

Abstract
We propose the hypothesis that small high-density lipoprotein (HDL) particles reduce the risk of Alzheimer’s disease (AD) by virtue of their capacity to exchange lipids, affecting neuronal membrane composition and vascular and synaptic functions. Concentrations of small HDLs in cerebrospinal fluid (CSF) and plasma were measured in 180 individuals ≥60 years of age using ion mobility methodology. Small HDL concentrations in CSF were positively associated with performance in three domains of cognitive function independent of apolipoprotein E (APOE ε4 status, age, sex, and years of education. Moreover, there was a significant correlation between levels of small HDLs in CSF and plasma. Further studies will be aimed at determining whether specific components of small HDL exchange across the blood, brain, and CSF barriers, and developing approaches to exploit small HDLs for therapeutic purposes.
Keywords: APOE, cerebrospinal fluid, cognition, HDL, lipidomics, proteomics
1 |. NARRATIVE
Brain autopsy studies have long demonstrated the accumulation of lipid droplets1 in Alzheimer’s disease (AD), starting with Alois Alzheimer’s initial description of “adipose saccules” in the brain in 1907.2 Interest in peripheral cholesterol metabolism in relation to the brain was sparked by an association of greater plasma cholesterol levels in midlife with an increase in AD risk.3,4 Moreover, there were reports that lower plasma HDL cholesterol (HDL-C) levels were associated with increased severity of AD,5 and conversely that higher HDL-C levels were associated with better cognitive function.6 Overall, however, results of studies examining the associations between plasma HDL-C levels and AD risk have been inconsistent, such that a metaanalysis of 100 studies did not demonstrate this relationship to be significant.7 Similarly, measures of brain cholesterol content had inconsistent associations with AD, with reports of reduced, increased, and no changes in brain cholesterol levels in patients with AD dementia compared to controls.8
A limitation of studies based on measurement of HDL-C levels in plasma is that this measure does not capture the functional properties of HDL particles that transport cholesterol. Moreover, HDL particles comprise a diverse spectrum of lipid-protein complexes of differing size, structure, and function.9,10 The brain makes HDL-like particles, but to date these have been incompletely characterized. Although they carry fewer lipids compared to larger HDLs, small HDLs have functional properties such as cholesterol efflux capacity.11,12 Herein we present data supporting the hypothesis that measurement and characterization of small HDL particles in cerebrospinal fluid (CSF) provides novel information bearing on AD risk that better captures their functions than cholesterol levels.
1.1 |. Genetic associations of AD with genes affecting cholesterol and HDL metabolism
Among genetic variants associated with AD risk in genome-wide association studies are a number at loci for proteins that impact HDL structure and function. These include apolipoprotein E (APOE) and apolipoprotein J (APOJ) (encoding apolipoprotein components of HDL), the ATP binding cassettes 7 (ABCA7), and Subfamily G Member 1 (ABCG1) (encoding membrane proteins that transport lipids onto HDL), and Sortilin Related Receptor 1(SORL1), Bridging Integrator 1 (BIN1), Phosphatidylinositol Binding Clathrin Assembly Protein (PICALM) (encoding proteins involved in the cellular uptake and recycling of HDL).13–15 In addition, gene sequencing has identified associations of rare variants of the ATP-binding cassette 1 (ABCA1) (mediates cellular efflux of cholesterol and phospholipids to form HDL) with AD risk.16,17 These associations support a key role for proteins affecting HDL structure and function in the pathogenesis of AD.
1.2 |. Brain lipoprotein particles
Compared to plasma, much less is known about brain lipoprotein particle composition and structure.10 Plasma HDL, predominantly made of apolipoprotein A-I (apoA-I), is comprised of multiple size subspecies ranging in particle diameter from 7 to 14.5 nm, of which five can be discriminated by non-denaturing gradient gel electrophoresis.18 These have been grouped into two major categories corresponding to the subclasses classically defined by ultracentrifugation: HDL 3 (3a, 3b, and 3c) and HDL 2 (2a and 2b). Ion mobility (IM) can discriminate the largest of these (HDL2b, 10.5-14.5 nm) from the smaller species.19 In CSF, some of these particles are in the density and size range of plasma HDL, although there are major compositional differences between these species.20,21 The major brain-derived apolipoproteins include apoE, apoJ, and apoD. Although astrocytes are the predominant source of apoE, microglia also produce a limited amount of lipid-poor apoE.22 The contributions of microglial apoE to AD pathology are currently not clear.23
1.3 |. ABCA1, small HDL particles, and brain amyloid deposition
Cellular lipid efflux mediated by ABCA1 results in the formation of nascent small HDL particles.11,12 In the brain, ABCA1 mediates the lipidation of apoE to form apoE HDLs.24 Nascent lipid-poor discoidal HDL particles rapidly acquire lipids to form small HDLs.25 ABCG1 completes the lipidation of small HDLs (by transferring more cholesterol and lipids) to form larger HDLs.26 ApoE HDL promotes amyloid beta (Aβ) degradation in astrocytes and microglia.27,28 In animal models, knocking out ABCA1 has been shown to decrease brain apoE levels and increase lipid-poor apoE and brain amyloid deposition.29,30 Furthermore, overexpression of ABCA1 increases apoE HDL and decreases brain amyloid deposition.31 Lipid-poor apoE protein aggregates seem to play an important role in reducing ABCA1 function.28 ApoE4, due to its greater potential to self-assemble or aggregate, traps ABCA1 in endosomes, hindering its membrane recycling and its ability to lipidate apoE.28 Restoring ABCA1 activity by an ABCA1 agonist peptide in ApoE4 targeted replacement mice allows the formation of apoE-containing HDL. Among the various HDLs, small HDLs have the most efficient ABCA1 cholesterol efflux capacity.11,12 Notably, ABCA1-mediated cholesterol efflux by CSF in patients with AD is reduced compared with non-demented controls.32 Therefore, measuring small HDL particles in CSF may provide a surrogate marker of ABCA1 function and apoE lipidation in the brain.
1.4 |. HDL, membrane lipids, and synaptic plasticity
Glial cholesterol and other lipids delivered to neurons by apoE-containing HDLs are necessary to support the high lipid demands imposed by the synthesis of new membranes that influence synaptic connectivity following brain damage and neurodegenerative insults.33,34 Notably, ABCA1 is actively involved in the brain’s plastic response to neurodegeneration and deafferentation for making new synaptic connections.35 In a model of entorhinal cortex lesioning that leads to deafferentation of the hippocampus followed by reinnervation, upregulation of ABCA1, but not ABCG1, resulted in a significant increase in the acetylcholine esterase activity in the ipsilateral hippocampus.35 The co-expression of apoE with ABCA1 by the liver X receptor and retinoid X receptor (LXR/RXR) system36 in astrocytes confirms the importance of apoE lipidation after its synthesis to support neuronal connectivity. HDLs have additional effects on the structure and function of the neuronal membrane lipid bilayer, where insertion of cholesterol into the membrane has profound effects on membrane-embedded proteins such as the amyloid precursor protein (APP).37 By controlling cholesterol biosynthesis and efflux, cholesterol content of neurons is kept low, thereby inhibiting Aβ accumulation.38
1.5 |. Small HDLs and inflammation
In addition to interacting with ABCA1, apoE-containing small HDLs may also activate ABCA739 and promote its recycling to the plasma membrane, similar to ABCA1.28 ABCA7 promotes the efflux of phospholipids from plasma membranes.40 The function of this transporter protects vulnerable cells from deleterious lipids that accumulate during normal function and aging. Similar to atherosclerosis, where lipid-loaded macrophages or “foam cells” accumulate in arterial walls and elicit a chronic inflammatory state,41 the accumulation of lipid droplets in astrocytes and microglia can promote an inflammatory response.1 Small HDLs can mitigate the effects of lipid accumulation through clearing cellular membranes from neurodegenerative lipids for exchange within brain cells or outside the brain. The accumulation of lipids in an inflammatory state creates a milieu that can damage the integrity of the blood-brain barrier (BBB),42,43 impair synaptic plasticity, and result in cognitive decline.
