

Abstract
Rationale
The small airway epithelium (beyond the sixth generation), the initiation site of smoking-induced airway disorders, is highly sensitive to the stress of smoking. Because of variations over time in smoking habits, the small airway epithelium transcriptome is dynamic, fluctuating not only among smokers but also within each smoker.
Objectives
To perform accurate assessment of the smoking-related dysregulation of the human small airway epithelium despite the variation of smoking within the same individual and of the effects of smoking cessation on the dysregulated transcriptome.
Methods
We conducted serial sampling of the same smokers and nonsmoker control subjects over time to identify persistent smoking dysregulation of the biology of the small airway epithelium over 1 year. We conducted serial sampling of smokers who quit smoking, before and after smoking cessation, to assess the effect of smoking cessation on the smoking-dysregulated genes.
Measurements and Main Results
Repeated measures ANOVA of the small airway epithelium transcriptome sampled four times in the same individuals over 1 year enabled the identification of 475 persistent smoking-dysregulated genes. Most genes were normalized after 12 months of smoking cessation; however, 53 (11%) genes, including CYP1B1, PIR, ME1, and TRIM16, remained persistently abnormally expressed. Dysregulated pathways enriched with the nonreversible genes included xenobiotic metabolism signaling, bupropion degradation, and nicotine degradation.
Conclusions
Analysis of repetitive sampling of the same individuals identified persistent smoking-induced dysregulation of the small airway epithelium transcriptome and the effect of smoking cessation. These results help identify targets for the development of therapies that can be applicable to smoking-related airway diseases.
Keywords: transcriptome, repeated measures, cessation, common versus individual dysregulation
At a Glance Commentary
Scientific Knowledge on the Subject
There is a large variation in smoking habits over time for each individual smoker. To identify the genes that are consistently dysregulated by smoking despite these variabilities, we repetitively sampled the small airway epithelium, the initial site of smoking-related lung pathology, four times over 1 year, permitting assessment of variable responses of the small airway epithelium transcriptome over time. Sampling and transcriptome assessment of the small airway epithelium in smokers before and after smoking cessation demonstrated the effect of smoking cessation and identified the genes that were not reversible and remained persistently dysregulated despite 12 months of smoking cessation.
What This Study Adds to the Field
These data provide insights into the dynamic response of the human genome to environmental stress and a roadmap to identify genes commonly dysregulated in the population, as well as genes responsive and nonresponsive to cessation of the environmental stress.
In the United States, cigarette smoking is the primary cause of chronic obstructive pulmonary disease (COPD), the third leading cause of death; and lung cancer, the most common lethal cancer (1–3). Although smoking eventually causes pathology throughout the lung, the pathogenesis of COPD and most lung cancers start in the small airway epithelium (SAE) (4–9).
The cigarette smoking–SAE interaction is an example of the complexity of deciphering environment–gene interactions in humans. Identification of the biologic responses to the environment is dictated, in part, by individual-to-individual differences in the inherited genome, variability in the stress imposed on the cell population exposed to the environment, and variations in the methodology of sampling and technology used to quantify biologic changes (10–15). Single time point assessment of the effect of cigarette smoking on the airway epithelium has demonstrated that both the SAE and the large airway epithelium are highly sensitive to the stress of smoking, and exposure to even low concentrations of cigarette smoke evokes significant changes in the SAE transcriptome (11, 16–22). However, the transcriptome of the cell population is dynamic, fluctuating over time in response to varying exogenous signals such as smoking; that is, single time point assessment provides only a snapshot of the SAE transcriptome, based on the smoking status at the time of sampling. Not only are there individual-to-individual genome differences that dictate the SAE transcriptome responses, but the environmental stress on humans cannot be controlled. Because smoking habits vary over time for each individual smoker, there must be significant individual fluctuations in smoking-induced SAE gene dysregulation over time. Adding complexity to this assessment, exposure to pollution contributes to the time-dependent dysregulation of the SAE transcriptome (21).
On the basis of recognition of these variables, we hypothesized that serial sampling of the same smokers and nonsmoker control subjects over time would provide a more accurate assessment of the effect of smoking on the human SAE. Integrating the variation of smoking within the same individuals over time and between individual smokers would identify the persistent smoking-induced dysregulation of the biology of the SAE over 1 year. To accomplish this, fiberoptic bronchoscopy was used to repetitively sample the SAEs of 37 healthy smokers and 23 nonsmokers four times over 1 year (baseline and 3, 6, and 12 mo), permitting assessment of the variable smoking habits and responses of the SAE transcriptome over 1 year. This strategy permitted identification of the genes that are persistently dysregulated in response to smoking. Confirmation of the persistent smoking-dysregulated genes was based on assessment of two replication cohorts. Finally, in a separate cohort of 17 healthy smokers who were sampled while smoking and then repetitively over 1 year after smoking cessation (3, 6, and 12 mo), we assessed the effect of smoking cessation on the SAE dysregulated genes and determined which genes do not revert to normal expression levels after smoking cessation. The identification of the genes that are persistently dysregulated by smoking, and the effect of smoking cessation on the regulation of these genes, helps identify targets for the development of therapies that can be applicable to smoking-related airway diseases. Some of the results of these studies were previously reported in the form of abstracts (23–28).
Methods
Study Population
Four cohorts were evaluated: 1) the primary cohort of nonsmokers (n = 23) and smokers (n = 37) underwent serial sampling of the SAE four times over 1 year (baseline and 3, 6, and 12 mo); 2) replication cohort 1 of nonsmokers (n = 60) and smokers (n = 74), sampled once; 3) replication cohort 2 of nonsmokers (n = 20) and smokers (n = 23), sampled once; and 4) cessation cohort of smokers (n = 17) who agreed to stop smoking (quitters), sampled before (baseline) and after smoking cessation (3, 6, and 12 mo). All subjects were recruited from New York City, and their smoking status was confirmed by urine tobacco metabolites at each time point (see the Supplemental Methods section in the online supplement for recruitment and inclusion and exclusion criteria).
SAE Sampling, Processing, and Analysis
SAE samples were collected from 10th- to 12th-order bronchi using flexible bronchoscopy as previously described (18) and as detailed in the Supplemental Methods section. For the primary cohort, replication cohort 1, and the cessation cohort, transcriptome data were collected using Affymetrix HG-U133 Plus 2.0 Microarray GeneChips (Affymetrix), as previously described (11, 19), and for replication cohort 2, transcriptome data were collected by RNA sequencing using the Illumina HiSeq 2500 platform, as previously described (29). See the Supplemental Methods section for details on data processing protocols and methods. For all cohorts, the raw data are publicly available from the National Center for Biotechnology Information Gene Expression Omnibus website (www.ncbi.nlm.nih.gov/geo/) under accession number GSE133924.
