

Graphical abstract
Keywords: NGS reproducibility, NGS data quality, NGT (New Genomic Techniques), GE (Gene Editing), GMO (Genetically Modified Organism), Detection
Highlights
-
•
Targeted Next-Generation-Sequencing (NGS) data reproducibility is very high, even between independent external service providers, if a sufficient amount of reads is provided.
-
•
PacBio HiFi long read technology, producing reads of 15 kb length, significantly improves targeted sequencing in a complex genome structure (e.g. numerous highly similar DNA stretches).
-
•
PacBio HiFi long read Whole Genome Sequencing is not suited for standard workflows in a regulatory setting due to a limited reproducibility caused by a high inter-laboratory variance.
-
•
An oilseed rape (OSR) gene edited genetically modified organism (GE GMO) DNA admixture of 0.1% (copy/copy) is well detectable in a wild type OSR DNA mixture.
Abstract
Next-generation-sequencing (NGS) becomes increasingly important for laboratories tasked with the detection of genetically modified organisms (GMOs) in food, feed and seeds. Its implementation into standardized workflows demands reliable intra- and inter-laboratory reproducibility. Here, we analyze the reproducibility of short- and long-read targeted NGS and long-read whole genome sequencing (WGS) data between three independent laboratories. Replicate samples were submitted for sequencing and comparatively analyzed. The targeted-NGS-samples consisted of oil seed rape (OSR) sampled from a commodity shipment spiked with a genome edited (GE) OSR and the WGS-samples consisted of leaf material from the GMOs’ parental line. All laboratories delivered highly reproducible high-quality targeted NGS data with little variation. The detection of GMO-related sequences works well regardless of the facility, while the mapping to the complex genome is superior using long read data. Long read WGS is currently not suitable for routine use in enforcement laboratories, due to a large inter-laboratory variation.
1. Introduction
Genetic modifications (GM) resulting from “classical” genetic engineering are detectable using well established methods like PCR-based systems (Holst-Jensen et al., 2012) as the stably integrated transgenes and constructs make them mostly unique and thus identifiable. Genome editing techniques (also known as New Genomic Techniques (NGTs)) like CRISPR/Cas (CRISPR), Transcription Activator-Like Effector Nuclease (TALEN) and others (European Commission. Joint Research Centre., 2021), have changed the scenario for detection considerably (Bortesi and Fischer, 2015, Grohmann et al., 2019). NGTs allow for relatively precise changes of the host’s genetic material, which can impact single bases, potentially not leaving obvious traces of their use (Bessoltane et al., 2022, Martínez-Fortún et al., 2022). The detection of small changes, like single nucleotide variants (SNVs), while still possible with established PCR systems, ideally utilizes technologies with a much higher resolution. A group of technologies, which is able to precisely decode nucleic acid sequences with base level resolution and at very high throughput, is collectively called Next Generation Sequencing (NGS). Due to its continually decreasing cost per sequenced base, NGS promises the possibility to screen for numerous GM base changes and line-specific markers simultaneously. It is thus increasingly becoming an important tool for the detection, identification and possibly quantification of genetically modified organisms (GMOs) in general (Arulandhu et al., 2016, Chen et al., 2021, Debode et al., 2019, Fraiture et al., 2023, Fraiture et al., 2017, Fraiture et al., 2017, Fraiture et al., 2018, Grohmann et al., 2019, Košir et al., 2017, Saltykova et al., 2022, Wahler et al., 2013).
In an official inspection setting, along with other performance parameters the robustness and reproducibility of a method, both inter- and intra-laboratory, are crucial for the acceptance of the validity of its results (European Network of GMO Laboratories (ENGL), 2015). Most standard methods applied in official GMO inspection and control in the European Union (EU) rely on PCR analyses (Regulation No. 641/2004). PCR-based methods, however, can no longer be the only technology to be used, particularly for the detection and identification of GMOs developed by NGTs (GE GMO) (Fraiture et al., 2023, Grohmann et al., 2019). Other methodologies are needed in the routine processes of control laboratories, empowering them to fulfil their obligations. NGS, with its high resolution and generalized character, is uniquely suited for these demands because of its enhanced informative analytical output. However, limiting factors for a broad dissemination of this approach are the NGS capacities and downstream bioinformatics analysis at official control laboratories. NGS capacities can be broadened, if purchased from external NGS service providers. Little experience exists regarding the quality of NGS data received from external service providers. To enlighten this step in the analysis workflow, we designed a study to assess the intra- and inter-laboratory reproducibility of NGS data from different sequencing facilities and different NGS platforms.
We focus on three NGS applications, which are of importance for the challenging GE GMO analyses:
-
i.
Targeted short read sequencing using a sequencing-by-synthesis approach: It is the most common application. Specific DNA areas of interest are enriched using different methods (e.g. target-specific biotinylated probes, PCR). The enriched DNA segments around the specific SNV or InDel present in the GE GMO are sequenced. This approach is limited to comparatively short sequences of up to 2x300 bp (paired-end).
-
ii.
Targeted long read sequencing based on a synthesis-by-binding approach: Longer read sizes (up to 15 kb) and thus larger areas are analyzed without the need for read assembly. This is of particular interest if, for instance, the SNV is in a repetitive region or neighboring SNVs are sequenced to help identify the GE GMO.
-
iii.
Whole genome sequencing (WGS) using a sequencing-by-binding system as above: Here, the complete DNA of a sample is sequenced. It offers the advantage of long reads spanning regions of low complexity or repetitive regions in a genome, which would cause problems while assembling short reads. It is a possible NGS application for the molecular characterization of an unknown GE GMO or the screening for GM markers without a priori knowledge of the modification.
