

Abstract
Genetic underpinnings of diseases are appropriately recognized to be vast and complex. However, a substantial proportion of disease risk is attributable to environmental exposures. Specifically, environmental pollutants cause one in six deaths worldwide and are one of the largest risk factors for premature death in the world. An appreciation of how environmental pollutants interact with our cells to produce deleterious health effects has led to advances in our understanding of the molecular mechanisms underlying the pathogenesis of chronic diseases. Here, we discuss emerging research on interactions of environmental exposures with the human genome and review evidence of the environmental impact on epigenetic mechanisms including DNA methylation, histone modification, and non-coding RNAs. We introduce emerging related areas of investigation including extracellular vesicles, mitochondrial genomics, and epitranscriptomics. Finally, we discuss current challenges and reflect on the exposome and its integration into future environmental health research.
Introduction
Environmental exposures are influential determinants of human health. The modern expansion of industrialization, fossil fuel combustion, and mechanized agriculture has led to a rising global burden of air, water, chemical, and metal pollution.1 As populations around the world are exposed to rising levels of environmental contaminants,2 pollution-related deaths continue to rise. Pollution causes one in six deaths worldwide and is the largest environmental risk factor for premature death in the world.3 Pollution is also the second leading worldwide cause of non-communicable diseases after smoking, contributing to a rising pandemic of respiratory diseases, cardiovascular disease, neurodegenerative diseases, and cancers.4 These health consequences emphasize the importance of understanding environmental drivers of human health and elucidating how environmental exposures induce deleterious health effects.
Environmental exposures are often linked to disease pathogenesis through molecular pathways that disrupt epigenetic regulators of gene expression.2 Epigenetic mechanisms including DNA methylation, histone modification, and non-coding RNAs can modulate gene expression levels without changing the underlying DNA sequence. Many epigenetic modifications are dynamic, reflecting cumulative environmental exposures throughout the lifespan and correlating with aging-related diseases and outcomes.5 As a result, beyond providing a mechanistic link between environmental stressors and disease pathogenesis, epigenetic modifications can also function as reliable markers of accelerated aging and subclinical disease.6,7 Understanding the ways in which environmental exposures induce epigenetic alterations is therefore critical for the mitigation and prevention of environmentally driven diseases.
In addition to classic epigenetic mechanisms, recent studies have expanded to examine the molecular processes that interact with epigenetic pathways. For example, recent advancements have enabled the isolation and detection of molecular cargo within extracellular vesicles (EVs). EVs are membrane-bound vesicles that are released from multiple organs in the body in response to environmental insults that mediate intercellular communication through complex molecular cargo including microRNA (miRNA).8 Similarly, environmental pollutants trigger chemical modification of RNA molecules,9 collectively known as the epitranscriptome, which alters phenotypic expression.10 Environmental insults additionally impact mitochondrial gene activity in a manner that can confer increased disease risk.11 The study of EVs, epitranscriptomics, and mitochondrial genomics offers promising and novel insights into the mechanisms underlying environmental diseases.
In this review, we critically examine the molecular mechanisms linking environmental exposures to human diseases. We discuss the interplay of environmental insults with the human genome and explore how epigenetic modifications underlie the association of environmental exposures with disease pathogenesis. We explore how emerging research related to EVs, epitranscriptomics, and mitochondrial genomics can inform our understanding of environmental contributions to disease risk. Finally, we offer perspectives on new approaches that can further elucidate environmental influences on human health.
Gene-Environment Interactions
Most human diseases arise from the interplay of the human genome and the surrounding environment.12 Gene-environment interactions reflect the complex ways in which genes interact with environmental factors to influence human traits. When gene-environment interactions exist, environmental exposure-related disease susceptibility differs for individuals with different genotypes.13 As a result, evaluating genetic or environmental factors separately may fail to identify high-risk genotypes or vulnerable populations. Gene-environment interaction studies are critical to elucidate biological mechanisms of human disease, risk-stratify patients based on their individual genotypes, and understand the public health implications of prevalent environmental exposures.
Genomic research has demonstrated that certain gene variants predispose individuals to complex diseases. However, genome-wide association studies (GWAS) may fail to identify genotypes with weak or modest effects on disease prevalence but comparatively strong effects on carriers with certain environmental exposures. For example, while asthma is a heritable disease linked to several genetic loci,14 prior GWAS did not identify an association between glutathione S-transferase (GST) genes and asthma prevalence.15 In contrast, gene-environment interaction studies showed that carriers of GST null genotypes had increased susceptibility to indoor air pollution and experienced higher rates of asthma compared to participants with functional GST alleles (Figure 1).16–18 The GST genes encode detoxifying enzymes that protect against pollution-related oxidative stress and may interact with environmental insults to moderate asthma risk.19
Figure 1. Gene-environment interactions impact disease phenotypes.
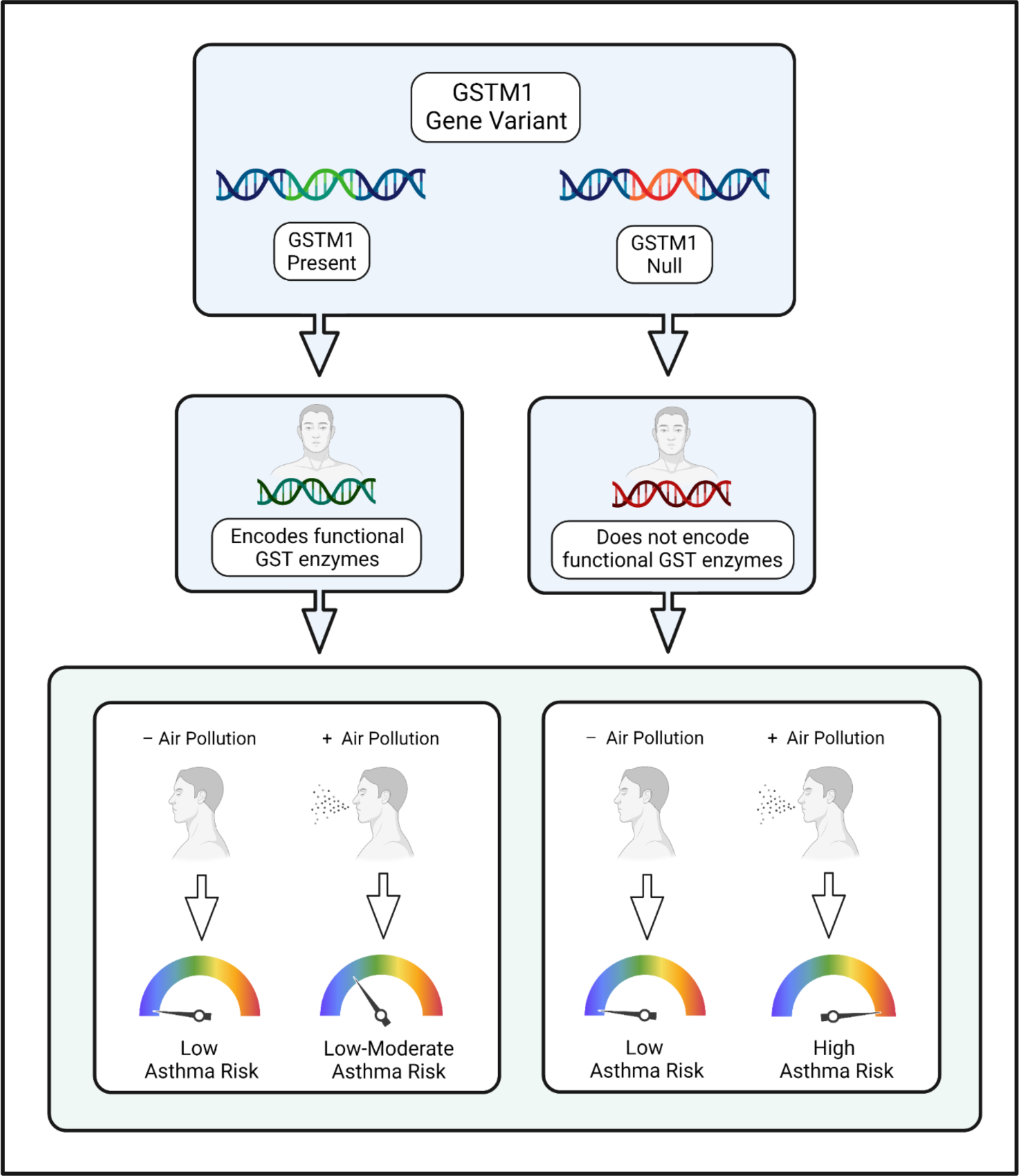
When gene-environment interactions exist, environmental exposures confer differing levels of disease risk for individuals with different genotypes. In this example, the glutathione S-transferase gene (GSTM1) encodes detoxifying enzymes that defend against oxidative stress. The gene has null alleles that cause loss of enzyme activity. Recent studies have shown that carriers of GSTM1 null alleles who were exposed to indoor air pollution experienced increased risk of asthma and lung function impairment compared to participants with functional GSTM1 alleles. This example highlights the importance of evaluating gene-environment interactions to identify SNPs that alter disease risk in the presence of prevalent environmental insults.
Beyond identifying novel genomic variants, gene-interaction studies can also strengthen causal inference derived from observational studies.20,21 For example, a recent population-based study showed that military personnel who were exposed to a nerve agent and carried a paraoxonase-1 (PON1) Q192R polymorphism had increased susceptibility to Gulf War Illness,20 a debilitating syndrome that afflicted military personnel deployed in the Persian Gulf War. Importantly, the study showed that it is possible to demonstrate compositional epistasis in gene-environment interaction studies and to provide stronger evidence for a causal relationship. The study also showed that gene-environment interaction studies may not be impacted by recall bias or confounding by ancestry, which often limit the validity of environmental epidemiologic studies and GWAS. Moving forward, gene-environment interaction studies are essential to identify genotypes that modify disease risk in the presence of key environmental exposures.
While elucidating gene-environment interactions is critical to understanding the genetic liability of human diseases, traditional tests of gene-environment interactions have relatively low power and require strict corrections when comparing multiple genotypes.22 However, evolving analytical frameworks can increase power to detect gene-environment interactions,23,24 and resources such as the UK Biobank, the Environmental influences on Child Health Outcomes (ECHO) consortium, and the All of Us project may help overcome sample size limitations.25,26 With the advent of cutting-edge tools and resources, gene-environment interaction studies have the potential to identify novel genetic loci that contribute to human disease, improve diagnostic methods, optimize prevention strategies, and revolutionize targeted preventive measures in susceptible populations.27
Epigenome
DNA Methylation
DNA methylation occurs when DNA methyltransferase enzymes transfer a methyl group to the C5 position of a cytosine nucleotide forming 5-methylcytosine.28 In contrast, DNA demethylation occurs through a series of deamination and oxidation reactions that remove the methyl group from the cytosine base.29 DNA methylation levels regulate gene expression and exert distinct influences in different genomic regions. In intergenic regions, DNA methylation suppresses potentially harmful genetic elements that can trigger mutation events.30 Similarly, methylation of gene promoter regions often leads to transcriptional silencing.31 In contrast, gene body methylation can increase gene expression.32 Given the intimate connection between DNA methylation and gene activity, understanding how environmental insults impact DNA methylation is critically important.
