

Abstract
Under stress conditions, mitochondria release low levels of reactive oxygen species (ROS), which triggers a cytoprotective response, called “mitohormesis”. It still remains unclear how mitochondria respond to stress‐derived stimuli and release a low level of ROS. Here, we show that N‐acetyl‐l‐tyrosine (NAT) functions as a plausible intrinsic factor responsible for these tasks in stressed animals. NAT is present in the blood or hemolymph of healthy animals, and its concentrations increase in response to heat stress. Pretreatment with NAT significantly increases the stress tolerance of tested insects and mice. Analyses using Drosophila larvae and cultured cells demonstrate that the hormetic effects are triggered by transient NAT‐induced perturbation of mitochondria, which causes a small increase in ROS production and leads to sequential retrograde responses: NAT‐dependent FoxO activation increases in the gene expression of antioxidant enzymes and Keap1. Moreover, we find that NAT represses tumor growth, possibly via the activation of Keap1. In sum, we propose that NAT is a vital endogenous molecule that could serve as a triggering factor for mitohormesis.
Keywords: hormesis, insects, mammals, N‐acetyl‐l‐tyrosine, stress
Subject Categories: Metabolism, Molecular Biology of Disease, Organelles
The metabolite N‐acetyl‐l‐tyrosine functions as an endogenous triggering factor for mitohormesis in mammals and insects.
Introduction
The broad distribution of organisms over diverse ecological niches owes much to the evolution of multiple mechanisms to protect them against environmental stresses because they can maintain homeostasis and viability only when stress levels do not exceed the defense capacity 1. The tolerance of organisms to stresses varies, however, depending on their intrinsic and extrinsic backgrounds 2. It is known that organisms can acquire an adaptive tolerance to lethal stress when they have experienced certain mild stresses shortly before the lethal stress. Such beneficial alterations in response to transient sublethal stresses affect their viability when they are exposed to severe stressors 3, 4, 5, 6. This physiological plasticity has been observed in a variety of organisms 7, and various aspects of its mechanisms, such as metabolic controls 8 and cellular signaling pathways 9, have been investigated. In recent years, particular attention has been paid to the contribution of mitochondria to a hormetic state 10. Reactive oxygen species (ROS) produced during oxidative phosphorylation in mitochondria have long been considered undesirable by‐products for a long time 11. However, there is growing evidence in the literature to suggest that ROS transiently produced within the mitochondria by mild stresses cause an adaptive response that appears to induce a wide‐ranging cytoprotective state resulting in long‐lasting metabolic and biochemical changes 12 for which the term “mitohormesis” has been proposed. Although such salutary effects of mild mitochondrial distress have been reported in multiple organisms including nematodes 13, 14, fruit flies 15, and mice 16, 17, we do not know the detailed mechanisms by which mitochondria respond to stress‐derived stimuli and release a low level of ROS. We hypothesized the presence of an intrinsic factor that transmits stress stimuli to mitochondria and leads mitochondria to subsequently release an appropriate level of ROS in order to modulate various biologically relevant pathways. To substantiate this hypothesis, we initially focused on parasitism, which subjects host insects to severe stress.
Results and Discussion
Parasitization increased the intrinsic factor that induces thermotolerance in insects
Parasitoids often impose very harmful stress on their hosts. Once a gregarious parasitoid wasp parasitizes a host insect, the many parasitoid larvae absorb much nutrition from the host to survive inside. However, as long as they stay inside the host, they never exert the crucial pressure that kills their host; otherwise, they would perish with the host 18. For example, a female parasitoid wasp of Cotesia kariyai lays approximately 100 eggs inside the host armyworm Mythimna separata larva, and they take about 10 days to grow large enough to emerge from the host, even as they impose extreme stress on the host 19, 20. The host's body is almost completely occupied by many parasitoid larvae the last several days before the parasitoids emergence (Fig 1A), but the host can still respond to stimuli and move to escape dangers until the very end 21. This implies that host larvae can maintain their homeostasis by entering a special physiological mode such as hormesis. Based on this interpretation, we hypothesized that the presence of internal parasitoid larvae must gradually induce a hyper‐resistant mode in the host larva during the growth of the parasites. Comparison of survivals of unparasitized and parasitized host armyworm larvae after heat stress substantiated a parasitization‐induced increase in heat stress resistance (Fig 1B), indicating the possibility that a factor required for induction of thermotolerance is accumulated in the hemolymph of parasitized host larvae. We assessed this hypothesis by examining whether injection of the parasitized host hemolymph induces heat stress tolerance. The results showed that survival rates of test armyworm larvae injected with hemolymph of parasitized host larvae were significantly higher than those of larvae injected with unparasitized larval hemolymph (Fig 1C). These observations led us to seek the factor in the parasitized larval hemolymph that induced the heat stress tolerance.
Figure 1. Parasitization induces stress tolerance state in host insects.
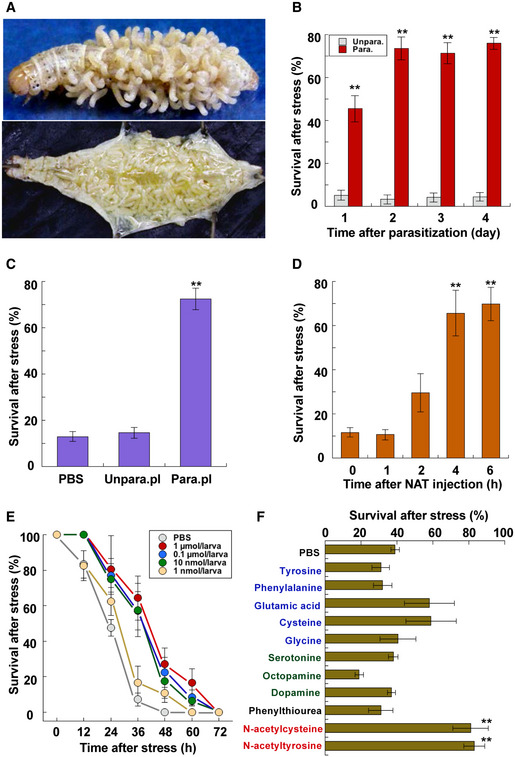
- Images of host armyworm Mythimna separata larvae. Upper: emergence of Cotesia kariyai parasitoid wasps. Lower: inside of host body extensively occupied by parasitoid larvae before their emergence.
