

Abstract
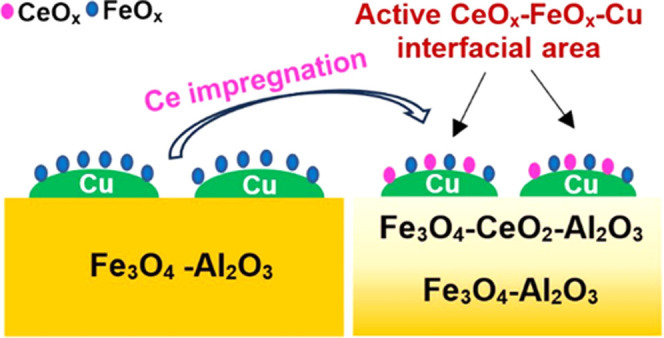
The effect of Ce addition to the Cr-free Al-promoted Cu–Fe oxide-based catalysts is investigated. Catalyst characterization (X-ray diffraction (XRD), in situ Raman spectroscopy, high-sensitivity low-energy ion scattering (HS-LEIS), Brunauer–Emmett–Teller (BET) analysis), CO-temperature-programmed reduction chemical probing, and steady-state WGS activity reveal that (i) in the absence of Al, Ce addition via coprecipitation has a detrimental effect on the catalytic activity related to the poor thermostability and formation of less active Ce–Cu–O NPs, (ii) the addition of Ce via coprecipitation also does not improve the performance of the CuAlFe catalyst because of the formation of a thick CeOx overlayer on the active Cu–FeOx interface, and (iii) impregnation of Ce onto the CuAlFe catalyst exhibits significant improvement in catalytic performance due to the formation of a highly active CeOx–FeOx–Cu interfacial area. In summary, Al does not surface-segregate and serves as a structural promoter, while Ce and Cu surface-segregate and act as functional promoters in Ce/CuAlFe mixed oxide catalysts.
Keywords: high-temperature, water−gas shift, iron oxide-based catalysts, Cr-free catalysts, cerium oxide, aluminum oxide
1. Introduction
The water−gas shift (WGS) reaction (CO + H2O ↔ CO2 + H2) is an important step in the production of hydrogen from hydrocarbons and generally follows the steam reforming reaction for hydrogen enrichment and carbon monoxide elimination.1−3 Industrially, the WGS reaction is performed in two steps: a high-temperature shift (HTS) (∼320–450 °C) reaction, followed by a low-temperature shift (LTS) (∼180–250 °C) reaction to achieve maximum equilibrium conversion for this equilibrated exothermic reaction.4−8 Iron oxide-based catalysts containing 80–90 wt % Fe2O3, 8–10% Cr2O3, and the balance promoters such as copper oxide are used for the high-temperature water−gas shift (HT-WGS) reaction, and Cu-based catalysts (Cu–ZnO–Al2O3) are used for the low-temperature water−gas shift reaction.5
In situ/operando bulk and surface structural characterization of the HT-WGS iron–chromium oxide-based catalysts revealed the molecular structure and nature of the active site during the HT-WGS reaction. In the freshly calcined chromium–iron oxide-based HT-WGS catalysts, prepared by coprecipitation of iron and chromium oxide precursors, followed by calcination, iron oxide is present as the bulk hematite (α-Fe2O3 or γ-Fe2O3) phase.5In situ Raman and high-sensitivity low-energy ion scattering (HS-LEIS) measurements reveal that Cr6+ oxide is mostly present on the surface of the bulk Fe2O3 phase in the fresh catalyst.9,10 After activation of the catalyst under HT-WGS reaction conditions, the bulk Fe2O3 phase transforms to the bulk Fe3O4 (magnetite) phase, which is the bulk catalytic active phase of iron oxide during the HT-WGS reaction.7 During the HT-WGS reaction, the surface Cr6+ oxide also reduces to Cr3+ oxide and forms a solid solution with the Fe3O4 lattice (Fe3–xCrxO4).11−13 The Fe3–xCrxO4 solid solution also stabilizes the iron oxide phase from becoming reduced to metallic Fe0, which can form undesired hydrocarbons, showing that chromium oxide serves as a textural promoter.9,10
The chromium–iron oxide-based HT-WGS catalysts possess several wt % Cr6+ when exposed to air in the fresh and spent catalysts, which is problematic since hexavalent Cr6+ is a carcinogen that threatens human health and the environment.8,14 Consequently, rigorous investigations have been performed to find a suitable nontoxic replacement of chromium exhibiting equivalent or better HT-WGS catalytic performance. Some of the elements investigated as potential replacements for Cr in the iron oxide-based HT-WGS catalysts are Al, Zn, Ca, Ni, Co, Ge, Mg, Ti, Si, Mo, Ce, Zr, and Nb.8,15−28
Many of these studies use copper oxide as a chemical promoter for iron oxide-based HT-WGS catalysts. Recent in situ characterization studies show that in the fresh catalyst, Cu is present as Cu2+ oxide in the lattice of the Fe2O3 phase. After activation of the catalyst under the HT-WGS reaction environment, Cu oxide reduces and segregates as metallic copper nanoparticles on the surface of the Fe3O4 phase (in situ X-ray absorption fine structure (XAFS), near-ambient-pressure X-ray photoelectron spectroscopy (NAP-XPS), X-ray absorption near-edge spectroscopy (XANES)).10,29−31 Interestingly, a fraction of the metallic copper nanoparticle surface was found to have been covered by an FeOx overlayer (HS-LEIS and transmission electron microscopy (TEM)).10 The copper promotion is realized by the introduction of new catalytic active sites, present as an interfacial area of the FeOx overlayers and Cu nanoparticles that increases the activity of HT-WGS. It was observed that the turnover frequency (TOF) value of chromium–iron oxide catalyst (1.2 × 10–3 s–1) increased by ∼3× (3.5 × 10–3 s–1) upon the addition of copper.32
Recent findings from Cr-free iron oxide-based HT-WGS studies15,21,22,33−38 confirm that aluminum is a very promising substitute for the Cr promoter. The Al promoter exhibits comparable structural stability, thermostability, and catalytic activity as that of the Cr-promoted HT-WGS catalysts.19 Cerium, a rare earth metal, is another promising promoter33,39−43 due to its high oxygen storage capability and excellent redox ability to change oxidation states between Ce4+ ↔ Ce3+.44 The combination of the Ce–Al33 and Ce–Zr39 promoters was found to enhance the thermal stability of iron oxide-based catalysts, even after several HT-WGS reaction cycles. Ceria promotion has also been suggested to have an electronic effect on catalytic activity.40 Unfortunately, these studies lacked critical direct in situ spectroscopic details about the dynamics of the catalyst structure to determine the actual promotion mechanism of Ce oxide, which is the motivation for the current study.
The objective of the current work is to understand the combined promotional effect of ceria and alumina promoters on Cr-free copper–iron oxide-based HT-WGS catalysts. For this purpose, coprecipitation and incipient-wetness impregnation of the Ce oxide promoter in copper–aluminum–iron oxide catalysts with varying Ce and Al loadings were done. The Brunauer–Emmett–Teller (BET) surface areas of the fresh and activated catalysts were measured in situ, without exposing the pyrophoric catalyst to the atmosphere. The bulk structure of the catalyst was determined with X-ray diffraction (XRD). In situ Raman spectroscopy measurements provided information about the bulk as well as surface structure of various metal oxides present in the catalysts under different environmental conditions. Additionally, the topmost surface layer of the catalysts was characterized with HS-LEIS spectroscopy. The catalyst reducibility was chemically probed by CO-temperature-programmed reduction (TPR) experiments, and the catalytic performance of various catalysts was compared via the steady-state HT-WGS reaction. The current findings offer an insight into the design of chromium-free iron oxide-based HT-WGS catalysts.
