

SUMMARY
Paired single-cell RNA and T cell receptor sequencing (scRNA/TCR-seq) has allowed for enhanced resolution of clonal T cell dynamics in cancer. Here, we report a scRNA/TCR-seq analysis of 187,650 T cells from 31 tissue regions, including tumor, adjacent normal tissues, and lymph nodes (LN), from three patients with non-small cell lung cancer after immune checkpoint blockade (ICB). Regions with viable cancer cells are enriched for exhausted CD8+ T cells, regulatory CD4+ T cells (Treg), and follicular helper CD4+ T cells (TFH). Tracking T cell clonotypes across tissues, combined with neoantigen specificity assays, reveals that TFH and tumor-specific exhausted CD8+ T cells are clonally linked to TCF7+ SELL+ progenitors in tumor draining LNs, and progressive exhaustion trajectories of CD8+ T, Treg, and TFH cells with proximity to the tumor microenvironment. Finally, longitudinal tracking of tumor-specific CD8+ and CD4+ T cell clones reveals persistence in the peripheral blood for years after ICB therapy.
In brief
Pai et al. report a single T cell lung cancer dataset allowing for the lineage tracing of T cells across tumor regions, lymph nodes, and peripheral blood. This resource reveals clonal linkage of antigen-specific TCF7+ SELL+ progenitor exhausted cells in the lymph node and their exhausted counterparts in the tumor, and long-term peripheral persistence of these cells after checkpoint blockade.
Graphical abstract
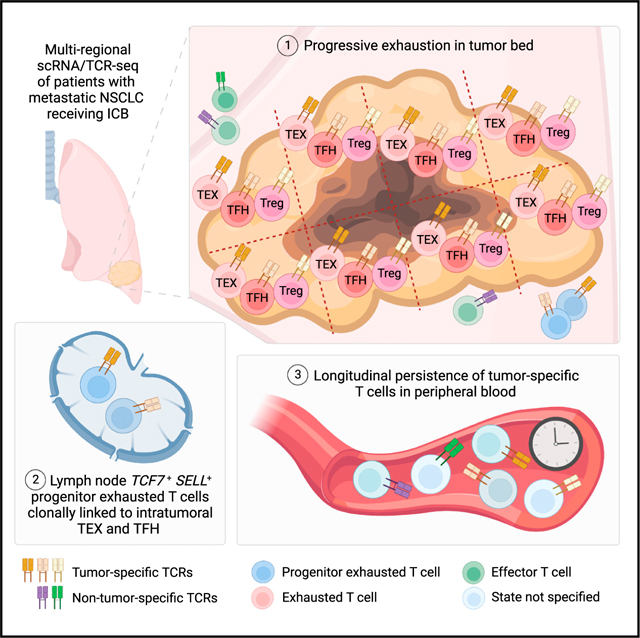
INTRODUCTION
Immune checkpoint blockade (ICB) has been a remarkable clinical advance in the treatment of cancer. Nonetheless, the majority of patients do not benefit from ICB therapy, and many of those who do eventually succumb to the disease. Recent data have demonstrated that ICB can operate via activation, expansion, and recruitment of CD8+ T cells from the peripheral circulation.1,2 Unfortunately, isolated tumor biopsies at the time of resistance to ICB are limited in their ability to capture T cell dynamics at a systemic level.
The development of paired single-cell RNA and T cell receptor sequencing (scRNA/TCR-seq) has enabled deep profiling of T cells in the context of their clonal lineage, phenotypic heterogeneity, tissue distribution, and peripheral persistence.3 However, compared with murine systems, the analysis of T cell responses to cancer in humans has been limited by the challenge of simultaneously achieving sufficient scale of scRNA/TCR-seq T cell profiles that is linked to multi-regional and longitudinal sampling within the same patient. For example, we previously generated a scRNA/TCR-seq dataset in basal cell carcinoma, which revealed that ICB can function to expand a new clonal repertoire of T cells; however, this dataset was limited by its lack of assessment of multiple tumor regions, healthy tissue, and longitudinal peripheral blood samples.4 Recent studies have analyzed either large patient cohorts5 or regional tumor heterogeneity with scRNA-seq6; however, these studies were limited by the depth of per-patient T cell clone sampling. Because of these challenges, several open questions remain. First, are there phenotypic differences between clonally related T cells in different anatomical sites (i.e., lymph node [LN] vs tumor) in humans? Second, while TCF-1+ progenitor exhausted T cells have been described in murine systems,7–13 whether progenitor exhausted T cells that are clonally linked to intratumoral exhausted CD8+ T cells are present in human tumor-draining LN remains understudied.14 Finally, the temporal persistence of tumor-specific T cell clones, including exhausted CD8+ T cells, in the circulation after ICB also remains unknown.
To help address these gaps, we performed scRNA/TCR-seq on 187,650 T cells from 31 tumor, adjacent normal tissue, and regional LNs from three patients undergoing ICB, representing substantially greater depth of profiling for human T cell clonal dynamics per patient than achieved in prior studies. This enabled the analysis of region-dependent transcriptional programs of thousands of T cell clonotypes, as well as the identification of progenitor exhausted T cells in the LNs that were clonally linked to intratumoral exhausted populations. In addition, we combined scRNA/TCR-seq analysis with functional assays for neoantigen specificity to empirically define tumor-specific T cells and systematically profiled the regional distribution, region-dependent cell states, and long-term persistence of tumor- and viral-specific T cells. Overall, we provide a high-depth, clonally resolved view of the tumor-specific T cell response during ICB therapy.
RESULTS
Clinical and pathological characteristics of lung cancer resections after ICB
We profiled three patients (MSK 1263, 1302, and 1344) with metastatic non-small cell lung cancer (NSCLC) who were treated with anti-programmed cell death (PD-1) monotherapy at Memorial Sloan Kettering Cancer Center (Table S1). All three patients had mixed responses, with most metastatic sites demonstrating response, but at least one site showing persistence or progression during treatment (Figures 1A and S1A). In these cases, the resistant site of disease was surgically resected, and multiple regions from each lesion were collected for analyses. After resection, two patients (MSK 1302 and 1344) remain alive for more than 2 years afterward, while one patient (MSK 1263) quickly developed systemic disease recurrence and died. From the three patients, we obtained four tumor resections that underwent sectioning into eight 1- to 2-cm2 sections per tumor and were subjected to pathological evaluation, regional bulk RNA sequencing, flow cytometry, and scRNA/TCR-seq of sorted CD3+ T cells (Figure 1B). We also obtained adjacent normal tissue and regional LNs (not involved by tumor on pathological analysis) from MSK 1263 and 1302. Serial peripheral blood samples were collected up to 216, 452, and 1,013 days after the start of anti-PD-1 therapy in MSK 1263, 1302, and 1344, respectively.
Figure 1. Exhausted CD8+, Treg, and TFH cells are enriched in proximity to viable cancer cells.

(A) Quantification of surface area of individuals lesions on radiographical studies over time in three patients. Red lines indicate lesions that were resected and analyzed in this study.
(B) Schematic of time interval from start of anti-PD-1 therapy to the time of resections and clinical outcome across the three patients. Timeline of associated peripheral blood collections are indicated in red text below. Purple triangle indicates a change in systemic therapy from anti-PD-1 monotherapy. Cross indicates patient death. AWD, alive with disease; NED, no evidence of disease.
(C) Uniform manifold approximation and projection (UMAP) of cell clusters obtained from scRNA/TCR-seq of sorted CD3+ T cells, which are further defined in (D).
(D) Heat map of differentially expressed genes found in each T cell cluster.
(E) UMAP overlaid with TCRαβ clone size as assessed from scTCR-seq data.
(F and G) Proportion of cells from each region type in each CD8+ (F) and CD4+ (G) T cell cluster. Heatmap colors show proportions scaled per cluster.
(H) Scatter plot of exhaustion scores among CD8+ T cells ordered along diffusion pseudotime (DPT), colored by anatomical region.
(I) Box and whisker plot of exhaustion score per cell in the indicated region types. Statistical testing by two-sided t test (****p < 0.0001).
(J and K) Flow cytometric quantification of %CD39 (J) or PD-1 MFI (K) on CD8+ T cells across the indicated region types. Statistical testing by two-sided t test (**p < 0.01). Error bars represent standard error of the mean.
For (I–K), only thoracic resection regions from MSK 1263 and 1302 were included in this analysis due to concomitant availability of adjacent normal, no viable tumor, viable tumor, and LN regions.
(L) CD39 and PD-1 flow cytometry plots from two adrenal regions involved with tumor in MSK 1263 gated on CD8+ T cells.
(M) Paired box and whisker plots of average exhaustion score per clonotype that is matched between regions without viable tumor and regions with viable tumor. Statistical testing by paired two-sided t test. Error bars represent standard error of the mean.
(N–O) Scatter plot of exhaustion scores among Treg (N) or TFH (O) cells ordered along DPT. Points are colored by region type as in (H).
(P) Comparison and overlap of top genes correlated with DC1 (top 20th percentile) for CD8+, Treg, and TFH. Numbers indicate the number of genes in each set. Select genes in each category are shown.
All box and whisker plots are defined as follows: center line, median; box, interquartile range; upper whisker limit, maximum without outliers; lower whisker limit; minimum without outliers; points, outliers.
See also Figures S1–S6, Data S1, and Table S1.
Pathological analysis revealed substantial tumor heterogeneity among the various tissue regions (Table S1). MSK 1263 and 1302 each had four regions containing varying amounts of cancer cells and four regions without evident viable cancer cells (Data S1); MSK 1344 had viable cancer cells in all regions but with varying involvement (Data S1). Bulk RNA sequencing of tumor regions demonstrated inter-regional heterogeneity, particularly in MSK 1263 and 1302 (Figures S1B–S1E, Table S1). The tumor regions with viable cancer cells showed enrichment for pathways that indicated an ongoing immune response, including an inflammatory response and an interferon gamma response (Figure S1E, Table S1). In line with this observation, CD8+ immunohistochemistry (IHC) revealed that areas with viable tumor cells consistently showed an immune-infiltrated pattern, as opposed to immune-desert or immune-excluded patterns (Data S1).15 Since intra- and inter-patient heterogeneity can be obscured by bulk analysis, we hypothesized that applying scRNA/TCR-seq to CD3+ T cells (Figure S1F) in these heterogeneous regions could yield important insights into the systemic anti-tumor T cell response during ICB.
Multi-regional scRNA/TCR-seq of the T cell response to NSCLC
Recent work has reported scRNA/TCR-seq datasets across a number of different cancer types at relatively low per-patient depth. To obtain greater resolution into the intra-patient T cell response during ICB, we performed scRNA/TCR-seq on sorted CD3+ T cells from 32 tumor, adjacent normal, and LN regions (31 passing quality control) (Figures S2A–S2C, Table S1, STAR Methods). T lymphocytes were clustered into seven CD8+ T, six CD4+ T, and one mucosal-associated invariant T cell clusters (Figure 1C). These clusters were annotated by comparing differentially expressed cluster markers with previously published cluster definitions in scRNA-seq datasets4,16,17 (Figures 1D, S2D–S2F, and S3A–S3C, Table S1, STAR Methods). We next used scTCR-seq data to identify and link T cell clones to their cellular phenotypes. As expected, there was minimal TCR overlap among the three patients (Figure S4A). By pairing TCR information with phenotypes, we observed that CD8+ T cell clusters contained clones with substantially larger clone sizes relative to CD4+ T cell clusters (Figures 1E, S4B, and S4C). We observed that the TCR composition within the three adrenal regions from MSK 1263 were more similar to each other than to the primary tumor or adjacent normal regions (Figure S4D). As expected, we found that the TCR repertoire was most diverse in the LN regions; in contrast, there was greater clonal enrichment in the tumor regions (Figure S4E). We hypothesized that integration of T cell clonotypic features with cell state and pathological features would yield informative insights into the clonal T cell response during ICB.
