

Abstract
Chikungunya virus (CHIKV) is a mosquito-transmitted alphavirus that causes epidemics of acute and chronic musculoskeletal disease. Here, we analyzed the human B cell response to a chikungunya virus-like particle (VLP) adjuvanted vaccine (PXVX0317) from samples obtained from a phase 2 clinical trial in humans (NCT03483961). Immunization with PXVX0317 induced high levels of neutralizing antibody in serum against CHIKV and circulating antigen-specific B cells up to 6 months post-immunization. Monoclonal antibodies generated from peripheral blood B cells of three PXVX0317-vaccinated subjects on day 57 post-immunization potently neutralized CHIKV infection, and a subset of these inhibited multiple related arthritogenic alphaviruses. Epitope mapping and cryo-electron microscopy defined two broadly neutralizing mAbs that uniquely bind to the apex of the B domain of the E2 glycoprotein. These results demonstrate the inhibitory breadth and activity of the human B cell response induced by the PXVX0317 vaccine against CHIKV and potentially other related alphaviruses.
One sentence summary:
Antibodies generated from B cells of human subjects immunized with a chikungunya vaccine inhibited infection of multiple alphaviruses.
INTRODUCTION
Alphaviruses are arthropod-transmitted enveloped viruses that cause arthritis, musculoskeletal disease, or encephalitis (1, 2). The arthritogenic alphaviruses include Mayaro (MAYV), O’nyong’nyong (ONNV), Ross River (RRV), and chikungunya (CHIKV) viruses, the latter of which has caused epidemics with substantial morbidity in Asia, Africa, Oceania, and the Americas. Although the encephalitic alphaviruses, Eastern equine encephalitis (EEEV), Venezuelan equine encephalitis (VEEV), and Western equine encephalitis (WEEV) viruses, cause fewer infections than the arthritogenic alphaviruses, by virtue of their spread to the brain, they have greater potential to cause fatal disease.
CHIKV causes a self-limiting febrile illness associated with joint pain, myalgia, and rash. Some patients experience prolonged arthralgia or arthritis lasting months to years (3, 4). The explosive outbreaks of CHIKV across several continents highlight the need for an effective vaccine; indeed, several candidates are in preclinical and clinical development (5). One advanced candidate is a recombinant virus-like particle (VLP)-based vaccine, generated by expression of CHIKV capsid and envelope structural proteins in cells. The non-infectious, secreted VLPs are structurally indistinguishable from wild-type virions (6) and highly immunogenic in animal models, including non-human primates (7). Phase 1 and 2 clinical trials in humans of the CHIKV VLP vaccine demonstrated favorable safety and tolerability profiles with strong humoral responses (8, 9). A recently published phase 2 trial for PXVX0317, an aluminum hydroxide-adjuvanted formulation of the CHIKV VLP vaccine, showed the vaccine generated a robust and durable (≥ 2 years) immune response (NCT03483961) (10).
Several animal studies have established the protective activity of polyclonal or neutralizing monoclonal antibodies (mAbs) against CHIKV infection (11–14). In humans, a majority of identified neutralizing mAbs against CHIKV and other alphaviruses target the surface-exposed domains of the E2 glycoprotein, which forms a heterodimer with E1 (15–18). The alphavirus E2 protein has three domains, two of which are primary targets of neutralizing antibodies, including the B domain and the adjacent A domain (19). The E1-E2 heterodimer forms a trimeric spike on the surface of mature virions (6). The dominant immunogenicity of E2 can be explained at least in part by its positioning atop the spike with most segments of E1 buried beneath. Nonetheless, while E1 is poorly exposed on the surface of virions, protective, non-neutralizing anti-E1 mAbs have been isolated that function by blocking viral egress and interacting with innate immune cells to promote clearance (17, 20, 21). Importantly, the structural proteins of arthritogenic alphaviruses share substantial sequence similarity (~70 to 90%), suggesting that the induction of broadly protective antibodies by vaccination is possible (20, 22).
Here, we characterize the B cell response after a two-dose vaccination series with the PXVX0317 vaccine from samples obtained during a phase 2 clinical trial (NCT03483961). Immunized individuals developed strongly neutralizing serum antibodies against representative members of the three different CHIKV genotypes in all subjects and other related arthritogenic alphaviruses in a subset of vaccine recipients. CHIKV-specific B cells were detected in blood 6 months after vaccination, the latest time-point sampled. A panel of 121 mAbs generated from B cells of vaccine recipients included type-specific and broadly neutralizing clones. Several of these mAbs demonstrated protective activity in mouse models of CHIKV infection, and some vaccine-elicited mAbs conferred protection against MAYV and RRV. High-throughput epitope mapping of the protective mAbs identified residues in the A and B domains of E2 as key determinants of binding. Structural analysis of two cross-reactive mAbs revealed proximal epitopes atop the apex of the B domain, highlighting a unique site for antibody-mediated neutralization of multiple arthritogenic alphaviruses.
RESULTS
Serological response to adjuvanted CHIKV VLP vaccine.
We first assessed the humoral response of serum from individuals who received the CHIKV VLP PXVX0317 vaccine as part of a phase 2 clinical trial (NCT03483961) (10). The cohort of 20 subjects from this clinical trial selected for the current studies received 20 μg of Alhydrogel® (alum)-adjuvanted VLPs followed by homologous boosting 28 days later (Fig. 1A). Sera collected at Days 1, 29, and 57 were assessed for antibody binding to CHIKV VLPs. We detected minimal binding to CHIKV VLPs at Day 1 (Fig. 1B) except for one sample (13–513), which suggested prior infection with CHIKV or a related alphavirus in this subject. To quantify CHIKV-specific serum IgG, we calculated end-point binding ELISA titers, which showed a geometric mean titer (GMT) at Day 29 of ~11,600 that increased to ~54,400 on Day 57 (28 day post-boost) with similar results obtained using half-maximal titer analysis (Fig. 1C and Fig. S1A) (23).
Figure 1. Humoral and B cell response to PXVX00317 Vaccination.

A, Schematic of vaccination regimen and specimen collection. B-C, IgG ELISA (B) against CHIKV VLPs (strain 37997) from serum of immunized subjects from (A) obtained at the indicated time points with interpolated endpoint titers in (c). D-F, EC50 values from FRNT with serum of immunized subjects against CHIKV-37997 (D), CHIKV-LR 2006 (E), and CHIKV RSU1 (F); red circles denote subject 513 who demonstrated pre-existing antibody titers. Data are shown as the mean of 2 experiments performed in technical duplicate. Dotted lines indicate the limit of detection (LOD) of the assay. G, Table of percent of subjects whose Day 57 serum samples demonstrated EC80 or EC50 values by FRNT at a dilution of 1/40 against the indicated arthritogenic alphaviruses. Data are representative of 2 experiments. H-I, Flow cytometry plots (H) of a representative subject showing expression of CD38 and CD71 and binding of VLP to CD19+IgD− B cells at the indicated time points following vaccinations, summarized in (I). Bars in (I) represent mean values. ** p < 0.01, ****p < 0.0001 by ANOVA with Sidak’s post-test.
We evaluated the neutralizing capacity of serum by performing focus reduction neutralization tests (FRNT) with infectious isolates from the three CHIKV genotypes: East Central South African [ECSA] (La Reunion OPY, 2006 [LR 2006]), West African (37997 Senegal), and Asian (RSU1). As expected, we did not observe neutralization of CHIKV with sera obtained at Day 1 (Fig. 1D–F), except for sample 13–513, which also showed VLP-specific IgG. Serum obtained at Day 29 had a GMT of half-maximal inhibition (EC50) value against CHIKV-37997 of ~6,800, which increased after boosting to ~19,000 on Day 57, with patient 13–513 exhibiting the highest titers likely due to boosting (Fig. 1C–F, red dots). Similar levels of inhibitory antibodies were detected against CHIKV-LR 2006 or CHIKV-RSU1 (Fig. 1D–F) suggesting that PXVX0317 induces neutralizing antibodies against all CHIKV genotypes. We still observed serum neutralizing activity against all three CHIKV strains in samples collected at day 182 post-immunization, with GMTs of ~1,000 against CHIKV-37997 (Fig. 1D–F), consistent with prior results (10).
Given the sequence identity (54 to 83%) of the E2 glycoprotein among Semliki Forest complex alphaviruses (Fig. S1B–C), we evaluated the neutralizing capacity of serum from vaccine recipients against some related arthritogenic viruses. We assessed whether serum obtained at Day 57 at a 1/40 dilution demonstrated greater than 50% or 80% neutralization against ONNV, MAYV, RRV, and Una virus (UNAV); these cutoff values were selected based on prior in vitro correlate of protection studies of serum neutralizing antibodies against alphaviruses in humans (24, 25). At a 1/40 serum dilution, 100% of samples showed 50 and 80% neutralization (FRNT50 and FRNT80) of ONNV, which is most closely related to CHIKV (Fig. 1G and Fig. S1B). Less cross-neutralization was observed against MAYV, with 80 or 25% of samples showing 50 or 80% inhibition, respectively. In contrast, less than 10% of samples neutralized infection by UNAV or RRV, which are more distant genetically to CHIKV (Fig. 1G and Fig. S1B). Overall, these data suggest that the PXVX0317 vaccine can induce some degree of cross-protection against related alphaviruses.
CHIKV-specific B cell responses after VLP vaccination.
In general, antibodies derived from circulating plasma cells wane over time; in contrast, antigen-specific memory B cells (MBCs) persist and can differentiate rapidly into antibody-secreting cells in response to subsequent cognate antigen exposure (26, 27). To assess the induction and persistence of CHIKV-specific B cells, we isolated peripheral blood mononuclear cells at Days 1, 29, 57 (28 days post-boost), and 182 (153 days post-boost). We identified CHIKV-specific B cells by incubating them with CHIKV VLPs followed by a fluorochrome-conjugated mAb, CHK-265 (11, 28), which binds the B domain of CHIKV E2. By using CHIKV VLPs as the detection reagent, we identified B cells with surface antigen receptors that can bind quaternary epitopes on viral particles in addition to ones that engage linear or tertiary epitopes on soluble envelope proteins.
While B cells that bind CHIKV VLPs were absent from blood in samples at Day 1, we detected an appreciable frequency (~0.2 to 0.6%) of VLP-binding CD19+IgD− B cells in all samples at Day 29 with a similar frequency detected on Day 57 (28 days post-boost) (Fig. 1H–I and Fig. S1D). We also detected circulating VLP-binding CD19+IgD− B cells on Day 182 in blood, indicating long-term persistence of antigen-specific cells. In contrast to samples from Day 29, the vast majority of VLP-specific CD19+IgD− B cells at Day 182 lacked expression of the activation markers CD38 and CD71, consistent with a memory phenotype (29) (Fig. 1H–I).