The primary cohort was used to identify genes persistently dysregulated by smoking over 1 year using repeated measures ANOVA with a repeated factor for measurement time and factors for smoking status, age, and sex, applied using R (https://svn.r-project.org/R-packages/trunk/nlme) (see the Supplemental Methods section for code details). Smokers were compared with nonsmokers genome-wide (n = 14,676 genes), and significance was based on P < 0.05 (adjusted using Benjamini-Hochberg correction [30]) and fold difference of expression ⩾1.5. An additional characterization was applied to the persistently smoking-dysregulated genes identified by comparing the distribution of expression in smokers with that in nonsmokers for each gene. See the Supplemental Methods section for analysis details. Replication cohorts 1 and 2, consisting of samples obtained at a single time point, were used to confirm the smoking-dysregulated genes identified in the primary cohort. To identify the smoking-dysregulated genes in each of the replication cohorts, ANOVA was applied, in which a gene was considered to be replicated if expression was significant at a Benjamini-Hochberg (30) P value adjusted for multiple testing <0.05 and fold change of gene expression ⩾1.5, and the directionality of the dysregulation was the same in the replication cohort as in the primary cohort. See the Supplemental Methods section for additional details.
The cessation cohort was used to assess the effect of smoking cessation on the persistent smoking-dysregulated genes identified in the primary cohort. The expression of the smoking-dysregulated genes was compared in the cessation cohort and nonsmokers at each time point after cessation (Months 3, 6, and 12), and genes were defined as “nonreversible” if the Benjamini-Hochberg corrected P value was <0.05 at all three time points after cessation); “reversible” if the P value was ⩾0.05 compared with nonsmokers at Month 3, Month 6, or Month 12 and all subsequent time points; or “intermittent reversible” if the Benjamini-Hochberg corrected P value was ⩾0.05 at Month 3 or 6 but not at all subsequent time points (Month 6 and/or Month 12, respectively). For the reversible genes, the speed of reversibility was defined on the basis of the first time point at which the P value was ⩾0.05 (after 3, 6, or 12 mo of smoking cessation). As an additional evaluation of the effect of smoking cessation on the expression of the smoking-dysregulated genes identified in the primary cohort, the cessation cohort was compared with smokers at each time point (Months 3, 6, and 12). Genes with a Benjamini-Hochberg corrected P value <0.05 were considered significantly different between the groups, and the overlap of these genes with the nonreversible genes identified by comparing the cessation cohort with the nonsmokers at each time point was calculated. See Figure E1 in the online supplement for a description of the analysis.
Gene Ontology and the Human Protein Reference Database (www.hprd.org) were used for functional annotation, and Ingenuity Pathway Analysis (QIAGEN 2019) was used to identify enriched pathways. See the Supplemental Methods section for additional analysis details.
Results
The primary cohort included 23 nonsmokers and 37 smokers who underwent bronchoscopy for sampling of the SAE four times over 1 year (baseline and 3, 6, and 12 mo). All subjects had normal spirometry, DlCO, and alpha-1 antitrypsin concentrations (Table 1). As expected, cough scores and sputum scores were higher and FEV1/FVC ratio was lower in the smokers and quitters than in the nonsmokers. There was large variability in the baseline smoking habits among the smokers in the primary cohort and within each smoker over 1 year (Figure 1, Figure E2, and the Supplemental Results section). The expression level of each gene varied over time for each individual where the variability across individuals correlated with the sample mean expression across all time points (Figure E3). The variability of genes differed among genes, and there were subtle differences in the variability between smokers and nonsmokers.
Table 1.
Demographics of Primary Cohort and Cessation Cohort*
| Parameters | Primary Cohort |
Cessation Cohort |
P Value† |
|||
|---|---|---|---|---|---|---|
| Nonsmokers | Smokers | S vs. NS | Quitters vs. NS | Quitters vs. S | ||
| No. of subjects | 23 | 37 | 17 | — | — | — |
| Sex, male/female | 13/10 | 31/6 | 10/7 | <0.03 | >0.8 | <0.05 |
| Age, yr | 36 ± 12 | 44 ± 8 | 45 ± 10 | <0.003 | <0.02 | >0.8 |
| Ethnicity, AA/E/H/O | 9/6/8/0 | 20/7/10/0 | 10/2/3/2 | >0.9 | >0.5 | >0.5 |
| BMI, kg/m2 | 27 ± 5 | 26 ± 4 | 30 ± 4 | >0.3 | >0.06 | <0.002 |
| Smoking history‡ | ||||||
| Pack-year | N/A | 25 ± 13 | 19 ± 8 | N/A | N/A | >0.08 |
| Packs per day | N/A | 0.9 ± 1.0 | 0.6 ± 0.2 | N/A | N/A | <0.01 |
| Age of initiation, yr | N/A | 16 ± 4 | 16 ± 3 | N/A | N/A | >0.9 |
| Urine cotinine, ng/ml | N/A | 1,636 ± 799 | 1,376 ± 953 | N/A | N/A | >0.3 |
| Cough score§ | 0.5 ± 0.6 | 1.7 ± 1.0 | 1.7 ± 1.4 | <10−3 | <0.006 | >0.9 |
| Sputum score§ | 0.5 ± 0.5 | 1.5 ± 1.0 | 1.1 ± 1.1 | <10−3 | <0.04 | >0.2 |
| Alpha-1 antitrypsin, mg/dl‖ | 149 ± 26 | 147 ± 20 | 144 ± 16 | >0.7 | >0.5 | >0.6 |
| Lung function¶ | ||||||
| FVC, % predicted | 106 ± 11 | 110 ± 10 | 113 ± 14 | >0.1 | >0.1 | >0.7 |
| FEV1, % predicted | 106 ± 12 | 109 ± 10 | 105 ± 16 | >0.3 | >0.7 | >0.8 |
| FEV1/FVC, % observed | 83 ± 54 | 81 ± 4 | 76 ± 6 | <0.03 | <0.03 | >0.3 |
| TLC, % predicted | 97 ± 17 | 96 ± 13 | 96 ± 10 | >0.8 | >0.8 | >0.9 |
| DlCO, % predicted | 92 ± 12 | 87 ± 9 | 90 ± 6 | >0.1 | >0.5 | >0.3 |
Definition of abbreviations: AA = African American; BMI = body mass index; E = European; H = Hispanic; NS = nonsmokers (primary cohort); N/A = not applicable; O = Other; S = smokers.
Subjects were followed for 1 year. Detailed data are from the baseline visit.
Quitters were the cessation cohort.
Smoking status was verified at each time point by urine nicotine and its derivative cotinine; baseline smoking was defined as minimum urine cotinine concentration of 104 ng/ml (11); active smokers and nonsmokers maintained their smoking status at each time point, quitters quit smoking immediately after the baseline visit. See the Supplemental Methods for detailed criteria of smoking status definition.
Cough and sputum scores were each evaluated on a scale of 0–4: 0 = not at all; 1 = only with chest infections; 2 = a few days per month; 3 = several days per week; 4 = most days of the week (68).
All subjects had negative test results for HIV and had normal concentrations of alpha-1 antitrypsin.
Lung function parameters are presented as prebronchodilator values and as percent predicted, except the FEV1/FVC ratio, which is presented as percentage observed. The DlCO was corrected for hemoglobin and carboxyhemoglobin (69).
Figure 1.

Variability of smoking over 1 year. All data are from the primary cohort. (A–C) Smoking history at the baseline of the 1-year study of 37 smokers based on self-reported history. Data are presented for each smoker (circles), using a box plot for the median for all smokers. (D and E) Examples of variability in urine cotinine concentrations over 1 year (baseline and 3, 6, and 12 mo). (A) Packs per day. (B) Pack-year history. (C) Age of smoking initiation. (D) Examples of smokers with constant levels. (E) Examples of smokers with variable levels.