Exhausted CD8+ T cells, regulatory CD4+ T cells, and follicular helper CD4+ T cells are enriched in tumor regions with viable cancer cells
We next evaluated whether specific T cell phenotypes were enriched in regions containing viable tumor cells. We focused on 20 regions from MSK 1263 and 1302 resection samples that included all representative region types. Among CD8+ T cell clusters, we observed that LNs were enriched for CD8-NAIVE and CD8-TCF1 cells, while adjacent normal regions were enriched in CD8-EFF cells (Figure 1F). The two exhausted CD8+ clusters (TEX) were enriched in the tumor regions relative to adjacent normal regions, and this effect was more pronounced in the tumor bed regions with viable cancer cells, which is consistent with prior reports in lung cancer.16,18 Among CD4+ T cell clusters, we observed an enrichment of CD4-NAIVE, CD4-regulatory CD4+ T cell (TREG)1, and CD4-follicular helper CD4+ T (TFH) cells in LNs, while CD4-EFF1 and CD4-EFF2 were enriched in adjacent normal regions (Figure 1G). CD4-TREG1, CD4-TREG2, and CD4-TFH cells were enriched in tumor regions relative to adjacent normal regions, and these cells were further enriched in the regions with viable tumor. Furthermore, by tracing T cell clones across regions, we observed that clones enriched among regions with viable tumor were over-represented by CD4-TREG1 and CD4-TFH phenotypes (Figures S4F–S4G). In summary, the regional distribution of T cell sub-types is non-random, and TEX, Treg, and TFH are coordinately enriched in regions with viable tumor.
Transcriptional signatures of progressive CD8+ T cell exhaustion across organs
We systematically characterized gene expression changes of CD8+ T cells with respect to their anatomical locations to identify cell fate transitions during the T cell response to ICB. We first used diffusion maps to reconstruct developmental relationships between CD8+ T cell subsets using pseudotime19 (Table S1). We found that cells were ordered along the diffusion pseudotime (DPT) according to phenotype cluster, with CD8-NAIVE, CD8-EFF, and CD8-PROLIF-EXH cells each at one of the three ends of the diffusion map (Figure S5A). Using DPT to identify potential differentiation branch points, we observed that CD8-NAIVE T cells transitioned through the CD8-GZMK and CD8-TRM populations in branch 1 before diverging into an exhaustion branch or effector branch (Figures S5B and S5C). Cells along the exhaustion branch showed preferential localization to the viable tumor and adrenal regions, whereas cells along the effector branch were preferentially found in the adjacent normal tissue and tumor regions without viable tumor (Figure S5D).
We next calculated a CD8+ T cell exhaustion score20 (Table S2) along the DPT, which revealed that CD8+ T cells in viable tumor areas displayed the highest level of exhaustion (Figures 1H, 1I, S5E, and S5F). Notably, this increase in exhaustion was maintained even when genes in the exhaustion signature were removed from the DPT calculation (Figure S5G), suggesting that the DPT is capturing a broader biological program independently associated with CD8+ T cell exhaustion. To verify this finding, we performed flow cytometry on CD8+ T cells and found that cells from viable tumor regions expressed higher levels of the exhaustion markers, CD39 and PD-1, compared with T cells from other regions (Figures 1J–1L and S5H). Finally, clone-matched analysis of 851 CD8+ T cell clones (612 from MSK 1263; 239 from MSK 1302) present in both non-viable and viable tumor regions demonstrated that CD8+ T cells exhibited a marginally greater exhaustion score in viable tumor regions than in non-viable tumor regions (Figure 1M), indicating that cells within a clone may take on distinct cell states depending on positioning within the tumor.
Treg and TFH have distinct clonal repertoires and acquire exhaustion-associated transcriptional programs
Since Treg and TFH also showed similar regional enrichment as CD8+ TEX, we further interrogated their regional gene expression patterns. We first identified 52 Treg- and 51 TFH-predominant clonotypes (clone size >10 cells) based on the majority phenotype among cells within each clonotype (Figures S6A and S6B). Notably, Treg and TFH-predominant clonotypes were largely non-overlapping (Figure S6C). Next, we performed DPT analysis on the Treg- and TFH-predominant clonotypes to examine transitional states across anatomical regions. For both Treg- and TFH-predominant clonotypes, DPT correlated with the anatomical region of the tumor, similar to CD8+ T cells (Figures S6D and S6E). Since DPT was associated with anatomical region, we examined the genes that correlated with DC1 to discover region-dependent transcriptional patterns of Treg and TFH (Table S1). Despite minimal clonal overlap, Treg and TFH cells shared region-associated gene expression changes, including ENTPD1, PDCD1, TNFRSF18 (GITR), TNFRSF4 (OX-40) as genes that positively correlated with DC1 and CXCR4, KLF6, and IL7R as genes that negatively correlated with DC1 (Figures S6F and S6G, Table S1). To validate these findings, we performed flow cytometry on regional samples and found that the expression of CD39, PD-1, and GITR was greater in the tumor regions with viable tumor (relative to the normal and LN regions), while CXCR4 expression was lower (Figures S6H–S6O).
IL32 and CXCL13 were observed to be the top positively correlated gene for DC1 in Treg and TFH, respectively (Figures S6F and S6G, Table S1). Since gene variation from bulk diffusion component analysis could be explained by intra-clonotypic regional heterogeneity or differential regional prevalence of clonotypes with distinct gene expression programs, we evaluated the regional variation in IL32 and CXCL13 in Treg and TFH, respectively, at the clonal level. To this end, we examined 40 Treg-predominant clones and 42 TFH-predominant clones that were present across at least two region types and observed that the expression of IL32 and CXCL13 varied regionally even when controlling for clonotype (Figures S6P and S6Q).
We observed that the genes associated with CD8+ T cell exhaustion were also associated with region-correlated DPT for Treg and TFH (e.g. LAG3, CTLA4, PDCD1, TIGIT, HAVCR2, ENTPD1). Although T cell exhaustion is best described for CD8+ lymphocytes, it has been proposed that an analogous pathway may exist in CD4+ T cells.21–23 To assess whether CD4+ T cells also demonstrate region-dependent progressive exhaustion, we evaluated the exhaustion signature score for Treg and TFH clones and observed that the exhaustion signature increased in both CD4+ populations along DPT (Figures 1N and 1O). Notably, there was no rise in DPT-associated exhaustion score for effector CD4+ clones (Figures S6R and S6S). Common genes associated with region-associated exhaustion in TEX, Treg, and TFH included the activation/exhaustion markers TNFRSF18, CD38, HAVCR2, TIGIT, and HLA-DRA, the chemokine receptor CXCR3, and the proliferation marker TUBB (Figure 1P).
CXCL13 was highly expressed in thoracic regions containing viable tumor relative to regions of no viable tumor (Figure S6T). Since CXCL13 from CD8+ T cells has been associated with the recruitment of TFH and B cells to tertiary lymphoid structures (TLS),24 we assessed whether TLSs are also enriched in regions of viable tumor. We quantified CD3+ CD20+ TLSs by IHC across the various thoracic and adrenal regions. While there was a correlation between TLS number and the amount of stroma in a particular region, there was no correlation with the level of viable tumor in a region (Figures S6U–S6Z). This suggests that the enrichment of exhausted CD8+ T cells and TFH in regions of viable tumor may occur independently of TLSs, which has been previously described for human tumor-infiltrating lymphocytes.25,26 Thus, our combined clonal, phenotypic, and regional analysis reveals that Treg and TFH undergo gene expression changes that resemble CD8+ T cell exhaustion, suggesting that tumor antigen-specific signaling may also drive Treg and TFH differentiation.
Intratumoral exhausted CD8+ T cells are clonally linked to LN progenitors
Recent studies have demonstrated that exhausted CD8+ T cells in the tumor are derived from LN TCF-1+ progenitor exhausted CD8+ T cells.11,13,27 We next asked whether human exhausted CD8+ T cells in the tumor have a clonally linked TCF-1+ counterpart in the draining LN, and whether the cell states among clonally linked CD8+ T cells in the two compartments are distinct. To improve the resolution of analyzing exhausted cell states, we re-clustered cells from 115 clones (comprising 7,116 cells) exhibiting high intratumoral exhaustion scores (>0, exhaustionhi) that were expanded (>2 cells) and present in both the LN and tumor regions of MSK 1263 and 1302, the two patientsfromwhich we had sequenced uninvolved draining LN T cells (20.4% of 564 expanded exhaustionhi clones) (Figures 2A and S7A, Table S2). This yielded 7 clusters, ranging from central memory-like and progenitor exhausted clusters to 4 exhausted populations expressing varying levels of inhibitory receptors. We observed that cells from the LN regions were enriched for progenitor exhausted cluster 2 (Figure 2B) (41.2% of LN cells; 1.0% of tumor compartment), which expressed TCF7 and other memory T cell markers such as SELL (encoding CD62L, L-selectin) and IL7R, albeit at slightly lower levels than the central memory cluster (Figure 2C). Cells in this progenitor exhausted cluster also expressed high levels of TOX and moderate levels of PDCD1, HAVCR2, and LAG3 in comparison to the more terminally exhausted clusters 4–7. Differential expression analysis of this LN-dominant cluster identified several additional less well-described genes that mark these clonally linked LN cells, including CD27, LAMP1, ITGB2, GZMA, IL32, KLF12, and KLRG1 (Figure 2C).
Figure 2. Intratumoral CD8+ T cells can be found in a TCF-1+ CD62L+ progenitor exhausted state in the regional LN.

(A) Uniform manifold approximation and projection (UMAP) of re-clustered cells from CD8+ T cell clones with high exhaustion scores (exhaustionhi) that were expanded (more than 2 cells) and found in both LN and tumor regions. Cells are colored according to phenotype cluster.
(B) UMAP of re-clustered cells from (A) split by region type: LN (left), tumor (right). Bar plots of phenotypic cluster distribution among cells from LN or tumor regions (bottom).
(C) Heat map showing expression of select memory, exhaustion, and progenitor exhausted cluster markers among the clusters from (A).
(D) Volcano plot of differentially expressed genes between clone-matched cells in the LN and tumor from exhaustionhi CD8+ T cell clones.
(E and F) Pie chart of CD8+ T cell clones in the CD8-EXH and CD8-PROLIF-EXH clusters (E) or exhaustionhi clones (F) in the tumor that could be matched to a clonotype in the LN (medium blue and dark blue, “TCR match in LN”). Dark blue slice indicates that the matched clone could be found with a progenitor score of greater than 0 in the LN.