To gain additional insight into the B cell response to VLP vaccination, we performed single-cell RNA sequencing of bulk CD19+ IgD− B cells from blood of three subjects at Day 57. To identify VLP-specific cells, we used oligonucleotide-conjugated probes (streptavidin-TotalSeqC to label biotinylated-CHK-265) and B cell receptor sequencing, also termed LIBRA-Seq (30). Uniform manifold approximation and projection (UMAP) analysis of the B cells from each patient (between 3,276 and 6,874 cells) revealed two main clusters: activated (CD71hi) and memory (CD71lo) B cells in one cluster and plasmablasts [defined by PRDM1 (BLIMP-1), CD38, and MKI-67 expression] in another (Fig. 2A and Fig. S2A). Our strategy identified cells with barcoded streptavidin sequence reads above background, which correspond to VLP-binding B cells (Fig. 2B and Fig. S2B). Approximately 30 to 200 of the recovered B cells from each subject bound to VLPs and were mapped almost exclusively to non-activated B cells (CD71lo) (Fig. S2B), which is consistent with our flow cytometry data.
Figure 2. Isolation of anti-CHIKV mAbs from vaccine recipients.

A, Merged uniform manifold approximation and projection (UMAP) feature plots of single-cell RNA-seq of 13,764 cells from 3 subjects showing expression of the indicated markers. B, UMAP plots of CD19+ B cells (from subjects 506–3276, 516–6874, and 520–3614) with productive V-D-J contigs showing normalized SAV-TotalSeqC reads corresponding to VLP binding. C, Heatmap of variable heavy chain usage corresponding to each individual from mAbs expressed recombinantly. D, Quantification of mutations in the variable region of the heavy chain from recombinantly expressed mAbs from each subject. E, Heat map of ELISA binding to CHIKV VLP, p62-E1, or E2, and neutralization of CHIKV-LR 2006 by recombinantly expressed mAbs from VLP-binding B cells identified from (A). Intensity of heatmap corresponds to endpoint titer (N = no detectable binding) for ELISA and binned EC50 values for neutralization. Data in (E) are representative of 2 experiments.
We focused our genetic and functional analyses on B cells with the highest level of CHIKV VLP binding, as determined by the number of streptavidin-TotalSeqC feature barcode reads in each sample. We chose the top 48 VLP-binding cells for subjects 516 (of ~150) and 520 (of ~50), and all 33 cells with signal above background for subject 506 (Fig. S2B). We did not observe clonotype overlap as defined by V-D-J usage and identical CDR3 length with >90% sequence identity. This finding may be due to a sampling limitation due to the relatively low total number of sequenced B cells or reflect a high diversity of CHIKV-reactive clones in the B cell repertoire. When we examined antibody VH gene segment usage, we observed enrichment of specific VH genes between subjects, such as IGHV3–30 and IGHV5–51, that did not share CDR3 homology, suggesting features of heavy chains encoded by some VH gene segments may have intrinsic affinity for CHIKV (Fig. 2C) (31). We also assessed the extent of somatic hypermutation in VLP-binding cells as a gauge of affinity maturation: 72% of clones harbored mutations in their VH regions, whereas 28% showed no mutations compared to their inferred germline gene sequences (Fig. 2D), a distribution similar to antigen-specific cells identified in other contexts (32).
Functional properties of mAbs.
We next generated human IgG1 plasmid constructs from assembled V-D-J contig sequences for recombinant mAb expression (Fig. 2C–D). We successfully isolated 121 of 127 mAbs from the 3 subjects and initially assessed binding to VLPs by ELISA. Nearly all (120 of 121) expressed mAbs bound to VLPs (Fig. 2E and Fig. S2C–E), and many bound at concentrations below 100 ng/mL. We also tested binding to the constituent E1-E2 heterodimer proteins using recombinant p62-E1 protein derived from the CHIKV-LR 2006 strain (19). Although the majority of VLP-binding mAbs bound p62-E1, a few (e.g., 520.F01 and 520.G03) did not bind even at 10 μg/mL, potentially indicating engagement of quaternary epitopes or a genotype-dependent (5% amino acid difference in E1-E2 compared to strain 37997) loss in p62-E1 binding. Most of the VLP-binding antibodies also bound to soluble recombinant E2 (Fig. 2E and Fig. S2C–E). We note that some mAbs (520.G02, 520.G03, 520.G05, 516.H11) bound E2 but not p62-E1 appreciably; however, these mAbs generally bound weakly, and this finding could reflect differential limits of detection with the ELISA-based assays.
We screened mAbs for inhibitory activity by performing FRNTs at selected (100 ng/mL, 1 μg/mL, and 10 μg/mL) concentrations against the CHIKV-LR 2006 strain. Notably, 54 of 121 mAbs neutralized CHIKV-LR 2006 infection at the highest tested concentration (10 μg/mL), and 20 of 121 mAbs inhibited infection at concentrations below 100 ng/mL (Fig. 2E). Subject 13–516 demonstrated a higher proportion of neutralizing mAbs (73%) than subjects 13–506 and 13–520 (18 and 37%, respectively), which could reflect functional differences in immune responses or technical bias of limited clone sampling.
Neutralization and epitope binning of vaccine-induced mAbs.
To assess the neutralizing potency of our mAbs more quantitatively, we performed full dose-response FRNTs against the immunizing strain CHIKV-37997 and CHIKV-LR 2006 for the subset of mAbs that were the most potently inhibitory in our initial screen. Several mAbs from all three subjects showed strong inhibitory activity with EC50 values < 100 ng/mL (Fig. 3A–C). Two mAbs (506.C01 and 520.D02) showed ‘elite’ activity against both CHIKV strains with EC50 values < 10 ng/mL (Fig. 3A–C).
Figure 3. Characterization of vaccine-elicited mAbs.
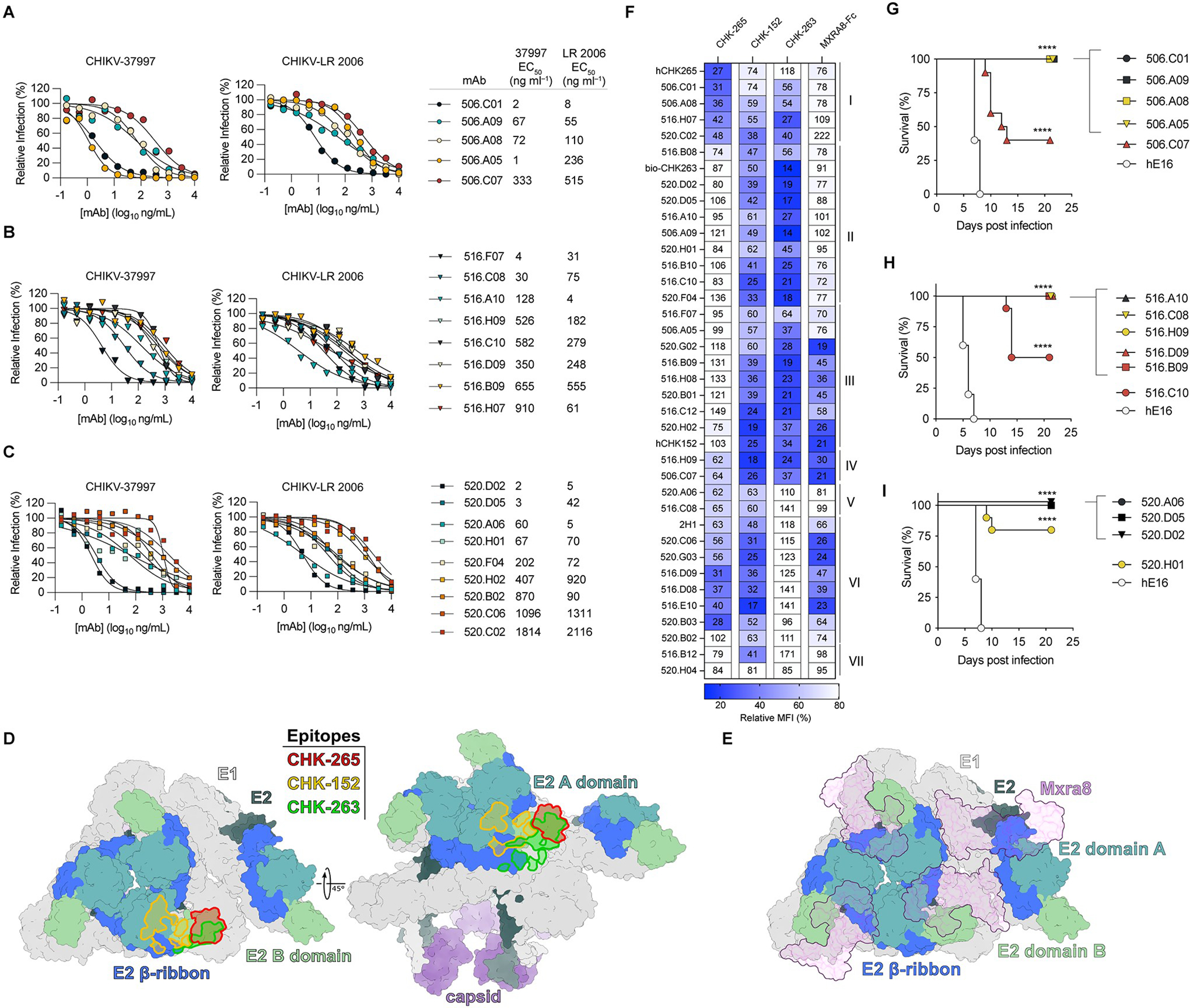
A-C, Dose-response curves and table of EC50 values of indicated mAbs from subjects 506 (A), 516 (B), and 520 (C) against CHIKV-37997 and CHIKV-LR 2006 by FRNT. Plots in A-C are representative of 4 experiments with EC50 values generated from the mean of all experiments. D, CHIKV asymmetric unit (PDB: 6NK5) with E1 shown in light gray and capsid shown in purple. E2 is colored by domain with B domain in pale green, A domain in teal, and β-ribbon in royal blue, with the remainder of E2 colored dark gray. The footprints of indicated mAbs are highlighted in red (CHK-265), green (CHK-263), and yellow (CHK-152). E, CHIKV asymmetric unit (PDB: 6NK6), colored as in (e). Bound MXRA8 is shown in purple. F, Heatmap of relative binding of each mAb (rows) in the presence of reference mouse mAbs or MXRA8-mouse Fc (columns). Binding to Expi293 cells expressing CHIKV structural proteins (capsid-E3-E2–6k-E1) was assessed by flow cytometry by measuring the mean fluorescence intensity (MFI). Numbers indicate % MFI in the presence of competitor (mAb or MXRA8-mouse Fc) compared to no competitor. G-I, Survival of 4-week-old male C57BL/6J mice treated with 500 μg of anti-IFNAR1 and indicated anti-CHIKV mAb (100 μg, ~5 mg/kg) one day prior to subcutaneous inoculation of 10 FFU of CHIKV-LR 2006. MAbs were isolated from subjects 506 (G), 516 (H), and 520 (I). Data are combined from 2 experiments with n = 10 mice per group. Kaplan-Meier survival curve analysis (log-rank test) with Bonferroni correction compared to isotype control mAb (anti-WNV, hE16); **** (p < 0.0001).