Particulate matter (PM) air pollution is one of the most ubiquitous pollutants in the world and has been associated with an array of adverse health effects. In particular, PM has been associated with genome-wide differences in DNA methylation in lung and blood cells. Experimental models have shown that when PM is inhaled, small particles disrupt epithelial cell integrity in the lungs, leading to neutrophil chemotaxis and production of reactive oxygen species (ROS).33,34 Ultrafine particles pass into the systemic circulation and generate endothelial injury and oxidative stress.35 ROS catalyze DNA demethylation through oxidation of 5-methylcytosine to 5-hydroxymethylcytosine, after which DNA methylation is passively depleted over serial cellular divisions (Figure 2).36 In vitro models have also shown that air pollution reduces DNA methyltransferase activity, which is required to maintain DNA methylation levels.37 In addition, pollution-related ROS reduce expression of methionine adenosyltransferase 1A (MAT1A), which decreases availability of biologic methyl donors.38 Together, these pollution-related effects have been associated with genome-wide differences in DNA methylation levels in lung epithelial cells and nucleated blood cells.39
Figure 2. Air pollution alters DNA methylation in genes that regulate expression of inflammatory cytokines.
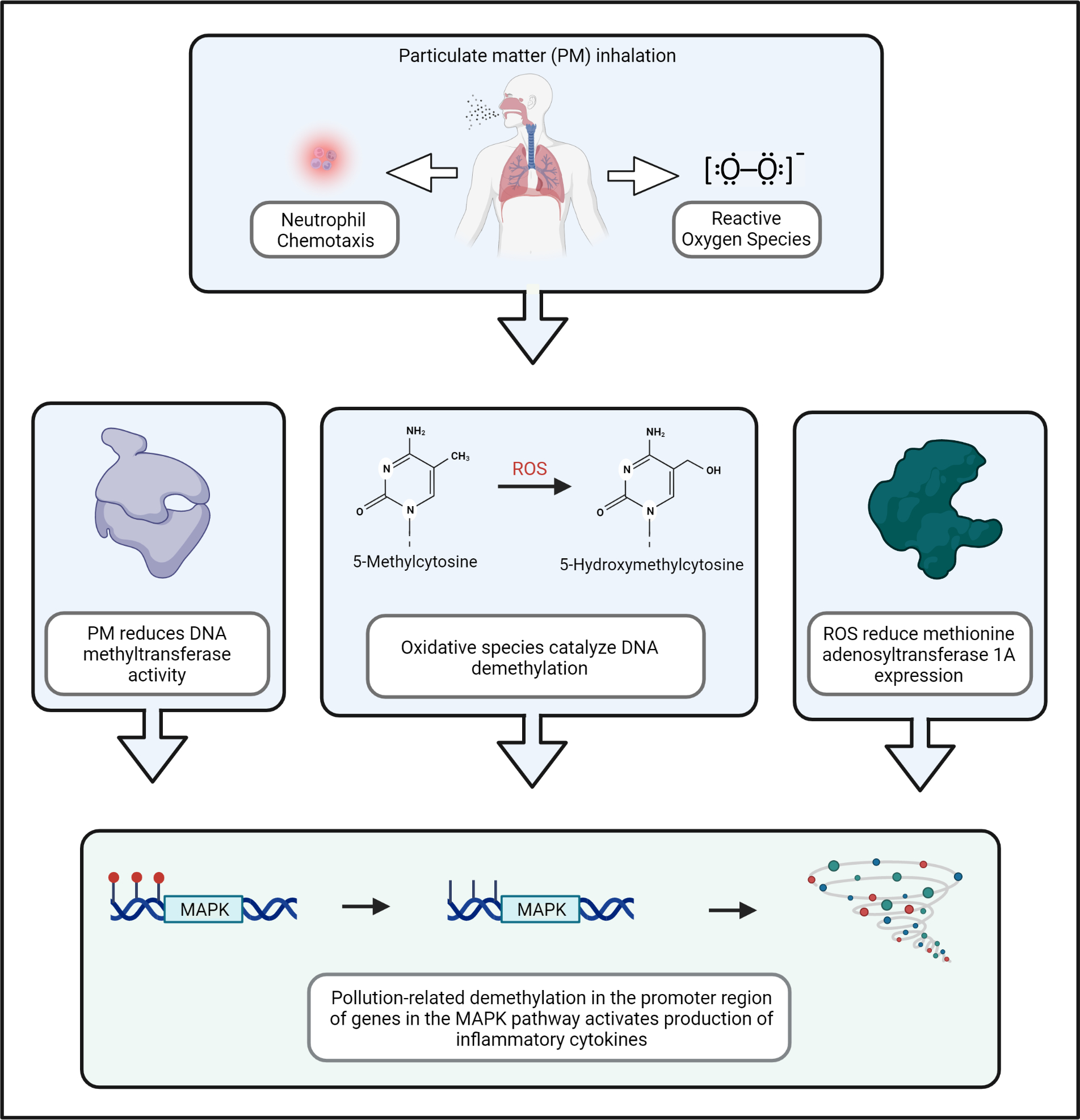
When particulate matter (PM) air pollution is inhaled into the lungs, small particles disrupt epithelial cell integrity in the lungs and trigger neutrophil chemotaxis and production of reactive oxygen species (ROS). Oxidative species reduce DNA methyltransferase activity, catalyze oxidation of 5-methylcytosine to 5-hydroxymethylcytosine, and decrease expression of methionine adenosyltransferase 1A, thereby reducing availability of biologic methyl donors. These effects lead to pollution-related alterations in DNA methylation, including demethylation in the promoter region of genes in the mitogen-activated protein kinase (MAPK) pathway. These alterations hinder DNA methylation-mediated suppression of inflammatory genes and link air pollution exposure to the production of pro-inflammatory cytokines.
Several population-based studies have confirmed that air pollution exposure was associated with differences in DNA methylation levels in blood leukocytes. One of these studies showed that air pollution exposure was associated with demethylation in the promoter regions of genes in the mitogen-activated protein kinase (MAPK) pathway.40 These alterations may hinder DNA methylation-mediated suppression of inflammatory genes and link air pollution exposure to increased cytokine production.41 In a cross-over trial, PM exposure was associated with reduced methylation in pro-coagulant genes, which may link PM exposure to vascular thrombosis and cardiovascular health.42 In a study of non-diabetic men, differential methylation of intercellular adhesion molecule-1 (ICAM-1), an inflammatory gene, mediated the relationship between short-term PM exposure and increased diabetes risk.43 A fourth study showed PM was associated with hypomethylation of the aryl hydrocarbon receptor repressor (AHRR) gene, which is heavily implicated in the pathogenesis of obstructive lung diseases.44 Tobacco smoking is associated with similar differences in DNA methylation patterns,45,46 suggesting that these inhaled environmental insults modify respiratory and cardiovascular disease risk through similar mechanistic pathways.
DNA Methylation-Based Biomarkers
Epigenetic changes can function as biomarkers of environmental exposures and disease predisposition. For example, in epigenome-wide association studies (EWAS), tobacco smoking was associated with differences in DNA methylation at thousands of CpG sites in nucleated blood cells.46 While smoking cessation was associated with reversion to normative methylation levels at some CpG sites, other sites annotated to genes associated with lung and heart diseases do not return to normative levels even decades after smoking cessation.47,48 This is largely because DNA methylation patterns can be maintained in the DNA of daughter leukocyte cells released in the bloodstream through the activity of DNA methyltransferase enzymes. In this context, blood-based biomarkers that index cumulative smoking-related differences in DNA methylation levels can detect smoking exposures and classify smoking behaviors.49 Epigenetic biomarkers can also assess environmentally mediated disease risk in applications that mirror the use of polygenic risk scores to evaluate heritable disease risk.50 In a recent population-based study, a DNA methylation-based classifier of tobacco smoke exposure identified former smokers at increased risk of obstructive lung disease and death.7 Compared to self-reported smoking exposures, which are subject to recall bias and misreporting,51,52 DNA methylation-based smoking indices may capture true smoking-related biologic effects and help identify smokers with increased risk of adverse smoking-related health outcomes.
Beyond reflecting specific environmental insults such as smoking, epigenetic changes can also serve as a proxy for accelerated biological aging. Prior research in different types of human tissue identified age-related changes in DNA methylation that were leveraged to develop epigenetic aging clocks.53 Aging clocks use a collection of CpG sites conserved across mammalian tissue to estimate the “biological age” of human tissue and can reflect age acceleration when the calculated DNA methylation age is higher than the chronological age.54 Using these proxy measures, environmental insults have been shown to accelerate aging. In population-based studies, individuals exposed to air pollution and tobacco smoke demonstrated advanced DNA methylation age in blood and lung tissue (Figure 3).55–57 Similarly, organochlorine pesticide exposure was associated with accelerated epigenetic aging in peripheral blood leukocytes.58 In contrast, another epidemiologic study showed that improved diet and educational attainment were associated with decelerated epigenetic aging in blood leukocytes.59,60
Figure 3. Environmental stressors impact DNA methylation age.
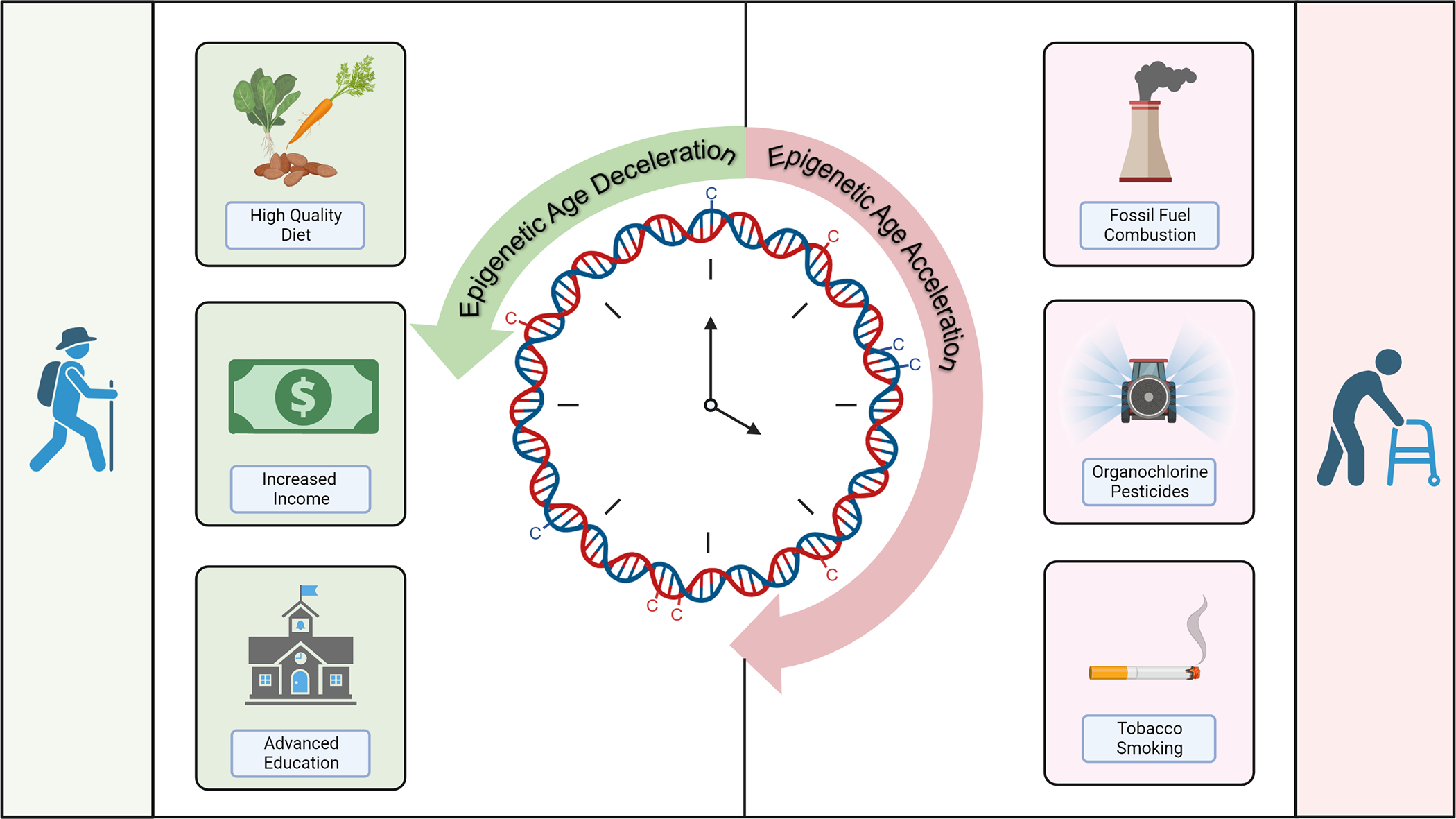
Epigenetic aging clocks encompass CpG sites that estimate DNA methylation age of human tissue. Harmful environmental exposures including fine particulate matter air pollution, organochlorine pesticides, and polycyclic aromatic hydrocarbons are associated with epigenetic age acceleration. In contrast, improved diet quality and higher socioeconomic status are associated with epigenetic age deceleration. Epigenetic age acceleration leads to genomic instability and aberrant gene expression and is associated with aging-related diseases and functional decline.