(G) Paired box and whisker plots of average progenitor score per CD8+ T cell clone in the CD8-EXH and CD8-PROLIF-EXH clusters in thoracic regions of MSK 1263 and 1302 (left) or adrenal regions of MSK 1263 (right) that is matched among the LN, regions without viable tumor, and regions with viable tumor. Statistical testing by paired two-sided t test. Error bars represent standard error of the mean.
(H) Paired box and whisker plots of average progenitor score per exhaustionhi CD8+ T cell clone in thoracic regions of MSK 1263 and 1302 (left) or adrenal regions of MSK 1263 (right) that is matched among the LN, regions without viable tumor, and regions with viable tumor. Statistical testing by paired two-sided t test. Error bars represent standard error of the mean.
(I and J) Pie chart of exhaustionhi CD8+ T cell clones in two external datasets that could be matched to a clonotype in the LN (medium blue and dark blue, “TCR match in LN”). Dark blue slice indicates that the matched clone could be found with a progenitor score of greater than 0 in the LN.
(K and L) Paired box and whisker plot of average progenitor score per clone that is matched among the LN and tumor regions in five separate patients from two external datasets. Statistical testing by paired two-sided t test. Error bars represent standard error of the mean.
All box and whisker plots are defined as follows: center line, median; box, interquartile range; upper whisker limit, maximum without outliers; lower whisker limit; minimum without outliers; points, outliers.
By comparing the phenotype of cells within the LN and tumor compartment at the clonal level, we found that 63 of 115 clones (54.8%; 44.4% of MSK 1263 clones; 92.0% of MSK 1302 clones) contained LN cells present in the progenitor exhausted cluster (Figure S7B). Furthermore, a higher proportion of the LN compartment per clone was found in the progenitor exhausted state (average percent of clone in progenitor exhausted cluster 2: LN, 7.9%; tumor, 1.1%) (Figures S7C and S7D). Conversely, a higher fraction of tumor CD8+ T cells was present in the terminally exhausted clusters as compared with their clonally linked LN counterparts. Analogous analysis using the original total T cell population-based clustering yielded similar results (Figure S7D). To probe for finer transcriptional differences between LN and tumor cells, we performed clone-matched differential expression analysis of these exhausted CD8+ T cell clones that could be found in both the LN and tumor. As expected, we observed a higher level of TCF7 in the LN (Figure 2D), as well as increased expression of SELL, CD27, GZMK, and heat shock proteins (HSPA1A, HSPA1B, and HSPA6). Conversely, CD8+ T cells within the tumor regions overexpressed DUSP4, ZFP36, CCL4, CXCR4, and exhaustion-related markers such as CXCL13 and ZNF683.
To validate these findings through alternative transcriptional signature-based approaches, we enumerated the frequency of LN progenitor exhausted CD8+ T cell clones by surveying clones that could be found in the exhausted CD8+ T clusters (CD8-EXH or CD8-PROLIF-EXH) in the tumor tissue of MSK 1263 and 1302 and identifying clone-matched cells in the regional LN. We then assessed the percentage of the matched clones that had a TCF7 transcript of greater than 0 (Figure S7E). We observed that 16.7% and 21.4% of intratumoral exhausted CD8+ T cell clones with paired representation in the LN of MSK 1263 and 1302, respectively, were TCF-1+ (5.7% and 7.3% of total exhausted CD8+ T cell clones) (Figure S7F). Since TCF-1 expression may also mark naive CD8+ T cells rather than progenitor exhausted populations, and since gene dropout might result in undercounting of TCF-1+ progenitors, we repeated this analysis with a progenitor signature that was derived from antigen-specific TCF-1+ Tim-3– PD-1+ CD8+ T cells from a murine melanoma model28 and validated in human lung cancer,29 which included TCF7, SLAMF6, IL7R, and XCL1 (Table S2). Using a progenitor score cutoff of greater than 0 (Figure S7G), we noted that 24.3% and 35.7% of exhausted CD8+ clones that could be found in the LN of MSK 1263 and 1302, respectively, could be found in a progenitor exhausted state (8.4% and 12.2% of total exhausted CD8+ T cell clones) (Figure 2E). We also performed the same analysis using exhaustionhi CD8+ T cell clones, which yielded a similar proportion of clones found in a LN progenitor state (Figures 2F and S7H). To assess how clonal CD8+ T cell states vary across regions, we evaluated the extent to which the progenitor phenotype of exhausted CD8+ T cell clones could be found in LNs, regions of no viable tumor, and regions of viable tumor. As expected, the progenitor score of CD8+ T cell clones was decreased in cells from the tumor relative to the LN (Figures 2G and 2H).
To confirm these findings in independent cohorts, we examined scRNA/TCR-seq data from five patients across two cohorts with resection of primary tumor and regional LNs after receiving anti-PD-1 treatment for lung cancer.30,31 We found that 27.6%–45.1% of CD8+ T cell clones with high exhaustion scores that were present in both the LN and tumor could be found in a LN progenitor state(12.8%–21.6% of total exhausted CD8+ T cell clones) (Figures 2I, 2J, S7I, and S7J). In these patients, there was also a decrease in progenitor score when comparing clone-matched CD8+ T cells between the LN and tumor (Figures 2K and 2L).
We have recently characterized a TCF-1+ PD-1+ precursor of TFH in a murine lymphocytic choriomeningitis virus (LCMV) model.32 To evaluate for a putative human LN progenitor of CD4+ T cell populations, we examined TFH and Treg clones that were present in both the LN and tumor compartments. This clone-matched analysis revealed greater expression of TCF7 and PDCD1 in the LN for TFH, but not Treg clones (Figures S7K and S7L). This transcriptional difference between LN and tumor cells from TFH clones was observed even though cells from both compartments were designated as TFH based on clustering (Figures S7M and S7N). Altogether, these results point to the presence of TCF-1+ LN progenitor populations that are clonally linked to exhausted CD8+ T cells and TFH in the tumor microenvironment as a feature of T cell responses in human lung cancer.
Tumor-specific CD8+ T cells are enriched in viable tumor regions
To identify tumor-specific T cells, we first used a tumor-reactivity signature score based on published features of tumor-specific CD8+ T cells33 (Table S2). This tumor-reactivity signature had high concordance with three other recently published signatures derived from single-cell sequencing of neoantigen- and tumor antigen-specific tumor-infiltrating lymphocytes30,34,35 and, as expected, had minimal signature overlap with viral-specific CD8+ T cells (Figures S8A and S8B, Table S2). Consistent with prior reports that exhausted T cells comprise the tumor-specific population and that they are enriched in tumor regions,24,30,33,34,36–38 we found that the CD8+ TEX clusters displayed the highest tumor-reactivity score (Figure S8C), and that CD8+ T cells in viable tumor regions had the highest tumor-reactivity score (Figure S8D). Additionally, the top 40 most expanded CD8+ T cell clones with high tumor-reactivity scores (TRhi) (Figure S8E) were preferentially found in viable tumor regions, whereas the top 40 most expanded CD8+ T cell clones with low tumor-reactivity scores (TRlo) were more enriched in the LN, adjacent normal lung, and tumor regions without viable cancer (Figure 3A). Furthermore, TRhi CD8+ T cell clones were often found in the CD8-TRM and TEX clusters, whereas TRlo CD8+ clones were enriched in effector CD8+ clusters (Figure 3B), suggesting that T cell clones with tumor-specific features are preferentially present in an exhausted state within regions with viable cancer.
Figure 3. Phenotypic and regional enrichment of tumor-specific CD8+ T cell clones.

(A) Bar plots of the proportion of cells in the indicated region type among the top 40 most expanded TRhi (left) or TRlo (right) CD8+ clones.
(B) Bar plots of the proportion of cells in the indicated phenotype clusters among the top 40 most expanded TRhi (left) or TRlo (right) CD8+ clones.
(C) Venn diagram of overlap between TCRβ sequences from MSK 1263 identified by empirical tumor-specific methods and the tissue sorted CD3+ scRNA/TCR-seq dataset (yellow). Numbers indicate the number of TCRβ sequences in each intersection. Numbers colored in red represent TCRβ clones identified by at least two empirical methods (designated as high-confidence neopeptide-specific clones).
(D) Box and whisker plot of tumor-reactivity scores33 among CD8+ T cells with the indicated TCR specificity.
(E) Box and whisker plots of the proportion of cells in clones within each specificity belonging to the indicated CD8+ T cell clusters. Each point represents one TCR clone. Statistical testing by two-sided Wilcoxon-test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
(F and G) Bar plots of the proportion of cells in the indicated phenotype cluster (F) or region type (G) among the top most expanded high-confidence neopeptide-specific clones (left), viral-specific clones (middle), or clones with unknown specificity (right).
(H) Paired box and whisker plot of average progenitor scores per high-confidence neopeptide-specific clone in MSK 1263 that is matched among the LN and tumor regions. Statistical testing by paired two-sided t test.
(I) Box and whisker plots of gene signature scores for CD8+ tumor-reactivity33 among clones with the indicated TCR regional pattern.
(J) Bar plots of the proportion of clones with each TCR specificity colored by TCR tumor regional pattern.
All box and whisker plots are defined as follows: center line, median; box, interquartile range; upper whisker limit, maximum without outliers; lower whisker limit; minimum without outliers; points, outliers.
See also Figures S8–S11 and Table S3.
We performed a similar analysis with a 40-parameter tumor-reactivity score for CD4+ T cells35 and observed that the Treg and TFH clusters exhibited the highest CD4+ tumor-reactivity score (Figure S8F, Table S2). Concordant with CD8+ T cells, the top 40 most expanded TRhi CD4+ T clones were more enriched in viable tumor regions relative to TRlo CD4+ clones (Figures S8G and S8H). Moreover, the top 40 most expanded TRhi CD4+ T clones were enriched in the Treg and TFH cell states (Figure S8I). Overall, these results show that clonally expanded CD8+ and CD4+ T cells with tumor-specific features are enriched in regions of viable tumor.
Validation of tumor-specific T cell responses with empirically defined TCR clones
Next, to confirm these features of tumor-specific clones, we computationally predicted neoantigens from tumor whole exome sequencing of each patient using NetMHC, a neural network-based algorithm trained on a large dataset of peptide binding to human leukocyte antigens (HLAs)39,40 (Figure S9A, Table S3, STAR Methods). Predicted candidate neoantigens were then tested for empiric HLA binding capacity by flow cytometry (Figure S9B, STAR Methods). In total, we identified six, six, and eight neoantigen peptide candidates that could bind the cognate HLA for MSK 1263, 1302, and 1344, respectively. We then employed three parallel methods to identify T cell clones that recognize these neoantigens: (1) bulk TCR sequencing of multimer+ CD8+ tumor-infiltrating lymphocytes (TILs), (2) mutation-associated neoantigen functional expansion of specific T cells (MANAFEST) assay,41 and (3) scRNA/TCR-seq of multimer+ CD8+ TILs.
First, we generated multimers against predicted neoantigen peptides (neopeptides) and performed bulk TCR sequencing of sorted multimer+ CD8+ TILs (Figures S9C–S9E). For MSK 1263, we observed that approximately 4%–38% of CD8+ T cells were specific for neopeptides (Figure S9C), which indicated that our neoantigen prediction pipeline could identify bona fide neopeptides. Since the multimer+ population was negligible for MSK 1302 and 1344 (Figures S9D and S9E), we restricted subsequent analyses to MSK 1263.
