

Summary
The major cause of treatment failure and mortality among medulloblastoma patients is metastasis intracranially or along the spinal cord. The molecular mechanisms driving tumor metastasis in Sonic hedgehog-driven medulloblastoma (SHH-MB) patients, however, remain largely unknown. In this study we define a tumor suppressive role of KMT2D (MLL2), a gene frequently mutated in the most metastatic β-subtype. Strikingly, genetic mouse models of SHH-MB demonstrate that heterozygous loss of Kmt2d in conjunction with activation of the SHH pathway causes highly penetrant disease with decreased survival, increased hindbrain invasion and spinal cord metastasis. Loss of Kmt2d attenuates neural differentiation and shifts the transcriptional/chromatin landscape of primary and metastatic tumors toward a decrease in differentiation genes and tumor suppressors and an increase in genes/pathways implicated in advanced stage cancer and metastasis (TGFβ, Notch, Atoh1, Sox2, and Myc). Thus, secondary heterozygous KMT2D mutations likely have prognostic value for identifying SHH-MB patients prone to develop metastasis.
Subject areas: Genetics, Cancer, Model organism
Graphical abstract
Highlights
-
•
Heterozygous and homozygous loss of Kmt2d decreases survival in SHH-MB
-
•
Kmt2d mutations increase spinal cord metastasis in two mouse models of SHH-MB
-
•
Tumors lacking Kmt2d have decreased expression of neural differentiation genes
-
•
Tumors lacking Kmt2d upregulate stem cell maintenance and metastasis genes
Genetics; Cancer; Model organism
Introduction
The cerebellar tumor medulloblastoma (MB) is the most common malignant pediatric brain tumor, and encompasses 4 major disease subgroups: Wingless-related integration site (WNT), Sonic hedgehog (SHH), group 3, and group 4.1,2,3,4,5 Metastasis is the major cause of morbidity and mortality among MB patients and occurs most commonly intracranially or along the spinal cord, as a result of hematogenous or leptomeningeal dissemination (LMD).6,7,8,9,10,11,12 SHH-MB accounts for ∼30% of all MB cases and has an intermediate prognosis with a metastasis rate of 15–35%.4,13,14,15,16,17,18 Of the four subgroups of MB, mouse models of SHH-MB are the most advanced and have been used to implicate over-expression of several genes including Atoh1 in promoting metastasis with LMD throughout the brain and spinal cord.8,11,19,20 However, the human correlates of the mutations in SHH-MB disease are not clear. Given the lifelong neurological sequalae suffered by patients that survive surgery and treatments for SHH-MB, identification of prognostic markers and potential druggable targets are essential to improve survival and quality of life, especially for treating metastatic disease.
SHH-MB arises from granule cell precursors (GCPs) that cover the surface of the cerebellum21,22,23,24 during the third trimester and first year of life25 and tumors form preferentially in the lateral cerebellar hemispheres.26,27,28,29 GCPs are dependent on SHH-signaling and ATOH1 to maintain a proliferative progenitor state.30,31,32 Most patients within this subgroup harbor mutations at one of four genes that lead to constitutive activation of the SHH pathway: loss-of-function mutations in PTCH1 or SUFU, gain-of-function mutations in SMO and TP53 mutations associated with amplification of GLI2 or MYCN.33,34,35,36 SHH-MB has been deconstructed into several subtypes based on patient demographics, genomic and epigenomic landscapes, tumor histology, metastatic status, and clinical outcomes.37,38,39,40,41 A widely accepted subtyping categorizes SHH-MB into 4 subtypes: α, β, γ, and δ.38,41 The β subtype occurs most frequently in infants and has the highest incidence of metastasis (30–35%) and the worst 5-year survival rate (65–70%).34,41,42 Mutations in PTCH1 and SUFU that activate SHH signaling are prevalent in the β-subtype of SHH-MB.34,41,42 In addition, recurrent mutations in KMT2D are preferentially seen in this subtype of SHH-MB;34,41,42 however, the consequences of such mutations on disease progression and metastasis have not been explored in animal models.
Somatic mono-allelic mutations in KMT2D (also known as MLL2 in humans and Mll4 in mouse) that are predicted to result in a truncated protein are seen in 8–15% of SHH-MB patients.34,36,42,43,44,45,46 KMT2D is a SET-domain containing histone-lysine N-methyltransferase that mono-, di-, or tri-methylates (me1, me2, me3) lysine 4 on histone H3 (H3K4) preferentially at gene enhancers or promoters to generate a mark associated with active transcription or a poised bivalent state of genes.47,48,49,50 The roles described for KMT2D during development and tumor progression are context-dependent,50,51 and little is known about its role in medulloblastoma. Mice with a nervous system-specific floxed conditional mutation in Kmt2d (exons 16–19; Nestin-Cre/+; Kmt2df/f) were reported to be viable and have a morphologically normal cerebellum at one month of age, although granule neurons and Purkinje cells appeared to lack some mature marker proteins.50 Nonetheless, 35% of male homozygous mutants went on to develop MB-like tumor growths in the cerebellum by 7 months. Based on transcriptional profiling of three lesions, the tumors were described to best resemble group 3 MB.50 This result indicates that KMT2D is a tumor suppressor in MB. However, a role in SHH-MB or in metastasis has not been addressed.
Given the prevalence of KMT2D mutations in SHH-MBs and enrichment in the β-subtype (12–30% in Garcia-Lopez et al. and Skowron et al.),34,42 we tested the impact of heterozygous and homozygous Kmt2d mutations in the context of mouse models of SHH-MB. Using two sporadic genetic models, Smo activation (SmoM2) and Ptch1 loss (Ptch1fl/fl) combined with N- and C-terminal floxed truncating alleles of Kmt2d we show that heterozygous or homozygous loss of Kmt2d in rare postnatal GCPs greatly accelerates SHH-MB tumorigenesis and penetrance compared to mice with only a SHH pathway activating mutation. Furthermore, loss of Kmt2d drives tumor cell invasion into the surrounding brain. Most notably, heterozygous loss of Kmt2d is sufficient to drive fully penetrant leptomeningeal metastasis to the spinal cord, a hallmark of advanced stage disease. Transcriptomic and chromatin analyses revealed major differences between primary tumors with intact or mutant Kmt2d in the context of SmoM2 expression but the metastatic landscape of Kmt2d mutant cells is similar to primary Kmt2d mutant tumors. Of likely significance, tumors lacking Kmt2d have upregulation of genes involved in pathways implicated in stem cell maintenance and cellular processes associated with tumor metastasis including cell migration and epithelial to mesenchymal transition (EMT). The data indicate the mechanism involves KMT2D normally augmenting expression of neural differentiation genes and repressors of Notch and TGF signaling. Our findings thus reveal that Kmt2d is a potent suppressor of SHH-MB tumor growth, invasion, and metastasis, and when reduced changes the transcriptional/chromatin landscape of SHH-MB into an aggressive disease increasing oncogenic programs including TGFβ,52 Notch,50,53 MYC,54,55 ATOH1,11 and SOX2/9.56,57,58,59,60
Results
KMT2D mutations are prevalent in SHH-MB and co-occur with mutations that activate SHH signaling
To gain insight into the significance of KMT2D mutations in SHH-MB, we determined the frequency of mutations in KMT2D and its homolog KMT2C, as well as mutations that activate SHH-signaling in 127 patient samples considered to be SHH-MB36 using the cBioPortal.61,62 Like previous studies, we found that KMT2D is mutated in ∼14% of the samples (Figure 1A), whereas KMT2C mutations are less prevalent (∼6%) (Figure 1A). Furthermore, in 66% of the samples the mutations in KMT2D occur in conjunction with mutations in genes that lead to activation of the SHH pathway (SMO, PTCH1, SUFU, and TP53) (Figure 1A). Thus, KMT2D mutations are recurrent in SHH-MB and frequently co-occur with SHH pathway activating mutations, indicating KMT2D mutations are secondary mutations that might impact tumor progression.
Figure 1.

Two mutations in Kmt2d delay early cerebellum development but Kmt2d loss does not drive SHH-MB tumorigenesis
(A) An oncoprint of 127 patient samples considered to be SHH-MB (Kool et al., 2014) using the cBioPortal (Cerami et al., 2012; Gao et al., 2013a) showing the frequency of mutations in KMT2C/D and genes that activate the SHH pathway.
(B) Schematic of mouse Kmt2dNf floxed allele with loxP sites surrounding exons 16–19.63
(C) Schematic of mouse Kmt2dCf floxed allele with loxP sites surrounding exons 50 and 51.64
(D and E) H&E-stained mid-sagittal sections from E18.5 animals of the indicated genotypes with the EGL length in black and convex cerebellar length in red. Scale bars: 500 μm.
(F–I) Quantification at E18.5 of the cerebellar area (mm2) (F and H) and Folding Index ((1-[convex length/EGl length]) x 100) in control (Kmt2dNf/+ and Kmt2dNf/Nf or Kmt2dCf/+ and Kmt2dCf/Cf embryos (black dots) or Nes-Cre/+; Kmt2dNf/+ or Nes-Cre/+; Kmt2dCf/+ (blue dots)) and conditional mutants (red dots) (G and I).
(J–M) Images of H&E-stained mid-sagittal sections from a control and Kmt2dCf/Cf conditional mutants using the three Cre drivers at adult stages (P250-P300). Scale bars: 1 mm. Statistical significance was determined using an unpaired t-test. Error bars: SD. Also see Figure S1.
N-terminal and C-terminal mutations in Kmt2d transiently disrupt cerebellum development
Before testing whether mutations in Kmt2d impact disease severity in sporadic mouse models of SHH-MB with low penetrance, we revisited whether Kmt2d alone plays a role in cerebellum development. Since several mouse conditional mutant alleles of Kmt2d have been generated, we first tested the consequences of conditional deletion of Kmt2d using the Nestin-Cre transgene utilized previously by Dhar et al. (2018) to delete the same N-terminal exons (16–19) in the brain (floxed allele referred to as Nf;63 [Figure 1B]) or C-terminal exons 50–51 (floxed allele referred to as Cf;64 [Figure 1C]). Surprisingly, we found that both Nes-Cre/+; Kmt2dNf/Nf and Nes-Cre/+; Kmt2dCf/Cf conditional mutants do not survive past birth (n > 85 and n > 50 animals, respectively examined from crosses of Nes-Cre/+; Kmt2df/+ X Kmt2df/f mice). Embryonic day (E) 18 brains from both homozygous mutants had similar sized cerebella to littermate controls but surface folding of the cerebella was significantly reduced in both mutants suggesting a decrease in expansion of the GCPs in the external granule layer (EGL)65 (Figures 1D–1I). Consistent with this, the area of the EGL was reduced in E18.5 Nes-Cre/+; Kmt2dCf/Cf mutants compared to controls (Figure S1A) and the proportion of the EGL made up of differentiating cells was reduced (Figure S1B), although the proliferation rate of GCPs was not reduced at E18.5 in Nes-Cre/+; Kmt2dCf/Cf mutants (Figure S1C). These results indicates Kmt2d normally promotes differentiation of GCPs.
As a means to circumvent the lethality seen in our Nes-Cre conditional mutants, we deleted Kmt2d in three lineages of the embryonic cerebellum which together comprise all the cell types in the cerebellum: (1) the rhombic lip excitatory neuron lineage (including GCPs) using an Atoh1-Cre transgene,66 (2) the ventricular zone inhibitory neuron lineage (Purkinje cells and interneurons) using a Ptf1aCre allele,67 and (3) the ventricular zone-derived Bergmann glia, astrocytes, late born interneurons and rhombic-lip-derived GCPs and unipolar brush cells using a hGFAP-Cre transgene.68 We found that the Atoh1-Cre; Kmt2dCf/Cf, Ptf1aCre/+; Ktm2dCf/Cf, and hGFAP-Cre; Ktm2dCf/Cf conditional mutants survived to adulthood, and moreover when aged to >P200 none had cerebellar tumors or major foliation or size defects (n = 25, 25, and 15, respectively; Figures 1J–1M). Furthermore, in P30 mutants, granule cell and Purkinje cell marker protein expression (NeuN and Calbindin, respectively) appeared normal compared to littermate controls (n = 3; Figures S1D–S1E′). Thus, in our mouse genetic background nervous system homozygous conditional mutation of Kmt2d using two distinct alleles of Kmt2d cause similar cerebellar developmental defects at E18.5 and lethality at birth, whereas lineage-specific Ktm2dCf/Cf homozygous deletion alone does not result in lethality or lead to cerebellar tumor formation.