In order to resemble a real-life situation and to test the applicability of NGS in routine GMO analysis, we combined this study with a GE GMO detection and quantitative estimation with two different GE GMO content levels (1% and 0.1%) in the short and long read targeted NGS. To do so, we spiked a GE GMO oilseed rape (OSR; Brassica napus) into a B. napus sample taken from a commercial shipment at a port of entry. The WGS analysis utilized a wild type OSR variety. OSR has a complex genome, often resulting in complications when running GMO analyses. It is an amphidiploid species (2n = 38, AACC genome) derived from the hybridization of the two closely related species Brassica rapa (contributing the AA genome) and Brassica oleracea (CC). Several alleles of each gene are typically present, with a very high sequence identity of the A- and C-gene copies (Braatz et al., 2017). The GE GMO used in this study has a small heterozygous modification (1 bp insertion, 3 bp deletion) in one of its four CRT1A (calreticulin) alleles resulting in the loss of function of the CRT1A gene. The sequence similarity in the four different CRT1A loci can cause false mappings of short reads, making this a very good example case for the comparison of long read and short read targeted sequencing. Running WGS on the parent variety of a GE GMO, producing a specific reference genome to align data from targeted sequencing against, allows the direct comparison of the GE GMO to its origin rather than the publicly available reference genome of a different variety.
2. Materials and methods
2.1. Plant material
Three different plant materials were used in this work: i) a seed sample from wild type (wt) B. napus provided by the Hamburg Institute for Hygiene and Environment (HU) from a standard sampling at a port of entry performed in 2015. Since the sample was taken from a commodity shipment it must be considered genetically heterogeneous; ii) frozen leaf material of GE GMO C3E4; and iii) freshly frozen leaf material of B. napus wt variety Mozart, collected from eight individual plants. C3E4 is based on the variety Mozart and genetically modified using the CRISPR technology (Pröbsting et al., 2020). Leaf material was kindly provided by the Christian-Albrechts-University Kiel (CAU).
2.2. Targeted NGS
36 spiked samples with mixtures of DNA of the seed sample collected by HU (wt) and DNA from the GE GMO C3E4 were prepared. First, two stock mixtures with specified ratios of unmodified and modified target sequence copies (cp) were prepared, one with 0.1% (cp/cp) and the other with 1.0% GMO spike (cp/cp) (see supplementary materials for sample preparation details). Each mixture was divided into two sets, each set comprising of 18 replicate samples. One of these sets was used to construct short amplicons (∼250 bp) while the other was used to construct long amplicons (∼3 kb) around the genetic modification with the corresponding primer combination (Table 1). Three randomly selected samples of each of the sets were given to three independent NGS service providers for sequencing (Fig. 1). Very few specifications were imposed to the laboratories other than that the short amplicons were to be sequenced with an Illumina instrument using either 250 bp or 300 bp paired-end sequencing (Zhang et al., 2022) and the long amplicons using the Pacific Bioscience (PacBio) Sequel II system producing high fidelity (HiFi) circular consensus sequence (CCS) reads in the range of about 3,000 bp. This was deliberate to allow each laboratory to utilize their respective standard and well established workflows. The protocols used by the appointed laboratories are unknown to the authors as is the standard case in outsourced sequencing.
Table 1.
The primers for amplifications as designed by CAU and the corresponding target regions within the four homoeologous alleles of CRT1A gene in the B. napus genome (GCA_020379485.1) (Chr. = chromosome) are listed. Lowercase letters represent mismatches between the reference and the C01 and A01 regions. Regions flanked by primers without nucleotide mismatches are shown in bold.
| Application | Primer |
Targets |
|||
|---|---|---|---|---|---|
| Direction | Sequence | Chr. | Chr.-Position | Insert Size | |
| Short Amplicon |
Forward | TGACAACTAGATaTGACGTGTA | A01 | 17,891,589–17,891,824 | 236 bp |
| Reverse | CCTCGAAGATAACACTAGCAG | A09 | 12,023,963–12,024,221 | 259 bp | |
| C01 | 29,845,940–29,846,188 | 249 bp | |||
| C09 | 18,586,605–18,586,863 | 259 bp | |||
| Long Amplicon |
Forward | TGACAACTAGATaTGACGTGTA | A01 | 17,889,194–17,891,824 | 2,631 bp |
| Reverse | TTTATCAAGTCTAAAAcAAGCTG | A09 | 12,023,963–12,029,170 | 5,208 bp | |
| C01 | 29,843,335–29,846,188 | 2,854 bp | |||
| C09 | 18,586,605–18,589,560 | 2,956 bp | |||
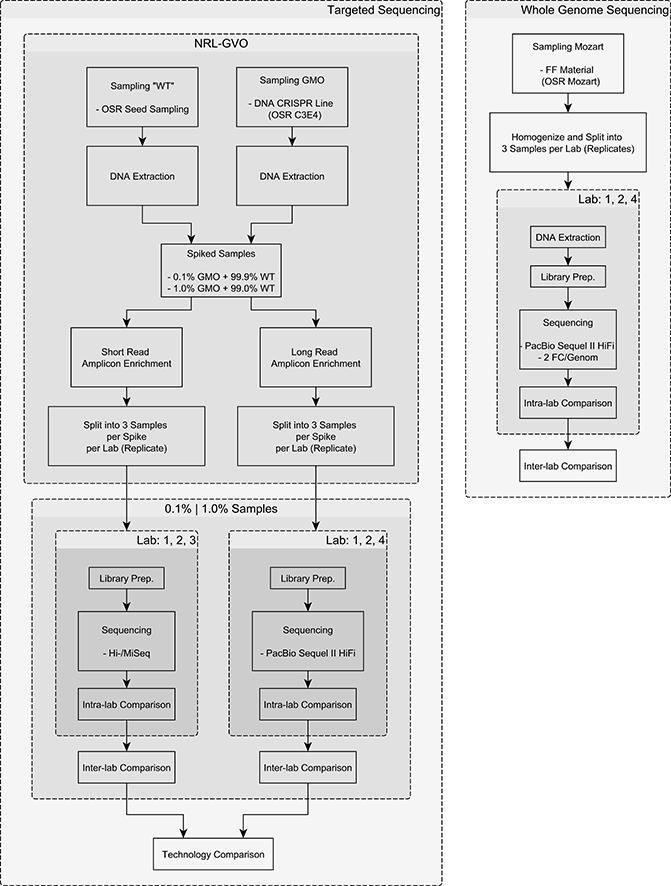
Fig. 1.

