

Abstract
Purpose of Review:
This article reviews recent advances in imaging and fluid biomarkers for Alzheimer disease (AD) and their application to newly proposed diagnostic criteria across the continuum of AD.
Recent Findings:
There have been remarkable developments in neuroimaging markers for AD over the past decade, most notably the advent of positron emission tomography (PET) amyloid imaging using radiotracers that label fibrillar forms of amyloid-β (Aβ). Similarly, new research in CSF markers suggests CSF levels of Aβ1−42 and phosphorylated tau may be useful in the early diagnosis of AD and prediction of cognitive decline. The National Institute on Aging and the Alzheimer’s Association recently convened three workgroups to develop joint recommendations for new diagnostic guidelines across the spectrum of AD. These recommendations incorporate biomarkers and propose updated criteria for the previously recognized stage of AD dementia, the evolving definition of mild cognitive impairment, and a newly proposed concept of stages of preclinical AD.
Summary:
Recent advances in AD biomarkers have increased the ability to detect evidence of early AD pathology in vivo. These biomarkers have been incorporated into new diagnostic recommendations, but a number of challenges remain for the biomarkers to become widely applied in clinical practice.
Diagnostic criteria for Alzheimer disease (AD) were first proposed more than a quarter of a century ago.1 The original criteria served the neurologic field well, both for diagnosis in clinical practice and for inclusion of appropriate populations into clinical research; over the past few years, however, it has become evident that the diagnostic criteria should be revisited and updated to reflect recent advances in biomarker research and the recognition that AD represents a continuum across several stages of disease. This review will cover new developments in AD biomarkers and the application of these biomarkers to the proposed criteria for the spectrum of AD.
BIOMARKERS
The quest for accurate and useful in vivo biomarkers for AD has evolved rapidly over the past decade, with advances in CSF markers and multiple types of neuroimaging. Biomarkers for AD can be categorized in many ways, but in terms of the new diagnostic criteria, the biomarkers were conceptualized into two broad categories: (1) markers of amyloid-β (Aβ) accumulation and (2) markers of neuronal injury or neurodegeneration.
Markers of Amyloid-β Accumulation
Markers of Aβ accumulation currently include CSF assays of Aβ1–42 and positron emission tomography (PET) amyloid imaging. Paradoxically, AD is associated with a decrease in CSF Aβ1–42 that is generally thought to represent evidence that Aβ is polymerizing and depositing in the brain as fibrillar plaques. PET imaging of Aβ utilizes derivatives of histopathologic stains, such as thioflavins, that bind to fibrillar forms of Aβ.
In general, CSF and PET markers of Aβ accumulation have yielded similar findings. CSF assays have been available for 20 years, and therefore more longitudinal data are available, but the more recent advent of PET amyloid imaging has facilitated larger multicenter studies. The majority of studies published to date have utilized carbon-11 (11C)–based Pittsburgh Compound B (PiB) imaging. Recently, however, several tracers using fluorine-18 (18F)—including 18F-flutemetamol, 18F-florbetaben, and 18F-florbetapir—have been developed. Florbetapir was recently approved by the US Food and Drug Administration (FDA) for detection of amyloid as a diagnostic aid in evaluating causes of dementia, and it is likely that additional approvals will follow. At this point, it has not been determined whether insurance, in particular Medicare, will cover PET amyloid imaging as part of the diagnostic workup.
The few studies combining CSF and PET markers of Aβ published to date suggest a fairly tight correlation between these two markers.2 There is some suggestion that decreases in CSF Aβ could precede evidence of Aβ deposition in PET amyloid imaging, but this may reflect the thresholds for positivity that are utilized in PET studies. It is also important to note that both CSF and PET amyloid imaging markers are thought to reflect amyloid accumulation primarily into fibrillar forms and may not reveal other forms of Aβ, such as smaller soluble species—so-called oligomeric species of Aβ—that some researchers believe represent the toxic forms of Aβ. There is intense, ongoing research to develop stable assays for oligomeric Aβ; however, these assays have not been validated in clinical studies to date.
The ability to detect Aβ deposits during life with PET imaging has transformed clinical AD research, permitting direct visualization of one of the defining neuropathologic features of AD. Recent studies suggest that amyloid PET is likely equivalent to demonstration of amyloid plaque pathology at autopsy.3 Aβ deposits contain a characteristic pattern of protein structure known as beta-sheet, and numerous PET ligands have been developed that bind specifically to this type of structure and permit either visual or quantitative assessment of amyloid positivity. A negative amyloid PET study thus signifies few or no amyloid deposits and indicates that the likelihood of cognitive impairment due to AD is low. In contrast, a positive amyloid PET has been shown to correspond to moderately to severely elevated levels of Aβ deposits (Figure 1-1). The most important concept to recognize in considering the high image-to-pathology correlation is that amyloid positivity does not reliably distinguish clinical diagnoses, so that neurologically normal people as well as those with mild cognitive impairment (MCI), AD dementia, and other neurodegenerative diseases including Lewy body dementia can all be “amyloid positive.” In other words, positivity on amyloid markers alone does not establish a diagnosis of AD dementia and must be considered in a full clinical context.
Figure 1-1.