Elucidating environmental factors that impact DNA methylation age is critical because DNA methylation age is predictive of age-related health outcomes. Age-related changes in DNA methylation generate genomic instability and aberrant gene expression that contribute to disease risk. In a population-based study in young adults, epigenetic age acceleration was associated with increased risk of incident type 2 diabetes.61 In a second prospective cohort study, increased epigenetic age acceleration was associated with increased incidence of coronary artery disease, peripheral artery disease, and heart failure independently of traditional cardiovascular risk factors.62 In a third study, epigenetic age acceleration was associated with truncated life expectancy.63 These findings suggest that epigenetic markers serve as an important proxy of aberrant biological aging. Further research is required to determine if epigenetic biomarkers can be applied in clinical settings to facilitate disease risk stratification and enable early diagnosis of environmentally mediated diseases.
Chromatin Remodeling and Histone Modifications
In most cells, chromatin consists of repeating units of nucleosomes. Each nucleosome comprises ~147 DNA base pairs wrapped around eight histone proteins. Each of these “octamer” beads contains two copies each of H2A, H2B, H3, and H4 histones while a single H1 “linker” histone binds the DNA to the octamer.64 These core histones have several variants, many of which are tissue-specific, have particular functions, and have been associated with genetic disorders and cancers.64 Histones, along with a suite of post-translational modifications including acetylation and phosphorylation, are a critical layer of epigenetic regulation that interacts with DNA methylation and non-coding RNAs to regulate gene expression.65,66 These modifications help define the transcriptional state of the chromatin toward the more transcriptionally active euchromatin or less transcriptionally active heterochromatin. Nucleosome positioning and histone modifications are critical for essentially all biological processes and can affect both the health of the individual67 and ensuing generations.68,69 Thus, it is critically important to understand how environmental exposures alter histone biology.
Exposure to arsenic, a heavy metal and common environmental pollutant, has been observed to influence several histone modification patterns.70,71 It is well-established from both observational and experimental evidence that arsenic alters histone methylation, including global methylation of H2B, H3, and H4. 70,71 Arsenic also alters methylation at specific sites including H3K4me3, H3K9me3, and H3K27me3.70 While the exact mechanisms underlying arsenic-induced changes in histone methylation are unknown, there is some evidence that arsenic activates specific methyltransferase enzymes including G9a.72,73 Arsenic may thereby affect the bivalent status of the chromatin.71 Specifically, simultaneous presence of activating (e.g. H3K4me3) and repressing (H3K27me3) modifications, both of which have been observed with arsenic exposure, is a hallmark of bivalency. This bivalent state may lead to the upregulation of several oncogenes74 and may underlie the oncogenic effect of arsenic. Arsenic-induced histone modifications have also been shown to generate oxidative stress, DNA damage, and regulate the cell cycle.71
Aside from altering histone methylation, arsenic has also been shown to affect the abundance of histone variants via polyadenylation of H3 histone mRNA. H3 polyadenylation disrupts the physiological balance of histone variants and is suspected to be carcinogenic, though confirmatory experimental evidence is necessary.75,76 Emerging evidence shows other environmental exposures can trigger posttranslational histone modifications including trace metals,77 air pollution,78–83 polycyclic aromatic hydrocarbons (PAHs),84 pesticides,85–87 dioxins,88 and plasticizers89,90.
Non-coding RNAs
Non-coding RNAs are key regulators of gene expression at the post-transcriptional level via direct interactions with target genes and coordinated responses with other epigenetic machinery. MicroRNAs (miRNAs) are one class of small non-coding RNAs91. MiRNAs can directly bind to 3’UTR, 5’UTR and coding sequences to inhibit translation or to promoter regions to induce transcription.91 Acting in a controlled epigenetic “circuit”, miRNAs can also affect the chromatin state to promote transcription92 and are themselves regulated by DNA methylation and histone modifications.93
Despite early evidence that miRNAs are responsive to a range of environmental exposures,94 details of the underlying mechanisms have been historically limited.95 While oxidative stress and inflammation have long been suspected to play important roles in air pollution-induced miRNA dysregulation, only a few studies have provided experimental evidence.96–98 In a cross-over trial, diesel exhaust was shown to affect expression of several miRNA and mRNAs, leading to inflammatory cell recruitment and epithelial cell shedding.96 In a recent in vitro study, PM-induced ROS downregulated hsa-miR-137, resulting in greater expression of IL-6 and COX-II. This sequence effectively linked air pollution exposure to a pro-inflammatory state that contributed to the pathogenesis of rheumatoid arthritis.97 Another in vitro showed that PM led to differential expression of miRNAs that were taken up by alveolar macrophages, leading to increased inflammation in the lungs and pulmonary epithelial cell damage.98 In the same study, PM2.5 increased levels of Peroxiredoxin 6 (Prdx6) via downregulation of mmu-mir-467c-5p, potentially as a protective response to regulate inflammatory injuries.98 The connection between PM2.5 and miRNA dysregulation has also been implicated in the pathogenesis of Alzheimer’s disease.
While miRNAs have historically been the most studied non-coding RNA in environmental health, there is mounting evidence that other non-coding RNAs are modifiable by environmental threats. Circular RNA are single-stranded RNA in a closed continuous loop that derive from protein-coding regions.99 Circular RNAs regulate gene expression by acting as miRNA “decoys” or by interacting directly with proteins. Recently, in vivo rodent studies showed that air pollutants lead to differentially expressed circular RNAs in the lung100,101 and in rodent embryos102.
Environmental insults likely alter most RNA regulators of gene expression. For example, tRNA fragments are responsive to heat103, ultraviolet radiation103,104, and oxidative stress.103,105,106 While tRNAs have traditionally been viewed in the context of translation, tRNAs can also regulate gene expression via translation repression and gene silencing.107,108 In addition, our growing understanding of how long non-coding RNAs regulate gene expression through modulation of chromatin structure and direct interactions with target genes109 is accompanied by emerging evidence that air pollution,101,110 metals,111,112 and other environmental contaminants113–115 alter long non-coding RNA expression. These trends affirm that further research elucidating how environmental insults impact expression of non-coding RNAs is critical to expand our understanding of how environmental insults affect human health.
Emerging Areas of Molecular Investigation
Epitranscriptomics
Epitranscriptomics evaluates post-transcriptional modifications of RNA. There are over 100 post-transcriptional modifications across all RNA types that play a critical role in RNA folding, splicing, stability, localization, and translation. The location and quantity of RNA modifications help determine how modifications affect RNA function.116 Similar to epigenetic modifications, RNA modifications are controlled and maintained by a group of “reader”, “writer”, and “eraser” proteins (RWEs).116 The most well-studied and common epitranscriptomic modification is the addition of a methyl group on the sixth nitrogen atom of adenine, also known as N6-methyladenosine (m6A). The m6A modification can be found in all types of RNA117 and helps regulate RNA folding, splicing, stability, and translation.118
Epitranscriptomic changes related to environmental exposures are an underexplored but potentially important pathway through which environmental insults impact human health. For example, m6A has been shown to regulate the inflammatory response, activate the adaptive and cellular immune systems, and modulate T-cell homeostasis and differentiation.119,120. Similarly, METTL3, an m6A methyltransferase (“writer”), is essential for the production of inflammatory cytokines.121 Experimental research has shown that m6A is responsive to external stressors in vitro, especially environmental pollutants associated with oxidative stress including cigarette smoke and PM2.5.80,122–125 Accordingly, oxidative stress has been shown to affect m6A at hundreds of mRNA transcripts.126,127
Environment-induced alterations in global m6A levels may stem in part from altered expression of RWEs. PM2.5 exposure leads to hypomethylation of the promoter regions of RWE genes128, resulting in differential expression of METTL380,123,124,128 and METTL14.80 METTL3 and METTL14 are two “writers” that form the m6A methyltransferase complex.129 Similarly, cigarette smoke has been shown to alter METTL3 and METTL14 expression via promoter hypomethylation in vitro,130,131 resulting in higher levels of m6A on select miRNAs and long non-coding RNAs. In experimental models, PM2.5-related alterations in METTL3 also induce m6A modification of Oxidative Stress-Induced Growth Inhibitor 1 (OSGIN1)124 and cadherin 1 (CHD1) mRNAs.132 The CHD1 m6A modification is part of a mechanism that spans across multiple different biological regulatory networks. Specifically, environmentally-related METTL3 promoter hypomethylation leads to more abundant m6A modification on CHD1 that is then recognized by YTHDF2, an m6A “reader” that is upregulated via PM2.5-induced downregulation of hsa-miR-494–3p. YTHDF2 in turn inhibits E-cadherin, which can trigger an epithelial-mesenchymal transition characteristic of pulmonary fibrosis (Figure 4).132 In human studies, the relationship between air pollution and m6A is less clear.123,133
Figure 4. One example of how air pollution can trigger coordinated epigenetic and epitranscriptomic responses that impact human health.
