

Abstract
Purpose of Review:
This review serves as an overview of neurologic conditions associated with alcohol abuse or withdrawal, including epidemiology, clinical symptoms, diagnostic approach, and treatment.
Recent Findings:
Frequent alcohol abuse and frank alcoholism are very common among adults in the United States. Although rates decline with each decade, as many as 10% of the elderly drink excessively. Given the ubiquitous nature of alcoholism in society, its complications have been clinically recognized for generations, with recent advances focusing on improved understanding of ethanol’s biochemical targets and the pathophysiology of its complications.
Summary:
The chronic effects of alcohol abuse are myriad and include neurologic complications through both direct and indirect effects on the central and peripheral nervous systems. These disorders include several encephalopathic states related to alcohol intoxication, withdrawal, and related nutritional deficiencies; acute and chronic toxic and nutritional peripheral neuropathies; and myopathy. Although prevention of alcoholism and its neurologic complications is the optimal strategy, this article reviews the specific treatment algorithms for alcohol withdrawal and its related nutritional deficiency states.
Introduction
Chronic alcohol abuse is a common disorder, with nearly one-third of the adult population in the United States drinking excessively on a routine basis, including 10% of the elderly.1 The neurologic complications of alcohol abuse are numerous, including immediate effects of intoxication and withdrawal and delayed effects such as acute and chronic cognitive disorders and neuropathy, which may be related to the direct effects of ethanol abuse or related nutritional deficiencies. In addition, alcoholism has the potential to take its toll on the offspring of alcoholics through the fetal neurotoxic effects of alcohol. This review serves as an overview of each condition associated with alcohol abuse or withdrawal, including epidemiology, clinical symptoms, diagnostic approach, and treatment. Other aspects of alcoholism, including secondary neurologic effects through its medical complications such as cirrhosis, as well as prevention, are reviewed elsewhere.
CENTRAL NERVOUS SYSTEM COMPLICATIONS OF ALCOHOLISM
Acute Effects
Intoxication. Alcohol (principally consumed as ethanol) has multiple effects on the CNS, although no endogenous ethanol receptors are known. Ethanol is thought to primarily act through facilitation of inhibitory γ-aminobutyric acid (GABA) receptors and inhibition at excitatory glutamate receptors. Ethanol follows zero-order kinetics, with 70 mg to 150 mg of ethanol being metabolized per kilogram of the drinker’s body weight per hour, equivalent to 10 mg/dL to 25 mg/dL blood ethanol per hour.2 Acute ethanol intoxication induces CNS depression, perhaps initially in the reticular formation, followed by cerebral cortical involvement. Clinical intoxication is related to the rate of blood ethanol increase and the individual’s ethanol tolerance. Examination during intoxication may demonstrate saccadic visual pursuit, dysarthric speech, and ataxia. Behavioral changes during intoxication can be idiosyncratic; individuals can be unpredictable from one bout of intoxication to the next, including exhibiting extremes of irrational, violent, or psychotic behavior termed “pathologic intoxication.” These episodes are often associated with amnesia but are distinct from “alcoholic blackouts,” which lack extremes of aberrant behavior and may relate to impaired memory encoding, perhaps due to direct hippocampal effects.3 With moderate alcohol intoxication, EEG demonstrates increased beta (fast) activity, whereas higher levels induce slowing. Alcohol intoxication reduces sleep-onset latency, sleep efficiency, and duration of REM sleep and is associated with diminished perceived sleep quality; these effects may be more evident in women than in men.4 Central respiratory drive may substantially decline to a potentially deadly degree at blood ethanol levels of 400 mg/dL, although levels as high as 700 mg/dL may be required in some chronic alcoholics. Treatment of acute ethanol intoxication is largely supportive, but appropriately intensive monitoring, including intensive care unit–level care, may be needed; sedation may progress after presentation, depending upon amount and timing of the last drink. The differential diagnosis for profoundly intoxicated patients should remain broad, given that such patients may be prone to injury including intracerebral trauma, infection including meningitis, and metabolic disarray including hypoglycemia, ketoacidosis, and hepatorenal toxicity.2 Parenteral thiamine should always precede IV dextrose to avoid precipitating thiamine deficiency syndromes described below.
Ethanol withdrawal syndromes. Ethanol withdrawal is thought to occur only in persons with ethanol dependence. The likelihood of withdrawal in an individual alcoholic is difficult to predict but becomes more likely after prolonged binges using escalating amounts of alcohol. Following the described effects of ethanol on GABA and glutamate receptors, the presumed effect of abstinence on this system is thought to be glutamate receptor up-regulation and GABA-receptor down-regulation, leading to a host of neurologic complications as described here. Seizures, occurring within 48 hours of last intoxication, are thought to be the earliest manifestation of alcohol withdrawal, but seizures may occur while the person remains inebriated with decreasing blood ethanol levels.5 Ethanol may additionally increase seizure risk among epileptics, typically following either brief use or overt intoxication. Alcoholic withdrawal tremors may occur in a similar time frame following, but seldom during, intoxication. Tremors may occur after shorter bouts of heavy ethanol abuse, typically requiring less chronic ethanol exposure to develop this symptom than needed to develop the more profound withdrawal syndromes, but tremors may last for days to weeks if no further alcohol is used. Alcoholic hallucinosis includes spontaneous and complex visual, auditory, and/or tactile phenomena and can last several days. Delirium tremens represents the most advanced, deadly, and prolonged form of alcohol withdrawal and includes features of alcoholic hallucinosis and agitation, delirium, and autonomic disarray (including tachycardia, fever, hypertension, and diaphoresis). The syndrome typically begins 72 to 96 hours following intoxication and is far more likely to occur in persons with alcohol withdrawal seizures or hallucinosis. Thus, persons at risk should be monitored closely and treated early and aggressively according to standardized protocols such as the revised Clinical Institute Withdrawal Assessment of Alcohol Scale (CIWA-Ar) (Figure 7-1).6 At the authors’ institution, patients are followed closely in a formal protocol after being triaged within three treatment categories: initial stabilization, ongoing stabilization/tapering, and discontinuation/discharge. In all categories, CIWA-Ar is assessed every 4 hours, or with additional frequency as clinically indicated. Patients are categorized based on CIWA-Ar scores into one of five categories: mild (<10), moderate (11–15), severe (16–20), very severe (>21), and delirium tremens and/or treatment resistant (persistently >25). Patients in the initial phase of care and particularly those with high CIWA-Ar scores are often triaged to areas treating diseases with intense or high acuity (ie, intensive care unit or step-down-level care). All patients are assessed for concomitant disorders and receive IV thiamine, folate, and multivitamins upon admission, and these medications are transitioned to oral regimens thereafter. Benzodiazepine selection is at the discretion of the treating physician, but generally patients are treated with IV benzodiazepines (eg, diazepam or lorazepam) in severe cases. In moderate cases oral chlordiazepoxide is added to minimize IV benzodiazepine medication reliance and toxicity. Patients with mild CIWA-Ar scores are treated only with oral chlordiazepoxide. Depending on the level of alertness while being monitored in the protocol, oral feeding may need to be restricted. Once a patient has achieved CIWA-Ar score of less than 8 for more than 24 hours, patients are transitioned into the discontinuation/discharge phase of care. Case 7-1 details a typical presentation.