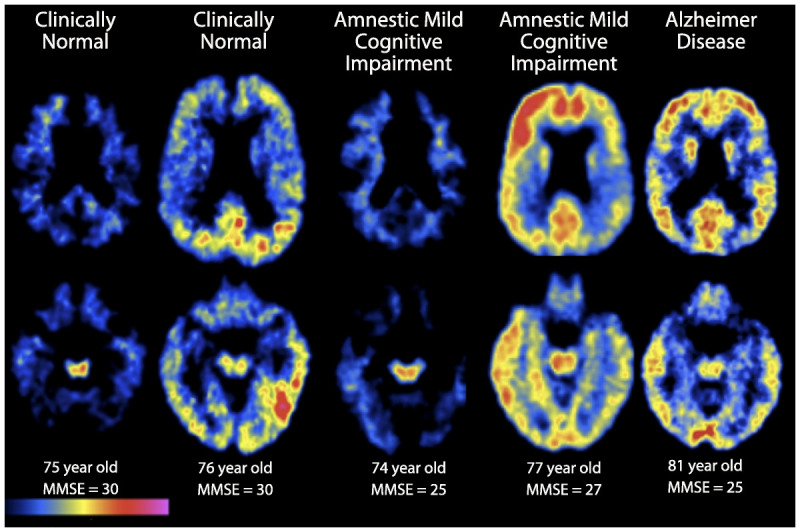
Carbon-11 Pittsburgh Compound B positron emission tomography (PET) amyloid images show two transaxial slice levels in six older individuals, with age and Mini-Mental State Examination (MMSE) score listed at the bottom. Regions of red and yellow indicate high Pittsburgh Compound B retention, indicating presence of fibrillar amyloid deposition. From left to right: a 75-year-old clinically normal woman who is amyloid negative; a 76-year-old clinically normal man with evidence of amyloid deposition in parietal and temporal cortices; a 74-year-old woman with amnestic mild cognitive impairment (aMCI) and an MMSE of 25 who is amyloid negative; a 77-year-old man with aMCI and an MMSE of 27 demonstrating amyloid deposition in frontal, temporal, and parietal cortices; and an 81-year-old woman with mild Alzheimer disease dementia with an MMSE of 25 who demonstrates evidence of amyloid deposition in frontal, temporal, and parietal cortices.
The amyloid-positive PET brain images have for the first time permitted a detailed view of the specific anatomy of amyloid accumulation during life. The exploration of this anatomy coincided with important developments in functional imaging, which converged over the past decade to focus on highly interconnected brain regions organized into large-scale cortical networks, in particular a set of functionally connected brain regions known as the default network, which has been implicated in memory and other complex cognitive processes. The components of the default network include precuneus, posterior cingulate, inferior parietal, lateral temporal, and superior frontal cortices, which are particularly vulnerable to amyloid deposition.4,5,6 The earliest and most heavily affected brain regions are the middle frontal gyri (part of the cognitive control network) and parietal/precuneus/posterior cingulate regions, thought to be key nodes of the default-mode network. The anatomy is highly stereotyped, but substantial variability has been observed in early phases of amyloid accumulation regarding the specific regions involved and the bilateral symmetry of involvement over time. Typically, by the time a person manifests objectively measurable cognitive impairment, amyloid PET is strongly positive in these regions, and eventually, as has been noted in the neuropathologic literature, even primary cortices become involved at the end stages of AD dementia.
The majority of data available on amyloid PET comes from the 11C agent PiB.7 Because of the short physical half-life of 11C-PiB (20 minutes), it is rapidly being replaced in most clinical settings and for multicenter studies by amyloid-binding ligands labelled with 18F, which has a 110-minute half-life.3,8,9 The rates of amyloid positivity reported for amyloid PET according to traditional clinical classifications are approximately as follows: for clinically diagnosed AD dementia, 90% positive; for MCI, approximately 60% positive; and for cognitively intact older people, approximately 30% to 40% positive, depending on the age cohort. Similar proportions have been reported using CSF biomarkers of Aβ.10 In longitudinal clinical follow-up of patients with MCI with baseline amyloid PET, approximately half were positive on amyloid imaging (versus only 10% of patients who were negative for MCI) convert to a clinical diagnosis of AD dementia over the course of 1 to 3 years.11 Of particular interest is the substantial fraction of apparently healthy elderly with normal cognition who are amyloid positive, whose condition is termed preclinical AD on the hypothesis that the presence of amyloid puts them at higher risk for developing AD dementia. The precise level of increased risk is being actively evaluated in research settings.
The clinical utility of amyloid PET in dementia care is likewise a topic of investigation about which data are only now being gathered and evaluated. Substantial clinical value may lie in differential diagnosis of patients with established and documented impairment (Case 1-1), particularly in younger patients for whom a more certain pathologic basis for their diagnosis could guide management. The underlying pathology of frontotemporal lobar degeneration (FTLD), for example, is not thought to involve Aβ, and a small but growing body of evidence confirms amyloid negativity in autopsy-confirmed FTLD.12 For disorders in which the histopathology is heterogeneous (such as posterior cortical atrophy, progressive aphasia, and corticobasal syndrome) and for Lewy body dementia, amyloid PET may not provide definitive diagnostic utility (Case 1-2).
Case 1-1
A 74-year-old, right-handed professor of linguistics was referred for a second opinion regarding progressive cognitive decline. The patient and his wife reported changes in cognition dating back approximately 2 years, when he had increased difficulty in giving lectures without consulting his notes. He also described word-finding difficulty and occasional problems with getting lost while driving in familiar places. There was no report of behavioral changes except that he seemed a bit more withdrawn in social situations, which they attributed to decreased confidence in his conversational abilities. He had been diagnosed with Alzheimer disease by another academic neurologist approximately 6 months ago and was started on a cholinesterase inhibitor. The patient and his wife wanted another opinion because they felt he was continuing to worsen, and they were interested in enrolling in an antiamyloid antibody trial.