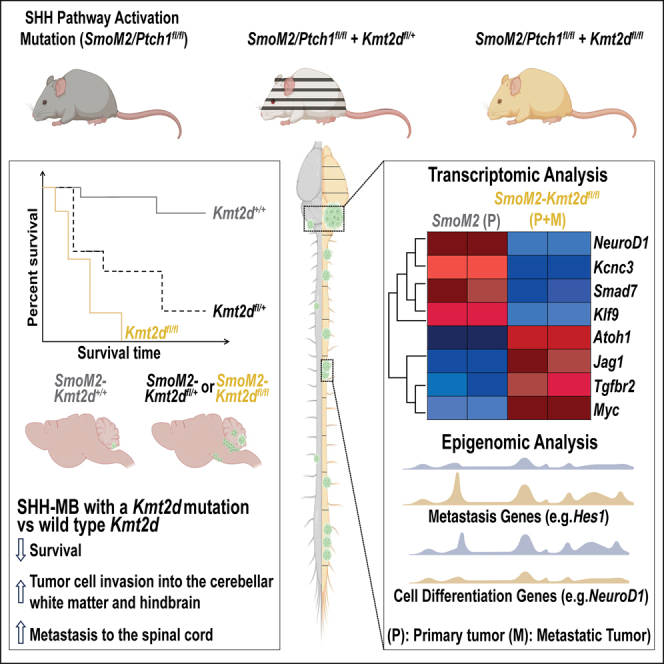
Secondary loss-of-function mutations in Kmt2d greatly increase SHH-MB tumor penetrance
We next tested whether loss of Kmt2d in two sporadic mouse models of SHH-MB with low penetrance enhance tumor formation. To closely model human SHH-MBs we used a Flp-inducible eGFP-Cre approach (MASTR: mosaic analysis with spatial and temporal control of recombination) in which Atoh1-FlpoER and tamoxifen induces simultaneous expression of SmoM2 or deletion of both Ptch1 alleles along with a heterozygous or homozygous deletion of Kmt2d in ∼10,000 scattered GCPs at P2 (Figures 2A and 2B).29,69 Strikingly, with the SmoM2 model and C-terminal exon deletion of Kmt2d, we found that compared to control animals with intact Kmt2d (Atoh1-FlpoER/+; R26MASTR/SmoM2 called SmoM2; n = 25), Atoh1-FlpoER/+; R26MASTR/SmoM2; Kmt2dCf/+ (SmoM2-Kmt2dCf/+; n = 31) and Atoh1-FlpoER/+; R26MASTR/SmoM2; Kmt2dCf/Cf (SmoM2-Kmt2dCf/Cf; n = 38) mice had significantly different survival curves (Figure 2C). Whereas only 56% of SmoM2 animals showed signs of tumor burden by P150, 87% of SmoM2-Kmt2dCf/+ and 100% of SmoM2-Kmt2dCf/Cf did. We similarly found that a heterozygous or homozygous deletion of N-terminal exons of Kmt2d in the SmoM2 model (SmoM2-Kmt2dNf/+ or SmoM2-Kmt2dNf/Nf, respectively) resulted in significantly different survival curves compared to SmoM2 (Figure 2D). Penetrance by P150 was increased from 54% in SmoM2 (n = 13) to 95% in SmoM2-Kmt2dNf/+ (n = 17) and 100% in SmoM2-Kmt2dNf/Nf (n = 7) animals (Figure 2D). Since SHH-MB tumor biology is influenced by the type of mutation that activates SHH signaling,29,36,42 we tested whether the Cf mutation in Kmt2d in a Ptch1 model accelerates SHH-MB tumorigenesis. Indeed, Ptch1-Kmt2dCf/+ (Atoh1-FlpoER/+; R26MASTR/+; Ptch1f/f; Kmt2dCf/+; n = 19) and Ptch1-Kmt2dCf/Cf (Atoh1-FlpoER/+; R26MASTR/+; Ptch1f/f; Kmt2dCf/Cf; n = 14) animals had significantly altered survival curves compared to Ptch1 (Atoh1-FlpoER/+; R26MASTR/+; Ptch1f/f) (n = 41) (Figure 2E), with an increase in tumor penetrance by P200 from 27% in Ptch1 mutants to 71% in Kmt2d heterozygotes and 93% in homozygotes. Of note, mutations in Kmt2d did not alter tumor histology—all genotypes had tumors with classic histology (Figures 2F–2N).
Figure 2.

Two Kmt2d deletion mutations enhance tumor progression in SmoM2-driven SHH-MB and Kmt2d loss also enhances tumor progression when Ptch1 is deleted
(A and B) Schematic showing the two sporadic mouse models of SHH-MB used, SmoM2 (A) and Ptch1fl/fl (B) with and without a heterozygous or homozygous loss of Kmt2d (S: Stop of transcription/translation sequence).
(C–E) Kaplan-Meier curves (left) and statistics (right) for SHH-MB survival using a SmoM2-driven mouse model with a C-terminal (C) or N-terminus (D) heterozygous or homozygous mutation in Kmt2d, and a Ptch1 loss-of-function model with C-terminal mutations of Kmt2d. Statistical significance was calculated using the log-rank test.
(F–N) Representative images of H&E-stained sagittal sections through the hemispheres from end stage SmoM2, SmoM2-Kmt2dCf/+, and SmoM2-Kmt2dCf/Cf animals (F–H), SmoM2, SmoM2-Kmt2dNf/+, and SmoM2-Kmt2dNf/Nf animals (I–K), and Ptch1, Ptch1-Kmt2dCf/+,and Ptch1-Kmt2dCf/Cf animals (L–N). Scale bar: 1 mm.
(O) Overlay of RNA-seq data at the Kmt2d locus in SmoM2 tumors (n = 5), SmoM2-Kmt2dCf/+ tumors (n = 2), and SmoM2-Kmt2dCf/Cf tumors (n = 4). Also see Figure S2.
To confirm that Kmt2d was effectively deleted in homozygous mutant tumor cells and determine if a wild type (WT) allele was present in heterozygotes, we performed bulk RNA-sequencing (RNA-seq) of GFP+ tumor cells isolated by fluorescent activated cell sorting (FACS) from SmoM2, SmoM2-Kmt2dCf/+, and SmoM2-Kmt2dCf/Cf animals (see details in the following; Table S1). Analysis of RNA-seq data confirmed a decrease in read counts for exons 50 and 51 in SmoM2-Kmt2dCf/+ animals compared to other exons and compared to SmoM2 animals, and an absence of sequence reads for exons 50 and 51 in SmoM2-Kmt2dCf/Cf animals (Figure 2O). To test the consequences of loss of Kmt2d on chromatin modification, we performed western blot analysis of whole tumor protein for H3K4me1 and H3K4me3 from end stage animals. The results showed a significant decrease in H3K4me1 and H3K4me3 in SmoM2-Kmt2dCfl/Cf tumors compared to SmoM2 and significant decrease in H3K4me3 in SmoM2-Kmt2dCf/+ animals (Figures S2A–S2C). Taken together, the survival data and molecular results demonstrate that KMT2D has a tumor suppressive role in SHH-MB. Furthermore, in SmoM2-Kmt2dCf/+ mice the decreased survival is due to haploinsufficiency, not loss of heterozygosity during tumor progression.
The pro-tumorigenic effect of loss of Kmt2d can be seen at P21
To assess how early the pro-tumorigenic effects of Kmt2d loss are initiated, tumor histology was analyzed at early stages of tumor development (P12 and P21) using the SmoM2-Kmt2dCf model. GFP staining of mutant cells on sagittal cerebellar hemisphere sections at P12 revealed the EGL was composed almost entirely of mutant GCPs in SmoM2, SmoM2-Kmt2dCf/+ and SmoM2-Kmt2dCf/Cf animals, with no statistically significant differences in the percentage of the cerebellum that contains the EGL in animals with or without Kmt2d in the hemispheres or the vermis (Figures 3A–3E). Quantification of the density of dying cells (#TUNEL+ particles per EGL area) (Figures 3F–3I) and the proliferation rate (#EdU+ cells per EGL area) (Figures 3J–3M) in the thickened EGL in the posterior hemispheres also detected no significant difference in either measurement at P12. Thus, loss of Kmt2d at P2 in GCPs with co-activation of SHH signaling does not lead to an immediate increase in the number of GCPs. At P21, however, when no EGL is present in normal mice, there was a significant increase in the EGL/lesion areas in the hemispheres and vermis of SmoM2-Kmt2dCf/+ and SmoM2-Kmt2dCf/Cf mice compared to in SmoM2 mice (Figures 3N–3R). Thus, the increase in tumor progression seen after loss of Kmt2d in SHH-MB tumors can be seen by P21, a preneoplastic stage of tumorigenesis.
Figure 3.

Heterozygous and homozygous mutations in Kmt2d promote tumor growth by P21
(A–C) Images of GFP stained (DAB) P12 sagittal sections from the hemispheres of SmoM2, SmoM2-Kmt2dCf/+, and SmoM2-Kmt2dCf/Cf animals. Scale bar: 1 mm. Green boxes indicate where the images in (A′–C′) were taken and black boxes where the images in (F–L′) are from. (A′–C″) Images of overlayed Hoechst (blue) and GFP (green) staining (A′–C′) and GFP (white) staining (A″–C″). Scale bar: 100 μm.
(D and E) Quantification of the percentage of the cerebellum sectional area taken up by the EGL in the hemispheres (D) and vermis (E) of P12 SmoM2, SmoM2-Kmt2dCf/+, and SmoM2-Kmt2dCf/Cf animals (n = 3 samples per genotype).
(F–H) Images of overlayed Hoechst (blue) and TUNEL (green) staining (or single channel TUNEL staining (F′– H′) in the EGL in the area indicated by the black box in (A–C). Scale bar: 250 μm.
(I) Quantification of the density of TUNEL+ cells in the EGL in the area of the hemispheres shown in (F–H) of SmoM2, SmoM2-Kmt2dCf/+, and SmoM2-Kmt2dCf/Cf mice (n = 3 samples per genotype).
(J′–L′) Representative images of overlayed Hoechst (blue) and EdU (red) staining or single channel EdU (red) staining in the EGL in the area indicated by the black box in (A–C). Scale bar: 250 μm.
(M) Quantification of the density of EdU+ cells in the EGL in the area of the hemispheres shown (J–L) of SmoM2, SmoM2-Kmt2dCf/+, and SmoM2-Kmt2dCf/Cf mice (n = 3 samples per genotype).
(N–P′) Images of GFP stained (DAB) P21 sagittal sections from the hemispheres (N–P) and vermis (N′–P′) of SmoM2, SmoM2-Kmt2dCf/+, and SmoM2-Kmt2dCf/Cf animals. Scale bar: 1 mm.
(Q and R) Quantification of the percentage of the cerebellum sectional area taken up by the EGL/lesion in the hemispheres (Q) and vermis (R) of P21 SmoM2, SmoM2-Kmt2dCf/+, and SmoM2-Kmt2dCf/Cf animals (n = 3 samples per genotype). All statistical significance was determined using an unpaired t-test comparing Kmt2d wildtype tumors to each of the Kmt2d mutants. Error bars: SD.
Loss of Kmt2d in SHH-MB leads to reduced expression of neural differentiation genes and repressors of Notch- and TGFβ-signaling and increased expression of genes associated with progenitor behaviors
To address the molecular mechanisms by which loss of Kmt2d promotes SHH-MB tumor progression, we compared the transcriptome and chromatin accessibility profiles of SmoM2, SmoM2-Kmt2dCf/+, and SmoM2-Kmt2dCf/Cf primary tumors using bulk RNA-seq and ATAC-seq (Figure 4A; Table S1). The primary tumors were dissected from the brain and tumor cells were enriched for by isolation of GFP+ cells by FACS. Hierarchical clustering of bulk RNA-seq data (p <= 0.05; fold change [FC] >= 1.5) demonstrated that SmoM2 tumors clustered separately from SmoM2-Kmt2dCf/Cf and SmoM2-Kmt2dCf/+ tumors (Figure S3A; Table S2). Furthermore, SmoM2-Kmt2dCf/+ tumor cells could be separated from SmoM2-Kmt2dCf/Cf because they had enhanced expression of the gene signatures specific to both SmoM2 and SmoM2-Kmt2dCf/Cf tumors.
Figure 4.

Bulk RNA-seq analysis reveals downregulation of neural differentiation genes and upregulation of Notch/TGFβ signaling and stem cell genes in Kmt2d mutant SHH-MB
(A) Experimental strategy to define the transcriptomic and chromatin accessibility landscape of tumors from SmoM2, Smom2-Kmt2dCf/+, and SmoM2-Kmt2dCf/Cf mice.
(B) PCA analysis of RNA-seq data showing two distinct clusters of tumors from SmoM2 (black oval) and SmoM2-Kmt2dCf/Cf (red oval) animals.
(C) Heatmap analysis of RNA-seq data showing differentially expressed genes between SmoM2 and SmoM2-Kmt2dCf/Cf tumors (padj ≤ 0.05; FC ≥ 1.5).
(D and E) GO term analysis of the RNA-seq data showing the top downregulated (D) and upregulated (E) pathways in SmoM2-Kmt2dCf/Cf tumors compared to SmoM2 tumors.
(F) RT-qPCR analysis of indicated genes in GFP+ cells isolated by FACS from SmoM2-Kmt2dCf/Cf tumors (n = 6) relative to SmoM2 tumors (n = 5). Statistics were determined using the Mann-Whitney test. See also Tables S3 and S4, and Figures S3–S5.
In order to focus on differences in gene expression due to loss of Kmt2d, all further analysis was performed using the SmoM2 and SmoM2-Kmt2dCf/Cf tumor samples. As expected, principal component analysis and heatmaps with hierarchical clustering (p <= 0.05; FC >= 1.5) of bulk RNA-seq data from SmoM2 and SmoM2-Kmt2dCf/Cf tumor samples revealed two distinct clusters (Figures 4B and 4C; Table S3). Gene Ontology (GO) term analysis of the 1,004 genes downregulated in SmoM2-Kmt2dCf/Cf tumors (p <= 0.05; FC >= 1.5) showed enrichment for genes associated with late stages of neuron differentiation, such as synapse organization, axonogenesis and regulation of membrane potential in the top 20 categories (Figure 4D). In contrast, GO term analysis of the 516 genes upregulated in SmoM2-Kmt2dCf/Cf tumors (p <= 0.05; FC >= 1.5) showed enrichment for genes associated with cell processes involved in organ development including stem cell maintenance, epithelial cell proliferation, and cell migration (including ameboidal-type cell migration, extracellular matrix organization, or EMT) (Figure 4E). Our results indicate that KMT2D normally promotes expression of genes involved in neural differentiation and indirectly inhibits genes that promote a progenitor state (possibly through activating inhibitors), thus KMT2D can be considered to function as a tumor suppressor.