Figure 7-1.

The revised Clinical Institute Withdrawal Assessment of Alcohol Scale (CIWA-Ar).
Chronic Complications
Wernicke syndrome. Wernicke syndrome, also known as Wernicke encephalopathy, occurs due to thiamine deficiency, develops in an acute to subacute manner over the course of days to weeks, and is characterized by a cognitive disorder, gait ataxia, and ophthalmoparesis. Although often associated with chronic alcohol abuse (where, in addition to alcoholism-related nutritional deficiency, there may be synergistic effects on the clinical syndrome related to alcohol-related toxicity),7,8 Wernicke syndrome can occur in association with any disorder leading to deficiency of thiamine (see the article “Neurologic Complications of Bariatric Surgery” by Neeraj Kumar, MD, in this issue of CONTINUUM). Chronic Korsakoff syndrome is clinically distinct from the acute Wernicke syndrome and is discussed below.
Cognitive manifestations of Wernicke syndrome include restricted attention, impaired memory, disorientation, and diminished spontaneous speech output. Neurobehavioral symptoms may initially be erroneously ascribed to a mood disorder such as alcoholic depression; patients often experience some degree of social decline but typically have frank depression as a relatively minor clinical component. Depending on the degree of cognitive impairment influencing accurate history taking, patients may report various neuro-ophthalmic concerns, including diplopia or more subtle visual symptoms. Examination often reveals horizontal nystagmus, which may accompany rotatory or vertical nystagmus, and bilateral but typically asymmetric lateral rectus palsy. Horizontal and lesser vertical gaze palsies are often present. Truncal and gait ataxia are found in most patients; symptoms may be profound and impair gait or even the ability to sit. As described later in this article, gait ataxia of Wernicke syndrome may be masked by thiamine neuropathy. In its most pronounced form, Wernicke syndrome can produce coma without significant neuro-ophthalmic findings.
The diagnosis of Wernicke syndrome is often suspected based on clinical grounds, and laboratory testing may not be additionally useful. Treatment with thiamine repletion, currently recommended at 1 gram of IV thiamine per 24 hours for alcoholics with suspected Wernicke encephalopathy,9 should not be delayed while awaiting diagnostic laboratory results such as blood transketolase levels, which may be normal unless assessed prior to treatment. Death occurs in nearly 20% of patients with delayed treatment.9 EEG and CSF analysis may exclude other explanatory or concomitant conditions, but these tests are generally unrevealing in central thiamine deficiency states. MRI may reveal restricted diffusion or fluid-attenuated inversion recovery (FLAIR) signal abnormalities in areas implicated in the syndrome, including the thalamus, hypothalamus (mammillary bodies), midbrain (periaqueductal gray and oculomotor regions), and pons (abducens and medial vestibular nuclei). Imaging abnormalities outside of these areas are more typical of Wernicke encephalopathy cases in nonalcoholic patients.10 Despite appropriate treatment, measureable memory deficiency may persist for up to 2 years in patients with Wernicke encephalopathy.9 Case 7-2 describes MRI findings detailing specific changes in a patient with Wernicke encephalopathy.
Korsakoff confabulatory amnestic syndrome. Korsakoff syndrome is another CNS syndrome resulting from thiamine deficiency in alcoholics. As with Wernicke encephalopathy, its epidemiology, including the proportion with antecedent Wernicke encephalopathy, is not well known; cases of undiagnosed Wernicke syndrome can progress to Korsakoff syndrome. Korsakoff syndrome is distinguished from acute Wernicke syndrome by prominent anterograde and retrograde amnesia without substantially impaired alertness and attention or extraocular movement disturbance. Manifestations include anterograde amnesia, the impaired ability to acquire or retain new information; prominent confabulation is produced because of the inability to recall even a brief, simple story or recent information. Retrograde amnesia is identified by the inability to recall elements of both recent and distant biographical information, often in an unpredictable manner. Patients may have a significant degree of vacuous spontaneous speech and abulia that may be mistaken for depressive symptoms when inadequately explored. Current evidence suggests the syndrome may result when both the thalamus (particularly the anterior thalamic nucleus) and hypothalamus (medial mammillary nucleus) are injured, but other cortical and subcortical areas modulate this process.11 With appropriate therapy, which includes parenteral thiamine, confabulatory elements of the syndrome may resolve, although the amnestic syndrome persists. The syndrome has devastating effects, and up to 25% of patients require institutionalization.9
Alcoholic-related dementia. With advancing age, patients who are chronic alcoholics may develop cognitive impairment or dementia without demonstrable micronutrient deficiency. Neuropathologic hallmarks are few and thus contribute to an underappreciation of chronic alcohol effects in autopsy series of patients with dementia and likely to controversy about whether this disorder exists at all or is instead simply synergistic in the expression of other cognitive disorders of aging. Nonetheless, at least one study of middle-aged adult men suggested excessive alcohol intake (≥36 g/d ethanol) was associated with 2 to 6 years of additional cognitive decline beyond changes of aging alone.12 Cognitive changes are thought to relate to recurrent alcohol binges impacting GABA-mediated (GABAergic) inhibition, resulting in neurotoxic glutamatergic excitation that leads to hippocampal and neocortical neuronal loss.13 Some studies suggest reduced cortical and subcortical brain volumes in chronic alcoholics, which may be more likely to occur among aged alcoholics but may recover partially during periods of sobriety.14 Additional challenges in establishing the diagnosis include the effects of micronutrient deficiencies, hepatic insufficiency, and trauma, which often coexist in patients with dementia associated with alcoholism. Patients with alcoholic dementia may have symptoms that overlap with other common neurodegenerative cognitive disorders such as Alzheimer disease, frontotemporal dementia, dementia with Lewy bodies, or vascular dementias. Clinically, patients with alcohol-related dementia typically present at an earlier age than other acquired or late-onset forms of dementia. Moreover, current neuropathologic criteria may bias patients with primarily alcohol-related dementia to be diagnosed with neurodegenerative disease when some changes of the latter are evident at autopsy.