On examination, he was alert and oriented except for day of the week. He demonstrated mild inattention with months of the year backward and on serial 7’s. He encoded five words, had four of the five at 30 seconds without distraction, and recalled two of the five after 5 minutes. He correctly recognized one additional word on multiple-choice testing. His speech was fluent but he had several pauses for word-finding, and his vocabulary was less rich than might have been expected for a person of his background. His repetition and basic grammatical comprehension were intact, but his clock drawing was impaired, with particular difficulty in placing the hands on a clock to indicate 11:10. He scored 24 in the Mini-Mental State Examination and 1.0 on the Clinical Dementia Rating Scale. The remainder of his neurologic examination was otherwise unremarkable except for mild saccadic intrusions.
Both he and his wife were anxious to get him enrolled in research studies, particularly clinical trials for potential disease-modifying therapies. He underwent MRI and positron emission tomography (PET) amyloid imaging as part of a research protocol. The MRI revealed mild atrophy in both frontal and parietal cortices, with relatively intact medial temporal lobe structures. The PET amyloid imaging was negative on visual inspection and by quantitative threshold. Because this patient was strongly considering enrolling in an antibody trial, the PET amyloid imaging results were felt to be clinically relevant. The PET findings were discussed with the patient and his wife, since it was felt that the negative amyloid result made the diagnosis of Alzheimer disease less likely. Other neurodegenerative diseases, including frontotemporal lobar degeneration, were also discussed.
A few weeks later, the patient’s wife called to say that his niece (his brother’s daughter, who was living in another country) had been diagnosed with Pick disease, but no imaging or genetic testing was available. At the family’s request, genetic testing was performed on the patient and revealed a progranulin mutation, confirming a diagnosis of frontotemporal lobar degeneration. The patient was taken off cholinesterase inhibitors and was started on a serotonin-selective reuptake inhibitor and memantine (also used for treatment in frontotemporal dementia by some neurologists).
Comment. This case illustrates the potential utility of a negative PET amyloid imaging study in a patient with dementia. The negative PET scan substantially decreased the likelihood that this patient’s clinical syndrome was due to Alzheimer disease, prompting further diagnostic workup.
Case 1-2
A 76-year-old man was evaluated for memory loss and a gait disorder with the onset associated with a fall approximately 4 years ago. He reported a false belief about the presence of a nonexistent family member living in the house. His Mini-Mental State Examination score was 25/30, and his neuropsychological evaluation was notable for mild episodic memory difficulties. A fluorodeoxyglucose positron emission tomography (PET) scan showed bilateral temporoparietal associated with left frontal hypometabolism, consistent with Alzheimer disease (AD). A neurologist noted parkinsonism and began treatment with dopaminergic medication, which was thought to be ineffective; the neurologist then made a diagnosis of Lewy body dementia.
His motor function, memory, and delusions worsened. Two years later, he underwent amyloid PET with Pittsburgh Compound B (PiB), which revealed a pattern of marked positivity in widespread cortical regions. He died of complications from a traumatic subdural hematoma; an autopsy revealed Lewy body dementia with marked congophilic angiopathy, and a positive correlation between amyloid-β levels and regional PiB retention.13
Comment. This case illustrates that a positive PET amyloid imaging may be associated with cerebral amyloid angiopathy and Lewy body dementia and therefore is not specific to a clinical diagnosis of AD dementia.
Markers of Neuronal Injury or Neurodegeneration
Researchers are pursuing multiple biomarkers of neuronal injury or neurodegeneration, which might be further subcategorized as (1) molecular markers of neuronal injury, (2) imaging markers of synaptic dysfunction, and (3) imaging markers of neuronal loss and atrophy. CSF phosphorylated tau (P-tau) 181 and total tau measures are thought to reflect neuronal injury and neuronal death, respectively. The combination of elevated CSF P-tau with low CSF Aβ1–42 is thought to indicate a biomarker signature of AD (Case 1-3). Although hyperphosphorylation of tau is thought to be a key contributor to intraneuronal neurofibrillary-tangle formation, it is not clear whether CSF tau measures fully reflect the burden of tangle pathology and whether these markers will remain dynamic at later stages of AD. Increased levels of CSF tau and P-tau have also been demonstrated to predict progression to dementia in subjects with MCI14 and are already elevated in clinically normal mutation carriers in autosomal dominant familial AD.15
Case 1-3
A 54-year-old, left-handed businesswoman was referred by her primary care physician for evaluation of her cognitive and behavioral changes. The patient’s husband and daughter reported that they had begun to notice the changes 3 years ago, with her decreased interest in hobbies and exercise, and increased difficulty preparing complicated meals. She had initially been diagnosed with depression and anxiety but continued to decline on antidepressants. Over the past year, she had gotten lost twice while driving, and her husband had taken over paying most of the bills.
The patient also described intermittent episodes of bilateral paresthesia involving her arms and legs, and sometimes her face. She was in otherwise excellent health and took no medications. Her mother had developed dementia late in life (ie, early eighties) but had not had an autopsy.
