how AMPK is regulated by eENT1 in CKD remains unclear. Notably, recent studies showed that extracellular adenosine promotes 2,3-BPG production and O2 delivery through ADORA2B-AMPK signaling to counteract hypoxia caused by high altitude and CKD.17,18 However, no difference in plasma adenosine levels was observed in CKD models between controls and eENT1-specific knockouts, implicating that eENT1-mediated AMPK-BPGM activation is independent of erythrocyte ADORA2B receptors. Our untargeted metabolomics and isotopically labeled adenosine tracing analyses revealed that the circulating adenosine uptaken rapidly by eENT1 is phosphorylated to AMP, which is not deaminated to IMP by inhibiting AMPD3. Altogether, our findings revealed an important protective role of eENT1-mediated extracellular adenosine uptake in CKD by inhibiting eAMPD3 activity, leading to the accumulation of AMP and activation of AMPK and subsequently inducing BPGM activity, 2,3-BPG production, and O2 delivery from erythrocytes.
There are three isoforms of AMPD encoded by three independent genes: muscle (AMPD1), liver (AMPD2), and erythrocyte (AMPD3). Although AMPD3 is highly expressed in erythrocytes,51 it is also expressed in various tissues and cells, such as heart, kidney, muscle, and tumors.23,52–56 For example, decreased AMPD3 expression results in the suppression of tumor cell migration and invasion,56 whereas overexpression of AMPD3 decreases ATP levels and mitochondrial protein synthesis rates in muscle cell, leading to the muscle atrophy.52 Given our unexpected discovery that eENT1-mediated uptake of adenosine is further metabolized to AMP but not IMP due to the inhibition of eAMPD3, which is responsible for eENT1-induced AMPK activation in CKD, we further conducted genetic, cellular, and metabolic studies. Functionally, we revealed that global AMPD3 ablation promotes erythrocyte adaptive metabolic reprogramming to promote erythrocyte O2 delivery and anti-ROS capacity to counteract renal hypoxia and CKD progression. Metabolomic screening confirmed early studies showing that global AMPD3-deficient mice have high ATP levels only in erythrocytes, while total AMPD enzyme activity does not change significantly in various tissue, including kidney, between wild type and Ampd3−/− mice.23,57 Consistently, we found that adenosine, AMP, ADP, ATP, and IMP/AMP did not change significantly in the kidneys of each group (Supplemental Figure 3, metabolites identified in mouse kidney). Thus, these findings indicate that global Ampd3−/− only affects erythrocytes but not renal total AMPD activity and purinergic components, such as ATP, AMP, and IMP. Because ATP can bind to Hb and promote O2 release, P50 is already significantly induced in Ampd3−/− mouse erythrocytes at the basal level. Intriguingly, erythroid glycolytic RLS is not induced at the basal levels, while Ang II infusion promotes AMPK-BPGM activity and thus 2,3-BPG production in Ampd3−/− mice. Similarly, human translational studies confirmed that eAMPD3 activity is significantly reduced in the erythrocytes of patients with CKD along with elevated AMPK-BPGM activity, 2,3-BPG, and P50. At this moment, global AMPD3-deficient mice might not rule out the potential role of AMPD3 in other tissues and cells in CKD. However, given the nature of high expression of AMPD3 in erythrocytes and in view of our findings from both genetic and human studies, we conclude that decreased eAMPD3, followed by increased AMPK-BPGM-2,3-BPG is a compensatory metabolic network to combat CKD by promoting increased adenine nucleotide pools, energy supply, and O2 delivery capacity.