- Survival of parasitized and unparasitized host larvae 2 days after heat stress at 44°C for 50 min (data are means ± SEM; n = 10). **P < 0.01 vs. unparasitized hosts. Day 4 last instar larvae were used for this assay. Test larvae were parasitized by the wasp on day 0 of last instar.
- Survival of armyworm larvae injected with PBS, unparasitized, and parasitized larval hemolymph 2 days after heat stress at 44°C for 50 min (data are means ± SEM; n = 12). **P < 0.01 vs. PBS. Test larvae were exposed to the heat stress 4 h after injection of each sample.
- Time‐dependent survival of armyworm larvae injected with 0.8 μmol/larva N‐acetyl‐l‐tyrosine (NAT): post‐NAT injection 2 days after heat stress at 44°C for 50 min (data are means ± SEM; n = 12). **P < 0.01 vs. zero time.
- Dose‐dependent survival of armyworm larvae injected with NAT after heat stress at 44°C for 50 min (data are means ± SEM; n = 12). Injection of more than 10 nmol NAT/larva caused a significant difference compared with zero (log‐rank test using R version 3.2.2).
- Survival of armyworm larvae injected with indicated chemical solution 1 day after heat stress at 44°C for 50 min (data are means ± SEM; n = 10). **P < 0.01 vs. PBS. Each chemical (0.5 μmol/larva) was dissolved in PBS; a minimal volume of dimethyl sulfoxide was used for poorly soluble chemicals such as tyrosine.
Identification and validation of the triggering factor of the thermotolerance
We purified the factor from the hemolymph of parasitized host larvae by reversed‐phase HPLC (Appendix Fig S1) and found it to be a non‐peptidergic molecule with a molecular mass of 223 by LC/MS (Appendix Fig S2). Amino acid analysis showed the presence of a tyrosine molecule (Appendix Fig S2), and subsequent analyses using LC‐MS/MS together with 1H and 13C MNR spectroscopy revealed that the active molecule is N‐acetyl‐l‐tyrosine (NAT) (Appendix Fig S3).
We confirmed that NAT induced the heat stress tolerance of armyworm larvae in a time‐dependent manner after injection: It did not increase the stress tolerance immediately after injection but significantly induced it over 4 h after injection (Fig 1D). Tracking plasma NAT concentrations in this experiment showed that NAT removal from the larval plasma was initiated soon after injection and that the initial NAT levels had decreased by approximately 98% 4 h post‐injection (Appendix Fig S4). Dose–response curves of NAT injection show that injection of more than 10 nmol NAT/larva induced a significant elevation in the thermotolerance (Fig 1E). NAT‐induced stress tolerance was demonstrated in other insect species such as silkworm Bombyx mori larvae and honey bee Apis mellifera workers. When silkworm larvae were injected with NAT 4 h before lethal heat stress, survivals were elevated as in the case of armyworm larvae (Appendix Fig S5). Furthermore, NAT supplementation also increased the survivals of A. mellifera workers transferred from their hive to a small cage: Most animals fed NAT‐free honey solution had died 48 h after the transfer, while more than a half survived for 48 h with 0.01–0.1 mM NAT solution (Appendix Fig S6). Heat stress tolerance of armyworm larvae was also elevated by N‐acetyl‐d‐tyrosine in a similar manner, indicating that there is no difference between the two enantiomeric isomers in activities (Appendix Fig S7). Various other chemicals including amino acids and biogenic amines were tested for their capacity to induce thermotolerance after injection into armyworm larvae. Except for NAT, only N‐acetyl‐l‐cysteine, which is known as a potent antioxidant 22, caused significant elevation of the stress tolerance (Fig 1F).
An increase in the plasma NAT concentration was observed in armyworm larvae that had been exposed to 42°C: The NAT concentration was more than quadrupled after heat stress at 42°C for over 90 min (Appendix Fig S8).
NAT is present in the blood of mammals and shows similar effects on them
Although there has been no report of the presence of NAT in the blood of healthy humans as far as we know, we demonstrated its presence in human serum (Fig 2A), indicating the possibility that NAT works as a hormesis inducer in mammals just like in insects. To assess this possibility, mice were used for more detailed characterization of NAT functions in mammals. We first measured NAT concentrations in the blood of mice under heat and restraint stress and detected the elevation of its concentration during the 45‐min observation (Fig 2B). Effects of pretreatment with NAT were evaluated by measuring concentrations of two blood components, corticosterone and peroxidized lipid. NAT pretreatment lowered corticosterone concentrations in the serum of mice that had been restrained for 1 h (Fig 2C). Serum‐peroxidized lipid concentrations also significantly decreased in NAT‐treated mice that had been forced to swim for 1 h: peroxidized lipid levels in NAT‐treated mice were almost equivalent to those in positive control mice treated with imipramine (Fig 2D).
Figure 2. Analyses of the NAT‐induced hormesis response in mammals.
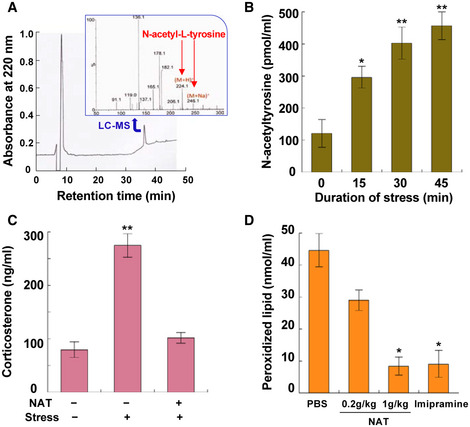
- HPLC chromatogram and LC‐MS spectrogram of N‐acetyltyrosine purified from pooled human serum (#12181201; Cosmo Bio Co., Japan). The peak fraction purified by HPLC was confirmed to be N‐acetyltyrosine by LC‐MS (inserted spectrogram).
- NAT concentrations in the serum of mice (Slc:ddY, 18‐week‐old male) exposed to heat (40°C) and restraint stress (data are means ± SEM; n = 5). *P < 0.05, **P < 0.01 vs. zero time.