Persistent Smoking-dysregulated Genes
The repeated measures ANOVA analysis identified 475 genes persistently dysregulated by smoking at each of the four time points. A correlation analysis of smoking intensity (cotinine concentration) with expression level demonstrated that there was no correlation for any of the 475 genes identified as persistently dysregulated (P value adjusted with Benjamini-Hochberg for multiple test correction >0.05, all genes), perhaps indicating that smoking intensity in this cohort had a threshold impact on these smoking-responsive genes such that slight differences in smoking intensity did not correlate with changes in the expression of these genes, as previously demonstrated by Strulovici-Barel and colleagues (11). Of the 475 persistent smoking-dysregulated genes, 236 (50%) were downregulated and 239 (50%) were upregulated in smokers versus nonsmokers (Figure 2A). The most common categories of smoking-dysregulated genes were metabolism related (n = 80; 17%) and signal transduction related (n = 59; 12%) (Figure 2B). Interestingly, for 74 (16%) persistent smoking-dysregulated genes, the function is unknown (Figures 2B and 2C, Table E1). The top 10 pathways enriched with smoking-dysregulated genes included antigen presentation pathway, B cell development, neuroinflammation signaling pathway, and nicotine degradation (Table E2). See Supplemental Results, Figures E4–E8, and Tables E6–E22 for additional results of the comparison of smoker versus nonsmoker distributions for these 475 genes.
Figure 2.
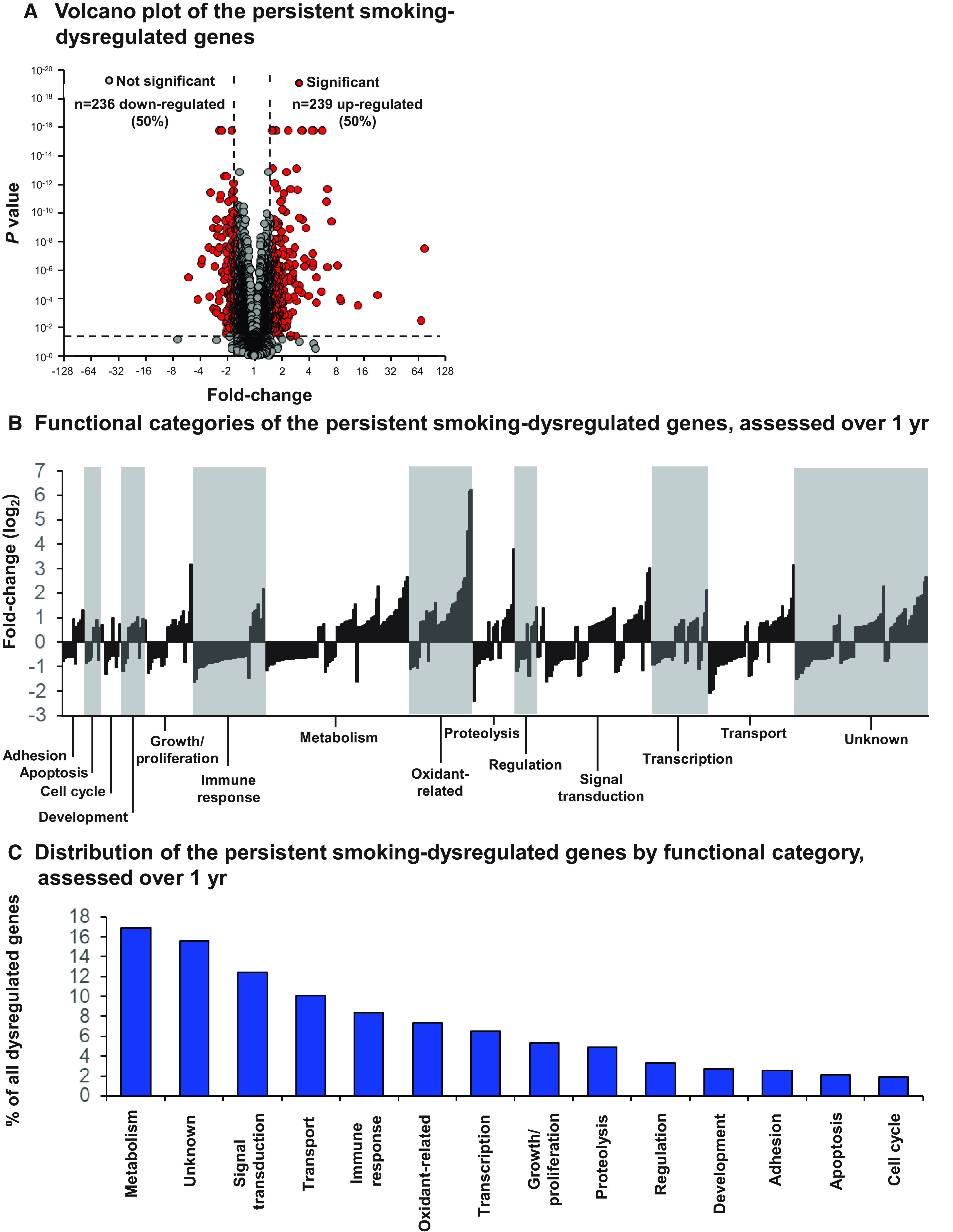
Persistent smoking-dysregulated genes in the small airway epithelium assessed over 1 year in the primary cohort of smokers and nonsmokers (baseline [M0], 3 [M3], 6 [M6], and 12 [M12] months) using repeated measures ANOVA. (A) Volcano plot of the persistent smoking-dysregulated genes, assessed over 1 year. Date is presented as fold change (x-axis) versus corrected P value (Benjamini-Hochberg [30]) over the four time points (y-axis). Each dot represents one gene. Gray dots represent nonsignificant genes; red dots represent the significant persistent smoking-dysregulated genes. Dashed lines represent significance threshold. (B and C) Functional categories of the persistent smoking-dysregulated genes, assessed over 1 year. (B) Each bar represents one gene. The data are presented as fold change (log2) of gene expression in smokers versus nonsmokers. White and shaded areas are used to separate different functional categories. (C) Distribution of all genes by functional category.
Replication
The persistent smoking-dysregulated genes were confirmed using two replication cohorts: 1) 60 nonsmokers and 74 smokers with SAE samples obtained at one time point (replication cohort 1) and 2) 20 nonsmokers and 23 smokers sampled once and processed on a different platform from the primary cohort (RNA sequencing rather than microarray, replication cohort 2). As in the primary cohort, all subjects in these two replication cohorts had normal spirometry, DlCO, and alpha-1 antitrypsin concentrations (Table E3). Three hundred seventy-three (79%) persistently dysregulated genes from the primary cohort were also dysregulated in the same direction in replication cohort 1, and 262 (55%) were also dysregulated in the same direction in replication cohort 2 (Table E4). The overlap of these genes in both replication cohorts in the same direction was for 195 genes (41%). This overlap was significantly higher than with previously identified smoking-dysregulated genes in the large airway epithelium and nasal epithelium sampled at one time point (6–10%) (17, 31–33). The pathways enriched with the 195 genes included nicotine degradation and xenobiotic metabolism signaling (Table E5).