Gene set enrichment analysis (GSEA) of genes upregulated in SmoM2-Kmt2dCf/Cf tumors further highlighted that SmoM2-Kmt2dCf/Cf tumors (indirectly) upregulate pathways associated with stem proliferation (Figure S3B), and genes/pathways that can regulate advanced stage cancers including TGFβ signaling, Notch signaling, and MYC (Figures S3C–S3E). Consistent with KMT2D normally being associated with activation of gene expression, Smad7 and Klf9, suppressors of TGFβ and Notch signaling, respectively,70,71,72 were significantly downregulated in SmoM2-Kmt2dCf/Cf tumors (Table S3). Many of the key changes in gene expression identified by RNA-seq were validated by quantitative RT-PCR of RNA from GFP+ tumor cells isolated by FACS. RNA in situ and protein immunostaining of sections from additional tumor samples also provided validation and assessment of gene expression patterns. qRT-PCR analysis showed significant upregulation of Atoh1, Sox2, Myc, Jag1, Hes1, Tgfbr2, and Smad3 and downregulation of Neurod1 in SmoM2-Kmt2dCf/Cf tumors (n = 6) compared to SmoM2 (n = 5) (Figure 4F; Table S4). Smad7 and Klf9 appeared downregulated, although not to statistical significance (p = 0.052 and 0.126, respectively). RNA in situ analysis of sagittal sections of primary tumors from SmoM2 and SmoM2-Kmt2dCf/Cf animals (n = 3 each) showed that Kcnc3, a gene associated with neural differentiation, was broadly expressed across SmoM2 tumors whereas in SmoM2-Kmt2dCf/Cf tumors expression was more patchy indicating less cells differentiate when Kmt2d is mutant (Figures S3F–S3I). Myc was expressed throughout the tumors of both genotypes and at higher levels in SmoM2-Kmt2dCf/Cf tumors (Figures S3J–S3M). Double immuno-staining for NueroD1 protein, a neural differentiation gene significantly reduced based on qRT-PCR analysis, and Ki67 (proliferation marker), showed as expected that Ki67 and Neurod1 staining was largely complementary although some cells were double labeled (Figure S4). Consistent with the tumors having a classic histology, Ki67 was broadly expressed in both genotypes. However, in SmoM2-Kmt2dCf/Cf tumors NeuroD1 high areas appeared reduced compared to in SmoM2 tumors. Quantification of the fluorescence pixels in nuclei (Dapi+) demonstrated that Neurod1 had a lower density in SmoM2-Kmt2dCf/Cf tumors compared to SmoM2 tumors whereas Ki67 was not changed. Thus, KMT2D normally promotes a more differentiated state in SHH-MB tumor cells.
Network analysis for genes significantly upregulated in SmoM2-Kmt2dCf/Cf tumors compared to SmoM2 (Figure S5) revealed upregulation of genes that could enhance tumor growth, including genes previously shown to positively regulate cerebellar neural progenitor proliferation; Sox2/9,56,57,58,59,60 Atoh1, and Myc/Mycl.54,55 In addition, genes/pathways implicated in metastasis were upregulated in SmoM2-Kmt2dCf/Cf tumors: TGFβ signaling (Tgfbr2, Tgf1i1, Smad3, and Epha3),52,73,74 and Notch signaling (Notch1, Hes1, and Jag1),50,53,75,76 the two pathways inhibited by Smad7 and Klf9 (p ≤ 0.05; FC ≥ 1.5).
In parallel to the RNA-seq data analysis, we performed ATAC-seq analysis of GFP+ cells from primary tumors, including a subset of the tumors in which RNA-seq was performed (Table S1). A comprehensive QC assessment of the ATAC-seq data showed a high signal-to-noise ratio (Figure S6A). Differential analysis based on peak annotations identified two distinct clusters of SmoM2 and SmoM2-Kmt2dCf/Cf primary tumors with a sub-cluster of tumors seen within the SmoM2-Kmt2dCf/Cf cluster (Figure 5A). The differentially accessible chromatin regions displayed via a volcano plot further identified a separation of the SmoM2 and SmoM2-Kmt2dCf/Cf tumors and indicated two sub-clusters of SmoM2-Kmt2dCf/Cf tumors (Figure 5B). A more detailed analysis of the ATAC-seq data showed that the global transcriptomic trends found using bulk RNA-seq between GFP+ SmoM2 and SmoM2-Kmt2dCf/Cf cells were retained in the global chromatin accessibility trends and the two corroborated with one another (p <= 0.1; FC >= 1.5), i.e., the genes with the most significant changes in mRNA and DNA peaks are in the concordant quadrants (red dots; Figure 5C). Similar to the gene expression trends seen in the RNA-seq analysis, ATAC-seq analysis identified an enhancement of open chromatin domains within neuronal differentiation genes (e.g., NeuroD1) in SmoM2 tumors, whereas in SmoM2-Kmt2dCf/Cf tumor cells open chromatin domains were enhanced in genes associated with TGFβ (Smad3) and Notch (Hes1) pathways (Figures 5D–5F; Table S5). In support of our hypothesis that loss of Kmt2d indirectly results in upregulation of the TGFβ and Notch pathways via decreased expression of inhibitors, ATAC-seq analysis showed diminution of open chromatin domains within Klf9 and Smad7 genes in SmoM2-Kmt2dCf/Cf tumor cells, although not statistically significantly (Figures 5G and 5H; Table S5). Furthermore, motif enrichment analysis showed that motifs for pro-neural transcription factors (TFs) seen in differentiating neurons (e.g., NeuroD1 and ASCL1) were enriched in SmoM2 tumors (Figure S6B; Table S5). In contrast, in SmoM2-Kmt2dCf/Cf tumor cell DNA motifs bound by TFs associated with stem/progenitor cell populations (e.g., SOX2 and SOX9) were enriched (Figure S6B; Table S5), further validating our conclusions drawn from the RNA-seq data. SmoM2-Kmt2dCf/Cf tumors also were enriched for motifs bound by SMAD3, a downstream effector of the TGFβ pathway (Figure S6B; Table S5). Interestingly, DNA motifs bound by ATOH1, a TF shown to augment SHH-MB tumor growth and metastasis when over-expressed in Ptch1+/− GCPs,11 also were enriched in SmoM2-Kmt2dCf/Cf tumors (Figure S6B; Table S5). Furthermore, motif enrichment analysis showed enhancement of sequences bound by NEUROD1 and ASCL1 at the NeuroD1 gene in SmoM2 tumors and SMAD3 and MYC motifs in the Smad3 gene in SmoM2-Kmt2dCf/Cf tumors (Figures S6C and S6D; Table S5). Finally, network analysis of genes annotated at differentially accessible peaks enriched in SmoM2-Kmt2dCf/Cf tumors showed Notch signaling forms a central node for several pathway and many of the other nodes are similar to the RNA-seq network analysis (Table S6; Figure S6E). Taken together, the bulk RNA-seq and ATAC-seq data demonstrate that loss of Kmt2d in SHH-MB primary tumors promotes a pro-tumorigenic transcriptional/epigenetic signature that includes genes and pathways associated with neural progenitor states and advanced disease, likely as a result of down regulation of suppressors of these genes/pathways and neural differentiation genes.
Figure 5.

ATAC-seq data analysis reveals differences in DNA accessibility of pathways associated with neural differentiation and advanced cancer in SmoM2-Kmt2dCf/Cf vs. SmoM2 tumor
(A) Heatmap showing the clustering of differential peaks between SmoM2 and SmoM2-Kmt2dCf/Cf tumors (FC ≥ 1.5, padj ≤ 0.1).
(B) Volcano plots displaying the differentially accessible chromatin regions in SmoM2-Kmt2dCf/Cf compared to SmoM2 tumor cells (FC ≥ 1.5, padj ≤ 0.1).
(C) Comparison of the log2 fold changes (red indicates FC ≥ 1.5, padj ≤ 0.1) in global trends in SmoM2-Kmt2dCf/Cf tumor cells compared to SmoM2 in the RNA-seq and ATAC-seq datasets.
(D–H) ATAC-seq plots showing regions of increased and decreased chromatin accessibility at specific gene loci in SmoM2-Kmt2dCf/Cf (n = 12) compared to SmoM2 (n = 7) primary tumors. The number below indicates the FC in a single 500-bp peak. Fold changes with a padj ≤ 0.1 are indicated with a star. Also see Figure S6 and Tables S5 and S6.
Loss of Kmt2d promotes local invasion in SHH-MB
Given that the RNA-seq and ATAC-seq data from SHH-MB tumors lacking Kmt2d indicate upregulation of pathways associated with aggressive tumors, we next asked whether the tumors have LMD and invade the surrounding brain regions, particularly the cerebellar white matter (WM) and brainstem (BS) at end stage or P150 in mice that survive and have primary tumors (Figure 6A–6D″; Table 1). Tumors of all genotypes had some LMD, but it was restricted to the hindbrain region. Strikingly, compared to SmoM2 animals with 25% invasion into the WM (n = 4/16 mice) and 50% into the BS (8/16), significantly higher frequencies of local invasion were seen in SmoM2-Kmt2dCf/+ (WM: 12/15; BS:13/15) and SmoM2-Kmt2dCf/Cf (WM: 18/18; BS: 18/18) (Table 1). The N-terminal mutation similarly increased local invasion from 57% in the WM (4/7) and BS (4/7) of SmoM2 animals to 100% in the WM (8/8, p = 0.0192) and 87.5% into the BS (7/8, p = 0.0924) of SmoM2-Kmt2dNf/+ animals and 100% in the WM (6/6, p = 0.0337) and BS (6/6, p = 0.03337) of SmoM2-Kmt2dNf/Nf animals (Table 1). Furthermore, in the Ptch1 model of SHH-MB with a low amount of local invasion (WM: 4/10; BS: 3/10) a significantly higher percentage of Ptch1-Kmt2dCf/+ (WM: 9/10; BS: 8/10) and Ptch1-Kmt2dCf//Cf (WM: 7/9; BS: 7/9) animals had WM and/or BS invasion at end stage or P200 (Figures S7A–S7B″; Table 1). These results provide in vivo evidence across two different mouse models of SHH-MB that a heterozygous or homozygous loss of Kmt2d promotes invasion of tumor cells locally.
Figure 6.

Loss of Kmt2d in two models of SHH-MB promotes local cell invasion and spinal cord metastasis
(A–D″) Representative images of sagittal sections from end stage tumors stained with Hoechst (blue) and GFP (green) without tumor cell invasion into the white matter (WM) (A, A′, A″) or with tumor cell invasion into the WM (B, B′, B″), and without tumor cell invasion into the brainstem (BS) (C, C′, C″) or with tumor cell invasion into the BS (D, D′, D″). Overlayed Hoechst and GFP images of the whole cerebellum are shown in (A–D) (Scale bar: 1 mm). Overlayed Hoechst and GFP images in the specific regions indicated by rectangles in (A) are shown in (A′–D′) and single channel GFP-staining of the same regions in (A″–D″) (Scale bar: 250 μm).
(E–H) Representative sections at the rostral-caudal levels indicated of spinal cords stained with H&E from SmoM2 animals without metastasis (Scale bar: 500 μm).
(E′–H′) Adjacent sections stained with Hoechst, GFP (green) and Ki67 (red) (Scale bar: 500 μm).
(I–L) Representative sections of spinal cords stained with H&E from SmoM2-Kmt2dCf/Cf animals (Scale bar: 500 μm).
(I′–L′) Fluorescence images of adjacent sections stained with Hoechst, GFP (green) and Ki67 (red) (Scale bar: 500 μm). Single channel GFP (I″-L″) and Ki67 (I‴–L‴) staining of metastatic tumors in the spinal cord (Scale bar: 250 μm).
(M–O) Quantification of the percentage of sectional spinal cord area taken up by tumor in >30 sections per mouse in the SmoM2-driven model of SHH-MB with and without a Kmt2d C-terminal mutation (M) or Kmt2d N-terminal mutation (N), and in the Ptch1 model (O). Open dots indicate animals that did not show symptoms of tumor burden at P150 (SmoM2 models) or P200 (Ptch1 model) but had primary tumors. All statistical significance was determined using an unpaired t-test comparing Kmt2d wildtype tumors to each of the Kmt2d mutants. Also see Figure S7.
Table 1.
Summary of the phenotypes of mice with a heterozygous or homozygous Kmt2dCf or Kmt2dNf mutations in two models of SHH-MB
| Genotype | % White Matter invasion | % Brainstem invasion | % Spinal cord Metastasis |
|---|---|---|---|
| SmoM2 | 25 (n = 16) | 50 (n = 16) | 33.3 (n = 12) |
| SmoM2-Kmt2dCf/+ | 80 (n = 15) [0.0011] | 86.6 (n = 15) [0.0145] | 100 (n = 13) [0.0002] |
| SmoM2-Kmt2dCf/Cf | 100 (n = 18) [<0.0001] | 100 (n = 18) [0.0003] | 100 (n = 11) [0.0004] |
| SmoM2 | 57.1 (n = 7) | 57.1 (n = 7) | 14.2 (n = 7) |
| SmoM2-Kmt2dNf/+ | 100 (n = 8) [0.0192] | 87.5 (n = 8) [0.0924] | 62.5 (n = 8) [0.0286] |
| SmoM2-Kmt2dNf/Nf | 100 (n = 6) [0.0337] | 100 (n = 6) [0.0337] | 100 (n = 6) [0.0010] |
| Ptch1 | 40 (n = 10) | 30 (n = 10) | 33.3 (n = 6) |
| Ptch1-Kmt2dCf/+ | 90 (n = 10) [0.0095] | 80 (n = 10) [0.0123] | 70 (n = 10) [0.0762] |
| Ptch1-Kmt2dCf/Cf | 77.7 (n = 9) [0.0479] | 77.7 (n = 9) [0.0186] | 100 (n = 9) [0.0021] |
% White Matter invasion: the percentage of animals with a primary tumor (number give in brackets) that had tumor cell invasion in the white matter.
% Brainstem invasion: the percentage of animals with a primary tumor (number give in brackets) that had tumor cell invasion into the brainstem.
% Tumor metastasis: the percentage of animals with a primary tumor (number give in brackets) that had metastasis to the spinal cord.
All animals either were sacrificed when they showed signs or at P150 (SmoM2 models) or P200 (Ptch1 model) and only mice with primary tumors when analyzed.
Statistical significance [p value] for each parameter was calculated using Chi-square test for comparing proportions between Kmt2dfl/+ or Kmt2dfl/fl mutant tumors and Kmt2d+/+ tumors of the same model.
