

Abstract
Purpose of Review:
This article provides an overview of the evaluation and management of ischemic stroke in young adults, with an emphasis on cervical artery dissection, patent foramen ovale, and hypercoagulable states.
Recent Findings:
The incidence of ischemic stroke in young patients is increasing, although the reasons remain unclear. Patients with ischemic stroke at a young age are more likely to die at an early age than their peers. Well-established vascular risk factors are prevalent in young stroke patients. Recent studies have informed the treatment of dissection and patent foramen ovale among others. The utility of testing for hypercoagulable states in ischemic stroke is unclear.
Summary:
Ischemic stroke in young adults is a major public health problem. A wide range of etiologies of ischemic stroke is found in this age group. A careful history, thorough examination, and methodical workup are essential. Specific management is predicated on identification of the underlying etiology.
INTRODUCTION
Stroke is often considered a disease of older people, but an estimated 10% of patients with stroke are younger than 50 years. Recent evidence suggests that the incidence of ischemic stroke in young adults is rising,1 although the reasons for that increase are unclear. These trends are especially concerning in light of recent evidence that young stroke patients are at a greatly increased risk of early death compared with the general population.2 The purpose of this article is to provide an overview of some of the potential stroke etiologies to consider in patients under 50 years of age.
STANDARD RISK FACTORS
Classic vascular risk factors, including hypertension, dyslipidemia, diabetes mellitus, and tobacco abuse are prevalent in several studies of young stroke patients. In the largest study, Finnish investigators retrospectively evaluated 1008 consecutive ischemic stroke patients aged 15 to 49 in a single university center registry from 1994 to 2007. Conventional risk factors for stroke were highly prevalent in this young stroke population: hypertension (39%), dyslipidemia (60%), and smoking (44%).3 Investigators at a university center in the United States found similarly high rates of conventional risk factors.4 Given the prevalence of conventional risk factors in young stroke patients, initial evaluation should approximate that of older stroke patients. Further evaluation may be necessary pending initial results. A suggested initial evaluation is shown in Table 6-1.
Table 6-1.
Initial Evaluation of a Young Adult With Ischemic Stroke

As Table 6-25 demonstrates, there are multiple potential etiologies of ischemic stroke in young adults. Of particular interest for this review are three categories especially germane to a discussion of stroke in young adults: (1) cervical artery dissections, (2) patent foramen ovale (PFO), and (3) hypercoagulable syndromes, including inherited thrombophilias and the antiphospholipid antibody syndrome.
Table 6-2.
Uncommon Causes of Strokea

ARTERIAL DISSECTION
Dissection is the separation of the structural components of the connective tissue network of the arterial wall and can occur in the carotid, vertebral, and intracranial arteries. The second most common lesion of the cervical arteries after atherosclerosis, dissection has traditionally been characterized as either spontaneous or secondary to major trauma. Several connective tissue disorders have also been associated with dissection, including vascular Ehlers-Danlos syndrome (type IV), Marfan syndrome, and osteogenesis imperfecta, among others. Although dissections account for about 2% of all ischemic strokes,6 they account for a higher proportion in young adults.3 A population-based study in Olmsted County, Minnesota estimated the incidence as 1.72 per 100,000 per year for internal carotid artery dissections and 0.97 per 100,000 for vertebral artery dissections.7 The peak incidence occurs in the fifth decade of life. Intracranial dissections appear to comprise approximately 10% of published dissection cases. In 13% to 16% of cases of dissection, multiple arteries are involved.8 Recent analyses have noted significant differences in the clinical presentation, risk factors, and functional outcomes of internal carotid artery versus vertebral dissections.9,10
The classic teaching is that cervical artery dissections arise from an intimal tear and that blood enters the wall and forms an intramural hematoma. But the absence of a demonstrable intimal tear on some histologic examinations suggests that at least some dissections are caused by a primary intramural hematoma.11 One study found outer wall abnormalities in superficial temporal arteries of dissection patients and proposed that an underlying arteriopathy affecting the media and adventitia played a crucial role in dissection pathogenesis.12 Dissection is likely a multifactorial process, and both environmental and genetic risk factors have been implicated. The most important environmental risk factor appears to be trauma. Dissection occurs commonly with major trauma but is also associated with minor trauma, such as chiropractic manipulation, coughing, and sneezing, among others.13 Recent infection has also been associated with dissection. Several lines of evidence suggest a genetic contribution to dissection, including reports of familial dissection cases. Several candidate genes have been proposed, including the intercellular adhesion molecule 1 gene (ICAM1), the collagen, type III, alpha 1 gene (COL3A1), and the methylenetetrahydrofolate reductase (NAD[P]H) gene (MTHFR). The Cervical Artery Dissections and Ischemic Stroke Patients (CADISP) consortium is actively investigating these and other genes as well as gene-environment interactions.8
Clinical manifestations of cervical artery dissections include both local signs and symptoms and ischemic events. Local manifestations of internal carotid artery dissections include Horner syndrome (ipsilateral), neck pain, headache, tinnitus, facial pain, and cranial nerve palsies (IX to XII most commonly). Local manifestations in vertebral dissections include posterior headache or neck pain, cervical root involvement (most commonly C5-C6 level), and lower brainstem compression (in intracranial vertebral dissection). Ischemic manifestations of internal carotid artery dissection include stroke (most commonly in the middle cerebral artery distribution), amaurosis fugax, ischemic optic neuropathy, and retinal infarction. Ischemic manifestations in vertebral dissection include posterior circulation strokes, subarachnoid hemorrhage (in intracranial vertebral artery dissection), and spinal cord ischemia. Internal carotid artery or vertebral artery dissections resulting in arterial occlusion or stenosis are more likely to result in ischemic manifestations, while local manifestations are more likely to occur in the absence of luminal narrowing.14 Case 6-1 illustrates a common presentation of a young adult with carotid dissection.