- Effects of NAT on corticosterone concentrations in the serum of mice (Slc:ddY, 9‐ to 10‐week‐old male) exposed to restraint stress for 60 min (data are means ± SEM; n = 6). **P < 0.01 vs. without stress. Test mice drank 1.5 mg/ml NAT solution for 24 h before exposure to stress.
- Effect of NAT on stress‐induced peroxidized lipid in the serum of mice (Slc:ddY, 9‐week‐old male) forced to swim for 60 min (data are means ± SEM; n = 10). *P < 0.05 vs. PBS.
NAT supplementation affords thermotolerance in Drosophila larvae
The mechanisms by which NAT enhances thermotolerance were investigated using Drosophila larvae. We first confirmed that a pretreatment of mild heat stress at 35°C for 30 min significantly elevated survivals of test larvae after lethal heat stress at 38.5°C for 60 min (Appendix Fig S9). Enhancement of thermotolerance was also induced by feeding NAT to second‐instar larvae for 24 h: Feeding NAT increased survivals of test larvae almost five times 2 days after lethal heat stress (Fig 3A). Because our prior study showed enhanced expression of antioxidant enzyme genes such as catalase, SOD1, and SOD2 in acclimated armyworms exposed to mild heat stress 23, we examined whether NAT enhances such enzyme expression in Drosophila and found that supplementation with NAT for 12 h significantly elevated expression of all tested enzyme genes in Drosophila larvae (Fig 3B).
Figure 3. Analysis of NAT‐induced hormesis response in Drosophila larvae.
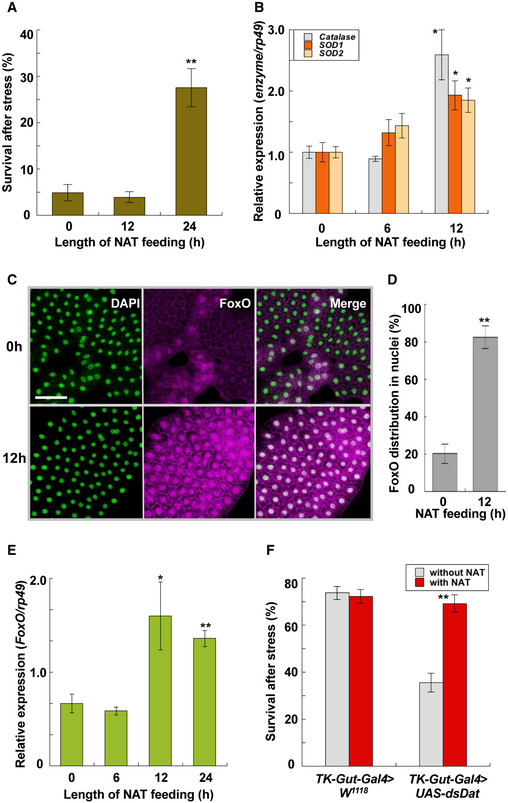
- Survival of Drosophila melanogaster (W 1118 ) larvae fed with 2 μmol NAT/g‐diet 2 days after heat stress at 38.5°C for 60 min (data are means ± SEM; n = 10). **P < 0.01 vs. zero time.
- Effects of NAT feeding on expression of antioxidant enzymes, catalase, SOD1, and SOD2 (data are means ± SEM; n = 10). *P < 0.05 vs. zero time.
- Representative images of nuclei (left) and FoxO (middle) in the fat body of Drosophila larvae before and after NAT feeding for 12 h. White signals in the merged image (right) show the overlap of both signals. Scale bar: 50 μm.
- Relative whiteness intensities before and after NAT feeding (data are means ± SEM; n = 15). **P < 0.01 vs. zero time.
- Effects of NAT feeding on FoxO expression in the fat body of Drosophila larvae (data are means ± SEM; n = 8). *P < 0.05, **P < 0.01 vs. zero time.
- Effects of N‐acetyltransferase (Dat) knockdown in gut enteroendocrine cells (TK‐Gut‐Gal4 > UAS‐dsDat) on survival of Drosophila larvae (data are means ± SEM; n = 15). **P < 0.01 vs. without NAT.
FoxO transcription factor was examined because expression of the antioxidant enzyme genes has been reported to be regulated by FoxO in many animals 24. Immunocytochemistry of FoxO showed its translocation into the nuclei of fat body cells of Drosophila larvae supplemented with NAT (Fig 3C and D). Observation of FoxO signals in these immunocytochemical studies allowed us to recognize the NAT‐induced increase in FoxO signal intensities in fat body cells, indicating the possibility that FoxO expression is also elevated by ingesting NAT. Quantitative measurement of FoxO expression levels verified its transcriptional enhancement by supplementing with NAT for over 12 h (Fig 3E).
The contribution of N‐acetyltransferase, which likely synthesizes NAT from tyrosine 25, to thermal acclimation in Drosophila larvae, was examined by using N‐acetyltransferase (CG3318) RNAi. When control strain (actin‐Gal4 > W 1118 ) larvae were pretreated by mild stress at 35°C for 30 min, they became acclimated to more severe heat stress at 38.5°C. An N‐acetyltransferase knockdown in gut enteroendocrine cells (TK‐Gut‐Gal4 > UAS‐dsDat) significantly decreased survivals of test larvae under the same heat treatment, while the RNAi‐induced decrease in the survivals was rescued by NAT supplementation (Fig 3F).
NAT induces thermotolerance by mitohormesis
To reveal how NAT activates the FoxO‐dependent gene expression, we used Drosophila S2 cells to analyze the detailed mechanism of NAT functions. We first confirmed the heat acclimation of S2 cells: Pre‐exposure to 37°C for 30 min significantly increased cellular survivals after subsequent lethal heat stress at 42°C for 60 min (Fig 4A). Treatment with NAT significantly increased cellular survivals without pre‐exposure to the sublethal heat condition (Fig 4A), indicating that NAT functions as an inducer of stress acclimation at the cellular level. The NAT‐induced thermotolerance was confirmed to be due to the enhancement of FoxO gene expression as well as its translocation into the nuclei (Appendix Fig S10) which was followed by increased expression of antioxidant enzyme genes (Appendix Fig S10). Addition of an amino acid transporter 1 inhibitor, 2‐aminobicyclo‐(2,2,1)‐heptane‐2‐carboxylic acid (BCH), abolished the NAT‐dependent increase in FoxO expression as well as the thermotolerance, indicating that NAT induces hormetic responses after its entry into cells via the amino acid transporter (Fig 4A and Appendix Fig S10). A subsequent finding that antimycin A eliminated both NAT‐induced and preheating‐induced stress acclimation responses in S2 cells (Fig 4B) implies that NAT requires mitochondrial function to serve as an acclimation inducer because antimycin A is known to inhibit mitochondrial electron transport chain complex III. However, the negative effect of antimycin A was dose‐dependent: Low concentration (about one‐thousandth of the inhibitory concentrations) of antimycin A induced slightly but significantly elevated survivals of S2 cells after heat stress (Appendix Fig S11). Similar concentration‐dependent effects were also observed with rotenone, a well‐described complex I inhibitor (Appendix Fig S11).