2. Experimental Section
2.1. Catalyst Synthesis
Cerium-, aluminum-, and copper-doped iron oxide catalysts were prepared by using cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O, Alfa Aesar), aluminum nitrate nonahydrate (AlN3O9·9H2O, Sigma-Aldrich 99.99% trace metal basis), copper(II) nitrate hemi(pentahydrate) (CuN2O6·2.5H2O, Aldrich 99.99% trace metal basis), and iron(III) nitrate nonahydrate (FeN3O9·9H2O, Sigma-Aldrich 99.99% trace metal basis) precursors. Two different methods were followed for incorporation of the Ce promoter.
In the coprecipitation method, calculated amounts of all of the metal oxide precursors were dissolved in 100 mL of distilled water and mixed well. To this solution, ammonium hydroxide solution (30%, m/v) was added dropwise under constant stirring at room temperature until the pH reached 8.5. At this point, the formation of a dark brown precipitate was observed and was further aged for 24 h at room temperature, followed by filtration. The filtered precipitate was then put in a temperature-programmable oven. It was dried at 80 °C for 12 h and finally calcined at 400 °C for 4 h in a static air environment. These catalysts contain 8 wt % Ce and/or Al promoters, 3 wt % Cu promoter, and 89 wt % iron, in the oxidized form. The catalysts synthesized using the coprecipitation method are denoted as follows: 3Cu8AlFe, 3Cu8CeFe, and 3Cu5Al3CeFe.
The Ce promoter was added via incipient-wetness impregnation to the copper–aluminum–iron oxide catalyst, prepared by the coprecipitation method (3Cu8AlFe). The calculated amount of Ce oxide precursor aqueous solution was added dropwise. The impregnated sample was then dried for 24 h at room temperature, followed by additional drying at 80 °C for 12 h and finally calcined at 400 °C for 4 h, under static air environment. These catalysts are denoted by 3Ce/3Cu8AlFe and 5Ce/3Cu8AlFe. In all preparation steps, the final calcined catalysts were always ground to make a fine powder.
2.2. BET Surface Area Measurement
The surface area values of both fresh and WGS reaction mixture-treated catalysts were measured by a 3-point flow BET method in an Altamira Instruments system (AMI-200) equipped with a TCD detector. After preconditioning of the catalysts and prior to the BET measurements, all samples were degassed under He flow at ∼150 °C. The N2 adsorption/desorption amounts were measured at three different partial pressures (P/P0 = 0.14, 0.22, and 0.30) for the calculation of surface area values. The fresh catalysts were always dehydrated at 400 °C, under flowing 10% O2/Ar (Praxair, for 1 h), prior to BET measurements. For catalyst activation under HT-WGS reaction conditions, 10% CO/Ar (Praxair, 10 mL/min) and He (Airgas, UHP certified gas, 30 mL/min) gas mixture was passed through a water saturator at room temperature to maintain a CO/H2O ratio ∼1. The catalysts were activated at 330 °C for 1 h and 500 °C for 2 and 5 h. Any residual gases were flushed with He after the HT-WGS reaction. The surface areas of the activated catalysts were measured without exposing the catalysts to the ambient atmosphere (in situ BET measurements).
2.3. X-ray Diffraction (XRD) Spectroscopy
Powder X-ray diffraction patterns of fresh calcined catalysts were measured by the Panalytical Empyrean X-ray Diffraction Unit under ambient conditions. The Cu attenuator mask was utilized for alignment. Full scans between 20 and 80° (2Θ) at a rate of 5°/min were utilized. Additionally, the Fe2O3 (110) peak in the range of 34.5–36.5° was scanned with a rate of 1°/min.
2.4. In Situ Raman Spectroscopy
The Raman spectroscopic studies were performed with a Horiba-Jobin Yvon LabRam High-Resolution instrument equipped with a confocal microscope (50× long working distance objective, Olympus BX-30-LWD). A laser excitation of 442 nm (generated by a He–Cd laser, 110 mW maximum power) and single-stage monochromator with 900 grooves/mm grating were utilized for spectra collection. The scattered light from the sample was passed through the monochromator grating and collected with a visible sensitive liquid N2-cooled CCD detector (Horiba-Jobin Yvon CCD-3000 V). Catalysts were placed in a high-temperature reactor cell (Linkam CCR1000) with a temperature controller (Linkam TMS94). Spectra of the dehydrated samples were collected after removal of any possible organic impurities and adsorbed moisture by flowing of 10% O2/Ar (Praxair) with 30 mL/min flow rate at 400 °C for 1 h. For the collection of in situ spectra of the activated catalysts during the reverse-WGS (RWGS) reaction, the catalysts were further treated with a mixture of 30 mL/min 5%CO2/Ar (GTS-WELCO) and a 15 mL/min 10%H2/Ar (Airgas, UHP certified gas) mixture at 400 °C for 1 h.
2.5. High-Sensitivity Low-Energy Ion Scattering (HS-LEIS) Spectroscopy
The HS-LEIS surface analysis of the catalysts was performed in an ION-TOF Qtac100 spectrometer. Powder catalyst samples were compressed in a sample holder for heat treatments within the HS-LEIS spectrometer. The design of the HS-LEIS instrument allows one to directly transfer the sample from the pretreatment chamber to the analysis chamber without exposing to the ambient atmosphere. The fresh catalyst samples were dehydrated in a ∼10 mbar 10% O2/Ar static gas environment at 400 °C for 1 h. The RWGS reaction mixture-activated catalysts were conditioned in the ∼10 mbar static gas mixture environment of CO2 + H2 (CO2/H2 ratio ∼1) at 400 °C for 1 h. After the desired pretreatment, the topmost surface of the catalyst was analyzed using 3 keV He+ (for lighter atomic weight elements like O and Al) and 5 keV Ne+ (for heavier elements like Fe, Cu, and Ce) ion probes. Further, to gain information about the composition of subsurface layers, the topmost layer was sputtered with 0.5 keV Ar+ ions allowing for depth profiling. Charge neutralization was invoked during spectral acquisition as well as sputtering steps. The probes employed were 3 keV He+ (1 × 1014 ions/cm2·cycle) and 5 keV Ne+ (1 × 1014 ions/cm2·cycle), 1.5 × 1.5 mm2. For sputtering, 0.5 keV Ar+ (1.5 × 1014 ions/cm2·cycle, 2.0 × 2.0 mm2) was applied. A dose of 1 × 1015 ions/cm2 corresponds to removal of approximately one atomic layer from the catalyst surface.45
2.6. CO-Temperature-Programmed Reduction (TPR)
The CO-TPR experiments were performed using an Altamira Instruments system (AMI-200) equipped with a Dycor Dymaxion DME100MS online quadrupole mass spectrometer. Dehydrated catalysts (under flowing 10% O2/Ar, Praxair, at 350 °C for 1 h) were activated via the HT-WGS reaction (10 mL/min 10% CO/Ar (Praxair), 30 mL/min He (Airgas, UHP certified gas), flow through bubbler at room temperature) at 350 °C for 90 min. After the activation process, the reactor was flushed by He and cooled down to 80 °C. The CO-TPR experiments were then carried out by ramping up the temperature under 10% CO/Ar (Praxair) at a rate of 10 °C/min with simultaneous online gas analysis by a mass spectrometer. The resulting CO2/CO-TPR spectra were normalized by the MS signal for Ar that was used as the internal standard.
2.7. Steady-State HT-WGS Reaction
The steady-state forward water−gas shift reaction activity measurements were carried out by using an Altamira AMI-200 instrument equipped with a Dycor Dymaxion DME100MS online quadrupole mass spectrometer. Approximately 10 mg of the powder catalyst was loaded into a U-type quartz tube and held in place with quartz wool. First, 10% O2/Ar (Praxair) was passed with a flow rate of 40 mL/min at 400 °C for 1 h to remove any possible adsorbed organics. Then, the system was flushed with He for 10 min. Afterward, the reaction mixture was introduced (10% CO/Ar (Praxair, 10 mL/min), He (Airgas, UHP certified gas, 30 mL/min), and water vapor introduced by flowing the gas through a water bubbler at room temperature). The total flow rate was 40 mL/min for approximately 10 mg catalyst. The GHSV equals 240,000 mL/h·gcat. This high GHSV maintains low conversion with minimal transport limitations.46 The HT-WGS reaction was performed at different temperatures (330, 350, 370 °C) for 90 min to ensure steady-state conditions. The following m/z values were recorded during the experiment: H2, m/z = 2; H2O, m/z = 18; CO (corrected for CO2 cracking), m/z = 28; CO2, m/z = 44; and Ar, m/z = 40. Ar was used as an internal standard.