Flowchart of the NGS experiments.
The individual laboratories (L1, L2 and L3) had different requirements regarding the sample amount of genomic DNA for the short read sequencing (supplementary Table 5), with L2 requiring the largest amount (around 3 µg) of DNA, up to ten times more than L1 and six times more than L3. The sample specifications for the long read sequencing (supplementary Table 6) were equal across all sequencing laboratories (L1, L2 and L4).
2.3. WGS
The leaf material from eight non-GE GMO Mozart individual plants was homogenized and portioned into nine replicate samples. Each sequencing laboratory received three randomly selected test portions and was instructed to treat each sample independently (Fig. 1). The three laboratories (L1, L2 and L4) were instructed to use the PacBio Sequel II and deliver HiFi-CCS reads. These reads have a target length of about 15 kb and, due to a repeated sampling (at least 10 times) of the same circularized DNA molecule, are of high quality. This high read length requires the extraction of high molecular weight DNA with fragments of at least 40 kb. As is the case with the targeted NGS, very few specifications were imposed on the performing laboratories in order to allow them the use of their respective well-established workflows, other than that a sufficient amount of reads had to be provided to reach a coverage of the sample genome of at least 25×.
The sample specifications are listed in supplementary Table 7 and show that L4 required more than twice the amount of plant material compared to the other two laboratories.
2.4. Data analyses
The data analysis focuses on two parts (see supplementary materials for more information including references). In the first part general per sample metrics are collected, namely the read amount, the read length and the read quality per delivered sample, respectively. Further metrics for the targeted NGS are the alignment rate and the subsequent rate of reads mapped to the targeted area, allowing for the detection of reads showing the GMO specific variants. An additional measure for the evenness of the coverage is also included. For the WGS application further analyses are based on assembly measures (e.g. N50, L50) followed by a measure of completeness of the assemblies. This information is given in the supplementary materials (Table 8 through 10). The second part of the analyses compares the afore gathered measures of single samples and groups of samples between one another. For this purpose, statistical tests are used comparing groups (i.e. samples per laboratory) using a MANOVA test and single samples between one another using multiple t-tests with Bonferroni correction for multiple testing. The cut-off for statistical significance was chosen to be p-value <0.05. In order to visualize the principal characteristics of the data sets and their relation to one another a PCA analysis shows the nine (targeted approach) and eight (WGS approach) dimensional samples projected onto two dimensions. An overview of the definitions of the used measures can be found in the supplementary information.
3. Results and discussion
All three NGS service providers delivered NGS data suitable for the intra- and inter-laboratory comparison.
3.1. Targeted NGS analyses of OSR DNA samples spiked with DNA of GE GMO event C3E4
For this NGS application, a real-life situation with traces of a GE GMO in a large shipment of seeds is resembled, by using DNA extracted from the wt sample provided and negatively tested for known GMO traces and spiked with low levels of the DNA extracted from GE GMO C3E4. C3E4 possesses a heterozygous modification on chromosome A09, either a 3 bp deletion at base position 12,024,160 or a 1 bp insertion at base position 12,024,161, respectively.
3.1.1. Comparison of amplicon short read data sets based on sequencing-by-synthesis (Illumina)
In this section, the data sets of all three NGS service providers from the short read paired end sequencing of the short amplicons are compared. The amplicons were generated based on primers that target CRT1A regions in the amphidiploid B. napus genome (GCA_020379485.1; Table 1). The forward primer has a one base pair mismatch in the 01 chromosomes. The targeted regions share a high similarity of up to over 98% (Table 2). This high similarity results in identically sized amplicons in A09 and C09 with 259 bp, while the amplicon sizes in C01 and A01, being less similar, are smaller with 236 bp and 249 bp, respectively. The locus of genetic modification (GMO locus) is located 197 nucleotides upstream of the 3′ end of the amplicons.
Table 2.
The sequence similarity of the short read and long read targets in percent, respectively. The target region within which the GMO modification is located is shown in bold.
| Short Read Targets |
Long Read Targets |
|||||||
|---|---|---|---|---|---|---|---|---|
| Target Chr. | A09 | C09 | C01 | A01 | A09 | C09 | C01 | A01 |
| A09 | 100.00 | 98.46 | 81.70 | 81.58 | 100.00 | 73.96 | 65.42 | 64.58 |
| C09 | 100.00 | 81.28 | 81.14 | 100.00 | 77.44 | 76.65 | ||
| C01 | 100.00 | 95.22 | 100.00 | 93.73 | ||||
| A01 | 100.00 | 100.00 | ||||||
3.1.1.1. Evaluation and comparison of delivered sequence data
The part 1 analyses resulted in the following per sample metrics (supplementary Table 8). L1 and L2 provided similar amounts of reads of around 450,000 reads per sample, while L3 provided significantly fewer reads (150,000 reads per sample). Furthermore, L1 delivered strongly varying read amounts for the three samples, while the read amounts from L2 and L3 are much more even. A theoretical read length of two times 250 bp or two times 300 bp is possible based on the NGS system used. A read length between 236 bp and 259 bp is expected based on the amplicon design (Table 1). L2 and L3 delivered reads within the expected length range. L1, however, delivered reads nearly half the expected size (120 bp); this remarkable difference might be due to different library preparation approaches used. The length is very even among the samples per laboratory. The quality per laboratory is evenly high for all laboratories (Phred between 41 and 42) (Petrackova et al., 2019). The highest read quality was reached by L1 while the lowest and most even quality was achieved by L2. The absolute alignment rate is very high for all laboratories (between 95% and 100% of all reads could be aligned, at least once). This is a clear sign that little to no contamination with foreign material has occurred. L2 and L1 show consistently high rates (99.9% and 97.3% respectively), while L3 is slightly lower with less consistency across its samples (∼95.2%).