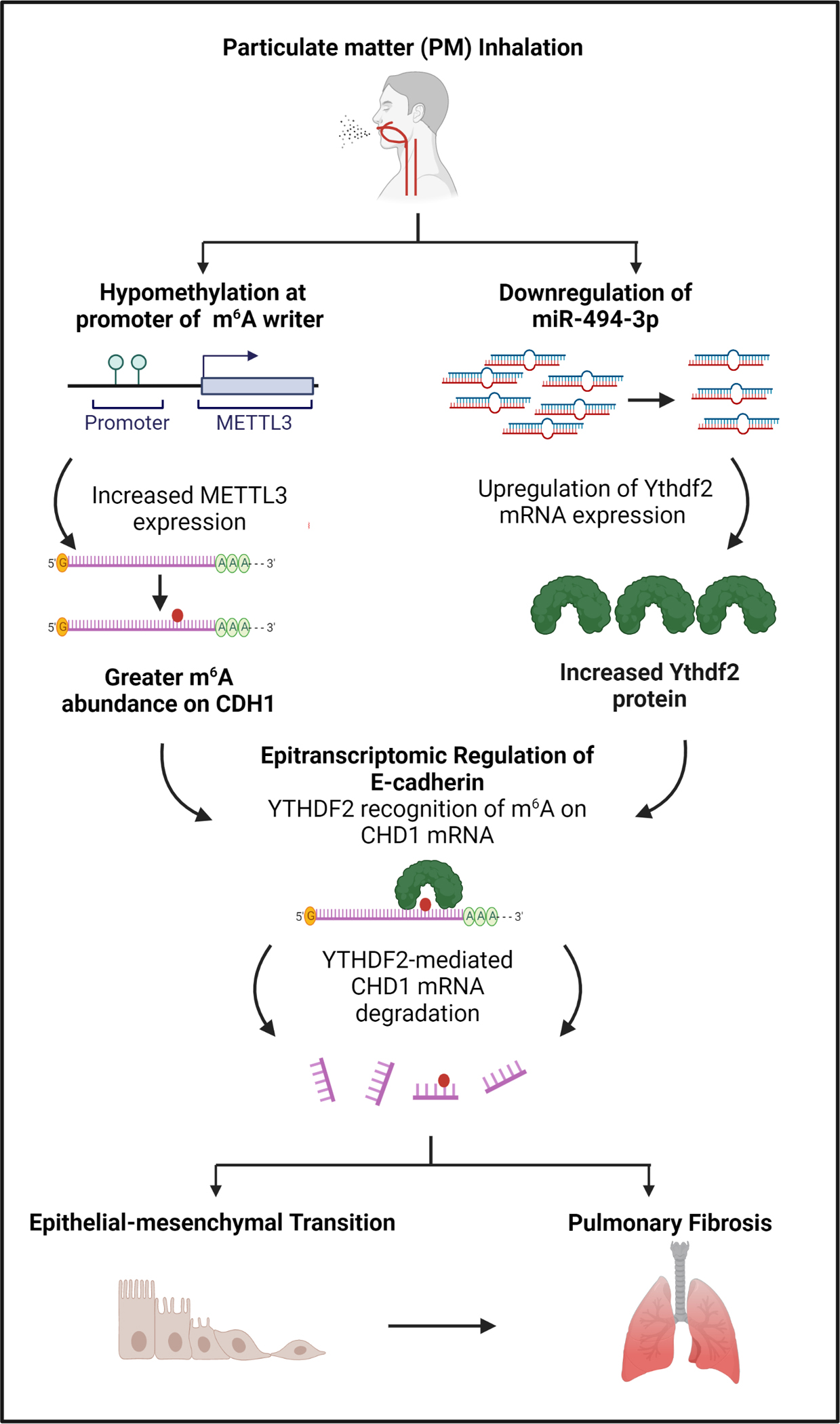
Exposure to particulate matter air pollution can trigger alterations in epitranscriptomic machinery and multiple other regulatory mechanisms that interact to generate a systemic response. In this example, air pollution exposure triggers changes in DNA methylation, small non-coding RNA, and epitranscriptomic machinery that may influence risk of pulmonary fibrosis.
Heavy metals and chemical toxicants have also been shown to alter m6A and RWE expression in vitro. High levels of arsenic altered global m6A levels123,134,135 via upregulation of the m6A methyltransferase complex and downregulation of FTO, a demethylase.134,136,137 This effect was also observed in vivo as mice treated with arsenic showed higher global m6A levels and downregulation of FTO.138 Other metals and chemical toxicants including cobalt,139 manganese,140 chromium,141 cadmium142, polychlorinated biphenyls (PCBs),143,144 PAHs,145 and other common environmental pollutants123,146–148 were also associated with m6A modifications and RWE expression levels.
Overall, there is compelling and mounting evidence that RNA modifications are responsive to environmental stimuli in patterns specific to different RNA subtypes. These effects may be driven by changes in RWE expression and activity, which may be mediated by changes in DNA methylation in the promoter regions of these genes. Environmental exposure-related oxidative stress also leads to altered expression of RWEs including m6A demethylase FTO138 and m5C methyltransferase NSUN2.149 At present, few studies have examined components of the epitranscriptome beyond m6A. However, m6A is a small part of the epitranscriptomic landscape. As the field evolves, it is imperative to examine other influential RNA abundant modifications.150
Extracellular Vesicles
EVs are nano-sized membranous vesicles that are released from multiple cell types in the body under physiologic conditions and in response to environmental insults.151 The physiologic state of the parent cell regulates packaging of EV cargo, which includes proteins, nucleic acids, lipids, and metabolites. EVs are released from the parent cell and are endocytosed by recipient cells and alter cell biology through their molecular cargo. For example, EV-encapsulated microRNAs (EV-miRNAs) regulate cellular function by degrading complementary mRNA transcripts.152 EVs convey molecular signals between different organs throughout the body and thereby function as a mechanistic link between environmental insults and transcriptional activity.
In population-based studies, exposure to inhaled pollutants triggers lung epithelial cells and alveolar macrophages to release large quantities of EVs into the blood (Figure 5).153 For example, it has been shown that in humans, EVs can generate a pro-inflammatory signaling cascade that causes endothelial injury, hypercoagulability, and end-organ dysfunction.154 Subsequent in vitro studies showed that PM-related EVs also triggered hyperresponsiveness of bronchial smooth muscle cells,155 accelerate vascular thrombosis,156 and precipitate neurotoxic signaling.157 EVs thereby mediate the toxic effects of air pollution exposures and contribute to pollution-related risk of chronic lung, heart and neurologic diseases.
Figure 5. Inhaled environmental exposures trigger EV signaling that mediates systemic inflammation and disease.
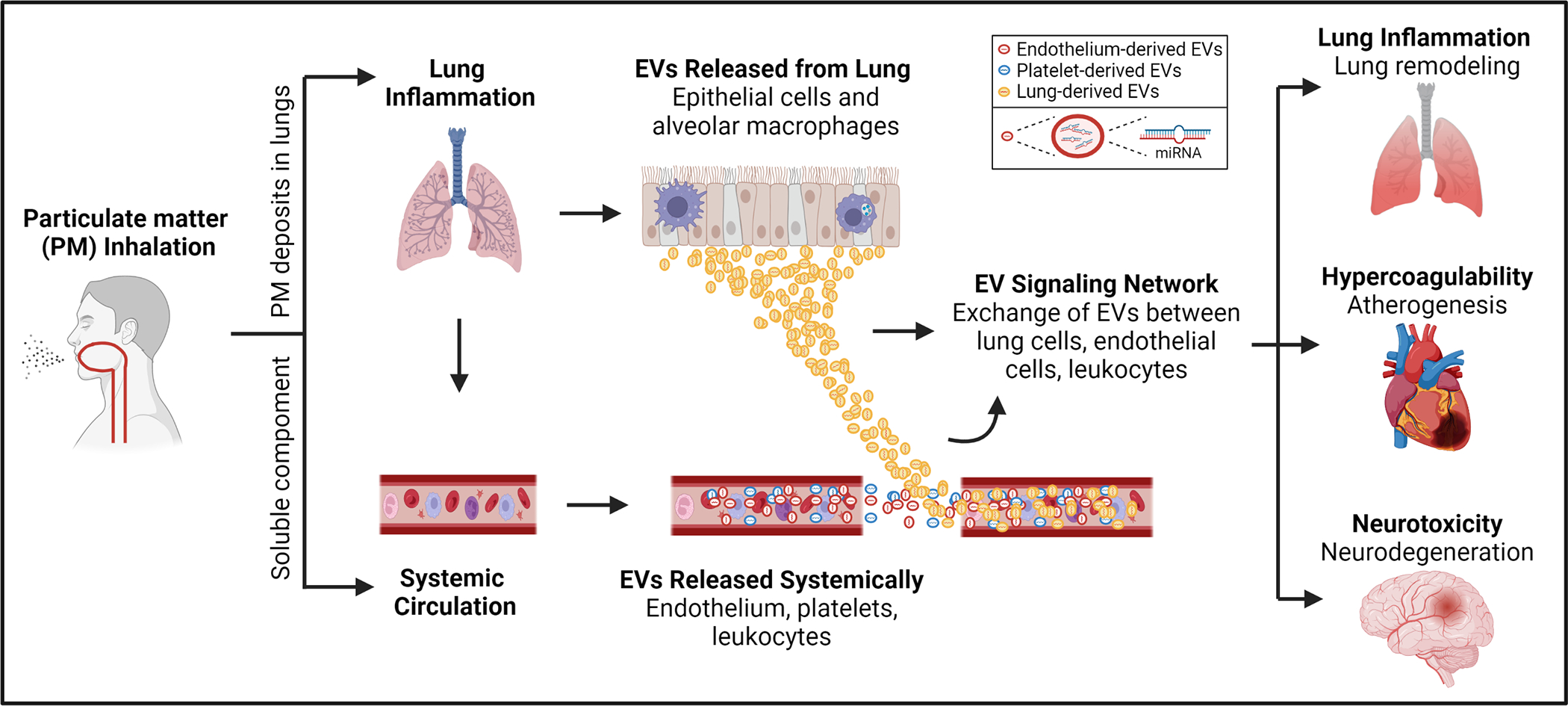
Particulate matter inhalation triggers alveolar macrophages and airway epithelial cells to release pro-inflammatory EVs. Ultrafine particles also enter the systemic circulation and trigger EV release from endothelial cells, platelets, and circulating blood leukocytes. EVs amplify production of inflammatory cytokines and enhance recruitment of inflammatory cells. As a result, circulating EVs create a cycle that intensifies inflammation and can lead to end organ dysfunction including lung function impairment, atherogenesis, and neurodegeneration.
Heavy metals158–160 and plasticizers including bisphenol-A (BPA) and di(2-ethylhexyl) phthalate (DEHP) have been shown to modulate EV biology and adversely affect the reproductive system. BPA has been detected in the follicular fluid that surrounds oocytes and contributes to oocyte development in women.161 Recent in vitro studies showed that BPA altered EV-miRNA expression in primary granulosa cells.162 Population-based studies in women undergoing in vitro fertilization confirmed that phenol and phthalate exposure altered EV-miRNA expression in follicular fluid.163,164 In addition, human studies showed that BPA exposure altered protein expression in placenta-derived EVs and impacted pathways known to modulate placental cellular injury.165 Together, these findings suggest that EVs and their molecular cargo may function as a mechanistic link between chemical exposures and reproductive toxicity.
EVs translate environmental exposures into substantive disease risk through synergistic interactions with miRNAs and other epigenetic mechanisms.166 EVs may thereby represent viable biomarkers of subclinical disease in humans. For example, in a prospective cohort study, plasma EV-miRNAs functioned as viable biomarkers of lung function impairment.167 Recent efforts to isolate plasma EVs derived from specific tissue types may further enable accessible liquid biopsies that are predictive of future health outcomes.
Mitochondrial Genomics
Mitochondria facilitate energy metabolism, redox signaling, fat homeostasis, and metabolic regulation.168 Accordingly, mitochondrial impairments or dysregulation contribute to numerous disease states.168 Mitochondria are particularly vulnerable to the effects of environmental exposures. The mitochondrion thereby functions both as a sentinel for exposure-induced damage and a central mechanism through which environmental exposures impact human health.169 As an example, a recent study using a direct assessment of mitochondrial function showed that the neurodevelopmental outcomes associated with prenatal PM2.5 exposure are partially mediated by long-term changes in mitochondrial respiration.170
The number of mitochondrial genomes per cell or the mitochondrial DNA copy number is a commonly used marker of mitochondrial damage. Numerous environmental exposures have been shown to alter mitochondrial DNA copy number including PM air pollution,171 PAHs,172 heavy metals,169 and other chemical173–175 and occupational exposures.176–178 However, the relationship between environmental stressors and mitochondrial DNA copy number varies depending on the type and duration of the exposure169 and it is difficult to discern the underlying mechanisms. However, recent work using known mitochondrial toxicants has elucidated more precise molecular mechanisms underlying environment-induced mitochondrial impairment.