3. Results
3.1. BET Surface Area
The BET surface area values of freshly calcined and activated catalyst samples exposed to the WGS reaction are listed in Table 1. To compare the thermostability of the catalyst samples, the HT-WGS reaction treatment was conducted at multiple temperatures (330 °C for mild reaction conditions and 500 °C for more rigorous reaction conditions) and durations (1–5 h).
Table 1. BET Surface Area of Various Cu–Al–Ce–Fe Oxide Catalysts after Dehydration: 10% O2/Ar at 400 °C for 1 h and WGS Reaction: 10% CO/Ar (10 mL/min), He (30 mL/min), and Water Vapor (H2O/CO ∼1) at 330 (for 1 h) and 500 °C (for 2 and 5 h).
| BET
surface area (m2/g) |
|||||
|---|---|---|---|---|---|
| sample | composition (wt %) | fresh | 330 °C, 1 h | 500 °C, 2 h | 500 °C, 5 h |
| 3Cu8AlFea | 3% CuO – 8% Al2O3 – 89% Fe2O3 | 86 | 44 | 25 | 23 |
| 3Cu8CeFea | 3% CuO – 8% CeO2 – 89% Fe2O3 | 74 | 34 | 23 | 20 |
| 3Cu5Al3CeFea | 3% CuO – 5% Al2O3 – 3% CeO2 – 89% Fe2O3 | 97 | 43 | 27 | 25 |
| 3Ce/3Cu8AlFeb | 3% CeO2/(3% CuO – 8% Al2O3 – 89% Fe2O3) | 79 | 50 | 29 | 28 |
| 5Ce/3Cu8AlFeb | 5% CeO2/(3% CuO – 8% Al2O3 – 89% Fe2O3) | 73 | 48 | 28 | 26 |
Coprecipitated.
Coprecipitation, followed by impregnation of Ce.
3.1.1. Fresh Catalysts
The coprecipitated 3Cu8AlFe catalyst has a BET surface area of 86 m2/g. Replacement of Al oxide with equivalent weight loading of a Ce oxide (3Cu8CeFe) catalyst results in a decrease in the surface area to 74 m2/g. The mixture of coprecipitated Al and Ce oxides, with equivalent total promoter loading of 8 wt % (3Cu5Al3CeFe), however, increases the BET surface area of the catalyst to 97 m2/g. In contrast, impregnation of the 3 and 5% Ce onto the coprecipitated calcined 3Cu8AlFe catalysts showed an opposite trend in BET surface area values with the surface areas decreasing to 79 and 73 m2/g, respectively. Overall, the initial BET values only modestly varied (73–97 m2/g) with the coprecipitated catalysts possessing slightly higher BET values.
3.1.2. HT-WGS-Activated Catalysts
After activation of the catalyst samples under the HT-WGS reaction conditions, the surface area of all of the samples decreased significantly (Table 1). Even after mild activation (330 °C, 1 h treatment), a 35–55% decrease in the surface area of the catalyst samples occurred. Under the milder HT-WGS conditions (330 °C, 1 h treatment), the 3Cu8CeFe catalyst exhibited the lowest BET surface area of 34 m2/g, and all of the other catalysts possessed surface area values in a close range (44–50 m2/g) with the 3Ce/3Cu8AlFe catalyst having the highest surface area. After the harsher treatment of the catalyst samples (at 500 °C for 5 h), all of the catalysts showed a significant decrease of ∼60–75% in the surface area compared to their respective fresh catalysts. Although the surface area values are close to each other, the 3Cu8CeFe exhibits slightly poorer thermostability and the catalysts that were impregnated with ceria or coimpregnated with Ce and Al exhibited slightly better thermostability. The more harshly activated catalysts, however, possess very similar BET surface areas that are independent of the composition and synthesis method.
3.2. XRD
The XRD diffractograms of the freshly calcined catalysts under ambient conditions are presented as full scans, and the main Fe2O3(110) peaks are shown in Figure 1. The bulk phase of the initial promoted iron oxide catalysts is crystalline α-Fe2O3 (hematite).47 The intensity of the Fe2O3(110) peak decreases with Al and Ce addition, most probably from a decrease in particle size and/or greater disorder. The X-ray diffraction patterns of all catalysts do not show any observable distinct nanoparticles of crystalline Al2O3,48 CeO2,49 CuOx, or any compounds between Cu–Ce–Al–Fe oxides. This observation suggests that these NPs are either absent or are too small (<3 nm) to be detected by XRD. The (110) hematite peak can be used to examine for possible solid solutions of the promoters and the iron oxide catalyst. The slight shift of (110) hematite peak in the XRD indicated solid solution formation between Cr oxide and Fe2O3 in excellent agreement with prior studies.10 The slight, yet opposite, shift of the Fe2O3(110) peak for both 3Cu8AlFe and 3Cu8CeFe catalysts, compared to the unpromoted Fe2O3 catalyst, indicates that a solid solution is formed between Al oxide and Fe2O3 as well as Ce oxide and Fe2O3, respectively. This is consistent with the smaller ionic radii of Al3+ (53.5 pm)50 than the ionic radius of Fe3+ (64.5 pm)50 and the larger ionic radii of Ce4+ (87 pm)50/Ce3+ (101 pm)50 than the ionic radius of Fe3+ (64.5 pm).50 Incorporation of ceria into the alumina-containing catalysts has different effects. Coprecipitation of alumina and ceria promoters (3Cu5Al3CeFe catalyst) shows the dominant effect of larger ionic radii of the cerium ion as the 2θ position of the main (110) peak slightly decreases compared to the unpromoted Fe2O3 phase. On the other hand, the amount of ceria loading determines the 2θ position of the main (110) peak of the Fe2O3 phase of ceria-impregnated 3Cu8AlFe catalysts. For the impregnated 3Ce/3Cu8AlFe catalyst, the Fe2O3 110 peak remains the same as that of the 3Cu8AlFe catalyst. This suggests that 3 wt % loading of Ce oxide by impregnation has a very limited effect on the solid solution of Al with the Fe2O3 phase. An increase in the ceria loading to 5 wt % reflects the effect of Ce incorporation into the Al–Fe2O3 solid solution phase and is indicated by the shift of the Fe2O3(110) peak compared to the 3Cu8AlFe catalyst. Although the XRD spectra were not collected for the WGS-activated catalysts, previous in situ XRD studies showed that the bulk iron oxide Fe2O3 (hematite) phase transforms to the Fe3O4 (magnetite) phase (see the Section 3.3 for confirmation).19 Although the promoters formed solid solutions with the Fe oxide phases, they did not affect the bulk structure of the Fe oxide phase.
Figure 1.

XRD diffractograms of fresh calcined catalysts collected under ambient conditions as a function of 2Θ (A) full scan and (B) the Fe2O3 (110) peak region.
3.3. In Situ Raman Spectroscopy
The in situ Raman spectra of the calcined catalysts are presented in Figure 2. The Raman spectra of all of the dehydrated freshly calcined catalysts exhibit the characteristic bands of the bulk α-Fe2O3 phase (hematite: 226, 245, 292, 411, 497, 612 cm–1).51 The weak and broad new band at 620–700 cm–1 has been attributed to crystal disorder caused by the solid solution of Fe2–xMxO3 (M: Al and/or Ce) with small crystalline dimensions.52
Figure 2.