1.6 |. Small HDLs and vascular functions
There is abundant evidence that cardiovascular disease and cardiovascular risk factors play an important role in the etiology of AD.44 Plasma HDLs have protective effects on vascular functions, which include protecting against cerebral amyloid angiopathy.45 One of HDL’s functions involve scavenging of oxidized lipids,46 which protects the vascular wall from atherosclerosis. LDL oxidation increases LDL uptake by macrophage scavenger receptors, forming fatty streaks. HDL particles, primarily small HDLs,47 provide potent protection of LDL from oxidative damage by free radicals. It is plausible that the ability of small HDLs to scavenge oxidized lipids in the cerebral blood vessels can reduce AD risk, particularly before the onset of dementia. Vasoprotective HDLs provide one explanation for the association of lower LDL and greater HDL cholesterol levels in midlife with lower AD risk decades later.48
1.7 |. Small HDLs as a therapeutic target
In amyloid mouse models, several interventions targeting apoE lipids have shown promising anti-AD effects, reducing Aβ deposition, and improving cognitive functions. These included the oral administration of the RXR agonist bexarotene,49 the Peroxisome Proliferator Activated Receptor Gamma (PPARγ) agonist pioglitazone combined with the LXR agonist GW3965,50 and the Acyl-CoA: cholesterol acyltransferase 1 (ACAT1) enzyme inhibitor.51 A functional polymorphism in CETP, encoding the cholesteryl ester transfer protein (CETP), has been shown to be associated with decreased AD risk,52 and CETP inhibitors are being explored for AD. It is interesting to note that these drugs promote ABCA1-mediated cholesterol efflux to form small HDLs.53 Treatment with ABCA1 agonist peptide CS6253 increases the formation of small HDLs.54 APOE ε2 homozygotes are protected from AD,55 and overexpression of APOE ε2 allele increases apoE lipidation,56 and reduces insoluble Aβ42 in the hippocampus.57 Therefore, accurate measurement of HDL in CSF in relation to cognitive outcomes can guide the development of newer interventions focused on mechanisms that enhance apoE lipidation.
1.8 |. Exchangeability of HDL across the blood-brain and blood-CSF barriers
There is limited information regarding the exchangeability of HDLs and their components across the BBB and the blood-CSF barrier (BCSFB). There are several apolipoproteins found in CSF that are not made in the brain, including apoA-I,58 apoA-II,59 and apoC-III.60 Some evidence supports that small HDLs containing these apolipoproteins can traverse the BBB and BCSFB. For example, discoidal, lipid-poor apoA-I-HDL, which constitutes a small fraction of plasma apoA-I,25 is able to cross the BBB in vitro.61 Moreover, radiolabeled small HDLs can bind to the luminal membrane of brain capillary endothelial cells with high affinity and high specificity.62 In vivo imaging studies have identified a route of entry for circulating lipid-free apoA-I into the brain via the choroid plexus (CP) epithelium.63 Peripheral apoA-I is suspected to aid in the transport of other plasma proteins across the BCSFB.59,63,64 Upon entry into the CSF, plasma-derived apoA-I and apoA-II form lipoprotein complexes with brain-derived apoE,20,59 potentially limiting self-aggregation of apoE65 that alters its binding to its receptors.65 Correlations between plasma and CSF apolipoprotein concentrations further support a mechanism of exchange between the plasma and CSF compartments.64,66 In contrast to apoA-I, apoE exchange across the BBB and BCSFB appears to be limited. In human liver transplant recipients, serum apoE phenotypes match that of their respective liver donors, whereas CSF apoE phenotypes remain unchanged.67 Similarly, human apoE3 is not detected in CSF after intravenous injection into C57BL/6J mice.68 It remains unclear, however, whether apoE isoforms differ in their ability to cross the BBB or BCSFB. A model for small HDL exchange across the blood-CSF barrier with implications for neuronal functions is provided in Figure 1. The observation of both ABCA1 and ABCG1 on the CP epithelium supports the hypothesis that they mediate lipidation of apolipoproteins derived from the peripheral circulation following entry into the CSF compartment.69,70
FIGURE 1.
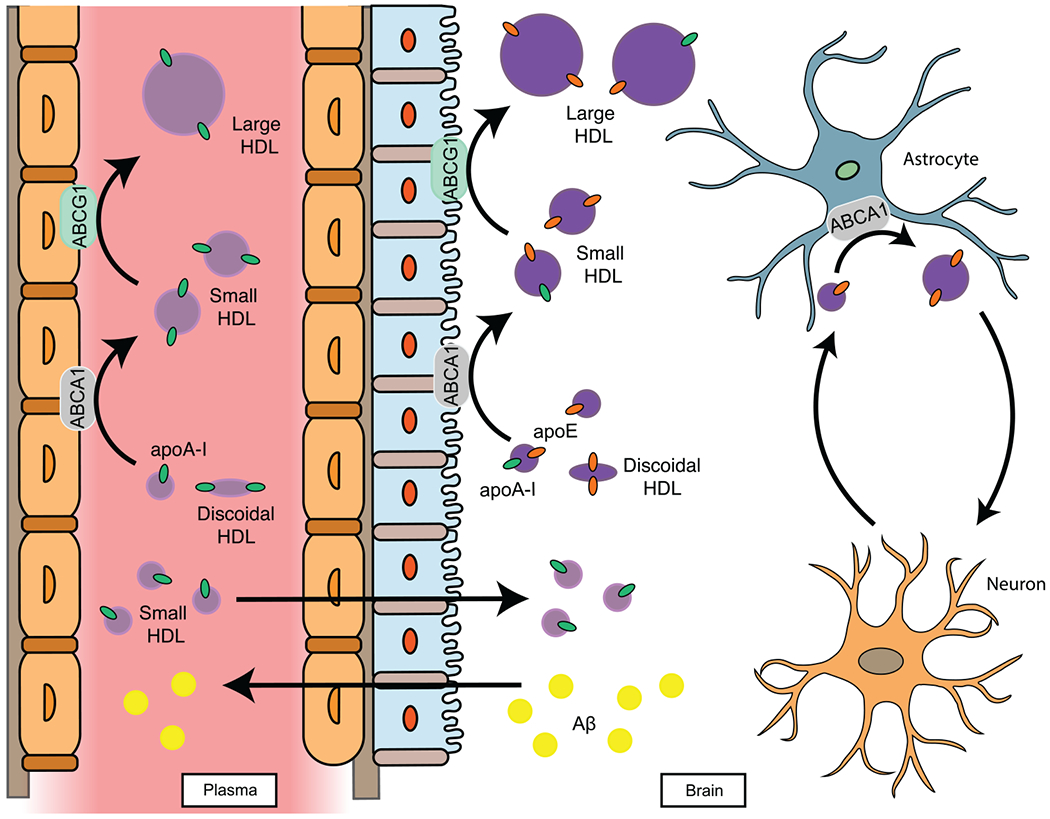
A model for exchangeable apolipoproteins across the blood-CSF (cerebrospinal fluid) and brain barriers. The small HDL hypothesis proposes that small HDL components can exchange into the brain and that the interaction between plasma-derived and brain-derived apolipoproteins stabilize ATP-binding cassette 1 (ABCA1) function to form small HDLs in the brain. The brain-systemic circulation crosstalk is illustrated here by an interaction of plasma-derived apolipoprotein A-I (apoA-I) with brain-derived apolipoprotein E (apoE) to form apoE/apoA-I small HDLs. The formation of small HDLs in the brain promotes membrane lipid remodeling, clearance of Aβ, and synaptic plasticity
The purpose of the present study was to test the hypothesis that greater levels of small HDLs in CSF are associated with lower AD risk, as assessed by better performance on measures of cognition, and a positive correlation with greater CSF Aβ42 levels as a surrogate of lower cerebral amyloidosis. Concentrations of small (7-10.5 nm) and large (10.5-14.5 nm) HDLs in matched CSF and plasma samples were measured using IM in 180 individuals aged ≥60 years from the University of Southern California (USC) Alzheimer Disease Research Center (ADRC) and Huntington Memorial Research Institute (HMRI) Aging Program (Table 1), and apolipoprotein and lipid concentrations were measured in a representative subset of 80 samples. Concentrations of small HDLs in CSF were positively correlated with better performance in cognitive functions independent of APOE ε4 status, age, sex, and education years. These associations were stronger in persons without cognitive impairment, implicating a role for small HDLs in prevention. Small HDL levels in CSF significantly correlated with greater CSF Aβ42 levels. We observed that small, and not large HDL particles, correlated with CSF lipid levels, potentially representing novel targets for treatment. Moreover, significant correlations of levels of small HDLs as well as specific protein and lipid HDL components were observed between CSF and plasma, supporting common mechanisms that promote their formation across the BCSFB that likely involve apoE.
TABLE 1.
Study participant demographics
| Variable | Mean (range) | |
|---|---|---|
| Age | 76.6 (58-94) | |
| Body Mass Index (BMI) | 26.8 (17.5-42.0) | |
| HDL-C | 62.1 (25.0-111.0) | |
| LDL-C | 107.4 (35.0-275.0) | |
| Total-C | 183.8 (87.0-297.0) | |
| Variable | Count(%) | |
| Sex | Male | 69 (38.3) |
| Female | 111 (61.7) | |
| Ethnicity | Hispanic/Latino | 15 (8.3) |
| Non-Hispanic/Latino | 165 (91.7) | |
| APOE Genotype | ε2/ε3 | 24 (13.3) |
| ε3/ε3 | 102 (56.7) | |
| ε2/ε4 | 4 (2.2) | |
| ε3/ε4 | 42 (23.3) | |
| ε4/ε4 | 7 (3.9) | |
| NA | 1 (0.6) | |
| Clinical Dementia Rating Score (CDR) | 0 | 102 (56.7) |
| 0.5 | 48 (26.7) | |
| >0.5 | 8 (4.4) | |
| NA | 22(12.2) | |
| Domain Impairment | 0 | 101 (56.1) |
| 1 | 31 (17.2) | |
| 2+ | 9 (5.0) | |
| NA | 39(21.7) | |
| Statins | Yes | 59 (32.8) |
| No | 112 (62.2) | |
| NA | 9(5.0) | |
n = 180.