Previous studies reported that 2,3-BPG inhibits the activity of AMPD3,21,41 while ROS activates AMPD3.42 However, how erythrocyte AMPD3 activity is inhibited and in turn promotes O2 delivery in CKD is unknown. Intriguingly, in both WT mice and patients with CKD, we observed elevated 2,3-BPG and O2 delivery along with the inhibition of AMPD3 activity. Genetic experiments with mice show that eENT1 ablation significantly reduces 2,3-BPG and in turn increases ROS along with AMPD3 activity in Ang II-induced severe CKD model. By contrast, AMPD3 ablation significantly induces AMPK-BPGM activity, 2,3-BPG production, and O2 delivery and anti-ROS capacity to mitigate renal impairment in the Ang II-induced CKD model. These findings prompted us to further discover that hypoxia but not Ang II directly inhibits AMPD3 activity in cultured primary erythrocytes isolated from WT murine and normal humans. Mechanistic studies further revealed that hypoxia-mediated rapid O2 delivery and subsequent reduction of ROS underlies hypoxia-mediated inhibition of AMPD3 activity in cultured primary murine and human erythrocytes. As such, AMPD3 inhibition underlies AMPK-BPGM activation, elevated 2,3-BPG production, and enhanced O2 delivery, thereby further reduction of ROS and additional inhibition of AMPD3 is a positive feedforward loop to counteract CKD progression.
In conclusion, we have solved multiple puzzles regarding how erythrocytes sense hypoxia and in turn enhance their O2 delivering and antioxidative capacity to counteract CKD. We demonstrated the function of erythrocytes and the metabolic basis of maintaining normal renal oxygen supply, metabolism, and function to protect against CKD. Our human and mouse genetic studies coupled with in vitro studies support a new but compelling working model: In patients and mice with CKD, hypoxia rapidly induces O2 release, leading to reduced ROS and subsequently suppressed AMPD3 activity. As such, eENT1 transported extracellular adenosine is rapidly converted to AMP but not IMP due to repressed AMPD3 activity, leading to increased AMPK-BPGM activity, more 2,3-BPG production, and further O2 delivery and additional ROS reduction as a positive feedforward loop (Figure 9F). Thus, ENT1-AMPD3 signaling network is a critical “erythrocyte hypoxia sensor” to preferentially promote intracellular adenosine convert to AMP which induce AMPK-BPGM–mediated elevation of 2,3-BPG, enhanced O2 delivery and anti-ROS capacity to combat renal hypoxia, damage and progression of CKD (Figure 9F). Our findings have provided functional, molecular, and metabolic new insight into erythrocyte as a hypoxia sensor to offset renal hypoxia and progression of CKD and highlighted multiple erythrocyte-based early diagnostic and therapeutic targets.
Supplementary Material
Disclosures
A. D'Alessandro reports Consultancy: Hemanext Inc., Macopharma Inc.; and Ownership Interest: Omix Technologies Inc. All remaining authors have nothing to disclose.
Funding
This work was supported by Feifan Scholar Fund of Xiangya Hospital of Central South University (Y.X.), NSFC82230023 (Y.X.), HL136969 (Y.X.), HL137990 (Y.X.), McGovern Fund (Y.X.), NSFC82100788 (T.T.X.), and the Bob and Hazel Casey Endowed Chair of University of Texas Health Science Center-McGovern Medical School (R.E.K.); R01HL146442, R01HL149714, R01HL148151, R21HL150032 (A.D.).
Author Contributions
Conceptualization: Angelo D’Alessandro, Yang Xia, Xin Zhang.
Data curation: Benjamin C. Brown, Changhan Chen, TingTing Xie, Weiru Zhang, Yujin Zhang.
Formal analysis: Changhan Chen, Angelo D’Alessandro, TingTing Xie, Yujin Zhang.
Funding acquisition: Angelo D’Alessandro, Rodney E. Kellems, Yang Xia, Tingting Xie.
Investigation: Changhan Chen.
Methodology: Benjamin C. Brown, Angelo D’Alessandro, Rodney E. Kellems, Lizhen Lin, Yiyan Wang, Fang Yu, Weiru Zhang, Xin Zhang, Yujin Zhang.
Supervision: Angelo D’Alessandro, Rodney E. Kellems, Yang Xia.
Visualization: Changhan Chen.
Writing – original draft: Changhan Chen.
Writing – review & editing: Yang Xia.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/E496, http://links.lww.com/JSN/E497, http://links.lww.com/JSN/E498.
Supplemental Figure 1. Generation of mice with deletion of Ent1 on erythroid cells.