Figure 4. Analysis of NAT‐induced hormesis in Drosophila S2 cells.
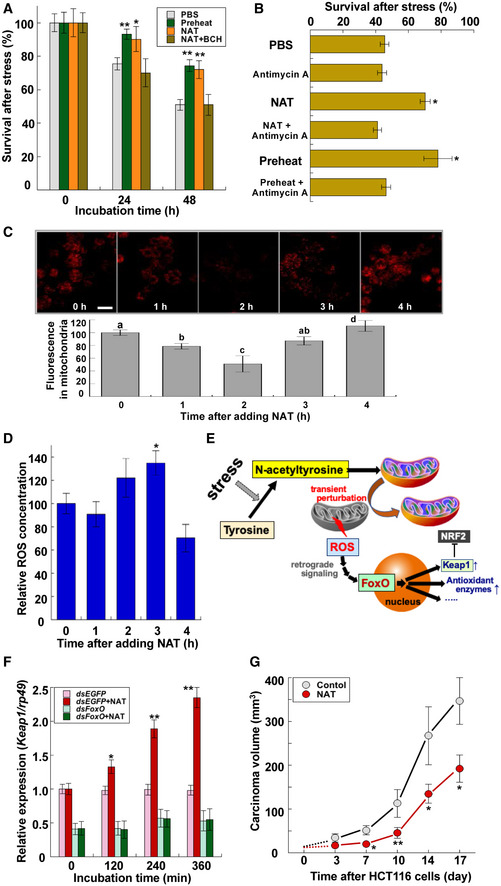
- Survival of Drosophila S2 cells treated with preheating, 100 μM NAT, and 100 μM NAT with 10 mM 2‐aminobicyclo‐(2,2,1)‐heptane‐2‐carboxylic acid (BCH) after heat stress at 42°C for 60 min (data are means ± SEM; n = 10). After pretreatment with chemicals for 12 h, they were removed by changing the medium. *P < 0.05, **P < 0.01 vs. PBS.
- Survival of Drosophila S2 cells pretreated with 100 μM NAT, 100 μM NAT with 30 μg/ml antimycin A, and preheating with or without 30 μg/ml antimycin A 48 h after heat stress at 42°C for 60 min (data are means ± SEM; n = 10). After pretreatment with chemicals for 12 h, they were removed by changing the medium. *P < 0.05 vs. PBS.
- Effects of 100 μM NAT on MitoRed localization in mitochondria of S2 cells. Upper: representative images of time‐dependent changes in MitoRed localization in mitochondria after adding NAT. Scale bar: 20 μm. Lower: Relative intensities of MitoRed fluorescent signals in mitochondria were quantified by microplate reader. Different letters above bars represent significant differences [P < 0.05 (data are means ± SEM, n = 12)].
- ROS concentrations in S2 cells after adding 100 μM NAT (data are means ± SEM; n = 12). *P < 0.05 vs. zero time.
- Graphic depicting the participation of NAT in triggering mitohormesis in animal cells.
- Effects of dsRNA targeting of FoxO on NAT‐dependent expression of Keap1 (data are means ± SEM; n = 12). *P < 0.05, **P < 0.01 vs. without NAT. Note that NAT feeding did not increase Keap1 expression in FoxO knockdown flies.
- Effect of NAT on growth of HCT116 colon cancer cells in mice (BALB/cSlc‐nu/nu, 6‐week‐old female) (data are means ± SEM; n = 5). *P < 0.05, **P < 0.01 vs. without NAT. Test mice began to freely drink 5 mg/ml NAT solution for 7 days before transplantation of the tumor cells on day 0: The average amount of NAT consumed by each mouse was about 8.8 mg/day (0.4–0.5 g/kg weight/day). HCT116 cells (5.0 × 106 cells) were transplanted into each test mouse using a 1‐ml syringe with a 27G needle.
To characterize the contribution of mitochondria to the NAT‐induced thermotolerance, we monitored the mitochondria status of S2 cells using MitoRed, whose staining is dependent on mitochondrial membrane potential 26. The relative intensity of MitoRed‐derived fluorescent signals had significantly declined 2 h after adding NAT, while the signals were totally recovered 2 h later, indicating the transient NAT‐induced depolarization of mitochondria (Fig 4C). In contrast, MitoRed signals were constantly decreased by applying carbonyl cyanide m‐chlorophenyl hydrazone (CCCP), a typical mitochondrial uncoupler, or antimycin A to S2 cells without any significant recovery of the signals (Appendix Fig S12), which emphasized the specific transient effect of NAT on mitochondria. This interpretation was substantiated by quantification of mitochondrial ROS (mROS) in S2 cells after treatments with these chemicals: NAT induced a transient small peak of mROS concentrations in S2 cells 2–3 h after adding NAT (Fig 4D), while such an mROS peak was not observed after adding CCCP or antimycin A (Appendix Fig S13). Therefore, it is reasonable to assume that NAT‐dependent elevation of thermotolerance is due to the transient perturbation of mitochondria, which is generally called “mitohormesis” (Fig 4E). The interpretation was enforced by the observation that N‐acetylcysteine also exerted similar effects on mitochondria in S2 cells: It transiently induced depolarization of mitochondria and elevated mROS in S2 cells (Appendix Fig S14). These data are consistent with the above observation that both N‐acetylcysteine and NAT induced thermotolerance in armyworm larvae by injection prior to lethal heat stress (Fig 1F).