To begin to define the epitopes of our neutralizing human mAbs generated after vaccination, we performed competition-binding assays with reference mAbs derived from mice and included several non-neutralizing mAbs for comparison. We developed a flow cytometry assay with cells expressing the structural proteins (capsid-E3-E2–6K-E1) on the plasma membrane following transfection. We first stained cells with reference mouse mAbs: CHK-265 (E2, B domain), CHK-263 (E1, E2 B domain), and CHK-152 (E2, A/B domain) (6, 11, 28, 33) (Fig. 3D). We also performed competition binding with MXRA8, a mammalian cell surface receptor for CHIKV (Fig. 3E), using recombinant MXRA8-mouse Fc protein. After binding of the reference mAbs or MXRA8-Fc, we incubated transfected cells with mAbs from our panel followed by detection with A647-conjugated anti-human IgG (Fig. S3A–B). A majority of tested mAbs (20 of 34) competed with CHK-152 and CHK-263 but not with CHK-265, designated here as clusters II and III (Fig. 3F). Given that CHK-263 makes contacts with E1 (33), we tested binding of cluster II and III mAbs to purified E1; however, none demonstrated binding (Fig. S3C). Cluster I contained fewer mAbs (e.g., 506.A08, 506.C01, and 516.H07), which competed with CHK-265, a broadly-neutralizing E2 B domain mAb (Fig. 3D, F). Even though a majority of the mAbs in our competition panel were neutralizing, only a subset (clusters III and VI) competed (>50% reduction) with MXRA8-Fc binding (Fig. 3E–F). Thus, based on competition-binding data, neutralizing mAbs derived from humans following VLP vaccination appear to bind multiple antigenic sites on CHIKV E2, an important feature that decreases the likelihood of viral escape.
As an orthogonal method, we performed a competition-binding assay to recombinant CHIKV p62-E1 in the solid-phase by ELISA. We down-selected our panel of mAbs (including both neutralizing and non-neutralizing) to those with the highest level of binding to p62-E1 protein. We biotinylated 15 mAbs and performed binding assays with pre-bound non-biotinylated versions of each mAb. We also included previously characterized CHIKV-specific human mAbs (15) to provide context for our results. Consistent with our cell-based competition, mAbs in cluster I (506.C01 and 506.A08) competed with each other and with CHK-265 (Fig. S3D). However, the majority of the other mAbs formed one large competition group that included antibodies recognizing epitopes in both the A and B domain (see below). The non-neutralizing mAbs 520.B02 and 520.H04 did not compete with any other mAb, suggesting they engage discrete epitopes (Fig. S3D).
Protection of mAbs in vivo.
Given that multiple mAbs showed neutralizing activity and formed unique competition groups, we tested 4 to 6 mAbs from different competition clusters and from each vaccinated subject for protective activity in vivo. We first used a pathogenesis model in immunodeficient mice in which all animals succumb to CHIKV infection (11). We passively administered a single 100 μg (~5 mg/kg) dose of anti-CHIKV mAb as well as 500 μg (~25 mg/kg) of anti-IFNAR1 mAb (MAR1–5A3) one day prior to subcutaneous inoculation with 10 focus-forming units (FFU) of CHIKV-LR 2006. The most potently neutralizing mAbs completely protected mice against lethality, whereas animals administered an isotype control anti-West Nile virus [WNV]) mAb (hE16) (34) succumbed to infection within 6 to 8 days (Fig. 3G–I). The anti-CHIKV mAbs (520.H01, 506.C07, and 516.C10) with less neutralizing capacity demonstrated variable (40 to 80% survival) protection (Fig. 3G–I). For mAbs conferring complete protection at 10 mg/kg, we repeated experiments with lower doses to identify those with the greatest activity in vivo. At a 20 μg-dose (1 mg/kg), only 516.A10 conferred 100% protection. Most mAbs demonstrated >50% protection at this dose including: 520.D02, 520.D05, 506.A08, 506.A09, and 506.C01 (Fig. S4A–C). In contrast, 506.A05 and 516.D09 lost protection, which correlated with their poorer neutralization potency.
We next tested a subset of mAbs (506.A08, 506.A09, 506.C01, 516.B09, 516.D09, 516.C10, 520.D02, 520.D05, 520.A06) in an immunocompetent mouse model of CHIKV infection (35). C57BL/6 mice were inoculated subcutaneously in the rear footpad, which results in foot and ankle swelling, immune cell (monocyte and CD4+ T cell) infiltration, and viral burden in adjacent and distant musculoskeletal tissues. When administered as prophylaxis at a dose of 10 mg/kg, all anti-CHIKV mAbs reduced foot swelling compared to animals treated with an isotype control mAb (Fig. S4D). Viral burden in all analyzed tissues including ankle and calf showed substantial reductions by all tested anti-CHIKV mAbs compared to the isotype control mAb (Fig. S4E–H). Histological analysis of the ipsilateral feet of CHIKV-infected mice that had been treated with a subset of the mAbs (506.A08, 506.C01, or 520.D02) showed marked reductions in myositis compared to animals administered an isotype control mAb (Fig S4I).
Epitope mapping by alanine-scanning mutagenesis and neutralization escape.
To better define the epitopes engaged by mAbs that demonstrated in vivo protection, we performed alanine-scanning mutagenesis coupled with cell-surface display and high-throughput flow cytometry to identify residues in the E2 glycoprotein required for mAb binding. Cells were transfected with capsid-E3-E2–6K-E1 expression plasmids encoding individual proteins with alanine (or serine for alanine) substitutions (residues 1–270) in the E2 gene (7). We defined critical interaction residues as those with <25% binding to a given mAb that retained >90% binding to an anti-CHIKV oligoclonal antibody mixture. A majority of the inhibitory mAbs mapped to the B domain of E2 (Fig. 4A–B). 506.C01 and 506.A08 mapped to the apex of the B domain (28) (Fig. 4A, C); 520.D02 and 520.A06 mapped to the flank of the B domain (Fig. 4A, D); and 516.C10, 516. H09, and 506.C07 mapped to residues in both the B domain and the β-ribbon connecting the A and B domains (Fig. 4A, E). We also identified residues in the A domain as critical for binding of 516.D09, 516.C08, and 520.D05 mAbs (Fig. 4A, F). We confirmed our alanine-scanning mutagenesis results with charge reversal substitutions, which resulted in even greater loss-of-binding than alanine mutations (Fig. S5A). Moreover, we mapped 516.A10 to R198/G209/L210 and 506.A05 to K233 by charge-reversal mutations, which did not meet criteria as critical residues by alanine scanning mutagenesis (Fig. S5A). We also performed epitope mapping by passaging CHIKV in the presence of 10 μg/mL of each mAb and assessing for neutralization escape. Sequencing of the structural genes revealed mutations in the same or neighboring residues identified by alanine/arginine mutagenesis for all mAbs (Fig. 4A and Fig. S5B). Thus, neutralizing mAbs in humans induced by PXVX0317 vaccination target both the A and B domains of CHIKV E2, which are the same targets of potently neutralizing mAbs isolated after natural infection (15, 17, 31).
Figure 4. Epitope mapping of protective mAbs.

A, Table showing percent relative binding of each mAb (columns) against indicated residues mutated to alanine (rows). The value in each cell correspond to % binding compared to WT and are the mean of 2 experiments. Cells colored in blue are residues also identified by neutralization escape. B, CHIKV E1-E2 trimer (PDB: 6NK5) surface representation, with E1 shown in light gray, E2 B domain in pale green, E2 A domain in teal, E2 β-ribbon in royal blue, and the remainder of E2 colored dark gray. Alanine scanning hits are colored red. A single E1-E2 heterodimer is outlined in magenta. C-F, Ribbon diagrams highlighting alanine scanning hits on E1-E2 heterodimer. Structural proteins are colored as in (B). Antibodies are clustered by site, including those binding B domain sites (apex and flank), both B domain and β-ribbon, or A domain. Alanine scanning mutagenesis loss-of-binding residues shared by all mAbs in each cluster are shown in yellow, with others reducing binding of subsets of mAbs shown in red.
Reactivity of protective mAbs against related alphaviruses.
Some human anti-E2 mAbs produced during natural alphavirus infection (e.g., mAbs RRV-12 and DC2.M357) can neutralize multiple arthritogenic alphaviruses (16, 31). To assess the cross-reactivity of our protective mAbs against other alphaviruses, we evaluated binding to the surface of virus-infected Vero cells by flow cytometry and included additional mAbs from our panel as comparators. A majority of our mAbs cross-reacted ONNV, consistent with its high sequence identity with CHIKV (Fig. 5A, Fig. S1B, S6A–C). A smaller number of mAbs cross-reacted with MAYV, and a few bound to cells infected with more distantly related alphaviruses including UNAV, RRV, or Bebaru virus (BEBV) (Fig. S1C). The neutralizing mAbs 506.C01 and 506.A08, both from cluster I, showed broad reactivity and bound to the cell surface of ONNV, MAYV, and RRV-infected cells, similar to the binding pattern of CHK-265 (Fig. 5A–B). One potently neutralizing mAb, 516.A10, showed cross-reactivity with ONNV, BEBV, and also EEEV, as shown using a chimeric Sindbis virus (SINV) expressing the EEEV structural proteins (36) (Fig. 5A–B).
Figure 5. Vaccine-induced mAbs protect against related arthritogenic alphaviruses.

A, Heatmap displaying normalized MFI of cell-surface binding of mAbs in Vero cells inoculated with the indicated alphaviruses. Normalized MFI was calculated by dividing geometric MFI of each mAb by geometric MFI of hE16 (isotype control). B, Flow cytometry plots showing binding of indicated mAbs to infected Vero cells. Data in A-B are representative of 3 experiments. C-D, Dose-response curves (C) and mean EC50 values (D) of mAbs against indicated arthritogenic alphaviruses by FRNT. Data are representative (C) or the mean values (D) of 3 experiments. E-F, Viral titers in indicated tissues in 4-week-old C57BL/6 mice administered mAb one day prior to inoculation with 103 FFU of MAYV (E) or RRV (F). Data are pooled from 2 experiments with n = 7–10 mice per group. * (p< 0.05), ** (p < 0.01), *** (p <0.001), **** (p < 0.0001) by Kruskal-Wallis with Dunn’s post-test correction.