Loss of Kmt2d promotes spinal cord metastasis in SHH-MB
We next asked whether loss of Kmt2d leads to increased metastatic disease in the spinal cord since it is prevalent in SHH-MB patients and since LMD was not seen anterior to the hindbrain in our models. A series of coronal sections of the entire spinal cord of each tumor bearing mouse was examined for cytology (H&E staining), and tumor cells were confirmed by expression of GFP and Ki67 (Figures 6E–6L″ and S6C–C″). Strikingly, we found that whereas only 33.3% of SmoM2 (n = 12) animals had metastatic lesions along the spinal cord, SmoM2-Kmt2dCf/+ (n = 13) and SmoM2-Kmt2dCf/Cf (n = 11) mice had a significant increase in tumors to 100% (Table 1). Furthermore, the percentage of the spinal cord containing metastatic tissue was significantly increased in Kmt2d mutants (Figure 6M). Clearing of a whole spinal cord from a SmoM2-Kmt2dCf/Cf animal stained with antibodies to GFP (tumor cells) and Yoyo1 dye (nuclei) confirmed that metastatic tumors in SmoM2-Kmt2dCf/Cf animals can form throughout the length of the spinal cord (Figures S7D–S7D‴). The N-terminal mutation of Kmt2d also significantly increased the percentage of mice with metastatic tumors (14.2% SmoM2 vs. 62.5% SmoM2-Kmt2dNf/+ and vs. 100% SmoM2-Kmt2dNf/Nf) (Table 1) and the relative sizes of the metastatic lesions of homozygous mutants (Figure 6N). In the Ptch1 model of SHH-MB the frequency of animals with metastatic disease was increased in the Ptch1-Kmt2dCf/Cf animals compared to the Ptch1 animals (33.3% Ptch1 vs. 70% Ptch1-Kmt2dCf/+, p = 0.076 and vs. 100% Ptch1-Kmt2dCf/Cf, p = 0.0021) (Table 1). Similarly, the relative sizes of metastatic lesions were significantly increased in Ptch1-Kmt2dCf/Cf (p = 0.019) but not Ptch1-Kmt2dCf/+ (p = 0.58) mutants (Figure 6O). In summary, we found that reduction of, or loss of Kmt2d in two mouse models of SHH-MB greatly promotes metastatic disease to the spinal cord.
Metastatic and primary tumors have similar transcriptomic and chromatin accessibility profiles
Given that the deletion of Kmt2d in mouse models of SHH-MB results in invasive and metastatic disease, we compared the transcriptomes of primary and metastatic GFP+ tumor cells from the same SmoM2-Kmt2dCf/Cf animals (Tables S1 and S7). Surprisingly, there were only 41 differentially expressed genes between the primary and metastatic tumors (Figure 7A; Table S7) (p <= 0.05; FC >= 1.5). Furthermore, 19 of the 41 differentially expressed genes might have been from contaminating infiltrating immune cells or spinal cord neurons/glia based on the genes over-expressed, low read counts and/or high variability between samples (bold font genes in Table S7). Interestingly, among the remaining genes, a subset of the 16 genes over-expressed in metastatic cells has been shown to be associated with poor prognosis in cancer (Sparc, Nr4a1, Itgb5, Fos, and Fosb).77,78,79,80,81
Figure 7.

Transcriptional profiling of SMoM2-Kmt2dCf/Cf primary and metastatic tumors compared to SmoM2 reveals upregulation of genes/pathways associated with aggressive cancer
(A) Heatmap analysis of bulk RNA-seq showing differentially expressed genes between SmoM2-Kmt2dCf/Cf primary and metastatic tumors. (p <= 0.05; FC >= 1.5). See also Table S7.
(B) Heatmap analysis of bulk RNA-seq showing differentially expressed genes between SmoM2 primary tumors and SmoM2-Kmt2dCf/Cf primary and metastatic tumors. (p <= 0.05; FC >= 1.5). See also Table S8.
(C) Network enrichment analysis of genes upregulated in SmoM2-Kmt2dCf/Cf primary and metastatic tumors compared to SmoM2 primary tumors. Also see Figure S8.
A similarity and stability of the transcriptional landscapes of the primary and metastatic compartments of SmoM2-Kmt2dCf/Cf animals was also seen when comparing RNA-seq data to SmoM2 primary tumors using PCA analysis (Figure S8A) and hierarchical clustering (Figure 7B; Table S8), as the SmoM2-Kmt2dCf/Cf primary and metastatic tumors clustered together and were separated from the SmoM2 primary tumors. Pathway analysis similarly confirmed that SmoM2-Kmt2dCf/Cf tumor cells from the primary and metastatic compartments both had downregulation of genes associated with neuronal differentiation and upregulation of genes associated with organ morphogenesis and EMT compared to primary SmoM2 tumor cells (Figures S8B and S8C). Furthermore, network enrichment analysis comparing SmoM2 mutant primary tumors to SmoM2-Kmt2dCf/Cf primary and metastatic tumors showed upregulation of previously identified genes and pathways that can drive progenitor states and tumor progression: Sox2/9, Myc, Atoh1, and TGFβ and Notch pathway genes (Figure 7C). Given that some of the upregulated genes identified by the RNA-seq data analysis are not specific to the SHH-MB subgroup, we compared the SmoM2-Kmt2dCf/Cf primary and metastatic tumor cell expression profiles to genes associated with the 4 subgroups of MB (Figure S8D). Although some primary and metastatic SmoM2-Kmt2dCf/Cf tumor samples express genes associated with subgroups of MB other than SHH, there was enrichment for an SHH-MB signal in the tumors (Figure S8D).
A QC assessment of the ATAC-seq data from SmoM2-Kmt2dCf/Cf metastatic tumors showed a high signal-to-noise ratio (Figure S6A). ATAC-seq analysis of SmoM2-Kmt2dCf/Cf metastatic tumors in addition to SmoM2 and SmoM2-Kmt2dCf/Cf primary tumors showed the predicted decrease in chromatin accessibility in neural differentiation associated genes (e.g., Kcnc3) and in Klf9 and Smad7 in metastatic as well as primary SmoM2-Kmt2dCf/Cf tumors and an increase in accessibility of tumor genes associated with the Notch pathway (Jag1), TGFβ pathway (Smad3), and progenitor cell populations (Sox2) compared to SmoM2 (Figures S8E–S8J; Tables S5 and S9). Thus, our transcriptomic and epigenetic data reveal that loss of Kmt2d in SmoM2 tumors promotes MB tumor progression and metastasis through the (indirect) upregulation of the TGFβ and Notch oncogenic pathway genes, as well as Myc, Sox2/9, and Atoh1, all of which have been previously implicated as key drivers of tumor growth and metastasis in MB.
Discussion
Our study demonstrates that in the context of mouse models of SHH-MB, Kmt2d is a tumor suppressor that inhibits tumor growth, invasion into the hindbrain and metastatic spread to the spinal cord, in part through promoting neural differentiation. Furthermore, heterozygous loss-of-function mutations in Kmt2d, as seen in patients, are sufficient to drive SHH-MB to advanced stages of the disease via haploinsufficiency. Metastasis is a hallmark of late-stage disease and responsible for the highest number of cancer-related fatalities. However, the molecular drivers of metastatic disease, especially in brain cancers, remain largely unknown in part due to a lack of robust animal model systems. One approach used to identify candidate genes that drive SHH-MB dissemination into the leptomeninges and spinal cord metastasis was an insertional mutagenesis screen that mobilizes a Sleeping Beauty transposon81 in cerebellar GCPs of Ptch+/− animals.20 Follow up studies showed that over-expression of Eras, Lhx1, Ccrk, Arnt, Gdi2 or activated AKT are sufficient to increase dissemination and metastasis when combined with forced expression of Shh by viral infection of GCPs and transplantation.20,82,83 Another study identified circulating tumor cells in the blood using this and other mouse transplantation models and analysis of human tumor cells. The CCL2-CCR2 axis was identified as upregulated in disseminated cells and shown to be sufficient to enhance metastasis of GCPs expressing SHH.8 In a separate model, over-expression of Atoh1 in GCPs of Ptch1+/− mice using a conditional transgene approach demonstrated ATOH1 can promote tumor penetrance and spinal cord metastasis to 100%.11 While these papers identified genes that when over-expressed in mouse models can facilitate metastasis of GCPs with constitutive SHH signaling, their relevance to the normal genetic changes seen in SHH-MB are not clear.
Loss of Kmt2d in SHH-MB leads to downregulation of neural differentiation and upregulation of genes/pathways associated with a progenitor state
Bulk RNA-seq analysis and concordant ATAC-seq results revealed that SmoM2-Kmt2dCf/Cf primary tumors are distinct from SmoM2 tumors, and that SmoM2-Kmt2dCf/+ tumors share upregulated genes with both tumor genotypes. Whether SmoM2-Kmt2dCf/+ tumors upregulate the genes seen in Kmt2d homozygous mutant tumors and maintain the genes expressed in Kmt2d WT tumors or instead have a mixture of both cell phenotypes is not clear from our bulk RNA-seq data. Given the function of KMT2D in augmenting gene expression, loss of Kmt2d is predicted to downregulate genes. Of likely relevance, the differentially expressed genes significantly downregulated based on RNA-seq analysis in SmoM2-Kmt2dCf/Cf primary tumors are greatly enriched for genes/pathways involved in neural differentiation, and two genes were validated with qRT-PCR and/or analysis of protein/RNA in sections. Kcnc3 RNA or NeuroD1 protein were detected broadly across SmoM2 tumors, as was Ki67, whereas in SmoM2-Kmt2dCf/Cf tumors the Kcnc3/NeuroD1 domains were diminished. These results indicate KMT2D promotes expression of neural differentiation genes, even in progenitors within the tumors and therefore that KMT2D promotes the transition of GCPs from a progenitor to differentiated state. Loss of Kmt2d therefore promotes a progenitor state. Consistent with this, in E18.5 embryos lacking Kmt2d in GCPs, the proportion of differentiated cells in the EGL (inner EGL) is reduced. Furthermore, in tumors with a partial or complete loss of Kmt2d, genes that regulate a GCP progenitor state are upregulated. In addition, some of these genes have been associated with advanced cancer in SHH-MB, including Sox256,57,58,59,60 and Atoh1.11 However, none of the genes identified by Sleeping Beauty enabled mutagenesis20,82,83 were found to be differentially expressed in SmoM2-Kmt2dCf/Cf tumors compared to SmoM2 although some genes (Akt2, Gdi2, Arnt, and Lhx1) were expressed at high levels in both genotypes (Table S10).
In SmoM2-Kmt2dCf/Cf tumors lacking Kmt2d, genes in the TGFβ52,73,74 and Notch pathway50,53,75,76 which can increase oncogenesis are significantly upregulated in the RNA-seq data compared to SmoM2 primary tumors. Interestingly, suppressors of TGFβ and Notch signaling, Smad770 and Klf9,71,72 respectively, are significantly downregulated in SmoM2-Kmt2dCf/Cf tumor cell RNA-seq data. While Smad7 was close to significantly decreased based on qRT-PCR (p = 0.052) in SmoM2-Kmt2dCf/Cf tumors compared to SmoM2, the reduction in Klf9 (p = 0.126) was not. Nevertheless, a significant reduction of Klf9 in a specific progenitor pool might lower Notch signaling. Thus, Smad7 and Klf9 expression might normally be enhanced by KMT2D in a particular cell type and a decrease in mutant tumor cells could contribute to the increase in expression of Notch and TGFβ pathway genes.
Key candidate up- and downregulated genes were confirmed by qRT-PCR analysis of GFP+ cells isolated from SmoM2 and SmoM2-Kmt2dCf/Cf tumors. Furthermore, ATAC-seq data from SmoM2 and SmoM2-Kmt2dCf/Cf primary tumors revealed trends in motif enrichment and open chromatin in concordance with the RNA-seq data. While the trends observed in the ATAC-seq data are subtle, we hypothesize that the overall effects are bolstered by the concomitant changes across a number of genes within the same pathway and across different pathways (Tables S5, S6, and S9). Furthermore, the analysis of RNA/protein expression on tumor sections of differentiation and proliferation markers showed that the tumors are heterogeneous, with SmoM2-Kmt2dCf/Cf tumors having a reduction in the areas expressing high Neurod1 and Kcnc3 compared to SmoM2. We predict that heterozygous mutations in KMT2D shift the transcriptional and epigenetic landscape of human SHH-MBs to a more aggressive tumor gene signature that enhances metastasis through promoting a progenitor state and enhancing Notch/TGFβ signaling, likely in a subset of cells that go on to form the bulk of the tumor.
Primary and metastatic tumors share a transcriptional gene signature
Differential transcriptomic and epigenomic analysis of SmoM2 primary tumors and SmoM2-Kmt2dCf/Cf primary and metastatic spinal cord tumors showed little segregation between the SmoM2-Kmt2dCf/Cf primary and metastatic tumors using supervised on unsupervised clustering techniques. Moreover, the metastatic and primary SmoM2-Kmt2dCf/Cf tumor cells (GFP+) from the same mice had very similar transcriptional landscapes, with only ∼20 genes being clearly differentially expressed (Tables S7 and S8). Some genes involved in handling hypoxia84,85,86 were expressed in both primary and metastatic tumors (Tables S8 and S10). This result is consistent with SHH-MB primary tumors in our mouse models growing on the surface of the brain and thus being in a hypoxic environment as in spinal cord metastases. Unlike our finding of largely similar transcriptional profiles in primary and metastatic SmoM2-Kmt2dCf/Cf tumor cells, when Atoh1 alone was over-expressed in a Ptch1 model of SHH-MB, bulk RNA-seq comparing primary and metastatic tumors identified extensive differential gene expression.11,20 Additional mutational and epigenetic analysis and clinical outcomes data of limited human MB samples support a similar conclusion.11,20 Nevertheless, studies have shown matched MB primary and metastatic tumors maintain their subgroup-specific global transcriptional profiles.87 One possible explanation for our finding is that mutations in genes like KMT2D that encode epigenetic modifiers can result in global transcriptional changes that are sufficient to drive metastasis as well as enhanced tumor growth in most primary tumor cells. Finally, of note, although SmoM2-Kmt2dCf/Cf primary and metastatic tumors preferentially express genes associated with SHH-MB (Figure S8D), they have upregulation of Notch pathway genes and MYC, as well as upregulation of Bcl2 (Table S3) which are associated with group 3 MB and have been implicated in mouse models of Group 3 MB that express MYC.50,53,88,89 Thus, mutations in chromatin remodeling genes might partially blur the lines between the transcriptional profiles of MB subgroups.