Conventional angiography, the diagnostic mainstay of cervicocephalic dissection for many years, has now been largely replaced by noninvasive means such as CT angiography (CTA) and MR angiography (MRA). Conventional angiography is limited both by its invasiveness and its inability to visualize intramural hematoma. The luminal contour can also at times appear normal on conventional angiography. MRA can be combined with axial T1-weighted cervical MRI with fat suppression to better identify small intramural hematomas and does not require radiation. CTA, on the other hand, might be superior to MRA with regard to diagnosis of intimal flaps, pseudoaneurysms, and high-grade stenosis15 in addition to superior identification of vertebral artery dissections. Figure 6-1 demonstrates the superiority of CTA in the setting of high-grade stenosis. High-resolution 3-tesla MRI is also emerging as a potential tool for detection of cervicocephalic dissection and delineation of intramural thrombus versus intraluminal thrombus.16
Figure 6-1.
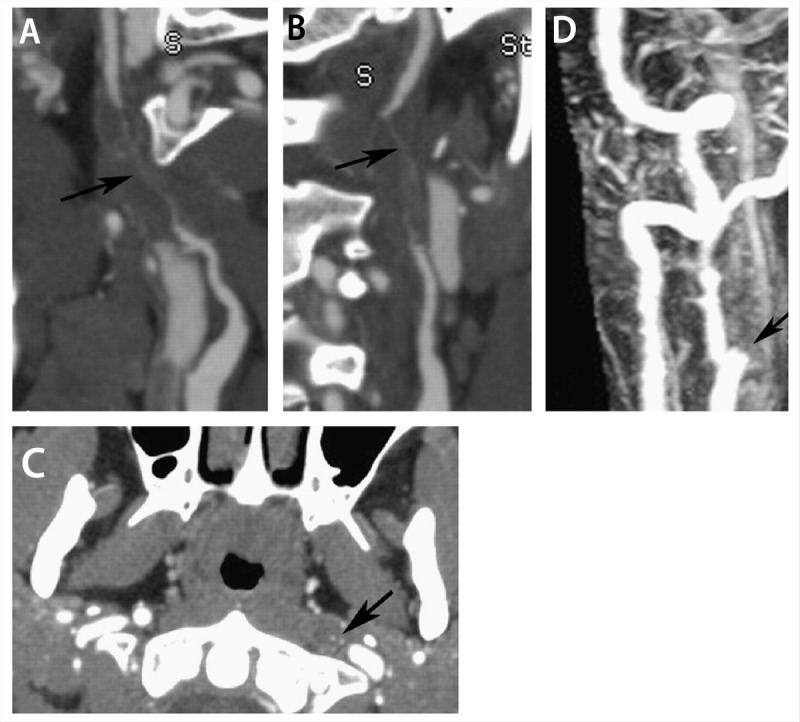
Example of a long-segment high-grade stenosis (string sign) from dissection identified on CT angiography (CTA), which appeared occluded on MRI/magnetic resonance angiography (MRA). A, B, Curved planar reformatted images of the left internal carotid artery demonstrate flamelike tapering of the proximal vessel with wall thickening and long-segment high-grade stenosis (arrow). C, Axial plane from CTA shows the tiny residual lumen of the vessel (arrow). D, Contrast-enhanced MRA acquired on the same day demonstrates apparent occlusion just distal to the carotid bifurcation (arrow). Modified with permission from Vertinsky AT, et al, AJNR Am J Neuroradiol.15 © 2008 American Society of Neuroradiology. www.ajnr.org/content/29/9/1753.long.
The treatment of cervicocephalic dissections is controversial owing to a lack of randomized controlled trials. In the hyperacute setting, there is no clear contraindication to thrombolysis. A recent meta-analysis found that thrombolysis in patients with stroke related to dissection had similar safety and outcome profiles compared with patients with strokes unrelated to dissection,17 although other studies have questioned the efficacy of thrombolysis for dissection.18 Patients are commonly prescribed anticoagulants given concerns about recurrent events. A common practice is to initiate an anticoagulant in the acute setting and then to repeat angiography in 3 months. If the dissection has resolved, the patient is typically transitioned to an antiplatelet agent. If irregularities persist, anticoagulation is generally continued until the arterial dissection resolves, completely occludes, or stabilizes, at which time an antiplatelet agent is initiated. Recent work, however, has questioned whether this approach is warranted. A large nonrandomized study found that ischemic recurrence rates were very low both in patients treated with antiplatelet agents and those treated with anticoagulants.19 An ongoing randomized clinical trial compares antiplatelet agents and anticoagulation in acute dissection20; the nonrandomized portion of this study found no evidence of superiority for anticoagulation.21
Recurrence rates of both ischemic events and dissections are generally reported as low. One-year ischemic recurrence rates range from 0% to 13%,8 and recurrences generally occur in the first few weeks following initial presentation. Dissection recurrence rates are low in population-based studies.7 Academic centers report higher rates but may be subject to referral bias.8
Case 6-1
A 34-year-old man presented with a right posterior headache, neck pain, and left-sided weakness that he noted when he awakened in the morning. He had recently started a yoga routine. He had no prior significant medical history, although he was a smoker. General examination was unremarkable. Neurologic examination was remarkable for a mild left hemiparesis. Brain MRI demonstrated restricted diffusion in the right middle cerebral artery distribution. CT angiography of the head and neck demonstrated dissection of the right internal carotid artery. Echocardiography was unremarkable. The patient was started on a heparin drip and warfarin 5 mg daily. A follow-up CT angiogram at 3 months showed resolution of the dissection, and anticoagulation was transitioned to aspirin 325 mg daily. He has had no further complications in 5 years of follow-up.
Comment. This patient sustained a right middle cerebral artery stroke due to dissection apparently related to minor trauma. Both his arterial healing time and the absence of recurrence are common in carotid artery dissection.