On examination, the patient was alert but disoriented to the date. She could not give any details of recent current events. She had marked difficulty with attention and even simple calculations. On memory testing, she encoded three words but recalled only one of the three at 5 minutes. She recognized two out of three on multiple-choice but also picked one false answer. Her speech was fluent, but she had mild naming difficulty for body parts and low-frequency items. Her affect was not depressed, but she described being “petrified” that she was losing hermind. HerMini-Mental State Examination score was 25. The remainder of her neurologic examination was only significant for mild symmetric hyperreflexia.
Her MRI revealed mild hippocampal atrophy for her age, slight atrophy in left greater than right temporal and parietal cortices, and no evidence of white matter lesions. She had a previous spine MRI to work up the paresthesia, which was read as normal.
Her husband was particularly anxious to have more diagnostic workup done because some of the family members and other physicians still thought the symptoms were psychiatric or possibly related to Lyme disease, although her serum titers were negative for both immunoglobulin G and immunoglobulin M. She underwent a lumbar puncture, which revealed no white blood cells, a slightly elevated protein level of 64 mg/dL, low CSF amyloid-β, and elevated phosphorylated tau on a commercial CSF assay, thought to be consistent with a diagnosis of Alzheimer disease.
Comment. This case illustrates the potential utility of CSF markers in aiding diagnosis in young patients and those who may present with unusual clinical symptoms.
Several imaging markers are available that are presumed to reflect synaptic dysfunction. The most widely studied is 18F-fluorodeoxyglucose (FDG)-PET, which has been used since the 1980s. FDG-PET measures glucose metabolism in the brain and is thought to reflect synaptic activity. The characteristic pattern of FDG abnormalities associated with AD is bilateral temporoparietal hypometabolism, although frontal hypometabolism has also been reported in later stages of AD. Single-photon emission computed tomography (SPECT) imaging has shown a very similar pattern of functional abnormality but is not widely used now because of the increased availability of PET imaging with superior resolution.
FDG-PET is a widely available technology that may aid clinicians in making a correct diagnosis, particularly when deciding whether early stages of cognitive impairment are due to AD pathology or FTLD. However, further work is needed to determine the diagnostic value of FDG when assessing preclinical stages of AD. Like other imaging biomarkers, FDG does not indicate clear spatial patterns that distinguish between normal aging and MCI or AD.16 As with these other modalities, the observed heterogeneity in FDG patterns of metabolism may be due to individual differences (particularly in cognitive reserve)17 or to comorbid conditions of aging such as vascular disease, as well as to idiosyncratic factors. While FDG hypometabolism does seem to be somewhat more sensitive than structural imaging biomarkers for monitoring cognitive transition from preclinical to established AD,18 it is still unknown which variable is best associated with the cognitive symptoms and how it could be translated into clinical use. The availability of FDG-PET has increased dramatically over the past decade and the standardization of methods has improved substantially; however, costs of PET technology remain high, and cost effectiveness in dementia diagnosis has been difficult to demonstrate.
Functional MRI (fMRI) is a less developed method for assessing synaptic function but has the potential advantage of not requiring any contrast agent or radiation exposure. Functional MRI can assess brain function during cognitive tasks, which typically requires special stimulus equipment, or during the resting state, sometimes referred to as “task-free” fMRI using blood oxygen level–dependent (BOLD) imaging. In task-free fMRI, the correlation between activity within intrinsic networks or “functional connectivity” is assessed. Functional connectivity MRI (fc-MRI) studies have demonstrated similar patterns of dysfunction as those reported in FDG-PET, preferentially affecting the connectivity in a network of cortical regions known as the default-mode network (defined above).4 Another MRI technique for assessing brain function and blood flow is arterial spin labeling MRI (ASL-MRI), which is also noninvasive and has demonstrated similar patterns of abnormalities to regional FDG hypometabolism.19 Both fc-MRI and ASL-MRI have yet to be validated in multicenter studies but may prove useful in assessing therapeutic response in clinical trials.
Structural imaging (particularly volumetric MRI) has been extensively documented in AD. Progressive cerebral atrophy is a characteristic feature of neurodegeneration that can be assessed with structural imaging, in particular with volumetric MRI. AD is characterized by an insidious onset and inexorable progression of atrophy in the medial temporal lobe structures (including the hippocampus and entorhinal cortex) and by cortical thinning, particularly in heteromodal cortices including the posterior cingulate, precuneus, lateral parietal, temporal, and frontal regions.20,21 Patients with AD with atypical clinical presentations tend to demonstrate patterns of atrophy concordant with their deficits, such as visual variants demonstrating posterior cortical atrophy and those with prominent language deficits demonstrating left temporal atrophy.
Convergent studies suggest that atrophy begins years before the diagnosis of dementia. Even in very mildly impaired individuals (ie, those with Mini-Mental State Examination scores greater than 24), the volumes of medial temporal lobe structures are reduced by 15% to 30%.22,23 This is consonant with autopsy studies suggesting that patients with MCI have already lost 50% of neurons in the perforant pathway.24 Several studies have also demonstrated evidence of atrophy in amyloid-positive, clinically normal older individuals, in a pattern similar to that observed in MCI and AD.25,26 It should be noted that the majority of these research studies employ computer software to calculate quantitative measurements. It is likely to be more difficult to detect subtle atrophy in early AD with visual inspection, as would be available in most clinical situations. MRI may also be useful for ruling out other relatively less common but potentially treatable causes of dementia and cognitive impairment, including cerebrovascular disease and neoplasm. Also, it is important to note that atrophy is not specific to AD. Indeed, hippocampal volume loss is seen in normal aging as well as in hippocampal sclerosis and other neurodegenerative diseases that can mimic AD. Nevertheless, the combination of volumetric MRI and molecular markers of AD may prove valuable in improving both diagnostic accuracy and prediction of future clinical course.