NAT stimulates FoxO‐Keap1 signaling
Characterization of NAT‐induced stimulation of signaling pathways revealed that NAT treatment also elevated transcription levels of Kelch‐like ECH‐associated protein 1 (Keap1). This elevation was not observed in FoxO RNAi S2 cells (Fig 4F), confirming that FoxO mediates the NAT‐induced expression of Keap1. Moreover, Keap1 RNAi abolished NAT‐induced thermotolerance of Drosophila larvae (Appendix Fig S15). Since Keap1 is known as a negative regulator of the transcription factor NF‐E2‐related factor 2 (Nrf2) that regulates a set of antioxidant and detoxifying enzyme genes, we examined the effects of Nrf2 RNAi in Drosophila larvae. Nrf2 RNAi larvae also did not show NAT‐induced thermotolerance (Appendix Fig S15). Furthermore, the NAT‐dependent elevation of thermotolerance was not observed in FoxO RNAi S2 cells (Appendix Fig S16) although a transient decrease in MitoRed fluorescent signals was normally observed in the mitochondria of these cells 2 h after adding NAT (Appendix Fig S16). We therefore interpreted these results to indicate that NAT‐induced mitohormesis requires enhanced FoxO‐Keap1 signaling downstream (Fig 4E).
The finding that NAT stimulated FoxO‐Keap1 signaling axis in Drosophila S2 cells also led us to assess the possibility of repressive effects of NAT on tumor progression because a tumor suppressor function of FoxO3 through constraining Nrf2 signaling has recently been reported 24. To examine the possibility that NAT affects tumor progression, we focused on human colorectal cancer HCT116 cells because expression of Nrf2 and its target genes has been recently reported to be increased in HCT116 colonospheres 27. We transplanted HCT116 cells into nude mice with or without NAT treatment and compared the tumor volumes in control and NAT‐treated animals for 17 days. NAT supplementation significantly suppressed the growth of HCT116 cells, indicating that NAT supplementation has a suppressive activity on growth of HCT116 cancer cells perhaps through activation of FoxO‐Keap1 signaling axis (Fig 4G). Moreover, treatment with NAT had a significant negative impact on the weight gain of test mice (Appendix Fig S17).
Concluding comments
NAT has been reported to increase in the urine of patients with tyrosyluria 28, 29 and tyrosinosis 30, 31, indicating the possibility that NAT is synthesized in human bodies and that its concentrations fluctuate depending on internal conditions. Although some physiological analyses such as quantification of urine NAT concentrations and its retention times after administration to human bodies have been reported 32, 33, basic information on NAT is still quite limited. The present study showed that NAT is normally present in the blood of all tested animals including insects and mammals and that its concentrations are elevated by stresses, which induce hormesis in mice as well as insects. These observations indicated the possibility that stress‐induced NAT works as a hormesis inducer in all animals including humans.
Analyses using Drosophila and the culture cell S2 demonstrated that NAT transiently distressed mitochondria, which produced a low level of mROS. Although we have no solid evidence to explain how mitochondria ROS production is mediated by NAT at present, we propose the mechanism by which NAT serves as an essential intrinsic factor that can transmit stress‐derived stimuli to mitochondria in the graphic model (Fig 4E). The NAT‐induced activation of the FoxO–Keap1 signaling axis negatively regulates Nrf2 functions and enforces ROS release from mitochondria because Nrf2 regulates mitochondrial homeostasis 34. However, this inhibition must be relaxed soon after ROS is released from mitochondria by losing the prominent regulator Nrf2, which allows Nrf2 to evade Keap1‐mediated repression because ROS‐induced Keap1 conformational changes liberate Nrf2 35. Therefore, it is reasonable to presume that both FoxO and Nrf2 sequentially stimulate expression of their target genes with anti‐stress abilities because NAT‐induced elevation of Keap1 expression would not cause long‐lasting inhibition of Nrf2. This interpretation was at least partly confirmed by the observation that neither Keap1 RNAi nor Nrf2 RNAi Drosophila larvae showed NAT‐induced thermotolerance.
One of the primary changes via these signaling pathways is enhanced expression of antioxidant enzyme genes by FoxO transcription factor. Moreover, the fact that FoxO is also an upstream transcription factor of Keap1, the negative regulator of Nrf2 35, led us to examine the tumor‐suppressive activity of NAT because abnormal activation of Nrf2 has been reported in many types of cancer cells such as cholangiocarcinoma and lung adenocarcinoma 24, 36, 37, 38, 39. The results showed significant NAT‐induced suppression of colorectal cancer cell growth in nude mice. Taken together, this study proposes that NAT serves as an intrinsic trigger of mitohormesis, which contributes to maintaining homeostasis in animals, and highlights the value of exploiting both basic and functional features of NAT.
Materials and Methods
Animals
Mythimna separata larvae were reared on an artificial diet (10% kidney beans, 10% wheat bran, 4.2% dried yeast, 0.5% ascorbic acid, 0.3% antiseptic reagents, and 1.3% agar) at 25°C with a photoperiod of 16‐h light:8‐h dark 40, and Drosophila melanogaster stocks were reared on cornmeal–glucose–yeast medium at 23°C with a photoperiod of 16‐h light:8‐h dark 41. The UAS‐dsNat, TK‐Gut‐Gal4, UAS‐dsKeap1, and UAS‐dsNrf2 strains were supplied by NIG‐FLY (National Institute of Genetics, Japan). Actin‐Gal4 and hs‐Gal4 were previously described 41. Drosophila S2 cells were maintained in Schneider's Drosophila medium (Gibco, USA) supplemented with 5% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin at 25°C. Mice, Slc:ddY and BALB/cSlc‐nu/nu, were purchased from Japan SLC, Inc (Japan). The mice were housed at 23 ± 2°C with a photoperiod of 12‐h light: 12‐h dark and had ad libitum access to food and water. Experiments on mice were conducted with the approval of Saga University Animal Care and Use Committee according to the National Institutes of Health guidelines.