The distribution of aligned reads across the target regions is similar among the laboratories (Fig. 2B). Nearly 100% of all aligned reads fall evenly within the target regions (Table 1). Between 20% and 30% of reads align against chromosome A09, which closely resembles the expected even distribution of reads among the four targets. In the L2 and L3 sample sets no significant difference between the count of reads aligned to A09 and the count of reads spanning the GMO locus can be detected. This is not the case in the L1 sample set, where significantly fewer reads span the GMO locus than those aligning to A09 (21% to A09 and 15% spanning the GMO locus). This is to be expected, considering the read lengths in the L2 and L3 sets were approximately the same size as the amplicons targeted, however, the reads from L1 are significantly shorter than the target amplicons, resulting in a number of reads not spanning the GMO locus, even though they do align to chromosome A09.
Fig. 2.

Shows the post alignment results of the short read (B and C) and long read (D and E) data sets, respectively. (A) depicts the read groupings for the subsequent analyses, starting with the total aligned reads (dark blue), to the subgroup of reads aligned to the targeted regions (light blue) and finally the reads spanning the GMO locus (green) within the group of reads aligning to chromosome A09 (orange). (B) and (D) show the alignment distribution of the short read and long read data sets, respectively. Showing the relative amount of reads aligning to the target regions, aligning to A09 within the target region and spanning the GMO locus per laboratory respectively as boxplots. (C) and (E) show the percentage of reads with a GMO modification versus all reads spanning the GMO locus using 0.1% spiked samples and 1.0% spiked samples per laboratory respectively as boxplots. The red dotted lines show the targeted spike (0.1% and 1.0% of all reads spanning the GMO locus). The boxplots show, from lowest to highest bar, the minimum, the 25th percentile, the median, the 75th percentile and maximum values per sample set respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The observed median coverage, i.e. the actual read count mapped to a given reference base, comes close to the expected median coverage with a maximum divergence of 8%. Looking at the coverage inter quartile range (IQR), the coverage evenness varies significantly among the laboratories. Broadly speaking L2 and L3 provide a high and even coverage, while the coverage provided by L1 seems to be normally distributed, suggesting an uneven coverage with most bases having a medium coverage (supplementary Fig. 4).
The theoretical required minimum read count (RMRC) is, as shown in detail in the supplementary material, 120,000 reads per sample. Only L3 is close to this expected value (130,000). L1, due to the significant differences in coverage and alignment ratio (i.e. the amount of reads spanning the GMO locus relative to the total aligned reads), needs to provide more reads (200,000), since fewer reads over all cover the GMO locus. L2 needs to deliver fewer reads than expected (100,000). L2 shows a relatively high even coverage. However, due to the way the RMRC is calculated (see formula in supplementary materials), having a lower than expected RMRC can only be achieved when the targeted regions are not covered equally, i.e. A09 and C09 have a higher coverage than C01 and A01 (30% of aligned reads map to A09 rather than approximately 25%). Each laboratory, on average, exceeded its corresponding minimum read count (L1: 469,922; L2: 475,669.50; L3: 174,439.67) and provided a sufficient amount of very high quality reads to theoretically detect a 0.1% GMO spike with at least 30 reads. It is remarkable, that L1 needs to deliver more than twice as many reads to reach the RMRC for GMO detection compared to L2. This is caused by the apparent “read loss” between reads aligning to A09 and those spanning the GMO locus, due to the lower read length and the resulting coverage distribution in L1 samples.
The results for the GMO detection (Fig. 2C) are very similar across all samples, independent of the laboratory. However, regarding relative quantification of the GMO percentage, there is a significant bias between the admixed relative content (0.1% and 1.0%) and the actual observed percentages (∼0.07% and ∼ 0.65%). Both the 0.1% level and the 1.0% level were underestimated, with similar relative biases from the expected values. Considering the short read length and relative similarity of the CRT1A loci at the four target regions on A01, A09, C01 and C09, this might be due to some reads being aligned erroneously and thus not being counted during the analysis of the specific GMO locus. This is further underlined by the fact that all spiked samples show a smaller than expected number of GMO reads.
The part 2 comparison analyses were done on the basis of the calculated per-sample metrics as categorical data.
Three important information can be drawn from the comparison of short read analyses (Table 3):
-
1.
The three laboratories are significantly different in most categories, except coverage ratio, and coverage IQR, where only one laboratory diverges significantly, and the GMO detection, where no laboratory diverges significantly.
-
2.
The intra-laboratory reproducibility is very high as can be seen in the principal component analysis (PCA) (Fig. 3A). This results in significant p-values between laboratories even when the apparent difference between the laboratories is very small, e.g. in read quality (41.9 and 41.3) and in the alignment rate (99.9% and 97.2%).
-
3.
The laboratories perform equally well regarding GMO detection. With a p-value greater than 0.05, no significant inter-laboratory difference could be found, although the laboratories are significantly different in most other categories.