PATENT FORAMEN OVALE
PFO, an embryonic defect in the atrial septum, has been postulated as a source for paradoxical embolism for decades. The proportion of the general population with PFO is approximately 20%, but in young stroke patients this proportion is about 50%. Co-occurrence with atrial septal aneurysm (greater than 10-mm septal excursion during a cardiac cycle) seems to confer additional risk.22,23 The presence of PFO in the setting of an inherited or acquired thrombophilia is especially important because it provides a potential mechanism for venous material to pass into the arterial circulation (ie, paradoxical embolization). Dozens of observational studies have suggested a higher rate of recurrent stroke in medically treated patients compared with closure,24 but randomized data were not available.
Three randomized controlled trials, each investigating the role of PFO for secondary stroke prevention, have now been published. The first of these, the CLOSURE-I trial, investigated the STARFlex device in patients 60 years old or younger with stroke or TIA and a PFO.25 The primary end point was a composite of stroke or TIA in 2 years of follow-up, 30-day all-cause mortality, and death from neurologic cause between 31 days and 2 years. More than 900 patients were enrolled. The primary end point occurred in 5.5% of the closure group and 6.8% of the medical group (hazard ratio [HR] 0.78; 95% confidence interval [CI], 0.45 to 1.35; P=.37). Major periprocedural complications occurred in 13 (3.2%) patients in the closure group. The authors found plausible alternative explanations to a PFO-related cause for stroke or TIA in a majority of patients in both the closure and medical groups.
The PC trial investigated a different device, the Amplatzer PFO occluder, in patients younger than 60 years with stroke, TIA, or peripheral thromboembolic event who had a PFO.26 More than 400 patients were enrolled. The primary end point (a composite of nonfatal stroke, TIA, peripheral embolism, and death) occurred in 3.4% of the closure group and 5.2% of the medical group (HR 0.63; 95% CI, 0.24 to 1.62; P=.34). The authors reported three (1.5%) minor procedural complications in the closure group.
The Randomized Evaluation of Recurrent Stroke Comparing PFO Closure to Established Current Standard of Care Treatment (RESPECT) trial was published concurrently with the PC trial and also investigated the PFO Amplatzer occluder.27 The investigators enrolled nearly 1000 patients with cryptogenic strokes and PFO. The primary end point was a composite of nonfatal and fatal ischemic stroke and early death. Nine patients with recurrent stroke were in the closure group and 16 in the medical group (HR 0.49; 95% CI, 0.22 to 1.11; P=.08). Procedure-related adverse events occurred in 21 (4.2%) patients in the closure group.
Taken individually, these trials did not demonstrate the superiority of PFO closure over medical management; however, a recent meta-analysis of the trials suggests potential benefit.28 Given the relatively large statistical effect sizes in the trials, further studies are likely forthcoming in an attempt to identify the patient population most likely to benefit.
The optimal management of cryptogenic stroke in a young person with PFO remains unclear at this point. Given the negative trial data, for now closure should be limited to the setting of a clinical trial. The optimal long-term antithrombotic agent for the stroke patient with PFO is also unclear. No specific guidelines have been developed for the co-occurrence of PFO and atrial septal aneurysm. Given the lack of definitive data, the decision regarding antiplatelet versus anticoagulant therapy should be informed by patient risk stratification and risk tolerance, although in the absence of a deep vein thrombosis or known hypercoagulable state antiplatelet therapy is typically recommended. Modifiable risk factors should be aggressively addressed.
HYPERCOAGULABLE CAUSES OF STROKE
Up to 4% of ischemic strokes are thought to be due to coagulation disorders, and young patients in particular are often screened for hypercoagulable syndromes. Hypercoagulable syndromes may include inherited deficiencies and acquired conditions. The coagulation cascade is a complex system with multiple sites in which alteration could result in thrombophilia.
Several conditions are purported to predispose patients to thromboembolism and, by extension, acute ischemic stroke. Most of these conditions are primarily associated with venous thrombosis, although a few conditions such as homocystinuria and antiphospholipid antibody syndrome are associated with arterial thrombosis. The presumed mechanism for arterial stroke in a patient with a venous thrombophilia is paradoxical embolus, most commonly through a PFO. A pressure gradient promoting passage of venous material to the arterial circulation may be generated via the Valsalva maneuver. The inherited thrombophilias with the best-established evidence base for venous thromboembolism are factor V Leiden and activated protein C resistance, prothrombin gene mutation, protein S deficiency, protein C deficiency, and antithrombin III deficiency. The most common acquired thrombophilia is antiphospholipid antibody syndrome. Table 6-329 and Table 6-429 list the most common inherited and acquired thrombophilic states, respectively.
Table 6-3.
Primary/Inherited Hypercoagulable Statesa

Table 6-4.
Secondary/Acquired Hypercoagulable Statesa

Factor V Leiden
Approximately 90% to 95% of patients with activated protein C resistance have the identical mutation, which is a single point mutation in the factor V gene (factor V R506Q). Factor V Leiden increases the risk of thrombosis by increasing thrombin production. Factor V Leiden is by far the most common genetic risk factor for thrombophilia. The prevalence of factor V Leiden heterozygosity varies within American populations; the frequency is 5.3% in whites, 2.2% in Hispanics, 1.3% in Native Americans, 1.2% in African Americans, and 0.5% in Asian Americans.30 Factor V Leiden is not fully penetrant, and only 5% to 10% of factor V Leiden heterozygotes will experience a symptomatic venous thromboembolic event. Homozygotes are much rarer but have a markedly elevated thrombotic risk (the relative risk for first episode of venous thrombosis is 7 in heterozygotes and 80 in homozygotes).31 Activated protein C resistance is associated with several other factors, including elevated factor VIII levels, oral contraceptives, pregnancy, hormone replacement, cancer, antiphospholipid antibody syndrome, and smoking. Patients with factor V Leiden frequently have other thrombophilias,32,33,34 and these patients appear to be at increased risk for thrombosis. Factor V Leiden has not been convincingly linked to arterial thrombosis. Case 6-2 is a typical example of a patient with factor V Leiden.