While no specific neuropsychological profile associated with alcohol-related dementia exists, in contrast to the more common neurodegenerative dementias that have focal impairment in one or more of the cognitive domains (ie, attention, processing speed, memory, language, and/or visuospatial function), patients with alcohol-related dementia more typically have a rather globally impaired neuropsychological profile, with similar impairment in most or all domains even in early disease stages. Further clouding the clinical syndrome may be depression or apathy blamed on the alcoholism itself but which does not appreciably resolve once abstinence is achieved. As is often the case with neuropsychological testing in neurodegenerative disorders, once dementia is clinically moderate to severe, the reliability of neuropsychological testing to distinguish one form of dementia from another declines. The evaluation of patients should be the same as an evaluation of any patient with dementia, including excluding reversible causes and attempting to provide specific diagnoses whenever possible.
Marchiafava-Bignami disease. Marchiafava-Bignami disease is a rare acute to subacute disorder that most often occurs in chronic alcoholics. The syndrome consists of altered mental status, impaired gait, loss of consciousness, dysarthria, amnesia, and cortical disconnection syndrome that all relate to specific corpus callosum involvement, with particular involvement of the splenium. Its earliest descriptions were pathologic and thus likely represented disease extremes. The disease is strongly associated with thiamine deficiency (and recovery may therefore occur with early recognition and repletion) and can be misidentified as other syndromes. Involvement of the splenium may be related to myriad toxic-metabolic etiologies of splenium injury independent of alcohol.15 MRI hallmarks more suggestive of Marchiafava-Bignami disease include diffusion-weighted imaging changes in multifocal regions or the entirety of the corpus callosum (Figure 7-316).17
Figure 7-3.
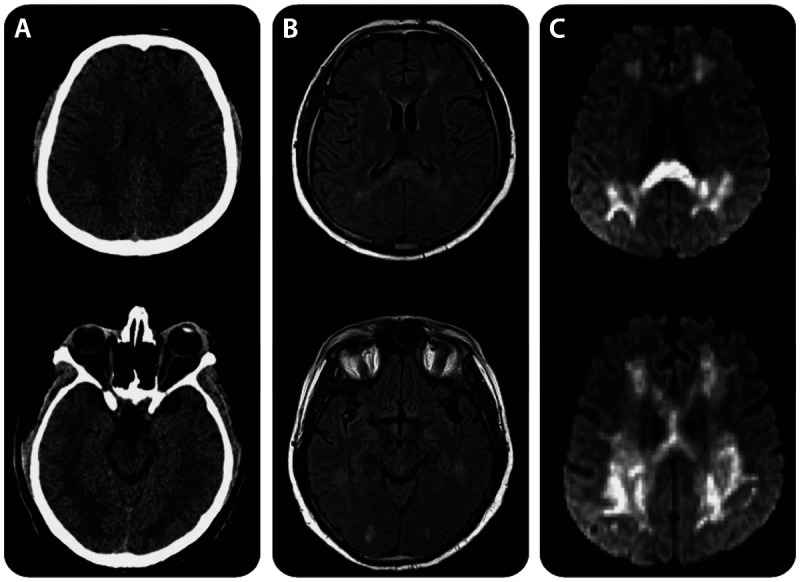
CT and MRI features of Marchiafava-Bignami disease. A, Axial CT showing symmetric hypodense white matter lesions. B, Fluid-attenuated inversion recovery (FLAIR)–weighted MRI depicting hyperintense white matter lesions predominantly involving the splenium of the corpus callosum. C, Diffusion-weighted images revealing marked restriction with corresponding low apparent diffusion coefficient values. The mammillary bodies and the periaqueductal region appear normal. Reprinted with permission from Tozakidou M, et al, Neurology.16 © 2011, American Academy of Neurology. www.neurology.org/content/77/11/e67.long.
Cerebellar degeneration. Cerebellar degeneration may occur in alcoholics with or without micronutrient deficiency states, such as thiamine deficiency. The process is thought to be the most common CNS complication of alcoholism, affecting 10% to 25% of alcoholics.18,19 The distinction between acute thiamine deficiency and this syndrome is made on clinical grounds; both Wernicke syndrome and cerebellar degeneration may produce acute to subacute manifestations, but recovery is less likely in cerebellar degeneration despite treatment. Some studies have suggested that a relative thiamine deficiency may be necessary to cause clinical cerebellar dysfunction in alcoholics.20 Most typically, the superior cerebellar vermis is principally involved, with multilayer neuronal loss (particularly affecting Purkinje cells) and cerebellar white matter loss.20 The number of years of heavy alcohol abuse may be the strongest single determinant of alcoholic ataxia development.21 Treatment of this disorder, aside from thiamine and other micronutrient repletion, is largely supportive once developed.
Case 7-1
A 48-year-old right-handed man presented to the local emergency department with a seizure 1 day after his last drink. This generalized tonic-clonic seizure was the third of his lifetime, and each one had occurred in the context of abrupt attempts at sobriety following 30 years of alcoholism. Although he was no longer seizing upon arrival of emergency medical services, he was given a short-acting benzodiazepine in the field. Several hours later, upon assessment in the emergency department, his neurologic examination was normal aside from mild fatigue. He was given parenteral thiamine, diagnosed with a symptomatic urinary tract infection, and triaged to a general hospital bed. His first 3 hospital days were rather uneventful aside from mild tremor in the hands. On the morning of his fourth hospital day, he was found to appear internally preoccupied and minimally conversant with hospital staff. Swings in blood pressure and pulse then developed. A treatment protocol for alcohol withdrawal, including a titrating schedule of benzodiazepines, was implemented, and over the next 4 days he had close cardiac and neurologic ICU monitoring. Subsequently, benzodiazepines were slowly tapered, counseling and outpatient preventive care were arranged, and long-term alcohol abstinence encouraged.