We tested the human mAbs 506.A08, 506.A09, 506.C01, 516.A10, 520.D05, 520.A06, and 516.H07 that cross-reacted with three or more alphaviruses for breadth of cross-neutralizing activity. Except for 506.A08, all of these mAbs neutralized ONNV infection potently, with EC50 values < 50 ng/mL (Fig. 5C–D). 506.A08 and 506.C01 also inhibited infection of MAYV and RRV to lesser degrees. The mAb 506.C01 demonstrated the greatest neutralization breadth, as it also blocked infection of UNAV and the distantly related Getah virus (GETV), a pattern similar to that of CHK-265 (28). However, 516.A10 did not neutralize infection of SINV-EEEV (Fig. S6D) despite binding to the surface of infected cells. Because the broadly neutralizing murine mAb CHK-265 can inhibit viral egress (28), we determined whether 506.A08 and 506.C01 also had this activity. When 506.A08 and 506.C01 mAbs were added to Vero cells after virus entry and infection, we detected substantially reduced viral RNA in the supernatant 6 h later compared to cells treated with isotype control mAb (Fig. S6F). These data establish that B cells that produce broadly neutralizing antibodies against alphaviruses are induced in humans after immunization with the PXVX0317 vaccine.
To test whether the cross-reactive mAbs could protect against related alphavirus infections, we used established models of MAYV and RRV pathogenesis in mice (28, 37, 38). We evaluated 506.C01 and 506.A08, as they demonstrated the greatest breadth of binding to cell-surface antigen and cross-neutralization. When administered as prophylaxis, both 506.A08 and 506.C01 reduced MAYV viral burden at 3 dpi in the ipsilateral ankle and calf and completely prevented joint swelling (Fig. 5E, Fig. S6E). However, only 506.A08 reduced viral burden in the contralateral ankle and calf with tissues from several mice below the limit of detection (Fig. 5E), consistent with its greater neutralization potency than 506.C01 against MAYV. In contrast, when mice were inoculated with RRV, we observed marked protection only with 506.A08, with reduced viral burden in all musculoskeletal tissues; 506.C01 exerted a lesser level of protection, consistent with its poorer neutralizing activity against RRV (Fig. 5F). Given that prophylaxis with 506.A08 and 506.C01 protected against infection by multiple arthritogenic alphaviruses, we tested whether these mAbs also had activity as post-exposure therapy. Indeed, when administered 24 h after CHIKV infection, both mAbs reduced viral burden in all musculoskeletal tissues examined (Fig. S6G).
Structural analysis of broadly neutralizing and protective mAbs.
Competition-binding analysis, alanine-scanning mutagenesis, and neutralization escape studies suggested that 506.A08 and 506.C01 target similar epitopes on the B domain of E2. However, given their differing cross-reactivity profiles (Fig. 5A), we hypothesized that these mAbs might target overlapping yet distinct epitopes, providing an opportunity to map structurally the exact regions on the B domain targeted by broadly-neutralizing antibodies. We and our collaborators previously generated a low-resolution (~16 Å) cryo-electron microscopy (cryo-EM) reconstruction of the broadly-neutralizing murine mAb CHK-265 bound to CHIKV and intermediate-resolution (6.3, 5.3, and 5.3 Å) reconstructions of the broadly-neutralizing human mAb RRV-12 bound to RRV, CHIKV, and MAYV, respectively (16, 28). These structures showed similar mAb binding orientations and footprints in the B domain, with potential crosslinking of A domains in adjacent trimers via framework region contacts. Here, we generated higher-resolution cryo-EM reconstructions of CHIKV VLPs complexed with 506.A08 or 506.C01 Fabs, or CHK-265 Fab for reference (Fig. 6A–C, Fig. S7 and S8A–F, Table S1). Following icosahedral reconstruction of Fab-bound VLPs, asymmetric units (n = 60 per VLP) were extracted for independent classification and focused refinement to account for deviations from ideal icosahedral geometry (Fig. S7). The resulting reconstructions of CHIKV structural proteins in complex with 506.A08, 506.C01, or CHK-265 Fabs achieved resolutions of 3.13 Å, 3.27 Å, or 3.18 Å, respectively (Fig. 6D–F and Fig. S8G–O).
Figure 6. Cryo-EM reconstructions of broadly-neutralizing vaccine-induced mAbs and CHK-265 bound to CHIKV VLPs.

A-C, CHIKV VLP reconstructions with bound Fab fragments colored green (506.A08) (A), yellow (506.C01) (B), or pink (CHK-265) (C), and structural proteins colored radially. Equatorial cross-sections are shown as round insets. Axes of symmetry are designated in (A) by a pentagon (5-fold; i5), triangles (3-fold; i3), three-pointed stars (quasi-3-fold; q3), and a diamond (2-fold; i2), with axial orientations displayed in the inset. D-F, Asymmetric unit reconstructions with Fabs colored dark green/lime green (506.A08 heavy/light chain), dark gold/yellow (506.C01 heavy/light chain), or crimson/pink (CHK-265 heavy/light chain). CHIKV E1 is shown in white, and capsid is shown in purple. E2 is colored by domain, with the B domain in light green, A domain in teal, and β-ribbon in royal blue, with the remainder of E2 colored dark gray. G, Magnified region from black boxes in D-F and colored as in D-F. A surface representation of E1-E2, with superimposed ribbon diagrams of 506.A08, 506.C01, and CHK-265 Fv fragments. H, Side (left, right) and top (center) views of magnified region from D-F, with mAb footprints outlined in green (506.A08), yellow (506.C01), and red (CHK-265). Residue hits from scanning mutagenesis or neutralization escape are labeled, with impacted mAb(s) indicated by colored stars. I, Alignment of E2 B domains from arthritogenic alphaviruses, including CHIKV-37997, CHIKV-LR 2006, ONNV, GETV, BEBV, RRV, MAYV, and UNAV. Viruses that escape cross-recognition by 506.A08 or 506.C01 are designated by green or yellow diamonds, respectively. Antibody contact residues (determined by PISA solvent exclusion analysis) are delineated below the alignment, with scanning mutagenesis or neutralization escape residues designated by stars. Within the alignment, residues implicated in neutralization escape and correlating with loss of cross-recognition are colored magenta.
All three Fabs bound to the B domain of CHIKV E2. However, each demonstrated a different angle of engagement, with 506.A08 positioned upright on the apex of B domain, 506.C01 positioned at a 30° incline on the apex, and CHK-265 binding the lateral tip of B domain nearly parallel to the plane of the viral membrane (Fig. 6D–F). Each Fab likewise established a discrete footprint on the B domain, with 506.A08 centered on the apex, CHK-265 centered on the lateral tip, and 506.C01 spanning both regions (Fig. 6G–I and Fig. S9A–C, Table S2). 506.C01 achieved this recognition by using an unusually long, disulfide-stabilized CDR-H3, which folded over the lateral tip of B domain and extended toward the viral membrane (Fig. S9B). Despite their different approaches, 506.C01 and CHK-265 had similar footprints on the surface of E2 (Fig. 6H–I). These structural results may explain the cross-reactivity patterns for 506.A08 and 506.C01. Although these mAbs could accommodate a variety of substitutions within their respective epitopes, divergence from CHIKV at particular E2 residues resulted in loss of reactivity. Given their central positions within each epitope and our mutagenesis findings, polymorphisms N187D (BEBV) and T213S/V (GETV/UNAV) likely contribute to the different binding patterns of 506.C01 and 506.A08, respectively (Fig. 6I). We did not observe cross-linking of the A domain by any of the three mAbs, including CHK-265 (Fig. S9D), indicating that binding to the B domain alone is sufficient for broadly neutralizing activity. 506.A08 and 506.C01 define unique neutralizing epitopes atop the apex of B domain, whereas other previously characterized broadly-neutralizing mAbs (CHK-265 and RRV-12) approach the lateral tip of the B domain from the side (Fig. S10).
DISCUSSION
Despite the substantial morbidity associated with CHIKV-induced acute and chronic musculoskeletal disease, approved countermeasures do not exist. To address this unmet clinical need, several therapeutic antibodies and vaccines have advanced to human clinical studies (5). Two completed clinical trials in humans of the unadjuvanted CHIKV VLP vaccine demonstrated favorable safety and tolerability profiles with strong humoral responses (8, 9). The adjuvanted CHIKV VLP (PXVX0317) vaccine has been tested in three phase 2 trials (NCT03992872, NCT05065983, and NCT03483961) and currently is being evaluated in two phase 3 trials (NCT05072080 and NCT05349617). PXVX0317 is well-tolerated and induces a robust and durable (≥ 2 years) immune response (10). Our results show that two doses of PXVX0317 vaccine induce high levels of neutralizing antibody against all CHIKV genotypes, and its breadth of response extends to several other arthritogenic alphaviruses, including ONNV and MAYV. We also profiled the B cell response to vaccination, which revealed the induction and persistence of antigen-specific B cells in blood with type-specific and broadly-neutralizing capacity.
While the gold-standard for assessment of vaccine efficacy is protection from clinical disease, the execution of such trials for mosquito-transmitted viruses is challenging, given that the epidemiology of infection varies on an annual basis. Whereas in vitro correlates of protection are established for several viral infections, limited data exist for CHIKV, with one study in humans suggesting that an 80% plaque reduction neutralization titer of 10 is associated with protection against infection (24, 25). Although the CHIKV VLP vaccine with or without adjuvant was shown in phase 1 and 2 studies to induce serum neutralizing antibodies (8, 9), the functional diversity of antibodies and dynamics of the B cell response were not characterized. Prior studies have isolated neutralizing mAbs from CHIKV-infected subjects during convalescence that map to multiple epitopes in the E2 A and B domain (15, 17, 21, 31). Our work corroborates and extends these findings to vaccine-induced B cell responses, as all neutralizing mAbs isolated from immunized individuals also bound to the A or B domain of the CHIKV E2 glycoprotein.
The identification of potently inhibitory mAbs likely stemmed from both the immunogenicity of the VLP-based vaccine and our use of VLPs as bait for identifying CHIKV-specific B cells. Prior studies have used soluble protein to isolate B cells, which could bias against clones that exclusively engage quaternary epitopes (17, 31). A limitation of our methodology is the relative inability to identify E1-specific mAbs, given the lack of exposure of E1 epitopes on CHIKV VLPs (20, 39). Additional studies are required to determine the extent to which E1-specific clones are elicited by the VLP vaccine. Moreover, our strategy may not have isolated all CHIKV-reactive B cells, some of which may bind to VLPs below our detection threshold, which may constitute a substantial fraction of B cells (40). Nevertheless, our work establishes that the PXVX0317 vaccine can induce B cell clones encoding potently neutralizing antibodies whose inhibitory activity is on par with those isolated from naturally-infected humans (15, 17, 21).