Case 6-2
A 42-year-old woman without a significant prior medical history presented with a 2-day history of difficulty seeing to the left. She did not take oral contraceptives, smoke, or use illicit drugs. She had one previous miscarriage but no history of deep venous thromboses (DVTs), pulmonary emboli, or other bleeding or clotting issues. She did, however, have a strong family history of factor V Leiden mutation in her mother and brother, and her maternal grandmother died of blood clots. Neurologic examination demonstrated a left homonymous hemianopia. Brain MRI demonstrated restricted diffusion in the right occipital lobe. CT angiography of the head and neck was unremarkable. Transesophageal echocardiography demonstrated a small right-to-left shunt via foramen ovale on the saline contrast study, and an atrial septal aneurysm. She was heterozygous for the factor V Leiden mutation. The remainder of her hypercoagulable workup was unremarkable. Lower extremity Doppler ultrasound demonstrated no DVT in the legs. Magnetic resonance venography of the pelvis demonstrated DVT in the left common iliac vein due to May-Thurner syndrome. She was initially treated with aspirin 325 mg/d and then transitioned to anticoagulation with warfarin.
Comment. This case emphasizes the importance of a methodical approach in cryptogenic stroke. A strong family history prompted an investigation into thrombophilia, transesophageal echocardiography demonstrated a shunt, and further evaluation yielded a venous source. May-Thurner syndrome (iliocaval compression) (Figure 6-235) is an important consideration in the setting of cryptogenic stroke, thrombophilia, and patent foramen ovale.
Figure 6-2.
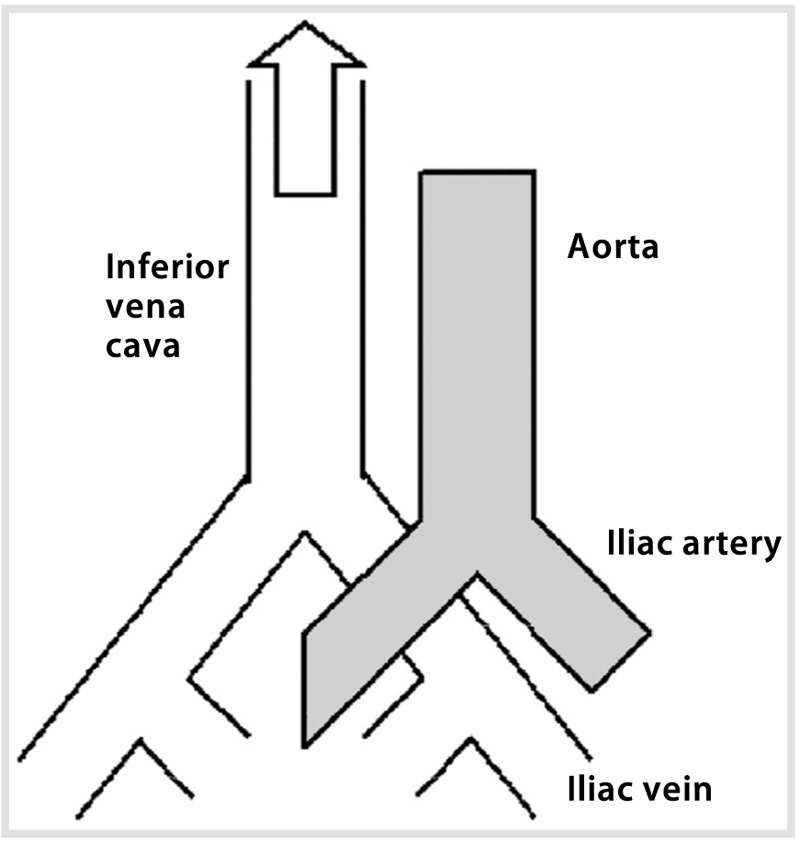
Anatomic diagram of May-Thurner syndrome with the right common iliac artery overlying the left common iliac vein. Iliocaval compression can lead to thrombus formation.Reprinted with permission from Kiernan TJ, et al, Stroke.35 © 2009 American Heart Association. stroke.ahajournals.org/content/40/4/1502.long.
Prothrombin Gene Mutation
Prothrombin deficiency is the second most common inherited thrombophilia after factor V Leiden, and the source of the thrombophilia is a single point mutation (G20210A). Carriers have higher prothrombin levels and are at higher risk of thrombosis. Prevalence of the mutation varies in populations; up to 7% of the white population carries the mutation, while it is very rare in black or Asian populations.36 Homozygotes are at increased risk for DVT and cerebral venous sinus thrombosis, especially in patients taking oral contraceptives. Conclusive evidence of increased risk for arterial thrombosis in patients with prothrombin deficiency has not been established.
Protein S Deficiency
Protein S is a vitamin K-dependent protein that enhances fibrinolysis, inhibits prothrombin activation, and reduces thrombin production. Protein S circulates in both free and protein-bound forms. Deficiencies of protein S can be genetic or acquired. In genetic cases, inheritance is predominantly autosomal dominant. Heterozygotes are at risk for recurrent venous thromboembolism; homozygous protein S deficiency is usually incompatible with life. Acquired protein S deficiency is associated with pregnancy, oral contraceptive pill use, disseminated intravascular coagulation, HIV, nephrotic syndrome, systemic lupus erythematosus (SLE), liver disease, and asparaginase therapy. Protein S deficiency has not been convincingly linked to arterial thrombosis.