Comment. Alcohol withdrawal syndromes can be insidious and often ascribed to alternative diagnoses if alcohol histories are not fully divulged upon initial hospital presentation. In particular, delirium tremens can begin several days into hospitalization, at a time when the initial, perhaps incorrect, diagnoses warranting hospitalization are either partially or completely treated. Careful attention to early autonomic signs, which would be distinct from urosepsis (ie, which may occur with inadequately treated urinary tract infections) may assist in establishing a timely diagnosis of delirium tremens. A careful, open-ended alcohol history is very important, particularly among patients who may be unable to provide complete histories upon initial presentation.
Case 7-2
A 51-year-old man with a history of chronic alcoholism and colon adenocarcinoma with local intraabdominal metastases presented to the emergency department following several days of blurred vision followed by confusion and somnolence. Review of additional medical history revealed that he had had three prolonged hospitalizations over the past 6 months for ileus and failure to thrive. In retrospect, a nationwide shortage of parenteral multivitamins limited his micronutrient intake to the rare days when he either briefly tolerated a liquid diet or received total parenteral nutrition, and he did not receive IV thiamine. Examination demonstrated disorientation to time and place, shortened attention span including impaired registration of unrelated words intended for subsequent recall, and amnesia without confabulation. Additionally, he had horizontal and vertical gaze–evoked nystagmus and weakness of right eye abduction and weakness of left eye elevation, as well as a distal symmetric sensory loss and diminished patellar and Achilles reflexes. MRI revealed T2 and FLAIR hyperintensities in the periaqueductal gray (Figure 7-2A and B), midbrain tectum (Figure 7-2C), and mammillary bodies (Figure 7-2D). Brain MRI performed 6 weeks previously (for an episode of transient confusion in the context of urosepsis) revealed no abnormalities. No signal changes were evident on contrast-enhanced brain MRI; EEG revealed mild slowing, and CSF analyses were normal. High-dose IV thiamine repletion was begun. Follow-up brain MRI performed 10 days after presentation (and 10 days of IV thiamine repletion) supported imaging resolution of the syndrome. His cognition gradually improved, and was unremarkable 1 month after thiamine repletion; however, fixed neuro-ophthalmic deficits, including right abducens paresis, remained at that time.
Figure 7-2.
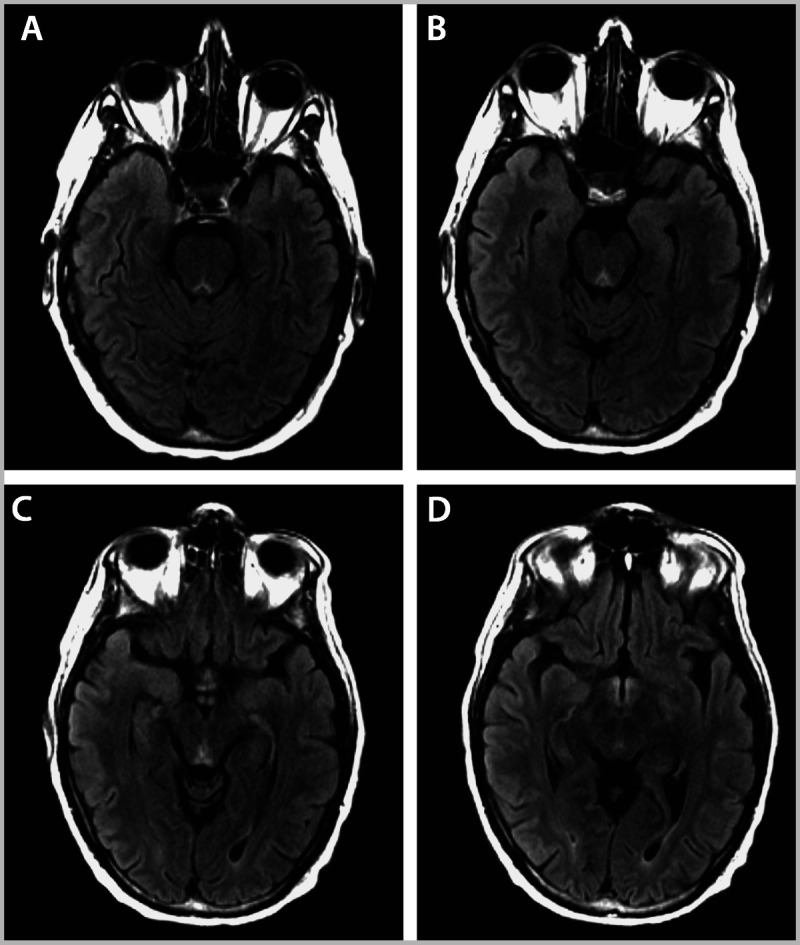
Brain MRI of a patient with Wernicke syndrome, as detailed in Case 7-2. MRI reveals T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the periaqueductal gray (A, B), midbrain tectum (C), and mammillary bodies (D).
Comment. This case underscores the often complex factors, including medical comorbidities and prolonged, obfuscated, or underappreciated histories, that may cloud or delay the diagnosis of Wernicke encephalopathy, which is easily treated once recognized. Given minimal risk associated with empiric thiamine treatment, intravenous thiamine should be given immediately upon first clinical suspicion of Wernicke encephalopathy and should not await imaging or laboratory confirmation.