Exposure-related excess ROS is a ubiquitous mechanism underlying mitochondrial dysfunction related to PM air pollution179, heavy metals,180 PAHs172, and select pesticides181–183. ROS are typically produced in the matrix of mitochondria in the form of superoxide (O2−) and hydrogen peroxide (H2O2). However, alterations in this process related to environmental insults can lead to accumulation of electrons and increased ROS production. The accumulation of ROS can lead to altered permeability of mitochondrial membranes,184 imbalance in calcium homeostasis,185 increased mitochondrial DNA mutations,186 and damage to the mitochondrial respiratory chain and ATP production.185 Mitochondrial dysfunction then triggers systemic effects including inflammasome and inflammatory cytokine release.172,179
Recent research has also identified several specific pollutant-specific pathways. For example, air pollution can lead to aberrant mitochondrial DNA methylation and DNA strand breaks.179 Dioxins and PAHs can bind aryl hydrocarbon receptors (AhR)187, which leads to mitochondrial AhR degradation and alterations in cellular respiration.188 Finally, cyanide and rotenone can inhibit cellular respiration via interactions with complex IV and complex I of the electron transport chain.189
An area of growing interest in environmental health research is mitochondrial heteroplasmy. Each cell comprises mixed copies of mtDNA genomes and heteroplasmies are mutations that are present in a subset of mtDNA within a cell.190 Mitochondrial genomes are naturally more susceptible to mutations compared to nuclear genomes and heteroplasmy has been shown to alter mitochondrial gene expression and contribute to several chronic diseases.190 There is mounting evidence that environmental influences are associated with increased mutation load and mitochondrial heteroplasmy,169 making mitochondrial heteroplasmy a promising biomarker of both mitochondrial damage and cumulative exposure to environmental exposures.
Current Limitations and Future Perspectives
Need for Multi-Omic Approaches
In order to interpret genomic influences and epigenetic changes in the context of environmental exposures and their impact on human health, it is essential to improve our understanding of the downstream biological consequences of these molecular modifications. At present, most investigations are limited to either a single mechanism (or –omic layer) or use limited sample sizes. However, environmental exposures trigger alterations in multiple regulatory mechanisms that interact to create a systemic response. For example, air pollution triggers changes in DNA methylation, small non-coding RNAs, and epitranscriptomic machinery that collectively influence risk of downstream pulmonary fibrosis.191 There is also an established interplay between non-coding RNA and m6A modifications in the context of environmental exposures.130,136,142 Given the well-established crosstalk between epigenetic layers,192,193 studies should aim to capture multiple biological mechanisms to better elucidate how environmental pollutants affect human health. This is especially true of major environmental threats including air pollution and heavy metals because they are known to have wide-ranging systemic effects that impact multiple biological systems. Technical limitations related to the analysis of high-dimensional datasets and use of relatively small samples have hindered our ability to conduct multi-omic research. However, emerging analytical tools have facilitated impactful multi-omic research.194–196
High-dimensional genomic and epigenomic discovery studies rely heavily on ontological pathway analyses to elucidate the ways in which molecular changes impact cellular function. While pathway analyses are useful, they are imprecise and carry substantial uncertainty. Currently, these knowledge-based analyses (e.g. KEGG197, GO198) are curated from evolving knowledge base, but must make strong assumptions to postulate how epigenomic changes impact cellular function. For example, DNA methylation sites or non-coding RNAs associated with environmental exposures are matched to genes that they putatively regulate. These genes are then used for ontological and pathway-based analyses without explicit evidence that shows how certain molecular changes impact gene expression. The pitfalls of such assumptions are illustrated by the complicated relationship between mRNA and protein abundance: although there is often a correlation between the two, changes in one layer (e.g. mRNA) do not always lead to changes in the other (e.g. proteins).199 To minimize false discoveries and conclusions from observed epigenetic changes, it is vital that population-based studies start to pair epigenomic changes with downstream functional changes. Such studies, which could be multi-omic or targeted in nature, will facilitate more substantiated interpretation of epigenetic and epitranscriptomic changes. Additionally, it would be helpful to include other epigenetic or molecular layers to accommodate the known interactions between epigenetic machinery.
Tissue Specificity
Due to logistical constraints, human studies are often limited to broad interrogations using heterogeneous cell populations and/or non-specific biomatrices such as blood. While blood-based investigations may reflect systemic changes, these studies often lack the specificity required to understand the pathogenesis of specific diseases. Furthermore, not all cell types will respond to environmental stimuli identically, which makes generalizations difficult. Given the existing restraints and our desire to understand the environmental impact on multiple organ systems, we need to adopt a combination of approaches to address this challenge. Adoption of single-cell sequencing may be one solution to this challenge. Single-cell techniques and approaches can resolve and characterize responses from individual cell types that is commonly found in biospecimen of typical epidemiologic studies. With technical advances and declining costs, this may be the next major step in molecular environmental health studies. Alternatively, deconvolution of multi-tissue samples200 is a useful strategy to estimate the composition of different cell types in a heterogeneous mixture. However, deconvolution alone does not allow us to examine specific cell or tissue types and complementary approaches are necessary. In the context of EVs, experimental strategies can be developed to isolate targets from specific tissues.201,202 This approach may allow us to isolate tissue-specific effects that can more clearly convey the impact of environmental exposures on key organ systems. Future research should take these considerations into account to more clearly elucidate the impact of the environment on human health (Box 1).
Box 1. Keys for Future Molecular Environmental Health Studies.
Future studies, particularly epidemiologic investigations, should acknowledge the complex interplay across different biological mechanisms (e.g., interactions and feedbacks across histones, DNA methylation, and non-coding RNAs) that are often considered in isolation.
If possible, studies should strive to expand beyond one single biological mechanism of interest to capture multiple connected systems using the growing number of multi-omic analysis approaches. This is necessary to show, rather than assume, that more “upstream” changes (e.g. DNA methylation) have downstream effects on transcriptional activity and protein expression.
Investigators should take advantage of studies and programs that have already collected data on multiple molecular mechanisms to conduct both hypothesis-driven and hypothesis-free investigations.
Studies should consider adopting single cell-based technologies to enrich or purify sample types and capture differential responses from each cell type.
Studies should consistently acknowledge the uncertainties inherent in ontological and pathway analyses.
Given recent advancements in exposure assessment and statistical mixture analyses, there is now an opportunity for investigators to consider the environment more holistically, rather than to evaluate discrete exposures independently.
Beyond better detection of harmful pollutants, there is a need to identify natural exposure patterns and concomitant exposures in a way that would directly address potential for co-exposure confounding.
While animal studies have demonstrated transgenerational effects of certain environmental pollutants transmitted through epigenetic mechanisms, human studies thus far have been lacking and future studies may consider leveraging multi-generational cohorts to address this gap.
Untargeted Discovery and the Exposome
Historically, exposure assessment has lagged behind genetic and epigenetic research with respect to our ability to comprehensively capture an individual’s environmental exposures. This has restricted our ability to monitor and understand environmental impacts on human health. However, just as genome-wide approaches have spurred tremendous advancements in our understanding of complex-trait genetics, implementing a more expansive and comprehensive view of environmental exposures will help elucidate the complex ways in which the environment impacts human health.203 In practice, we should consider the environment as a high dimensional –omic layer, otherwise known as the exposome.204 Performing untargeted environmental exposure assessments may produce a wealth of data that will more deeply inform our understanding of the complex mechanisms through which the environment shapes human health.
Conclusions
Environmental insults influence epigenetic modifications and related biological systems that interactively shape gene expression. Together, these pathways translate environmental exposures into substantive disease risk. Understanding the influence of environmental stressors on DNA methylation, histone modification, non-coding RNAs, epitranscriptomics, EVs, and mitochondrial genomics is critical to recognize and mitigate the effect of environmental exposures on human health.
Figure 6. Exposomics and multi-omic responses.

The exposome is the cumulative measure of environmental influences including the external environment, lifestyle, behavior, and diet and the resulting biological and endogenous processes. The exposome induces biological responses at every level and should be integrated into multi-omic studies. Ultimately, the complex non-linear interactions between the environment and our genome, epigenome, transcriptome, metabolome, and proteome drive the aging process and the pathogenesis of chronic diseases.
Glossary
- Biological age
The biological age is impacted by living conditions and lifestyle and refers to the physiological and functional status of an individual; the biological age may be older or younger than the chronological age and serves as a reflection of health and aging.
- Compositional Epistasis
Compositional epistasis is a central requirement to a statistical interaction is a mechanistic interaction and requires a study to show that some individuals have the disease of interest if both environmental and genetic exposures are present but will not have the disease of interest if just one exposure is present.
- Epigenome-Wide Association Studies
An epigenome-wide association study (EWAS) is a genome-wide study of epigenetic changes such as DNA methylation and their association with a health outcome of interest.
- Liquid Biopsy
A liquid biopsy is a peripheral blood test that can detect cells derived from specific types of tissue in the body.
- Particulate Matter
Particulate matter consists of microscopic particles of solid or liquid matter that are suspended in the air; fine particles have a diameter of ≤ 2.5 μm and are designated PM2.5.
- Polygenic Risk Score
A polygenic risk score is an estimate of a person’s genetic liability for a disease of interest based upon their genotype.
- Crossover trial
A longitudinal study where all participants receive two or more treatments, often in random order and separated by a washout period.