In situ Raman spectra of (A) 3Cu8AlFe, (B) 3Cu8CeFe, (C) 3Ce/3Cu8AlFe, (D) 5Ce/3Cu8AlFe, and (E) 3Cu5Al3CeFe catalysts under dehydrated (10% O2/Ar) and RWGS reaction conditions (30 mL/min 5%CO2/Ar and 15 mL/min 10%H2/Ar) at 400 °C. RWGS_1 and RWGS_2 denote spectra collection from two different points on the catalyst surface.
Under the RWGS reaction conditions, the bulk α-Fe2O3 (hematite) phase reduces to the bulk Fe3O4 (magnetite) phase (306, 538, 668 cm–1).53 This is in agreement with literature reports that the Fe3O4 phase is the dominant bulk phase of iron oxide under HT-WGS reaction conditions.7 No additional NPs from the Ce, Cu, and Al are observed for any of the Al-containing catalysts. A new broad Raman band at ∼427 cm–1, however, is observed for the 3Cu8CeFe catalyst and can be assigned to the mixed oxide phase of Ce with Cu (Ce–Cu–O solid solution).54 The spectra of the 3Cu8CeFe catalyst collected from different spots, however, indicate that the Cu–Ce–O mixed oxide phase, formed during the RWGS reaction, does not uniformly cover the catalyst surface. This Raman band, however, was not detected in the Raman spectra of any of the Cu–Ce–Al–Fe oxide catalysts. The in situ Raman spectra confirm that the same bulk Fe oxide phases are present among all of the freshly calcined catalysts and among all of the activated catalysts. The presence of a bulk CuxCe1–xO2 mixed oxide phase was also detected for the activated 3Cu8CeFe catalyst.
3.4. High-Sensitivity Low-Energy Ion Scattering (HS-LEIS) Spectroscopy
Elemental compositional analysis of the outermost surface layer and subsequent subsurface layers by depth profiling of the synthesized catalysts, after dehydration and activation under RWGS reaction conditions, were measured with HS-LEIS and are presented in Figure 3. The Fe signal is not shown in any of the HS-LEIS spectra because previous investigations showed a significant increase in the Fe signal intensity due to preferential sputtering of oxygen from iron oxides.55,56 The intensity of the Al signal is relatively lower than the intensity of the Cu and Ce signals because the Al signals were collected with the lighter He+ ions and Cu/Ce signals were collected with the heavier Ne+ ions.
Figure 3.

HS-LEIS depth profile of O2 calcined (solid symbols) and RWGS reaction (CO2 + H2) activated (open symbols) catalysts after pretreatments. The He+ ion is the probe for Al and the Ne+ ion is the probe for Ce and Cu (Cu: blue rectangle; Ce: black sphere; Al: red square).
3.4.1. 3Cu8AlFe Catalyst
In the dehydrated freshly calcined catalyst (see Figure 3 solid symbols), the intensity of the Al and Cu signals is relatively the same on the outermost surface layer and throughout the subsurface region of the catalyst, indicating that Al and Cu are uniformly distributed in the outermost surface layers of this dehydrated catalyst. Upon catalyst activation in the RWGS reaction environment, the intensity of the Al signal remained constant and the intensity of the Cu signal increased in the outermost surface layer (see Figure 3 open symbols). After activation of the catalyst, the intensity of the Al signal remains constant, suggesting uniform distribution of Al with depth, but the intensity of the Cu signal monotonically increased with depth profiling, reflecting the covering of the Cu nanoparticles most likely with an FeOx overlayer.19
3.4.2. 3Cu8CeFe Catalyst
For the dehydrated freshly calcined catalyst (see Figure 3 solid symbols), the intensity of the Ce signal remains relatively the same from the outermost surface layer to the subsurface layers reflecting its uniform distribution with depth. The corresponding intensity from Cu mildly decreases with sputtering depth, suggesting that Cu is slightly surface-enriched. Upon catalyst activation under RWGS (see Figure 3 open symbols), the intensity of the Cu signal decreased and the intensity of the Ce signal slightly decreased at the outermost surface layer. Furthermore, the intensity of both Cu and Ce signals decreases with catalyst activation, reflecting surface enrichment of both Cu and Ce.
3.4.3. 3Ce/3Cu8AlFe Catalyst
For the dehydrated catalyst (see Figure 3 solid symbols), the intensity of the Al and Ce signals remains constant from the outermost surface layer and subsurface layers of the catalyst. The Cu intensity, however, decreases slightly with depth profiling. After activation under RWGS (see Figure 3 open symbols), the intensity at the outermost surface layer for Al slightly increases, Cu significantly increases, and Ce remains constant. During depth profiling, the intensity of the Al signal is constant, the Ce signal slightly decreases and Cu signal increases up to 1–2 atomic layers and stays nearly constant after that, reflecting the surface enrichment of Ce and surface depletion of Cu. Furthermore, the Ce oxide NPs decorating the Cu surface are ∼3–4 nm in size.
3.4.4. 5Ce/3Cu8AlFe Catalyst
For the dehydrated catalyst (see Figure 3 solid symbols), during sputtering, the intensities of the Al and Ce signals are relatively constant, indicating the uniform concentration of Al and Ce from the outermost surface layer to the subsurface layers. The intensity of the Cu signal, however, initially increases and then decreases reflecting the concentration of Cu peaking in the subsurface region. After RWGS treatment of the catalyst (see Figure 3 open symbols), the intensity at the outermost surface layer of Al does not change, Ce increased, and Cu decreased. With depth profiling, the intensity of Al remained the same, Cu increased, and Ce monotonically decreased. The Cu signal intensity first increased until ∼2 atomic layers and then remained constant. These trends suggest that the Al makes a solid solution with the Fe3O4 phase, and Cu NPs present on the catalyst surface are decorated by ∼0.5 nm size Ce oxide NPs.
3.4.5. 3Cu5Al3CeFe Catalyst
In the dehydrated freshly calcined catalyst, the Al signal has the same intensity for the outermost surface layer and subsurface region. In contrast, the intensity from Ce increases slightly with depth profiling. The signal intensity from Cu first increases up to about a monolayer and then decreases with depth profiling. After treatment of the catalyst in the RWGS environment, no change in the Al signal intensity is observed between the outermost surface layer and the subsurface of the catalyst. The Ce signal intensity is observed to decrease monotonically with depth profiling. Finally, the signal from Cu increased for the initial 2–3 atomic layers and then remained nearly constant. These trends suggest the formation of a thicker Ce oxide overlayer on the Cu NP surface.
3.5. CO-TPR
The reducibility of the WGS reaction-activated catalysts was examined with CO-TPR since CO oxidation represents a key reduction step in the WGS reaction. The normalized CO2 MS signals, obtained by using Ar as an internal standard, as a function of the catalyst bed temperature, are presented in Figure 4. The formation of CO2 at the lower temperatures is assigned to the removal of oxygen from the surface region of the catalyst, and the CO2 formed at higher temperatures is associated with the reaction of CO with oxygen from the bulk lattice.10 The strong consumption of CO starts at ∼260 °C due to the deep reduction of the bulk Fe oxide phase.
Figure 4.
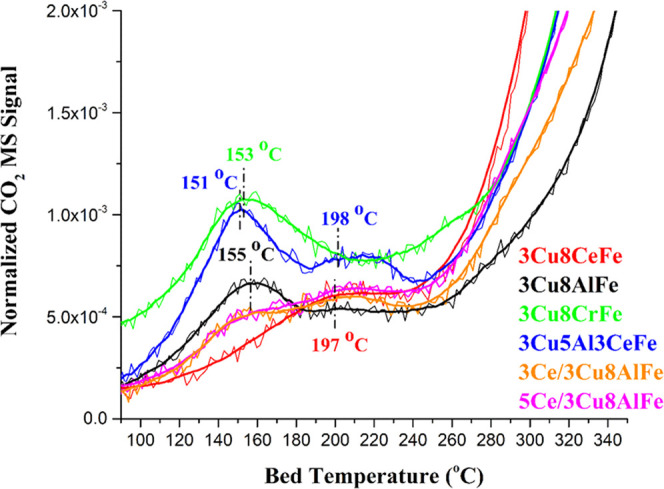
CO-TPR spectra of catalysts activated under the WGS reaction conditions at 350 °C.