Supplemental Figure 2. HIF-1α immunohistochemistry staining.
Supplemental Figure 3. Renin–angiotensin system–related gene expression in the kidney.
Supplemental Figure 4. Mouse complete blood count.
Supplemental Figure 5. Flow cytometry analysis of hematopoietic progenitor cells of bone marrow and the life span of erythrocytes.
Supplemental Figure 6. Mouse kidney glycolysis, TCA cycle, and PPP pathway heatmap.
Supplemental Figure 7. GSH, GSSG, GSSG/GSH, and ATP levels in mouse kidney.
Supplemental Figure 8. Mouse plasma, kidney, and urine amino acids heatmap.
Supplemental Figure 9. Labeled adenosine in supernatant.
Supplemental Figure 10. Mouse complete blood count.
Supplemental Figure 11. Renin–angiotensin system–related gene expression in the kidney.
Supplemental Figure 12. Mouse erythrocyte AMPD3 activity with or without Ang II under normoxia and hypoxia.
Supplemental Figure 13. Mouse and human erythrocyte ROS level, AMPD3 activity, and AMPKα activity with different concentration of H2O2 treatment.
Supplemental Table 1. Labeled metabolites identified in wild type mouse red blood cells.
Supplemental Table 2. Complete blood count of healthy controls and patients with CKD.
Supplemental Dataset 1. Metabolites identified in mouse red blood cells, plasma, kidney, and urine (Excel File).
Supplemental Dataset 2. Metabolites identified in mouse red blood cells, plasma, and kidney (Excel File).
References
- 1.Lv JC, Zhang LX. Prevalence and disease burden of chronic kidney disease. Adv Exp Med Biol. 2019;1165:3–15. doi: 10.1007/978-981-13-8871-2_1 [DOI] [PubMed] [Google Scholar]
- 2.Ren Z, Fan Y, Li A, Shen Q, Wu J, Ren L. Alterations of the human gut microbiome in chronic kidney disease. Adv Sci (Weinh). 2020;7(20):2001936. doi: 10.1002/advs.202001936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalantar-Zadeh K, Jafar TH, Nitsch D, Neuen BL, Perkovic V. Chronic kidney disease. Lancet. 2021;398(10302):786–802. doi: 10.1016/s0140-6736(21)00519-5 [DOI] [PubMed] [Google Scholar]
- 4.Klinkhammer BM, Lammers T, Mottaghy FM, Kiessling F, Floege J, Boor P. Non-invasive molecular imaging of kidney diseases. Nat Rev Nephrol. 2021;17(10):688–703. doi: 10.1038/s41581-021-00440-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fine LG, Orphanides C, Norman JT. Progressive renal disease: the chronic hypoxia hypothesis. Kidney Int Suppl. 1998;65:S74–S78. PMID: 9551436. [PubMed] [Google Scholar]
- 6.Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int. 2008;74(7):867–872. doi: 10.1038/ki.2008.350 [DOI] [PubMed] [Google Scholar]
- 7.Zhang W, Zhang Y, Wang W, Dai Y, Ning C, Luo R. Elevated ecto-5'-nucleotidase-mediated increased renal adenosine signaling via A2B adenosine receptor contributes to chronic hypertension. Circ Res. 2013;112(11):1466–1478. doi: 10.1161/circresaha.111.300166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension. 2012;59(1):136–144. doi: 10.1161/hypertensionaha.111.173328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai Y, Zhang W, Wen J, Zhang Y, Kellems RE, Xia Y. A2B adenosine receptor-mediated induction of IL-6 promotes CKD. J Am Soc Nephrol. 2011;22(5):890–901. doi: 10.1681/ASN.2010080890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brewer GJ. 2,3-DPG and erythrocyte oxygen affinity. Annu Rev Med. 1974;25(1):29–38. doi: 10.1146/annurev.me.25.020174.000333 [DOI] [PubMed] [Google Scholar]
- 11.Valeri CR, Yarnoz M, Vecchione JJ, Dennis RC, Anastasi J, Valeri DA. Improved oxygen delivery to the myocardium during hypothermia by perfusion with 2,3 DPG-enriched red blood cells. Ann Thorac Surg. 1980;30(6):527–535. doi: 10.1016/s0003-4975(10)61725-0 [DOI] [PubMed] [Google Scholar]