Models of Biomarker Trajectories in Alzheimer Disease
Accumulating data from multiple biomarker and imaging modalities—acquired by the Alzheimer’s Disease Neuroimaging Initiative (ADNI); the Australian Imaging, Biomarkers and Lifestyle (AIBL) study; and several other longitudinal cohort studies at leading academic centers—facilitated the development of hypothetical models of the temporal course of these biomarkers across the spectrum of AD.18 These models proposed a specific sequence of biomarker abnormalities that began before any evidence of clinical symptoms, and a series of sigmoid curves to characterize the temporal dynamics of these biomarkers (Figure 1-2).27 These models postulated that markers of amyloid accumulation typically become abnormal first but, importantly, suggested that amyloid might be necessary but not sufficient to result in AD dementia. These models also acknowledged some gaps in understanding and data that did not fully fit these models, including evidence of early synaptic dysfunction present in some genetic at-risk groups that might precede evidence of amyloid accumulation. It is also important to acknowledge that demographic factors might influence the temporal trajectory of these hypothetical curves, including age, genetics, socioeconomic factors such as education, and other indicators of cognitive reserve.
Figure 1-2.
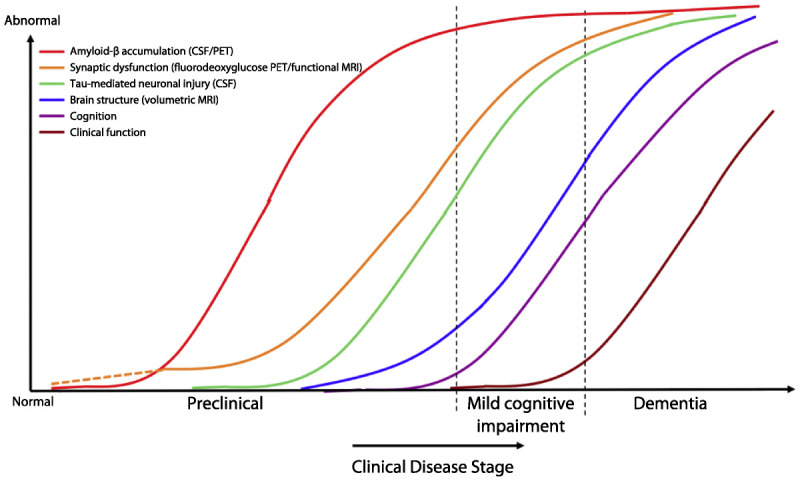
Hypothetical model of dynamic biomarkers of Alzheimer disease. Biomarkers: Amyloid-β (Aβ) as identified by CSF Aβ42 assay or positron emission tomography (PET) amyloid imaging (red); synaptic dysfunction evidenced by fluorodeoxyglucose PET or functional MRI (orange), with a dashed line to indicate that synaptic dysfunction may be detectable in carriers of the *E4 allele of the APOE gene prior to detectable amyloid-β deposition; neuronal injury evidenced by CSF tau or phosphorylated tau (green); volumetric MRI (blue). Biomarkers change from normal to maximally abnormal (y axis) as a function of disease stage (x axis). The temporal trajectories of two key clinical indicators used to stage the disease, cognition (purple) and clinical function (maroon), are also illustrated.
Adapted from Jack CR Jr, et al, Lancet Neurol.18 © 2010, with permission from Elsevier. www.sciencedirect.com/science/article/pii/S1474442209702996.
Reprinted from Sperling RA, et al, Alzheimers Dement.27 © 2011 The Alzheimer’s Association. All rights reserved. www.alzheimersanddementia.com/article/S1552-5260(11)00099-9/fulltext.
NEW DIAGNOSTIC CRITERIA
Largely informed by the advances in biomarker research discussed above and by increasing recognition of early stages of clinical impairment, a number of expert groups have worked over the past decade to develop new diagnostic criteria. Petersen and colleagues28 began to define and validate criteria for MCI. An international group of experts led by Dubois put forth the concept of a prodromal stage of AD, which suggested that amnestic MCI plus evidence of positive AD biomarkers might be considered very mild AD. In 2010, the National Institute on Aging and the Alzheimer’s Association (NIA-AA) established three working groups to develop new criteria for AD. Each group was asked to focus on one of the three phases of AD: (1) the presymptomatic or preclinical phase, (2) the symptomatic predementia phase, often referred to as MCI, and (3) the dementia phase. The working groups were international in composition and included people with a range of expertise, including neurologists, psychiatrists, neuropsychologists, radiologists, epidemiologists, and biostatisticians from both academia and the pharmaceutical industry. The groups were asked to review the literature and develop consensus on new diagnostic criteria that could be used by clinicians and researchers. There was also a small workgroup charged with harmonizing the discussion of biomarkers across the three workgroups, which helped to develop a common diagnostic framework across the continuum.