The mAbs were isolated from blood samples obtained at Day 57 post-prime (29 days after boosting), which likely reflects the repertoire of circulating cells with a memory-like phenotype (predominantly CD71lo). An advantage of our approach is the increased likelihood of isolating cross-reactive clones, which are thought to be enriched in the MBC compartment (41). The broadly neutralizing mAbs 506.C01 and 506.A08 bound to the apex of the B domain, partially overlapping with previously identified cross-reactive human mAb, RRV-12 (16). Whereas RRV-12 only partially neutralized RRV and CHIKV infection and had resistant virus fractions at high concentrations in neutralization tests, 506.C01 and 506.A08 completely inhibited infection of CHIKV-37997 or CHIKV-LR 2006. The cross-neutralizing human mAb DC2.M357, which maps to residues K189 and N218 in the B domain, also completely inhibits CHIKV infection, albeit less potently (EC50 of ~800 ng/mL) than 506.C01 or 506.A08 (31). Given that a large percentage of serum samples from vaccine recipients also neutralized infection of ONNV and MAYV, immunization with PXVX0317 might confer protection against infection caused by multiple alphaviruses.
Our structural analysis of vaccine-elicited mAbs expands on studies that defined domain B of E2 as a binding site for alphavirus cross-neutralizing antibodies. Previous structural analyses of CHK-265 and RRV-12 suggested that cross-linking of neighboring trimers via framework region contacts to A domain may be required for broad neutralization (16, 28). However, neither 506.A08 nor 506.C01 contact neighboring A domains, and we observe little evidence for such cross-linking by CHK-265 on CHIKV VLPs in our higher-resolution structure. Although it is possible that CHK-265 contacts E3 of neighboring trimers in mature virions (versus our VLPs, which lack E3), the binding modes of 506.A08 and 506.C01 are incompatible with such contacts. Our analysis suggests that E2 trimer cross-linking is not required for neutralization of related arthritogenic alphaviruses. Furthermore, CHK-265 and RRV-12 both bind the lateral tip of E2 B domain nearly parallel to the viral membrane, whereas 506.A08 and 506.C01 engage the apex of B domain. Within these epitopes, we identify key residue changes in CHIKV E2 that possibly explain the loss of binding for 506.C01 to BEBV (N187D), 506.A08 to GETV (T213S), and 506.A08 to UNAV (T213V).
An important limitation of our study is that we did not interrogate the repertoire of long-lived plasma cells in the bone marrow, which are the primary producers of serum antibody at late time points following vaccination. The antibodies secreted by long-lived plasma cells are at least in part clonally-related to activated and MBCs but may include specificities that we did not characterize. While we showed protection of mAbs in two mouse models, all mAbs were produced as human IgG1, which could impact protection mediated by Fc effector functions (42). Moreover, non-neutralizing mAbs or T cell responses against alphaviruses are induced following infection and also might contribute to protection in vivo (20, 43, 44), which we did not assess.
In summary, these studies show induction of protective type-specific and broadly neutralizing antibodies against arthritogenic alphaviruses following PXVX0317 vaccination of individuals from a phase 2 clinical trial, demonstrating that vaccination with CHIKV VLP induces B cell immunity similar to natural infection.
METHODS
Study Design.
The goal of this study was to evaluate the B cell response to PXVX0317 vaccination in adults. Peripheral blood collected post-vaccination was used to isolate antigen-specific B cells and used to generated recombinant monoclonal antibodies which were characterized by a variety of methods. All data collected was included without exclusion of outliers. Mice were randomly assigned to treatments, and investigators performing the immunological analyses were not blinded. Sample sizes were chosen based on prior experience in evaluating differences in viral infection in mice.
Subject cohort and sample collection.
Subject samples were obtained from Group 9 of NCT03483961, a Phase 2 trial evaluating the immune response to and safety profile of PXVX0317 in healthy adults aged 18–45. The ages of the 20 participants were: 19–30 years old (n = 16), 30–45 years old (n = 4). Trial design and outcome is summarized elsewhere (10).The participants in Group 9 received 20 μg of PXVX0317 (alum-adjuvanted CHIKV VLP) at Day 1 and Day 28. Informed written consent was obtained by Emergent BioSolutions prior to initiation of the study. Peripheral blood was collected into serum separator tubes followed by centrifugation and freezing of serum at −80°C. Peripheral blood mononuclear cells were isolated from anticoagulated leukopaks by density-gradient centrifugation (Ficoll) and frozen in liquid nitrogen. De-identified samples were received at Washington University for further processing.
Virus and cell culture.
Vero cells (CCL-81, ATCC) were maintained at 37°C in DMEM supplemented with 10% fetal bovine serum, 100 U/mL of penicillin, 100 μg/mL of streptomycin, 10 mM HEPES, non-essential amino acids, and Gluta-MAX (Thermo Fisher). The following alphaviruses were obtained from the World Reference Center for Emerging Viruses and Arboviruses and propagated in Vero CCL-81 cells: CHIKV (AF15561, LR 2006, 37997, RSU1, and 181/25), ONNV (MP30), MAYV (BeH407), UNAV (BeAr 2380), RRV (T48), GETV (AMM 2021), and BEBV (MM 2354). SINV-EEEV (FL93–939) and SINV-VEEV (TrD) have been described previously (36, 45).
Expression of recombinant mAbs.
Selected heavy and light chain contig sequences were extracted from the cell ranger output file “filtered_contig.fasta” from each subject. The V-D-J region for each was codon-optimized, synthesized, and cloned into a mammalian expression vector (GenBank FJ475055 and FJ475056) containing a CMV promoter and the human IgG1 constant regions (Genscript). For antibody expression, plasmids (1:1 ratio of heavy and light chain vectors) were diluted into Opti-MEM, complexed with Expifectamine transfection reagent (Thermo Fisher), and introduced into Expi293 cells (Thermo Fisher) in deep-well 96-well plates using (for micro-scale production) and 250 mL to 1000 mL baffled flasks (for midi-scale production).
For micro-scale purification, supernatant was harvested 5 days post-transfection, centrifuged to remove cellular debris, and incubated with Magne® Protein G beads (Promega) for 4 h at room temperature with shaking. Antibodies were purified using a Kingfisher instrument (Thermo Fisher) by sequential washing with PBS and elution with 0.2 M citric acid pH 3.0 followed by neutralization with 2 M Tris pH 7.4 and immediate buffer exchange into PBS with a 96-well Zeba desalting plate (Thermo Fisher). Antibody concentrations were determined with NanoOrange Protein Quantitation kit (Thermo Fisher) and interpolated from a human IgG1 standard curve.
For midi-scale antibody purification, supernatants (50 to 200 mL) were harvested 5 to 6 days post-transfection, centrifuged to remove cell debris, and purified using Protein A Sepharose 4B (Thermo Fisher) with the same washing, elution and neutralization conditions as described above. Antibodies were desalted into PBS with PD-10 columns (Cytiva). Protein concentration was assessed by Nanodrop (Thermo Fisher), and purity was confirmed using SDS-PAGE analysis.
Mouse experiments.
Animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (Assurance number A3381–01).
Virus inoculations were performed under anesthesia that was induced and maintained with isoflurane, and all efforts were made to minimize animal pain and discomfort. 3-week-old male C57BL/6J mice were purchased from Jackson Laboratories (catalog #00664). For the lethal challenge model of CHIKV infection, 4–5 week-old male mice were treated with 500 μg of anti-IFNAR1 (46) (MAR1–5A3, Leinco) by intraperitoneal injection and either 100 μg (~5 mg/kg) or 20 μg (~1 mg/kg) of the indicated mAb one day prior to subcutaneous inoculation of the footpad with 101 FFU of CHIKV-LR 2006 (11, 15). For immunocompetent models of alphavirus infection, 3- to 4-week-old male mice were administered 200 μg (~10 mg/kg) of the indicated mAb one day prior to inoculation in the footpad with 103 FFU of CHIKV-LR 2006, MAYV (strain BeH407), or RRV (strain T48). Foot swelling in this model was determined using digital calipers (Fowler) by measurement of the height and width of feet.
Viral burden analysis.
At 3 dpi, mice were euthanized, and tissues were collected. Tissues were homogenized with a MagNA Lyser (Roche), and viral RNA was extracted with a MagMAX Viral RNA extraction kit using a Kingfisher Flex instrument. Viral load was determined using a TaqMan RNA-to-Ct 1-Step Kit on a QuantStudio 6 (Thermo Fisher) using previously established CHIKV-LR 2006, MAYV, and RRV primer and probe sets (28, 37, 38). FFU equivalents were determined by parallel processing of a viral stock with known titer.
Histology.
Hair was removed using Nair™. Feet were rinsed in PBS followed by fixation for 24 h in 4% paraformaldehyde (PFA). Feet were submitted to the Washington University Musculoskeletal Histology and Morphometry Core for decalcification, paraffin embedding, sectioning, and hematoxylin and eosin staining. Slides were imaged with an Echo Revolve microscope (Echo/BICO).
Virus-like particle production.
Chikungunya VLPs of the 37997 strain were prepared as described previously (47). Briefly, suspension-adapted, human embryonic kidney 293 cells were transfected in serum-free media with an expression plasmid containing the structural genes. Following centrifugation and filtration of the transfected harvest, VLPs were purified by tangential flow filtration followed by anion exchange chromatography. The purified VLPs were concentrated and buffer exchanged into 10 mM potassium phosphate, 218 mM sucrose, and 25 mM sodium citrate, sterile filtered and stored at −80°C.
Recombinant proteins.
Recombinant mammalian cell expressed CHIKV E2 and E1 were purchased from Sino Biologicals (40440-V08B) or Native Antigen (CHIKV-E1–100), respectively. CHIKV p62-E1 (19) was expressed transiently in Expi293 cells with Expifectamine 293. Four days after transfection, supernatants were harvested, centrifuged, filtered, and concentrated, and dialyzed overnight into 20 mM Tris pH 8.0, 200 mM NaCl using a Millipore Ultra-Cel 30 kDa membrane. Dialyzed supernatant was passed over Ni-NTA agarose column (Thermo Fisher), and purified protein was eluted with 250 mM imidazole (pH 8.0). Eluted protein was exchanged into 1× PBS with PD-10 columns followed by concentration with Amicon centrifugal filter with a 30 kDa cutoff. Protein purity was assessed by SDS-PAGE, and proteins were stored frozen at −80°C.
ELISA.
(a) VLPs. Maxisorp plates (Thermo Fisher) were coated with 1 μg/mL of alphavirus cross-reactive mAbs with MAY-117 and MAY-119 (37) overnight in 0.1 M NaHCO3 buffer pH 9.3 at 4°C. After washing, CHIKV VLPs were added in blocking buffer (2% BSA in PBS) at a concentration of 1 μg/mL. (b) E2 and p62-E1. For CHIKV E2 and CHIKV p62-E1 ELISA, plates were coated with 2 μg/mL of purified recombinant protein. MAbs were diluted in blocking buffer and added to plates for 1 h at 25°C. Plates were washed and incubated with horseradish peroxidase conjugated goat-anti human IgG (H+L) (1:10,000 dilution, Jackson ImmunoResearch) for 30 min at 25°C. After washing, plates were developed with 1-Step™ Ultra TMB-ELISA substrate (Thermo Fisher), stopped with 2 N H2SO4, and read at 450 nM using a Synergy H1 plate reader or Cytation 7 (Biotek).