- 12.Finch CA, Lenfant C. Oxygen transport in man. N Engl J Med. 1972;286(8):407–415. doi: 10.1056/nejm197202242860806 [DOI] [PubMed] [Google Scholar]
- 13.Morpurgo G, Battaglia P, Carter ND, Modiano G, Passi S. The Bohr effect and the red cell 2-3 DPG and Hb content in Sherpas and Europeans at low and at high altitude. Experientia. 1972;28(11):1280–1283. doi: 10.1007/bf01965295 [DOI] [PubMed] [Google Scholar]
- 14.Subudhi AW, Bourdillon N, Bucher J, Davis C, Elliott JE, Eutermoster M. AltitudeOmics: the integrative physiology of human acclimatization to hypobaric hypoxia and its retention upon reascent. PLoS One. 2014;9(3):e92191. doi: 10.1371/journal.pone.0092191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woodson RD, Torrance JD, Shappell SD, Lenfant C. The effect of cardiac disease on hemoglobin-oxygen binding. J Clin Invest. 1970;49(7):1349–1356. doi: 10.1172/JCI106351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bersin RM, Kwasman M, Lau D, Klinski C, Tanaka K, Khorrami P. Importance of oxygen-haemoglobin binding to oxygen transport in congestive heart failure. Heart. 1993;70(5):443–447. doi: 10.1136/hrt.70.5.443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng Z, Luo R, Xie T, Zhang W, Liu H, Wang W. Erythrocyte adenosine A2B receptor-mediated AMPK activation: a missing component counteracting CKD by promoting oxygen delivery. J Am Soc Nephrol. 2019;30(8):1413–1424. doi: 10.1681/ASN.2018080862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu H, Zhang Y, Wu H, D'Alessandro A, Yegutkin GG, Song A. Beneficial role of erythrocyte adenosine A2B receptor-mediated AMP-activated protein kinase activation in high-altitude hypoxia. Circulation. 2016;134(5):405–421. doi: 10.1161/circulationaha.116.021311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song A, Zhang Y, Han L, Yegutkin GG, Liu H, Sun K. Erythrocytes retain hypoxic adenosine response for faster acclimatization upon re-ascent. Nat Commun. 2017;8(1):14108. doi: 10.1038/ncomms14108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sayama S, Song A, Brown BC, Couturier J, Cai X, Xu P. Maternal erythrocyte ENT1-mediated AMPK activation counteracts placental hypoxia and supports fetal growth. JCI Insight. 2020;5(10):e130205. doi: 10.1172/jci.insight.130205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sasaki R, Ikura K, Katsura S, Chiba H. Regulation of human erythrocyte AMP deaminase by ATP and 2,3-bisphosphoglycerate. Agric Biol Chem. 1976;40(9):1797–1803. doi: 10.1080/00021369.1976.10862294 [DOI] [Google Scholar]
- 22.Heinrich AC, Pelanda R, Klingmüller U. A mouse model for visualization and conditional mutations in the erythroid lineage. Blood. 2004;104(3):659–666. doi: 10.1182/blood-2003-05-1442 [DOI] [PubMed] [Google Scholar]
- 23.Daniels IS, O′Brien WG, Nath V, Zhao Z, Lee CC. AMP deaminase 3 deficiency enhanced 5'-AMP induction of hypometabolism. PLoS One. 2013;8(9):e75418. doi: 10.1371/journal.pone.0075418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu P, Chen C, Zhang Y, Dzieciatkowska M, Brown BC, Zhang W. Erythrocyte transglutaminase-2 combats hypoxia and chronic kidney disease by promoting oxygen delivery and carnitine homeostasis. Cell Metab. 2022;34(2):299–316.e6. doi: 10.1016/j.cmet.2021.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie T, Chen C, Peng Z, Brown BC, Reisz JA, Xu P. Erythrocyte metabolic reprogramming by sphingosine 1-phosphate in chronic kidney disease and therapies. Circ Res. 2020;127(3):360–375. doi: 10.1161/circresaha.119.316298 [DOI] [PubMed] [Google Scholar]