Table 3.
P-values calculated by statistical testing on the short read and long read data sets, respectively. MANOVA tests were run all against all, while the t-tests where run one against one utilizing Bonferroni compensation for multiple testing. Values where no significant p-value was calculated (p < 0.05) are shown in bold.
| Category | Short Read |
Long Read |
||||||
|---|---|---|---|---|---|---|---|---|
| MANOVA |
t-Test |
MANOVA |
t-Test |
|||||
| All vs. All | L3 vs. L2 | L1 vs. L2 | L1 vs. L3 | All vs. All | L4 vs. L2 | L1 vs. L2 | L1 vs. L4 | |
| Read Length | 1.2 e−21 | 2.8 e−03 | 4.4 e−21 | 9.3 e−21 | 5.5 e−05 | 3.1 e−03 | 1.3e−01 | 4.5 e−05 |
| Read Quality | 4.4 e−24 | 8.9 e−22 | 3.9 e−24 | 2.1 e−16 | 1.5 e−21 | 2.7 e−16 | 4.6 e−18 | 9.8 e−22 |
| Alignment Rate | 3.6 e−13 | 2.4 e−13 | 3.5 e−10 | 1.2 e−07 | 5.0 e−06 | 1.8 e−05 | 1.8 e−05 | 1.0e+00 |
| Alignment Ratio | 6.3 e−18 | 1.8 e−13 | 3.9 e−18 | 7.3 e−14 | 1.1 e−03 | 1.1 e−02 | 8.6 e−01 | 1.2 e−03 |
| Coverage Ratio | 4.6 e−08 | 1.1 e−07 | 4.2 e−07 | 9.5 e−01 | 6.3 e−01 | 1.0 e+00 | 1.0 e+00 | 1.0 e+00 |
| Coverage IQR | 2.2 e−04 | 4.4 e−01 | 4.5 e−03 | 2.2 e−04 | 3.0 e−09 | 1.4 e−07 | 2.1 e−02 | 3.5 e−09 |
| RMRC | 4.7 e−15 | 4.7 e−07 | 4.7 e−15 | 5.2 e−13 | 1.2 e−03 | 1.2 e−02 | 9.0 e−01 | 1.3e−03 |
|
GMO 0.1% Detection |
8.7e−01 | 1.0 e+00 | 1.0 e+00 | 1.0 e+00 | 2.9 e−01 | 4.0 e−01 | 1.0 e+00 | 8.0 e−01 |
|
GMO 1.0% Detection |
2.1 e−01 | 1.0 e+00 | 2.5 e−01 | 8.9 e−01 | 3.0 e−01 | 6.8 e−01 | 4.5 e−01 | 1.0 e+00 |
Fig. 3.

The chart shows principal components 1 vs. 2 of principal component analyses using read length, read quality, alignment rate, alignment ratio, coverage ratio, coverage IQR, required minimum read count, GMO detection for 0.1% and 1.0% per laboratory respectively for the short read data sets (A) and the long read data sets (B), respectively. The important information is the relative distance of a point to any other point; the axes dimensions have no relevance. The individual component proportion is stated in parenthesis in the axes titles.
The PCA was performed on the categorical data (Table 3), and principal components (PC) 1 and 2, jointly accounting for 77% of the data variance, were plotted in Fig. 3A. Distinct point clouds and a relatively tight grouping can be observed, supporting the notion that there is a low intra-laboratory variance and a higher inter-laboratory variance. The tightest grouping can be seen for the L2 samples and the largest spread is visible in the L1 samples. In general, the data sets can be clearly differentiated by laboratory, speaking once more to the high intra-laboratory reproducibility.
3.1.2. Comparison of amplicon long read data sets based on sequencing-by-binding (PacBio)
While technologies are well-established providing longer reads, the drawback has long been a considerably lower read quality and a much higher price per base. The higher error rate in long read technologies has long been limiting their use in many fields where high base quality is paramount (e.g. SNV detection). In order to solve this problem, the “circular consensus sequence” (CCS) system has been developed. The nucleic acid is circularized and this circle is than sequenced multiple times, resulting in multiple reads based on the same molecule. The fundamental idea behind this approach is, that, while any kind of biological abnormality (e.g. SNVs) is static and would therefore be found in every single CCS read, sequencing errors are random and will not be sequenced at the same locus multiple times. CCSs that consist of at least ten reads are HiFi reads and have a very high quality, due to the repeated pass removing many sequencing errors. Using this system read lengths of approximately 15 kb can be achieved (Wenger et al., 2019).
The long amplicons (A01: 2,631 bp, A09: 5,208 bp, C01: 2,854 bp and C09: 2,956 bp) encompass the complete CRT1A genes in the B. napus genome (GCA_020379485.1 (Table 1)). The primers used for amplification have one mismatch to the A01 and C01 chromosome reference sequence (7 and 10 nucleotides 3′ respectively). The target regions on chromosome A01 and C01 are very similar with up to 93% (Table 2), however, the region on A09, where the GMO specific variants are located, is the most dissimilar (<74%), resulting in a significantly larger amplicon. The remarkable difference in the size of the A09 amplicon compared to the other three loci may well be an error in the used assembly. All reads aligning to the A09 target region show a ∼ 2.3 kb gap at the same location. Similarly, reads matching the smallest amplicon size (A01: 2,631 bp) could not be found in any data set, while numerous reads did map to the targeted A01 region, also pointing to a possible error in the reference sequence. These errors, however, have no impact on the analyses, since all alignments (sequence similarity and read alignment) are based on seeded local alignments. The GMO locus is located 197 bases upstream of the 3′ end of the amplicons.