Protein C Deficiency
Activated protein C inactivates factors Va and VIIIa, which are important for thrombin generation. Protein C deficiency can be either genetic or acquired. Heterozygosity occurs in 0.2% to 0.5% of the general population. Acquired protein C deficiency is associated with severe infection (especially meningococcemia), liver disease, disseminated intravascular coagulation, acute respiratory distress syndrome, sepsis, and several medications, including methotrexate, 5-fluorouracil, and cyclophosphamide. A convincing link to arterial thrombosis has not been demonstrated.
Antithrombin Deficiency
Antithrombin (formerly called antithrombin III) is an inhibitor of thrombin. Antithrombin deficiency can be both genetic and acquired. The inheritance pattern is usually autosomal dominant. The prevalence in the general population may be 1 in 500. Acquired antithrombin deficiency is associated with disseminated intravascular coagulation, sepsis, liver disease, nephrotic syndrome, oral contraceptives, estrogen, and asparaginase. Patients with antithrombin deficiency appear to be at no increased risk for arterial thrombosis.
Antiphospholipid Antibody Syndrome
The antiphospholipid antibody syndrome (APS) is an autoimmune condition associated with both venous and arterial thrombosis. APS often but not always occurs with SLE. The most common thrombotic event in patients with APS is DVT; stroke is the most common arterial event. The diagnostic criteria for APS are found in Table 6-537. Any patient with rheumatologic disease and stroke should be screened for APS; it may also be reasonable to screen any patient with stroke younger than age 45 for APS.38 Treatment generally consists of long-term anticoagulation. Case 6-3 is an example of SLE-associated APS manifesting with multiple posterior circulation strokes.
Table 6-5.
Revised Classification Criteria for the Antiphospholipid Syndromea

Catastrophic antiphospholipid antibody syndrome occurs in a small subset of patients with antiphospholipid antibody syndrome and is characterized by three or more new organ thromboses within 1 week. Definitive diagnosis requires demonstration of microthrombus on biopsy.39 Treatment of catastrophic antiphospholipid antibody syndrome includes treatment of any underlying predisposing condition, heparin, and steroids, with the addition of plasma exchange if warranted.40
Case 6-3
A 24-year-old woman with systemic lupus erythematosus (SLE) and a history of migraine headaches presented with headache and difficulty walking. Neurologic examination was remarkable for somnolence, dysarthria, and a central right facial droop. No focal weakness or dystaxia was noted. Brain MRI demonstrated acute infarcts in the bilateral cerebellar hemispheres, the bilateral pons, and the left thalamus. CT angiogram of the head and neck demonstrated diminished flow in the right vertebral artery and occlusion or near occlusion of the distal basilar artery but no dissection. Transesophageal echocardiography showed no left atrial appendage abnormality, valvular abnormality, or right-to-left shunt. Anticardiolipin and anti–β2-glycoprotein titers were markedly elevated. Antiphospholipid antibody syndrome was suspected. She was started by a rheumatologist on steroids, rituximab, and an antiplatelet agent (aspirin 325 mg/d) initially and was subsequently transitioned to anticoagulation (warfarin). Follow-up studies 4 months after the initial event confirmed the diagnosis of antiphospholipid antibody syndrome.
Comment. The presumed mechanism of stroke in this young woman was antiphospholipid antibody syndrome associated with SLE. Antiphospholipid antibody syndrome can affect both the venous and arterial circulations, and the most common arterial manifestation is ischemic stroke.
Diagnostic Approach
Factor V Leiden diagnostic testing includes functional activated protein C assays or direct mutation testing via PCR. Prothrombin mutation is diagnosed via G20210A PCR testing. The reliability of nongenetic tests for protein S, protein C, and antithrombin deficiency depends on the clinical scenario. Definitive diagnosis can be challenging given testing limitations, timing considerations in the acute phase, and potential confounding due to medications (especially anticoagulants). For protein S deficiency, both free and total protein S antigen and functional protein S assays are commonly used. The diagnosis of protein C deficiency is made somewhat difficult by the wide range of normal values in the population. Normal protein C antigen ranges from 70% to 140% of normal; levels less than 55% are likely due to a genetic abnormality. Protein C functional levels are the preferred screening test. Antithrombin deficiency is diagnosed using the antithrombin-heparin cofactor assay.