Quantitative RT–PCR analysis
To quantify gene expression, RT–PCR was conducted essentially according to the previously described procedure as follows 42. Total RNAs were prepared from larvae or cells. First‐strand cDNA was synthesized with oligo(dT)12–18 primer using ReverTra Ace RT–PCR Kit (Toyobo, Japan) according to the manufacturer's protocol. The cDNAs for target genes were amplified with a specific primer pair indicated in Table EV1. PCR was conducted under the following conditions: 20–28 cycles at 95°C for 30 s, 55–58°C for 30 s, and 72°C for 45 s. Quantitative real‐time PCR was carried out with the cDNAs in a 20 μl reaction volume of LightCycler Fast DNA Master SYBR Green I (Roche Applied Science, USA), using the LightCycler 1.2 instrument and software (Roche Applied Science). The PCR cycling conditions were denaturation at 95°C for 10 min, followed by 45 cycles of 95°C for 10 s, annealing at 55°C for 5 s, and extension at 72°C for 15 s. Using the second derivative maximum method provided in the LightCycler software (version 3.5), a standard curve was generated by plotting the external standard concentration against threshold cycle. The software automatically calculated the PCR product concentration for each sample. All samples were analyzed in duplicate, and assay variation was typically within 10%. Data were normalized according to the expression level of rp49 determined in duplicate by reference to a serial dilution calibration curve. All primers used in this study are listed in Table EV1.
Confocal immunofluorescence
Fat body and S2 cells were fixed in 4% paraformaldehyde in PBS at 25°C for 30 min and extensively washed in phosphate‐buffered saline containing 0.1% Triton X‐100 (PBT). Tissues were then blocked for 1 h in PBT containing 5% normal donkey serum and subsequently incubated with anti‐FoxO antibody (1:200; Cosmo Bio Co., Japan) at 4°C for 24 h. After washing in PBT, tissues were incubated at 25°C for 3 h with secondary antibody (1:500, goat anti‐rabbit IgG Alexa Fluor 555; Life Technologies Co., USA). Tissues were imaged using a laser scanning confocal microscope (EZ‐Ti System; Nikon, Japan). FoxO fluorescence was recorded from a confocal Z series with 0.5‐μm steps, using identical laser power and scan settings. Signal quantification was performed with ImageJ 43.
Purification procedures of plasma N‐acetyltyrosine
Hemolymph was collected from parasitized larvae of armyworm M. separata 10 days after parasitization on ice and immediately centrifuged at 4°C for 3 min at 500 g. The supernatant was mixed with −20°C acetonitrile (final concentration: 50%), and following centrifugation at 4°C for 10 min at 20,000 g, the collected supernatant was concentrated by lyophilization. The concentrated sample was dissolved in 0.05% TFA and separated by three HPLC runs. The active fraction from the first HPLC column (Mightysil RP‐18 GP, 250 × 4.6 mm (5 μm); Kanto Chemical Co., Japan) was purified sequentially by a second column (Mightysil NH2, 250 × 4.6 mm (5 μm); Kanto Chemical Co., Japan) and a third column (J‐Pak Symphonia C18, 250 × 4.6 mm (5 μm); Jasco Engineering Co., Japan).
Identification of N‐acetyltyrosine by LC‐MS, MC‐MS/MS, and NMR
N‐acetyltyrosine was identified by analyzing the purified active peak fraction of HPLC using LC‐MS/MS (Waters ACQUITY UPLC I‐Class/SYNAPT G2‐2 HDMS LC‐MS/MS System, USA) and NMR spectrometer with cryo‐probe (Avance III‐500; Bruker BioSpin Inc., Germany).
Viability test of larvae and cells
After lethal heat stresses, test larvae were reared with a normal diet and their movement after gentle poking with forceps was periodically observed. Motility was assessed to determine the vital status of test larvae. Survival of S2 cells was measured by using a Cell Counting Kit‐8 (Dojindo Molecular Technology Inc., Japan) according to the manufacturer's instructions.
Double‐stranded RNA‐mediated interference in vitro
Individual DNA fragments approximately 700 bp in length, containing coding sequences for the proteins to be “knocked down”, were amplified by using PCR. Each primer used in the PCR contained a 5′ T7 RNA polymerase binding site (GAATTAATACGACTCACTATAGGGAGA) followed by sequences specific for the targeted genes, FoxO and EGFP (see Table EV1). The PCR products were purified and were used as templates by using a MEGAscript T7 Transcription Kit (Ambion Co., USA) to produce dsRNA according to the manufacturer's protocol. Ten μg of dsRNA was analyzed by 1.5% agarose gel electrophoresis to ensure that the majority of the dsRNA existed as a single band of approximately 700 bp. S2 cells were incubated at 1 × 105 cells/well in 96‐well plates in 1× Schneider's Drosophila media with 5% FBS at 25°C. dsRNA was added directly to the media to a final concentration of 37 nM. The cells were incubated for 3 days to allow for turnover of the target protein.
Quantification of plasma (or serum) N‐acetyltyrosine concentration
Eighty microliters of hemolymph collected from test armyworm larvae was collected into a microtube containing 450 μl of 100 mM citrate buffer (pH 5.8) and immediately boiled for 3 min. The boiled hemolymph was mixed with 200 μl of 1,2‐dichloroethane, centrifuged at 4°C for 20 min at 20,000 g, and the supernatant was analyzed by three sequential HPLC runs using a Mightysil RP‐18 GP Column (250 × 4.6 mm (5 μm); Kanto Chemical Co., Japan), J‐Pak Vario ASB C18 Column (250 × 4.6 mm (5 μm); Jasco Engineering Co., Japan), and Unison UK‐C18 Column (250 × 4.6 mm (3 μm); Imtakt Co., Japan). The single peak eluted from the third Unison UK‐C18 Column was identified as an N‐acetyl‐l‐tyrosine (NAT) peak, and its volume was quantified to calculate NAT concentration in the hemolymph. NAT concentrations in the blood of mice were measured using the serum preparations according to the same procedures described above.
Quantification of blood corticosterone and peroxidized lipids
Blood collected from each test mouse was stored at 4°C for 12 h, centrifuged at 4°C for 10 min at 500 g, and the supernatant was used for quantification of corticosterone and peroxidized lipid concentrations. Corticosterone and peroxidized lipid analyses were performed by using corticosterone and ARK Checker CORT EIA (Ark Resource Co., Japan) and a lipid hydroperoxide assay kit (Cayman Chemical Co., USA), respectively, according to the manufacturer's instructions.