PERIPHERAL NERVOUS SYSTEM COMPLICATIONS OF ALCOHOLISM
Chronic Complications
Chronic alcoholic neuropathy. The association of peripheral nerve disease and ethanol use has been recognized for centuries, and painful sensory neuropathy in association with alcoholism was recognized in the late 1700s.22 However, the question of the cause—due to ethanol as a putative direct nerve toxin or due to nutritional deficiency or both explanations—was debated until recently. Malnutrition and vitamin deficiency are common in chronic alcoholics, and thiamine has been the primary focus of investigation. Unlike Wernicke syndrome, a clear association between reduction of thiamine levels or thiamine-mediated enzyme activity (transketolase) and alcoholic peripheral neuropathy has not been conclusively established. Ethanol does appear to directly interfere with retrograde axonal transport in some animal models,23 although demonstration of such changes in primate models has been limited. Differences in thiamine levels or enzyme activity between alcoholics with and without neuropathy have not been consistently identified, even though alcoholic neuropathy patients often do have reduced levels of various vitamins. However, the vitamin-deficient syndromes are now recognized to be distinct, on the basis of clinical and laboratory features, from alcoholic neuropathy patients with normal thiamine levels. In short, chronic, well-fed alcoholics without vitamin deficiency primarily develop slowly progressive sensory loss affecting small-fiber-mediated functions, especially nociception;24 pain and burning paresthesia are common in this group, but not ataxia or weakness from neuropathy. Primary nonalcoholic thiamine deficiency–associated neuropathy, in contrast, more typically produces prominent subacute weakness and sensory ataxia from large-diameter more than small-diameter fiber sensory neuropathy. Patients with both alcohol exposure and thiamine deficiency demonstrate a mixture of findings that lead to similar treatment paradigms addressing both states.25
Clinical and diagnostic features. Peripheral distal sensorimotor neuropathy is a distal axonopathy that is a common finding in alcoholic patients (in upward of 90% of alcoholics if both electrodiagnostic criteria and clinical symptoms are combined).26 Because of additional medical and neurologic complications related to alcoholism, clinical signs and symptoms of neuropathy may be overlooked by the patient and clinician; likewise, patients may disregard minor paresthesia or anesthetic areas until significant pain or gait difficulties evolve. When identified, alcoholic neuropathy is indistinguishable from other distal sensorimotor axonal processes. As with many other etiologies, symptoms typically begin with distal paresthesia in the feet and slowly progress proximally. In most cases, the onset is typically slow and insidious and may begin to affect the hands once leg symptoms ascend well above ankle level, thus yielding the classic symmetric stocking-glove sensory pattern. Ankle deep tendon reflexes are typically lost at a relatively early stage. Distal weakness and atrophy are usually late findings following sensory disturbance and are less profound, with weakness that may be limited to toe extensors. Gait may become unstable from sensory ataxia once proprioception is significantly affected. Gait disturbance due to sensory ataxia may be difficult to distinguish from, or be concomitant with, alcoholic cerebellar degeneration. Paresthesia is usually mild to moderate in severity but can become quite unpleasant or even frankly painful. Although patients may initially present with hand dysesthesia, more commonly hand symptoms follow anesthesia in the legs, which may be otherwise unrecognized or overlooked until more bothersome symptoms evolve. Autonomic symptoms, including orthostatic hypotension, impotence, incontinence, hyper- or hypohidrosis, and peripheral vasomotor dysfunction, are not uncommon among alcoholics.27 Trophic skin changes, such as thinning, glossiness, hair loss, hyperpigmentation, and impaired sweating are common in affected distributions. Neuropathic “Charcot” joints, a traumatic arthropathy typically of the ankle, may develop in advanced cases with severe loss of nociception as the patient forcefully strikes the ground to perceive placement of footing leading to joint destruction. Autonomic signs are difficult to demonstrate at the bedside unless frank orthostatic hypotension is present. Case 7-3 details a typical presentation.
Evaluation includes identifying laboratory abnormalities supporting alcohol abuse when the history is not otherwise clear; these findings may include abnormal liver function tests and red cell macrocytosis. Thiamine levels are not consistently reduced, but the thiamine-mediated enzyme transketolase is measured in some laboratories. CSF is typically normal or shows a mildly raised total protein. Electrodiagnostic testing shows typical evidence of an axonal sensorimotor neuropathy. Sensory distal amplitudes are reduced, or potentials are unrecordable. Motor evoked amplitude may be reduced, but to a lesser degree. Distal latency, conduction velocity, and minimum F-wave latency (when present) are normal or consistent with the degree of axonal loss and show no signs of demyelination. H reflexes are absent at an early stage and correlate with absent ankle reflexes. Patients with alcoholism may have behaviors, such as prolonged immobility or adverse body positions, that put them at an increased risk of compression neuropathy, and electrodiagnostic findings can be complicated if superimposed traumatic or compressive mononeuropathies are present. Autonomic testing of parasympathetic and sympathetic reflexes is often abnormal, including analysis of heart rate variability, Valsalva maneuver, handgrip, tilt table, and standing maneuvers.
Methods of demonstrating small-diameter fiber neuropathy, such as skin biopsy for epidermal nerve fiber density, can support the sensory involvement and infer autonomic small fiber involvement in alcoholic neuropathy.26,28 Sural nerve biopsy shows evidence of generalized distal axonal loss affecting both large and small fibers but without distinctive pathologic features and is not typically indicated.22 Open nerve biopsy is not recommended in this setting unless reasonable suspicion of a separate process with distinctive pathologic features, such as vasculitis, inflammatory neuropathy, or amyloidosis, exists.
Treatment and prognosis. Cessation from ethanol is paramount to improvement, as it is for disorders of CNS involvement. Despite apparently adequate nutrition, multivitamin supplements and thiamine are indicated for all alcoholic neuropathy patients; however, vitamin supplementation alone in the setting of ongoing ethanol use has not been convincingly shown to be sufficient for improvement in most patients. Long-term follow-up of reformed alcoholics demonstrates that significant improvement of alcoholic neuropathy is possible, although often incomplete. Patients with mild to moderate neuropathy can significantly improve,27 but the improvement is usually incomplete in those with severe findings.
Disulfiram (Antabuse) neuropathy. Disulfiram blocks the oxidation of alcohol at the acetaldehyde stage, leading to accumulation of acetaldehyde and the characteristic disulfiram-alcohol reaction after ethanol ingestion. Although disulfiram has been largely replaced by the non-neurotoxic agents naltrexone and acamprosate for treating alcohol dependence,29 it is still used as a drinking deterrent in many countries outside the United States. A minority of patients receiving chronic disulfiram develop an axonal neuropathy,30 which appears to be dose-related; higher doses cause both a shorter-onset latency and more severe findings. The pathophysiology of disulfiram neurotoxicity remains uncertain. Onset is usually within weeks to several months, and the majority occur within 3 months. Acute or fulminant cases are described following an overdose, especially if combined with ethanol. Neuropathy may progress quickly, especially at higher doses, and progression is typically more rapid than alcoholic polyneuropathy. A stocking-glove pattern of distal paresthesia and numbness predominantly affects large fiber modalities. Weakness ensues and is much more prominent than that for alcoholic neuropathy; reflexes are diminished or lost distally depending on severity. Improvement after cessation of disulfiram is typically seen, but prognosis is related to the severity and the degree of axonal loss.