- Circular RNAs
Single-stranded RNA in a closed continuous loop that are most often derived from protein-coding regions.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Landrigan PJ et al. The Lancet Commission on pollution and health. Lancet 391, 462–512 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Peters A, Nawrot TS & Baccarelli AA Hallmarks of environmental insults. Cell 184, 1455–1468 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuller R et al. Pollution and health: a progress update. Lancet Planet Health 6, e535–e547 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Prüss-Ustün A et al. Environmental risks and non-communicable diseases. BMJ 364, l265 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sen P, Shah PP, Nativio R & Berger SL Epigenetic Mechanisms of Longevity and Aging. Cell 166, 822–839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rutledge J, Oh H & Wyss-Coray T Measuring biological age using omics data. Nat Rev Genet (2022) doi: 10.1038/s41576-022-00511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eckhardt CM et al. Predicting risk of lung function impairment and all-cause mortality using a DNA methylation-based classifier of tobacco smoke exposure. Respir Med 200, 106896 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alibhai FJ et al. Cellular senescence contributes to age-dependent changes in circulating extracellular vesicle cargo and function. Aging Cell 19, e13103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cayir A, Byun H-M & Barrow TM Environmental epitranscriptomics. Environ Res 189, 109885 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Teng P-C et al. RNA Modifications and Epigenetics in Modulation of Lung Cancer and Pulmonary Diseases. Int J Mol Sci 22, 10592 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunst KJ, Baccarelli AA & Wright RJ Integrating mitochondriomics in children’s environmental health. J Appl Toxicol 35, 976–991 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunter DJ Gene-environment interactions in human diseases. Nat Rev Genet 6, 287–298 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Hartiala JA, Hilser JR, Biswas S, Lusis AJ & Allayee H Gene-Environment Interactions for Cardiovascular Disease. Curr Atheroscler Rep 23, 75 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Almoguera B et al. Identification of Four Novel Loci in Asthma in European American and African American Populations. Am J Respir Crit Care Med 195, 456–463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minelli C et al. Glutathione-S-transferase genes and asthma phenotypes: a Human Genome Epidemiology (HuGE) systematic review and meta-analysis including unpublished data. Int J Epidemiol 39, 539–562 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee S-Y et al. Modification of additive effect between vitamins and ETS on childhood asthma risk according to GSTP1 polymorphism: a cross -sectional study. BMC Pulm Med 15, 125 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muñoz B et al. The relationship among IL-13, GSTP1, and CYP1A1 polymorphisms and environmental tobacco smoke in a population of children with asthma in Northern Mexico. Environ Toxicol Pharmacol 33, 226–232 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Dai X et al. Do Glutathione S-Transferase Genes Modify the Link between Indoor Air Pollution and Asthma, Allergies, and Lung Function? A Systematic Review. Curr Allergy Asthma Rep 18, 20 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Hoskins A, Wu P, Reiss S & Dworski R Glutathione S-transferase P1 Ile105Val polymorphism modulates allergen-induced airway inflammation in human atopic asthmatics in vivo. Clin Exp Allergy 43, 527–534 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haley RW, Kramer G, Xiao J, Dever JA & Teiber JF Evaluation of a Gene-Environment Interaction of PON1 and Low-Level Nerve Agent Exposure with Gulf War Illness: A Prevalence Case-Control Study Drawn from the U.S. Military Health Survey’s National Population Sample. Environ Health Perspect 130, 57001 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weisskopf MG Response to ‘Comment on “Evaluation of a Gene-Environment Interaction of PON1 and Low-Level Nerve Agent Exposure with Gulf War Illness: A Prevalence Case-Control Study Drawn from the U.S. Military Health Survey’s National Population Sample”‘. Environ Health Perspect 130, 68005 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith PG & Day NE The design of case-control studies: the influence of confounding and interaction effects. Int J Epidemiol 13, 356–365 (1984). [DOI] [PubMed] [Google Scholar]
- 23.Gauderman WJ et al. A Unified Model for the Analysis of Gene-Environment Interaction. Am J Epidemiol 188, 760–767 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang P, Lewinger JP, Conti D, Morrison JL & Gauderman WJ Detecting Gene-Environment Interactions for a Quantitative Trait in a Genome-Wide Association Study. Genet Epidemiol 40, 394–403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H et al. Genotype-by-environment interactions inferred from genetic effects on phenotypic variability in the UK Biobank. Sci Adv 5, eaaw3538 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlsten C et al. Genes, the environment and personalized medicine: We need to harness both environmental and genetic data to maximize personal and population health. EMBO Rep 15, 736–739 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore LD, Le T & Fan G DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhutani N, Burns DM & Blau HM DNA demethylation dynamics. Cell 146, 866–872 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinberg DN et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee K et al. Integrated Analysis of Tissue-Specific Promoter Methylation and Gene Expression Profile in Complex Diseases. Int J Mol Sci 21, E5056 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26, 577–590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huff RD, Carlsten C & Hirota JA An update on immunologic mechanisms in the respiratory mucosa in response to air pollutants. J Allergy Clin Immunol 143, 1989–2001 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Cui A et al. VCAM-1-mediated neutrophil infiltration exacerbates ambient fine particle-induced lung injury. Toxicol Lett 302, 60–74 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Pope CA et al. Exposure to Fine Particulate Air Pollution Is Associated With Endothelial Injury and Systemic Inflammation. Circ Res 119, 1204–1214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kohli RM & Zhang Y TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leclercq B et al. Genetic and epigenetic alterations in normal and sensitive COPD-diseased human bronchial epithelial cells repeatedly exposed to air pollution-derived PM2.5. Environ Pollut 230, 163–177 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Rider CF & Carlsten C Air pollution and DNA methylation: effects of exposure in humans. Clin Epigenetics 11, 131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang SK et al. Effect of concentration and duration of particulate matter exposure on the transcriptome and DNA methylome of bronchial epithelial cells. Environ Epigenet 7, dvaa022 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carmona JJ et al. Short-term airborne particulate matter exposure alters the epigenetic landscape of human genes associated with the mitogen-activated protein kinase network: a cross-sectional study. Environ Health 13, 94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang Y et al. TET2 promotes IL-1β expression in J774.1 cell through TLR4/MAPK signaling pathway with demethylation of TAB2 promoter. Mol Immunol 126, 136–142 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Chen R et al. DNA hypomethylation and its mediation in the effects of fine particulate air pollution on cardiovascular biomarkers: A randomized crossover trial. Environ Int 94, 614–619 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Peng C et al. Particulate Air Pollution and Fasting Blood Glucose in Nondiabetic Individuals: Associations and Epigenetic Mediation in the Normative Aging Study, 2000–2011. Environ Health Perspect 124, 1715–1721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tantoh DM et al. AHRR cg05575921 methylation in relation to smoking and PM2.5 exposure among Taiwanese men and women. Clin Epigenetics 12, 117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jhun MA et al. Modeling the Causal Role of DNA Methylation in the Association Between Cigarette Smoking and Inflammation in African Americans: A 2-Step Epigenetic Mendelian Randomization Study. Am J Epidemiol 186, 1149–1158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joehanes R et al. Epigenetic Signatures of Cigarette Smoking. Circ Cardiovasc Genet 9, 436–447 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernández-Sanlés A et al. DNA methylation biomarkers of myocardial infarction and cardiovascular disease. Clin Epigenetics 13, 86 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kodal JB, Kobylecki CJ, Vedel-Krogh S, Nordestgaard BG & Bojesen SE AHRR hypomethylation, lung function, lung function decline and respiratory symptoms. Eur Respir J 51, 1701512 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Bollepalli S, Korhonen T, Kaprio J, Anders S & Ollikainen M EpiSmokEr: a robust classifier to determine smoking status from DNA methylation data. Epigenomics 11, 1469–1486 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Park SK, Zhao Z & Mukherjee B Construction of environmental risk score beyond standard linear models using machine learning methods: application to metal mixtures, oxidative stress and cardiovascular disease in NHANES. Environ Health 16, 102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benowitz NL et al. Prevalence of smoking assessed biochemically in an urban public hospital: a rationale for routine cotinine screening. Am J Epidemiol 170, 885–891 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsieh SJ et al. Biomarkers increase detection of active smoking and secondhand smoke exposure in critically ill patients. Crit Care Med 39, 40–45 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horvath S DNA methylation age of human tissues and cell types. Genome Biol 14, R115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Horvath S & Raj K DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet 19, 371–384 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Cardenas A et al. Epigenome-wide association study and epigenetic age acceleration associated with cigarette smoking among Costa Rican adults. Sci Rep 12, 4277 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nwanaji-Enwerem JC et al. Long-term ambient particle exposures and blood DNA methylation age: findings from the VA normative aging study. Environ Epigenet 2, dvw006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu X et al. Effect of tobacco smoking on the epigenetic age of human respiratory organs. Clin Epigenetics 11, 183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lind PM, Salihovic S & Lind L High plasma organochlorine pesticide levels are related to increased biological age as calculated by DNA methylation analysis. Environ Int 113, 109–113 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Fiorito G et al. Socioeconomic position, lifestyle habits and biomarkers of epigenetic aging: a multi-cohort analysis. Aging (Albany NY) 11, 2045–2070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim Y et al. Higher diet quality relates to decelerated epigenetic aging. Am J Clin Nutr 115, 163–170 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim K et al. DNA Methylation GrimAge and Incident Diabetes: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. Diabetes 70, 1404–1413 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roetker NS, Pankow JS, Bressler J, Morrison AC & Boerwinkle E Prospective Study of Epigenetic Age Acceleration and Incidence of Cardiovascular Disease Outcomes in the ARIC Study (Atherosclerosis Risk in Communities). Circ Genom Precis Med 11, e001937 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen BH et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY) 8, 1844–1865 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martire S & Banaszynski LA The roles of histone variants in fine-tuning chromatin organization and function. Nat Rev Mol Cell Biol 21, 522–541 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bannister AJ & Kouzarides T Regulation of chromatin by histone modifications. Cell Res 21, 381–395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cedar H & Bergman Y Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10, 295–304 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Greer EL & Shi Y Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13, 343–357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lismer A, Siklenka K, Lafleur C, Dumeaux V & Kimmins S Sperm histone H3 lysine 4 trimethylation is altered in a genetic mouse model of transgenerational epigenetic inheritance. Nucleic Acids Res 48, 11380–11393 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lismer A et al. Histone H3 lysine 4 trimethylation in sperm is transmitted to the embryo and associated with diet-induced phenotypes in the offspring. Dev Cell 56, 671–686.e6 (2021). [DOI] [PubMed] [Google Scholar]