Measurement of MitoRed localization in mitochondria of Drosophila S2 cells
S2 cells were treated with PBS, 10 μM CCCP, 18 μM antimycin A, or 100 μM N‐acetyl‐l‐tyrosine for indicated periods and washed three times with PBS. Cells were immediately incubated with 200 nM MitoRed (Dojindo, Japan) for 30 min at 37°C and were fixed in 4% paraformaldehyde in PBS containing 0.3% Triton X‐100 for 5 min at 25°C. Fluorescence was visualized using a laser scanning confocal microscope (EZ‐Ti System; Nikon, Japan). Images were acquired using a 563‐nm He–Ne laser to excite MitoRed fluorescence. Fluorescence was recorded from a confocal Z series with 0.5‐μm steps, using identical laser power and scan settings. Relative intensities of MitoRed fluorescent signals in S2 cells were measured at Ex/Em = 535/595 nm using a Multimode Detector DTX880 (Beckman Coulter).
Measurement of mitochondrial ROS levels of Drosophila S2 cells
S2 cells were treated with PBS, 10 μM CCCP, 18 μM antimycin A, or 100 μM N‐acetyl‐l‐tyrosine for indicated periods and washed three times with PBS. Cells were incubated with 15 μM 2,7‐dichlorodihydrofluorescein diacetate (Cayman Chemical, USA) for 30 min at 37°C and were fixed in 4% paraformaldehyde in PBS containing 0.3% Triton X‐100 for 5 min 25°C. Fluorescence was visualized using a laser scanning confocal microscope (EZ‐Ti System; Nikon, Japan). Images were acquired using a 488‐nm argon laser to excite dichlorofluorescein fluorescence. Fluorescence was recorded from a confocal Z series with 0.5‐μm steps using identical laser power and scan settings. Relative intensities of fluorescent signals in S2 cells were measured at Ex/Em = 485/530 nm using a Multimode Detector DTX880 (Beckman Coulter).
Growth assay of colon cancer cells
Following administration of 5 mg/ml N‐acetyl‐l‐tyrosine for 7 days, 5.0 × 106 HCT116 colon cancer cells were transplanted into each test mouse (BALB/cSlc‐nu/nu) using a 1‐ml syringe with a 27G needle. We periodically measured dimensions of tumors using a caliper and calculated the volume using the formula V = (W(2) × L)/2, where V is tumor volume, W is tumor width, and L is tumor length.
Statistics
P values were calculated by analysis of variance with Tukey's honestly significant difference (HSD) post hoc test unless otherwise stated. A normality test, the Shapiro–Wilk test, showed that data sets do not deviate from normality. In some experiments, data were analyzed using Kruskal–Wallis ANOVA on ranks following Tukey's HSD. Significant differences among measurements (P < 0.05) were represented by different letters. All statistical analyses were performed using JMP 9.0.2 (SAS Institute). Numbers and/or technical replicates are stated in each figure legend. No statistical method was used to predetermine sample size, and experiments were not randomized.
Author contributions
YH conceived the study and wrote the paper. TM, OU, FM, HM, and YH designed the experiments. TM, OU, KT, HM, and YH performed the experiments. TM, OU, FM, HM, and YH analyzed the data. TM, OU, FM, and HM provided the tools. TM, OU, and YH supervised the execution of the experiments. All authors read and approved the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Acknowledgements
This research was supported by a Grant‐in‐Aid for Scientific Research (A) (Grant Number: 16H0259) from JSPS (Y.H.).
EMBO Reports (2020) 21: e49211
See also: F Fischer & M Ristow (May 2020)
References
- 1. Teets NM, Denlinger DL (2014) Surviving in a frozen desert: environmental stress physiology of terrestrial Antarctic arthropods. J Exp Biol 217: 84–93 [DOI] [PubMed] [Google Scholar]
- 2. Rattan SI (2008) Hormesis in aging. Ageing Res Rev 7: 63–78 [DOI] [PubMed] [Google Scholar]
- 3. Calabrese EJ, Baldwin LA (2003) Hormesis: the dose‐response revolution. Annu Rev Pharmacol Toxicol 43: 175–197 [DOI] [PubMed] [Google Scholar]
- 4. Gems D, Partridge L (2008) Stress‐response hormesis and aging: “that which does not kill us makes us stronger”. Cell Metab 7: 200–203 [DOI] [PubMed] [Google Scholar]
- 5. Le Bourg E (2009) Hormesis, aging and longevity. Biochim Biophys Acta 1790: 1030–1039 [DOI] [PubMed] [Google Scholar]
- 6. Mattson MP (2008) Hormesis defined. Ageing Res Rev 7: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaffee R, Roberts J (1971) Temperature acclimation in birds and mammals. Annu Rev Physiol 33: 155–202 [DOI] [PubMed] [Google Scholar]
- 8. Hayes DP (2007) Nutritional hormesis. Eur J Clin Nutr 61: 147–159 [DOI] [PubMed] [Google Scholar]
- 9. Sonna LA, Fujita J, Gaffin SL, Lilly CM (2002) Invited review: Effects of heat and cold stress on mammalian gene expression. J Appl Physiol 92: 1725–1742 [DOI] [PubMed] [Google Scholar]
- 10. Yun J, Finkel T (2014) Mitohormesis. Cell Metab 19: 757–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300 [DOI] [PubMed] [Google Scholar]
- 12. Ristow M, Zarse K (2010) How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp Gerontol 45: 410–418 [DOI] [PubMed] [Google Scholar]
- 13. Dillin A, Hsu A‐L, Arantes‐Oliveira N, Lehrer‐Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002) Rates of behavior and aging specified by mitochondrial function during development. Science 298: 2398–2401 [DOI] [PubMed] [Google Scholar]
- 14. Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS (2005) A systematic RNAi screen for longevity genes in C. elegans . Genes Dev 19: 1544–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Copeland JM, Cho J, Lo T Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW (2009) Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol 19: 1591–1598 [DOI] [PubMed] [Google Scholar]
- 16. Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG (1999) The p66 shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402: 309 [DOI] [PubMed] [Google Scholar]