The 3Cu8AlFe and 3Cu8CeFe catalysts possess only one low-temperature CO2/CO-TPR reduction peak at 155 and 197 °C, respectively. For reference, the 3Cu8CrFe catalyst exhibits only one low-temperature CO2/CO-TPR reduction peak at 153 °C that is similar to that of the 3Cu8AlFe catalyst. The 3Cu5Al3CeFe mixed oxide catalyst, however, has two distinct low-temperature CO2/CO-TPR reduction peaks at 151 and 198 °C. The ceria-impregnated xCe/3Cu8AlFe catalysts show two CO2/CO-TPR reduction peaks in the similar temperature ranges as that of the 3Cu5Al3CeFe catalyst. This observation suggests that the origin of the CO2/CO-TPR reduction peak at ∼150–155 °C is related to the effect of Al, while the 195–200 °C CO2/CO-TPR reduction peak is associated with the Ce promoter.
The areas under the low-temperature CO2/CO-TPR curves were used to quantify the number of redox catalytic active sites (Ns) present in each catalyst, and the values are listed in Table 2. It is assumed that the overall catalytic activity is dominated by the active sites available at the lowest temperature for CO oxidation.20 An Al-free 3Cu8CeFe catalyst has the highest Ns value. While coprecipitation of the Ce promoter in the Cu–Al–Fe catalyst slightly increases the Ns value, the Ns value of the 3Cu8AlFe catalyst is slightly decreased by the impregnation of the Ce promoter. The numbers of reducible active sites of the two ceria-impregnated catalysts are similar but somewhat lower than those of the other catalysts. Impregnation of cerium oxide apparently decreases the number of the catalytic active sites, whereas the cerium-coprecipitated catalysts have a higher number of active sites.
Table 2. Number of Active Sites (Ns), Catalytic Activities Based on the CO Consumption Rate [(10–6 mol/s/g) (10% CO/Ar (10 mL/min), He (30 mL/min), and Water Vapor (H2O/CO ∼ 1)) (T = 330 °C)], Turnover Frequencies (TOFs), and Activation Energies of Cu–Al–Ce–Fe Oxide Catalysts.
| catalyst | catalytic activity (10–6 mol/s·g) | number of active sites from CO-TPR (Ns) (10–3 mol/g) | TOF (10–3 s–1) | activation energy (Ea) (kJ/mol) |
|---|---|---|---|---|
| 3Cu8AlFe | 4.1 ± 0.2 | 0.87 | 4.7 | 56 |
| 3Cu8CeFe | 3.7 ± 0.2 | 1.05 | 3.5 | 63 |
| 3Cu5Al3CeFe | 4.3 ± 0.3 | 0.93 | 4.6 | 56 |
| 3Ce/3Cu8AlFe | 6.5 ± 0.4 | 0.80 | 8.1 | 41 |
| 5Ce/3Cu8AlFe | 5.2 ± 0.3 | 0.79 | 6.6 | 41 |
3.6. Steady-State WGS Reaction
The steady-state catalytic activity values of the catalysts (based on the CO conversion) at 330 °C are presented in Table 2. Although the catalytic activity of 3Cu8CeFe is lower than the activity of 3Cu8AlFe, replacement of some amount of alumina with ceria by the coprecipitation method (3Cu5Al3CeFe catalyst) increases the activity. The main effect of ceria promotion is seen on the catalytic activity of the catalysts that are prepared by impregnation of ceria into 3Cu8AlFe catalysts. Impregnation of 3 wt % ceria dramatically increases the activity of the 3Cu8AlFe catalyst. Higher loadings of impregnated ceria result in a decrease in activity, which is still higher than the activity of the 3Cu8AlFe catalyst. The 3Ce/3Cu8AlFe catalyst is the most active, and the 3Cu8CeFe catalyst is least active. The steady-state activity follows the order 3Ce/3Cu8AlFe > 5Ce/3Cu8AlFe > 3Cu5Al3CeFe ∼ 3Cu8AlFe > 3Cu8CeFe.
The TOF values (Table 2) were calculated by dividing the values of the steady-state catalytic activity by the number of participating surface redox oxygen atoms. The TOF values vary from 3.5 × 10–3 to 8.1 × 10–3 s–1 with the order of 3Ce/3Cu8AlFe > 5Ce/3Cu8AlFe > 3Cu5Al3CeFe ∼ 3Cu8AlFe > 3Cu8CeFe, which is the same as the steady-state catalytic activity trend.
Differences in the catalytic activities are further examined by generating Arrhenius plots, shown in Figure 5. The corresponding activation energy (Ea) values are listed in Table 2. The Ea values follow the trend: 3Cu8CeFe > 3Cu8AlFe ∼ 3Cu5Al3CeFe > 3Ce/3Cu8AlFe ∼ 5Ce/3Cu8AlFe. The general trend is that catalysts with higher Ea values correspond to lower TOF values.
Figure 5.

Arrhenius plots for steady-state WGS reaction rates for Cu–Ce–Al–Fe oxide catalysts.
4. Discussion
4.1. Catalyst Structure
4.1.1. Fresh Catalysts
The freshly calcined catalysts possess the bulk Fe2O3 (hematite) phase (XRD and Raman), which is in agreement with previous literature reports.47,57 The Al and Ce promoters form solid solutions with the Fe2O3 phase (shifts in the XRD and Raman band positions). Additional crystalline NPs of promoters are not observed (XRD and Raman). The Al is uniformly distributed between the outermost surface layer and subsurface region of the catalysts (HS-LEIS). Except for the 3Cu5Al3CeFe catalyst, all of the other catalysts also exhibit uniform distributions of Ce between the outermost surface layer and subsurface region of the catalysts. For the 3Cu5Al3CeFe catalyst, the Ce is more concentrated in the subsurface region. In contrast, the Cu promoter is surface-enriched or segregated in all catalysts (HS-LEIS). A schematic of the initially calcined catalyst structures is shown in Scheme 1 (left).
Scheme 1. Structures of (A) 3Cu8AlFe, (B) 3Cu8CeFe, (C) 3Ce/3Cu8AlFe, (D) 5Ce/3Cu8AlFe, and (E) 3Cu5Al3CeFe Catalysts before (Left) and during (Right) the WGS Reaction.

4.1.2. RWGS-Activated Catalysts
Dynamic changes are observed after treatment of the catalysts with the RWGS reaction mixture. The bulk Fe2O3 phase of iron oxide transforms to the bulk Fe3O4 phase (XRD and Raman). This bulk phase transformation is associated with a significant decrease in the catalyst surface area (Table 1). Except for the 3Cu8CeFe catalyst, the other catalysts do not exhibit any crystal phase other than Fe3O4 (XRD and Raman). The Al is still uniformly distributed between the outermost surface layer and subsurface region of the catalysts after the RWGS treatment, whereas Ce surface-segregated (HS-LEIS). The Cu has been shown to form metallic Cu0 NPs on the catalyst surface during the WGS reaction.10 With the exception of the 3Cu8CeFe catalyst, the metallic Cu0 NPs are decorated by secondary metal oxides (HS-LEIS). For the 3Cu8CeFe catalyst, the formation of the Ce–Cu–O mixed oxide phase is observed after the RWGS treatment (in situ Raman). Furthermore, it is observed that the Ce–Cu–O phase is present near the surface region of the catalyst (HS-LEIS), but it is not uniformly distributed (in situ Raman). A schematic of the activated catalyst structures is shown in Scheme 1 (right).