The NIA-AA workgroups that dealt with the symptomatic phases of AD (ie, MCI and dementia) developed sets of core clinical diagnostic criteria that were intended to serve as a guide for a wide range of clinicians confronted with the typical patient presenting for evaluation of cognitive decline. In these groups, biomarkers were considered to be a potential adjunct to diagnosis, primarily for research purposes, that would serve to increase the certainty of the etiology of the clinical diagnosis. The NIA-AA workgroup on preclinical AD and the international workgroup led by Dubois took a slightly different approach, using biomarker criteria to better identify patients who were on the early AD trajectory for research studies.
Clinical Criteria for Alzheimer Dementia and Mild Cognitive Impairment Due to Alzheimer Disease
The clinical criteria for AD dementia in the NIA-AA criteria are similar to the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria from 1984. The core clinical criteria for AD dementia29 include an insidious onset over months to years with a clear-cut history of progression of cognitive decline, usually obtained from an informant. Cognitive decline can be affirmed by neuropsychological testing. The most common presentation of AD dementia is the amnestic form, which involves impairment of episodic memory (ie, the ability to learn and retain new information). However, some patients have initial involvement of other cognitive domains, such as language or visuospatial or executive functioning.
The term mild cognitive impairment applies to patients who have some evidence of cognitive impairment but have not progressed to dementia. The core clinical criteria in the NIA-AA criteria30 are similar to those listed for AD dementia but differ in the degree of functional impairment. These criteria include (1) concern regarding a change in cognition, which may be identified by the patient, an informant, or a clinician; (2) impairment in one or more cognitive domains that is greater than one would expect for the patient’s age or educational background and may be confirmed with neuropsychological testing; and (3) preservation of independence in functional activities. There may be some mild problems with more complex activities, but those with MCI maintain their independence in daily life, requiring only minimal assistance. The degree of decline in cognition and function is not yet consistent with dementia.
It is important to note that AD is increasingly recognized as a continuum. There are no exact transition points that define when a patient has progressed from the MCI phase to the dementia phase (nor from the preclinical phase to MCI). These transitions are typically a matter of clinical judgment, relying on multiple pieces of information, and the division into discrete stages may be somewhat arbitrary.
Although the focus of these workgroups was on diagnostic criteria for AD, it is important to acknowledge that there are causes of MCI and dementia other than AD pathology. It is necessary to consider other systemic and brain diseases that can alter cognitive function, such as vascular disease, other neurodegenerative diseases, and neuropsychiatric diseases, including depression. AD is less likely to be the underlying cause of impairment when there are atypical symptoms or a clinical course suggestive of vascular disease, frontotemporal dementia, or Lewy body dementia. Biomarkers may play an important role in the differential diagnosis of AD as the underlying etiology of clinical impairment.
Research Criteria for Preclinical Alzheimer Disease
The most controversial aspect of the NIA-AA criteria was the proposal to recognize a new presymptomatic or preclinical stage of AD.27 This stage was conceptualized to represent the “silent” phase of AD when the pathophysiologic process has begun in the brain but there is not yet clear evidence of clinical impairment. The preclinical phase of the disease is primarily defined by changes in biomarkers, but the last stage of preclinical AD may be characterized by evidence of cognitive or behavioral change from the patient’s baseline status or poor performance on challenging cognitive tests. After much debate, the term preclinical was selected, as opposed to presymptomatic, to recognize that patients may demonstrate very subtle changes in cognition and behavior that can precede MCI by several years but would not typically be brought to a clinician’s attention. The recommendations for the study of preclinical AD were intended purely for research purposes, since they do not have any clinical utility at the present time.
Impact of Biomarkers on Diagnostic Criteria
As mentioned above, the NIA-AA workgroups conceptualized biomarkers as falling into two major categories: (1) biomarkers that reflect the accumulation of the Aβ peptide—either CSF Aβ1–42 or PET amyloid imaging, and (2) biomarkers of neuronal injury, which include CSF tau, FDG-PET, and volumetric MRI.
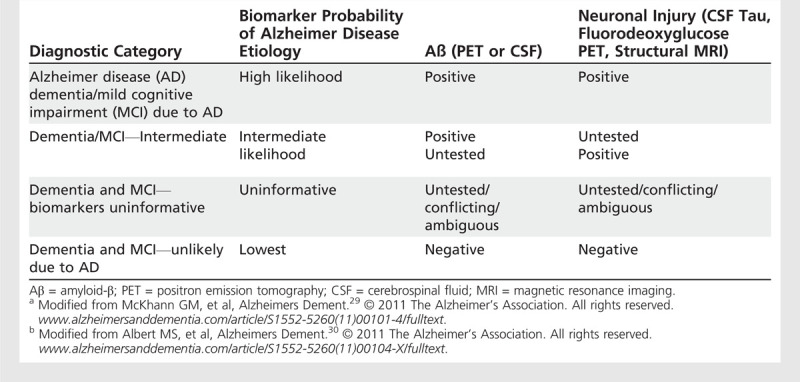
For the diagnostic criteria pertaining to the symptomatic phases of disease, a set of algorithms were proposed by which biomarkers from the two categories mentioned above could be examined individually (because often only one biomarker test is available) or in combination, to enhance the certainty that the underlying etiology of the symptoms is AD (Table 1-1). It was also recognized that biomarkers might be useful to estimate the likelihood of progression to the next stage of the disease (eg, from MCI to dementia).
Table 1-1.