Effect of Smoking Cessation on the Expression of SAE Persistently Dysregulated Genes
Seventeen smokers agreed to quit smoking immediately after the initial sampling of the SAE; the “cessation cohort”). All had normal spirometry, DlCO, and alpha-1 antitrypsin concentrations (Table 1). The smoking status of all subjects was confirmed at each time point using tobacco urine metabolites (Figure 3A, Supplemental Results).
Figure 3.

The effect of smoking cessation over 1 year. Seventeen smokers who quit smoking immediately after the baseline bronchoscopy and refrained from smoking for 1 year (cessation cohort) were compared with 37 smokers who continued smoking and 23 nonsmokers who remained nonsmokers for 1 year (primary cohort). All subjects were sampled four times over 1 year (baseline and 3, 6, and 12 mo). (A) Urine cotinine concentrations over 1 year. Data are presented as the average of all subjects at each time point ±2 SD. (B and C) PCA collapsing the expression levels of all persistent smoking-dysregulated genes. (B) Baseline small airway epithelium expression of the persistent smoking-dysregulated genes. (C) Small airway epithelium expression after 1 year. (D) Speed of reversibility for all reversible genes by functional category. PC = Principal component; PCA = Principal component analysis.
Repetitive sampling of the SAE of these individuals before (baseline) and after smoking cessation (3, 6, and 12 mo) demonstrated that smoking cessation significantly affected the SAE transcriptome as measured by the expression level of the 475 persistent smoking-dysregulated genes. Principal component analysis demonstrated the differences and similarities in gene expression of all 475 genes among the smokers before and after quitting compared with nonsmokers. Although at baseline the quitters (before quitting) overlapped with the smokers, separately from the nonsmokers (Figure 3B), after 12 months of smoking cessation, the quitters overlapped with the nonsmokers, separately from the active smokers (Figure 3C).
Analysis of the effect of smoking cessation on the expression of each persistent smoking-dysregulated gene showed that, over 12 months, the expression of 320 (67%) genes reverted to that of the nonsmokers, and they were considered “reversible,” whereas the expression of 102 (21%) genes reverted to that of the nonsmokers at Month 3 or Month 6, but not at all subsequent time points, and they were considered “intermittent reversible.” However, the expression of 53 (11%) genes never reverted and remained persistently dysregulated after 12 months of smoking cessation, and they were considered “nonreversible” (Table 2, Table E1). Pathways enriched with the nonreversible genes included xenobiotic metabolism signaling, bupropion degradation, and nicotine degradation (Table E23). As expected, there was a significant overlap between genes not found to be significant in the cessation versus nonsmoker analysis and genes found to be significant in the cessation versus smoker analysis at each time point, where the overlap was 95% at 3 months, 98% at 6 months, and 100% at 12 months, further confirming the reversibility or irreversibility of these genes.
Table 2.
Small Airway Epithelium Genes Persistently Dysregulated Despite 1 Year of Smoking Cessation
| Gene | Gene Name | Functional Category | P Value | BH-Corrected P Value*† |
Fold Change*‡ |
|---|---|---|---|---|---|
| ME1 | Malic enzyme 1 | Oxidant related | <1 × 10−16 | <1 × 10−16 | 4.34 |
| PIR | Pirin | Oxidant related | 1.11 × 10−16 | 8.92 × 10−14 | 2.93 |
| SLC2A1 | Solute carrier family 2 member 1 | Transport | 1.11 × 10−16 | 8.92 × 10−14 | 1.59 |
| TRIM16 | Tripartite motif-containing 16 | Transcription | 2.22 × 10−16 | 1.53 × 10−13 | 2.27 |
| NPAS3 | Neuronal PAS domain protein 3 | Transcription | 4.44 × 10−16 | 2.79 × 10−13 | −2.12 |
| CYP4X1 | Cytochrome P450 family 4 subfamily X member 1 | Oxidant related | 1.33 × 10−15 | 8.03 × 10−13 | −1.70 |
| WNK4 | WNK lysine-deficient protein kinase 4 | Signal transduction | 1.33 × 10−14 | 5.67 × 10−12 | −2.34 |
| RTN4RL1 | Reticulon 4 receptor-like 1 | Metabolism | 5.13 × 10−14 | 1.85 × 10−11 | −1.93 |
| GALNT6 | Polypeptide N-acetylgalactosaminyl transferase 6 | Metabolism | 2.03 × 10−13 | 6.11 × 10−11 | 2.07 |
| S100A10 | S100 calcium binding protein A10 | Signal transduction | 2.39 × 10−12 | 4.94 × 10−10 | 1.82 |
| NAV1 | Neuron navigator 1 | Unknown | 3.11 × 10−12 | 5.78 × 10−10 | 1.84 |
| NOL3 | Nucleolar protein 3 | Metabolism | 3.57 × 10−12 | 6.22 × 10−10 | 1.51 |
| CLDN10 | Claudin 10 | Transport | 9.96 × 10−12 | 1.45 × 10−9 | 2.45 |
| KDELR3 | KDEL endoplasmic reticulum protein retention receptor 3 | Transport | 1.56 × 10−11 | 2.18 × 10−9 | 2.00 |
| FAM110C | Family with sequence similarity 110, member C | Cell cycle | 6.26 × 10−11 | 6.97 × 10−9 | 1.68 |
| CHST15 | Carbohydrate sulfotransferase 15 | Metabolism | 9.70 × 10−11 | 1.02 × 10−8 | 1.58 |
| GALNT7 | GalNAc transferase 7 | Metabolism | 2.25 × 10−10 | 2.17 × 10−8 | 1.55 |
| FHOD3 | Formin homology 2 domain containing 3 | Growth/proliferation | 3.31 × 10−10 | 2.75 × 10−8 | −1.89 |
| CYP1B1 | Cytochrome P450, family 1, subfamily B, polypeptide 1 | Oxidant related | 4.04 × 10−10 | 3.25 × 10−8 | 76.06 |
| PLAG1 | PLAG1 zinc finger | Transcription | 4.62 × 10−10 | 3.59 × 10−8 | −1.51 |
| SLC29A1 | Solute carrier family 29, member 1 | Transport | 1.02 × 10−9 | 6.79 × 10−8 | −1.72 |
| KCNK6 | Potassium two pore domain channel subfamily K member 6 | Signal transduction | 1.47 × 10−9 | 9.18 × 10−8 | 1.64 |
| LOC102724094 | LOC102724094 | Unknown | 1.87 × 10−9 | 1.13 × 10−7 | 2.22 |
| LOC105378604 | Uncharacterized LOC105378604 | Unknown | 3.66 × 10−9 | 2.00 × 10−7 | −1.68 |
| MSRB1 | LDL receptor-related protein 4 | Oxidant related | 4.13 × 10−9 | 2.17 × 10−7 | 1.62 |
| PEG10 | Paternally expressed 10 | Apoptosis | 4.20 × 10−9 | 2.19 × 10−7 | −1.74 |
| S100A14 | S100 calcium binding protein A14 | Apoptosis | 5.89 × 10−9 | 2.65 × 10−7 | 1.63 |
| LRP4 | Low-density lipoprotein receptor-related protein 4 | Development | 1.35 × 10−8 | 5.63 × 10−7 | 1.86 |
| TMCC3 | Transmembrane and coiled-coil domain family 3 | Unknown | 1.45 × 10−8 | 5.85 × 10−7 | 2.01 |