4.2. Structural Promotion
The uniform distribution of Al in the surface region, as opposed to surface segregation of Ce after the RWGS treatment (compare 3Cu8AlFe and 3Cu8CeFe catalysts in Figure 3), results in higher thermostability (Table 1). The Al promotion is also apparent in other Cu–Al–Ce–Fe catalysts (uniform Al distribution, Figure 3), exhibiting a higher surface area than the 3Cu8CeFe catalyst without Al promotion. Furthermore, the thermostability is even greater (lower decrease in surface area, Table 1) when Ce is added via impregnation of the precalcined 3Cu8AlFe catalyst, as opposed to coprecipitation in the 3Cu5Al3CeFe catalyst. These observations indicate that Al acts as a better structural promoter than Ce.
4.3. Functional Promotion
The Al is distributed uniformly throughout the surface and subsurface regions of the iron oxide-based catalysts after activation with the RWGS treatment (HS-LEIS) and, consequently, does not promote the catalytic active sites (FeOx–Cu interfacial sites). In contrast, Ce is surface-segregated after RWGS treatment (HS-LEIS). Surface segregation is enhanced for the Ce-impregnated catalysts, where a higher ceria loading results in a thicker overlayer formation on the Cu NPs (compare Cu HS-LEIS signals in RWGS-treated 3Ce/3Cu8AlFe and 5Ce/3Cu8AlFe catalysts). This observation suggests that some CeOx is most probably also decorating the surface of the Cu NPs forming a CeOx–FeOx–Cu interface. Thus, CeOx serves as a functional promoter that is better than AlOx.
4.4. Catalyst Structure–Activity Relationships and Role of Promoters
4.4.1. 3Cu8AlFe Catalyst
The Al promoter provides good structural stability, and the Cu–FeOx interface provides very good functional promotion, resulting in moderate Ea and SS activity.
4.4.2. 3Cu8CeFe Catalyst
In the absence of the Al promoter, the catalyst exhibits poorer thermostability and the presence of the Ce–Cu–O mixed oxide phase hinders the formation of a highly active Cu–Fe3O4 interfacial sites.10,19 Hence, despite possessing the highest number of the reducible active sites, the catalyst exhibits the highest Ea, low SS activity, and lowest TOF.
4.4.3. 3Cu5Al3CeFe Catalyst
The combination of Al with Ce promoters provides good structural stability, and the absence of the Ce–Cu–O mixed oxide phase allows for the formation of the CeOx–FeOx–Cu interfacial sites. The thick oxide overlayer on the Cu NPs, however, results in only minor improvement in SS activity and similar TOF and activation energy values.
4.4.4. 3Ce/3Cu8AlFe Catalyst
The Al provides excellent thermostability, and impregnation of a small amount of the Ce promoter results in a greater amount of the CeOx–FeOx–Cu interfacial sites. This results in the lowest Ea and the highest catalytic activity. This catalyst shows the synergistic effect of Ce and Al promotion toward the WGS/RWGS reaction.
4.4.5. 5Ce/3Cu8AlFe Catalyst
The higher loading of the Ce promoter results in a slightly thicker CeOx overlayer on the Cu NPs (lower number of active sites) compared to the 3Ce/3Cu8AlFe catalyst. Overall, low Ea, very good catalytic activity, and higher TOF are observed.
5. Conclusions
Several Cr-free copper–iron oxide-based high-temperature water−gas shift catalysts were prepared with the Al promoter (3Cu8AlFe), Ce promoter (3Cu8CeFe), and copresence of both promoters (3Cu5Al3CeFe, 3Ce/3Cu8AlFe, and 5Ce/3Cu8AlFe). The 3Cu8CeFe catalyst exhibits the additional presence of a Ce–Cu–O mixed oxide phase in the RWGS-treated sample that was not detected from the Cu–Ce–Al–Fe oxide catalysts due to low ceria loading.
The Al promoter uniformly distributes through the outermost surface layer and subsurface layers of the catalyst and serves as a structural promoter that provides improved catalyst thermostability. The Ce promoter is surface-segregated and appears to decorate the Cu NPs to form a CeOx–FeOx–Cu interfacial area. The copresence of the Al and Ce promoters results in higher catalytic activity than the corresponding individual promotion because of the presence of a highly active Ce–Cu–Fe oxide interface. The impregnation of Ce onto the 3Cu8AlFe catalyst, as opposed to coprecipitation, results in the formation of a CeOx overlayer on the Cu NPs with a higher Cu–CeOx interfacial area leading to ∼50% increase in catalytic activity. The molecular level understanding from this study can guide the rational design of Cr-free HT-WGS catalysts.
Acknowledgments
Professor Jih-Mirn Jheng is thanked for assisting with the collection of the CO-TPR data. Dr. Henry Luftman and Dr. Ryan Thorpe of Lehigh University are thanked for collecting the HS-LEIS data. The authors acknowledge financial support from the National Science Foundation Grant CBET-1511689.
Author Contributions
O.Y.: writing—original draft, conceptualization, investigation; S.S.: writing—review and editing; I.E.W.: funding acquisition, supervision, writing—review and editing.
The authors declare no competing financial interest.
References
- Smith R J B.; Loganathan M.; Shantha M. S. A Review of the Water Gas Shift Reaction Kinetics. Int. J. Chem. React. Eng. 2010, 8 (1), 1–32. 10.2202/1542-6580.2238. [DOI] [Google Scholar]
- Gradisher L.; Dutcher B.; Fan M. Catalytic Hydrogen Production from Fossil Fuels via the Water Gas Shift Reaction. Appl. Energy 2015, 139, 335–349. 10.1016/j.apenergy.2014.10.080. [DOI] [Google Scholar]
- Levalley T. L.; Richard A. R.; Fan M. The Progress in Water Gas Shift and Steam Reforming Hydrogen Production Technologies - A Review. Int. J. Hydrogen Energy 2014, 39 (30), 16983–17000. 10.1016/j.ijhydene.2014.08.041. [DOI] [Google Scholar]
- Ladebeck J. R.; Wagner J. P.. Catalyst Development for Water-Gas Shift. In Handbook of Fuel Cells; John Wiley & Sons, Inc., 2003; Vol. 3, pp 190–201. [Google Scholar]
- Ratnasamy C.; Wagner J. P. Water Gas Shift Catalysis. Catal. Rev. 2009, 51 (3), 325–440. 10.1080/01614940903048661. [DOI] [Google Scholar]
- Mendes D.; Mendes A.; Madeira L. M.; Iulianelli A.; Sousa J. M.; Basile A. The Water-Gas Shift Reaction: From Conventional Catalytic Systems to Pd-Based Membrane Reactors - a Review. Asia-Pac. J. Chem. Eng. 2010, 5, 111–137. 10.1002/apj.364. [DOI] [Google Scholar]
- Rhodes C.; Hutchings G. J.; Ward A. M. Water-Gas Shift Reaction: Finding the Mechanistic Boundary. Catal. Today 1995, 23 (1), 43–58. 10.1016/0920-5861(94)00135-O. [DOI] [Google Scholar]
- Lee D. W.; Lee M. S.; Lee J. Y.; Kim S.; Eom H. J.; Moon D. J.; Lee K. Y. The Review of Cr-Free Fe-Based Catalysts for High-Temperature Water-Gas Shift Reactions. Catal. Today 2013, 210, 2–9. 10.1016/j.cattod.2012.12.012. [DOI] [Google Scholar]
- Keturakis C. J.; Zhu M.; Gibson E. K.; Daturi M.; Tao F.; Frenkel A. I.; Wachs I. E. Dynamics of CrO3-Fe2O3 Catalysts during the High-Temperature Water-Gas Shift Reaction: Molecular Structures and Reactivity. ACS Catal. 2016, 6 (7), 4786–4798. 10.1021/acscatal.6b01281. [DOI] [Google Scholar]
- Zhu M.; Rocha T. C. R.; Lunkenbein T.; Knop-Gericke A.; Schlogl R.; Wachs I. E. Promotion Mechanisms of Iron Oxide-Based High Temperature Water-Gas Shift Catalysts by Chromium and Copper. ACS Catal. 2016, 6 (7), 4455–4464. 10.1021/acscatal.6b00698. [DOI] [Google Scholar]