- 26.Issaian A, Hay A, Dzieciatkowska M, Roberti D, Perrotta S, Darula Z. The interactome of the N-terminus of band 3 regulates red blood cell metabolism and storage quality. Haematologica. 2021;106(11):2971–2985. doi: 10.3324/haematol.2020.278252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stefanoni D, Shin HKH, Baek JH, Champagne DP, Nemkov T, Thomas T. Red blood cell metabolism in Rhesus macaques and humans: comparative biology of blood storage. Haematologica. 2020;105(8):2174–2186. doi: 10.3324/haematol.2019.229930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nemkov T, Stefanoni D, Bordbar A, Issaian A, Palsson BO, Dumont LJ. Blood donor exposome and impact of common drugs on red blood cell metabolism. JCI Insight. 2021;6(3):e146175. doi: 10.1172/jci.insight.146175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nemkov T, Reisz JA, Gehrke S, Hansen KC, D'Alessandro A. High-throughput metabolomics: isocratic and gradient mass spectrometry-based methods. Methods Mol Biol. 2019:1978:13–26. doi: 10.1007/978-1-4939-9236-2_2 [DOI] [PubMed] [Google Scholar]
- 30.Pertoft H, Laurent TC, Låås T, Kågedal L. Density gradients prepared from colloidal silica particles coated by polyvinylpyrrolidone (Percoll). Anal Biochem. 1978;88(1):271–282. doi: 10.1016/0003-2697(78)90419-0 [DOI] [PubMed] [Google Scholar]
- 31.Clasquin MF, Melamud E, Rabinowitz JD. LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr Protoc Bioinformatics. 2012;Chapter 14:Unit14.11. doi: 10.1002/0471250953.bi1411s37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaney AL, Marbach EP. Modified reagents for determination of urea and ammonia. Clin Chem. 1962;8(2):130–132. doi: 10.1093/clinchem/8.2.130 [DOI] [PubMed] [Google Scholar]
- 33.Cicerchi C, Li N, Kratzer J, Garcia G, Roncal-Jimenez CA, Tanabe K. Uric acid-dependent inhibition of AMP kinase induces hepatic glucose production in diabetes and starvation: evolutionary implications of the uricase loss in hominids. FASEB J. 2014;28(8):3339–3350. doi: 10.1096/fj.13-243634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Dai Y, Wen J, Zhang W, Grenz A, Sun H. Detrimental effects of adenosine signaling in sickle cell disease. Nat Med. 2011;17(1):79–86. doi: 10.1038/nm.2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mikdar M, González-Menéndez P, Cai X, Zhang Y, Serra M, Dembele AK. The equilibrative nucleoside transporter ENT1 is critical for nucleotide homeostasis and optimal erythropoiesis. Blood. 2021;137(25):3548–3562. doi: 10.1182/blood.2020007281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Berka V, Song A, Sun K, Wang W, Zhang W. Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. J Clin Invest. 2014;124(6):2750–2761. doi: 10.1172/jci74604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo R, Zhang W, Zhao C, Zhang Y, Wu H, Jin J. Elevated endothelial hypoxia-inducible factor-1α contributes to glomerular injury and promotes hypertensive chronic kidney disease. Hypertension. 2015;66(1):75–84. doi: 10.1161/hypertensionaha.115.05578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zwifelhofer NM, Cai X, Liao R, Mao B, Conn DJ, Mehta C. GATA factor-regulated solute carrier ensemble reveals a nucleoside transporter-dependent differentiation mechanism. PLoS Genet. 2020;16(12):e1009286. doi: 10.1371/journal.pgen.1009286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Everds NE. Chapter 5—Hematology of the Laboratory mouse. In: Fox JG, Davisson MT, Quimby FW, Barthold SW, Newcomer CE, Smith AL, editors. The Mouse in Biomedical Research, 2nd Edition. Academic Press; 2007:133–XVIII. [Google Scholar]