Second, we performed a MANAFEST assay on the peripheral blood of MSK 1263 to identify neoantigen- and viral antigen-specific clones (Figures S9F and S9G). Briefly, CD8+ T cells were cultured with no peptide, a pool of neoantigen peptides, or a pool of viral peptides. Enrichment of TCRs in each culture condition was then assessed by bulk TCR sequencing to determine reactivity to neoantigen or viral peptides. Nine TCRs were found to be reactive to neoantigens, while 12 TCRs were reactive to viral antigens.
Third, we performed scRNA/TCR-seq on sorted multimer+ CD8+ T cells from tumor and LN regions from MSK 1263 (Figure S10A, Table S3). From this approach, we obtained 25,588 multimer+ CD8+ T cells, of which 22,440 (87.7%) had paired TCRab chains captured (Figure S10B). To examine the concordance of multimer+ CD8+ phenotypes with those in the original tissue regional dataset (Figure 1C), we projected the multimer+ cells onto the original data using label transfer and observed that most of the multimer+ cells mapped to the CD8-GZMK, CD8-TRM, and TEX clusters (Figures S10C–S10E).
We examined the extent of overlap between the three independent methods. Fifty-four TCR clones were identified as tumor specific by at least two methods, of which 53 were present in the original tissue scRNA/TCR-seq dataset (Figure 3C, Figure S10F). These clones are referred to as tumor-specific high-confidence clones, while all other clones identified as tumor-specific by one method are categorized as low-confidence. Next, we assessed the concordance between empirically defined tumor-specific T cells and those inferred based on the tumor-reactivity signature score. We observed high concordance between the two definitions, as 11,818 of 12,935 (91.3%) high-confidence tumor-specific T cells were also categorized as TRhi (Figure S10G). Notably, all viral antigen-specific T cells identified by the MANAFEST assay were categorized as TRlo. Additionally, high-confidence tumor-specific T cells displayed the highest CD8+ T cell tumor-reactivity score relative to low-confidence tumor-specific, viral-specific, or unknown-specificity clones (Figure 3D). In comparison with viral-specific clones, we observed that tumor-specific clones were composed mainly of CD8-TRM and TEX cells (Figures 3E and 3F), which was in line with the phenotypes of TRhi clones. Conversely, viral-specific clones and clones with unknown specificity were dominated by CD8-EFF and CD8-GZMK clusters, which mirrored TRlo clones.
We next examined the regional presence of tumor-specific clones. In line with our observations on regional skewing of TRhi clones, we observed that tumor-specific clones were preferentially present in viable tumor regions (Figure 3G). We also evaluated whether tumor-specific clones could be found in a progenitor exhausted state in the LN. Indeed, among tumor-specific T cell clones that could be found in both LN and tumor, we found that the LN CD8+ T cells had a higher clonal progenitor score relative to their intratumoral counterparts (Figure 3H). These tumor-specific LN cells expressed TCF7, CCR7, IL7R, and GZMK, while their clone-matched counterparts in the tumor expressed DUSP4, CCL4, CD52, CXCR6, HLA-DRB1, HLA-DPA1, and GZMB (Figure S10H). We also compared tumor-specific CD8+ T cells within regions with or without viable tumor and observed that tumor-specific CD8+ T cells in regions with viable tumor expressed higher levels of GZMB, CD27, CD38, and GZMK, as well as markers associated with tumor-reactivity such as ENTPD1 and TNFRSF9 (Figure S10I). Altogether, these findings demonstrate that empirically defined tumor-specific T cells display region-dependent transcriptional states and are clonally linked to LN progenitors.
Tumor-specific clones display pan-tumor, but not ubiquitous, regional distribution
We next investigated the regional distribution of the tumor-specific T cell clones. By assigning TCR clones into mutually exclusive regional categories (Figures S11A–S11C, STAR Methods), we observed that tumor-specific clones were most frequently observed in the pan- and oligo-regional tumorenriched distribution (Figure 3I), suggesting that they move throughout the tumor and are not restricted to a single region. We observed similar distributions for empirically defined tumor-specific clones, as nearly all expanded high-confidence tumor-specific clones were present in multiple or all tumor regions (Figure 3J).
A comparison of tumor-enriched and ubiquitous CD8+ T cell clones revealed higher expression of DUSP4, CXCL13, TIGIT, TOX, ENTPD1, and CTLA4 in tumor-enriched clones (Figure S11D, Table S3), supporting the notion that these clones likely recognize tumor antigen. Conversely, ubiquitous CD8+ T cell clones differentially expressed cytotoxic genes such as NKG7, PRF1, GZMA, GZMB, and other markers of activation, such as CCL4. Similarly, tumor-enriched CD4+ T cell clones also overexpressed many of the same genes as tumor-enriched CD8+ T cell clones, including DUSP4, CXCL13, TIGIT, TOX, ENTPD1, and CTLA4 (Figure S11E). Altogether, characterization of regional clonotype distribution patterns revealed that tumor-reactive CD8+ and CD4+ T cell clones are preferentially found in a tumor-enriched distribution.
Tumor-specific T cell clones persist throughout the course of ICB
We next assessed whether the different T cell clusters in resected tumors were differentially represented in the peripheral blood. To do this, we performed bulk TCRβ sequencing from the peripheral blood of each patient at multiple time points after ICB, which included the time period before, during, and after resection. The latest blood collection ranged from 216 to 1,013 days after the start of ICB (Table S1). For both CD8+ and CD4+ T cell clusters, we noted substantial heterogeneity in the representation of each cluster in the peripheral blood (Figure 4A). Among CD8+ T cells, the variability was striking: TCR clones associated with the CD8-TCF1 cluster were the least prevalent in the peripheral blood, whereas clones associated with the CD8-EFF cluster were the most prevalent, with an almost 100-fold difference between the two (Figure 4A). Among CD4+ T cells, TFH clones in the tissue were the least prevalent in the peripheral blood, while CD4+ effector clones were the most prevalent (Figure 4A). Furthermore, the tumor-reactivity score of both CD8+ and CD4+ T cells from the tissue was inversely proportional to their frequency in the peripheral blood at the phenotypic cluster level (Figure 4B).
Figure 4. Peripheral persistence of tumor-specific CD8+ T cell clones.

(A) Circulating frequency of clonotypes with the indicated CD4+, CD8+, or mucosal-associated invariant T (MAIT) phenotypes designated by tissue scRNA/TCR-seq in MSK 1263, 1302, and 1344. Each clonotype was counted once based on majority phenotype. Box and whisker plots are defined as follows: center line, median; box, interquartile range; upper whisker limit, maximum without outliers; lower whisker limit; minimum without outliers.
(B) Spearman correlation of mean tumor-reactivity score and peripheral blood frequency per CD8+ (left) or CD4+ (right) T cell cluster.
(C) Circulating frequency over time of TRhi (top) and TRlo (bottom) CD8+ clones from patients MSK 1263, MSK 1302, and MSK 1344.
(D) Circulating frequency over time of CD8+ T cell clones with the indicated empirical antigen specificity from patient MSK 1263.
See also Figures S9–S11.
Because of the logistical difficulty of tracking a multitude of tumor-specific CD8+ T cell clones, the importance of the persistence of endogenous tumor-specific T cell clones is not well established. We evaluated the persistence of tumor-specific clones defined by transcriptional features in the peripheral blood during ICB using bulk TCR sequencing. We tracked 287, 137, and 438 CD8+ T cell clones that could be found at all time points analyzed from MSK 1263, 1302, and 1344, respectively, and observed that both TRhi and TRlo CD8+ T cell clones persist over time in the peripheral blood (Figure 4C). For MSK 1344, we were able to track these clones for nearly 3 years after the start of ICB. Additionally, empirically validated tumor-specific CD8+ T cell clones were traced in the peripheral blood of MSK 1263 and were found to persist over time (Figure 4D). We also longitudinally tracked 427, 189, and 957 CD4+ T cell clones that could be found at all time points analyzed from MSK 1263, 1302, and 1344, respectively (Figure S11F) and observed persistence of these clones in the periphery.
DISCUSSION
In this study, we performed paired scRNA/TCR-seq of 187,650 T cells from 31 tissue regions, including matched tumor, adjacent normal tissues, and LNs from 3 patients. This dataset yielded several insights into the tissue distribution, persistence, and differentiation trajectories of T cells in patients receiving ICB therapy. We first demonstrated that regions with viable cancer cells were enriched for exhausted CD8+ T cells, Treg, and TFH. These findings confirm prior reports demonstrating the correlation of Treg and exhausted CD8+ T cells in tumor regions (relative to adjacent normal) in lung cancer16,18,42 and further strengthen the findings by including analyses of tumor bed regions that were not involved with viable cancer cells. These findings are also consistent with a recent pan-cancer analysis which revealed that exhausted CD8+ T, TNFRSF9+ Treg, and TFH are tumor-enriched meta-clusters, indicating that this enrichment may not be specific to NSCLC.5 Beyond enrichment, we used DPT analyses to demonstrate that CD8+ T cells, Treg, and TFH undergo progressive exhaustion in proximity to viable cancer cells, suggesting that tumor antigen recognition may drive similar transcriptional programs in each cell type. Concordantly, intratumoral ICOS+ PD-1+ CD4+ TFH cells preferentially recognize tumor-derived neoantigens compared with other CD4+ subsets.43
The depth of paired scRNA/TCR-seq data across tissue regions enabled the identification of a population of LN progenitors that was clonally linked to intratumoral exhausted CD8+ T cells. Clonally linked cells in the LN were marked by higher expression of TCF7, SELL, CD27, and KLRG1 in comparison with their intra-tumor counterparts, supporting recent reports of a stem-like progenitor exhausted sub-population that expresses higher levels of SELL.44,45 Functional tumor-reactivity assays to empirically define tumor-specific T cells confirmed neoepitope specificity of clones containing progenitor exhausted T cells. Moreover, by comparing TFH clones between the LN and tumor tissues, we also observed clonally linked LN counterparts for TFH that expressed higher levels of PD-1 and TCF-1, which is consistent with a recent report of PD-1+ TCF-1+ progenitors of TFH cells in LCMV infection.32 Hence, our data substantiate the concept that progenitors in the draining LNs fuel the intratumoral T cell response in human cancers, and that similar progenitor cell states may underlie CD8+ and CD4+ T cell responses to persistent tumor antigens. However, it is important to note that while we infer a unidirectional movement from the LN to the tumor microenvironment, we cannot rule out the possibility that T cells may egress from the tumor and seed the LN, which has been demonstrated in murine models.46
We assessed the regional localization of tumor-specific T cells, compared with bystander or virus-specific T cells. We demonstrated that CD8+ T cell clones with a tumor-enrichedpan pattern score the highest for tumor-reactivity relative to other regional distributions, which suggests that tumor-specific T cells frequently distribute broadly throughout the tumor bed. We confirmed this by tracking empirically defined tumor-specific CD8+ T cell clones and showing that they are infrequently found in only a single tumor region. Thus, the regional resolution of our dataset revealed the anatomical distribution of tumor-specific T cells and highlights their ability to permeate various tumor regions simultaneously, likely through a combination of preferential migration and local expansion.