Heterozygous mutations in Kmt2d are sufficient to cause metastatic disease
Whereas we did not find that two distinct homozygous Kmt2d mutations in all cerebellar lineages drive tumorigenesis, we did find that in two mouse models of SHH-MB (SmoM2 expression or Ptch1 homozygous loss) these same mutations in the heterozygous state are sufficient to dramatically accelerate tumor growth, and augment local invasion into the hindbrain and metastasis to the spinal cord. While there might be subtle differences in the extent of promotion of tumor growth rate, local invasion and spinal cord metastasis between the two mutations (Table 1), both mutations significantly increase the percentage of mice that develop primary tumors and metastatic disease when SmoM2 is expressed in GCPs. Comparison of the effect of one mutation (C-terminal) in our sporadic SmoM2 and Ptch1 models further showed that heterozygous (or homozygous) loss of Kmt2d greatly increases incidence of primary tumors and spinal cord metastasis, even when disease progression and incidence are different in the two models. We predict that KMT2D mutations will drive aggressive disease in the context of all the SHH pathway activating mutations seen in human SHH-MB. The clinical relevance of the mouse models developed in this study is highlighted by two aspects of patient tumor biology that are recapitulated in these models: (a) mono-allelic loss-of-function mutations in KMT2D are associated with SHH-MB tumorigenesis34,43,45,46 and (b) metastatic disease in SHH-MB patients occurs most frequently intracranially or along the spine.8 Of note, while the tumors of all genotypes had LMD around the hindbrain/cerebellum, the dissemination did not spread to more anterior brain regions, unlike other models of SHH-MB metastasis.8,11,83,90 This result could indicate that the kind metastasis-promoting gene undergoing a secondary mutation will influence the site of LMD, brain invasion and metastasis. Our findings thus highlight the importance of testing the involvement of genes in addition to KMT2D that are preferentially mutated in the β-subtype of SHH-MB, as well as other subtypes, to identify likely prognostic markers for patients at high risk of recurrent metastasis.
Limitations of the study
One limitation of the study is that for practical reasons the transcriptomic and epigenomic analyses were performed in only one of the 3 mouse models of SHH-MB, although we expect similar results with the N-terminal Kmt2d deletion mutation and in the Ptch1 model. In addition, the sample sizes for the SmoM2-Kmt2dNf and Ptch1-Kmt2dCf SHH-MB models might not be large enough to detect slight differences in tumor biology caused by the N- and C-terminal deletions of Kmt2d or by aberrantly activating SHH signaling through expression of SmoM2 versus loss of Ptch1. Beyond the scope of the study, functional studies to test the impact of removing the candidate genes identified by the transcriptomic/epigenomic analyses will be important. An additional possible long-term study would be to validate candidate direct and indirect targets of KMT2D, as this could contribute to both our understanding of the genetics of SHH-MB tumor metastasis and the molecular functions of KMT2D.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Monoclonal anti-Calbindin D-28K | Swant | 300 |
| Mouse Monocloanl anti-NeuN | EMD Millipore | MAB377 |
| Mouse Monoclonal anti-p27 | Thermo Fisher Scientific | 610241 |
| Purified Mouse anti-Ki-67 | BD Pharmingen | 556003 |
| Mouse Monoclonal anti-NeurodD1 | Abcam | ab60704 |
| Rabbit Polyclonal anti-PH3 | EMD Millipore | 06–570 |
| Donkey anti-Mouse 488 IgG, Alexa Fluor | Invitrogen | A-21202 |
| Donkey anti-Mouse 647 IgG, Alexa Fluor | Invitrogen | A-31571 |
| Donkey Anti-Rabbit IgG, Alexa Fluor 555 | Invitrogen | A-31572 |
| Chicken Polyclonal anti-GFP | Aves Lab | GFP-1010 |
| Goat Anti-Chicken 647 IgG, Alexa Fluor | Invitrogen | A-21449 |
| Yoyo1 Iodide (491/509) | Thermo Fisher Scientific | Y3601 |
| Streptavidin Alexa Fluor 647 Conjugate | Invitrogen | S-32357 |
| Click-iT™ EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 647 dye | Invitrogen | C10340 |
| Click-it EdU assay with Sulfo-Cyanine5 azide | Lumiprobe Corporation | A3330 |
| Rabbit Monoclonal Histone H3 (D1H2) | Cell Signaling Technology | 4499 |
| Rabbit Monoclonal H3K4me1 (D1A9) | Cell Signaling Technology | 5326p |
| Rabbit Monoclonal H3K4me3 (C42D8) | Cell Signaling Technology | 9751s |
| Goat Anti-Rabbit Cross-Adsorbed Secondary Antibody, HRP | Thermo Fisher Scientific | G-21234 |
| Thermo Scientific Richard-Allan, Eosin-Y Stain | Thermo Fisher Scientific | 22-050-110 |
| Thermo Scientific Richard-Allan, Hematoxylin 2 | Thermo Fisher Scientific | 22-050-201 |
| Hoechst 33258 (bis-Benzimide) | Thermo Fisher Scientific | H3569 |
| Chemicals, peptides, and recombinant proteins | ||
| Tagment DNA Enzyme 1 (TDE1) | Illumina | 15027865 |
| Tagment DNA Buffer | Illumina | 15027866 |
| Qiagen MinElute Reaction Cleanup Kit | Qiagen | 28204 |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | M0541S |
| SYBR Green I | Thermo Fisher Scientific | S7563 |
| Agencourt AMPure XP magnetic beads | Beckman Coulter | A63880 |
| Agilent High Sensitivity DNA Bioanalysis Kit | Agilent Technologies | 5067–4626 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32851 |
| Tamoxifen | Sigma Aldrich | T5648-1G |
| BSA | Sigma Aldrich | A2153-100G |
| EGTA | Sigma Aldrich | E0396 |
| Sodium Dodecyl Sulfate | Thermo Fisher Scientific | BP166-100 |
| Triton X-100 | Thermo Fisher Scientific | BP151-100 |
| NP-40 10% | Sigma/Roche | 11332473001 |
| Tween-20 | Sigma/Roche | 11332465001 |
| Digitonin | Promega | G9441 |
| Biotin-16-dUTP | Sigma Aldrich | 11093070910 |
| Terminal Transferase | Roche | 3333574001 |
| EdU | Invitrogen | E10187 |
| Mounting, Cryo-OCT | VWR | 25608-930 |
| 10% SDS | Thermo Fisher Scientific | NP0002 |
| 20% Glycerol | Sigma Aldrich | G1724 |
| Protease/phosphatase inhibitor | Thermo Fisher Scientific | 78440 |
| 4X Loading Buffer | Thermo Fisher Scientific | LDS0007 |
| SeeBlue Plus2 Pre-stained Protein Standard | Invitrogen | LC5925 |
| MES SDS running buffer | Thermo Fisher Scientific | NP0002 |
| NUPAGE 20X Transfer Buffer | Thermo Fisher Scientific | NP0006-1 |
| NUPAGE Antioxidant | Thermo Fisher Scientific | NP0005 |
| NuPAGE™ 12%, Bis-Tris, 1.0 mm, Mini Protein Gels | Thermo Fisher Scientific | NP0341PK2 |
| Thermo Scientific Pierce ECL Western Blotting Substrate | Thermo Fisher Scientific | 32106 |
| Dichloromethane | Sigma Aldrich | 270997-100ML |
| Dibenzyl ether (98%) | Sigma Aldrich | 108014-1KG |
| Critical commercial assays | ||
| Rneasy Plus Mini Kit | Qiagen | 74134 |
| iScript cDNA Synthesis Kit | Bio-Rad | 170-8891 |
| Papain Dissociation System | Worthington Biochemical Corporation | LK003150 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE211030 |
| Experimental models: Organisms/strains | ||
| Nestin-Cre | The Jackson Laboratory Tronche et al.,91 Giusti et al.92 |
Strain #003771 |
| Atoh1-Cre | Matei et al.66 | Strain #011104 |
| hGFAP-Cre | Zhuo et al.68 | Strain #004600 |
| Ptf1aCre | Kawaguchi et al.67 | MGI:2387804 |
| Kmt2dCf/Cf or Kmt2d(Exon50-51)flox/flox | Jang et al.64 | |
| Kmt2dNf/Nf or Kmt2d(Exon16-19)flox/flox | Lee et al.63 | Strain #032152 |
| Atoh1-FlpoER/+ | Wojcinski et al.,93 | Available through the Joyner Lab |
| R26LSL-eGFP-Cre | Lao et al.69 | Strain 019013 |
| R26LSL-SmoM2-Yfp | The Jackson Laboratory Mao et al.94 |
Strain #005130 |
| Ptch1fl/fl | The Jackson Laboratory Ellis et al.95 |
Strain #030494 |
| Oligonucleotides | ||
|
Kcnc3 Forward: ACCCAACTACTGCAAGCCTGAC Kcnc3 Reverse: CACTTGTCCTTTCTGTCTGCTG |
Allen Brain atlas | In situ hybridization |
|
Myc Forward: GTCCGAGTGCATTGACCC Myc Reverse: TACAGTCCCAAAGCCCCA |
Allen Brain atlas | In situ hybridization |
|
Gapdh Forward: AGGTCGGTGTGAACGGATTTG Gapdh Reverse: TGTAGACCATGTAGTTGAGGTCA |
IDT DNA | qPCR |
|
Atoh1 Forward: GGTCTGTGGTGATCGTTGTTA Atoh1 Reverse: TACAGAGGAAGGAGAAGGTAGG |
IDT DNA | qPCR |
|
Sox2 Forward: CAGGAGTTGTCAAGGCAGAG Sox2 Reverse: GCTCCAAACTTCTCTCCTTTCT |
IDT DNA | qPCR |
|
Myc Forward: CGCCTACATCCTGTCCATTC Myc Reverse: AAGCTGTTCGAGTTTGTGTTTC |
IDT DNA | qPCR |
|
NeuroD1 Forward: AGGCACGTCAGTTTCACTATTC NeuroD1 Reverse: GCACTTTGCAGCAATCTTAGC |
IDT DNA | qPCR |
|
Jag1 Forward: ATGGGTCAGAACTGTGACATAAA Jag1 Reverse: GGTGGACAGATACAGCGATAAC |
IDT DNA | qPCR |
|
Hes1 Forward: CCAGCCAGTGTCAACACGA Hes1 Reverse: AATGCCGGGAGCTATCTTTCT |
IDT DNA | qPCR |
|
Klf9 Forward: TAGGTTAGGCTGCCCATTTC Klf9 Reverse: CCCAAACTCCTCACTCACTAAA |
IDT DNA | qPCR |
|
Tgfbr2 Forward: GTTCGTGAGCATGGAGAGATAG Tgfbr2 Reverse: CAGGGCTGAGATGATAAGAGTG |
IDT DNA | qPCR |
|
Smad3 Forward: CACGCAGAACGTGAACACC Smad3 Reverse: GGCAGTAGATAACGTGAGGGA |
IDT DNA | qPCR |
|
Smad7 Forward: GCCCTCCCTGGATATCTTCTAT Smad7 Reverse: GATCTTGCTCCGCACTTTCT |
IDT DNA | qPCR |
| Software and algorithms | ||
| ImageJ | Schneider et al.96 | https://ImageJ.nih.gov/ij/ |
| DESeq2 | Love et al.97 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MACS2 | Zhang et al.98 | https://github.com/macs3-project/MACS |
| BEDTools | Quinlan et al.99 | http://bedtools.readthedocs.io |
| Homer | Heinz et al.100 | http://homer.ucsd.edu/ |
| deepTools | Ramírez et al.101 | https://deeptools.readthedocs.io/en/develop/ |
| Graphpad Prism | GraphPad | www.graphpad.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Alexandra L. Joyner (joynera@mskcc.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Animal work and breeding
All animal experiments were performed with the approval of the Institutional Animal Care and Use Committees at Memorial Sloan Kettering Cancer Center. The following mouse lines were used: Atoh1-Cre,66 Atoh1-FlpoER,93 Rosa26MASTR(R26frt-STOP-frt-GFPcre),69 R26LSL-SmoM2-YFP,94 Ptch1flox/flox,95 Ptf1aCre,67 hGFAP-Cre,68 Nestin-Cre,91,92 Kmt2dCf/Cf or Kmt2d(Exon50-51)flox/flox,64 Kmt2dNf/Nf or Kmt2d(Exon16-19)flox/flox.63 All mouse lines were maintained on an outbred Swiss Webster background, except R26LSL-SmoM2-YFP which was maintained on a C57BL/6 inbred background. Both sexes were used for the analysis. Animals were housed on a 12 hr light/dark cycle and were given access to food and water ad libitum. All experiments were performed using mice from embryonic stages to adult (ages E18–P300).
Animals for tumor studies were bred to have littermates that were either homozygous and heterozygous for the Kmt2d floxed allele or heterozygous and wild type for a Kmt2d floxed allele. For example, Atoh1-FlpoER/+; R26SmoM2/SmoM2; Kmt2dCf/+ male mice were bred with R26MASTR/+; Kmt2dCf/Cf females and Atoh1-FlpoER/+; R26SmoM2/SmoM2 male mice were bred with R26MASTR/MASTR; Kmt2dCf/+ females or Atoh1-FlpoER/+; Ptch1flox/flox; Kmt2dCf/+ male mice were bred with R26MASTR/+; Kmt2dCf/CF; Ptch1flox/flox females and Atoh1-FlpoER/+; Ptch1flox/flox male mice were bred with R26MASTR/+; Kmt2dCf/+; Ptch1flox/flox females. Animals for developmental studies were bred to have animals with no Cre or heterozygous and homozygous conditional mutants all from the same litter. The breeding schemes are described in the key resources table.
To induce genetic recombination using FlpoER, one 200ug/g dose of Tamoxifen (Tm) (Sigma-Aldrich) was injected subcutaneously into the back of P2 mice. Tm was dissolved and stored in corn oil (Sigma-Aldrich) at 20 mg/mL.