3.1.2.1. Evaluation and comparison of delivered sequence data
The number of delivered HiFi reads (read count) is very heterogeneous between the three laboratories, spanning from about 130,000 (L1) to 630,000 (L4) reads per sample. Most consistent read amounts were reached by L2. A theoretical read length of 3 kb was aimed for. However, this read length was limited by the amplicon size and should be between 2,631 bp and 3,000 bp (Table 1). L1 delivered the shortest reads (2,911), while L2 delivered the longest (2,917), yet most inconsistently sized reads. The reads delivered by L4 showed a very small variance. The absolute difference between the median read length between the laboratories is very small with 6 bp in ∼ 2,915 bp (maximum median difference of 0.2%). The quality is generally very high (Phred 91–93) and consistent within each laboratory. L1 is with around 91.2 slightly below the other two laboratories and the sample set shows a higher variance.
The absolute alignment rate is very high, meaning nearly all reads could be aligned to the reference. Only L2 shows a slightly lower rate of around 98% and is the only laboratory that shows a minor variance among its samples. Regarding the distribution of aligned reads (see Fig. 2D) across the target region, the laboratories show a very similar behavior. Close to 100% of aligned reads can be mapped to either chromosome A09 or C09. About 40% of the aligned reads are located in chromosome A09 and most of those reads also span the GMO locus. The intra-laboratory variance is very small. The aligned reads are not evenly distributed among the four targeted regions (Table 1). This is most likely, at least in part, caused by the primers matching perfectly to AC09 but having one mismatch towards AC01, respectively.
The observed median coverage varies significantly from the expected coverage (observed coverage is almost twice the expected coverage), while the IQR is very large, pointing to an uneven coverage. The coverage spread varies significantly between the three laboratories. While L1 shows an even coverage with either few, medium or very high read counts, the other two laboratories have a significant number of areas with a low coverage (supplementary Fig. 5). Since the four loci are not targeted equally, an uneven coverage of the loci is observable independent of the laboratory. The calculated RMRCs are very different from the expected ones (∼50.000) and very similar between the laboratories. Each laboratory exceeded its corresponding RMRC (L1: 127,661; L2: 526,383; L4: 633,066).
Concerning the GMO detection, the assigned contents are detected by all laboratories, though the 1.0% level is comparatively over-estimated by L1, as is the 0.1% level by L2 (Fig. 2E). Further, the variance between the 1.0% spike samples is remarkably higher than for the 0.1% spike samples. While the median observed for the 1.0% level in the sets sequenced by L2 and L4 matches the assigned 1.0% exactly, a quantitative result cannot be provided with a significant degree of certainty due to the sets variance.
The comparison analyses of the long read sequencing data were done on the basis of the per-sample metrics as categorical data and the following information can be summarized (Table 3):
-
1.
The three laboratories are significantly different in read length, read quality, alignment rate and coverage IQR.
-
2.
The intra-laboratory reproducibility is very high since even when the apparent inter-laboratory difference is very small, as stated above, a significant p-value (smaller 0.05) is calculated.
-
3.
No significant difference could be found for the GMO detection results, and laboratories perform equally well.
A PCA was performed on the categorical data, and PC1 and PC2, jointly accounting for 69% of the data variance, were plotted showing all samples to be well distinguishable (Fig. 3B). The results drawn from L1 samples often show a higher difference from the other two laboratories and the L4 samples are scattered further apart, speaking to a comparatively high intra-laboratory variance.
3.2. Whole genome sequencing comparison based on long read sequencing-by-binding (PacBio)
This section reports on the intra- and inter-laboratory reproducibility of WGS using the long read CCS HiFi technology (Fig. 1) and compares the data obtained by three independent laboratories L1, L2 and L4 using wt OSR (variety Mozart) leaf material (sample specifications, supplementary Table 7).
3.2.1. Evaluation and comparison of delivered sequence data
All three laboratories achieved results for the median read counts well above the minimum requirement of 1.66e6 (L1: 2.5e6; L2: 2.5e6; L4: 3e6). A comparatively high variance among the samples can be observed for L1. The targeted read length of 15 kb was surpassed by L2, while L1 provided reads close to 10 kb and L4 close to 13 kb. The read quality is high with Phred scores over 80, however, L1 and L4 show the highest scores with the median Phred score being close to 90. This higher quality is, in part, due to the smaller provided read size.
Reads were then assembled (Sharma et al., 2022) and the resulting contig amount varies greatly within the L1 and L4 sample sets, ranging in number from 1,800 to 4,000 and 4,000 to 7,000, respectively (supplementary Table 10). L2 shows the lowest variance (3,000–3,800). The total assembly length is close to the expected 1 Gb for all L1 samples. L4 and L2 produce similarly sized assemblies of 1.1 Gb. The N50 values show with 2 Mb to 4 Mb low intra-laboratory variance, L1 has the least favorable results in this category. L2 with values between 11 Mb and 13 Mb is in the middle and L4 with 12 Mb to 16 Mb has the best results, though the sample producing the low N50 seems to be an outlier in the L4 sample set in every category. The BUSCO statistics are based on the brassicales_odb10 data set and show that the assemblies are very similar between the laboratories and show an equally high completeness of 98.5% to 98.6% (supplementary Table 11). All samples have a high rate of duplicates of roughly 80%. This is to be expected, due to the bigenomic amphidiploid character of B. napus. On average, the duplicate BUSCOs appear 2.1 times, further underlining the specific genome structure.
The multiple t-test performed on the contig amount, the largest contig, the assembly length, the N50, the N75, the L50, the L75 and the BUSCO score categories revealed statistically significant differences between L1 and the other two laboratories, specifically in regard to assembly length, N50 and concordantly L50, while L2 and L4 show no significant differences in any of the categories (see Table 4).
Table 4.