Abbreviation: NA, not available.
The study has some limitations. Lipoprotein particles in plasma and CSF have differences in protein and lipid composition and may not be properly defined by their size. Even within the size range of small HDLs in CSF, several subpopulations exist from poorly lipidated to lipidated HDL particles, and it is still not clear which subpopulation is driving neuroprotective properties. It is important to note that lipid poor apoE protein aggregates are not likely to be in the size range or shape of small HDLs.28,65 Ongoing studies at our labs are examining the structure and composition of these particles, including their effects on neuronal/vascular membranes. More work is also needed to determine if there is exchange of specific small HDL constituents between the brain and the systemic circulation, and to test the possibility that systemically derived HDL components affect brain lipidation and ultimately the risk of neurodegenerative diseases. We acknowledge that the cohort studied is heterogenous and that the results may not be generalizable. We intend to expand the present cohort to better characterize the association between small CSF HDLs and cognitive function in distinct populations that differ by disease stage, ethnicity, sex, APOE genotype, and cognitive outcomes. We recognize that with cross-sectional study designs, it is not possible to infer causality. Here, greater levels of small HDLs were associated with some measures of lower AD risk, captured by the performance on cognition measures and CSF Aβ42. Studies in longitudinal cohorts will assess whether measurements of small HDLs in plasma and/or CSF are predictive of cognitive outcomes. In addition, strategies that can increase small HDLs, for example, treatment with ABCA1 agonists (eg, CS625328) are now being tested in monkeys and humans, and could lead to new therapeutic options for AD prevention.
In conclusion, our current findings, together with the known functions of small HDLs, form the basis for proposing the small HDL hypothesis of AD. Confirmation of the role of small HDLs in the early stages of AD has important implications for AD prevention and management. Measurements of neuroprotective small HDLs or its components in plasma and CSF could serve as biomarkers for guiding future clinical trials of AD prevention, and determination of their exchange between plasma and CSF could aid in the development of novel therapeutic agents.
2 |. CONSOLIDATED STUDY DESIGN AND RESULTS
One hundred eighty men and women with a mean age of 76.6 years and body mass index (BMI) of 26.8 were enrolled in the University of Southern California (USC) Alzheimer Disease Research Center (ADRC) and Huntington Memorial Research Institute (HMRI) Aging Program. Table 1 presents participant demographics. Of the sample population, 111 were female and 69 were male. In addition, 165 of the sample population identified as non-Hispanic/Latino, and 15 identified as Hispanic/Latino. There were 53 (30%) APOE ε4 carriers and 126 (70%) non-ε4 carriers. Among a subset of n = 141 in whom cognition was assessed, 101 had no domain impairment, 31 had one domain impaired, and 9 had two or more domains impaired and were not included in the cognitive analyses.
A significant positive correlation was observed between levels of small HDL particles in plasma and CSF (Figure 2A). There was a trend for a stronger correlation between CSF and plasma small HDLs among non-APOE ε4 carriers (interaction P = .077). In contrast, there was no correlation between large HDL particle numbers in plasma and CSF (Figure S1A). Protein and lipid concentrations were measured in a subset of matched CSF and plasma samples (n = 80, Table S1) that are representative of the whole cohort. Significant correlations were observed between CSF and a number of plasma HDL-associated proteins and lipids (CSF proteins and lipid levels were adjusted to albumin) (Figure 2B and C). Among HDL proteins in CSF, only apoE levels were significantly correlated with small HDL concentrations (Figure 2D). This correlation was not modified by APOE genotype (Figure S1B). Major CSF lipids such as sphingomyelin (SM) and phosphatidylcholine (PC) were significantly associated with CSF small HDL concentrations (Figure 2E and F). This finding is not surprising given that the majority (95%) of HDL particles in CSF are small in size (Figure S2A–E). Stratification by APOE ε4 status had no effect on the statistical significance of these associations. Finally, there was a significant positive correlation between CSF small HDL concentrations and the ability of CSF to activate ABCA1-mediated cholesterol efflux in cells ex vivo (rho = 0.65, P = .0058, n = 16, Figure 2G).
FIGURE 2.
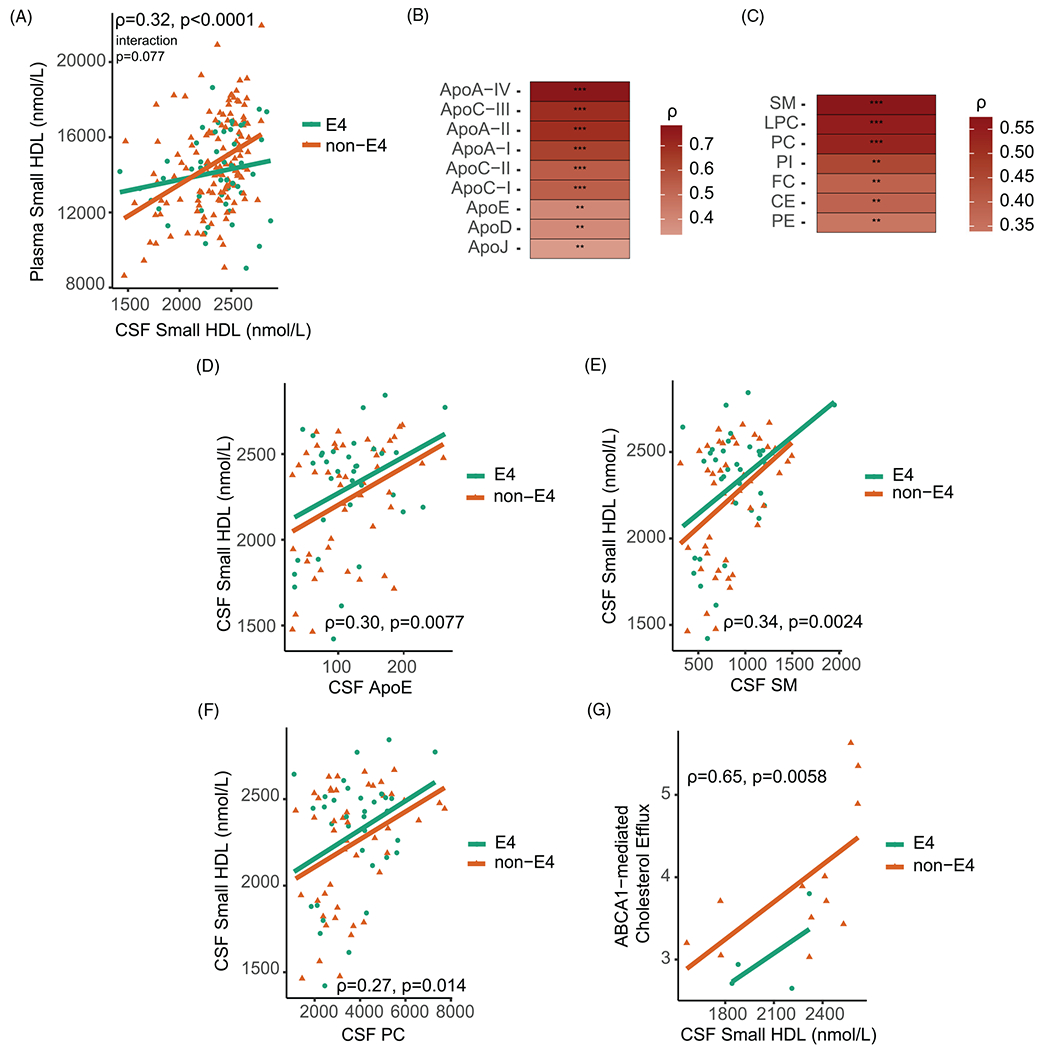