Method details
Tissue processing
E18 animals were collected after euthanizing the pregnant mouse by cervical dislocation after they were deeply anesthetized by inhalation of carbon dioxide and brains from embryos were harvested and fixed in 4% paraformaldehyde for 24-48 hr at 4°C. P4 animals were euthanized by decapitation and tissue harvested and fixed in 4% paraformaldehyde for 24-48 hr at 4°C. Animals P12-P300 were anesthetized and transcardially perfused with PBS followed by cold 4% paraformaldehyde (PFA). The brains and spinal cords with the vertebral column intact were dissected and post-fixed in 4% PFA at 4°C separately for 48 hr or overnight, respectively. Brains were then cryoprotected in 30% sucrose before freezing in Cryo-OCT. Frozen brains were sectioned in the sagittal plane at 14 μm. Micro-scissors were used to remove the spinal cords from the vertebral column and fixed in 4% PFA for another 24 hr and then cryoprotected in 30% sucrose before freezing in Cryo-OCT. Frozen spinal cords were coronally sectioned at 20 μm.
Microscopy
Images were collected either on a NanoZoomer 2.0 HT slide scanner (Hamamatsu Photonics) with a 20x objective using NDP.scan software or on a DM6000 Leica microscope using Zen software (Zeiss) using a 20x objective or on a Zeiss LSM 880 confocal microscope system with a 40X objective using Zen software. All images were processed using NDP.view2 or ImageJ Fiji software.96 3-dimensional images of spinal cord tumors were collected on a LCS SPIM Light Sheet Microscope from Luxendo with a 20x objective and processed using Imaris Software.
Histology, Immunofluorescence and Immunohistochemistry
To analyze the histopathology of brain and spinal cord tumors, sections were stained with Hematoxylin and Eosin (Thermo Fisher Scientific) (H&E) according to the protocol from the manufacturer. Images were collected on a Nanozoomer 2.0 HT slide scanner.
For immunohistochemical (IHC) analysis sections were blocked for at least 1 hr in 5% BSA (Sigma-Aldrich) and 0.3% Triton X-100 (Fischer Scientific) and incubated overnight at 4°C in antibody diluted in a blocking solution as described in the key resources table. Rabbit anti-PH3, Mouse anti-Ki67, Mouse anti-p27, Alexa fluor 555 Mouse anti-Ki-67, Mouse anti-NeurodD1 and Mouse anti-NeuN required a 40-minute incubation in antigen retrieval solution (10mM Sodium Citrate, 0.05% Tween-20, pH 6.0) at 95°C prior to blocking. Sections were then incubated with a species-specific secondary antibody for 2 hr at room temperature (RT) as described in the key resources table. Nuclei were counterstained with Hoechst 33258 (Invitrogen). Images were collected on a DM6000 Leica microscope or Nanozoomer 2.0 HT slide scanner.
TUNEL staining
Slides were permeabilized with 0.5% TritonX-100, and pre-incubated with Tdt buffer (30 mM Tris·HCl, 140 mM sodium cacodylate and 1 mM CoCl2) for 15 min at RT. Slides were then incubated for 1 hr at 37°C in a TUNEL reaction solution containing Terminal Transferase (Roche) and Biotin-16-dUTP (Sigma-Aldrich), following which slides were incubated with a Streptavidin Alexa Fluor 647 conjugate (Invitrogen S-32357) for 1 hr.
EdU (5-ethynyl-2’-deoxyuridine) Injection and staining
To assess cell proliferation, EdU (Invitrogen) was injected intraperitoneally at 100 mg/g 1 hr before euthanasia. A Click-it EdU assay (Invitrogen C10340) with Sulfo-Cyanine5 azide (Lumiprobe Corporation A3330) was used per the manufacturer’s protocol to stain sections.
Clearing and light sheet imaging
To image and construct a 3-dimensional map of a tumor along the spinal cord of a SmoM2-Kmt2dCf/Cf animal, the spinal cord was macro-dissected and cleared using an adaptation of the Adipo-Clear method from Dr. ZhuHao Wu.102 Briefly, samples were post-fixed in 4% PFA overnight at 4°C, dehydrated in a methanol series and delipidated in dichloromethane (DCM) before rehydration in a methanol series. The spinal cords were stained with a primary and secondary antibody for GFP+ tumor cells and a Yoyo1 dye for nuclei as described in the key resources table for 5 days each. Samples were then fixed in 4% PFA at 4°C, dehydrated through a methanol series and cleared using dibenzylether (DBE). Imaging was done on a LCS SPIM Light sheet microscope from Luxendo with a 20x objective.
RNA in situ hybridization
RNA in situ hybridization was performed as described in Blaess et al.103 using antisense RNA probes for Kcnc3 and Myc. The templates for Kcnc3 and Myc were generated by PCR using primers containing T7 or SP6 polymerase promoters from postnatal cerebellum cDNA. The sequences for the primer pairs used to generate probes are listed in the key resources table.
qRT-PCR analysis of GFP+ tumor cell RNA
Approximately 3 million GFP+ cells from SmoM2 and SmoM2-Kmt2dCf/Cf tumors were sorted and frozen in TRI-reagent (Sigma-Aldrich) from tumor-bearing animals of ages P35-P50. Total RNA was extracted using the mRNeasy kit (Qiagen), and cDNA was synthesized using SuperScript IV reverse transcriptase (Invitrogen). For the real-time PCR, 20ng of cDNA was used per reaction and amplified using StepOnePlus™ Real-Time PCR System. The reactions were using a Step One Plus apparatus and software. The fold change was determined using the formula 2-ΔΔCT, where ΔΔCT= ΔCT sample – Mean(ΔCT control) with ΔCT= CT gene-CT GAPDH. Statistics were determined using the Mann-Whitney test. Sequence of primer pairs used are listed in the key resources table.
Western Blot
Whole tumor tissue from SmoM2, SmoM2-Kmt2dCf/+ and SmoM2-Kmt2dCf/Cf animals were snap frozen using 2-methyl butane and dry-ice. Samples were stored at -80°C until ready for protein extraction. Total protein was extracted by sonicating the cells in the appropriate amount of 2x Laemmeli Buffer (4% SDS, 100mM Tris-HCl, pH 6.8, 4% Glycerol, 200mM DTT, with DNase and Protease/Phosphatase inhibitors (Thermo Fischer Scientific) on ice for 30 min. Samples were spun down for 10 minutes at 16,000g and the pellet resuspended in NuPAGE LDS sample buffer (Thermo Fisher Scientific) before protein concentration determination. Samples were run on a 12% Bis-Tris Protein Gel, transferred onto PVDF membranes, stained with primary antibodies for Histone H3, H3K4me1 and H3K4me3 over night at 4°C, followed by species-specific secondary antibodies for 2 hrs. at room temperature all described in the key resources table. ECL Western Blotting Substrate (Thermo Fisher Scientific) was used to develop the membrane. H3K4me1/3 signal intensity was normalized to the signal intensity of Histone H3. Protein content was measured using ImageJ96 to measure the signal intensity in the appropriate protein band from 3 cell protein samples of each of the 3 genotypes and the intensity reported relative to the average of SmoM2 tumors.
Sample preparation for sequencing experiments
SmoM2 animals between 6-16 weeks, and SmoM2-Kmt2dCf/+ and SmoM2-Kmt2dCf/Cf animals between 5-8 weeks were used for sequencing experiments. The tumor tissue of the brain was macro-dissected from the surrounding normal brain tissue, dissociated and processed using a Papain Kit (Worthington Biochemical Corporation) and the manufacturer’s protocol. The GFP+ tumor cells were isolated by fluorescence activated cell sorting in the MSKCC Flow Cytometry Core. From each sample 70,000 cells were processed for ATAC-seq, and 1-5 million cells were frozen at -80°C for RNA-seq.
The spinal cords from the same SmoM2-Kmt2dCf/Cf animals were dissected and dissociated using a papain kit. The GFP+ metastatic tumor cells were isolated by FACS at the MSKCC Flow Cytometry Core. 70,000 cells were processed for ATAC-seq, and 100,000-3 million cells were frozen at 80°C for RNA-seq. Note, the proportion of tumor cells in the spinal cord samples was much lower than in the brain samples for primary tumors, thus contamination with spinal cord cells and immune cells is more likely in the spinal cord samples than primary tumor samples.
RNA-seq sample preparation
Frozen cell pellets of 100,000-3,000,000 cells from spinal cord metastatic tumors and 1-5 million cells from primary brain tumors prepared as described above were submitted to the MSKCC Integrated Genomics Operations (IGO) Core facility for RNA isolation and sequencing.
ATAC-seq sample preparation
DNA was isolated from the sorted GFP+ cells describe above and prepared according to the Omni-ATAC protocol.104,105,106 Briefly, 70,000 cells per sample were lysed using 0.1% NP-40, 0.1% Tween and 0.01% Digitonin to yield nuclei. The resulting chromatin was fragmented and tagmented using Tn5 transposase. DNA was purified using a Qiagen MinElute Reaction Cleanup Kit (Qiagen) and amplified using a NEBNext 2x MasterMix (NEB). Libraries were prepared using universal forward and reverse primers from Ad2.1-Ad2.24.104 The final libraries were purified using a single left-handed bead purification with AMPure beads (Beckman Coulter). Libraries were sent to the MSKCC IGO Core Facility for sequencing.
Quantification and statistical analysis
E18.5 GCP proliferation rate, P12/21 TUNEL, KI67, NeuroD1 and EdU quantification
Proliferation rate was determined by quantifying the number of PH3+ cells per area of the outer EGL (mm2) using the ImageJ software96 on images collected on the NanoZoomer 2.0 HT slide scanner with a 20x objective. 3 near adjacent hemisphere sections were selected from 3 mice per genotype to manually quantify the number of PH3+ cells and the area of the outer and total EGL.
TUNEL images were taken on a Nanozoomer2.0 HT slide scanner and exported at 20x. Quantification of the TUNEL+ cells on P12 pre-neoplastic lesions/EGL were performed using ImageJ Fiji Software.96 Similar fields between the paramedian and copula pyramis lobules in the lateral posterior hemispheres of 3 near adjacent sections were selected and the areas of the EGL (mm2) manually measured. The quantification of the TUNEL+ particles within that area was automated. The reported cell death density was measured by averaging the TUNEL+ particle density on 3 near adjacent sections per animal from 3 animals of each genotype.
Ki67 and NeuroD1 quantification were done on Zeiss LSM 880 confocal images. Using ImageJ software,96 Ki67-DAPI double positive or NeuroD1-Dapi double positive pixels were quantified and normalized to all DAPI positive pixels as a percentage. For every sample (n=3 per genotype), 3 sections and 6 40x images (212.55 X 212.55 μm) per section were quantified. On the graph, every point represents the averages of the 3 sections (18 images).
Images of EdU staining in the lateral posterior hemispheres of 3 near adjacent sections of the cerebellum of P12 animals were taken at 20x on a DM6000 Leica Microscope and processed using ImageJ Fiji Software.96 Slides were stained with Sulfo-Cyanine5 azide (Lumiprobe Corporation, A3330) and Hoechst. The area of the EGL (mm2) was measured manually and the number of EdU+ GCPs in the area between the paramedian and copula pyramis lobules were quantified using automation with ImageJ96 and cell density reported. The reported density of EdU+ cells was measured by averaging the EdU+ cells on 3 near adjacent sections per animal from 3 animals of each genotype.
Quantification of CB and Tumor area
The area of the CB (mm2) and the Folding Index ((1-[convex length/EGL length]) x 100) at E18.5 in the Nestin-Cre Kmt2d mutants were measured manually on NanoZoomer digitized images of sections stained with H&E and measured using the NDP.view2 software from 3 sections per animal in 3 animals for each genotype.
The areas of the EGL/lesions at P12 and P21 were calculated as a percentage of the total area of the cerebellum. The regions were outlined manually on NanoZoomer digitized images of sections stained with H&E and area measured using the NDP.view2 software from 3 sections per animal in 3 animals for each genotype.
The areas of the metastatic tumors were calculated as a percentage of the total spinal cord area along the spinal cord. Sections were stained with GFP and DAPI and NanoZoomer digitized images created. NDP.view2 software was used to outline and measure the area of the tumors and the area of the spinal cord plus tumor on a serial series of coronal sections (every 10th section; ∼30 sections/mouse). The number of animals per genotype are listed in the Figures.
Transcriptome analysis
RNA sequencing reads were 3’ trimmed for base quality 15 and adapter sequences using version 0.4.5 of TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore), and then aligned to mouse assembly mm10 with STAR v2.4 using default parameters. Data quality and transcript coverage were assessed using the Picard tool CollectRNASeqMetrics (http://broadinstitute.github.io/picard/). Read count tables were generated with HTSeq v0.9.1. Normalization and expression dynamics were evaluated with DESeq297 using the default parameters and the Wald test and outliers were assessed by sample grouping in principal component analysis. Gene set enrichment analysis (GSEA, http://software.broadinstitute.org/gsea) was run against MsigDB v6 using the pre-ranked option and log2 fold change for pairwise comparisons. Over-representation analysis was done in R using enrichGO in the clusterProfiler package. Network analysis was performed using enrichplot::cnetplot in R with default parameters. GEO ID: GSE211030.
Epigenome analysis
ATAC sequencing reads were trimmed and filtered for quality and adapter content using version 0.4.5 of TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore), with a quality setting of 15, and running version 1.15 of cutadapt and version 0.11.5 of FastQC. Reads were aligned to mouse assembly mm10 with version 2.3.4.1 of bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) and were deduplicated using MarkDuplicates in version 2.16.0 of Picard Tools. To ascertain regions of chromatin accessibility, MACS298 (https://github.com/taoliu/MACS) was used with a p-value setting of 0.001. The BEDTools suite (http://bedtools.readthedocs.io)99 was used to create normalized read density profiles. A global peak atlas was created by first removing blacklisted regions (http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm10mouse/mm10.blacklist.bed.gz) then taking 500 bp windows around peak summits for ATAC and counting reads with version 1.6.1 of featureCounts (http://subread.sourceforge.net). DESeq297 was used to normalize read density (median ratio method) and to calculate differential enrichment for all pairwise contrasts. Peak-gene associations were created by assigning all intragenic peaks to that gene, and otherwise using linear genomic distance to transcription start site. Peak intersections were calculated using bedtools99 v2.29.1 and intersectBed with 1 bp overlap. Gene set enrichment analysis (GSEA, http://software.broadinstitute.org/gsea) was performed with the pre-ranked option and default parameters, where each gene was assigned the single peak with the largest (in magnitude) log2 fold change associated with it. Motif signatures were obtained using Homer v4.5 (http://homer.ucsd.edu).100 Composite and tornado plots were created using deepTools v3.3.0101 by running computeMatrix and plotHeatmap on normalized bigwigs with average signal sampled in 25 bp windows and flanking region defined by the surrounding 3 kb. Over-representation analysis was done in R using enrichGO in the clusterProfiler package (Table S6). Network analysis was performed using the output from clusterProfiler with enrichplot::cnetplot in R with default parameters. The RNA-ATAC scatterplot was created using peak-gene assignments described above. GEO ID: GSE211030.