The p-values calculated by multiple t-test utilizing Bonferroni compensation for multiple testing based on the contig amount, the largest contig, the assembly length, the N50, the N75, the L50, the L75 and the BUSCO score categories. Categories and values showing a significant difference between two laboratories (i.e. p-value < 0.05) are shown in bold.
| Category | L1 vs L2 | L4 vs L2 | L1 vs L4 |
|---|---|---|---|
| Amount Contigs | 1.00 | 0.45 | 0.31 |
| largest Contig | 0.36 | 1.00 | 0.26 |
| Assembly Length | 0.0044 | 1.0000 | 0.0029 |
| N50 | 0.0306 | 0.4504 | 0.0053 |
| N75 | 1.00 | 0.50 | 0.14 |
| L50 | 0.035 | 1.000 | 0.012 |
| L75 | 0.134 | 1.000 | 0.041 |
| BUSCO Score | 1.00 | 1.00 | 1.00 |
The most obvious difference between L1 and the other two laboratories is the read length, which has a significant impact on nearly all assembly measures. Generally speaking, to achieve long contigs, long reads combined with a high read count are desirable. This effect can be observed since the laboratory with the shortest reads has the lowest N50 and the highest L50, even though the read amount and the read quality is similar or even higher than in the other laboratories. The read length alone, however, is not solely responsible for a good assembly, since L2, having the longest reads, does not produce the best assembly either. Rather, L4, with relatively long reads and a high read count delivers comparatively the best result in this set.
4. Conclusion
4.1. Targeted NGS of a genome edited locus
The results of the targeted NGS analyses show that the amplicon data is highly reproducible, both intra- and inter-laboratory. The observed difference between laboratories is small, despite statistically significant differences in most of the analyzed categories. All three laboratories delivered sufficient amounts of high quality reads and provided, therefore, very similar results in regard to GMO detection, which is qualitatively (binary) clearly possible. However, a quantitative conclusion cannot be drawn from the present data sets. We conclude that the inter-laboratory reproducibility is high, since, as long as a minimum read count is provided, the overall impact of differences in a given laboratory is neglectable. For example, the L1 short read sample set shows a notable difference in coverage and read length, but no discernible difference in GMO detection and quantification compared to the other sample sets.
The difference between the two technologies, i.e. short read vs. long read sequencing, is more pronounced. While the short read system produced a lower intra-laboratory variability, the long reads mapped significantly better to the high similarity target references of the four copies of the CRT1A gene in the B. napus genome. Fewer reads were lost to false alignments since the longer reads encompass more of the few differences in the targets resulting in a higher alignment accuracy. Thus, the RMRCs are significantly smaller for the targeted long read sequencing application.
Because CCS is a relatively new technology a limited intra- and inter-laboratory reproducibility was expected. However, the data does not support this expectation. In regard to the underlying sequencing technology (short read versus long read) we conclude that short read sequencing is very suitable in a lot of cases. For long read sequencing the considerably higher price as well as the much more extensive wet-lab work may be justified for difficult samples, for example for species with complex genome structures.
4.2. WGS of an OSR sample
The results of the WGS application show that the sequencing provider may have a significant impact on the results. While the inter-laboratory differences between two of the three laboratories are not significant, the difference to the third laboratory is high indicating that the inter-laboratory reproducibility may be questionable. The utilization of a relatively new technology with very high demands on the sample preparation, e.g. many manual steps and the need for difficult-to-handle high molecular weight DNA (required minimum fragment size >40 kb), present a significant challenge for any laboratory. In addition, the high price per run makes replicate runs financially difficult. However, a routine use strongly depends on robust and reproducible outcomes without the need for in depth corrections. These current challenges strongly contradict the applicability of CCS HiFi WGS as a standard application in an enforcement laboratory. However, this technology shows enormous potential in cases where little to no a priori knowledge about a GE GMO is accessible but its presence is suspected. The possibility to screen long high quality reads spanning even low complexity or repetitive regions for GM markers with little need for complex assembly steps is a very promising approach.
Funding
The project was financed by institutional resources of the BVL.
CRediT authorship contribution statement
Steffen Pallarz: Conceptualization, Methodology, Software, Formal analysis, Investigation, Data curation, Writing – original draft, Writing – review & editing, Visualization, Project administration. Stefan Fiedler: Conceptualization, Investigation, Resources, Writing – review & editing. Daniela Wahler: Writing – review & editing, Funding acquisition. Jörn Lämke: Conceptualization, Resources, Writing – review & editing. Lutz Grohmann: Conceptualization, Resources, Writing – review & editing, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank the team at CAU for providing the GE and Mozart plant material and HU for providing the wt seed sample. We further thank Mirco Strissel for his technical support during the study and Joachim Bendiek for carefully reading the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.fochms.2023.100182.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- Arulandhu A.J., van Dijk J.P., Dobnik D., Holst-Jensen A., Shi J., Zel J., Kok E.J. DNA enrichment approaches to identify unauthorized genetically modified organisms (GMOs) Analytical and Bioanalytical Chemistry. 2016;408:4575–4593. doi: 10.1007/s00216-016-9513-0. [DOI] [PubMed] [Google Scholar]
- Bessoltane N., Charlot F., Guyon-Debast A., Charif D., Mara K., Collonnier C.…Nogué F. Genome-wide specificity of plant genome editing by both CRISPR–Cas9 and TALEN. Scientific Reports. 2022;12:9330. doi: 10.1038/s41598-022-13034-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortesi L., Fischer R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnology Advances. 2015;33:41–52. doi: 10.1016/j.biotechadv.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Braatz J., Harloff H.-J., Mascher M., Stein N., Himmelbach A., Jung C. CRISPR-Cas9 targeted mutagenesis leads to simultaneous modification of different homoeologous gene copies in polyploid oilseed rape (Brassica napus) Plant Physiology. 2017;174:935–942. doi: 10.1104/pp.17.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Zhou J., Li T., Fang Z., Li L., Huang G.…Peng H. GmoDetector: An accurate and efficient GMO identification approach and its applications. Food Research International. 2021;149 doi: 10.1016/j.foodres.2021.110662. [DOI] [PubMed] [Google Scholar]
- Debode F., Hulin J., Charloteaux B., Coppieters W., Hanikenne M., Karim L., Berben G. Detection and identification of transgenic events by next generation sequencing combined with enrichment technologies. Scientific Reports. 2019;9:15595. doi: 10.1038/s41598-019-51668-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Commission. Joint Research Centre . Publications Office; LU: 2021. New genomic techniques: State of the art review. [Google Scholar]
- European Network of GMO Laboratories (ENGL), 2015. Definition of Minimum Performance Requirements for Analytical Methods of GMO Testing.