Chronic alcoholic myopathy. The detrimental effects of alcohol on skeletal muscle have been known for centuries but only formally described relatively recently.31 Alcoholic muscle disease is estimated to chronically affect roughly 2% of all adults in Western countries,32 which would make this entity arguably the most common disorder of skeletal muscle. In contrast to the unusual bouts of acute myoglobinuria and rhabdomyolysis in the alcoholic, development of chronic, painless, proximal weakness with atrophy is common. Biopsy-based studies of hospitalized patients suggest a prevalence upward of 60% in alcoholics with at least a 3-year history of heavy alcohol abuse.32 Female alcoholics may be more prone to chronic cardiac and skeletal muscle complications despite lower lifetime exposure.33
Histopathologically, muscle necrosis is not typically seen. The pathologic changes of chronic alcoholic myopathy can be clinically helpful, but they are not specific. The most prominent finding is atrophy of type II fibers, which contain a higher content of glycolytic enzymes. Type IIb fibers, which have the highest dependence on glycolysis, show the most prominent atrophy. Other nonspecific myopathic changes include “moth-eaten” fibers (ie, fibers with multiple patches of decreased or absent oxidative enzyme activity). Type II atrophy is not specific for the effects of ethanol and is also a feature of glucocorticoid excess, disuse atrophy, hypothyroidism, hypophosphatemia with or without osteomalacia, and some cases of critical illness myopathy. Loss of the contractile elements with preservation of other organelles is seen. Signs of neuropathic change, such as fiber type grouping, are not generally seen in proximal muscles unless another process is superimposed. Although the pathogenesis of alcoholic myopathy is still unknown, evidence points to separate and direct toxic effects of ethanol and possibly the metabolite acetaldehyde. While other important confounding factors, such as poor nutrition and hepatic impairment, play a role in individual patients, neither is necessary for myopathy development in clinical and animal studies. Several models of the effects of ethanol on muscle have been proposed (Table 7-1).32,34,35
Table 7-1.
Mechanisms of Alcoholic Muscle Injury

Clinical and diagnostic features. Myopathy in the presence of a chronic, high ethanol intake is needed for diagnosis. Because of similar challenges and factors relevant to chronic alcoholics with regard to peripheral neuropathy, symptoms and signs of myopathy may be overlooked. Proximal weakness develops in an indolent manner over many months. Muscle pain is typically minimal; occasional cramps are noted. Proximal weakness is seen, sometimes accompanied by severe muscle wasting and generalized loss of muscle mass (up to 30%). In chronic myopathy, myoglobinuria is absent, and creatine kinase (CK) is normal, reduced, or mildly elevated, unless an acute myopathy is superimposed. Cardiomyopathy is also commonly present when skeletal muscle is affected. Other associations with alcoholism, such as malnutrition, liver dysfunction, vitamin deficiencies, hormonal alterations, and phosphate deficiency, are independent factors for alcohol myopathy development. Recurrent bouts of acute myoglobinuria are not the cause of chronic myopathy.
Electrodiagnostic studies commonly, but not exclusively, show evidence of coexisting peripheral neuropathy. Proximal muscle needle EMG typically shows short-duration polyphasic motor unit potentials and “early” myopathic recruitment (full interference pattern in a weak muscle). Signs of proximal denervation have been reported, but abundant spontaneous activity typical of acute alcoholic myopathy is not prominent.
Treatment and prognosis. Prolonged abstinence can improve clinical weakness and is associated with significant improvement in pathologic hallmarks31; however, some evidence suggests that heaviest drinkers are unlikely to demonstrate significant improvement in strength. Some alcoholics are able to successfully limit but not eliminate alcohol despite their best efforts; such “controlled” drinking can still permit recovery of strength.36
Case 7-3
A 40-year-old woman with a history of hypothyroidism reported paresthesia in her feet for the past few months and described more noticeably painful sensations in her hands over the past few weeks. These hand symptoms were debilitating and affected typing and collating files at work. She admitted to moderate but consistent alcohol use for many years. She also noted trouble feeling the cold on bathroom tiles and nearly fell several times in the middle of the night when ambient lighting was low. She had occasional muscle cramps but no significant muscle pain. She denied bouts of muscle pain or swelling or a change in urine color. On examination her strength was normal except for Medical Research Council (MRC) grade 4/5 in her extensor hallucis longus muscles bilaterally. Sensory examination was notable for profound loss of vibration and temperature sensation in the feet and to a slight degree in her hands. She had no ataxia or dysmetria. Reflexes were 1+ throughout except for absent ankle jerks. Gait was normal, and she could tandem for several steps, although an examiner needed to catch her during the Romberg test. Laboratory tests revealed normal electrolytes and mildly increased transaminase levels. Creatine kinase was 145 IU/L (normal up to 195 IU/L). Screening laboratory testing for identifiable causes of neuropathy was negative. Electrodiagnostic studies supported axonal sensorimotor neuropathy.
Comment. The case highlights the often delayed nature of symptom reporting in alcoholic patients with profound neuropathy. Symptoms may only be reported when bothersome in the course of hobbies or employment. Although this patient does not have frank cerebellar involvement, the etiology of ataxia can be difficult to discern once both large fiber sensory neuropathic changes and cerebellar dysfunction are evident.
Acute Complications
Acute alcoholic neuropathy. The entity of an acute alcoholic neuropathy has been debated for years. Rare cases have been reported of alcoholics with severe acute or subacute neuropathy that mimics Guillain-Barré syndrome.37 Biopsy and electrodiagnostic data show an axonal pattern (not demyelinating) with normal CSF protein. A causal but unproven association with ethanol exists, and most cases have no report of thiamine levels.
Alcohol-associated compressive neuropathies. Susceptibility to compression is a feature of most axonal neuropathies, and acute radial neuropathy, or Saturday night palsy, in alcoholics is well popularized. In this scenario, the radial nerve at the spiral groove is compressed when the arm is draped over the back of a chair or otherwise compressed during a long, deep, drunken sleep. However, alcoholics with a generalized peripheral neuropathy are prone to compression neuropathy at many different sites, including ulnar neuropathy at the elbow, radial or axillary nerve injury in the axilla (crutch-type compression), and fibular (peroneal) neuropathy at the fibular head. Other examples include the superficial radial nerve of handcuffed alcoholics, yielding a patch of superficial radial numbness.