Dementia and Mild Cognitive Impairment Criteria Incorporating Biomarkersa,b
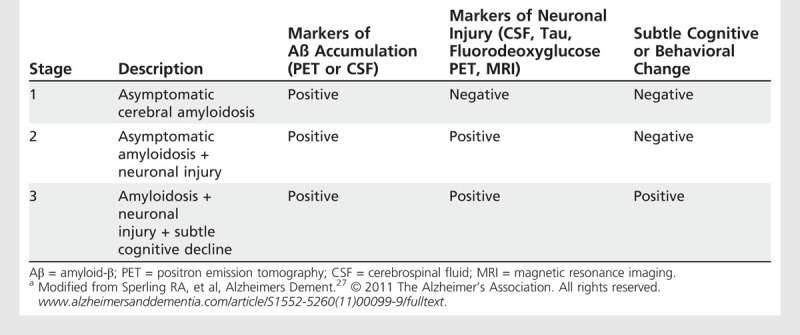
In the preclinical research criteria, a staging framework based on biomarkers was proposed to characterize patients at increasing risk of progression toward MCI and dementia. Stage 1 is characterized as asymptomatic cerebral amyloidosis; stage 2 is amyloidosis plus neurodegeneration; and stage 3 is amyloidosis, neurodegeneration, and evidence of very subtle change in cognition or behavior but is not sufficient to be diagnosed with MCI. Table 1-2 summarizes the categorization of biomarkers across the stages of preclinical AD.
Table 1-2.
Staging Categories for Preclinical Alzheimer Disease Researcha
INCORPORATING BIOMARKERS INTO CLINICAL PRACTICE
When the NIA-AA criteria for MCI and AD dementia were developed, it was noted that biomarkers were deemed to be primarily for research use. PET amyloid imaging was not yet clinically available, but one 18F PET agent has since been FDA approved. Several important issues about incorporating biomarkers into clinical practice were also raised in these guidelines—in particular, the need for standardization across centers. Although work has begun in standardizing CSF assays, PET imaging, and volumetric MRI measurements, there are not yet well-validated, established normative values and calibration metrics for any of the biomarkers. Additional work is required to make these biomarkers more easily interpretable to physicians in a variety of clinical settings.
SUMMARY AND FUTURE DIRECTIONS
The field of AD has seen many advances in diagnosis and biomarker development over the past decade. Hopefully these biomarkers will serve to improve understanding of the pathophysiologic processes of AD during life and to elucidate the link between the pathology and clinical manifestations of this terrible disease. Much work remains to be done to standardize currently available biomarkers to enable optimal use in the clinical setting, to develop more sensitive and specific biomarkers for early diagnosis, to track progression, and to monitor response to future disease-modifying therapy.
KEY POINTS
Biomarkers are conceptualized into two broad categories: (1) markers of amyloid-β accumulation and (2) markers of neuronal injury or neurodegeneration.
Paradoxically, Alzheimer disease is associated with a decrease in CSF amyloid-β1–42 that is generally thought to represent evidence that amyloid-β is polymerizing and depositing as fibrillar plaques. PET imaging of amyloid-β utilizes derivatives of histopathologic stains, such as thioflavins, that bind to fibrillar forms of amyloid-β.
Recent studies suggest that amyloid PET is likely equivalent to demonstration of amyloid plaque pathology at autopsy.
A negative amyloid PET study signifies few or no amyloid deposits and indicates that the likelihood of cognitive impairment due to Alzheimer disease is low.
The most important concept to recognize in considering the high image-to-pathology correlation is that amyloid positivity does not reliably distinguish clinical diagnoses.
Increased levels of CSF tau and phosphorylated tau have also been demonstrated to predict progression to dementia in subjects with mild cognitive impairment and are already elevated in clinically normal mutation carriers in autosomal dominant familial Alzheimer disease.
The characteristic pattern of fluorodeoxyglucose abnormalities associated with Alzheimer disease is bilateral temporoparietal hypometabolism.
Convergent studies suggest that atrophy begins years before the diagnosis of dementia.
Atrophy is not specific to Alzheimer disease.
Amyloid might be necessary but not sufficient to result in Alzheimer disease dementia.
The most common presentation of Alzheimer disease dementia is the amnestic form, which involves impairment in episodic memory (ie, the ability to learn and retain new information). However, some patients have initial involvement of other cognitive domains, such as language or visuospatial or executive functioning.
Alzheimer disease is increasingly recognized as a continuum.
Footnotes
Relationship Disclosure: Dr Sperling serves as a consultant for Bayer AG; Biogen Idec; Bristol-Myers Squibb Company; Eisai Co, Ltd; Janssen Pharmaceuticals, Inc; Eli Lilly and Company; Merck & Co, Inc; Neurophage Pharmaceuticals; Pfizer Inc; Hoffman-La Roche Inc; and Satori Pharmaceuticals. Dr Sperling receives research support from Alzheimer’s Association, American Health Assistance Foundation, Bristol-Myers Squibb Company, Fidelity Foundation, Massachusetts Alzheimer’s Disease Research Center, and National Institute on Aging. Dr Johnson serves as a consultant for Avid Radiopharmaceuticals, Bayer AG, Elan Corporation/Janssen Pharmaceuticals, Inc, GE Health Care, and Pfizer Inc. Dr Johnson has received honoraria and payment of travel expenses from Pfizer Inc; serves as an associate editor for the Journal of Neuroimaging; receives book royalties from Lippincott Williams & Wilkins; and receives research support from Alzheimer’s Association, Avid Pharmaceuticals, Bristol-Myers Squibb Company, Janssen Alzheimer Immunotherapy, NIH, and Pfizer Inc.