Associations of small HDLs and HDL protein and lipid components in plasma and CSF. (A) Spearman correlations of small HDL concentrations in plasma and CSF. The relationship between levels of CSF and plasma small HDLs was further examined by introducing an interaction term of CSF small HDLs by APOE ε4 status. The interaction between CSF small HDLs and APOE ε4 status on plasma small HDLs was marginally significant (P = .077). Spearman correlations of (B) HDL-associated protein and (C) lipid concentrations between plasma and CSF (n = 80). Strength of Spearman correlation coefficients are represented by variations of the color red as depicted in the legend. Spearman correlations of (D) CSF apoE levels, (E) CSF SMlevels, and (F) CSF PC levels with small HDL concentrations in CSF with the addition of APOE ε4 status as a covariate. (G) Spearman correlation of CSF small HDL particle numbers and ABCA1-mediated cholesterol efflux (n = 16). Relationships were further examined by introducing an interaction term of CSF protein or lipid variables by APOE ε4 status. The interaction between CSF apoE, CSF SM, or CSF PC and APOE ε4 status on CSF small HDL was not significant (P = .95, P = .85, P = .95, respectively). Apolipoprotein and lipid concentrations in the CSF were divided by CSF albumin concentrations and the ratio was multiplied by 1000. Abbreviations: FC, free cholesterol; CE, cholesteryl ester; PC, phosphatidylcholine; SM, sphingomyelin; PE, phosphatidylethanolamine; LPC, lysophosphatidylcholine; PI, phosphatidylinositol. ** P-value < .01, *** P-value < .001
To understand the relation with AD risk, protein, and lipid distributions along with HDL concentrations were analyzed in two groups: CSF Aβ42 ≤190 pg/mL (Aβ42+) or CSF Aβ42 >190 pg/mL (Aβ42−) groups. This CSF Aβ42 cut-off distinguished higher vs lower AD risk, respectively.71 In both CSF and plasma, there were greater levels of small HDLs in the Aβ42− group (Figure 3A and B, respectively). Surprisingly, APOE ε4 carriers had greater levels of CSF small HDLs in the Aβ42− group compared with the non-carriers (Figure 3A). This finding suggests that conditions that promote greater levels of brain small HDLs in individuals carrying the APOE ε4 allele may protect against the development of AD pathology. APOE ε4 carriers in the Aβ42+ group had greater concentrations of large CSF HDLs, whereas there was no difference for large plasma HDLs (Figure 3C and Figure S3A, respectively). APOE allele status had no significant association with levels of either small or large HDLs in plasma or CSF (Figure S3B–E). The association of APOE ε4 with larger sized particles in the Aβ42+ group is in agreement with a past report identifying greater affinity of apoE4 vs apoE3 and apoE2 to large lipid rich particles in CSF recovered in the fraction with a density of <1.006 kg/L.72 In plasma, apoE4 has a greater affinity than apoE3 or apoE2 to very-low-density lipoprotein (VLDL).73 This might be explained by the absence of cysteine-arginine interchanges at residues 112 and 158 of apoE3 and apoE2, respectively, that result in the formation apoE3-apoA-II and apoE2-apoA-II complexes.73 Lack of formation of these complexes may allow apoE4 the freedom to bind larger particles, and to promote faster clearance of apoE4-bound lipids in plasma as opposed to apoE2.74 Whether a similar mechanism operates in the brain is yet to be determined.
FIGURE 3.
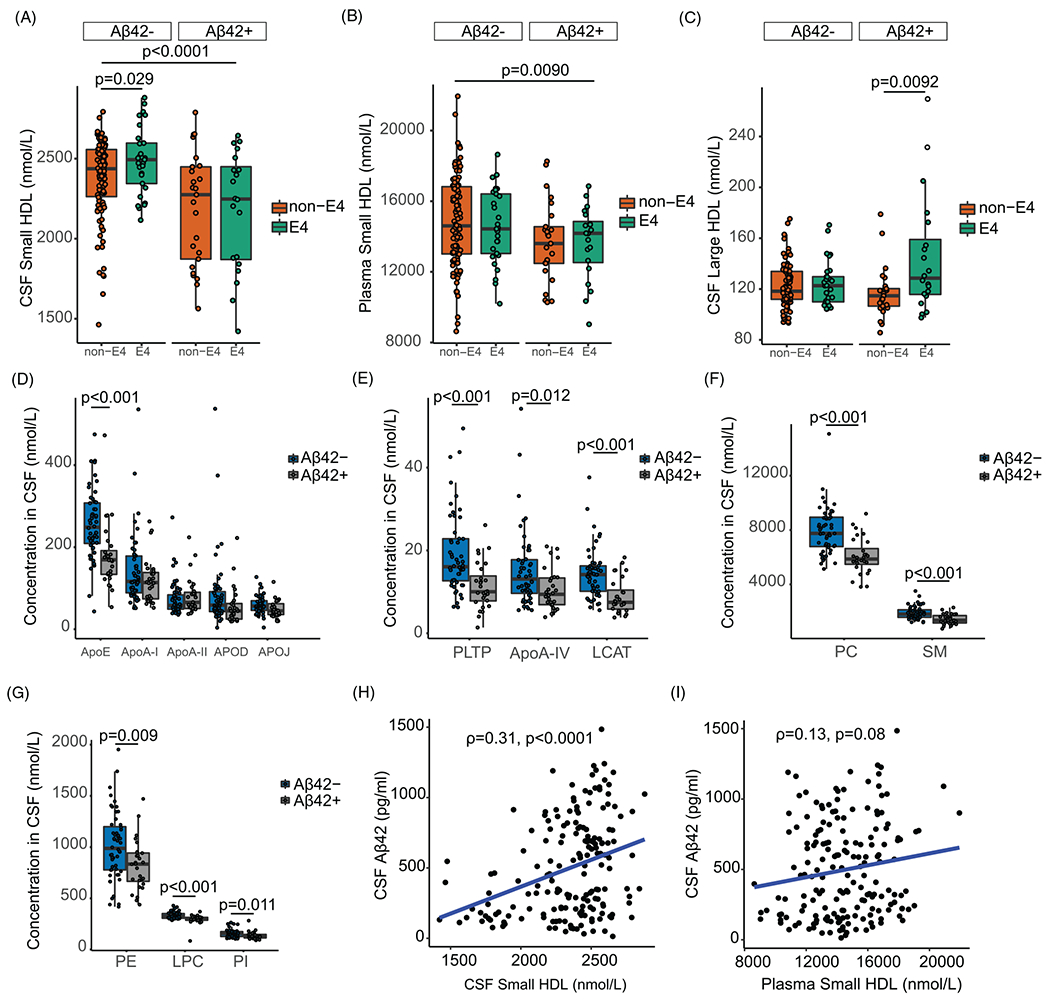
Distributions of HDL particles, CSF apoE, and lipid concentrations by Aβ, APOE, and CDR groups. Distribution of small HDLs in (A) CSF and (B) plasma by APOE status and Aβ groups (n = 180). Distribution (C) of large HDLs in CSF by Aβ groups (n = 180). Distribution of (D-E) protein and (F-G) lipid concentrations in CSF by Aβ groups (n = 80). (H) Correlation between levels of CSF Aβ42 and CSF small HDL. (I) Correlation between levels of CSF Aβ42 and plasma small HDLs. Participants were stratified as either Aβ42 positive (Aβ42+, <190 pg/mL) or Aβ42 negative (Aβ−, >190 pg/mL). Raw apolipoprotein and lipid concentrations in CSF were used. Relationships were assessed using linear regression models or Spearman correlation coefficient. Abbreviations: PLTP, phospholipid transfer protein; LCAT, lecithin cholesterol acyl transferase; PC, phosphatidylcholine; SM, sphingomyelin; PE, phosphatidylethanolamine; LPC, lysophosphatidylcholine; PI, phosphatidylinositol
Among CSF proteins, levels of apoE, apolipoprotein A-IV (apoA-IV), lecithin-cholesterol acyltransferase (LCAT), and phospholipid transfer protein (PLTP) were all higher in the Aβ42− group (Figure 3D and E). Among CSF lipids, there were greater concentrations of phosphatidylcholine (PC), sphingomyelin (SM), and lysophatidylcholine (LPC) in the Aβ42− group compared to the Aβ42+ group (Figure 3F and G). These findings underscore a possible role for proteins involved in the lipidation of small HDL particles in limiting cerebral amyloid accumulation. The corresponding proteins and lipids in plasma did not differ by Aβ42 groups (Figure S4A–E).
To control for BCSFB penetration, CSF/plasma ratios of albumin, small HDL, and HDL-associated protein variables were compared, and depicted in Figure S5. In this subset (Table S1), only one participant had a CSF/albumin ratio >10, suggestive of a compromise in BBB integrity.75 The correlation of small HDLs between CSF and plasma was not explained by the CSF/plasma albumin ratio. This finding resembled that for CSF/plasma apoE, which also was correlated with CSF/plasma small HDLs (Figure S5A–C). In contrast, there were strong positive correlations of CSF/plasma apoA-I and apoC-III with CSF/plasma albumin, supporting that apoA-I and apoC-III are transported from the blood into the CSF by similar mechanisms (Figure S5D and E, respectively). The correlation of small HDLs between plasma and CSF with apoE and not with albumin supports that their formation across the BCSFB shares common mechanisms and likely involves apoE.