In an acute compressive lesion, electrodiagnostic studies are helpful for both diagnosis and prognosis. Demonstration of a conduction block across the site of compression after 5 to 10 days is a favorable prognostic sign and implies a degree of neurapraxia and short-term improvement over days to weeks. If significant axonal injury occurs (axonotmesis), Wallerian degeneration follows, and the nerve must regenerate by collateral sprouting or axonal regrowth from the distal stump over many months. Older patients with coexisting peripheral neuropathy have a less favorable outcome in this setting.
Acute alcoholic myopathy. In addition to chronic myopathic changes, alcohol is associated with an acute myopathic syndrome with distinguishing clinical hallmarks, particularly for improvement. Alcohol is thought to have two acute effects on skeletal muscle including (1) inhibition of sarcolemmal calcium channels thus limiting influx of calcium and (2) compromise of sarcolemmal integrity. Increases in plasma CK follow alcohol ingestion and exercise. Muscle fiber necrosis and phagocytosis are seen, and the muscle fiber subsequently regenerates. This pattern is the same as with other causes of rhabdomyolysis; however, type I fibers appear to be particularly vulnerable38; in more severe cases, necrosis is widespread and affects all fiber types. The distribution of muscle breakdown should be diffuse and at the same stage of development. However, despite promising in vitro evidence, in vivo models have failed to elucidate these mechanisms,39 and the etiology of this form of muscle breakdown is not definitively known. Several mechanisms have been proposed (Table 7-2).
Table 7-2.
Mechanisms of Alcohol-Induced Muscle Necrosis

Clinical and diagnostic features and treatment. The spectrum of acute myopathy ranges from asymptomatic ultrastructural or electrodiagnostic changes and a raised serum CK to severe rhabdomyolysis and myoglobinuria leading to renal failure. A rise in serum myoglobin typically precedes the CK rise, and myoglobinuria usually occurs with a serum level over 250 µg/mL (normal <5 ng/mL). Estimates of the incidence vary widely depending on the criteria used and patient groups analyzed.32,34 When considering myoglobinuria, the incidence is roughly 1%,40 but when considering a transient rise in CK alone, incidence is upward of 78%. Ethanol is a common and important cause of rhabdomyolysis; estimates are that alcohol is the underlying cause of 20% or more of recognized cases of rhabdomyolysis in some series.
Acute alcoholic myopathy typically develops over hours to days following a recent binge. Most cases of the syndrome are asymptomatic, but when apparent, the syndrome consists of painful, swollen muscles, variable weakness, usually with myoglobinuria (as evidenced sometimes by tea- or cola-colored urine), and markedly elevated CK that normalizes within 1 to 2 weeks. The distribution of pain and weakness is typically proximal; however, regional or even focal involvement can occur, as detailed in Case 7-4. Local calf pain and swelling can be confused with thrombophlebitis. Bulbar muscle involvement can produce dysphagia. Muscle destruction may be enhanced by fasting, which commonly occurs in binge drinking. Attacks can be recurrent, correlating with additional episodes of heavy drinking. Recovery following cessation of drinking and repletion of electrolytes is usually rapid and dramatic. Despite repeated episodes, strength typically returns to normal unless a chronic myopathy or other complications are superimposed.
An elevated serum CK may be the only sign in subclinical cases. In more severely affected patients, myoglobinuria can lead to acute tubular necrosis and renal failure. Electrodiagnostic studies can corroborate an acute myopathic process but are not specific for alcohol-associated myopathy. These corroborating features include fibrillations and positive sharp waves due to segmental muscle necrosis, which isolates portions of muscle from the endplate zone after 10 to 21 days; myopathic motor unit changes; and early recruitment.
Case 7-4
A 48-year-old man with a history of hyperlipidemia and chronic ethanol abuse presented with 1 day of severe progressive muscle cramps and pain in both legs. Generally, he was relatively well nourished, but he had been on a recent binge and had not eaten for 2 days during the past week. He was attempting sobriety and had not had a drink in 2 days. On examination, his mental status was normal, including appropriate affect, orientation, and recall. He had mild tremulousness with his hands outstretched and difficulty holding a cup of water still enough to drink. His calves were mildly swollen and thighs tender; his right deltoid was noticeably sore, as well. Proximal leg strength was 4 to 4/5 and 4+ more distally. Reflexes were mildly depressed diffusely, and absent at the ankles. Urine was described as cola colored. Selected laboratory values included CK 7000 IU/L, potassium 3.7 mmol/L, creatinine 1.3 mmol/L, phosphate 3.1 mmol/L, and serum myoglobin 1250 μmol/L.
Comment. This acute, highly symptomatic syndrome is acute alcohol myopathy. Patients with alcohol-related neurologic problems often have several coincident conditions. In this case, absent deep tendon reflexes suggest a peripheral neuropathy, and tremulousness suggests either an alcohol withdrawal tremor or a more chronic tremor associated with chronic alcohol abuse; both of these are expressed independently of his acute myopathy.
Conclusions
Alcoholism is a common health problem with myriad central and peripheral neurologic complications, including neuropathic and myopathic disorders. Given the high prevalence of alcoholism and its complications and a significant morbidity and mortality often masked by other medical complications associated with aging or alcoholism itself, thorough knowledge of this disease and prompt recognition of its scope of complications is important in providing effective neurologic care.
KEY POINTS
Excessive alcohol use is a common disorder; clarifying specific use patterns during the social history can best inform risk stratification, prevention, and treatment strategies.
Delirium tremens is potentially deadly and may begin after alcoholic patients are hospitalized for conditions unrelated to alcoholism; all patients with significant alcohol histories should be followed under a Clinical Institute Withdrawal Assessment of Alcohol Scale protocol during at least the initial hospitalization period.
Alcohol withdrawal syndromes may follow a linear order of presentation, with seizures and tremor preceding hallucinosis and delirium tremens; however, all syndromes may co-occur, particularly seizures during refractory delirium tremens.
Even when in doubt of the diagnosis of thiamine deficiency, begin parenteral thiamine repletion, as it may importantly address or prevent early Wernicke syndrome or concomitant neuropathy associated with thiamine deficiency.
Early thiamine therapy likely decreases the chances of a patient developing Korsakoff syndrome, which can be devastating and leads to institutionalization in one-quarter of all such patients.
The evaluation of patients with alcohol-related dementia should be the same as an evaluation of any patient with dementia, including excluding reversible causes and attempting to provide specific diagnoses whenever possible.