Unlabeled Use of Products/Investigational Use Disclosure: Drs Sperling and Johnson discuss the unlabeled potential future use of amyloid imaging in preclinical and prodromal stages of Alzheimer disease.
REFERENCES
- 1.McKhann G,, Drachman D,, Folstein M, et al.. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984; 34 (7): 939–944. [DOI] [PubMed] [Google Scholar]
- 2.Fagan AM,, Mintun MA,, Shah AR, et al.. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med 2009; 1 (8–9): 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark C,, Pontecorvo M,, Beach T, et al., eds. Correspondence of florbetapir-PET and beta-amyloid pathology: analysis of 59 subjects who came to autopsy. Miami, FL: Human Amyloid Imaging, 2012. [Google Scholar]
- 4.Raichle ME,, MacLeod AM,, Snyder AZ, et al.. A default mode of brain function. Proc Natl Acad Sci U S A 2001; 98 (2): 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buckner RL,, Snyder AZ,, Shannon BJ, et al.. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci 2005; 25 (34): 7709–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sperling RA,, Laviolette PS,, O’Keefe K, et al.. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 2009; 63 (2): 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klunk WE,, Engler H,, Nordberg A, et al.. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 2004; 55 (3): 306–319. [DOI] [PubMed] [Google Scholar]
- 8.Barthel H,, Gertz HJ,, Dresel S, et al.. Cerebral amyloid-β PET with florbetaben (18F) in patients with Alzheimer’s disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet Neurol 2011; 10 (5): 424–435. [DOI] [PubMed] [Google Scholar]
- 9.Vandenberghe R,, Van Laere K,, Ivanoiu A, et al.. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann Neurol 2010; 68 (3): 319–329. [DOI] [PubMed] [Google Scholar]
- 10.Fagan AM,, Roe CM,, Xiong C, et al.. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 2007; 64 (3): 343–349. [DOI] [PubMed] [Google Scholar]
- 11.Klunk WE. Amyloid imaging as a biomarker for cerebral β-amyloidosis and risk prediction for Alzheimer dementia. Neurobiol Aging 2011; 32 (suppl 1): S20–S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabinovici GD,, Rosen HJ,, Alkalay A, et al.. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology 2011; 77 (23): 2034–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bacskai BJ,, Frosch MP,, Freeman SH, et al.. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol 2007; 64 (3): 431–434. [DOI] [PubMed] [Google Scholar]
- 14.Hansson O,, Zetterberg H,, Buchhave P, et al.. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol 2006; 5 (3): 228–234. [DOI] [PubMed] [Google Scholar]
- 15.Bateman RJ,, Xiong C,, Benzinger TL, et al.. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 2012; 367 (9): 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landau SM,, Harvey D,, Madison CM, et al.. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 2010; 75 (3): 230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stern Y,, Alexander GE,, Prohovnik I,, Mayeux R. Inverse relationship between education and parietotemporal perfusion deficit in Alzheimer’s disease. Ann Neurol 1992; 32 (3): 371–375. [DOI] [PubMed] [Google Scholar]
- 18.Jack CR, Jr,, Knopman DS,, Jagust WJ, et al.. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 2010; 9 (1): 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Musiek ES,, Chen Y,, Korczykowski M, et al.. Direct comparison of fluorodeoxyglucose positron emission tomography and arterial spin labeling magnetic resonance imaging in Alzheimer’s disease. Alzheimers Dement 2012; 8 (1): 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scahill RI,, Schott JM,, Stevens JM, et al.. Mapping the evolution of regional atrophy in Alzheimer’s disease: unbiased analysis of fluid-registered serial MRI. Proc Natl Acad Sci U S A 2002; 99 (7): 4703–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dickerson BC,, Bakkour A,, Salat DH, et al.. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex 2009; 19 (3): 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dickerson BC,, Goncharova I,, Sullivan MP, et al.. MRI-derived entorhinal and hippocampal atrophy in incipient and very mild Alzheimer’s disease. Neurobiol Aging 2001; 22 (5): 747–754. [DOI] [PubMed] [Google Scholar]
- 23.Schuff N,, Woerner N,, Boreta L, et al.. MRI of hippocampal volume loss in early Alzheimer’s disease in relation to ApoE genotype and biomarkers. Brain 2009; 132 (pt 4): 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomez-Isla T,, Price JL,, McKeel DW, Jr, et al.. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 1996; 16 (14): 4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schott JM,, Bartlett JW,, Fox NC,, Barnes JAlzheimer’s Disease Neuroimaging Initiative Investigators. Increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Aβ1–42. Ann Neurol 2010; 68 (6): 825–834. [DOI] [PubMed] [Google Scholar]
- 26.Becker JA,, Hedden T,, Carmasin J, et al.. Amyloid-β associated cortical thinning in clinically normal elderly. Ann Neurol 2011; 69 (6): 1032–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sperling RA,, Aisen PS,, Beckett LA, et al.. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7 (3): 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petersen RC,, Smith GE,, Waring SC, et al.. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999; 56 (3): 303–308. [DOI] [PubMed] [Google Scholar]
- 29.McKhann GM,, Knopman DS,, Chertkow H, et al.. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7 (3): 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albert MS,, DeKosky ST,, Dickson D, et al.. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7 (3): 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]