In CSF, greater levels of small HDLs were associated with significantly greater CSF Aβ42 concentrations (Figure 3H). This association remained significant after adjustment for age, sex, APOE status, and education years (Table 2). Levels of small HDLs in plasma were weakly associated with CSF Aβ42 levels (Figure 3I). There was no association between large plasma HDLs and CSF Aβ42 levels (P = .30, Figure S6A). Similarly, there were no significant correlations between plasma HDL-C and CSF Aβ42 levels (P = .22, Figure S6B). The inclusion of statin use as a potential cofounder did not change the association between levels of small HDLs and CSF Aβ42 (Table S2).
TABLE 2.
Multivariate association of CSF small HDLs with CSF Aβ42 and memory z scores
| CSF Aβ42, Estimate (SE), n = 180 |
Memory z score, Estimate (SE), n = 132 |
|||||
|---|---|---|---|---|---|---|
| Covariates | Model 1a | Model 2b | Model 3c | Model 1 | Model 2 | Model 3 |
| Small HDL | 0.382 (0.08)* | 0.366 (0.09)* | 0.584 (0.12)* | 0.823 (0.30)** | 0.796 (0.33)*** | 1.158 (0.35)** |
|
| ||||||
| Age | NA | 0.508 (3.26) | 6.240 (3.70) | NA | 0.033 (0.01)** | 0.014 (0.01) |
|
| ||||||
| Sex | 7.315 (52.73) | 34.311 (53.67) | 0.269 (0.18) | 0.196 (0.16) | ||
|
| ||||||
| APOE | −163.558 (53.25)** | −244.248 (58.09)* | −0.369 (0.19) | −0.293 (0.18) | ||
|
| ||||||
| Education years | −14.700 (8.91) | −11.713 (9.24) | 0.064 (0.03)*** | 0.051 (0.03) | ||
|
| ||||||
| Domain impairment | NA | 563.714 (501.79) | NA | 3.731 (1.52)*** | ||
|
| ||||||
| Small HDL by Domain Impairment Interaction | −0.274 (0.21) | −2.023 (0.65)** | ||||
Model 1 is a univariate model with CSF Aβ42 or memory as the dependent variable and CSF small HDL particle concentrations measured by ion mobility (IM) as the independent variable.
Model 2 is a multivariate model with CSF Aβ42 or memory as the dependent variable and CSF small HDL particle concentration as the independent variable, with age, sex, education years, and APOE as covariates.
Model 3 is a multivariate model where an interaction term of small HDLs by Domain Impairment was additionally regressed.
SE: Standard Error
P < .0001.
P < .01.
P < .05.
To investigate the relationship of HDL numbers in CSF with cognition, bivariate correlations were run between levels of HDL subspecies, Clinical Dementia Rating (CDR) scores (n = 150, Table S4), and cognitive domain z-scores (n = 132, Table S5). In assessing the relationship by CSF Aβ42 groups, individuals in the CDR = 0 group displayed greater levels of CSF small HDLs in the Aβ42− than the Aβ42+ group (Figure 4A). There was a positive correlation between levels of small HDLs in CSF and better performance on neuropsychological testing for the memory domain, global cognition, and attention/executive functioning domain (Figure 4B–D). However, after adjusting for age, sex, and APOE genotype in separate linear regression models with each cognitive domain as a dependent variable, only the association of small HDLs in CSF with the memory domain remained significant (Table 2). Sex did not affect the association of small HDLs with measures of cognition (Figure S7), and this association was not affected by statin use (Table S3). To determine whether the relationship of CSF small HDL concentrations with memory differed across domain impairment subgroups, we used a multivariate linear regression model including the interaction term of small HDLs by subgroup, and covarying for age, sex, education, and APOE status. The interaction was significant (P = .001, Table 2, Figure 4B). Follow-up bivariate analyses revealed a significant positive correlation between levels of CSF small HDLs and memory scores in participants with no domain impairment (n = 101, rho = 0.34, P = .005) but not in participants with one cognitive domain impaired (n = 31, rho = −0.30, P = .09). Neither large CSF HDLs nor small and large plasma HDL levels was significantly correlated with measures of cognition. In addition, no correlation was found between plasma HDL-C and measures of cognition. These findings highlight possible neuroprotective associations of small HDLs in persons without cognitive impairment.
FIGURE 4.
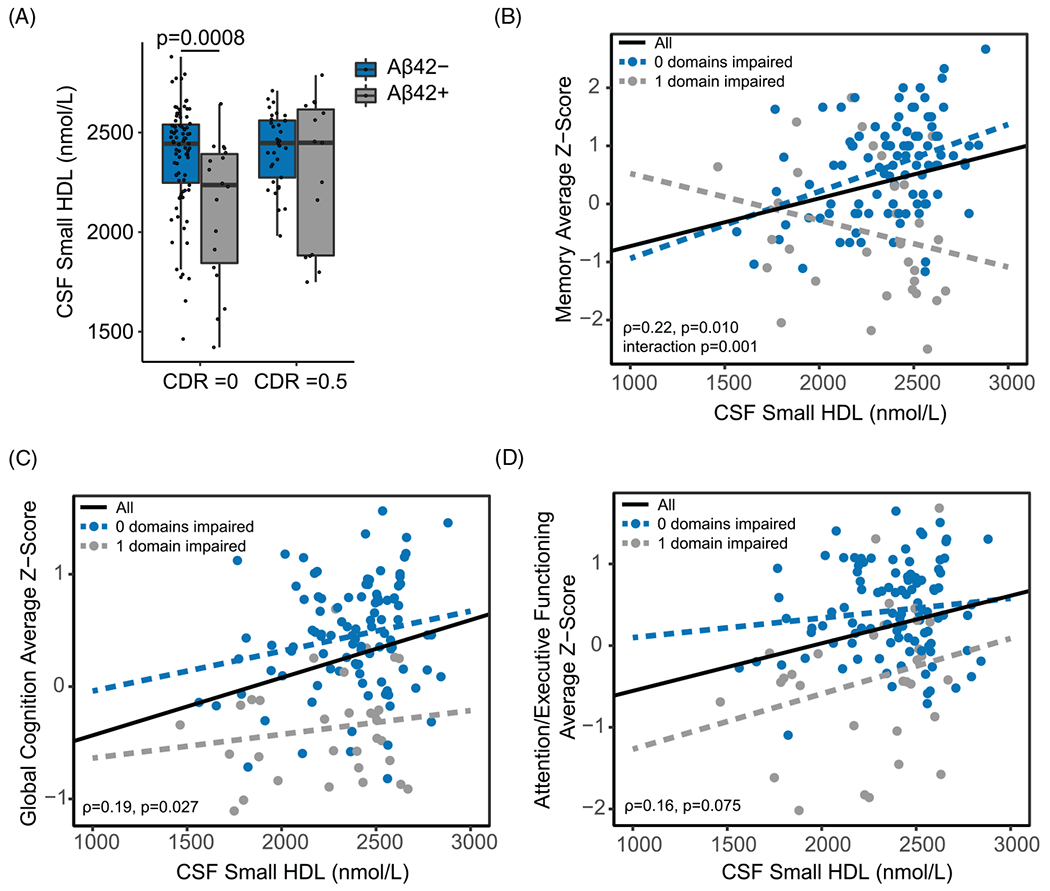
Relationship between CSF small HDL particle numbers and measures of cognition. (A) Distribution of small HDLs in CSF by Aβ and CDR subgroups (CDR = 0, n = 102, and CDR = 0.5, n = 48). (B) Relationship between CSF small HDLs and memory z-scores in the zero-domain impaired group (blue line; n = 101) and the one domain impaired group (gray line; n = 31). Associations of CSF small HDLs with (C) global cognition and (D) attention/executive functioning average z-scores. Participants were stratified as either Aβ42 positive (Aβ42+, ≤190 pg/mL) or Aβ42 negative (Aβ−, >190 pg/mL). Correlations were assessed using the Spearman correlation coefficient. Distributions of small CSF HDLs by Aβ and CDR subgroups were assessed using linear regression models. The relationship between CSF small HDLs and memory, global cognition, or attention/executive functioning was examined by introducing an interaction term of small HDLs by cognitive domain impairment group. Abbreviation: CDR, Clinical Dementia Rating
3 |. DETAILED METHODS
3.1 |. Clinical sample description
Participants were 180 healthy men and women age ≥60 who were enrolled in the University of Southern California (USC) Alzheimer Disease Research Center (ADRC) and Huntington Memorial Research Institute (HMRI) Aging Program. Inclusion criteria were enrichment with cardiovascular factors such as hypertension (blood pressure [BP] ≥140/90 mm Hg) and dyslipidemia (HDL-C <40 mg/mL for men and HDL-C <50 mg/mL for women). Exclusion criteria included no previous history of stroke and not receiving insulin. The study and procedures were approved by the institutional review board (USC IRB: HS-16-00888). All participants provided informed consent prior to enrollment in the study.