- 17. Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, Piccini D, Martin‐Padura I, Pelliccia G, Trinei M, Bono M (2004) The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans‐membrane potential. J Biol Chem 279: 25689–25695 [DOI] [PubMed] [Google Scholar]
- 18. Pennacchio F, Strand MR (2006) Evolution of developmental strategies in parasitic hymenoptera. Annu Rev Entomol 51: 233–258 [DOI] [PubMed] [Google Scholar]
- 19. Hayakawa Y (1990) Juvenile hormone esterase activity repressive factor in the plasma of parasitized insect larvae. J Biol Chem 265: 10813–10816 [PubMed] [Google Scholar]
- 20. Hayakawa Y (1991) Structure of a growth‐blocking peptide present in parasitized insect hemolymph. J Biol Chem 266: 7981–7984 [PubMed] [Google Scholar]
- 21. Hayakawa Y (1995) Growth‐blocking peptide: an insect biogenic peptide that prevents the onset of metamorphosis. J Insect Pysiol 41: 1–6 [Google Scholar]
- 22. Ezerina D, Takano Y, Hanaoka K, Urano Y, Dick TP (2018) N‐acetylcysteine functions as a fast‐acting antioxidant by triggering intracellular H2S and sulfane sulfur production. Cell Chem Biol 25: 447–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsumura T, Matsumoto H, Hayakawa Y (2017) Heat stress hardening of oriental armyworms is induced by a transient elevation of reactive oxygen species during sublethal stress. Arch Insect Biochem Physiol 96: e21421 [DOI] [PubMed] [Google Scholar]
- 24. Guan L, Zhang L, Gong Z, Hou X, Xu Y, Feng X, Wang H, You H (2016) FoxO3 inactivation promotes human cholangiocarcinoma tumorigenesis and chemoresistance through Keap1‐Nrf2 signaling. Hepatology 63: 1914–1927 [DOI] [PubMed] [Google Scholar]
- 25. Hintermann E, Grieder NC, Amherd R, Brodbeck D, Meyer UA (1996) Cloning of an arylalkylamine N‐acetyltransferase (aaNAT1) from Drosophila melanogaster expressed in the nervous system and the gut. Proc Natl Acad Sci USA 93: 12315–12320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lo CY, Chen S, Creed SJ, Kang M, Zhao N, Tang BZ, Elgass KD (2016) Novel super‐resolution capable mitochondrial probe, MitoRed AIE, enables assessment of real‐time molecular mitochondrial dynamics. Sci Rep 6: 30855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ryoo IG, Kim G, Choi BH, Lee SH, Kwak MK (2016) Involvement of NRF2 signaling in doxorubicin resistance of cancer stem cell‐enriched colonospheres. Biomol Ther (Seoul) 24: 482–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Coward R, Smith P (1968) Urinary excretion of 4‐hydroxyphenyllactic acids and related compounds in man, including a screening test for tyrosyluria. Biochem Med 2: 216–226 [Google Scholar]
- 29. Dubovsky J, Dubovska E (1965) The excretion of N‐acetyltyrosine in tyrosyluria. Clin Chim Acta 12: 118–119 [DOI] [PubMed] [Google Scholar]
- 30. Fairney A, Francis D, Ersser RS, Seakins JW, Cottom D (1968) Diagnosis and treatment of tyrosinosis. Arch Dis Child 43: 540–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kennaway NG, Buist NR (1971) Metabolic studies in a patient with hepatic cytosol tyrosine aminotransferase deficiency. Pediatr Res 5: 287 [DOI] [PubMed] [Google Scholar]
- 32. Druml W, Lochs H, Roth E, Hubl W, Balcke P, Lenz K (1991) Utilization of tyrosine dipeptides and acetyltyrosine in normal and uremic humans. Am J Physiol 260: E280–E285 [DOI] [PubMed] [Google Scholar]
- 33. Magnusson I, Ekman L, Wångdahl M, Wahren J (1989) N‐acetyl‐L‐tyrosine and N‐acetyl‐L‐cysteine as tyrosine and cysteine precursors during intravenous infusion in humans. Metabolism 38: 957–961 [DOI] [PubMed] [Google Scholar]
- 34. Dinkova‐Kostova AT, Abramov AY (2015) The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med 88: 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bollong MJ, Lee G, Coukos JS, Yun H, Zambaldo C, Chang JW, Chin EN, Ahmad I, Chatterjee AK, Lairson LL et al (2018) A metabolite‐derived protein modification integrates glycolysis with KEAP1‐NRF2 signalling. Nature 562: 600–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Namani A, Matiur Rahaman M, Chen M, Tang X (2018) Gene‐expression signature regulated by the KEAP1‐NRF2‐CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 18: 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pandey P, Singh AK, Singh M, Tewari M, Shukla HS, Gambhir IS (2017) The see‐saw of Keap1‐Nrf2 pathway in cancer. Crit Rev Oncol Hematol 116: 89–98 [DOI] [PubMed] [Google Scholar]
- 38. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sanchez‐Rivera FJ et al (2017) Keap1 loss promotes Kras‐driven lung cancer and results in dependence on glutaminolysis. Nat Med 23: 1362–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Taguchi K, Yamamoto M (2017) The KEAP1‐NRF2 system in cancer. Front Oncol 7: 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matsumura T, Nakano F, Matsumoto H, Uryu O, Hayakawa Y (2018) Identification of a cytokine combination that protects insects from stress. Insect Biochem Mol Biol 97: 19–30 [DOI] [PubMed] [Google Scholar]
- 41. Tsuzuki S, Ochiai M, Matsumoto H, Kurata S, Ohnishi A, Hayakawa Y (2012) Drosophila growth‐blocking peptide‐like factor mediates acute immune reactions during infectious and non‐infectious stress. Sci Rep 2: 210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tsuzuki S, Matsumoto H, Furihata S, Ryuda M, Tanaka H, Sung EJ, Bird GS, Zhou Y, Shears SB, Hayakawa Y (2014) Switching between humoral and cellular immune responses in Drosophila is guided by the cytokine GBP. Nat Commun 5: 4628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sung EJ, Ryuda M, Matsumoto H, Uryu O, Ochiai M, Cook ME, Yi NY, Wang H, Putney JW, Bird GS et al (2017) Cytokine signaling through Drosophila Mthl10 ties lifespan to environmental stress. Proc Natl Acad Sci USA 114: 13786–13791 [DOI] [PMC free article] [PubMed] [Google Scholar]