| LOC284825 | Long intergenic nonprotein coding RNA 1697 | Unknown | 1.60 × 10−8 | 6.36 × 10−7 | 6.35 |
| KCNB1 | Potassium voltage-gated channel subfamily B member 1 | Transport | 1.65 × 10−8 | 6.44 × 10−7 | −1.66 |
| CDC42EP5 | CDC42 effector protein 5 | Growth/proliferation | 2.13 × 10−8 | 7.88 × 10−7 | 1.87 |
| DPP10 | Dipeptidylpeptidase 10 | Proteolysis | 2.48 × 10−8 | 8.93 × 10−7 | 1.83 |
| FOXA2 | Forkhead box A2 | Transcription | 4.87 × 10−8 | 1.56 × 10−6 | −1.67 |
| LHX6 | LIM homeobox 6 | Growth/proliferation | 5.72 × 10−8 | 1.76 × 10−6 | 1.85 |
| SIX3 | SIX homeobox 3 | Regulation | 5.73 × 10−8 | 1.76 × 10−6 | 2.67 |
| GAD1 | Glutamate decarboxylase 1 | Metabolism | 1.16 × 10−7 | 3.20 × 10−6 | 4.79 |
| LFNG | LFNG O-fucosylpeptide 3-β-N-acetylglucosaminyltransferase | Signal transduction | 2.01 × 10−7 | 4.93 × 10−6 | 1.63 |
| DPP10-AS1 | DPP10 antisense RNA 1 | Unknown | 2.33 × 10−7 | 5.58 × 10−6 | 1.75 |
| MATN2 | Matrilin 2 | Growth/proliferation | 2.60 × 10−7 | 6.08 × 10−6 | 2.42 |
| NR4A3 | Nuclear receptor subfamily 4 group A member 3 | Transcription | 3.07 × 10−7 | 7.01 × 10−6 | 1.87 |
| LOC100507560 | Uncharacterized LOC100507560 | Metabolism | 2.85 × 10−6 | 4.56 × 10−5 | −1.73 |
| PRKCB | Protein kinase C, β | Signal transduction | 5.15 × 10−6 | 7.42 × 10−5 | −3.07 |
| PDGFRL | Platelet-derived growth factor receptor-like | Proteolysis | 5.29 × 10−6 | 7.59 × 10−5 | 1.80 |
| GSTM3 | Glutathione S-transferase mu 3 | Metabolism | 2.16 × 10−5 | 0.0002 | 1.80 |
| UCHL1 | Ubiquitin C-terminal hydrolase L1 | Proteolysis | 2.76 × 10−5 | ||
| CCEPR | Cervical carcinoma expressed PCNA regulatory LncRNA | Unknown | 3.99 × 10−5 | 0.0004 | 1.52 |
| KRT40 | Keratin 40 | Unknown | 6.32 × 10−5 | 0.0006 | 1.68 |
| DAPK1 | Death-associated protein kinase 1 | Apoptosis | 0.0002 | 0.001 | −1.62 |
| FAM3B | Family with sequence similarity 3, member B | Apoptosis | 0.0002 | 0.002 | 1.52 |
| DNAJC12 | DnaJ homolog, subfamily C, member 12 | Transport | 0.0003 | 0.002 | 1.70 |
| DEFB1 | Defensin, β1 | Immune response | 0.0007 | 0.004 | 2.29 |
| KIAA1211 | KIAA1211 | Unknown | 0.002 | 0.01 | 1.62 |
| CDH2 | Cadherin 2 | Adhesion | 0.004 | 0.02 | 1.92 |
Calculated in the primary cohort assessed over 1 year.
BH correction was adjusted using Benjamini-Hochberg (BH) correction (30).
A positive fold change represents upregulation in expression in smokers compared with nonsmokers; a negative fold change represents a downregulation in smokers compared with nonsmokers.
Of the reversible genes, 204 (64%) reverted to normal within 3 months, 263 (82%) within 6 months and 320 (100%) within 12 months. When analyzed per functional category, the most reversible genes were adhesion and immune response related (78% and 75% reversibility, respectively, or 100% and 98% reversibility or intermittent reversibility, after 12 mo of smoking cessation, respectively) (Figure 3D). The least reversible genes were apoptosis related and growth and proliferation related (50% and 16% of the genes remaining abnormal after 12 mo of smoking cessation, respectively). Among the reversible genes, regulation- and transcription-related genes had the fastest reversibility, with 92% and 83% of the genes normalized within 3 months and 100% and 96% of the genes within 6 months, respectively. In contrast, oxidant-, adhesion-, and metabolism-related genes had the slowest reversibility, with only 35%, 44%, and 47% of the genes normalized after 3 months, respectively.
Discussion
The effects of cigarette smoking on the human airways have been studies in vitro by continuously exposing cultured lung cells to cigarette smoke extract (CSE) under static conditions. Most in vitro studies followed the effects of a one-time or short-term exposure; however, some studies assessed the effects of chronic and repetitive exposure of epithelial lung cells to CSE, mimicking the repetitive exposure to smoke in cigarette smokers. These studies demonstrated cumulative and dose- and time-responsive damage to the exposed tissue (34–38). Assessment of the cultures after removal of the chronic exposure to CSE demonstrated either only a partial normalization of the dysregulated transcriptome or a full normalization only after a long period of time with no exposure (34, 36, 39).
Considerable attention has been given to in vivo fluctuations of the human transcriptome of various cell populations secondary to variability in the technology used to assess the transcriptome (31, 40, 41), and a variety of human studies have been performed to assess changes in the human transcriptome secondary to inherited variability, a single stress, or therapeutic interventions (42–52). However, to our knowledge, no human studies have evaluated in vivo the consequences to the transcriptome of a cell population exposed to variations of the same type of stress over time, a scenario similar to real life, where the environment is constantly changing. As an example, to quantify environment (smoking) and gene fluctuations (SAE) in the human transcriptome over time, we repetitively evaluated the SAE transcriptome over 1 year in smokers, nonsmokers, and quitters.
The SAE, covering the 6th to 23rd generations of the bronchial tree, is the cell population in which the pathogenesis of COPD and most lung cancers is initiated (1, 2, 4–9). The SAE transcriptome is impacted by smoking, where the dynamic impact of smoking over time may have a consequence for lung disease. To identify the persistent smoking-dysregulated genes, in a departure from the conventional single time point analysis of the airway epithelia of nonsmokers and smokers that we and others have used in prior studies (11, 13–22), we sampled the SAE of the same 37 smokers and 23 nonsmokers repetitively over 1 year at baseline and 3, 6, and 12 months. This approach demonstrated dynamic changes in the SAE transcriptome over time and enabled a robust identification of a persistent SAE transcriptome dysregulation in smokers versus nonsmokers integrated over 1 year. Also, in a separate cohort of 17 smokers who agreed to quit smoking, we used repetitive sampling of the SAE at baseline and over 1 year after smoking cessation to assess the effect of smoking cessation on the smoking-dysregulated genes.