3.2 |. Cognitive assessment
To assess cognitive status, we utilized both the CDR and the Neuropsychological Evaluation from Uniform Data Set (Versions 2 or 3) of the National Alzheimer’s Coordinating Center. The Neuropsychological Evaluation covered multiple cognitive domain considerations. Information regarding specific tests used in each study is provided in Table S3. Of the 180 participants included in this study, 141 underwent a full neuropsychological battery. For some analyses, participants were divided into those who had one impaired cognitive domain vs no impaired cognitive domains (n = 132, Table S5). Methods for determining cognitive domain impairment were roughly modeled after a previously published study.76 Domain impairment was determined using well-accepted criteria,77 and was defined as one or more test scores ≥1.5 standard deviations (SD) below normative means within the domain of memory, language, and/or attention/executive functioning. Having three or more impaired test scores across the eight tests was additionally classified as a domain impairment. Participants with two or more domains impaired were excluded from further analyses (n = 9), for a total of 132 participants included in cognitive analyses. (Additional details can be found in the Supplementary Material.)
3.3 |. Venipuncture and lumbar puncture
Whole blood was collected in EDTA tubes, and plasma was separated by standard low speed centrifugation and stored at −80°C. Lumbar puncture was performed by a neurologist or approved study staff. The spinal needle was inserted into the subarachnoid space at the L3/4 or L4/5 interspace using sterile technique and local anesthesia. About 22 mL of CSF was collected into sterile polypropylene tubes, processed, and stored in cryoprotective vials at −80°C until analysis.
3.4 |. APOE genotyping
DNA was isolated from leukocytes in the buffy coat layer of each participant’s blood sample. Polymerase chain reaction (PCR) amplification was conducted for three separate reactions, with each reaction designated by forward and reverse primers specific to one of the three common allelic variants of the APOE gene. Agarose gel electrophoresis was then performed to analyze PCR products and determine specific APOE genotype.
3.5 |. CSF Aβ42 measurement
CSF levels of Aβ42 were measured using Meso Scale Discovery (MSD)78 multiplex assay. The CSF Aβ42 cutoff level of 190 pg/mL was applied as reported previously for the MSD Aβ peptide assay.71
3.6 |. HDL particle measurements using ion mobility (IM)
Concentrations of HDL particles were measured by IM after treatment with dextran sulfate to remove non-lipid-bound proteins such as albumin from 30 μL of plasma or CSF as described previously.79 This procedure does not completely remove all of the albumin. (Additional details can be found in the Supplementary Material.) Representative IM particle concentration profiles in plasma and CSF are shown in Figure S3A. For the analyses described here, particles in the HDL size range were classified as small (7.0-10.5 nm) and large (10.5-14.5 nm) HDL. The majority of HDL particles in both CSF and plasma were found in the small size range, with a greater proportion of small to large HDLs present in CSF compared to plasma (Figure S3B).
3.7 |. Protein and lipid quantification in matched CSF and plasma samples by LC-MS/MS
Corresponding CSF and plasma samples from a subset of 80 individuals were employed to measure the major plasma protein and lipid class concentrations as described.80–82 CSF samples were analyzed without dilution while the matched plasma samples were diluted 100-fold. To minimize the effect of any potential blood contamination or varying CSF tap volumes on proteomic and lipidomic data quantification, CSF variables were adjusted (normalized) to CSF albumin concentration as described,83,84 and the ratio was multiplied by 1000.
3.8 |. Cell culture and cholesterol efflux assays
ABCA1-mediated cholesterol efflux facilitated by CSF was measured as described previously.32,85 BHK cells were plated at 4000 cells per well in high-glucose DMEM, 10% FBS in 96-well plates. Human CSF was used as the acceptor and cholesterol efflux was assessed using 15 μL CSF per well in triplicates.
3.9 |. Statistical analysis
Mean (range) and frequency were computed for demographic data. Spearman correlation was used to test the relationship between plasma and CSF protein concentrations after normalizing to plasma and CSF albumin concentrations, respectively. In addition, correlations were performed between HDL particle concentrations, CSF Aβ42, and CSF protein and lipid ratios. Participants were stratified based on CSF analysis as either Aβ42 positive (Aβ42+, <190 pg/mL) or Aβ42 negative (Aβ−, >190 pg/mL) based on accepted cutoff for AD risk.76 In the cognition association analysis, individuals with two impaired domains were excluded because the focus was on AD risk, and once dementia develops, interpreting the results becomes more complicated. If participants with dementia or mild cognitive impairment were included, then any observed associations might be driven by those extreme cases, or the result of dementia. We have previously used this approach.76,86 R coefficients were obtained by using Pearson if variables were normally distributed and Spearman if variables were not normally distributed. Welch two-sample t-test or linear regression models were used to compare two independent groups. Significant associations were further investigated using multiple linear regression analyses, with adjustment of potential cofounders including APOE ε4 status. P-value < .05 was defined as significant. Statistical analyses were performed using RStudio, version 1.2.5033.
Cognitive data analysis:
Spearman correlations were conducted between small and large HDLs and the three cognitive function considerations (memory, attention/executive functioning, and global cognition). Significant associations were further investigated using multiple linear regression analyses, with adjustment of potential cofounders including age at the time of sample collection, sex, education, and APOE ε4 status. The relationship between CSF small HDLs and memory was further examined by introducing an interaction term of CSF small HDL by cognitive impairment group (zero vs one domain impaired). Statistical analyses were performed using SAS software, version 9.4.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic Review: Lipid metabolism genes (apolipoprotein E [APOE], Apolipoprotein J [ApoJ], ATP-binding cassette 7 [ABCA7]) involved in forming high-density lipoprotein (HDL) particles associate with AD risk. A meta-analysis of 100 primary studies from 1986 to 2000 found non-significant associations between levels of HDL cholesterol (HDL-C) and AD risk. HDL-C measurements do not reflect the cholesterol transport function of HDL. Small HDL particles have neuroprotective properties, but their association with cognition is not known.
Interpretation: Concentrations of small HDL particles in cerebrospinal fluid (CSF) were positively associated with better performance in cognitive function before the onset of dementia, independent of age, sex, education, and APOE genotype.
Future Directions: Confirmation of the association of small HDL particles with cognitive outcomes in larger cohorts can lead to a novel AD biomarker and potential therapeutic targets for AD prevention.
ACKNOWLEDGEMENTS
The authors thank Dr. Caleb Finch for critically reviewing the manuscript and Thomas Urich for proof reading the final version of the manuscript.
HNY is supported by R21AG056518, R01AG055770, R01AG054434, R01AG067063 from the National Institute on Aging (NIA), and from the Batey Foundation (Ion Mobility Instrumentation). SDH was supported by R01AG055430 and RF1AG068166. XW was supported by R01AG033078, RF1AG054068, R01ES025888, P50AG005142, and P30AG066530. MGH was supported by the L.K. Whittier Foundation and HMRI Aging Research Program: RO1AG054434, and R01AG055770, and P01AG052350. RMK was supported by P50GM115318, the Dairy Research Institute, Quest Diagnostics, and Saliogen. GW received support from T32 AG000037. ZK received support from the Centers for Disease Control and Prevention allocated for Cardiovascular Biomarker Research. LSS is supported by P30 AG066530, grants/contracts from Eisai, Eli Lilly, Roche/Genentech, Biogen, Biohaven, Novartis, and Washington University/NIA DIAN-TU paid to the institution, and is also supported by the Della Martin Foundation. BVZ receives support from P01AG052350, P50AG005142, R01NS034467, R01AG039452, R01AG023084, Alzheimer’s Association strategic 509279 grant, Cure Alzheimer’s Fund, the Foundation Leducq Transatlantic Network of Excellence for the Study of Perivascular Spaces in Small Vessel Disease reference no.16 CVD 05, and Open Philanthropy. HCC receives support from P30AG066530, P01G052350, AG058162, R01AG055770, R01AG054434, ES025888, RF1AG054068, R56AG069130, and R01AG072490. Funders had no role in study design, data collection, data analysis, interpretation, or writing of the report.