- 70.Howe CG & Gamble MV Influence of Arsenic on Global Levels of Histone Posttranslational Modifications: A Review of the Literature and Challenges in the Field. Curr Environ Health Rep 3, 225–237 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bhattacharjee P, Paul S & Bhattacharjee P Understanding the mechanistic insight of arsenic exposure and decoding the histone cipher. Toxicology 430, 152340 (2020). [DOI] [PubMed] [Google Scholar]
- 72.Zhang X et al. Arsenic silences hepatic PDK4 expression through activation of histone H3K9 methylatransferase G9a. Toxicol Appl Pharmacol 304, 42–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suzuki T & Nohara K Long-term arsenic exposure induces histone H3 Lys9 dimethylation without altering DNA methylation in the promoter region of p16INK4a and down-regulates its expression in the liver of mice. Journal of Applied Toxicology 33, 951–958 (2013). [DOI] [PubMed] [Google Scholar]
- 74.Bernhart SH et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci Rep 6, 37393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brocato J et al. Arsenic Induces Polyadenylation of Canonical Histone mRNA by Down-regulating Stem-Loop-binding Protein Gene Expression *. Journal of Biological Chemistry 289, 31751–31764 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Costa M Review of arsenic toxicity, speciation and polyadenylation of canonical histones. Toxicology and Applied Pharmacology 375, 1–4 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Wu Y, Wang R, Liu R, Ba Y & Huang H The Roles of Histone Modifications in Metal-Induced Neurological Disorders. Biol Trace Elem Res (2022) doi: 10.1007/s12011-022-03134-5. [DOI] [PubMed] [Google Scholar]
- 78.Ryu YS et al. Particulate matter-induced senescence of skin keratinocytes involves oxidative stress-dependent epigenetic modifications. Exp Mol Med 51, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ji X et al. Histone modification in the lung injury and recovery of mice in response to PM2.5 exposure. Chemosphere 220, 127–136 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Li Z et al. The global DNA and RNA methylation and their reversal in lung under different concentration exposure of ambient air particulate matter in mice. Ecotoxicology and Environmental Safety 172, 396–402 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Gu L-Z, Sun H & Chen J-H Histone deacetylases 3 deletion restrains PM2.5-induced mice lung injury by regulating NF-κB and TGF-β/Smad2/3 signaling pathways. Biomed Pharmacother 85, 756–762 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Ding R et al. Dose- and time- effect responses of DNA methylation and histone H3K9 acetylation changes induced by traffic-related air pollution. Sci Rep 7, 43737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ding R et al. H3K9 acetylation change patterns in rats after exposure to traffic-related air pollution. Environ Toxicol Pharmacol 42, 170–175 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Zhang Z et al. Global H3K79 di-methylation mediates DNA damage response to PAH exposure in Chinese coke oven workers. Environ Pollut 268, 115956 (2021). [DOI] [PubMed] [Google Scholar]
- 85.Chiu K-C et al. Prenatal chlorpyrifos exposure in association with PPARγ H3K4me3 and DNA methylation levels and child development. Environ Pollut 274, 116511 (2021). [DOI] [PubMed] [Google Scholar]
- 86.Fraz S et al. Paternal Exposure to Carbamazepine Impacts Zebrafish Offspring Reproduction Over Multiple Generations. Environ Sci Technol 53, 12734–12743 (2019). [DOI] [PubMed] [Google Scholar]
- 87.Sánchez OF et al. Profiling epigenetic changes in human cell line induced by atrazine exposure. Environ Pollut 258, 113712 (2020). [DOI] [PubMed] [Google Scholar]
- 88.Yuan X et al. Histone acetylation is involved in TCDD‑induced cleft palate formation in fetal mice. Mol Med Rep 14, 1139–1145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li H et al. Bisphenol A Exposure Disrupts Enamel Formation via EZH2-Mediated H3K27me3. J Dent Res 100, 847–857 (2021). [DOI] [PubMed] [Google Scholar]
- 90.Escarda-Castro E, Herráez MP & Lombó M Effects of bisphenol A exposure during cardiac cell differentiation. Environ Pollut 286, 117567 (2021). [DOI] [PubMed] [Google Scholar]
- 91.O’Brien J, Hayder H, Zayed Y & Peng C Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Frontiers in Endocrinology 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xiao M et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol 14, 1326–1334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu X et al. Regulation of microRNAs by epigenetics and their interplay involved in cancer. Journal of Experimental & Clinical Cancer Research 32, 96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hou L, Wang D & Baccarelli A Environmental chemicals and microRNAs. Mutat Res 714, 105–112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ji X et al. MicroRNA-338–5p modulates pulmonary hypertension-like injuries caused by SO2, NO2 and PM2.5 co-exposure through targeting the HIF-1α/Fhl-1 pathway. Toxicol Res (Camb) 5, 1548–1560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rider CF et al. Controlled diesel exhaust and allergen coexposure modulates microRNA and gene expression in humans: Effects on inflammatory lung markers. J Allergy Clin Immunol 138, 1690–1700 (2016). [DOI] [PubMed] [Google Scholar]
- 97.Tsai M-H et al. Urban Particulate Matter Enhances ROS/IL-6/COX-II Production by Inhibiting MicroRNA-137 in Synovial Fibroblast of Rheumatoid Arthritis. Cells 9, E1378 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu G, Li Y, Zhou J, Xu J & Yang B PM2.5 deregulated microRNA and inflammatory microenvironment in lung injury. Environ Toxicol Pharmacol 91, 103832 (2022). [DOI] [PubMed] [Google Scholar]
- 99.Kristensen LS et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20, 675–691 (2019). [DOI] [PubMed] [Google Scholar]
- 100.Li M et al. Circular RNA circBbs9 promotes PM2.5-induced lung inflammation in mice via NLRP3 inflammasome activation. Environ Int 143, 105976 (2020). [DOI] [PubMed] [Google Scholar]
- 101.Zhong Y et al. Identification of long non-coding RNA and circular RNA in mice after intra-tracheal instillation with fine particulate matter. Chemosphere 235, 519–526 (2019). [DOI] [PubMed] [Google Scholar]
- 102.Li Z et al. Differentially expressed circular RNAs in air pollution-exposed rat embryos. Environ Sci Pollut Res Int 26, 34421–34429 (2019). [DOI] [PubMed] [Google Scholar]
- 103.Yamasaki S, Ivanov P, Hu G & Anderson P Angiogenin cleaves tRNA and promotes stress-induced translational repression. The Journal of Cell Biology 185, 35–42 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mishima E et al. Conformational change in transfer RNA is an early indicator of acute cellular damage. J. Am. Soc. Nephrol. 25, 2316–2326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dewe JM, Fuller BL, Lentini JM, Kellner SM & Fu D TRMT1-Catalyzed tRNA Modifications Are Required for Redox Homeostasis To Ensure Proper Cellular Proliferation and Oxidative Stress Survival. Mol Cell Biol 37, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Thompson DM, Lu C, Green PJ & Parker R tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA 14, 2095–2103 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fu Y, Lee I, Lee YS & Bao X Small Non-coding Transfer RNA-Derived RNA Fragments (tRFs): Their Biogenesis, Function and Implication in Human Diseases. Genomics Inform 13, 94–101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Anderson P & Ivanov P tRNA fragments in human health and disease. FEBS Lett 588, 4297–4304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Statello L, Guo C-J, Chen L-L & Huarte M Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol 22, 96–118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li Z et al. Aberrantly expressed long non-coding RNAs in air pollution-induced congenital defects. J Cell Mol Med 23, 7717–7725 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hussey MR et al. Placental lncRNA expression associated with placental cadmium concentrations and birth weight. Environ Epigenet 6, dvaa003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dong Z et al. LncRNA PU.1 AS regulates arsenic-induced lipid metabolism through EZH2/Sirt6/SREBP-1c pathway. J Environ Sci (China) 85, 138–146 (2019). [DOI] [PubMed] [Google Scholar]
- 113.Zhang H et al. LINC00173 Interacts With DNMT1 to Regulate LINC00173 Expression via Promoter Methylation in Hydroquinone-Induced Malignantly Transformed TK6 Cells and Benzene-Exposed Workers. Toxicol Sci 187, 311–324 (2022). [DOI] [PubMed] [Google Scholar]
- 114.Shi J, Deng H & Zhang M Whole transcriptome sequencing analysis revealed key RNA profiles and toxicity in mice after chronic exposure to microplastics. Chemosphere 304, 135321 (2022). [DOI] [PubMed] [Google Scholar]
- 115.Fan Z et al. A study on the roles of long non-coding RNA and circular RNA in the pulmonary injuries induced by polystyrene microplastics. Environ Int 163, 107223 (2022). [DOI] [PubMed] [Google Scholar]
- 116.Zaccara S, Ries RJ & Jaffrey SR Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 1–17 (2019) doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 117.Liu N & Pan T N6-methyladenosine–encoded epitranscriptomics. Nat Struct Mol Biol 23, 98–102 (2016). [DOI] [PubMed] [Google Scholar]
- 118.Jiang X et al. The role of m6A modification in the biological functions and diseases. Sig Transduct Target Ther 6, 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li H-B et al. m6A mRNA methylation controls T cell homeostasis by targeting IL-7/STAT5/SOCS pathway. Nature 548, 338–342 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Furlan M, Galeota E, de Pretis S, Caselle M & Pelizzola M m6A-Dependent RNA Dynamics in T Cell Differentiation. Genes (Basel) 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang H et al. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Baldridge KC & Contreras LM Functional implications of ribosomal RNA methylation in response to environmental stress. Critical Reviews in Biochemistry and Molecular Biology 49, 69–89 (2014). [DOI] [PubMed] [Google Scholar]
- 123.Cayir A, Barrow TM, Guo L & Byun H-M Exposure to environmental toxicants reduces global N6-methyladenosine RNA methylation and alters expression of RNA methylation modulator genes. Environmental Research 175, 228–234 (2019). [DOI] [PubMed] [Google Scholar]
- 124.Yuan Q et al. METTL3 regulates PM2.5-induced cell injury by targeting OSGIN1 in human airway epithelial cells. Journal of Hazardous Materials 415, 125573 (2021). [DOI] [PubMed] [Google Scholar]
- 125.Xia H et al. The aberrant cross-talk of epithelium–macrophages via METTL3-regulated extracellular vesicle miR-93 in smoking-induced emphysema. Cell Biol Toxicol 38, 167–183 (2022). [DOI] [PubMed] [Google Scholar]
- 126.Anders M et al. Dynamic m6A methylation facilitates mRNA triaging to stress granules. Life Science Alliance 1, e201800113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhou J, Wan J, Gao X, Zhang X & Qian S-B Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 526, 591–594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu H et al. Fine particulate matter induces METTL3-mediated m6A modification of BIRC5 mRNA in bladder cancer. J Hazard Mater 437, 129310 (2022). [DOI] [PubMed] [Google Scholar]
- 129.Liu J et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang J et al. Excessive miR-25–3p maturation via N6-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun 10, 1858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wu S et al. A Novel Micropeptide Encoded by Y-Linked LINC00278 Links Cigarette Smoking and AR Signaling in Male Esophageal Squamous Cell Carcinoma. Cancer Research 80, 2790–2803 (2020). [DOI] [PubMed] [Google Scholar]
- 132.Ning J et al. N6-Methyladenosine Modification of CDH1 mRNA Promotes PM2.5-Induced Pulmonary Fibrosis via Mediating Epithelial Mesenchymal Transition. Toxicol Sci 185, 143–157 (2022). [DOI] [PubMed] [Google Scholar]
- 133.Kupsco A et al. Associations of smoking and air pollution with peripheral blood RNA N6-methyladenosine in the Beijing truck driver air pollution study. Environment International 144, 106021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhao T et al. N6-methyladenosine mediates arsenite-induced human keratinocyte transformation by suppressing p53 activation. Environmental Pollution 259, 113908 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhao T, Li X, Sun D & Zhang Z Oxidative stress: One potential factor for arsenite-induced increase of N6-methyladenosine in human keratinocytes. Environmental Toxicology and Pharmacology 69, 95–103 (2019). [DOI] [PubMed] [Google Scholar]
- 136.Gu S, Sun D, Dai H & Zhang Z N6-methyladenosine mediates the cellular proliferation and apoptosis via microRNAs in arsenite-transformed cells. Toxicology Letters 292, 1–11 (2018). [DOI] [PubMed] [Google Scholar]
- 137.Cui Y-H et al. Autophagy of the m6A mRNA demethylase FTO is impaired by low-level arsenic exposure to promote tumorigenesis. Nat Commun 12, 2183 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bai L et al. m6A Demethylase FTO Regulates Dopaminergic Neurotransmission Deficits Caused by Arsenite. Toxicol Sci 165, 431–446 (2018). [DOI] [PubMed] [Google Scholar]