Repeated Measures of the Same SAE over 1 Year
Analysis of repeated measures of the same smokers over time demonstrated that the SAE transcriptome of these individuals is dynamic, varying considerably over time. There are several possible reasons for this, including variability in smoking habits over time, variability in air pollution and other environmental factors affecting the SAE transcriptome (21), and variability in sample collection and analytical methods. Despite this variability, comparing the smokers with nonsmokers using data from the same individual over 1 year on the basis of repeated measures ANOVA established an integrated assessment of the effects of smoking on the SAE transcriptome and identified genes persistently dysregulated by smoking. Many of these genes were metabolism, signal transduction, and transport related. Analysis of two replication cohorts that used different methodologies to assess the SAE transcriptome confirmed the dysregulation of most of the identified persistent genes.
Effects of Smoking Cessation
Epidemiologic studies have established that smoking significantly accelerates the rate of age-dependent loss in pulmonary function (2, 53, 54) and that smoking cessation slows down the smoking-related decline of lung function (55–57). Likewise, the increased risk for COPD and lung cancer declines with smoking cessation, but, even after years of smoking cessation, a previous smoker still has an increased risk for COPD and bronchogenic carcinoma (2, 53–59). Consistent with these epidemiologic observations, the data in the present study demonstrated that the smoking-induced dysregulation of the SAE is not completely reversible by smoking cessation.
Several studies comparing current smokers, ex-smokers, and nonsmokers have demonstrated that a subset of airway epithelial smoking-dysregulated genes is dysregulated in ex-smokers (16, 17, 58). However, to our knowledge, there have been no prior studies assessing the effect of smoking cessation comparing the airway transcriptome of the same individuals after smoking cessation with their baseline transcriptome before smoking cessation. In contrast to these prior studies of smoking cessation, the present study is unique in several aspects: 1) Although all prior studies assessed the large airway epithelium of current smokers versus ex-smokers, the cells sampled in the present study were from the SAE, the site of development of the smoking-related disease COPD and most types of lung cancer; 2) serial sampling of the SAE of the same quitters over 1 year, enabling the comparison of the SAE transcriptome in the same individual before and after smoking cessation; 3) all individuals underwent smoking cessation for the same period of time; and 4) this strategy identified genes persistently dysregulated by smoking over 1 year despite inter- and intraindividual variability.
The expression of 320 (67%) of the smoking-dysregulated genes changed in a manner consistent with reversal to that of nonsmokers within 12 months. Sixty-three percent of these genes were normalized within 3 months of smoking cessation, a time frame comparable with the renewal time of the cell population of the airway epithelium (60, 61). Of these genes, the regulation- and transcription-related genes had the fastest reversibility, whereas metabolism-, adhesion-, and oxidant-related genes had the slowest reversibility. An additional 102 (21%) genes demonstrated an intermittent reversibility. Importantly, a small subset (n = 53; 11%) of the smoking-dysregulated genes remained dysregulated even after 12 months of smoking cessation, including CYP1B1, a well-known smoking-related gene, involved in many oxidant-related pathways in various organs and linked to different types of carcinoma (62); ME1, involved in cell regulation and senescence mediated by p53 (63); TRIM16, associated with COPD (64); PIR, known to be overexpressed in response to smoking and involved in lung cancer (65); S100A10, associated with lung cancer (66); and PEG10, associated with carcinoma (67).
Because the human airway epithelium turns over within months (60, 61), identifying which genes and pathways are persistently dysregulated by smoking over 1 year and assessing the prevalence of abnormal expression of these genes among smokers can be used to develop drugs targeting a higher percentage of smokers or, specifically, individuals demonstrating less common genes that are persistently dysregulated by smoking despite this turnover. Identifying genes that remain dysregulated in the SAE long after smoking cessation suggests that the remnants of smoking are either in the stem cell population or, alternatively, in other cell populations (e.g., macrophages) that continue to stress the epithelium and may lead to COPD long after the original stress (smoking) has discontinued. Whatever the mechanism, identification of the genes that remain dysregulated despite smoking cessation may be used to develop candidates for drug development to prevent smoking-related lung disorders in former smokers.
Acknowledgments
Acknowledgment
The authors thank C. Williams, J. Salit, S. Shenoy, S. L. O’Beirne, J. Schymeinsky, K. QuAst, S. Visvanathan, J. S. Fine, and M. J. Thomas for useful discussion and advice and N. Mohamed for help with manuscript preparation.
Footnotes
Initially supported by NIH grants HL107882, HL113443, HL1189541, and UL1 TR000457 and Hoffmann-La Roche, Ltd, Nutley, New Jersey; and later by Boehringer Ingelheim Pharmaceuticals, Ridgefield, Connecticut.
Author Contributions: Y.S.-B.: design of the work, data analysis and interpretation, and drafting of the manuscript; M.R.R.: data analysis; R.J.K.: sample acquisition; J.G.M.: data analysis and drafting of the manuscript; R.G.C.: design of the work, data interpretation, and drafting of the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202204-0786OC on August 2, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet . 2007;370:765–773. doi: 10.1016/S0140-6736(07)61380-4. [DOI] [PubMed] [Google Scholar]
- 2.Global Initiative for Chronic Obstructive Lung Disease. 2019. https://goldcopd.org/archived-reports/
- 3.American Cancer Society. 2019. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html
- 4. Auerbach O, Stout AP, Hammond EC, Garfinkel L. Changes in bronchial epithelium in relation to cigarette smoking and in relation to lung cancer. N Engl J Med . 1961;265:253–267. doi: 10.1056/NEJM196108102650601. [DOI] [PubMed] [Google Scholar]
- 5. Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med . 1968;278:1355–1360. doi: 10.1056/NEJM196806202782501. [DOI] [PubMed] [Google Scholar]
- 6. Niewoehner DE, Kleinerman J, Rice DB. Pathologic changes in the peripheral airways of young cigarette smokers. N Engl J Med . 1974;291:755–758. doi: 10.1056/NEJM197410102911503. [DOI] [PubMed] [Google Scholar]
- 7. Cosio M, Ghezzo H, Hogg JC, Corbin R, Loveland M, Dosman J, et al. The relations between structural changes in small airways and pulmonary-function tests. N Engl J Med . 1978;298:1277–1281. doi: 10.1056/NEJM197806082982303. [DOI] [PubMed] [Google Scholar]
- 8. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med . 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 9. Michiels S, Koscielny S, Hill C. Prediction of cancer outcome with microarrays: a multiple random validation strategy. Lancet . 2005;365:488–492. doi: 10.1016/S0140-6736(05)17866-0. [DOI] [PubMed] [Google Scholar]