A reasonable approach for protein S, protein C, and antithrombin deficiency testing would be to repeat studies a few months after the stroke (and with the patient off anticoagulants for at least 2 weeks) before committing the patient to long-term anticoagulation. Interpretation can be challenging. One study found that approximately 30% of testing for hypercoagulable states in routine academic practice was performed in settings in which the results were not interpretable.41 Another study found that nearly 75% of the diagnosed thrombophilias were subsequently not confirmed on follow-up testing.42
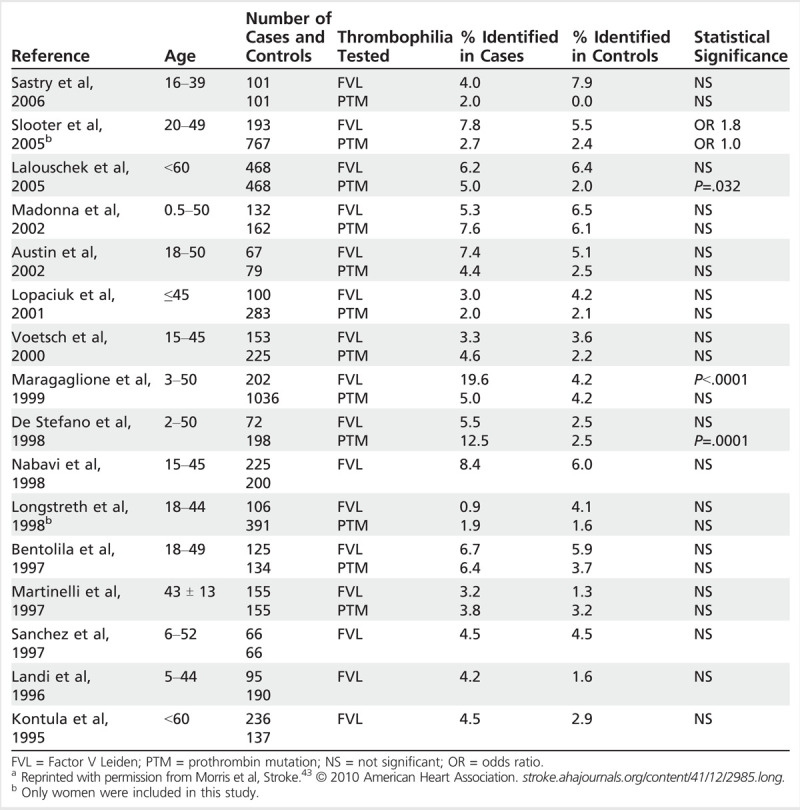
Even if a thrombophilia is reliably identified, it is unclear whether the thrombophilia is associated with an increased risk for ischemic stroke. An extensive review43 identified six case-control studies of 50 or more ischemic stroke cases with protein S deficiency, protein C deficiency, and/or antithrombin deficiency. In four of the six studies, the proportion of cases with the deficiency was greater than that of the controls, but this difference was not statistically significant in any of the studies (Table 6-6). One of these studies also stratified by presumed mechanism and inherited thrombophilia and did not find significant differences.44 This review also identified 16 case-control studies of 50 or more cases of factor V Leiden or prothrombin mutation. Fourteen of these studies found no statistical difference between the cases and controls with regard to factor V Leiden or prothrombin mutation (Table 6-7). The authors of that review also reviewed nine case-control studies (without limiting the number of cases) that evaluated the relationship between PFO, ischemic stroke, and inherited thrombophilias. None of the studies that evaluated protein C, protein S, or antithrombin deficiency found an association with PFO and stroke. The studies were mixed with regard to factor V Leiden and prothrombin mutation, with a few demonstrating a significant association.43
Table 6-6.
Case-Control Studies of Protein C Deficiency, Protein S Deficiency, and Antithrombin Deficiency in Patients With Ischemic Strokea
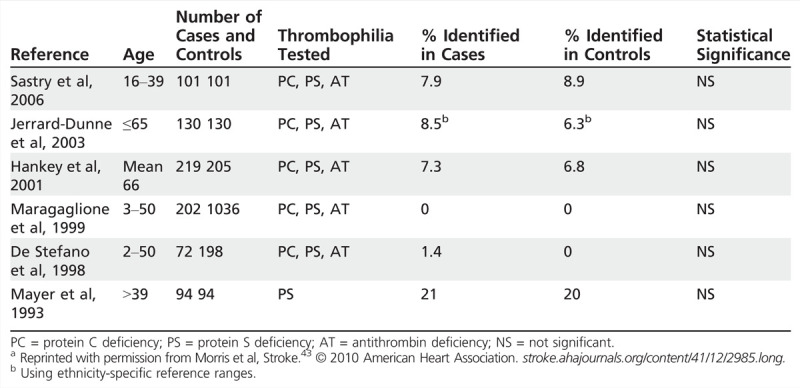
Table 6-7.
Case Control Studies of Factor V Leiden and Prothrombin Mutation in Young Patients (Younger Than 60 Years) With Ischemic Strokea
Most experts recommend a tailored approach to testing for hypercoagulable states, in which high-risk individuals (ie, younger, undetermined cause of stroke, history of venous thrombosis, multiple miscarriages, frequent thrombotic recurrences, family history of venous thromboembolic disease, etc) are selected for testing.45 Testing for factor V Leiden and prothrombin G20210A in nonwhite populations is likely to be very low-yield, given the very low prevalence of the mutations in these populations. A suggested approach to testing for thrombophilic states is shown in Table 6-8. In the absence of a right-to-left shunt, testing for thrombophilias (other than possibly antiphospholipid antibody syndrome) seems low yield. Young adults with stroke and a right-to-left shunt should be evaluated for deep venous thrombosis with compression ultrasonography of the lower extremities and venography of the pelvis.
Table 6-8.
Thrombophilia Screening Panel

Treatment
The best treatment regimen for most hypercoagulable states is unknown, and few large studies to direct treatment decisions are available. One exception is the Antiphospholipid Antibodies in Stroke Study/Warfarin-Aspirin Recurrent Stroke Study collaboration, which compared warfarin and aspirin for prevention of recurrent stroke in patients with antiphospholipid antibodies and found no statistical difference in outcomes.46
Many clinicians use long-term anticoagulation for patients with inherited coagulation disorders. The optimal duration of treatment is unclear. Whether the cost-benefit ratio is favorable over the long term for ischemic stroke patients identified with thrombophilias is also unclear. For acquired thrombophilias, therapy is targeted toward the specific predisposing condition. Many experts recommend discontinuing oral contraceptives and hormones in patients with thrombophilia. Smoking cessation is essential.
SUMMARY
The incidence of ischemic stroke in young adults is increasing. The list of potential stroke etiologies is extensive, and a thorough history, examination, and tailored workup are essential. Cervical artery dissection, PFO, and thrombophilias are potential causes of stroke in young adults.
KEY POINTS
The incidence of stroke in young adults appears to be increasing.
Stroke in young adults is associated with increased risk of early death.
Standard vascular risk factors are prevalent in young stroke patients.
CT angiography and magnetic resonance angiography (MRA) have supplanted conventional angiography in the diagnosis of dissection.
CTA appears to have some advantages over MRA in the diagnosis of dissection.
There is no contraindication to tissue plasminogen activator in strokes due to dissection.
Patent foramen ovale closure trials have yet to demonstrate superiority over medical management.
The best long-term antithrombotic agent for stroke patients with patent foramen ovale remains unclear.