CONFLICTS OF INTEREST
Hussein N. Yassine (HNY) is the 2019-2021 co-chair of the NMD PIA of the Alzheimer’s Association (non-paid). Michael G. Harrington (MGH) received honorarium as guest speaker at annual Headache Cooperative of the Pacific and NIH ZRG1 BDCN-W Study Section Meetings. Lon S. Schneider (LSS) receives consulting fees from Abbott, AC Immune, Avraham, Ltd, Boehringer Ingelheim, Cognition Therapeutics, Cortexyme, Eisai, FujiFilm, Immunobrain Checkpoint Ltd, Neurally Inc, Neurim Ltd, Neuronix Ltd, Novo-Nordisk, Samus, Takeda, and vTv. Helena C. Chui (HCC) is on the external advisory board to Oregon Health Science University Alzheimer’s Disease Research Center, and on the Alzheimer Los Angeles Board member and Chair of Medical Scientific Advisory Council (both non-paid). Timothy S. Collier (TSC) is an employee of Quest Diagnostics and has stocks in Quest Diagnostics. S. Duke Han (SDH) receives consulting fees from Rush University Medical Center and participates in the AARP Global Council on Brain Health. Ronald M. Krauss (RMK) reports receiving a grant from Quest Diagnostics, royalties from Lawrence Berkeley National Laboratory for patents for ion mobility methodology, and consulting fees from Saliogen Therapeutics and Merck. All the other authors declare that they have no competing interests.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
REFERENCES
- 1.Farmer BC, Walsh AE, Kluemper JC, Johnson LA. Lipid droplets in neurodegenerative disorders. Front Neurosci. 2020;14(742). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8(6):429–431. [DOI] [PubMed] [Google Scholar]
- 3.Whitmer RA, Sidney S, Selby J, Johnston SC, Yaffe K. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology. 2005;64(2):277–281. [DOI] [PubMed] [Google Scholar]
- 4.Kivipelto M, Helkala EL, Laakso MP, et al. Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ. 2001;322(7300):1447–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merched A, Xia Y, Visvikis S, Serot JM, Siest G. Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease. Neurobiol Aging. 2000;21(1):27–30. [DOI] [PubMed] [Google Scholar]
- 6.Bates KA, Sohrabi HR, Rainey-Smith SR, et al. Serum high-density lipoprotein is associated with better cognitive function in a cross-sectional study of aging women. Int J Neurosci. 2017;127(3):243–252. [DOI] [PubMed] [Google Scholar]
- 7.Sáiz-Vazquez O, Puente-Martínez A, Ubillos-Landa S, Pacheco-Bonrostro J, Santabárbara J. Cholesterol and Alzheimer’s disease risk: a meta-meta-analysis. Brain Sci. 2020;10(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wood WG, Li L, Müller WE, Eckert GP. Cholesterol as a causative factor in Alzheimer’s disease: a debatable hypothesis. J Neurochem. 2014;129(4):559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaisar T, Pennathur S, Green PS, et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117(3):746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Valkenburgh J, Meuret C, Martinez AE, et al. Understanding the exchange of systemic HDL particles into the brain and vascular cells has diagnostic and therapeutic implications for neurodegenerative diseases. Front Physiol. 2021;12:700847–700847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asztalos BF, de la Llera-Moya M, Dallal GE, Horvath KV, Schaefer EJ, Rothblat GH. Differential effects of HDL subpopulations on cellular ABCA1-and SR-BI-mediated cholesterol efflux. J Lipid Res. 2005;46(10):2246–2253. [DOI] [PubMed] [Google Scholar]
- 12.de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30(4):796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambert J-C, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beecham GW, Hamilton K, Naj AC, et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 2014;10(9):e1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reynolds CA, Hong MG, Eriksson UK, et al. A survey of ABCA1 sequence variation confirms association with dementia. Hum Mutat. 2009;30(9):1348–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nordestgaard LT, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement. 2015;11(12):1430–1438. [DOI] [PubMed] [Google Scholar]
- 18.Blanche PJ,Gong EL, Forte TM, Nichols AV. Characterization of human high-density lipoproteins by gradient gel electrophoresis. Biochim Biophys Acta. 1981;665(3):408–419. [DOI] [PubMed] [Google Scholar]
- 19.Rosenson RS, Brewer HB Jr., Chapman MJ,et al. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin Chem. 2011;57(3):392–410. [DOI] [PubMed] [Google Scholar]
- 20.Koch S, Donarski N, Goetze K, et al. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res. 2001;42(7):1143–1151. [PubMed] [Google Scholar]
- 21.Guyton JR, Miller SE, Martin ME, Khan WA, Roses AD, Strittmatter WJ. Novel large apolipoprotein E-containing lipoproteins of density 1.006-1.060 g/ml in human cerebrospinal fluid. J Neurochem. 1998;70(3):1235–1240. [DOI] [PubMed] [Google Scholar]
- 22.Shi Y, Manis M, Long J, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. 2019;216(11):2546–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henningfield CM, Arreola MA, Soni N, Spangenberg EE, Green KN. Microglia-specific ApoE knock-out does not alter Alzheimer’s disease plaque pathogenesis or gene expression. Glia. 2022;70(2):287–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wahrle SE, Jiang H, Parsadanian M, et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279(39):40987–40993. [DOI] [PubMed] [Google Scholar]
- 25.Nichols AV, Blanche PJ, Gong EL, Shore VG, Forte TM. Molecular pathways in the transformation of model discoidal lipoprotein complexes induced by lecithin:cholesterol acyltransferase. Biochim Biophys Acta. 1985;834(3):285–300. [DOI] [PubMed] [Google Scholar]
- 26.Vaughan AM, Oram JF. ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J Lipid Res. 2006;47(11):2433–2443. [DOI] [PubMed] [Google Scholar]
- 27.Lee CYD, Tse W, Smith JD, Landreth GE. Apolipoprotein E promotes β-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012;287(3):2032–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rawat V, Wang S, Sima J, et al. ApoE4 alters ABCA1 membrane trafficking in astrocytes. J Neurosci. 2019;39(48):9611–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280. [DOI] [PubMed] [Google Scholar]
- 30.Hirsch-Reinshagen V, Maia LF, Burgess BL, et al. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J Biol Chem. 2005;280(52):43243–43256. [DOI] [PubMed] [Google Scholar]
- 31.Wahrle SE, Jiang H, Parsadanian M, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118(2):671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yassine HN, Feng Q, Chiang J, et al. ABCA1-mediated cholesterol efflux capacity to cerebrospinal fluid is reduced in patients with mild cognitive impairment and Alzheimer’s disease. J Am Heart Assoc. 2016;5(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mauch DH, Nägler K, Schumacher S, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294(5545):1354. [DOI] [PubMed] [Google Scholar]
- 34.Posse de Chaves EI, Vance DE, Campenot RB, Kiss RS, Vance JE. Uptake of lipoproteins for axonal growth of sympathetic neurons *. J Biol Chem. 2000;275(26):19883–19890. [DOI] [PubMed] [Google Scholar]
- 35.Jasmin SB, Pearson V, Lalonde D, Domenger D, Théroux L, Poirier J. Differential regulation of ABCA1 and ABCG1 gene expressions in the remodeling mouse hippocampus after entorhinal cortex lesion and liver-X receptor agonist treatment. Brain Res. 2014;1562:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laffitte BA, Repa JJ, Joseph SB, et al. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc Natl Acad Sci U S A. 2001;98(2):507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eckert GP, Cairns NJ, Maras A, Gattaz WF, Müller WE. Cholesterol modulates the membrane- disordering effects of beta-amyloid peptides in the hippocampus: specific changes in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2000;11(4):181–186. [DOI] [PubMed] [Google Scholar]
- 38.Wang H, Kulas JA, Wang C, Holtzman DM, Ferris HA, Hansen SB. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc Natl Acad Sci. 2021;118(33):e2102191118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomioka M, Toda Y, Mañucat NB, et al. Lysophosphatidylcholine export by human ABCA7. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2017;1862(7):658–665. [DOI] [PubMed] [Google Scholar]
- 40.Aikawa T, Ren Y, Yamazaki Y, et al. ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc Natl Acad Sci. 2019;116(47):23790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bell RD, Winkler EA, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montagne A, Barnes SR, Sweeney MD, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Bruijn RFAG, Ikram MA. Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Med. 2014;12:130–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robert J, Button EB, Martin EM, et al. Cerebrovascular amyloid angiopathy in bioengineered vessels is reduced by high-density lipoprotein particles enriched in Apolipoprotein E. Mol Neurodegener. 2020;15(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brites F, Martin M,Guillas I, Kontush A. Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clin. 2017;8:66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kontush A, Chantepie S, Chapman MJ. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler Thromb Vasc Biol. 2003;23(10):1881–1888. [DOI] [PubMed] [Google Scholar]