- 10. Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet . 2010;11:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Strulovici-Barel Y, Omberg L, O’Mahony M, Gordon C, Hollmann C, Tilley AE, et al. Threshold of biologic responses of the small airway epithelium to low levels of tobacco smoke. Am J Respir Crit Care Med . 2010;182:1524–1532. doi: 10.1164/rccm.201002-0294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsang JS. Utilizing population variation, vaccination, and systems biology to study human immunology. Trends Immunol . 2015;36:479–493. doi: 10.1016/j.it.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu C, Cui H, Gu D, Zhang M, Fang Y, Chen S, et al. Genetic polymorphisms and lung cancer risk: evidence from meta-analyses and genome-wide association studies. Lung Cancer . 2017;113:18–29. doi: 10.1016/j.lungcan.2017.08.026. [DOI] [PubMed] [Google Scholar]
- 14. Wang J, Liu Q, Yuan S, Xie W, Liu Y, Xiang Y, et al. Genetic predisposition to lung cancer: comprehensive literature integration, meta-analysis, and multiple evidence assessment of candidate-gene association studies. Sci Rep . 2017;7:8371. doi: 10.1038/s41598-017-07737-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ragland MF, Benway CJ, Lutz SM, Bowler RP, Hecker J, Hokanson JE, et al. Genetic advances in chronic obstructive pulmonary disease: insights from COPDGene. Am J Respir Crit Care Med . 2019;200:677–690. doi: 10.1164/rccm.201808-1455SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, et al. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci USA . 2004;101:10143–10148. doi: 10.1073/pnas.0401422101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beane J, Sebastiani P, Liu G, Brody JS, Lenburg ME, Spira A. Reversible and permanent effects of tobacco smoke exposure on airway epithelial gene expression. Genome Biol . 2007;8:R201. doi: 10.1186/gb-2007-8-9-r201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harvey BG, Heguy A, Leopold PL, Carolan BJ, Ferris B, Crystal RG. Modification of gene expression of the small airway epithelium in response to cigarette smoking. J Mol Med (Berl) . 2007;85:39–53. doi: 10.1007/s00109-006-0103-z. [DOI] [PubMed] [Google Scholar]
- 19. Tilley AE, O’Connor TP, Hackett NR, Strulovici-Barel Y, Salit J, Amoroso N, et al. Biologic phenotyping of the human small airway epithelial response to cigarette smoking. PLoS One . 2011;6:e22798. doi: 10.1371/journal.pone.0022798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Steiling K, van den Berge M, Hijazi K, Florido R, Campbell J, Liu G, et al. A dynamic bronchial airway gene expression signature of chronic obstructive pulmonary disease and lung function impairment. Am J Respir Crit Care Med . 2013;187:933–942. doi: 10.1164/rccm.201208-1449OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O’Beirne SL, Shenoy SA, Salit J, Strulovici-Barel Y, Kaner RJ, Visvanathan S, et al. Ambient pollution-related reprogramming of the human small airway epithelial transcriptome. Am J Respir Crit Care Med . 2018;198:1413–1422. doi: 10.1164/rccm.201712-2526OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Staaf J, Jönsson G, Jönsson M, Karlsson A, Isaksson S, Salomonsson A, et al. Relation between smoking history and gene expression profiles in lung adenocarcinomas. BMC Med Genomics . 2012;5:22. doi: 10.1186/1755-8794-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Strulovici-Barel Y, Salit J, Hackett NR, Staudt MR, Fuller J, Tilley AE, et al. Memory of smoking: persistent dysregulation of expression of a subset of smoking-related genes in the small airway epithelium 3 months following smoking cessation [abstract] Am J Respir Crit Care Med . 2012;185:A2022. [Google Scholar]
- 24. Strulovici-Barel Y, Salit J, Kaner RJ, Fine J, Staudt MR, Crystal RG. “Memory of smoking”: persistent dysregulation of expression of a subset of smoking-related genes in the small airway epithelium and alveolar macrophages following cessation [abstract] Am J Respir Crit Care Med . 2015;191:A5993. [Google Scholar]
- 25. Strulovici-Barel Y, Staudt MR, Salit J, Kaner RJ, Tilley AE, Rogalski AM, et al. Precision transcriptomes: among a population of healthy smokers, what are the shared vs individual smoking-responsive genes in the small airway epithelium and alveolar macrophages? [abstract] Am J Respir Crit Care Med . 2016;193:A4620. [Google Scholar]
- 26. Strulovici-Barel Y, Salit J, Shenoy SA, Mezey JG, Kaner RJ, Staudt MR, et al. Human variability of smoking-induced dysregulation of the small airway epithelium transcriptome over 1 year [abstract] Am J Respir Crit Care Med . 2017;195:A3060. [Google Scholar]
- 27. Strulovici-Barel Y, Salit J, O’Beirne SL, Bitter H, Wolff G, Stevenson C, et al. Smoking-related small airway epithelium dysregulated genes that are shared by most smokers [abstract] Am J Respir Crit Care Med . 2018;197:A3577. [Google Scholar]
- 28. Strulovici-Barel Y, Salit J, O’Beirne SL, Kaner RJ, Thomas MJ, Schymeinsky J, et al. Persistence of the small airway epithelial dysregulation of gene expression despite smoking cessation [abstract] Am J Respir Crit Care Med . 2019;199:A1821. [Google Scholar]
- 29. Ryan DM, Vincent TL, Salit J, Walters MS, Agosto-Perez F, Shaykhiev R, et al. Smoking dysregulates the human airway basal cell transcriptome at COPD risk locus 19q13.2. PLoS One . 2014;9:e88051. doi: 10.1371/journal.pone.0088051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol . 1995;57:289–300. [Google Scholar]
- 31. Pierrou S, Broberg P, O’Donnell RA, Pawłowski K, Virtala R, Lindqvist E, et al. Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2007;175:577–586. doi: 10.1164/rccm.200607-931OC. [DOI] [PubMed] [Google Scholar]
- 32. Wang X, Chorley BN, Pittman GS, Kleeberger SR, Brothers J, II, Liu G, et al. Genetic variation and antioxidant response gene expression in the bronchial airway epithelium of smokers at risk for lung cancer. PLoS One . 2010;5:e11934. doi: 10.1371/journal.pone.0011934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang X, Sebastiani P, Liu G, Schembri F, Zhang X, Dumas YM, et al. Similarities and differences between smoking-related gene expression in nasal and bronchial epithelium. Physiol Genomics . 2010;41:1–8. doi: 10.1152/physiolgenomics.00167.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mertens TCJ, van der Does AM, Kistemaker LE, Ninaber DK, Taube C, Hiemstra PS. Cigarette smoke differentially affects IL-13-induced gene expression in human airway epithelial cells. Physiol Rep . 2017;5:e13347. doi: 10.14814/phy2.13347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ito S, Ishimori K, Ishikawa S. Effects of repeated cigarette smoke extract exposure over one month on human bronchial epithelial organotypic culture. Toxicol Rep . 2018;5:864–870. doi: 10.1016/j.toxrep.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amatngalim GD, Schrumpf JA, Dishchekenian F, Mertens TCJ, Ninaber DK, van der Linden AC, et al. Aberrant epithelial differentiation by cigarette smoke dysregulates respiratory host defence. Eur Respir J . 2018;51:1701009. doi: 10.1183/13993003.01009-2017. [DOI] [PubMed] [Google Scholar]