For epitope binning experiments, plates were coated with CHIKV p62-E1 as described above followed by incubation with 20 μg/mL of each mAb for 1 h at room temperature. Biotinylated mAbs were directly applied without washing at a concentration of 50 ng/mL and incubated for 30 min at room temperature. Following incubation, plates were washed and incubated with streptavidin-HRP (Jackson, 1:10,000) for 20 min at room temperature and developed with 1-Step Ultra-TMB.
Focus reduction neutralizing test.
Serially-diluted mAbs or sera were incubated with ~200 FFU of different alphaviruses for 1 h at 37°C. The antibody-virus complex was added to Vero cells for 60 min at 37°C followed by a 1% methylcellulose overlay in Minimal Essential Medium supplemented with 2% FBS. Cells were fixed at 16 h post-infection with 1% PFA in PBS. Cells were washed with PBS-T (0.05% Tween-20) and incubated for 2–4 h at room-temperature or overnight at 4°C with 1 μg/mL of CHK-48, CHK-265, or DC2.112 (11, 17, 20). After washing, cells were incubated for 1 h with HRP-conjugated goat anti-mouse or goat anti-human IgG (H+L) (1:2,000; Jackson ImmunoResearch). Plates were developed using TrueBlue substrate (KPL), foci were quantitated using a BioSpot plate reader, and neutralization data was analyzed with Prism 9 (GraphPad).
Flow cytometry of blood samples.
Samples were thawed rapidly at 37°C and stained with CHIKV VLPs at a concentration of 10 μg/mL in FACS buffer (0.1% BSA+ 0.05% NaN3 + 2 mM EDTA in PBS) for 30 min at 4°C. Cells were subsequently stained with the following mAbs and reagents from BioLegend: BV785-conjugated anti-CD19 (HIB19), BV421-conjugated anti-IgD (IA6–2), APC-Cy7-conjugated anti-CD38 (HIT-2), PE-Cy7-conjugated anti-CD71 (RI7217), BV605-conjugated anti-CD27 (O323), PE-Dazzle594-conjugated anti-CD4 (A161A1). CHK-265(11, 28) was conjugated to Alexa Fluor 488 and Alexa Fluor 647 with a Zip Antibody Labelling Kit (Thermo Fisher) according to the manufacturer’s instructions. Samples were analyzed in the presence of DAPI (0.1μg/mL) to exclude dead cells on a Cytek Aurora 3-laser (VBR) instrument. Data were analyzed with FloJo software version 10 (Treestar). An example of the gating strategy is provided (Fig. S1D).
Single-cell RNA sequencing.
Approximately 2 to 3 × 106 PBMCs were thawed and incubated sequentially with 10 μg/mL of CHIKV VLP for 30 min on ice and 1 μg/mL of biotinylated CHK-265. Cells then were stained with the following cocktail of reagents purchased from Biolegend unless otherwise noted: streptavidin-APC-TotalSeqC (Cat#405293), anti-CD19-TotalSeqC (Cat#302265), anti-CD71-TotalSeqC (Cat#334125) and anti-CD27-TotalSeqC (Cat# 302853), PE-conjugated anti-IgD (IA6–2) and PerCP-Cy5.5-conjugated anti-CD20 (2H7). Naïve B cells were depleted with anti-PE Mojo magnetic nanobeads, and B cells were enriched using EasySep™ pan B cell magnetic enrichment kit (STEMCELL). Approximately 20,000 to 30,000 non-naïve enriched B cells were obtained for downstream 10X Genomics analysis using this protocol.
V-D-J, 5′ gene expression, and probe feature libraries (TotalSeq-C) were prepared using the 10X Chromium System (10X Genomics). The Chromium Single Cell 5′ Library and Gel Bead v2 Kit, Human B Cell V(D)J Enrichment Kit, and Feature Barcode Library Kit were used. Libraries were pooled and sequenced using the NovaSeq (Illumina) 6000. Raw sequencing reads were processed using standard Cell Ranger (version 3.0.2) pipeline, including 5′ gene expression analysis, antigen probe analysis, and immunoprofiling analysis of B cells.
Cell Ranger output was processed using Seurat (an R package, for transcriptome, cell surface protein and antigen probe analysis). For transcriptome analysis, Seurat was used for cell quality control, data normalization, data scaling, dimension reduction (both linear and non-linear), clustering, and data visualization. Unwanted cells were removed according to the number of detectable genes (number of genes < 200 or > 2,500 were removed) and percentage of mitochondrial genes for each cell. Transcriptome data were normalized by a log-transform function with a scaling factor of 10,000, whereas cell surface proteins and antigen probes were normalized by a centered log-ratio (CLR) normalization. All computational analyses were performed in R.
Antibody binding to alphavirus-infected cells.
Vero cells were inoculated (multiplicity of infection [MOI] of 5) with alphaviruses in DMEM supplemented with 2% FBS. After allowing infection to proceed for 14 to 18 h, cells were detached using TrypLE (Thermo Fisher), washed with DMEM containing 2% FBS, and filtered through 100 μm nylon mesh (Corning). Cells then were incubated with 10 μg/mL of mAbs for 30 min at 4°C in FACS buffer. Cells were washed and incubated with Alexa Fluor 647 conjugated goat anti-human or anti-mouse IgG (1:2,000 dilution; Thermo Fisher) for 30 min at 4°C. Cells were washed, fixed with 2% paraformaldehyde (PFA), and subjected to flow cytometry analysis using an iQue3 flow cytometer (Sartorius).
Alanine-scanning and charge-reversal mutagenesis.
A pCAGGS plasmid encoding CHIKV VLP (capsid-E3-E2–6K-E1) was subjected to mutagenesis of all residues of CHIKV E2 to alanine except for alanine residues, which were mutated to serine (Genscript). Select residues identified by alanine-scanning were additionally mutated to arginine with arginine and lysine residues mutated to glutamate (Genscript). Plasmids were transfected in a 96-well format with Expifectamine 293 into Expi293 cells. 24 h post-transfection, cells were stained with pre-titrated sub-saturating concentrations of mAbs for 30 min at 4°C followed by secondary staining with anti-human IgG Alexa Flour 647 for 20 min at 4°C. Cells were resuspended in FACS buffer containing DAPI for exclusion of dead cells. Data were acquired on an iQue3 flow cytometer, and analyzed with ForeCyt software (Sartorius).
Epitope binning by flow cytometry.
CHIKV structural proteins (capsid-E3-E2–6k-E1) were expressed on the surface of Expi293 cells by transfection as described above. Cells were harvested 24 h after transfection and half were labelled with CellTrace™ CFSE (Thermo Fisher). Labelled cells were then pre-incubated with mouse mAbs or MXRA8-mouse Fc at a concentration of 10 μg/mL for 1 h at 4°C. Cells were extensively washed with FACS buffer and combined in a 1:1 ratio with CFSE-negative cells. The cell mixture was then incubated with mAbs at a concentration of 1 μg/mL for 30 min at 4°C, followed by incubation with anti-human IgG-A647 (Thermo Fisher) for 20 min at 4°C. Cells were washed, and resuspended in FACS buffer supplemented with DAPI and analyzed on an iQue3 flow cytometer. Percent binding was calculated for each mAb by geometric MFI in CFSE+ divided by CFSE− cells in each sample multiplied by 100.
Biolayer interferometry.
BLI experiments were performed using an Octet Red96 (ForteBio) at 25°C in 10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% P20 surfactant, and 1% BSA (w/v). MAbs were immobilized onto anti-human IgG Fc capture biosensors (ForteBio) and dipped into wells containing 1 μM CHIKV E1 (Native Antigen) to assess E1 reactivity of mAbs.
Neutralization escape.
To generate neutralization escape mutants, CHIKV 181/25 (105 FFU) was incubated with 10 μg/mL of anti-CHIKV mAbs for 1 h at 37°C. The virus–mAb complexes were added to Vero cells. One day post-infection, the virus supernatant was removed and then incubated with mAbs for 1 h at 37°C and added to new Vero cells. This process was repeated for 5 days. Viral RNA from bulk supernatant was extracted with MagMAX Viral RNA extraction kit using a Kingfisher Flex instrument (Thermo Fisher) according to the manufacturer’s instructions. cDNA was generated using ProtoScript® II First Strand cDNA Synthesis Kit (NEB) according to the manufacturer’s instructions using random hexamers. Viral structural genes were amplified with the following primers: 5′-TGCCATTCCAGTTATGTGCC-3′ and 5′-CACGCATAGCACCACGATTA-3′, purified with ChargeSwitch PCR clean-up kit (Thermo Fisher) and subject to long-read amplicon sequencing, Oxford Nanopore (Plasmidsaurus).
Egress inhibition assay
Vero cells were inoculated at an MOI of 1 with a chimeric virus, SINV-CHIKV-LR, which encodes for the non-structural proteins of SINV TR339 and the structural proteins of CHIKV LR 2006 (45). After a 2 h incubation at 37°C, cells were rinsed six times with PBS and incubated with mAbs (10 μg/mL) supplemented with 25 mM NH4Cl to prevent de novo infection. Supernatant was harvested at 1 and 6 h post infection, and viral RNA was recovered with a MagMAX Viral RNA isolation kit (Thermo Fisher) using a Kingfisher Flex instrument (Thermo Fisher). RNA was quantitated with a TaqMan RNA-to-Ct 1-Step Kit on a QuantStudio 6 (Thermo Fisher) using the following SINV primer and probe sets SINV nsP4 F: 50-AAGATCATC GACGCAGTCATC-30; SINV nsP4 R: 50-GCTGTGGAAGTAACCGAATCT-30; SINV nsP4 Probe: 50-/56-FAM/CCACCTTAC/ZEN/TTCTGCGGCGGATTTA/3IABkFQ/-30.
Cryo-EM sample preparation.
Fab was generated from mAbs 516.A08, 516.C01, and chimeric human CHK-265 using a FabALACTICA Fab kit (Genovis) according to manufacturer’s instructions. CHIKV VLPs were prepared at a nominal concentration of ~0.7 mg/mL in 10 mM potassium phosphate, 218 mM sucrose, and 25 mM sodium citrate. VLPs were incubated for 1 h on ice with approximately 2:1 molar excess of each Fab, applied to glow-discharged lacey carbon grids (Ted Pella #01895-F), and then flash-frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher).
Cryo-EM data collection.