Finally, we report that tumor-specific T cells defined by published transcriptional signatures and empiric validation persist in the blood after ICB therapy. The persistence of TRhi CD8+ T cell clones is consistent with prior reports longitudinally tracking individual tumor-specific T cells in metastatic melanoma.34 For MSK 1263, we did not find evidence that the overt loss of tumor-specific T cell clones coincided with this patient’s rapid clinical progression of cancer; however, the frequency of TRhi CD8+ T clones did decrease from the initial time point. Subsequent work in larger prospective cohorts will further investigate the clinical significance of partial decreases without overt loss of tumor-specific CD8+ T cells. Altogether, this work serves as a unique, comprehensive, single-cell resource with regional, clonal, and longitudinal resolution and provides insights into human T cell responses during ICB therapy.
Limitations of the current study include the inability to simultaneously profile the tumor cells and non-T immune cells from these patients as our focus was on T cell dynamics. We were unable to derive cell lines or patient-derived xenografts from these cases and, thus, do not have viable material to further interrogate the malignant cell compartment. The primary dataset included many discrete samples but were derived from a total of three patients. Moreover, since this study was not prospectively designed, there are patient-specific differences in the timing of tissue and blood collection. Future larger prospectively designed studies will help to overcome these limitations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ansuman T. Satpathy (satpathy@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All scRNA/TCR-seq and bulk RNA-seq data have been deposited to NIH GEO under accession number GSE185206. Bulk TCR-seq data have been deposited into the ImmuneACCESS database at Adaptive Biotechnologies. All other relevant data are available from the corresponding authors upon request.
Custom code used in this work is available at github.com/satpathylab/regional-lung-cancer.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human biospecimens
Resection materials and blood were obtained with informed consent from patients under protocol #06–107 approved by MSKCC. Regional clonal analyses comparing T cells from lymph node and primary tumor were performed for the MSK 1263 and 1302 lung resection samples. In these two samples, the mediastinal lymph nodes level 7 and 9, respectively, were evaluated as draining lymph nodes based on expected drainage patterns. These lymph nodes were not involved by tumor.
METHOD DETAILS
Pathologic review
Histologic review for extent of tumor viability was performed by J.L.S. and M.B. on H&E slides following the IASLC multidisciplinary recommendations for pathologic assessment of lung cancer resection specimens after neoadjuvant therapy.47 Briefly, the percent of the tissue section that was covered by the tumor bed (as opposed to uninvolved lung parenchyma, uninvolved adrenal tissue, or uninvolved lymph node) was noted by J.L.S. and M.B. Within the tumor bed region, sub-regions were defined as ‘viable tumor’, ‘necrosis’, or ‘stroma’. Thus, the sum of ‘viable tumor’, ‘necrosis’, ‘stroma’, and ‘uninvolved tissue’ is 100% (Data S1 and Table S1). ‘Non-viable tumor’ regions refer to the sum of all of the regions that are not ‘viable tumor’ regions (e.g. ‘necrosis’, ‘stroma’, and ‘non-viable’).
Determination of tumor-infiltrating lymphocyte pattern
CD8 IHC stain was performed by at the Precision Pathology Center at MSKCC. Tissue slides were stained with anti-human CD8 antibody (Clone C8/144B, Dako, catalog #M7103, 1:1000 dilution). IHC was performed on BOND RX platform (Leica Biosystems) using standard Protocol with the following steps: Heat epitope retrieval with ER2 for 30 min, incubation of primary antibody for 30 min, and BOND Polymer Refine Detection system (Leica, catalog # DS9800).
Within the regions with viable tumor (annotated on a separate slide as described above), the areas with viable tumor were analyzed by U.K.B. and M.J. for the dominant CD8+ tumor-infiltrating lymphocyte pattern: inflamed, excluded, or desert15 (Table S1).
Tertiary lymphoid structure immunohistochemistry
Histology was performed by Patrick Savickas at HistoWiz Inc. Brooklyn, NY using a GLP ready Standard Operating Procedure and a fully automated workflow. Samples were processed and embedded in paraffin followed by sectioning at 4mm. Immunohistochemistry (IHC) was performed on a Bond Rx autostainer (Leica Biosystems) with Citric Acid based retrieval buffer with a pH of 6.0 and Heat Induced Epitope Retrieval (HIER) for 20 min. Polyclonal CD20 antibody was used at a 1:2000 dilution and developed with DAB using the Leica Bond Refine Detection kit. CD3 antibody (clone SP7) was used at a 1:100 Dilution and developed with AEC Red using the Leica Bond Refine Red Detection Kit. This was followed by a hematoxylin counterstain and the stained slides were coverslipped with tape and imaged on a Leica Aperio AT2 line scanner.
The TLS were identified and quantified by HistoWiz Inc. using Halo software version 3.3.2541 (Indica Labs, USA) from Indica Labs and using the random forest classifier algorithm. The RF classifier was trained on a few representative slides by selecting a small number of ROIs as examples of TLS, tissue and glass. A minimum TLS size threshold of 60,000mm2 was set to exclude any TLS below this size threshold.
Bulk RNA sequencing
Approximately 200–500 ng of FFPE RNA extracted from FFPE slides with a DV200 range between 3–99 or 65–100 ng of fresh frozen RNA (DV200 98–99) per sample were used for RNA library construction using the KAPA RNA Hyper library prep kit (Roche, Switzerland). The number of pre-capture PCR cycles was adjusted based on the quality and quantity of RNA extracted from the samples. Customized adapters with 3bp unique molecular indexes (UMI) (Integrated DNA Technologies, USA) and sample-specific dual-index primers (Integrated DNA Technologies, USA) were added to each library. The quantity of libraries was measured with Qubit (Thermo Fisher Scientific, USA) and the quality was assessed by TapeStation Genomic DNA Assay (Agilent Technologies, USA). Approximately 500 ng of each RNA library were pooled for hybridization capture with IDT Whole Exome Panel V1 (Integrated DNA Technologies, USA) using a customized capture protocol modified from NimbleGen SeqCap Target Enrichment system (Roche, Switzerland). The captured DNA libraries were then sequenced on an Illumina HiSeq4000 in paired ends (2X100bp) to a target 50 million read pairs per sample. The demultiplexed FASTQ files were aligned to the human genome reference hg19/GRCh37 using STAR (v2.7.3a) and deduplicated from the combination of UMI sequence and alignment coordinate using UMI-tools (v1.0.1). Rsu-bread (v2.6.4) was used to extract the feature count matrix from alignments. We used edgeR (v3.34.1) for normalization, multidimensional scaling, differential expression, and gene ontology (GO) enrichment analyses. For GSEA, we used fgsea (v1.18.0) with MSigDB (v7.4) hallmark pathway gene set. Cell type deconvolution was performed using CIBERSORTx (https://cibersortx.stanford.edu) with reference matrix derived from one lung tumor sample (LUNG_T31) within previously published single-cell data.48
Fresh tumor preparation
Gross resection specimens were promptly sectioned within 1 hr of the resection and tumor pieces from the various regions were placed into human complete medium (RPMI +10% human serum albumin +1% penicillin with streptomycin +0.1% amphotericin + 1X sodium pyruvate + 1X GlutaMAX + 1X minimum essential amino acids) on ice. Human tissue from the various regions were minced with a razor blade and digested in GentleMACS enzyme mix in individual tubes per region for 30–60 min according to manufacturer’s recommendations. After centrifugation of a filtered single cell mix, the cell pellet was resuspended in human complete medium and underwent one round of ACK lysis. A subset of this cell pellet was cryopreserved for future use in Bambanker media (Wako Chemicals).
Flow cytometry and cell sorting
Cells were incubated with TruFCX (for human cells) to block nonspecific binding, and then stained (15 min, 4◦C) with appropriate dilutions of CD45-BV510 (clone 2D1), CD3-BV650 (clone UCHT1), CD8-PerCP-Cy5.5 (clone SK1), CD8-PerCP-Cy5.5 (clone BV510), CD4-Alexa700 (clone A161A1), CD39-APC (clone A1), CD39-PE-Cy7 (clone A1), PD-1-APC-Fire 750 (clone EH12.2H7), FOXP3-FITC (clone PCH101), CXCR5-PE (clone QA18A64), GITR-APC (clone 108–17), and CXCR4-PerCP-Cy5.5 (clone 12G5). TruFCX and all antibodies were purchased from BioLegend. DAPI− CD45+ CD3+ cells analyzed by a BD LSRII or were sorted by FACS Aria. Debris, doublets and dead cells were excluded on the basis of forward and side scatter and 4′,6-diamidino-2-phenylindole (DAPI, 1 mg/ml). Flow cytometry data was analyzed with FlowJo V10.8.1 (TreeStar). Representative gating strategy is depicted in Figure S1F.
Single-cell RNA sequencing
Sorted T cells were stained with Trypan blue and Countess II Automated Cell Counter (ThermoFisher) was used to assess both cell number and viability. Following QC, the single cell suspension was loaded onto Chromium Chip A (10X Genomics PN 230027) and GEM generation, cDNA synthesis, cDNA amplification, and library preparation of 2,700–11,000 cells proceeded using the Chromium Single Cell 5′ Reagent Kit (10X Genomics PN 1000006) according to the manufacturer’s protocol. cDNA amplification included 13–14 cycles and 11–50ng of the material was used to prepare sequencing libraries with 14–16 cycles of PCR. Indexed libraries were pooled equimolar and sequenced on a NovaSeq 6000 or NextSeq 500 in a PE26/92, PE28/91 or PE100 run using the NovaSeq 6000 SP, S1, or S2 Reagent Kit (100, 200, or 500 cycles) or TG NextSeq 500/550 High Output Kit v2.5 (150 cycles) (Illumina). An average of 179 million reads was generated per sample.
Single-cell TCR sequencing
An aliquot of cDNA generated using the methods described above was used to enrich for V(D)J regions using the Chromium Single Cell V(D)J Enrichment Kit Human T Cell (10X Genomics PN 1000005) according to the manufacturer’s protocol with 10 cycles of PCR during enrichment and 9 cycles during library preparation. Indexed libraries were pooled equimolar and sequenced on a NovaSeq 6000 in a PE150 run using the NovaSeq 6000 SP, S1, or S4 Reagent Kit (300 cycles) (Illumina). An average of 129 million paired reads was generated per sample.
Pre-processing of scRNA/TCR-seq libraries
Reads from 10x scRNA expression libraries were aligned to human genome assembly GRCh38 (hg19) and quantified using cellranger count (10x Genomics, v3.1.0). The filtered feature-barcode matrices containing only cellular barcodes were used for further analysis. Single cell gene expression matrices were imported into R (v3.6.1) and analyzed using Seurat (v3.1.4).49 Cells with >450 genes captured and <15,000 UMIs were included in downstream analyses. Additionally, cells with >15% mitochondrial RNA reads were excluded from subsequent analyses. Sequencing data from 31 of 32 regional samples passed initial quality control (Figure S2A, Table S1, STAR Methods). After removing the single region that did not pass QC, 63.5–89.9% of the individual cells per region (Figure S2B, Table S1) passed QC filtering, retaining 162,062 high-quality T cells for downstream analyses.