- 139.Tang J et al. Global N6-methyladenosine profiling of cobalt-exposed cortex and human neuroblastoma H4 cells presents epitranscriptomics alterations in neurodegenerative disease-associated genes. Environmental Pollution 266, 115326 (2020). [DOI] [PubMed] [Google Scholar]
- 140.Qi Z et al. Protective role of mRNA demethylase FTO on axon guidance molecules of nigro-striatal projection system in manganese-induced parkinsonism. Journal of Hazardous Materials 426, 128099 (2022). [DOI] [PubMed] [Google Scholar]
- 141.Wang Z et al. Chronic Hexavalent Chromium Exposure Upregulates the RNA Methyltransferase METTL3 Expression to Promote Cell Transformation, Cancer Stem Cell-Like Property, and Tumorigenesis. Toxicol Sci 187, 51–61 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yue Y et al. N6-methyladenosine-mediated downregulation of miR-374c-5p promotes cadmium-induced cell proliferation and metastasis by targeting GRM3 in breast cancer cells. Ecotoxicol Environ Saf 229, 113085 (2022). [DOI] [PubMed] [Google Scholar]
- 143.Klinge CM et al. Combined exposure to polychlorinated biphenyls and high-fat diet modifies the global epitranscriptomic landscape in mouse liver. Environ Epigenet 7, dvab008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aluru N & Karchner SI PCB126 Exposure Revealed Alterations in m6A RNA Modifications in Transcripts Associated With AHR Activation. Toxicol Sci 179, 84–94 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yin X et al. The intergenerational toxic effects on offspring of medaka fish Oryzias melastigma from parental benzo[a]pyrene exposure via interference of the circadian rhythm. Environ Pollut 267, 115437 (2020). [DOI] [PubMed] [Google Scholar]
- 146.Sun L et al. Triclosan-induced abnormal expression of miR-30b regulates fto-mediated m6A methylation level to cause lipid metabolism disorder in zebrafish. Sci Total Environ 770, 145285 (2021). [DOI] [PubMed] [Google Scholar]
- 147.Qi X et al. Comprehensive analysis of differences of N6-methyladenosine of lncRNAs between atrazine-induced and normal Xenopus laevis testis. Genes Environ 43, 49 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Su Q et al. Paraquat-induced oxidative stress regulates N6-methyladenosine (m6A) modification of long noncoding RNAs in Neuro-2a cells. Ecotoxicol Environ Saf 237, 113503 (2022). [DOI] [PubMed] [Google Scholar]
- 149.Rompala GR et al. Heavy Chronic Intermittent Ethanol Exposure Alters Small Noncoding RNAs in Mouse Sperm and Epididymosomes. Front Genet 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wiener D & Schwartz S The epitranscriptome beyond m6A. Nat Rev Genet 22, 119–131 (2021). [DOI] [PubMed] [Google Scholar]
- 151.Eckhardt CM, Baccarelli AA & Wu H Environmental Exposures and Extracellular Vesicles: Indicators of Systemic Effects and Human Disease. Curr Environ Health Rep 9, 465–476 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Valadi H et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9, 654–659 (2007). [DOI] [PubMed] [Google Scholar]
- 153.Bollati V et al. Microvesicle-associated microRNA expression is altered upon particulate matter exposure in healthy workers and in A549 cells. J Appl Toxicol 35, 59–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Emmerechts J et al. Air pollution-associated procoagulant changes: the role of circulating microvesicles. J Thromb Haemost 10, 96–106 (2012). [DOI] [PubMed] [Google Scholar]
- 155.Zheng R et al. Fine Particulate Matter Induces Childhood Asthma Attacks via Extracellular Vesicle-Packaged Let-7i-5p-Mediated Modulation of the MAPK Signaling Pathway. Adv Sci (Weinh) 9, e2102460 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Neri T et al. Particulate matter induces prothrombotic microparticle shedding by human mononuclear and endothelial cells. Toxicol In Vitro 32, 333–338 (2016). [DOI] [PubMed] [Google Scholar]
- 157.Chen X et al. Urban airborne PM2.5-activated microglia mediate neurotoxicity through glutaminase-containing extracellular vesicles in olfactory bulb. Environ Pollut 264, 114716 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ngalame NNO, Luz AL, Makia N & Tokar EJ Arsenic Alters Exosome Quantity and Cargo to Mediate Stem Cell Recruitment Into a Cancer Stem Cell-Like Phenotype. Toxicol Sci 165, 40–49 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen C et al. NF-kB-regulated exosomal miR-155 promotes the inflammation associated with arsenite carcinogenesis. Cancer Lett 388, 21–33 (2017). [DOI] [PubMed] [Google Scholar]
- 160.Xu Y et al. Exosomal miR-21 derived from arsenite-transformed human bronchial epithelial cells promotes cell proliferation associated with arsenite carcinogenesis. Arch Toxicol 89, 1071–1082 (2015). [DOI] [PubMed] [Google Scholar]
- 161.Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y & Taketani Y Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod 17, 2839–2841 (2002). [DOI] [PubMed] [Google Scholar]
- 162.Rodosthenous RS et al. Supraphysiological Concentrations of Bisphenol A Alter the Expression of Extracellular Vesicle-Enriched miRNAs From Human Primary Granulosa Cells. Toxicol Sci 169, 5–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Martinez RM et al. Urinary concentrations of phenols and phthalate metabolites reflect extracellular vesicle microRNA expression in follicular fluid. Environ Int 123, 20–28 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Barnett-Itzhaki Z et al. Association between follicular fluid phthalate concentrations and extracellular vesicle microRNAs expression. Hum Reprod 36, 1590–1599 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sheller-Miller S et al. Environmental pollutant induced cellular injury is reflected in exosomes from placental explants. Placenta 89, 42–49 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Miliotis S, Nicolalde B, Ortega M, Yepez J & Caicedo A Forms of extracellular mitochondria and their impact in health. Mitochondrion 48, 16–30 (2019). [DOI] [PubMed] [Google Scholar]
- 167.Eckhardt CM et al. Extracellular Vesicle-Encapsulated microRNAs as Novel Biomarkers of Lung Health. Am J Respir Crit Care Med (2022) doi: 10.1164/rccm.202109-2208OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Johannsen DL & Ravussin E The role of mitochondria in health and disease. Curr Opin Pharmacol 9, 780–786 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Reddam A, McLarnan S & Kupsco A Environmental Chemical Exposures and Mitochondrial Dysfunction: a Review of Recent Literature. Curr Envir Health Rpt (2022) doi: 10.1007/s40572-022-00371-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Frye RE et al. Prenatal air pollution influences neurodevelopment and behavior in autism spectrum disorder by modulating mitochondrial physiology. Mol Psychiatry 26, 1561–1577 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li Z et al. Air pollution and placental mitochondrial DNA copy number: Mechanistic insights and epidemiological challenges. Environ Pollut 255, 113266 (2019). [DOI] [PubMed] [Google Scholar]
- 172.Pardo M, Qiu X, Zimmermann R & Rudich Y Particulate Matter Toxicity Is Nrf2 and Mitochondria Dependent: The Roles of Metals and Polycyclic Aromatic Hydrocarbons. Chem Res Toxicol 33, 1110–1120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Huffman AM et al. Associations of urinary phthalate metabolites and lipid peroxidation with sperm mitochondrial DNA copy number and deletions. Environ Res 163, 10–15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen X et al. Prenatal exposure to benzotriazoles and benzothiazoles and cord blood mitochondrial DNA copy number: A prospective investigation. Environ Int 143, 105920 (2020). [DOI] [PubMed] [Google Scholar]
- 175.Giordano L et al. Cigarette toxicity triggers Leber’s hereditary optic neuropathy by affecting mtDNA copy number, oxidative phosphorylation and ROS detoxification pathways. Cell Death Dis 6, e2021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wang Y et al. The Association Between Polymorphisms in Cell-Cycle Genes and Mitochondrial DNA Copy Number in Coke Oven Workers. Front Public Health 10, 904856 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ji B et al. Gene-Environment Interactions Between Environmental Response Genes Polymorphisms and Mitochondrial DNA Copy Numbers Among Benzene Workers. J Occup Environ Med 63, e408–e415 (2021). [DOI] [PubMed] [Google Scholar]
- 178.Gaikwad AS, Mahmood R, Beerappa R, Karunamoorthy P & Venugopal D Mitochondrial DNA copy number and cytogenetic damage among fuel filling station attendants. Environ Mol Mutagen 61, 820–829 (2020). [DOI] [PubMed] [Google Scholar]
- 179.Sharma J et al. Emerging role of mitochondria in airborne particulate matter-induced immunotoxicity. Environ Pollut 270, 116242 (2021). [DOI] [PubMed] [Google Scholar]
- 180.Sun Q et al. Heavy metals induced mitochondrial dysfunction in animals: Molecular mechanism of toxicity. Toxicology 469, 153136 (2022). [DOI] [PubMed] [Google Scholar]
- 181.Heinz S et al. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci Rep 7, 45465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Won J-H, Park S, Hong S, Son S & Yu J-W Rotenone-induced Impairment of Mitochondrial Electron Transport Chain Confers a Selective Priming Signal for NLRP3 Inflammasome Activation. J Biol Chem 290, 27425–27437 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cochemé HM & Murphy MP Complex I is the major site of mitochondrial superoxide production by paraquat. J Biol Chem 283, 1786–1798 (2008). [DOI] [PubMed] [Google Scholar]
- 184.Rottenberg H & Hoek JB The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 16, 943–955 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Guo C, Sun L, Chen X & Zhang D Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res 8, 2003–2014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hollensworth SB et al. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic Biol Med 28, 1161–1174 (2000). [DOI] [PubMed] [Google Scholar]
- 187.Billiard SM et al. Binding of polycyclic aromatic hydrocarbons (PAHs) to teleost aryl hydrocarbon receptors (AHRs). Comp Biochem Physiol B Biochem Mol Biol 133, 55–68 (2002). [DOI] [PubMed] [Google Scholar]
- 188.Hwang HJ et al. Mitochondrial-targeted Aryl Hydrocarbon Receptor and the Impact of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin on Cellular Respiration and the Mitochondrial Proteome. Toxicol Appl Pharmacol 304, 121–132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Caito SW & Aschner M Mitochondrial Redox Dysfunction and Environmental Exposures. Antioxid Redox Signal 23, 578–595 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Stewart JB & Chinnery PF The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet 16, 530–542 (2015). [DOI] [PubMed] [Google Scholar]
- 191.Harari S et al. Fibrotic interstitial lung diseases and air pollution: a systematic literature review. Eur Respir Rev 29, 200093 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Vaissière T, Sawan C & Herceg Z Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutation Research/Reviews in Mutation Research 659, 40–48 (2008). [DOI] [PubMed] [Google Scholar]
- 193.Aure MR et al. Crosstalk between microRNA expression and DNA methylation drives the hormone-dependent phenotype of breast cancer. Genome Medicine 13, 72 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Uppal K, Ma C, Go Y-M, Jones DP & Wren J xMWAS: a data-driven integration and differential network analysis tool. Bioinformatics 34, 701–702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Peng C et al. A latent unknown clustering integrating multi-omics data (LUCID) with phenotypic traits. Bioinformatics 36, 842–850 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chen J & Nodzak C Statistical and Machine Learning Methods for eQTL Analysis. Methods Mol Biol 2082, 87–104 (2020). [DOI] [PubMed] [Google Scholar]
- 197.Kanehisa M & Goto S KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ashburner M et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Buccitelli C & Selbach M mRNAs, proteins and the emerging principles of gene expression control. Nat Rev Genet 21, 630–644 (2020). [DOI] [PubMed] [Google Scholar]
- 200.Avila Cobos F, Vandesompele J, Mestdagh P & De Preter K Computational deconvolution of transcriptomics data from mixed cell populations. Bioinformatics 34, 1969–1979 (2018). [DOI] [PubMed] [Google Scholar]
- 201.Mustapic M et al. Plasma Extracellular Vesicles Enriched for Neuronal Origin: A Potential Window into Brain Pathologic Processes. Front Neurosci 11, 278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kapogiannis D et al. Association of Extracellular Vesicle Biomarkers With Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol 76, 1340–1351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zhang P et al. Defining the Scope of Exposome Studies and Research Needs from a Multidisciplinary Perspective. Environ Sci Technol Lett 8, 839–852 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wild CP Complementing the Genome with an “Exposome”: The Outstanding Challenge of Environmental Exposure Measurement in Molecular Epidemiology. Cancer Epidemiol Biomarkers Prev 14, 1847–1850 (2005). [DOI] [PubMed] [Google Scholar]