Grids were loaded into a Cs-corrected FEI Titan Krios 300kV microscope equipped with a Falcon 4 direct electron detector and then imaged at a nominal magnification of 59,000×, resulting in a calibrated pixel size of 1.16 Å. Movies were collected in EER format with a total dose of 38.25 e−/Å2/movie (4.70 e−/Å2/s over 8.14 s acquisition).
Cryo-EM data processing.
EER movies were binned into 35 fractions (1.09 e−/Å2/f) and then subjected to patch motion and patch CTF corrections in cryoSPARC v3.1.0 (48). Particles were selected using a template picker then cleaned via two- and three-dimensional classification. Whole VLPs were reconstructed via homogeneous non-uniform refinement with I1 symmetry imposed. To extract asymmetric units as individual subparticles, we performed I1 symmetry expansion, applied particle shifts to center asymmetric units, and then re-extracted these particles with shifts applied. These asymmetric units were cleaned via three-dimensional classification without alignment in Relion 3.1 (49, 50), and the class of highest resolution for each Fab was refined via local non-uniform refinement in cryoSPARC.
Model building.
To facilitate model building, maps were sharpened using deep learning implemented via DeepEMhancer (51). Starting models for CHIKV structural proteins were adapted from a previous CHIKV VLP cryo-EM structure (PDB: 6NK5) (47), and initial Fv were modeled using AlphaFold2 implemented in ColabFold (52). These starting components were docked into the electron density maps and then refined iteratively using Coot v0.9.5(53), Isolde v1.1.0 (54), Phenix v1.19 (55), and Rosetta (56). Proteins, Interfaces, Structures, and Assemblies (PISA) solvent exclusion analysis was used to identify contact residues and calculate buried surface area (57). Structures were visualized using UCSF ChimeraX (58).
Phylogenetic inference.
E2 sequences were retrieved from the NCBI GenBank for each alphavirus: CHIKV (QKY67868.1), MAYV (QED21311.1), Una (UNAV, YP_009665989.1), ONNV (AAC97205.1), SFV (NP_463458.1), RRV (AAA47404.1), EEEV (ANB41743.1), VEEV (AGE98294.2), SINV (AAM10630.1), WEEV (QEX51909.1), GETV (ABK32032.1), and BEBV (AEJ36225.1). Sequences were aligned via Clustal Omega (59) with simple phylogeny inferred from pairwise distances. Results were visualized in R using the ggtree package (60).
Statistical analysis.
Statistical analysis was performed with Prism 9.0. Details of statistical tests are included in figure legends.
Supplementary Material
Figure S1. Serological and B cell responses to PXVX00317 vaccination.
Figure S2. Single-cell RNA-seq analysis and characterization of binding of recombinant mAbs.
Figure S3. Competition binding analysis by cell-surface expression and immobilization of soluble CHIKV structural proteins.
Figure S4. Protection by vaccine-elicited mAbs against CHIKV infection in immunodeficient and immunocompetent mice.
Figure S5. Mapping of epitopes by focused arginine scanning and neutralization escape.
Figure S6. Cross-neutralization of alphaviruses and therapeutic effect by vaccine-elicited mAbs.
Figure S7. Cryo-EM data processing pipeline.
Figure S8. Validation of cryo-EM reconstructions.
Figure S9. Structural features of broadly-neutralizing vaccine-induced mAbs and CHK-265.
Figure S10. Comparison of broadly-neutralizing anti-CHIKV mAbs with known structures.
Table S1. Cryo-EM data collection, processing, and model refinement statistics.
Table S2. Close contacts and buried surface area of CHIKV E2-Fab fragment interfaces.
Data Files. Raw data from Figures and Supplemental Figures.
Acknowledgements.
We thank Ali Ellebedy and Sarah Royalty Tredo for experimental and editorial advice. We thank Nicholas Borcherding and Bradley Whitener for technical assistance. We thank Michael Rau, Brock Summers, Katherine Basore, and James Fitzpatrick at the Washington University Center for Cellular Imaging (WUCCI) for cryo-EM sample preparation and data acquisition. We acknowledge Michael Lacy, Spencer Stonier, Christopher Morello, Jason Mendy and the Emergent BioSolutions CHIKV VLP vaccine team for the guidance and provision of clinical samples. The studies were supported by NIH Grants R01 AI141436, U19 AI142790, and contract AI201800001 awarded to M.S.D. and a Sponsored Research Agreement with Emergent BioSolutions.
Footnotes
Competing Financial Interests. M.S.D. is a consultant for Inbios, Vir Biotechnology, Senda Biosciences, Moderna, and Immunome. The Diamond laboratory has received unrelated funding support in sponsored research agreements from Moderna, Vir Biotechnology, and Immunome. J.E.C. has served as a consultant for Luna Labs USA, Merck Sharp & Dohme Corporation, and GlaxoSmithKline, is a member of the Scientific Advisory Board of Meissa Vaccines, a former member of the Scientific Advisory Board of Gigagen (Grifols) and is founder of IDBiologics. The laboratory of J.E.C. received unrelated sponsored research agreements from AstraZeneca, Takeda, and IDBiologics during the conduct of the study. K.L.W and L.V. are employees and shareholders of Emergent BioSolutions Inc.
Materials and Data availability. All data supporting the findings of this study are available within the paper and from the corresponding author upon request. Models of mAb complexes were generated from their respective PDB files with the following accession codes: CHK-265, PDB 8DWY; 506.A08, PDB 8DWW, and 506.C01, PDB 8DWX. RNA sequencing data has been deposited to NCBI GEO with the following accession code: GSE218843. Materials used in this paper (e.g., viruses, antibodies, cell lines, and proteins) are available through a Material Transfer Agreement with Washington University School of Medicine or Emergent BioSolutions.
REFERENCES
- 1.Suhrbier A, Jaffar-Bandjee M-C, Gasque P, Arthritogenic alphaviruses--an overview. Nat. Rev. Rheumatol. 8, 420–429 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Zacks MA, Paessler S, Encephalitic alphaviruses. Vet. Microbiol. 140, 281–286 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schilte C, Staikowsky F, Couderc T, Madec Y, Carpentier F, Kassab S, Albert ML, Lecuit M, Michault A, Chikungunya virus-associated long-term arthralgia: a 36-month prospective longitudinal study. PLoS Negl. Trop. Dis. 7, e2137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burt FJ, Chen W, Miner JJ, Lenschow DJ, Merits A, Schnettler E, Kohl A, Rudd PA, Taylor A, Herrero LJ, Zaid A, Ng LFP, Mahalingam S, Chikungunya virus: an update on the biology and pathogenesis of this emerging pathogen. Lancet Infect. Dis. 17, e107–e117 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Rezza G, Weaver SC, Chikungunya as a paradigm for emerging viral diseases: Evaluating disease impact and hurdles to vaccine development. PLoS Negl. Trop. Dis. 13, e0006919 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun S, Xiang Y, Akahata W, Holdaway H, Pal P, Zhang X, Diamond MS, Nabel GJ, Rossmann MG, Structural analyses at pseudo atomic resolution of Chikungunya virus and antibodies show mechanisms of neutralization. Elife 2, e00435 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akahata W, Yang Z-Y, Andersen H, Sun S, Holdaway HA, Kong W-P, Lewis MG, Higgs S, Rossmann MG, Rao S, Nabel GJ, A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat. Med. 16, 334–338 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goo L, Dowd KA, Lin T-Y, Mascola JR, Graham BS, Ledgerwood JE, Pierson TC, A Virus-Like Particle Vaccine Elicits Broad Neutralizing Antibody Responses in Humans to All Chikungunya Virus Genotypes. J. Infect. Dis. 214, 1487–1491 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen GL, Coates EE, Plummer SH, Carter CA, Berkowitz N, Conan-Cibotti M, Cox JH, Beck A, O’Callahan M, Andrews C, Gordon IJ, Larkin B, Lampley R, Kaltovich F, Gall J, Carlton K, Mendy J, Haney D, May J, Bray A, Bailer RT, Dowd KA, Brockett B, Gordon D, Koup RA, Schwartz R, Mascola JR, Graham BS, Pierson TC, Donastorg Y, Rosario N, Pape JW, Hoen B, Cabié A, Diaz C, Ledgerwood JE, VRC 704 Study Team, Effect of a Chikungunya Virus-Like Particle Vaccine on Safety and Tolerability Outcomes: A Randomized Clinical Trial. JAMA 323, 1369–1377 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett SR, McCarty JM, Ramanathan R, Mendy J, Richardson JS, Smith J, Alexander J, Ledgerwood JE, de Lame P-A, Royalty Tredo S, Warfield KL, Bedell L, Safety and immunogenicity of PXVX0317, an aluminium hydroxide-adjuvanted chikungunya virus-like particle vaccine: a randomised, double-blind, parallel-group, phase 2 trial. Lancet Infect. Dis. 22, 1343–1355 (2022). [DOI] [PubMed] [Google Scholar]
- 11.Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, Johnson S, Lee I, Akahata W, Nabel GJ, Richter MKS, Smit JM, Fremont DH, Pierson TC, Heise MT, Diamond MS, Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog. 9, e1003312 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Couderc T, Khandoudi N, Grandadam M, Visse C, Gangneux N, Bagot S, Prost J-F, Lecuit M, Prophylaxis and therapy for Chikungunya virus infection. J. Infect. Dis. 200, 516–523 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warter L, Lee CY, Thiagarajan R, Grandadam M, Lebecque S, Lin RTP, Bertin-Maghit S, Ng LFP, Abastado J-P, Desprès P, Wang C-I, Nardin A, Chikungunya virus envelope-specific human monoclonal antibodies with broad neutralization potency. J. Immunol. 186, 3258–3264 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Lee CY, Kam Y-W, Fric J, Malleret B, Koh EGL, Prakash C, Huang W, Lee WWL, Lin C, Lin RTP, Renia L, Wang C-I, Ng LFP, Warter L, Chikungunya virus neutralization antigens and direct cell-to-cell transmission are revealed by human antibody-escape mutants. PLoS Pathog. 7, e1002390 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith SA, Silva LA, Fox JM, Flyak AI, Kose N, Sapparapu G, Khomandiak S, Ashbrook AW, Kahle KM, Fong RH, Swayne S, Doranz BJ, McGee CE, Heise MT, Pal P, Brien JD, Austin SK, Diamond MS, Dermody TS, Crowe JE Jr, Isolation and Characterization of Broad and Ultrapotent Human Monoclonal Antibodies with Therapeutic Activity against Chikungunya Virus. Cell Host Microbe 18, 86–95 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powell LA, Miller A, Fox JM, Kose N, Klose T, Kim AS, Bombardi R, Tennekoon RN, Dharshan de Silva A, Carnahan RH, Diamond MS, Rossmann MG, Kuhn RJ, Crowe JE Jr, Human mAbs Broadly Protect against Arthritogenic Alphaviruses by Recognizing Conserved Elements of the Mxra8 Receptor-Binding Site. Cell Host Microbe 28, 699–711.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quiroz JA, Malonis RJ, Thackray LB, Cohen CA, Pallesen J, Jangra RK, Brown RS, Hofmann D, Holtsberg FW, Shulenin S, Nyakatura EK, Durnell LA, Rayannavar V, Daily JP, Ward AB, Aman MJ, Dye JM, Chandran K, Diamond MS, Kielian M, Lai JR, Human monoclonal antibodies against chikungunya virus target multiple distinct epitopes in the E1 and E2 glycoproteins. PLoS Pathog. 