- Edwards M. A.; Whittle D. M.; Rhodes C.; Ward A. M.; Rohan D.; Shannon M. D.; Hutchings G. J.; Kiely C. J. Microstructural Studies of the Copper Promoted Iron Oxide/Chromia Water-Gas Shift Catalyst. Phys. Chem. Chem. Phys. 2002, 4 (15), 3902–3908. 10.1039/b202347b. [DOI] [Google Scholar]
- Scariot M.; Francisco M. S. P.; Jordão M. H.; Zanchet D.; Logli M. A.; Vicentini V. P. An Investigation of the Activation Process of High Temperature Shift Catalyst. Catal. Today 2008, 133–135 (1–4), 174–180. 10.1016/j.cattod.2007.12.090. [DOI] [Google Scholar]
- Patlolla A.; Carino E. V.; Ehrlich S. N.; Stavitski E.; Frenkel A. I. Application of Operando XAS, XRD, and Raman Spectroscopy for Phase Speciation in Water Gas Shift Reaction Catalysts. ACS Catal. 2012, 2 (11), 2216–2223. 10.1021/cs300414c. [DOI] [Google Scholar]
- Zhu M.; Wachs I. E. Iron-Based Catalysts for the High-Temperature Water-Gas Shift (HT-WGS) Reaction: A Review. ACS Catal. 2016, 6 (2), 722–732. 10.1021/acscatal.5b02594. [DOI] [Google Scholar]
- Araujo G. C.; Rangel M. C.. An Environmental Friendly Catalyst for the High Temperature Shift Reaction. In Studies in Surface Science and Catalysis; Elsevier, 2000; Vol. 130, pp 1601–1606. [Google Scholar]
- Martos C.; Dufour J.; Ruiz A. Synthesis of Fe3O4-Based Catalysts for the High-Temperature Water Gas Shift Reaction. Int. J. Hydrogen Energy 2009, 34 (10), 4475–4481. 10.1016/j.ijhydene.2008.08.042. [DOI] [Google Scholar]
- Gawade P.; Mirkelamoglu B.; Tan B.; Ozkan U. S. Cr-Free Fe-Based Water-Gas Shift Catalysts Prepared through Propylene Oxide-Assisted Sol-Gel Technique. J. Mol. Catal. A: Chem. 2010, 321 (1–2), 61–70. 10.1016/j.molcata.2010.02.002. [DOI] [Google Scholar]
- Zhang L.; Wang X.; Millet J. M. M.; Matter P. H.; Ozkan U. S. Investigation of Highly Active Fe-Al-Cu Catalysts for Water-Gas Shift Reaction. Appl. Catal., A 2008, 351 (1), 1–8. 10.1016/j.apcata.2008.08.019. [DOI] [Google Scholar]
- Zhu M.; Yalçın Ö.; Wachs I. E. Revealing Structure-Activity Relationships in Chromium Free High Temperature Shift Catalysts Promoted by Earth Abundant Elements. Appl. Catal., B 2018, 232, 205–212. 10.1016/j.apcatb.2018.03.051. [DOI] [Google Scholar]
- Sourav S.; Wachs I. E. Cr-Free, Cu Promoted Fe Oxide-Based Catalysts for High-Temperature Water-Gas Shift (HT-WGS) Reaction. Catalysts 2020, 10 (3), 305 10.3390/catal10030305. [DOI] [Google Scholar]
- de Araújo G. C.; do Rangel M. C. An Environmental Friendly Dopant for the High-Temperature Shift Catalysts. Catal. Today 2000, 62 (2–3), 201–207. 10.1016/S0920-5861(00)00421-1. [DOI] [Google Scholar]
- Natesakhawat S.; Wang X.; Zhang L.; Ozkan U. S. Development of Chromium-Free Iron-Based Catalysts for High-Temperature Water-Gas Shift Reaction. J. Mol. Catal. A: Chem. 2006, 260 (1–2), 82–94. 10.1016/j.molcata.2006.07.013. [DOI] [Google Scholar]
- Lee J. Y.; Lee D.-W.; Lee K.-Y.; Wang Y. Cr-Free Fe-Based Metal Oxide Catalysts for High Temperature Water Gas Shift Reaction of Fuel Processor Using LPG. Catal. Today 2009, 146 (1–2), 260–264. 10.1016/j.cattod.2009.01.041. [DOI] [Google Scholar]
- Wang H.; Wang G.; Wang X.; Bai J. Hydrogen Production by Redox of Cation-Modified Iron Oxide. J. Phys. Chem. C 2008, 112 (14), 5679–5688. 10.1021/jp711587m. [DOI] [Google Scholar]
- Tabakova T.; Manzoli M.; Paneva D.; Boccuzzi F.; Idakiev V.; Mitov I. CO-Free Hydrogen Production over Au/CeO2-Fe2O3 Catalysts: Part 2. Impact of the Support Composition on the Performance in the Water-Gas Shift Reaction. Appl. Catal., B 2011, 101 (3–4), 266–274. 10.1016/j.apcatb.2010.11.021. [DOI] [Google Scholar]
- Zhang F.; Zheng Q.; Wei K.; Lin X.; Zhang H.; Li J.; Cao Y. Improved Performance of Au/Fe2O3 Catalysts Promoted with ZrO2 and Nb2O5 in the WGS Reaction under Hydrogen-Rich Conditions. Catal. Lett. 2006, 108 (3–4), 131–136. 10.1007/s10562-006-0047-5. [DOI] [Google Scholar]
- Meshkani F.; Rezaei M. High Temperature Water Gas Shift Reaction over Promoted Iron Based Catalysts Prepared by Pyrolysis Method. Int. J. Hydrogen Energy 2014, 39 (29), 16318–16328. 10.1016/j.ijhydene.2014.07.176. [DOI] [Google Scholar]
- Bouarab R.; Bennici S.; Mirodatos C.; Auroux A. Hydrogen Production from the Water-Gas Shift Reaction on Iron Oxide Catalysts. J. Catal. 2014, 2014, 1–6. 10.1155/2014/612575. [DOI] [Google Scholar]
- Kappen P.; Grunwaldt J. D.; Hammershoi B. S.; Troger L.; Clausen B. S. The State of Cu Promoter Atoms in High-Temperature Shift Catalysts - An in Situ Fluorescence XAFS Study. J. Catal. 2001, 198 (1), 56–65. 10.1006/jcat.2000.3071. [DOI] [Google Scholar]
- Puig-Molina A.; Cano F. M.; Janssens T. V. W. The Cu Promoter in an Iron-Chromium-Oxide Based Water-Gas Shift Catalyst under Industrial Conditions Studied by in-Situ XAFS. J. Phys. Chem. C 2010, 114 (36), 15410–15416. 10.1021/jp1035103. [DOI] [Google Scholar]
- Estrella M.; Barrio L.; Zhou G.; Wang X.; Wang Q.; Wen W.; Hanson J. C.; Frenkel A. I.; Rodriguez J. A. In Situ Characterization of CuFe2O4 and Cu/Fe3O4 Water - Gas Shift Catalysts. J. Phys. Chem. C 2009, 113, 14411–14417. 10.1021/jp903818q. [DOI] [Google Scholar]
- Zhu M.; Wachs I. E. Determining Number of Active Sites and TOF for the High-Temperature Water Gas Shift Reaction by Iron Oxide-Based Catalysts. ACS Catal. 2016, 6 (3), 1764–1767. 10.1021/acscatal.5b02961. [DOI] [Google Scholar]
- Ladebeck J.; Kochloefl K.. Cr-Free Iron-Catalysts for Water-Gas Shift Reaction. In Studies in Surface Science and Catalysis; Elsevier, 1995; Vol. 91, pp 1079–1083. [Google Scholar]
- Na H.-S.; Jeong D.-W.; Jang W.-J.; Shim J.-O.; Roh H.-S. The Effect of Preparation Method on Fe/Al/Cu Oxide-Based Catalyst Performance for High Temperature Water Gas Shift Reaction Using Simulated Waste-Derived Synthesis Gas. Int. J. Hydrogen Energy 2015, 40 (36), 12268–12274. 10.1016/j.ijhydene.2015.07.060. [DOI] [Google Scholar]
- Liu Q.; Ma W.; He R.; Mu Z. Reaction and Characterization Studies of an Industrial Cr-Free Iron-Based Catalyst for High-Temperature Water Gas Shift Reaction. Catal. Today 2005, 106 (1–4), 52–56. 10.1016/j.cattod.2005.07.150. [DOI] [Google Scholar]
- Meshkani F.; Rezaei M. High-Temperature Water-Gas Shift Reaction over Nanostructured Cr-Free Fe2O3-Al2O3-CuO-MO (M: Ba, Ca, Mg and Sr) Catalysts for Hydrogen Production. J. Ind. Eng. Chem. 2015, 30, 353–358. 10.1016/j.jiec.2015.05.039. [DOI] [Google Scholar]
- Jeong D.-W.; Subramanian V.; Shim J.-O.; Jang W.-J.; Seo Y.-C.; Roh H.-S.; Gu J. H.; Lim Y. T. High-Temperature Water Gas Shift Reaction Over Fe/Al/Cu Oxide Based Catalysts Using Simulated Waste-Derived Synthesis Gas. Catal. Lett. 2013, 143, 438–444. 10.1007/s10562-013-0981-y. [DOI] [Google Scholar]
- Meshkani F.; Rezaei M. Promoted Fe2O3-Al2O3-CuO Chromium-Free Catalysts for High-Temperature Water-Gas Shift Reaction. Chem. Eng. Technol. 2015, 38 (8), 1380–1386. 10.1002/ceat.201400668. [DOI] [Google Scholar]
- Yamaguchi D.; Tang L.; Burke N.; Trimm D.; Chiang K.; Nguyen K. In Redox Performance of Modified Iron Oxides for Hydrogen Production, Novel Gas Conversion Symposium 9; Elsevier: Lyon, France, 2011.