Single cell TCR reads were aligned to human genome assembly GRCh38 (hg19) and assembled into reconstructed TCR consensus sequences using cellranger vdj (10x Genomics, v3.1.0). Only productive TCRα and TCRβ sequences were considered for further analysis. At least one chain of the TCR was captured in 141,110 cells (87% of the cells that passed QC, 76.0–92.7% per region, Figure S2C, Table S1), and paired TCRαβ chains were captured in 103,181 cells in total. Cells with multiple TCRβ chains captured (ββ, αββ, ααββ) were excluded from further analysis. Only cells with conventional paired TCR chain combinations αβ or ααβ were included in downstream TCR clonal analyses. Cells sharing the same CDR3αβ nucleotide sequences were defined as belonging to the same TCR clone.
scRNA-seq data integration and clustering
scRNA-seq libraries from each region were log10-normalized individually and integrated with Seurat by identifying anchors between datasets using reciprocal PCA with 30 dimensions. TCR genes were excluded from the selection of integration anchors to prevent TCR chain driven biases. Dimensionality reduction of the integrated matrix was performed using Uniform Manifold Approximation and Projection (UMAP) with the first 30 principal components. Phenotypic clusters were defined by constructing a k-nearest neighbors graph and identifying groups of cells using the Louvain algorithm with resolution of 0.6.
Naive CD8+ T cells highly expressed SELL, CCR7, and IL7R. There were two effector CD8+ clusters: CD8-EFF highly expressed GNLY, NKG7, PRF1, and KLRG1, whereas CD8-GZMK highly expressed GZMK, CCL4, NKG7, GZMA, GZMH, PRF1, LAG3, and PDCD1. A CD8+ cluster that highly expressed GMZK, LAG3, NKG7, ENTPD1, HAVCR2, CD38, CD274, and TCF7 was annotated as CD8-TCF1. A CD8+ tissue resident memory (TRM) cluster highly expressed ITGAE, CD69, PDCD1, ZNF683, CXCR3, GZMA, and GZMB. Finally, two exhausted CD8+ T cells clusters distinguishable by their proliferative status were identified: CD8-EXH highly expressed TOX, GZMB, LAG3, NKG7, ENTPD1, HAVCR2, CXCL13, TNFRSF9, and IFNG, while CD8-PROLIF-EXH expressed high levels of these genes in addition to GZMA, CD38, and proliferation genes (TUBB, TUBA1, MKI67, AURKB). Similar to naive CD8+ T cells, naive CD4+ T cells expressed CCR7, SELL, IL7R, and LEF1. Among the two CD4+ T effector clusters, CD4-EFF1 highly expressed IL7R and CD69, while CD4-EFF2 highly expressed GZMA, PRDM1, and CXCR6. Two clusters expressing FOXP3 were annotated as Treg clusters; CD4-TREG1 and TREG2 were distinguished by lower and higher expression of FOXP3, ENTPD1, TNFRSF4, TNFRSF9, TNFRSF18, CD274, ICOS, CTLA4, and TIGIT, respectively. CD4-TFH highly expressed TOX, ICOS, PDCD1, BCL6, CXCR5, and CXCL13.
Categorization of CD4/CD8+ TCR clones
Clones with >75% cells within CD4+ T cell clusters were categorized as CD4+ clones (subcategorized into ‘CD4+ only’ clones with 100% CD4+ cells, or ‘CD4+ majority’ clones with 75–99% CD4+ cells). CD8+ T cell clones were similarly defined. Clones that were present in the MAIT cluster but none of the CD4+ or CD8+ clusters were categorized as MAIT clones. Clones that did not meet any of the above criteria were categorized as ‘mixed’ clones.
TCR clone regional pattern categorization
TCR clones were categorized into mutually exclusive regional patterns for each patient by assessing the combination of region types (i.e. LN, adjacent normal, or tumor regions) in which cells with shared CDR3αβ nucleotide sequences could be found. ‘Ubiquitous’ TCR clones were defined as those found in all LN, adjacent normal, and tumor regions sampled. ‘LN enriched’ and ‘normal enriched’ TCR clones were those found only in LN or adjacent normal regions, respectively. ‘Tumor enriched’ clones were found only in tumor regions, but not in LN nor adjacent normal regions, and were further sub-classified as ‘single region’ (found in only one tumor region), ‘oligo-regional’ (found in >1 but not all tumor regions), or ‘pan-regional’ (found in tumor regions).
TCR clone enrichment in viable/non-viable tumor
TCR clones were categorized as enriched in viable tumor regions or no viable tumor regions based on CDR3ab nucleotide sequence. For each clone, the number of cells found in viable tumor or no viable tumor regions was calculated and constructed into a 2×2 contingency table to test for enrichment by Fisher’s exact test. Clones with p-value <0.05 were considered enriched in viable or no viable tumor regions.
Gene signature scoring
To characterize cells according to previously reported gene signatures of tumor-reactivity, CD8+ T cell exhaustion, progenitor exhausted T cells, tumor- and viral-specificity, and expanded clones (Table S2), gene scores were calculated per cell using the AddModuleScore function from Seurat.
Diffusion pseudotime analysis
To investigate expression dynamics within CD8+ subsets, cells belonging to the CD8+ phenotype clusters were taken for diffusion component analysis. Diffusion maps were constructed with 40 principal components using destiny (v3.0.1).19 Diffusion pseudotime ordering was calculated with the DPT() function using a window width of 0.1 and specifying the top eigenvector-ranked cell as the root cell. Analogous diffusion component analyses were performed with Treg- and TFH-predominant clones expanded >10 cells to probe for gene expression dynamics within CD4+ T cell subsets across anatomical regions. Top genes that correlated with the primary diffusion component were analyzed further at the clonal level.
Clone-matched analysis
To compare cell state differences between CD8+ T cells in regions with no viable tumor vs. viable tumor, we performed clone-matched analysis of CD8+ T cell clones with at least one cell present in both no viable tumor and viable tumor regions. Clonal scores were calculated per region by averaging the scores of cells within each clone in each region.
To characterize T cell state transitions of CD8+ T cell clones between LN and tumor regions, CD8+ T cell clones in an exhausted state were defined in two ways: (1) clones with tumor cells belonging to the CD8-EXH or CD8-PROLIF-EXH phenotype cluster, or (2) clones displaying an average exhaustion score >0 among tumor cells (exhaustionhi). Clonal progenitor scores were calculated per region by averaging the scores of cells within each clone in each region.
External scRNA/TCR-seq dataset analysis
Single-cell data from Caushi et al.30 were obtained from GEO (GSE176021) and analyzed as described above. Only samples from patients with matched LN and tumor samples (MD01–004, MD01–005, MD043–011) were analyzed. Data from a second scRNA/TCR-seq dataset31 (DNA DataBank of Japan: JGAS000480), which included data from two lung cancer patients with matched LN and tumor samples (LC01 and LC03), were similarly analyzed.
Bulk TCR sequencing
gDNA was extracted from the peripheral blood utilizing the AllPrep DNA/RNA Kit (Qiagen) and was sent to Adaptive Biotechnologies for bulk TCRb sequencing. Data was processed using the ImmunoSEQ Analyzer (Adaptive Biotechnologies, v3.0).
Multimer assays
Neoantigens were predicted from whole exome sequencing data and bulk RNA sequencing data from the three patients. For neoantigen candidates that were expressed in the bulk RNA sequencing data (counts per million >0), the neopeptides were sorted by the difference (‘Rank Diff EL’ column in Table S3) between wild-type peptide binding rank (‘WT.rank_EL’ column) and mutant peptide binding rank (‘rank’ column) as predicted by NetMHC v4.0.39,50 For HLA alleles for which multimers were commercially available (e.g. HLA-A*01:01, A*02:01, A*03:01, C*07:01), the neoantigen candidates with the top 6 ‘Rank Diff EL’ scores were selected for empiric testing. In total, the top 10–12 neoantigen candidates per patient were custom synthesized by Genscript to ≥95% purity (highlighted in green in Table S3). Each candidate neopeptide was tested for stabilization of cognate MHC monomers (Immudex, Denmark) using a flow cytometry-based anti-human β2M-PE assay, per manufacturer’s recommendations. A mean fluorescence intensity ≥1000 was utilized as the cutoff for monomer stabilization. The 6–8 neopeptide candidates per patient that empirically stabilized the cognate MHC monomers were utilized for multimer assays and MANAFEST assay (below).
The initial multimer assays to identify tumor-specific TCRs were performed using U-Load monomers and PE-dextramers and APC-dextramers (Immudex), according to manufacturer’s instructions. Prepared dextramers specific for each patient were pooled prior to staining of thawed single cell suspensions from tissue regions. PE+ and APC+ CD8+ T cells were sorted on an Aria Sorter and the pellet was frozen. DNA was extracted from the frozen pellet and submitted for bulk TCRβ sequencing.
As a parallel method to identify tumor-specific TCRs for MSK 1263, we prepared pooled multimers from UV-cleavable Flex-T monomers (Biolegend). The monomers were linked in a peptide-specific manner by separated reactions with barcoded Streptavidin-PE reagents (Totalseq-C0951-C0955 and C0961, Biolegend) (Table S3). For MSK 1263, we thawed previously cryopreserved single cell suspensions and stained with TotalSeq C anti-human hashtag reagent with a unique barcode (Biolegend) to subsequently permit deconvolution of region from which the cell originated. After washing, these cells were then stained with pooled barcoded multimers, followed by staining for CD45, CD3, and CD8. The cells were then sorted on PE+ CD8+ T cells and submitted for single cell RNA/TCR/multimer sequencing. Subsequent analyses revealed that the barcoding by peptide could not deconvolve peptide specificity with the conditions utilized in this experiment. Although the specific neoantigen specificity could not be determined, the multimer+ cells, as assessed by presence of a barcode, could be inferred to be tumor-specific.
MANAFEST assay
This assay was carried out as previously described.30,41 Neoantigen peptide pools for MSK 1263 were prepared by mixing 1 mg/ml of the six neopeptides confirmed to stabilize the cognate HLA (as described above). The viral antigen peptide pool utilized was 1 mg/ml of the CEF (CMV, EBV, Flu) pool (jpt Peptide Technologies). In brief, on day 0, T cells were isolated from patient-specific thawed previously cryopreserved PBMC by EasySep Human T cell Isolation negative selection kit (STEMCELL Technologies). The T cell-negative fraction was irradiated in a Cesium source gamma irradiator at 30 Gy. 2×105 cells from this fraction were then co-cultured with an equal number of T cells in a 96 well plate in AIM V media with 50 μg/mL gentamicin with a neoantigen peptide pool, viral peptide pool, or no peptides. On day 3, half the medium was replaced with fresh medium containing cytokines for a final concentration of 50 IU mL−1 IL-2 (Peprotech), 25 ng ml−1 IL-7 (Peprotech) and 25 ng/ml IL-15 (Peprotech). On day 7, half the medium was replaced with fresh culture medium containing cytokines for a final concentration of 100 IU/ml IL-2 and 25 ng/ml IL-7 and IL-15. On day 10, cells were harvested, and the CD8+ fraction was isolated using a CD8+ EasySep negative enrichment kit (STEMCELL Technologies). Adaptive files were uploaded onto the publicly available MANAFEST analysis web app (http://www.stat-apps.onc.jhmi.edu/FEST) to bioinformatically identify tumor-specific T cell clonotypes.