- 48.Svensson T, Sawada N, Mimura M, Nozaki S, Shikimoto R, Tsugane S. The association between midlife serum high-density lipoprotein and mild cognitive impairment and dementia after 19 years of follow-up. Transl Psychiatry. 2019;9(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cramer PE, Cirrito JR, Wesson DW, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335(6075):1503–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skerrett R, Pellegrino MP, Casali BT, Taraboanta L, Landreth GE. Combined Liver X Receptor/Peroxisome Proliferator-activated Receptor γ agonist treatment reduces Amyloid β levels and improves behavior in Amyloid Precursor Protein/Presenilin 1 Mice. J Biol Chem. 2015;290(35):21591–21602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bryleva EY, Rogers MA, Chang CC, et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci U S A. 2010;107(7):3081–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanders AE, Wang C, Katz M, et al. Association of a functional polymorphism in the cholesteryl ester transfer protein (CETP) gene with memory decline and incidence of dementia. JAMA. 2010;303(2):150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nicholls SJ, Ruotolo G, Brewer HB, et al. Cholesterol Efflux Capacity and Pre-Beta-1 HDL Concentrations Are Increased in Dyslipidemic Patients Treated With Evacetrapib. J Am Coll Cardiol. 2015;66(20):2201–2210. [DOI] [PubMed] [Google Scholar]
- 54.Hafiane A, Bielicki JK, Johansson JO, Genest J. Novel Apo E-Derived ABCA1 Agonist Peptide (CS-6253) Promotes Reverse Cholesterol Transport and Induces Formation of prebeta-1HDL In Vitro. PLoS One. 2015;10(7):e0131997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reiman EM, Arboleda-Velasquez JF, Quiroz YT, et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11(1):667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu J, Liu C-C, Chen X-F, Zhang Y-w, Xu H, Bu G. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Aβ metabolism in apoE4-targeted replacement mice. Mol Neurodegener. 2015;10(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao L, Gottesdiener AJ, Parmar M, et al. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol Aging. 2016;44:159–172. [DOI] [PubMed] [Google Scholar]
- 58.Demeester N, Castro G, Desrumaux C, et al. Characterization and functional studies of lipoproteins, lipid transfer proteins, and lecithin: cholesterol acyltransferase in CSF of normal individuals and patients with Alzheimer’s disease. J Lipid Res. 2000;41(6):963–974. [PubMed] [Google Scholar]
- 59.Montine KS, Bassett CN, Ou JJ, Markesbery WR, Swift LL, Montine TJ. Apolipoprotein E allelic influence on human cerebrospinal fluid apolipoproteins. J Lipid Res. 1998;39(12):2443–2451. [PubMed] [Google Scholar]
- 60.Lenich C, Brecher P, Makrides S, Chobanian A, Zannis VI. Apolipoprotein gene expression in the rabbit: abundance, size, and distribution of apolipoprotein mRNA species in different tissues. J Lipid Res. 1988;29(6):755–764. [PubMed] [Google Scholar]
- 61.Dal Magro R, Simonelli S, Cox A, et al. The Extent of Human Apolipoprotein A-I Lipidation Strongly Affects the β-Amyloid Efflux Across the Blood-Brain Barrier in vitro. Front Neurosci. 2019;13(419). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin-Nizard F, Meresse S, Cecchelli R, Fruchart JC, Delbart C. Interactions of high-density lipoprotein 3 with brain capillary endothelial cells. Biochim Biophys Acta. 1989;1005(3):201–208. [DOI] [PubMed] [Google Scholar]
- 63.Stukas S, Robert J, Lee M, et al. Intravenously injected human apolipoprotein A-I rapidly enters the central nervous system via the choroid plexus. J Am Heart Assoc. 2014;3(6):e001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koch M, Furtado JD, Falk K, Leypoldt F, Mukamal KJ, Jensen MK. Apolipoproteins and their subspecies in human cerebrospinal fluid and plasma. Alzheimers Dement (Amst). 2017;6(1):182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hubin E, Verghese PB, van Nuland N, Broersen K. Apolipoprotein E associated with reconstituted high-density lipoprotein-like particles is protected from aggregation. FEBS Lett. 2019;593(11):1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu Y, Meuret C, Go S, Yassine H, Nedelkov D. Simple and fast assay for apolipoprotein E phenotyping and glycotyping: discovering isoform-specific glycosylation in plasma and cerebrospinal fluid. J Alzheimers Dis. 2020;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Linton MF, Gish R, Hubl ST, et al. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88(1):270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu M, Kuhel DG, Shen L, Hui DY, Woods SC. Apolipoprotein E does not cross the blood-cerebrospinal fluid barrier, as revealed by an improved technique for sampling CSF from mice. Am J Physiol Regul Integr Comp Physiol. 2012;303(9):R903–R908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fujiyoshi M, Ohtsuki S, Hori S, Tachikawa M, Terasaki T. 24S-hydroxycholesterol induces cholesterol release from choroid plexus epithelial cells in an apical- and apoE isoform-dependent manner concomitantly with the induction of ABCA1 and ABCG1 expression. J Neurochem. 2007;100(4):968–978. [DOI] [PubMed] [Google Scholar]
- 70.Cavelier C, Lorenzi I, Rohrer L, von Eckardstein A. Lipid efflux by the ATP-binding cassette transporters ABCA1 and ABCG1. Biochim Biophys Acta. 2006;1761(7):655–666. [DOI] [PubMed] [Google Scholar]
- 71.Pan C, Korff A, Galasko D, et al. Diagnostic Values of Cerebrospinal Fluid T-Tau and Aβ 42 using Meso Scale Discovery Assays for Alzheimer’s Disease. J Alzheimers Dis. 2015;45:709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamauchi K, Tozuka M, Hidaka H, Hidaka E, Kondo Y, Katsuyama T. Characterization of Apolipoprotein E-containing Lipoproteins in Cerebrospinal Fluid: Effect of Phenotype on the Distribution of Apolipoprotein E. Clin Chem. 1999;45(9):1431–1438. [PubMed] [Google Scholar]
- 73.Weisgraber KH. Apolipoprotein E distribution among human plasma lipoproteins: role of the cysteine-arginine interchange at residue 112. J Lipid Res. 1990;31(8):1503–1511. [PubMed] [Google Scholar]
- 74.Orth M, Wahl S, Hanisch M, Friedrich I, Wieland H, Luley C. Clearance of postprandial lipoproteins in normolipemics: role of the apolipoprotein E phenotype. Biochim Biophys Acta. 1996;1303(1):22–30. [DOI] [PubMed] [Google Scholar]
- 75.Skillbäck T, Delsing L, Synnergren J, et al. CSF/serum albumin ratio in dementias: a cross-sectional study on 1861 patients. Neurobiol Aging. 2017;59:1–9. [DOI] [PubMed] [Google Scholar]
- 76.Nation DA, Sweeney MD, Montagne A, et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25(2):270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303–308. [DOI] [PubMed] [Google Scholar]
- 78.Nair SB, Leung HY, Ince P, Ramsden RT, Wilson JA. Fibroblast growth factor receptor expression investibular schwannoma. Clin Otolaryngol Allied Sci. 2000;25(6):570–576. [DOI] [PubMed] [Google Scholar]
- 79.Mora S, Caulfield MP, Wohlgemuth J, et al. Atherogenic lipoprotein subfractions determined by ion mobility and first cardiovascular events after random allocation to high-intensity statin or placebo: the JUPITER trial. Circulation. 2015:CIRCULATIONAHA. 115.016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Toth CA, Kuklenyik Z, Jones JI, et al. On-column trypsin digestion coupled with LC-MS/MS for quantification of apolipoproteins. J Proteomics. 2017;150:258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gardner MS, Kuklenyik Z, Lehtikoski A, et al. Development and application of a high throughput one-pot extraction protocol for quantitative LC-MS/MS analysis of phospholipids in serum and lipoprotein fractions in normolipidemic and dyslipidemic subjects. J Chromatogr B. 2019;1118-1119:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gardner MS, McWilliams LG, Jones JI, Kuklenyik Z, Pirkle JL, Barr JR. Simultaneous Quantification of Free Cholesterol, Cholesteryl Esters, and Triglycerides without Ester Hydrolysis by UHPLC Separation and In-Source Collision Induced Dissociation Coupled MS/MS. J Am Soc Mass Spectrom. 2017;28(11):2319–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aasebo E, Opsahl JA, Bjorlykke Y, Myhr KM, Kroksveen AC, Berven FS. Effects of blood contamination and the rostro-caudal gradient on the human cerebrospinal fluid proteome. PLoS One. 2014;9(3):e90429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Opsahl JA, Vaudel M, Guldbrandsen A, et al. Label-free analysis of human cerebrospinal fluid addressing various normalization strategies and revealing protein groups affected by multiple sclerosis. Proteomics. 2016;16(7):1154–1165. [DOI] [PubMed] [Google Scholar]
- 85.Yassine HN, Belopolskaya A, Schall C, Stump CS, Lau SS, Reaven PD. Enhanced cholesterol efflux to HDL through the ABCA1 transporter in hypertriglyceridemia of type 2 diabetes. Metabolism. 2014;63(5):727–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Montagne A, Nation DA, Sagare AP, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.