- Ou X.-J.; Cheng J.-Y.; Wang H.-M.; Xiao Y. Effects of Metal Oxides on Stability and Activity of Iron-Based Chromia-Free Catalysts for Water-Gas Shift Reaction. J. Nat. Gas Chem. 1999, 231–237. [Google Scholar]
- Reddy G. K.; Boolchand P.; Smirniotis P. G. Unexpected Behavior of Copper in Modified Ferrites during High Temperature WGS Reaction-Aspects of Fe3+↔Fe2+ Redox Chemistry from Mossbauer and XPS Studies. J. Phys. Chem. C 2012, 116 (20), 11019–11031. 10.1021/jp301090d. [DOI] [Google Scholar]
- Reddy G. K.; Smirniotis P. G. Effect of Copper as a Dopant on the Water Gas Shift Activity of Fe/Ce and Fe/Cr Modified Ferrites. Catal. Lett. 2011, 141 (1), 27–32. 10.1007/s10562-010-0465-2. [DOI] [Google Scholar]
- Reddy G. K.; Gunasekara K.; Boolchand P.; Smirniotis P. G. Cr- and Ce-Doped Ferrite Catalysts for the High Temperature Water-Gas Shift Reaction: TPR and Mossbauer Spectroscopic Study. J. Phys. Chem. C 2011, 115 (4), 920–930. 10.1021/jp102959p. [DOI] [Google Scholar]
- Trovarelli A.; de Leitenburg C.; Dolcetti G. Design Better Cerium-Based Oxidation Catalysts. Chemtech 1997, 27, 32–37. [Google Scholar]
- Phivilay S. P.; Puretzky A. A.; Domen K.; Wachs I. E. Nature of Catalytic Active Sites Present on the Surface of Advanced Bulk Tantalum Mixed Oxide Photocatalysts. ACS Catal. 2013, 3 (12), 2920–2929. 10.1021/cs400662m. [DOI] [Google Scholar]
- Zhu M.; Chen J.; Shen L.; Ford M. E.; Gao J.; Xu J.; Wachs I. E.; Han Y. Probing the Surface of Promoted CuO-Cr2O3-Fe2O3 Catalysts during CO2 Activation. Appl. Catal., B 2020, 271, 118943 10.1016/j.apcatb.2020.118943. [DOI] [Google Scholar]
- Morris M. C.; McMurdie H. F.; Evans E. H.; Paretzkin B.; Parker H. S.; Panagiotopoulos N. C.; Hubbard C. R.. Standard X-Ray Diffraction Powder Patterns : Section 18- Data for 58 Substances; National Bureau of Standards: Gaithersburg, MD, 1981, p 37.
- Cava S.; Tebcherani S. M.; Souza I. A.; Pianaro S. A.; Paskocimas C. A.; Longo E.; Varela J. A. Structural Characterization of Phase Transition of Al2O3 Nanopowders Obtained by Polymeric Precursor Method. Mater. Chem. Phys. 2007, 103 (2–3), 394–399. 10.1016/j.matchemphys.2007.02.046. [DOI] [Google Scholar]
- Sudarsanam P.; Mallesham B.; Durgasri D. N.; Reddy B. M. Physicochemical Characterization and Catalytic CO Oxidation Performance of Nanocrystalline Ce-Fe Mixed Oxides. RSC Adv. 2014, 4 (22), 11322–11330. 10.1039/C3RA45778H. [DOI] [Google Scholar]
- Shannon R. D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr., Sect. A 1976, 32 (5), 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Cornell R. M.; Schwertmann U.. The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, 2003; p 147. [Google Scholar]
- Perez-Alonso F. J.; López Granados M.; Ojeda M.; Herranz T.; Rojas S.; Terreros P.; Fierro J. L. G.; Gracia M.; Gancedo J. R. Relevance in the Fischer–Tropsch Synthesis of the Formation of Fe-O-Ce Interactions on Iron-Cerium Mixed Oxide Systems. J. Phys. Chem. B 2006, 110 (47), 23870–23880. 10.1021/jp064575f. [DOI] [PubMed] [Google Scholar]
- Shebanova O. N.; Lazor P. Raman Spectroscopic Study of Magnetite (FeFe2O4): A New Assignment for the Vibrational Spectrum. J. Solid State Chem. 2003, 174 (2), 424–430. 10.1016/S0022-4596(03)00294-9. [DOI] [Google Scholar]
- Shan W.; Shen W.; Li C. Structural Characteristics and Redox Behaviors of Ce1-XCuxOy Solid Solutions. Chem. Mater. 2003, 15 (25), 4761–4767. 10.1021/cm0345974. [DOI] [Google Scholar]
- Anantharaman M. R.; Reijne S.; Jacobs J. P.; Brongersma H. H.; Smits R. H. H.; Seshan K. Preferential Exposure of Certain Crystallographic Planes on the Surface of Spinel Ferrites: A Study by LEIS on Polycrystalline Spinel Ferrite Surfaces. J. Mater. Sci. 1999, 34, 4279–4283. [Google Scholar]
- de Ridder M.; van de Ven P. C.; van Welzenis R. G.; Brongersma H. H.; Helfensteyn S.; Creemers C.; Van Der Voort P.; Baltes M.; Mathieu M.; Vansant E. F. Growth of Iron Oxide on Yttria-Stabilized Zirconia by Atomic Layer Deposition. J. Phys. Chem. B 2002, 106, 13146–13153. 10.1021/jp0211640. [DOI] [Google Scholar]
- Lyubutin I. S.; Lin C. R.; Korzhetskiy Y. V.; Dmitrieva T. V.; Chiang R. K. Mössbauer Spectroscopy and Magnetic Properties of Hematite/Magnetite Nanocomposites. J. Appl. Phys. 2009, 106, 034311 10.1063/1.3194316. [DOI] [Google Scholar]