- 40.O’Brien III WG, Berka V, Tsai AL, Zhao Z, Lee CC. CD73 and AMPD3 deficiency enhance metabolic performance via erythrocyte ATP that decreases hemoglobin oxygen affinity. Sci Rep. 2015;5(1):13147. doi: 10.1038/srep13147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lian CY, Harkness DR. The kinetic properties of adenylate deaminase from human erythrocytes. Biochim Biophys Acta. 1974;341(1):27–40. doi: 10.1016/0005-2744(74)90062-x [DOI] [PubMed] [Google Scholar]
- 42.Tavazzi B, Amorini AM, Fazzina G, Di Pierro D, Tuttobene M, Giardina B. Oxidative stress induces impairment of human erythrocyte energy metabolism through the oxygen radical-mediated direct activation of AMP-deaminase. J Biol Chem. 2001;276(51):48083–48092. doi: 10.1074/jbc.M101715200 [DOI] [PubMed] [Google Scholar]
- 43.Nemkov T, Sun K, Reisz JA, Song A, Yoshida T, Dunham A. Hypoxia modulates the purine salvage pathway and decreases red blood cell and supernatant levels of hypoxanthine during refrigerated storage. Haematologica. 2018;103(2):361–372. doi: 10.3324/haematol.2017.178608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldman N, Chen M, Fujita T, Xu Q, Peng W, Liu W. Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture. Nat Neurosci. 2010;13(7):883–888. doi: 10.1038/nn.2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5'-nucleotidase (CD73). Purinergic Signal. 2006;2(2):351–360. doi: 10.1007/s11302-005-5302-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Xia Y. Adenosine signaling in normal and sickle erythrocytes and beyond. Microbes Infect. 2012;14(10):863–873. doi: 10.1016/j.micinf.2012.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53(4):527–552. PMID: 11734617. [PMC free article] [PubMed] [Google Scholar]
- 48.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev. 2011;63(1):1–34. doi: 10.1124/pr.110.003285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509(7500):310–317. doi: 10.1038/nature13085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468(7324):653–658. doi: 10.1038/nature09571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conway EJ, Cooke R. The deaminases of adenosine and adenylic acid in blood and tissues. Biochem J. 1939;33(4):479–492. doi: 10.1042/bj0330479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller SG, Hafen PS, Law AS, Springer CB, Logsdon DL, O'Connell TM. AMP deamination is sufficient to replicate an atrophy-like metabolic phenotype in skeletal muscle. Metabolism. 2021;123:154864. doi: 10.1016/j.metabol.2021.154864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee S, Wang Y, Kim SO, Han J. AMPD3 is involved in anthrax LeTx-induced macrophage cell death. Protein Cell. 2011;2(7):564–572. doi: 10.1007/s13238-011-1078-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davis PR, Miller SG, Verhoeven NA, Morgan JS, Tulis DA, Witczak CA. Increased AMP deaminase activity decreases ATP content and slows protein degradation in cultured skeletal muscle. Metabolism. 2020;108:154257. doi: 10.1016/j.metabol.2020.154257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hsu CM, Chang SF, Tsai YT, Tsai MS, Chang GH, Chen HC. Down-regulation of AMPD3 is associated with poor survival in head and neck squamous cell carcinoma. In Vivo. 2022;36(2):704–712. doi: 10.21873/invivo.12756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong M, Funasaka K, Obayashi T, Miyahara R, Hirooka Y, Hamaguchi M. AMPD3 is associated with the malignant characteristics of gastrointestinal stromal tumors. Oncol Lett. 2017;13(3):1281–1287. doi: 10.3892/ol.2016.5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng J, Morisaki H, Toyama K, Ikawa M, Okabe M, Morisaki T. AMPD3-deficient mice exhibit increased erythrocyte ATP levels but anemia not improved due to PK deficiency. Genes Cells. 2012;17(11):913–922. doi: 10.1111/gtc.12006 [DOI] [PubMed] [Google Scholar]