- Fraiture M.-A., D’aes J., Guiderdoni E., Meunier A.-C., Delcourt T., Hoffman S.…Roosens N.H.C. Targeted high-throughput sequencing enables the detection of single nucleotide variations in CRISPR/Cas9 gene-edited organisms. Foods. 2023;12:455. doi: 10.3390/foods12030455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraiture M.-A., Herman P., De Loose M., Debode F., Roosens N.H. How can we better detect unauthorized GMOs in food and feed chains? Trends in Biotechnology. 2017;35:508–517. doi: 10.1016/j.tibtech.2017.03.002. [DOI] [PubMed] [Google Scholar]
- Fraiture M.-A., Herman P., Papazova N., De Loose M., Deforce D., Ruttink T., Roosens N.H. An integrated strategy combining DNA walking and NGS to detect GMOs. Food Chemistry. 2017;232:351–358. doi: 10.1016/j.foodchem.2017.03.067. [DOI] [PubMed] [Google Scholar]
- Fraiture M.-A., Saltykova A., Hoffman S., Winand R., Deforce D., Vanneste K.…Roosens N.H.C. Nanopore sequencing technology: A new route for the fast detection of unauthorized GMO. Scientific Reports. 2018;8:7903. doi: 10.1038/s41598-018-26259-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohmann L., Keilwagen J., Duensing N., Dagand E., Hartung F., Wilhelm R.…Sprink T. Detection and identification of genome editing in plants: Challenges and opportunities. Frontiers in Plant Science. 2019;10:236. doi: 10.3389/fpls.2019.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst-Jensen A., Bertheau Y., de Loose M., Grohmann L., Hamels S., Hougs L.…Wulff D. Detecting un-authorized genetically modified organisms (GMOs) and derived materials. Biotechnology Advances. 2012;30:1318–1335. doi: 10.1016/j.biotechadv.2012.01.024. [DOI] [PubMed] [Google Scholar]
- Košir A.B., Arulandhu A.J., Voorhuijzen M.M., Xiao H., Hagelaar R., Staats M.…van Dijk J.P. ALF: A strategy for identification of unauthorized GMOs in complex mixtures by a GW-NGS method and dedicated bioinformatics analysis. Scientific Reports. 2017;7:14155. doi: 10.1038/s41598-017-14469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Fortún J., Phillips D.W., Jones H.D. Natural and artificial sources of genetic variation used in crop breeding: A baseline comparator for genome editing. Frontiers in Genome Editing. 2022;4 doi: 10.3389/fgeed.2022.937853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrackova A., Vasinek M., Sedlarikova L., Dyskova T., Schneiderova P., Novosad T.…Kriegova E. Standardization of sequencing coverage depth in NGS: Recommendation for detection of clonal and subclonal mutations in cancer diagnostics. Frontiers in Oncology. 2019;9:851. doi: 10.3389/fonc.2019.00851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pröbsting M., Schenke D., Hossain R., Häder C., Thurau T., Wighardt L.…Cai D. Loss of function of CRT1a (calreticulin) reduces plant susceptibility toVerticillium longisporumin bothArabidopsis thalianaand oilseed rape (Brassica napus) Plant Biotechnol J. 2020;18:2328–2344. doi: 10.1111/pbi.13394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltykova A., Van Braekel J., Papazova N., Fraiture M.-A., Deforce D., Vanneste K.…Roosens N.H. Detection and identification of authorized and unauthorized GMOs using high-throughput sequencing with the support of a sequence-based GMO database. Food Chemistry: Molecular Sciences. 2022;4 doi: 10.1016/j.fochms.2022.100096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Masouleh A.K., Topp B., Furtado A., Henry R.J. De novo chromosome level assembly of a plant genome from long read sequence data. The Plant Journal. 2022;109:727–736. doi: 10.1111/tpj.15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahler D., Schauser L., Bendiek J., Grohmann L. Next-generation sequencing as a tool for detailed molecular characterisation of genomic insertions and flanking regions in genetically modified plants: A pilot study using a rice event unauthorised in the EU. Food Analytical Methods. 2013;6:1718–1727. doi: 10.1007/s12161-013-9673-x. [DOI] [Google Scholar]
- Wenger A.M., Peluso P., Rowell W.J., Chang P.-C., Hall R.J., Concepcion G.T.…Hunkapiller M.W. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature Biotechnology. 2019;37:1155–1162. doi: 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Zhang Y., Xu W., Li R., Zhang D., Yang L. Development and performance evaluation of whole-genome sequencing with paired-end and mate-pair strategies in molecular characterization of GM crops: One GM rice 114–7-2 line as an example. Food Chem. Mol. Sci. 2022;4 doi: 10.1016/j.fochms.2021.100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.