Statistical analysis
All statistical analyses of quantified data from sections were performed using Prism software (GraphPad) and significance was determined as P <= 0.05. The statistical analysis used for each figure/subfigure is described in the figure legends.
Acknowledgments
We are grateful to members of the Joyner lab for helpful comments throughout the work, in particular to I-Li Tan during the early stages of the work. We thank the Memorial Sloan Kettering Cancer Center Flow Cytometry Core, Integrated Genomics Operations Core Facility and the Center for Comparative Medicine and Pathology for technical support, and James Muller for assistance with 3D imaging. We also thank Daniel Medina-Cano from the Thomas Vierbuchen lab and Yanhong Yang from the Viviane Tabar lab at MSKCC for reagents. We are grateful to Lucia Grandison and Derek Lao for technical assistance and to Cara Monaco for helping prepare the manuscript. Some of the schematics in the graphical abstract were created using Biorender.com. The research was supported by grants to A.L.J. from the NCI (R01CA192176) and MSK Functional Genomics Initiative (GC24221), to S.E. from a Francois Wallace Monahan Fellowship and a National Cancer Institute Cancer Center Support Grant [P30 CA008748-48].
Author contributions
Conceptualization: R.M.S. and A.L.J.; Methodology: R.M.S. and A.L.J.; Investigation: R.M.S., H.M., D.N.S., S.E., Z.L., and N.S.B.; Formal Analysis: R.M.S., R.K., H.M., S.E., and A.L.J.; Resources: K.G.; Manuscript Preparation: R.M.S. and A.L.J.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse and equitable conduct of research.
Published: September 9, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107831.
Supplemental information
Data used to generate Figure S3A.
Data used to generate Figure 4F.
Data used to generate Figure 7A.
Data and code availability
-
•
All sequencing data have been deposited at GEO and are publicly available as of the date of the publication. The accession code is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze data reported in this paper is available from the lead contact upon request.
References
- 1.Taylor M.D., Northcott P.A., Korshunov A., Remke M., Cho Y.J., Clifford S.C., Eberhart C.G., Parsons D.W., Rutkowski S., Gajjar A., et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rossi A., Caracciolo V., Russo G., Reiss K., Giordano A. Medulloblastoma: from molecular pathology to therapy. Clin. Cancer Res. 2008;14:971–976. doi: 10.1158/1078-0432.CCR-07-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Northcott P.A., Dubuc A.M., Pfister S., Taylor M.D. Molecular subgroups of medulloblastoma. Expert Rev. Neurother. 2012;12:871–884. doi: 10.1586/ern.12.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Northcott P.A., Jones D.T.W., Kool M., Robinson G.W., Gilbertson R.J., Cho Y.J., Pomeroy S.L., Korshunov A., Lichter P., Taylor M.D., Pfister S.M. Medulloblastomics: the end of the beginning. Nat. Rev. Cancer. 2012;12:818–834. doi: 10.1038/nrc3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilbertson R.J., Ellison D.W. The origins of medulloblastoma subtypes. Annu. Rev. Pathol. 2008;3:341–365. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- 6.Meirson T., Gil-Henn H., Samson A.O. Invasion and metastasis: the elusive hallmark of cancer. Oncogene. 2020;39:2024–2026. doi: 10.1038/s41388-019-1110-1. [DOI] [PubMed] [Google Scholar]
- 7.Kciuk M., Gielecińska A., Budzinska A., Mojzych M., Kontek R. Metastasis and MAPK Pathways. Int. J. Mol. Sci. 2022;23:3847. doi: 10.3390/ijms23073847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garzia L., Kijima N., Morrissy A.S., De Antonellis P., Guerreiro-Stucklin A., Holgado B.L., Wu X., Wang X., Parsons M., Zayne K., et al. A Hematogenous Route for Medulloblastoma Leptomeningeal Metastases. Cell. 2018;173:1549. doi: 10.1016/j.cell.2018.05.033. [DOI] [PubMed] [Google Scholar]
- 9.Li M., Deng Y., Zhang W. Molecular Determinants of Medulloblastoma Metastasis and Leptomeningeal Dissemination. Mol. Cancer Res. 2021;19:743–752. doi: 10.1158/1541-7786.MCR-20-1026. [DOI] [PubMed] [Google Scholar]
- 10.Van Ommeren R., Garzia L., Holgado B.L., Ramaswamy V., Taylor M.D. The molecular biology of medulloblastoma metastasis. Brain Pathol. 2020;30:691–702. doi: 10.1111/bpa.12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grausam K.B., Dooyema S.D.R., Bihannic L., Premathilake H., Morrissy A.S., Forget A., Schaefer A.M., Gundelach J.H., Macura S., Maher D.M., et al. ATOH1 Promotes Leptomeningeal Dissemination and Metastasis of Sonic Hedgehog Subgroup Medulloblastomas. Cancer Res. 2017;77:3766–3777. doi: 10.1158/0008-5472.CAN-16-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhall G. Medulloblastoma. J. Child Neurol. 2009;24:1418–1430. doi: 10.1177/0883073809341668. [DOI] [PubMed] [Google Scholar]
- 13.Remke M., Ramaswamy V., Taylor M.D. Medulloblastoma molecular dissection: the way toward targeted therapy. Curr. Opin. Oncol. 2013;25:674–681. doi: 10.1097/CCO.0000000000000008. [DOI] [PubMed] [Google Scholar]
- 14.Kool M., Korshunov A., Remke M., Jones D.T.W., Schlanstein M., Northcott P.A., Cho Y.J., Koster J., Schouten-van Meeteren A., van Vuurden D., et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123:473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miranda Kuzan-Fischer C., Juraschka K., Taylor M.D. Medulloblastoma in the Molecular Era. J. Korean Neurosurg. Soc. 2018;61:292–301. doi: 10.3340/jkns.2018.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Northcott P.A., Korshunov A., Pfister S.M., Taylor M.D. The clinical implications of medulloblastoma subgroups. Nat. Rev. Neurol. 2012;8:340–351. doi: 10.1038/nrneurol.2012.78. [DOI] [PubMed] [Google Scholar]
- 17.Cho Y.J., Tsherniak A., Tamayo P., Santagata S., Ligon A., Greulich H., Berhoukim R., Amani V., Goumnerova L., Eberhart C.G., et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 2011;29:1424–1430. doi: 10.1200/JCO.2010.28.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Northcott P.A., Korshunov A., Witt H., Hielscher T., Eberhart C.G., Mack S., Bouffet E., Clifford S.C., Hawkins C.E., French P., et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011;29:1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fults D.W., Taylor M.D., Garzia L. Leptomeningeal dissemination: a sinister pattern of medulloblastoma growth. J. Neurosurg. Pediatr. 2019;23:613–621. doi: 10.3171/2018.11.PEDS18506. [DOI] [PubMed] [Google Scholar]
- 20.Wu X., Northcott P.A., Dubuc A., Dupuy A.J., Shih D.J.H., Witt H., Croul S., Bouffet E., Fults D.W., Eberhart C.G., et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012;482:529–533. doi: 10.1038/nature10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vladoiu M.C., El-Hamamy I., Donovan L.K., Farooq H., Holgado B.L., Sundaravadanam Y., Ramaswamy V., Hendrikse L.D., Kumar S., Mack S.C., et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature. 2019;572:67–73. doi: 10.1038/s41586-019-1158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schüller U., Heine V.M., Mao J., Kho A.T., Dillon A.K., Han Y.G., Huillard E., Sun T., Ligon A.H., Qian Y., et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson P., Tong Y., Robinson G., Thompson M.C., Currle D.S., Eden C., Kranenburg T.A., Hogg T., Poppleton H., Martin J., et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliver T.G., Read T.A., Kessler J.D., Mehmeti A., Wells J.F., Huynh T.T.T., Lin S.M., Wechsler-Reya R.J. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- 25.Haldipur P., Millen K.J., Aldinger K.A. Human Cerebellar Development and Transcriptomics: Implications for Neurodevelopmental Disorders. Annu. Rev. Neurosci. 2022;45:515–531. doi: 10.1146/annurev-neuro-111020-091953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao F., Li C., Zhou Q., Qu P., Wang B., Wang X., Zhang S., Wang X., Zhao C., Zhang J., et al. Distinctive localization and MRI features correlate of molecular subgroups in adult medulloblastoma. J. Neuro Oncol. 2017;135:353–360. doi: 10.1007/s11060-017-2581-y. [DOI] [PubMed] [Google Scholar]
- 27.Wefers A.K., Warmuth-Metz M., Pöschl J., von Bueren A.O., Monoranu C.M., Seelos K., Peraud A., Tonn J.C., Koch A., Pietsch T., et al. Subgroup-specific localization of human medulloblastoma based on pre-operative MRI. Acta Neuropathol. 2014;127:931–933. doi: 10.1007/s00401-014-1271-5. [DOI] [PubMed] [Google Scholar]
- 28.Perreault S., Ramaswamy V., Achrol A.S., Chao K., Liu T.T., Shih D., Remke M., Schubert S., Bouffet E., Fisher P.G., et al. MRI surrogates for molecular subgroups of medulloblastoma. AJNR. Am. J. Neuroradiol. 2014;35:1263–1269. doi: 10.3174/ajnr.A3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan I.L., Wojcinski A., Rallapalli H., Lao Z., Sanghrajka R.M., Stephen D., Volkova E., Korshunov A., Remke M., Taylor M.D., et al. Lateral cerebellum is preferentially sensitive to high sonic hedgehog signaling and medulloblastoma formation. Proc. Natl. Acad. Sci. USA. 2018;115:3392–3397. doi: 10.1073/pnas.1717815115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis P.M., Gritli-Linde A., Smeyne R., Kottmann A., McMahon A.P. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev. Biol. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 31.Corrales J.D., Blaess S., Mahoney E.M., Joyner A.L. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development. 2006;133:1811–1821. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- 32.Flora A., Klisch T.J., Schuster G., Zoghbi H.Y. Deletion of Atoh1 disrupts Sonic Hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science. 2009;326:1424–1427. doi: 10.1126/science.1181453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khatua S. Evolving molecular era of childhood medulloblastoma: time to revisit therapy. Future Oncol. 2016;12:107–117. doi: 10.2217/fon.15.284. [DOI] [PubMed] [Google Scholar]
- 34.Skowron P., Farooq H., Cavalli F.M.G., Morrissy A.S., Ly M., Hendrikse L.D., Wang E.Y., Djambazian H., Zhu H., Mungall K.L., et al. The transcriptional landscape of Shh medulloblastoma. Nat. Commun. 2021;12:1749. doi: 10.1038/s41467-021-21883-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Northcott P.A., Hielscher T., Dubuc A., Mack S., Shih D., Remke M., Al-Halabi H., Albrecht S., Jabado N., Eberhart C.G., et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011;122:231–240. doi: 10.1007/s00401-011-0846-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kool M., Jones D.T.W., Jäger N., Northcott P.A., Pugh T.J., Hovestadt V., Piro R.M., Esparza L.A., Markant S.L., Remke M., et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Northcott P.A., Buchhalter I., Morrissy A.S., Hovestadt V., Weischenfeldt J., Ehrenberger T., Gröbner S., Segura-Wang M., Zichner T., Rudneva V.A., et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–317. doi: 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osborn A.G., Louis D.N., Poussaint T.Y., Linscott L.L., Salzman K.L. The 2021 World Health Organization Classification of Tumors of the Central Nervous System: What Neuroradiologists Need to Know. AJNR. Am. J. Neuroradiol. 2022;43:928–937. doi: 10.3174/ajnr.A7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Archer T.C., Ehrenberger T., Mundt F., Gold M.P., Krug K., Mah C.K., Mahoney E.L., Daniel C.J., LeNail A., Ramamoorthy D., et al. Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 2018;34:396–410.e8. doi: 10.1016/j.ccell.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwalbe E.C., Lindsey J.C., Nakjang S., Crosier S., Smith A.J., Hicks D., Rafiee G., Hill R.M., Iliasova A., Stone T., et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol. 2017;18:958–971. doi: 10.1016/S1470-2045(17)30243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cavalli F.M.G., Remke M., Rampasek L., Peacock J., Shih D.J.H., Luu B., Garzia L., Torchia J., Nor C., Morrissy A.S., et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 2017;31:737–754.e6. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Lopez J., Kumar R., Smith K.S., Northcott P.A. Deconstructing Sonic Hedgehog Medulloblastoma: Molecular Subtypes, Drivers, and Beyond. Trends Genet. 2021;37:235–250. doi: 10.1016/j.tig.2020.11.001. [DOI] [PubMed] [Google Scholar]