Processing of multimer sorted single-cell sequencing data
Single-cell RNA, TCR, and antibody capture libraries from multimer sorted tissue CD8+ T cells were processed using cellranger multi (10x Genomics, v7.0.0). The dataset was filtered to only include cells with <10% mitochondrial content, number of genes captured within 2 standard deviations of the mean, <1,000 multimer tag counts. Additionally, only cells with TCRβ, TCRαβ, or TCRααβ were kept for further analysis. The 25,588 cells that passed these filter criteria were subsequently processed as describe above. To assess the correspondence of phenotypes between the multimer tissue T cell dataset and the total CD3+ tissue T cell dataset, multimer sorted cells (query) were mapped onto the total CD3+ tissue (reference) dataset by identifying anchors between the two datasets using Seurat’s FindTransferAnchors() function with 30 dimensions and projected onto the reference UMAP structure using MapQuery().49
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of bulk and single-cell sequencing data was performed in R (v3.6.1). Statistical analysis of flow cytometry data was performed in GraphPad Prism (v9.0).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| CD45-BV510 | Biolegend | Cat# 368526, RRID: AB_2687377 |
| CD3-BV650 | Biolegend | Cat# 304044, RRID: AB_2563812 |
| CD8-PerCP-Cy5.5 | Biolegend | Cat# 344710, RRID: AB_2044010 |
| CD8-BV510 | Biolegend | Cat# 344732, RRID: AB_2564624 |
| PD-1-APC/Fire 750 | Biolegend | Cat# 329954, RRID: AB_2616721 |
| CD39-APC | Biolegend | Cat# 328210, RRID: AB_1953234 |
| CD39-PE/Cy7 | Biolegend | Cat# 328212, RRID: AB_2099950 |
| CD4-Alexa Fluor 700 | Biolegend | Cat# 357418, RRID: AB_2616933 |
| FOXP3-FITC | Invitrogen | Cat# 11-4776-42, RRID: AB_1724125 |
| CXCR5-PE | Biolegend | Cat# 356904, RRID: AB_2561813 |
| GITR-APC | Biolegend | Cat# 371206, RRID: AB_2616843 |
| CXCR4-PerCP-Cy5.5 | Biolegend | Cat# 306516, RRID: AB_10642818 |
| Anti-β2M-PE | Biolegend | Cat# 316306, RRID: AB_492839 |
| Human TruStain FcX™ Blocking Buffer | Biolegend | Cat# 422302, RRID: AB_2818986 |
| CD8 antibody for IHC | Dako | Cat# M7103, RRID: AB_2075537 |
| CD20 antibody for IHC | Thermofisher | Cat# PA5-16701, RRID: AB_10980806 |
| CD3 antibody for IHC | Abcam | Cat# ab16669, RRID: AB_443425 |
|
| ||
| Biological samples | ||
|
| ||
| Tumor tissue from NSCLC patients | Biospecimens from patients with informed consent at Memorial Sloan Kettering Cancer Center | This study |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| RPMI | Corning | Cat# 10-041-CV |
| Human serum | Gemini Biosciences | Cat# 100-512-100 |
| Penicillin/streptomycin | Gibco | Cat# 15140122 |
| Amphotericin | Gibco | Cat# 15290026 |
| Glutamax | Gibco | Cat# 35050061 |
| Minimum essential amino acids | Millipore Sigma | Cat# M7145 |
| Sodium pyruvate | Gibco | Cat# 11360070 |
| Tumor Dissociation Kit, human | Miltenyi | Cat# 130-095-929 |
| EasySep™ Human CD8++ T Cell Isolation Kit | Stem Cell Technologies | Cat# 17953 |
| DNase I Solution (1 mg/mL) | Stem Cell Technologies | Cat# 100-0762 |
| AIM V Media | ThermoFisher Scientific | Cat# 12055083 |
| Gentamycin | Gibco | Cat# 15750078 |
| IL-2 | Peprotech | Cat# 200-02 |
| IL-7 | Peprotech | Cat# 200-07 |
| IL-15 | Peprotech | Cat# 200-15 |
| CEF MHC I Control Peptide Pool “Plus” | Jpt Peptide Technologies | Cat# PM-CEF-002 |
| Cell Trace Violet | Invitrogen | Cat# C34557 |
| Dynabeads™ Human T-Activator CD3/CD28 for T Cell Expansion and Activation | Invitrogen | Cat# 11132D |
| Bambanker Serum Free Cell Freezing Medium | Wako Chemicals | Cat# 101974-112 |
| Dapi, for nucleic acid staining | Sigma-Aldrich | Cat# D9542-50MG |
| LIVE/DEAD Violet | Invitrogen | Cat# L34955 |
| U-Load Dextramer® MHC I Kit (HLA-A*01:01, A*02:01, A*03:01, C*07:01) | Immudex | Custom Orders |
| Streptavidin Coated Polystyrene Particles, 6.0-8.0 μm | Spherotech | Cat# SVP-60-5 |
| Recombinant peptides (9-10mers) | Gensript | Custom Orders |
| Flex-T HLA-A*01:01 Monomer UVX | Biolegend | Cat# 280001 |
| Flex-T HLA-A*07:01 Monomer UVX | Biolegend | Cat# 280133 |
| TotalSeq™-C0951 PE Streptavidin | Biolegend | Cat# 405261 |
| TotalSeq™-C0952 PE Streptavidin | Biolegend | Cat# 405263 |
| TotalSeq™-C0953 PE Streptavidin | Biolegend | Cat# 405265 |
| TotalSeq™-C0954 PE Streptavidin | Biolegend | Cat# 405267 |
| TotalSeq™-C0955 PE Streptavidin | Biolegend | Cat# 405269 |
| TotalSeq™-C0961 PE Streptavidin | Biolegend | Cat# 405155 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Chromium Chip A | 10X Genomics | Cat# PN-230027 |
| Next GEM Chip K | 10X Genomics | Cat# PN-1000286 |
| Chromium Single Cell 5′ Reagent Kit | 10X Genomics | Cat# PN-1000006 |
| Next GEM Single Cell 5′ Kit v2 | 10X Genomics | Cat# PN-1000263 |
| Chromium Single Cell V(D)J Enrichment Kit Human T Cell | 10X Genomics | Cat# PN-1000005 |
| Chromium Single Cell 5′ Feature Barcode Library Kit | 10X Genomics | Cat# PN-1000080 |
| NovaSeq 6000 SP Reagent Kit v1.5 | Illumina | Cat# 20028401 |
| NovaSeq 6000 S1 Reagent Kit v1.5 | Illumina | Cat# 20028319 |
| NovaSeq 6000 S4 Reagent Kit v1.5 | Illumina | Cat# 20028312 |
| NovaSeq 6000 S2 Reagent Kit v1.5 | Illumina | Cat# 20028316 |
| aMPure XP beads | Beckman Coulter | Cat# A63882 |
| AllPrep DNA/RNA Mini Kits | Qiagen | Cat# 80204 |
|
| ||
| Deposited data | ||
|
| ||
| This work (bulk TCR sequencing of longitudinal blood samples, MANAFEST experiments, and sorted multimer+ TILs) | Adaptive Biotechnologies ImmuneAccess | https://doi.org/10.21417/JAP2023CC URL: clients.adaptivebiotech.com/pub/pai-2023-cc |
| This work (scRNA/TCR-seq of resected biospecimens, multimer+ TILs) | NIH GEO | GSE185206 |
| Single cell data from neoadjuvant lung cancer trial | NIH GEO | GSE176021 |
| Single cell data from resection sample | DNA Data Bank of Japan | JGAS000480 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo | TreeStar | Version 10.8.1 |
| Prism | Graphpad | Version 9.0.0 |
| Halo | Indica Labs | Version 3.3.2541 |
| CiberSortx | Stanford | https://cibersortx.stanford.edu |
| R | R | Version 3.6.1 |
| Seurat | https://github.com/satijalab/seurat | Version 3.1.1 |
| CellRanger | 10X Genomics | Version 3.1.0 |
| CellRanger Multi | 10X Genomics | Version 7.0.0 |
Highlights.
Single-cell RNA/TCR-seq with deep intra-patient, regional resolution in NSCLC
TFH, Treg, and CD8+ T cell subsets undergo progressive exhaustion with tumor proximity
LN TCF7+ progenitor exhausted T cells are clonally linked to tumor exhausted T cells
Tumor-specific T cells in aggregate persist after ICB
ACKNOWLEDGMENTS
We acknowledge the use of the Integrated Genomics Operation Core, which is funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-José e and Henry R. Kravis Center for Molecular Oncology. This research was funded in part through the NIH NCI Cancer Center Support Grant P30 CA008748, NCI R01 CA056821, U01 CA199215, U24 CA213274, P01 CA129243, R01 CA197936, R35 CA232130, R35 CA263816, K08 CA248723, and U01 CA260852; the Ludwig Collaborative and Swim Across America Laboratory; the Emerald Foundation, United States; the Parker Institute for Cancer Immunotherapy, MSKCC and Stanford; the Department of Medicine, MSKCC; Stand Up To Cancer (SU2C)-American Cancer Society Lung Cancer Dream Team Translational research grant (SU2C-AACR-DT17-15); the Mark Foundation for Cancer Research (Grant #19-029-MIA), the Kay-Stafford Fund, and the Gibbons-Scattone Family Fund. A.C. was supported by an MSKCC Investigational Cancer Therapeutics Training Program fellowship (T32 CA-009207) and Clinical Investigator Award from National Cancer Institute (K08 CA-248723). J.A.P. was supported by NIH Training Grant 5T32AI007290. A.T.S. was supported by a Career Award for Medical Scientists from the Burroughs Wellcome Fund, a Lloyd J. Old STAR Award from the Cancer Research Institute, a Pew-Stewart Scholars for Cancer Research Award, and the Donald and Delia Baxter Foundation. We are grateful to Clare Wilhem for help in editing this manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2023.03.009.