15, e1008061 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williamson LE, Gilliland T Jr, Yadav PK, Binshtein E, Bombardi R, Kose N, Nargi RS, Sutton RE, Durie CL, Armstrong E, Carnahan RH, Walker LM, Kim AS, Fox JM, Diamond MS, Ohi MD, Klimstra WB, Crowe JE Jr, Human Antibodies Protect against Aerosolized Eastern Equine Encephalitis Virus Infection. Cell (2020), doi: 10.1016/j.cell.2020.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voss JE, Vaney M-C, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA, Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468, 709–712 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Kim AS, Kafai NM, Winkler ES, Gilliland TC Jr, Cottle EL, Earnest JT, Jethva PN, Kaplonek P, Shah AP, Fong RH, Davidson E, Malonis RJ, Quiroz JA, Williamson LE, Vang L, Mack M, Crowe JE Jr, Doranz BJ, Lai JR, Alter G, Gross ML, Klimstra WB, Fremont DH, Diamond MS, Pan-protective anti-alphavirus human antibodies target a conserved E1 protein epitope. Cell 184, 4414–4429.e19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fong RH, Banik SSR, Mattia K, Barnes T, Tucker D, Liss N, Lu K, Selvarajah S, Srinivasan S, Mabila M, Miller A, Muench MO, Michault A, Rucker JB, Paes C, Simmons G, Kahle KM, Doranz BJ, Exposure of epitope residues on the outer face of the chikungunya virus envelope trimer determines antibody neutralizing efficacy. J. Virol. 88, 14364–14379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powers AM, Brault AC, Shirako Y, Strauss EG, Kang W, Strauss JH, Weaver SC, Evolutionary relationships and systematics of the alphaviruses. J. Virol. 75, 10118–10131 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartman H, Wang Y, Schroeder HW Jr, Cui X, Absorbance summation: A novel approach for analyzing high-throughput ELISA data in the absence of a standard. PLoS One 13, e0198528 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoon I-K, Alera MT, Lago CB, Tac-An IA, Villa D, Fernandez S, Thaisomboonsuk B, Klungthong C, Levy JW, Velasco JM, Roque VG Jr, Salje H, Macareo LR, Hermann LL, Nisalak A, Srikiatkhachorn A, High rate of subclinical chikungunya virus infection and association of neutralizing antibody with protection in a prospective cohort in the Philippines. PLoS Negl. Trop. Dis. 9, e0003764 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoon I-K, Srikiatkhachorn A, Alera MT, Fernandez S, Cummings DAT, Salje H, Pre-existing chikungunya virus neutralizing antibodies correlate with risk of symptomatic infection and subclinical seroconversion in a Philippine cohort. Int. J. Infect. Dis. 95, 167–173 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Pinna D, Corti D, Jarrossay D, Sallusto F, Lanzavecchia A, Clonal dissection of the human memory B-cell repertoire following infection and vaccination. Eur. J. Immunol. 39, 1260–1270 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frölich D, Giesecke C, Mei HE, Reiter K, Daridon C, Lipsky PE, Dörner T, Secondary immunization generates clonally related antigen-specific plasma cells and memory B cells. J. Immunol. 185, 3103–3110 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Fox JM, Long F, Edeling MA, Lin H, van Duijl-Richter MKS, Fong RH, Kahle KM, Smit JM, Jin J, Simmons G, Doranz BJ, Crowe JE Jr, Fremont DH, Rossmann MG, Diamond MS, Broadly Neutralizing Alphavirus Antibodies Bind an Epitope on E2 and Inhibit Entry and Egress. Cell 163, 1095–1107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanz I, Wei C, Jenks SA, Cashman KS, Tipton C, Woodruff MC, Hom J, Lee FE-H, Challenges and Opportunities for Consistent Classification of Human B Cell and Plasma Cell Populations. Front. Immunol. 10, 2458 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Setliff I, Shiakolas AR, Pilewski KA, Murji AA, Mapengo RE, Janowska K, Richardson S, Oosthuysen C, Raju N, Ronsard L, Kanekiyo M, Qin JS, Kramer KJ, Greenplate AR, McDonnell WJ, Graham BS, Connors M, Lingwood D, Acharya P, Morris L, Georgiev IS, High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell 179, 1636–1646.e15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malonis RJ, Earnest JT, Kim AS, Angeliadis M, Holtsberg FW, Aman MJ, Jangra RK, Chandran K, Daily JP, Diamond MS, Kielian M, Lai JR, Near-germline human monoclonal antibodies neutralize and protect against multiple arthritogenic alphaviruses. Proc. Natl. Acad. Sci. U. S. A. 118 (2021), doi: 10.1073/pnas.2100104118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng N-Y, Mays I, Garman L, Helms C, James J, Air GM, Capra JD, Ahmed R, Wilson PC, Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou QF, Fox JM, Earnest JT, Ng T-S, Kim AS, Fibriansah G, Kostyuchenko VA, Shi J, Shu B, Diamond MS, Lok S-M, Structural basis of Chikungunya virus inhibition by monoclonal antibodies. Proc. Natl. Acad. Sci. U. S. A. 117, 27637–27645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliphant T, Engle M, Nybakken GE, Doane C, Johnson S, Huang L, Gorlatov S, Mehlhop E, Marri A, Chung KM, Ebel GD, Kramer LD, Fremont DH, Diamond MS, Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 11, 522–530 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, Gunn BM, Elmore SA, Heise MT, A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am. J. Pathol. 178, 32–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim AS, Austin SK, Gardner CL, Zuiani A, Reed DS, Trobaugh DW, Sun C, Basore K, Williamson LE, Crowe JE Jr, Slifka MK, Fremont DH, Klimstra WB, Diamond MS, Protective antibodies against Eastern equine encephalitis virus bind to epitopes in domains A and B of the E2 glycoprotein. Nat Microbiol 4, 187–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Earnest JT, Basore K, Roy V, Bailey AL, Wang D, Alter G, Fremont DH, Diamond MS, Neutralizing antibodies against Mayaro virus require Fc effector functions for protective activity. J. Exp. Med. 216, 2282–2301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powell LA, Fox JM, Kose N, Kim AS, Majedi M, Bombardi R, Carnahan RH, Slaughter JC, Morrison TE, Diamond MS, Crowe JE Jr, Human monoclonal antibodies against Ross River virus target epitopes within the E2 protein and protect against disease. PLoS Pathog. 16, e1008517 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williamson LE, Reeder KM, Bailey K, Tran MH, Roy V, Fouch ME, Kose N, Trivette A, Nargi RS, Winkler ES, Kim AS, Gainza C, Rodriguez J, Armstrong E, Sutton RE, Reidy J, Carnahan RH, McDonald WH, Schoeder CT, Klimstra WB, Davidson E, Doranz BJ, Alter G, Meiler J, Schey KL, Julander JG, Diamond MS, Crowe JE Jr, Therapeutic alphavirus cross-reactive E1 human antibodies inhibit viral egress. Cell 184, 4430–4446.e22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Viant C, Weymar GHJ, Escolano A, Chen S, Hartweger H, Cipolla M, Gazumyan A, Nussenzweig MC, Antibody Affinity Shapes the Choice between Memory and Germinal Center B Cell Fates. Cell 183, 1298–1311.e11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purtha WE, Tedder TF, Johnson S, Bhattacharya D, Diamond MS, Memory B cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants. J. Exp. Med. 208, 2599–2606 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vidarsson G, Dekkers G, Rispens T, IgG subclasses and allotypes: from structure to effector functions. Front. Immunol. 5, 520 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Earnest JT, Holmes AC, Basore K, Mack M, Fremont DH, Diamond MS, The mechanistic basis of protection by non-neutralizing anti-alphavirus antibodies. Cell Rep. 35, 108962 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wauquier N, Becquart P, Nkoghe D, Padilla C, Ndjoyi-Mbiguino A, Leroy EM, The acute phase of Chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J. Infect. Dis. 204, 115–123 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun C, Gardner CL, Watson AM, Ryman KD, Klimstra WB, Stable, high-level expression of reporter proteins from improved alphavirus expression vectors to track replication and dissemination during encephalitic and arthritogenic disease. J. Virol. 88, 2035–2046 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheehan KCF, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo-Arthur C, Carrero JA, White JM, Hertzog PJ, Schreiber RD, Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J. Interferon Cytokine Res. 26, 804–819 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Basore K, Kim AS, Nelson CA, Zhang R, Smith BK, Uranga C, Vang L, Cheng M, Gross ML, Smith J, Diamond MS, Fremont DH, Cryo-EM Structure of Chikungunya Virus in Complex with the Mxra8 Receptor. Cell 177, 1725–1737.e16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Scheres SHW, RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJH, Lindahl E, Scheres SHW, New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanchez-Garcia R, Gomez-Blanco J, Cuervo A, Carazo JM, Sorzano COS, Vargas J, DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Communications Biology 4, 1–8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M, ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Croll TI, ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr D Struct Biol 74, 519–530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang RY-R, Song Y, Barad BA, Cheng Y, Fraser JS, DiMaio F, Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife 5 (2016), doi: 10.7554/eLife.17219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krissinel E, Henrick K, Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE, UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG, Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y, Ggtree : An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Serological and B cell responses to PXVX00317 vaccination.
Figure S2. Single-cell RNA-seq analysis and characterization of binding of recombinant mAbs.
Figure S3. Competition binding analysis by cell-surface expression and immobilization of soluble CHIKV structural proteins.
Figure S4. Protection by vaccine-elicited mAbs against CHIKV infection in immunodeficient and immunocompetent mice.
Figure S5. Mapping of epitopes by focused arginine scanning and neutralization escape.
Figure S6. Cross-neutralization of alphaviruses and therapeutic effect by vaccine-elicited mAbs.
Figure S7. Cryo-EM data processing pipeline.
Figure S8. Validation of cryo-EM reconstructions.
Figure S9. Structural features of broadly-neutralizing vaccine-induced mAbs and CHK-265.
Figure S10. Comparison of broadly-neutralizing anti-CHIKV mAbs with known structures.
Table S1. Cryo-EM data collection, processing, and model refinement statistics.
Table S2. Close contacts and buried surface area of CHIKV E2-Fab fragment interfaces.
Data Files. Raw data from Figures and Supplemental Figures.