- 37. Bodas M, Moore AR, Subramaniyan B, Georgescu C, Wren JD, Freeman WM, et al. Cigarette smoke activates NOTCH3 to promote goblet cell differentiation in human airway epithelial cells. Am J Respir Cell Mol Biol . 2021;64:426–440. doi: 10.1165/rcmb.2020-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xiong R, Wu L, Wu Y, Muskhelishvili L, Wu Q, Chen Y, et al. Transcriptome analysis reveals lung-specific miRNAs associated with impaired mucociliary clearance induced by cigarette smoke in an in vitro human airway tissue model. Arch Toxicol . 2021;95:1763–1778. doi: 10.1007/s00204-021-03016-0. [DOI] [PubMed] [Google Scholar]
- 39. Ito S, Matsumura K, Ishimori K, Ishikawa S. In vitro long-term repeated exposure and exposure switching of a novel tobacco vapor product in a human organotypic culture of bronchial epithelial cells. J Appl Toxicol . 2020;40:1248–1258. doi: 10.1002/jat.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tabassum R, Sivadas A, Agrawal V, Tian H, Arafat D, Gibson G. Omic personality: implications of stable transcript and methylation profiles for personalized medicine. Genome Med . 2015;7:88. doi: 10.1186/s13073-015-0209-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Straube J, Gorse AD, Huang BE, Lê Cao KA, PROOF Centre of Excellence Team A linear mixed model spline framework for analysing time course ‘omics’ data. PLoS One . 2015;10:e0134540. doi: 10.1371/journal.pone.0134540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science . 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 43. Raser JM, O’Shea EK. Noise in gene expression: origins, consequences, and control. Science . 2005;309:2010–2013. doi: 10.1126/science.1105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pedraza JM, Paulsson J. Effects of molecular memory and bursting on fluctuations in gene expression. Science . 2008;319:339–343. doi: 10.1126/science.1144331. [DOI] [PubMed] [Google Scholar]
- 45. Choi JK, Kim YJ. Intrinsic variability of gene expression encoded in nucleosome positioning sequences. Nat Genet . 2009;41:498–503. doi: 10.1038/ng.319. [DOI] [PubMed] [Google Scholar]
- 46. Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Chen R, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell . 2012;148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Agler AH, Crystal RG, Mezey JG, Fuller J, Gao C, Hansen JG, et al. Differential expression of vitamin E and selenium-responsive genes by disease severity in chronic obstructive pulmonary disease. COPD . 2013;10:450–458. doi: 10.3109/15412555.2012.761958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJ, Furman D, et al. Variation in the human immune system is largely driven by non-heritable influences. Cell . 2015;160:37–47. doi: 10.1016/j.cell.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van de Weg CA, van den Ham HJ, Bijl MA, Anfasa F, Zaaraoui-Boutahar F, Dewi BE, et al. Time since onset of disease and individual clinical markers associate with transcriptional changes in uncomplicated dengue. PLoS Negl Trop Dis . 2015;9:e0003522. doi: 10.1371/journal.pntd.0003522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature . 2017;552:362–367. doi: 10.1038/nature24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Silwal-Pandit L, Nord S, von der Lippe Gythfeldt H, Møller EK, Fleischer T, Rødland E, et al. The longitudinal transcriptional response to neoadjuvant chemotherapy with and without bevacizumab in breast cancer. Clin Cancer Res . 2017;23:4662–4670. doi: 10.1158/1078-0432.CCR-17-0160. [DOI] [PubMed] [Google Scholar]
- 52. Chen R, Xia L, Tu K, Duan M, Kukurba K, Li-Pook-Than J, et al. Longitudinal personal DNA methylome dynamics in a human with a chronic condition. Nat Med . 2018;24:1930–1939. doi: 10.1038/s41591-018-0237-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fletcher C, Peto R. The natural history of chronic airflow obstruction. BMJ . 1977;1:1645–1648. doi: 10.1136/bmj.1.6077.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med . 2011;365:1567–1575. doi: 10.1056/NEJMoa1106955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ . 2000;321:323–329. doi: 10.1136/bmj.321.7257.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Scanlon PD, Connett JE, Waller LA, Altose MD, Bailey WC, Buist AS, et al. Lung Health Study Research Group Smoking cessation and lung function in mild-to-moderate chronic obstructive pulmonary disease: the Lung Health Study. Am J Respir Crit Care Med . 2000;161:381–390. doi: 10.1164/ajrccm.161.2.9901044. [DOI] [PubMed] [Google Scholar]
- 57. Anthonisen NR, Connett JE, Murray RP. Smoking and lung function of Lung Health Study participants after 11 years. Am J Respir Crit Care Med . 2002;166:675–679. doi: 10.1164/rccm.2112096. [DOI] [PubMed] [Google Scholar]
- 58. Belinsky SA, Palmisano WA, Gilliland FD, Crooks LA, Divine KK, Winters SA, et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res . 2002;62:2370–2377. [PubMed] [Google Scholar]
- 59. Vineis P, Airoldi L, Veglia F, Olgiati L, Pastorelli R, Autrup H, et al. Environmental tobacco smoke and risk of respiratory cancer and chronic obstructive pulmonary disease in former smokers and never smokers in the EPIC prospective study. BMJ . 2005;330:277. doi: 10.1136/bmj.38327.648472.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Spencer H, Shorter RG. Cell turnover in pulmonary tissues. Nature . 1962;194:880. doi: 10.1038/194880a0. [DOI] [PubMed] [Google Scholar]
- 61. Stripp BR, Reynolds SD. Maintenance and repair of the bronchiolar epithelium. Proc Am Thorac Soc . 2008;5:328–333. doi: 10.1513/pats.200711-167DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Watanabe J, Shimada T, Gillam EM, Ikuta T, Suemasu K, Higashi Y, et al. Association of CYP1B1 genetic polymorphism with incidence to breast and lung cancer. Pharmacogenetics . 2000;10:25–33. doi: 10.1097/00008571-200002000-00004. [DOI] [PubMed] [Google Scholar]
- 63. Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature . 2013;493:689–693. doi: 10.1038/nature11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Araya J, Saito N, Hosaka Y, Ichikawa A, Kadota T, Fujita Y, et al. Impaired TRIM16-mediated lysophagy in chronic obstructive pulmonary disease pathogenesis. J Immunol . 2021;207:65–76. doi: 10.4049/jimmunol.2001364. [DOI] [PubMed] [Google Scholar]
- 65. Ji X, Qian J, Rahman SMJ, Siska PJ, Zou Y, Harris BK, et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene . 2018;37:5007–5019. doi: 10.1038/s41388-018-0307-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hou YL, Zhang JH, Guo JB, Chen H. Clinical significance of serum S100A10 in lung cancer. J Int Med Res . 2021;49:3000605211049653. doi: 10.1177/03000605211049653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kainz B, Shehata M, Bilban M, Kienle D, Heintel D, Krömer-Holzinger E, et al. Overexpression of the paternally expressed gene 10 (PEG10) from the imprinted locus on chromosome 7q21 in high-risk B-cell chronic lymphocytic leukemia. Int J Cancer . 2007;121:1984–1993. doi: 10.1002/ijc.22929. [DOI] [PubMed] [Google Scholar]
- 68. Heijdra YF, Pinto-Plata VM, Kenney LA, Rassulo J, Celli BR. Cough and phlegm are important predictors of health status in smokers without COPD. Chest . 2002;121:1427–1433. doi: 10.1378/chest.121.5.1427. [DOI] [PubMed] [Google Scholar]
- 69. Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J . 2005;26:720–735. doi: 10.1183/09031936.05.00034905. [DOI] [PubMed] [Google Scholar]