The thrombophilias with the best evidence for venous thromboembolism are factor V Leiden, prothrombin gene mutation, protein S deficiency, protein C deficiency, and antithrombin III deficiency.
Studies have not demonstrated a convincing association between hypercoagulable states and ischemic stroke.
Young adults with stroke and a right-to-left shunt should be evaluated for deep venous thrombosis with compression ultrasonography of the lower extremities and venography of the pelvis.
Footnotes
Relationship Disclosure: Dr Mackey has served as an expert medical record reviewer in vascular neurology for court cases and has received funding as the principal investigator for outcomes and processes of care in intracerebral hemorrhage for the Indiana University Health Values Fund and the Indiana Clinical and Translational Sciences Institute Project Development Team. He is also a recipient of a NIH Loan Repayment Program award.
Unlabeled Use of Products/Investigational Use Disclosure: Dr Mackey reports no disclosure.
REFERENCES
- 1.Kissela BM,, Khoury JC,, Alwell K, et al Age at stroke: temporal trends in stroke incidence in a large, biracial population. Neurology 2012; 79 (17): 1781–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rutten-Jacobs LC,, Arntz RM,, Maaijwee NA, et al Long-term mortality after stroke among adults aged 18 to 50 years. JAMA 2013; 309 (11): 1136–1144. [DOI] [PubMed] [Google Scholar]
- 3.Putaala J,, Metso AJ,, Metso TM, et al Analysis of 1008 consecutive patients aged 15 to 49 with first-ever ischemic stroke: the Helsinki young stroke registry. Stroke 2009; 40 (4): 1195–1203. [DOI] [PubMed] [Google Scholar]
- 4.Ji R,, Schwamm LH,, Pervez MA,, Singhal AB. Ischemic stroke and transient ischemic attack in young adults: risk factors, diagnostic yield, neuroimaging, and thrombolysis. JAMA Neurol 2013; 70 (1): 51–57. [DOI] [PubMed] [Google Scholar]
- 5.Caplan LR, ed. Uncommon causes of stroke. 2nd edition. Cambridge, UK: Cambridge University Press, 2008. [Google Scholar]
- 6.Giroud M,, Fayolle H,, André N, et al Incidence of internal carotid artery dissection in the community of Dijon. J Neurol Neurosurg Psychiatry 1994; 57 (11): 1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee VH,, Brown RD,, Mandrekar JN,, Mokri B. Incidence and outcome of cervical artery dissection: a population-based study. Neurology 2006; 67 (10): 1809–1812. [DOI] [PubMed] [Google Scholar]
- 8.Debette S,, Leys D. Cervical-artery dissections: predisposing factors, diagnosis, and outcome. Lancet Neurol 2009; 8 (7): 668–678. [DOI] [PubMed] [Google Scholar]
- 9.von Babo M,, De Marchis GM,, Sarikaya H, et al Differences and similarities between spontaneous dissections of the internal carotid artery and the vertebral artery. Stroke 2013; 44 (6): 1537–1542. [DOI] [PubMed] [Google Scholar]
- 10.Debette S,, Grond-Ginsbach C,, Bodenant M, et al Differential features of carotid and vertebral artery dissections: the CADISP study. Neurology 2011; 77 (12): 1174–1181. [DOI] [PubMed] [Google Scholar]
- 11.Schievink WI. Spontaneous dissection of the carotid and vertebral arteries. N Engl J Med 2001; 344 (12): 898–906. [DOI] [PubMed] [Google Scholar]
- 12.Völker W,, Dittrich R,, Grewe S, et al The outer arterial wall layers are primarily affected in spontaneous cervical artery dissection. Neurology 2011; 76 (17): 1463–1471. [DOI] [PubMed] [Google Scholar]
- 13.Engelter ST,, Grond-Ginsbach C,, Metso TM, et al Cervical artery dissection: trauma and other potential mechanical trigger events. Neurology 2013; 80 (21): 1950–1957. [DOI] [PubMed] [Google Scholar]
- 14.Baumgartner RW,, Arnold M,, Baumgartner I, et al Carotid dissection with and without ischemic events: local symptoms and cerebral artery findings. Neurology 2001; 57 (5): 827–832. [DOI] [PubMed] [Google Scholar]
- 15.Vertinsky AT,, Schwartz NE,, Fischbein NJ, et al Comparison of multidetector CT angiography and MR imaging of cervical artery dissection. AJNR Am J Neuroradiol 2008; 29 (9): 1753–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bachmann R,, Nassenstein I,, Kooijman H, et al High-resolution magnetic resonance imaging (MRI) at 3.0 Tesla in the short-term follow-up of patients with proven cervical artery dissection. Invest Radiol 2007; 42 (6): 460–466. [DOI] [PubMed] [Google Scholar]
- 17.Zinkstok SM,, Vergouwen MD,, Engelter ST, et al Safety and functional outcome of thrombolysis in dissection-related ischemic stroke: a meta-analysis of individual patient data. Stroke 2011; 42 (9): 2515–2520. [DOI] [PubMed] [Google Scholar]
- 18.Engelter ST,, Dallongeville J,, Kloss M, et al Thrombolysis in cervical artery dissection—data from the Cervical Artery Dissection and Ischaemic Stroke Patients (CADISP) database. Eur J Neurol 2012; 19 (9): 1199–1206. [DOI] [PubMed] [Google Scholar]
- 19.Georgiadis D,, Arnold M,, von Buedingen HC, et al Aspirin vs anticoagulation in carotid artery dissection: a study of 298 patients. Neurology 2009; 72 (21): 1810–1815. [DOI] [PubMed] [Google Scholar]