DECLARATION OF INTERESTS
M.D.H. reports advisory/consulting fees from Achilles, Adagene, Adicet, AstraZeneca, Blueprint Medicines, Bristol Myers Squibb, Da Volaterra, Eli Lilly, Genentech, Genzyme/Sanofi, Immunai, Instill Bio, Janssen, Mana Therapeutics, Merk, Mirati, Pact Pharma, Regeneron, Roche, and Shattuck Labs; research funding from Bristol Myers Squibb; stock interest with Arcus, Factorial, Immunai, and Shattuck Labs; a patent filed by Memorial Sloan Kettering related to the use of tumor mutation burden to predict response to immunotherapy (PCT/US2015/062208), which has received licensing fees from Personal Genome Diagnostics (PGDx); after the completion of this work, M.D.H.began as an employee (and equity holder) at AstraZeneca. J.D.W. has Equity in Apricity, Arsenal IO, Ascentage, Beigene, Imvaq, Linneaus, Georgiamune, Maverick, Tizona Pharmaceuticals, and Trieza. J.D.W. is a co-inventor on the following patent application: Xenogeneic (Canine) DNA vaccines, myeloid-derived suppressor cell (MDSC) assay, anti-PD1 antibody, anti-CTLA4 antibodies, anti-GITR antibodies and methods of use thereof, Newcastle disease viruses for cancer therapy, and prediction of responsiveness to treatment with immunomodulatory therapeutics and method of monitoring abscopal effects during such treatment. J.D.W. and T.M. are co-inventors on patent applications related to CD40 and in situ vaccination (PCT/US2016/045970). T.M. is a consultant for Immunos Therapeutics and Pfizer. T.M. is a cofounder of and equity holder in IMVAQ Therapeutics. T.M. receives research funding from Bristol-Myers Squibb, Surface Oncology, Kyn Therapeutics, Infinity Pharmaceuticals, Peregrine Pharmaceuticals, Adaptive Biotechnologies, Leap Therapeutics, and Aprea Therapeutics. T.M. is an inventor on patent applications related to work on oncolytic viral therapy, alpha virus-based vaccine, neoantigen modeling, CD40, GITR, OX40, PD-1, and CTLA-4. C.M.R. has consulted regarding oncology drug development with AbbVie, Amgen, Ascentage, AstraZeneca, BMS, Celgene, Daiichi Sankyo, Genentech/Roche, Ipsen, Loxo, and PharmaMar and is on the scientific advisory boards of Elucida, Bridge, and Harpoon. A.T.S. is a founder of Immunai and Cartography Biosciences and receives research funding from Allogene Therapeutics and Merck Research Laboratories. The remaining authors declare no competing interests.
REFERENCES
- 1.Yost KE, Chang HY, and Satpathy AT (2021). Recruiting T cells in cancer immunotherapy. Science 372, 130–131. 10.1126/science.abd1329. [DOI] [PubMed] [Google Scholar]
- 2.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. (2017). Systemic immunity is required for effective cancer immunotherapy. Cell 168, 487–502.e15. 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pai JA, and Satpathy AT (2021). High-throughput and single-cell T cell receptor sequencing technologies. Nat. Methods 18, 881–892. 10.1038/s41592-021-01201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25, 1251–1259. 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, Ren X, Wang L, Wu X, Zhang J, et al. (2021). Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474. 10.1126/science.abe6474. [DOI] [PubMed] [Google Scholar]
- 6.Krishna C, DiNatale RG, Kuo F, Srivastava RM, Vuong L, Chowell D, Gupta S, Vanderbilt C, Purohit TA, Liu M, et al. (2021). Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 39, 662–677.e6. 10.1016/j.ccell.2021.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. (2016). Follicular CXCR5-expressing CD8(+) T cells curtail chronic viral infection. Nature 537, 412–428. 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- 9.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427. 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 10.Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, Minnich M, Meckiff BJ, Wei Y, Hou Z, et al. (2016). CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol. 17, 1187–1196. 10.1038/ni.3543. [DOI] [PubMed] [Google Scholar]
- 11.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral tcf1(+)PD-1(+) CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10. 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 12.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, et al. (2019). Checkpoint blockade immunotherapy induces dynamic changes in PD-1(−)CD8(+) tumor-infiltrating T cells. Immunity 50, 181–194.e6. 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336. 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Connolly KA, Kuchroo M, Venkat A, Khatun A, Wang J, William I, Hornick NI, Fitzgerald BL, Damo M, Kasmani MY, et al. (2021). A reservoir of stem-like CD8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci. Immunol. 6, eabg7836. 10.1126/sciimmunol.abg7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hegde PS, and Chen DS (2020). Top 10 challenges in cancer immunotherapy. Immunity 52, 17–35. 10.1016/j.immuni.2019.12.011. [DOI] [PubMed] [Google Scholar]
- 16.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 24, 978–985. 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 17.Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJJ, Chitre AS, Chiang EY, Iftikhar H, O’Gorman WE, Au-Yeung A, et al. (2020). Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278. 10.1038/s41586-020-2056-8. [DOI] [PubMed] [Google Scholar]
- 18.Lavin Y, Kobayashi S, Leader A, Amir EAD, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. (2017). Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169, 750–765.e17. 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angerer P, Haghverdi L, Büttner M, Theis FJ, Marr C, and Buettner F. (2016). destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243. 10.1093/bioinformatics/btv715. [DOI] [PubMed] [Google Scholar]
- 20.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen J, Blank CU, et al. (2019). Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176, 775–789.e18. 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghorani E, Reading JL, Henry JY, Massy M.R.d., Rosenthal R, Turati V, Joshi K, Furness AJS, Ben Aissa A, Saini SK, et al. (2020). The T cell differentiation landscape is shaped by tumour mutations in lung cancer. Nat. Cancer 1, 546–561. 10.1038/s43018-020-0066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, and Wherry EJ (2014). Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 40, 289–302. 10.1016/j.immuni.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miggelbrink AM, Jackson JD, Lorrey SJ, Srinivasan ES, Waibl-Polania J, Wilkinson DS, and Fecci PE (2021). CD4 T-cell exhaustion: does it exist and what are its roles in cancer? Clin. Cancer Res. 27, 5742–5752. 10.1158/1078-0432.CCR-21-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C, et al. (2018). A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 24, 994–1004. 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, Bejnood A, Dionne D, Ge WH, Xu KH, et al. (2021). Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184, 4734–4752.e20. 10.1016/j.cell.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, Cardenas M, Wilkinson S, Lake R, Sowalsky AG, et al. (2019). An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470. 10.1038/s41586-019-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, van Nimwegen M, Lau SP, Latupeirissa K, Schetters S, et al. (2020). The PD-1/PD-L1-checkpoint restrains T cell immunity in tumor-draining lymph nodes. Cancer Cell 38, 685–700.e8. 10.1016/j.ccell.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 28.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud Y, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2020). Subsets of exhausted CD8+T cells differentially mediate tumor control and respond to checkpoint blockade. Cancer Immunol. Res. 8, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gueguen P, Metoikidou C, Dupic T, Lawand M, Goudot C, Baulande S, Lameiras S, Lantz O, Girard N, Seguin-Givelet A, et al. (2021). Contribution of resident and circulating precursors to tumor-infiltrating CD8(+) T cell populations in lung cancer. Sci. Immunol. 6, eabd5778. 10.1126/sciimmunol.abd5778. [DOI] [PubMed] [Google Scholar]
- 30.Caushi JX, Zhang J, Ji Z, Vaghasia A, Zhang B, Hsiue EHC, Mog BJ, Hou W, Justesen S, Blosser R, et al. (2021). Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 596, 126–132. 10.1038/s41586-021-03752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagasaki J, Inozume T, Sax N, Ariyasu R, Ishikawa M, Yamashita K, Kawazu M, Ueno T, Irie T, Tanji E, et al. (2022). PD-1 blockade therapy promotes infiltration of tumor-attacking exhausted T cell clonotypes. Cell Rep. 38, 110331. 10.1016/j.celrep.2022.110331. [DOI] [PubMed] [Google Scholar]
- 32.Xia Y, Sandor K, Pai JA, Daniel B, Raju S, Wu R, Hsiung S, Qi Y, Yangdon T, Okamoto M, et al. (2022). BCL6-dependent TCF-1(+) progenitor cells maintain effector and helper CD4(+) T cell responses to persistent antigen. Immunity 55, 1200–1215.e6. 10.1016/j.immuni.2022.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Leun AM, Thommen DS, and Schumacher TN (2020). CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232. 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, Forman J, Huang T, Li S, Zhang W, et al. (2021). Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature 596, 119–125. 10.1038/s41586-021-03704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowery FJ, Krishna S, Yossef R, Parikh NB, Chatani PD, Zacharakis N, Parkhurst MR, Levin N, Sindiri S, Sachs A, et al. (2022). Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science 375, 877–884. 10.1126/science.abl5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. (2014). PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 124, 2246–2259. 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, Goodall CP, Blair TC, Fox BA, McDermott JE, et al. (2018). Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 9, 2724. 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. (2018). Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579. 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, Brunak S, and Lund O. (2003). Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 12, 1007–1017. 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nielsen M, and Andreatta M. (2016). NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 8, 33. 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Danilova L, Anagnostou V, Caushi JX, Sidhom JW, Guo H, Chan HY, Suri P, Tam A, Zhang J, Asmar ME, et al. (2018). The mutation-associated neoantigen functional expansion of specific T cells (MANAFEST) assay: a sensitive platform for Monitoring antitumor immunity. Cancer Immunol. Res. 6, 888–899. 10.1158/2326-6066.CIR-18-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alvisi G, Brummelman J, Puccio S, Mazza EM, Tomada EP, Losurdo A, Zanon V, Peano C, Colombo FS, Scarpa A, et al. (2020). IRF4 instructs effector Treg differentiation and immune suppression in human cancer. J. Clin. Invest. 130, 3137–3150. 10.1172/Jci130426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duhen R, Fesneau O, Samson KA, Frye AK, Beymer M, Rajamanickam V, Ross D, Tran E, Bernard B, Weinberg AD, and Duhen T. (2022). PD-1 and ICOS coexpression identifies tumor-reactive CD4+ T cells in human solid tumors. J. Clin. Invest. 132, e156821. 10.1172/JCI156821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Q, Wu X, Wang Z, Chen X, Wang L, Lu Y, Xiong D, Liu Q, Tian Y, Lin H, et al. (2022). The primordial differentiation of tumor-specific memory CD8(+) T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell 185, 4049–4066.e25. 10.1016/j.cell.2022.09.020. [DOI] [PubMed] [Google Scholar]
- 45.Tsui C, Kretschmer L, Rapelius S, Gabriel SS, Chisanga D, Knöpper K, Utzschneider DT, Nüssing S, Liao Y, Mason T, et al. (2022). MYB orchestrates T cell exhaustion and response to checkpoint inhibition. Nature 609, 354–360. 10.1038/s41586-022-05105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z, Tuong ZK, Dean I, Willis C, Gaspal F, Fiancette R, Idris S, Kennedy B, Ferdinand JR, Peñalver A, et al. (2022). In vivo labeling reveals continuous trafficking of TCF-1+ T cells between tumor and lymphoid tissue. J. Exp. Med. 219, e20210749. 10.1084/jem.20210749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Travis WD, Dacic S, Wistuba I, Sholl L, Adusumilli P, Bubendorf L, Bunn P, Cascone T, Chaft J, Chen G, et al. (2020). IASLC multidisciplinary recommendations for pathologic assessment of lung cancer resection specimens after neoadjuvant therapy. J. Thorac. Oncol. 15, 709–740. 10.1016/j.jtho.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, Lee JI, Suh YL, Ku BM, Eum HH, et al. (2020). Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 11, 2285. 10.1038/s41467-020-16164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R. (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andreatta M, and Nielsen M. (2016). Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32, 511–517. 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All scRNA/TCR-seq and bulk RNA-seq data have been deposited to NIH GEO under accession number GSE185206. Bulk TCR-seq data have been deposited into the ImmuneACCESS database at Adaptive Biotechnologies. All other relevant data are available from the corresponding authors upon request.
Custom code used in this work is available at github.com/satpathylab/regional-lung-cancer.