- 43.Parsons D.W., Li M., Zhang X., Jones S., Leary R.J., Lin J.C.H., Boca S.M., Carter H., Samayoa J., Bettegowda C., et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rao R.C., Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer. 2015;15:334–346. doi: 10.1038/nrc3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones D.T.W., Jäger N., Kool M., Zichner T., Hutter B., Sultan M., Cho Y.J., Pugh T.J., Hovestadt V., Stütz A.M., et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pugh T.J., Weeraratne S.D., Archer T.C., Pomeranz Krummel D.A., Auclair D., Bochicchio J., Carneiro M.O., Carter S.L., Cibulskis K., Erlich R.L., et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guenther M.G., Levine S.S., Boyer L.A., Jaenisch R., Young R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Froimchuk E., Carey S.T., Edwards C., Jewell C.M. Self-Assembly as a Molecular Strategy to Improve Immunotherapy. Acc. Chem. Res. 2020;53:2534–2545. doi: 10.1021/acs.accounts.0c00438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ang S.Y., Uebersohn A., Spencer C.I., Huang Y., Lee J.E., Ge K., Bruneau B.G. KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Development. 2016;143:810–821. doi: 10.1242/dev.132688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dhar S.S., Zhao D., Lin T., Gu B., Pal K., Wu S.J., Alam H., Lv J., Yun K., Gopalakrishnan V., et al. MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol. Cell. 2018;70:825–841.e6. doi: 10.1016/j.molcel.2018.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Froimchuk E., Jang Y., Ge K. Histone H3 lysine 4 methyltransferase KMT2D. Gene. 2017;627:337–342. doi: 10.1016/j.gene.2017.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Massagué J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kahn S.A., Wang X., Nitta R.T., Gholamin S., Theruvath J., Hutter G., Azad T.D., Wadi L., Bolin S., Ramaswamy V., et al. Notch1 regulates the initiation of metastasis and self-renewal of Group 3 medulloblastoma. Nat. Commun. 2018;9:4121. doi: 10.1038/s41467-018-06564-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roussel M.F., Robinson G.W. Role of MYC in Medulloblastoma. Cold Spring Harb. Perspect. Med. 2013;3:a014308. doi: 10.1101/cshperspect.a014308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shrestha S., Morcavallo A., Gorrini C., Chesler L. Biological Role of MYCN in Medulloblastoma: Novel Therapeutic Opportunities and Challenges Ahead. Front. Oncol. 2021;11:694320. doi: 10.3389/fonc.2021.694320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vanner R.J., Remke M., Gallo M., Selvadurai H.J., Coutinho F., Lee L., Kushida M., Head R., Morrissy S., Zhu X., et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell. 2014;26:33–47. doi: 10.1016/j.ccr.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adolphe C., Millar A., Kojic M., Barkauskas D.S., Sundström A., Swartling F.J., Hediyeh-Zadeh S., Tan C.W., Davis M.J., Genovesi L.A., Wainwright B.J. SOX9 Defines Distinct Populations of Cells in SHH Medulloblastoma but Is Not Required for Math1-Driven Tumor Formation. Mol. Cancer Res. 2021;19:1831–1839. doi: 10.1158/1541-7786.MCR-21-0117. [DOI] [PubMed] [Google Scholar]
- 58.Selvadurai H.J., Luis E., Desai K., Lan X., Vladoiu M.C., Whitley O., Galvin C., Vanner R.J., Lee L., Whetstone H., et al. Medulloblastoma Arises from the Persistence of a Rare and Transient Sox2(+) Granule Neuron Precursor. Cell Rep. 2020;31:107511. doi: 10.1016/j.celrep.2020.03.075. [DOI] [PubMed] [Google Scholar]
- 59.Ahlfeld J., Favaro R., Pagella P., Kretzschmar H.A., Nicolis S., Schüller U. Sox2 requirement in sonic hedgehog-associated medulloblastoma. Cancer Res. 2013;73:3796–3807. doi: 10.1158/0008-5472.CAN-13-0238. [DOI] [PubMed] [Google Scholar]
- 60.Vong K.I., Leung C.K.Y., Behringer R.R., Kwan K.M. Sox9 is critical for suppression of neurogenesis but not initiation of gliogenesis in the cerebellum. Mol. Brain. 2015;8:25. doi: 10.1186/s13041-015-0115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee J.E., Wang C., Xu S., Cho Y.W., Wang L., Feng X., Baldridge A., Sartorelli V., Zhuang L., Peng W., Ge K. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jang Y., Broun A., Wang C., Park Y.K., Zhuang L., Lee J.E., Froimchuk E., Liu C., Ge K. H3.3K4M destabilizes enhancer H3K4 methyltransferases MLL3/MLL4 and impairs adipose tissue development. Nucleic Acids Res. 2019;47:607–620. doi: 10.1093/nar/gky982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lawton A.K., Engstrom T., Rohrbach D., Omura M., Turnbull D.H., Mamou J., Zhang T., Schwarz J.M., Joyner A.L. Cerebellar folding is initiated by mechanical constraints on a fluid-like layer without a cellular pre-pattern. Elife. 2019;8:e45019. doi: 10.7554/eLife.45019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matei V., Pauley S., Kaing S., Rowitch D., Beisel K.W., Morris K., Feng F., Jones K., Lee J., Fritzsch B. Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Dev. Dynam. 2005;234:633–650. doi: 10.1002/dvdy.20551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kawaguchi Y., Cooper B., Gannon M., Ray M., MacDonald R.J., Wright C.V.E. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 68.Zhuo L., Theis M., Alvarez-Maya I., Brenner M., Willecke K., Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 69.Lao Z., Raju G.P., Bai C.B., Joyner A.L. MASTR: a technique for mosaic mutant analysis with spatial and temporal control of recombination using conditional floxed alleles in mice. Cell Rep. 2012;2:386–396. doi: 10.1016/j.celrep.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan X., Liu Z., Chen Y. Regulation of TGF-beta signaling by Smad7. Acta Biochim. Biophys. Sin. 2009;41:263–272. doi: 10.1093/abbs/gmp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ying M., Sang Y., Li Y., Guerrero-Cazares H., Quinones-Hinojosa A., Vescovi A.L., Eberhart C.G., Xia S., Laterra J. Kruppel-like family of transcription factor 9, a differentiation-associated transcription factor, suppresses Notch1 signaling and inhibits glioblastoma-initiating stem cells. Stem Cell. 2011;29:20–31. doi: 10.1002/stem.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ying M., Tilghman J., Wei Y., Guerrero-Cazares H., Quinones-Hinojosa A., Ji H., Laterra J. Kruppel-like factor-9 (KLF9) inhibits glioblastoma stemness through global transcription repression and integrin alpha6 inhibition. J. Biol. Chem. 2014;289:32742–32756. doi: 10.1074/jbc.M114.588988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sarić N., Selby M., Ramaswamy V., Kool M., Stockinger B., Hogstrand C., Williamson D., Marino S., Taylor M.D., Clifford S.C., Basson M.A. The AHR pathway represses TGFbeta-SMAD3 signalling and has a potent tumour suppressive role in SHH medulloblastoma. Sci. Rep. 2020;10:148. doi: 10.1038/s41598-019-56876-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morabito M., Larcher M., Cavalli F.M., Foray C., Forget A., Mirabal-Ortega L., Andrianteranagna M., Druillennec S., Garancher A., Masliah-Planchon J., et al. An autocrine ActivinB mechanism drives TGFbeta/Activin signaling in Group 3 medulloblastoma. EMBO Mol. Med. 2019;11:e9830. doi: 10.15252/emmm.201809830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Adachi T., Miyashita S., Yamashita M., Shimoda M., Okonechnikov K., Chavez L., Kool M., Pfister S.M., Inoue T., Kawauchi D., Hoshino M. Notch Signaling between Cerebellar Granule Cell Progenitors. eNeuro. 2021;8 doi: 10.1523/ENEURO.0468-20.2021. ENEURO.0468-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Natarajan S., Li Y., Miller E.E., Shih D.J., Taylor M.D., Stearns T.M., Bronson R.T., Ackerman S.L., Yoon J.K., Yun K. Notch1-induced brain tumor models the sonic hedgehog subgroup of human medulloblastoma. Cancer Res. 2013;73:5381–5390. doi: 10.1158/0008-5472.CAN-13-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wei L., Shao N., Peng Y., Zhou P. Inhibition of Cathepsin S Restores TGF-beta-induced Epithelial-to-mesenchymal Transition and Tight Junction Turnover in Glioblastoma Cells. J. Cancer. 2021;12:1592–1603. doi: 10.7150/jca.50631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang L.Y., Guo Q., Guan G.F., Cheng W., Cheng P., Wu A.H. Integrin Beta 5 Is a Prognostic Biomarker and Potential Therapeutic Target in Glioblastoma. Front. Oncol. 2019;9:904. doi: 10.3389/fonc.2019.00904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kunigal S., Gondi C.S., Gujrati M., Lakka S.S., Dinh D.H., Olivero W.C., Rao J.S. SPARC-induced migration of glioblastoma cell lines via uPA-uPAR signaling and activation of small GTPase RhoA. Int. J. Oncol. 2006;29:1349–1357. https://www.ncbi.nlm.nih.gov/pubmed/17088972 [PMC free article] [PubMed] [Google Scholar]
- 80.Karki K., Li X., Jin U.H., Mohankumar K., Zarei M., Michelhaugh S.K., Mittal S., Tjalkens R., Safe S. Nuclear receptor 4A2 (NR4A2) is a druggable target for glioblastomas. J. Neuro Oncol. 2020;146:25–39. doi: 10.1007/s11060-019-03349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koso H., Tsuhako A., Lyons E., Ward J.M., Rust A.G., Adams D.J., Jenkins N.A., Copeland N.G., Watanabe S. Identification of FoxR2 as an oncogene in medulloblastoma. Cancer Res. 2014;74:2351–2361. doi: 10.1158/0008-5472.CAN-13-1523. [DOI] [PubMed] [Google Scholar]
- 82.Mumert M., Dubuc A., Wu X., Northcott P.A., Chin S.S., Pedone C.A., Taylor M.D., Fults D.W. Functional genomics identifies drivers of medulloblastoma dissemination. Cancer Res. 2012;72:4944–4953. doi: 10.1158/0008-5472.CAN-12-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jenkins N.C., Kalra R.R., Dubuc A., Sivakumar W., Pedone C.A., Wu X., Taylor M.D., Fults D.W. Genetic drivers of metastatic dissemination in sonic hedgehog medulloblastoma. Acta Neuropathol. Commun. 2014;2:85. doi: 10.1186/s40478-014-0085-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsai Y.P., Wu K.J. Hypoxia-regulated target genes implicated in tumor metastasis. J. Biomed. Sci. 2012;19:102. doi: 10.1186/1423-0127-19-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chi Y., Remsik J., Kiseliovas V., Derderian C., Sener U., Alghader M., Saadeh F., Nikishina K., Bale T., Iacobuzio-Donahue C., et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science. 2020;369:276–282. doi: 10.1126/science.aaz2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vengellur A., Woods B.G., Ryan H.E., Johnson R.S., LaPres J.J. Gene expression profiling of the hypoxia signaling pathway in hypoxia-inducible factor 1alpha null mouse embryonic fibroblasts. Gene Expr. 2003;11:181–197. doi: 10.3727/000000003108749062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang X., Dubuc A.M., Ramaswamy V., Mack S., Gendoo D.M.A., Remke M., Wu X., Garzia L., Luu B., Cavalli F., et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 2015;129:449–457. doi: 10.1007/s00401-015-1389-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jenkins N.C., Rao G., Eberhart C.G., Pedone C.A., Dubuc A.M., Fults D.W. Somatic cell transfer of c-Myc and Bcl-2 induces large-cell anaplastic medulloblastomas in mice. J. Neuro Oncol. 2016;126:415–424. doi: 10.1007/s11060-015-1985-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kawauchi D., Robinson G., Uziel T., Gibson P., Rehg J., Gao C., Finkelstein D., Qu C., Pounds S., Ellison D.W., et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell. 2012;21:168–180. doi: 10.1016/j.ccr.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Niesen J., Ohli J., Sedlacik J., Dührsen L., Hellwig M., Spohn M., Holsten T., Schüller U. Pik3ca mutations significantly enhance the growth of SHH medulloblastoma and lead to metastatic tumour growth in a novel mouse model. Cancer Lett. 2020;477:10–18. doi: 10.1016/j.canlet.2020.02.028. [DOI] [PubMed] [Google Scholar]
- 91.Tronche F., Kellendonk C., Kretz O., Gass P., Anlag K., Orban P.C., Bock R., Klein R., Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 92.Giusti S.A., Vercelli C.A., Vogl A.M., Kolarz A.W., Pino N.S., Deussing J.M., Refojo D. Behavioral phenotyping of Nestin-Cre mice: implications for genetic mouse models of psychiatric disorders. J. Psychiatr. Res. 2014;55:87–95. doi: 10.1016/j.jpsychires.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 93.Wojcinski A., Morabito M., Lawton A.K., Stephen D.N., Joyner A.L. Genetic deletion of genes in the cerebellar rhombic lip lineage can stimulate compensation through adaptive reprogramming of ventricular zone-derived progenitors. Neural Dev. 2019;14:4. doi: 10.1186/s13064-019-0128-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mao J., Ligon K.L., Rakhlin E.Y., Thayer S.P., Bronson R.T., Rowitch D., McMahon A.P. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66:10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ellis T., Smyth I., Riley E., Graham S., Elliot K., Narang M., Kay G.F., Wicking C., Wainwright B. Patched 1 conditional null allele in mice. Genesis. 2003;36:158–161. doi: 10.1002/gene.10208. [DOI] [PubMed] [Google Scholar]
- 96.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ramírez F., Dündar F., Diehl S., Grüning B.A., Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chi J., Crane A., Wu Z., Cohen P. Adipo-Clear: A Tissue Clearing Method for Three-Dimensional Imaging of Adipose Tissue. J. Vis. Exp. 2018;137:58271. doi: 10.3791/58271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Blaess S., Bodea G.O., Kabanova A., Chanet S., Mugniery E., Derouiche A., Stephen D., Joyner A.L. Temporal-spatial changes in Sonic Hedgehog expression and signaling reveal different potentials of ventral mesencephalic progenitors to populate distinct ventral midbrain nuclei. Neural Dev. 2011;6:29. doi: 10.1186/1749-8104-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B., et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data used to generate Figure S3A.
Data used to generate Figure 4F.
Data used to generate Figure 7A.
Data Availability Statement
-
•
All sequencing data have been deposited at GEO and are publicly available as of the date of the publication. The accession code is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze data reported in this paper is available from the lead contact upon request.