- 20.Cervical Artery Dissection in Stroke Study Trial Investigators. Antiplatelet therapy vs. anticoagulation in cervical artery dissection: rationale and design of the Cervical Artery Dissection in Stroke Study (CADISS). Int J Stroke 2007; 2 (4): 292–296. [DOI] [PubMed] [Google Scholar]
- 21.Kennedy F,, Lanfranconi S,, Hicks C, et al Antiplatelets vs anticoagulation for dissection: CADISS nonrandomized arm and meta-analysis. Neurology 2012; 79 (7): 686–689. [DOI] [PubMed] [Google Scholar]
- 22.Messé SR,, Silverman IE,, Kizer JR, et al Practice parameter: recurrent stroke with patent foramen ovale and atrial septal aneurysm: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2004; 62 (7): 1042–1050. [DOI] [PubMed] [Google Scholar]
- 23.Larrue V,, Berhoune N,, Massabuau P, et al Etiologic investigation of ischemic stroke in young adults. Neurology 2011; 76 (23): 1983–1988. [DOI] [PubMed] [Google Scholar]
- 24.Kitsios GD,, Dahabreh IJ,, Abu Dabrh AM, et al Patent foramen ovale closure and medical treatments for secondary stroke prevention: a systematic review of observational and randomized evidence. Stroke 2012; 43 (2): 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furlan AJ,, Reisman M,, Massaro J, et al Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med 2012; 366 (11): 991–999. [DOI] [PubMed] [Google Scholar]
- 26.Meier B,, Kalesan B,, Mattle HP, et al Percutaneous closure of patent foramen ovale in cryptogenic embolism. N Engl J Med 2013; 368 (12): 1083–1091. [DOI] [PubMed] [Google Scholar]
- 27.Carroll JD,, Saver JL,, Thaler DE, et al Closure of patent foramen ovale versus medical therapy after cryptogenic stroke. N Engl J Med 2013; 368 (12): 1092–1100. [DOI] [PubMed] [Google Scholar]
- 28.Kitsios GD,, Thaler DE,, Kent DM. Potentially large yet uncertain benefits: a meta-analysis of patent foramen ovale closure trials. Stroke 2013; 44 (9): 2640–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biller J, ed. Stroke in children and young adults. 2nd edition. Philadelphia, PA: Saunders, 2009. [Google Scholar]
- 30.Ridker PM,, Miletich JP,, Hennekens CH,, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997; 277 (16): 1305–1307. [PubMed] [Google Scholar]
- 31.Rosendaal FR,, Koster T,, Vandenbroucke JP,, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995; 85 (6): 1504–1508. [PubMed] [Google Scholar]
- 32.Koeleman BP,, Reitsma PH,, Allaart CF,, Bertina RM. Activated protein C resistance as an additional risk factor for thrombosis in protein C-deficient families. Blood 1994; 84 (4): 1031–1035. [PubMed] [Google Scholar]
- 33.Zöller B,, Berntsdotter A,, García de Frutos P,, Dahlbäck B. Resistance to activated protein C as an additional genetic risk factor in hereditary deficiency of protein S. Blood 1995; 85 (12): 3518–3523. [PubMed] [Google Scholar]
- 34.van Boven HH,, Reitsma PH,, Rosendaal FR, et al Factor V Leiden (FV R506Q) in families with inherited antithrombin deficiency. Thromb Haemost 1996; 75 (3): 417–421. [PubMed] [Google Scholar]
- 35.Kiernan JT,, Yan BP,, Cubeddu RJ, et al May-Thurner syndrome in patients with cryptogenic stroke and patent foramen ovale: an important clinical association. Stroke 2009; 40 (4): 1502–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosendaal FR,, Doggen CJ,, Zivelin A, et al Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost 1998; 79 (4): 706–708. [PubMed] [Google Scholar]
- 37.Miyakis S,, Lockshin MD,, Atsumi T, et al International consensus statement on an update of the classification criteria for definite antiphospholipid. J Thromb Haemost 2006; 4 (2): 295–306. [DOI] [PubMed] [Google Scholar]
- 38.Brey R. Neurologic manifestations of systemic lupus erythematosus and antiphospholipid antibody syndrome. Continuum (Minneap Minn) 2008; 14 (1): 94–119. [Google Scholar]
- 39.Erkan D,, Espinosa G,, Cervera R. Catastrophic antiphospholipid syndrome: updated diagnostic algorithms. Autoimmun Rev 2010; 10 (2): 74–79. [DOI] [PubMed] [Google Scholar]
- 40.Lockshin MD,, Erkan D. Treatment of the antiphospholipid syndrome. N Engl J Med 2003; 349 (12): 1177–1179. [DOI] [PubMed] [Google Scholar]
- 41.Bushnell C,, Siddiqi Z,, Morgenlander JC,, Goldstein LB. Use of specialized coagulation testing in the evaluation of patients with acute ischemic stroke. Neurology 2001; 56 (5): 624–627. [DOI] [PubMed] [Google Scholar]
- 42.Bushnell CD,, Siddiqi Z,, Goldstein LB. Improving patient selection for coagulopathy testing in the setting of acute ischemic stroke. Neurology 2001; 57 (7): 1333–1335. [DOI] [PubMed] [Google Scholar]
- 43.Morris JG,, Singh S,, Fisher M. Testing for inherited thrombophilias in arterial stroke: can it cause more harm than good? Stroke 2010; 41 (12): 2985–2990. [DOI] [PubMed] [Google Scholar]
- 44.Hankey GJ,, Eikelboom JW,, van Bockxmeer FM, et al Inherited thrombophilia in ischemic stroke and its pathogenic subtypes. Stroke 2001; 32 (8): 1793–1799. [DOI] [PubMed] [Google Scholar]
- 45.Seligsohn U,, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med 2001; 344 (16): 1222–1231. [DOI] [PubMed] [Google Scholar]
- 46.Levine SR,, Brey RL,, Tilley BC, et al Antiphospholipid antibodies and subsequent thrombo-occlusive events in patients with ischemic stroke. JAMA 2004; 291 (5): 576–584. [DOI] [PubMed] [Google Scholar]