Alcohol and thiamine deficiency neuropathies are distinct neuropathologic entities but bear similar treatment approaches, including alcohol abstinence and thiamine repletion.
Chronic alcoholic neuropathy can have prominent paresthesia or even frank pain; in advanced cases, autonomic dysfunction may develop.
Sensory ataxia, when evident, significantly clouds clinical examination; cerebellar dysfunction may be challenging to distinguish from proprioceptive loss.
Chronic alcoholic myopathy is common among alcoholics, with hallmarks of painless proximal weakness and normal serum creatine kinase.
In alcoholism, chronic myopathic and neuropathic syndromes have shared risk factors and often coexist.
Acute alcoholic myopathy usually follows recent binge drinking but in contrast to chronic alcoholic myopathy can be painful, with elevated creatine kinase.
Both acute and chronic alcoholic myopathies can improve with prolonged abstinence from alcohol.
Footnotes
Relationship Disclosure: Dr Noble’s institution receives grants from the National Institutes of Aging and the National Institute of Dental and Craniofacial Research. Dr Weimer reports no disclosure.
Unlabeled Use of Products/Investigational Use Disclosure: Drs Noble and Weimer report no disclosures.
REFERENCES
- 1.Dawson DA,, Grant BF,, Stinson FS,, Chou PS. Toward the attainment of low-risk drinking goals: a 10-year progress report. Alcohol Clin Exp Res 2004; 28 (9): 1371–1378. [DOI] [PubMed] [Google Scholar]
- 2.Brust JC. Ethanol. In: Neurologic aspects of substance abuse. Boston: Butterworth-Heinemann, 2004. [Google Scholar]
- 3.Lee H,, Roh S,, Kim DJ. Alcohol-induced blackout. Int J Environ Res Public Health 2009; 6 (11): 2783–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnedt JT,, Rohsenow DJ,, Almeida AB, et al. Sleep following alcohol intoxication in healthy, young adults: effects of sex and family history of alcoholism. Alcohol Clin Exp Res 2011; 35 (5): 870–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng SK,, Hauser WA,, Brust JC,, Susser M. Alcohol consumption and withdrawal in new-onset seizures. N Engl J Med 1988; 319 (11): 666–673. [DOI] [PubMed] [Google Scholar]
- 6.Sarff M,, Gold JA. Alcohol withdrawal syndromes in the intensive care unit. Crit Care Med 2010; 38 (9 suppl): S494–S501. [DOI] [PubMed] [Google Scholar]
- 7.Brust JC. Ethanol and cognition: indirect effects, neurotoxicity and neuroprotection: a review. Int J Environ Res Public Health 2010; 7 (4): 1540–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Todd KG,, Hazell AS,, Butterworth RF. Alcohol-thiamine interactions: an update on the pathogenesis of Wernicke encephalopathy. Addict Biol 1999; 4 (3): 261–272. [DOI] [PubMed] [Google Scholar]
- 9.Thomson AD,, Marshall EJ. The natural history and pathophysiology of Wernicke’s Encephalopathy and Korsakoff’s Psychosis. Alcohol Alcohol 2006; 41 (2): 151–158. [DOI] [PubMed] [Google Scholar]
- 10.Zuccoli G,, Santa Cruz D,, Bertolini M, et al. MR imaging findings in 56 patients with Wernicke encephalopathy: nonalcoholics may differ from alcoholics. AJNR Am J Neuroradiol 2009; 30 (1): 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kril JJ,, Harper CG. Neuroanatomy and neuropathology associated with Korsakoff’s syndrome. Neuropsychol Rev 2012; 22 (2): 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabia S,, Elbaz A,, Britton A, et al. Alcohol consumption and cognitive decline in early old age. Neurology 2014; 82 (4): 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vetreno RP,, Hall JM,, Savage LM. Alcohol-related amnesia and dementia: animal models have revealed the contributions of different etiological factors on neuropathology, neurochemical dysfunction and cognitive impairment. Neurobiol Learn Mem 2011; 96 (4): 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenbloom MJ,, Pfefferbaum AMD. Magnetic resonance imaging of the living brain: evidence for brain degeneration among alcoholics and recovery with abstinence. Alcohol Res Health 2008; 31 (4): 362–376. [PMC free article] [PubMed] [Google Scholar]
- 15.Doherty MJ,, Jayadev S,, Watson NF, et al. Clinical implications of splenium magnetic resonance imaging signal changes. Arch Neurol 2005; 62 (3): 433–437. [DOI] [PubMed] [Google Scholar]
- 16.Tozakidou M,, Stippich C,, Fischmann A. Teaching neuroimages: radiologic findings in Marchiafava-Bignami disease. Neurology 2011; 77 (11): e67. [DOI] [PubMed] [Google Scholar]
- 17.Hillbom M,, Saloheimo P,, Fujioka S, et al. Diagnosis and management of Marchiafava-Bignami disease: a review of CT/MRI confirmed cases. J Neurol Neurosurg Psychiatry 2014; 85 (2): 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yokota O,, Tsuchiya K,, Terada S, et al. Frequency and clinicopathological characteristics of alcoholic cerebellar degeneration in Japan: a cross-sectional study of 1,509 postmortems. Acta Neuropathol 2006; 112 (1): 43–51. [DOI] [PubMed] [Google Scholar]
- 19.Lindboe CF,, Loberg EM. The frequency of brain lesions in alcoholics. Comparison between the 5-year periods 1975–1979 and 1983–1987. J Neurol Sci 1988; 88 (1–3): 107–113. [DOI] [PubMed] [Google Scholar]
- 20.Harper C. The neuropathology of alcohol-related brain damage. Alcohol Alcohol 2009; 44 (2): 136–140. [DOI] [PubMed] [Google Scholar]
- 21.Fitzpatrick LE,, Jackson M,, Crowe SF. Characterization of cerebellar ataxia in chronic alcoholics using the International Cooperative Ataxia Rating Scale (ICARS). Alcohol Clin Exp Res 2012; 36 (11): 1942–1951. [DOI] [PubMed] [Google Scholar]
- 22.Lettsom JC. Mem Med Soc London, 1792; 1: 128. [Google Scholar]
- 23.Malatova Z,, Cizkova D. Effect of ethanol on axonal transport of cholinergic enzymes in rat sciatic nerve. Alcohol 2002; 26 (2): 115–120. [DOI] [PubMed] [Google Scholar]
- 24.Koike H,, Iijima M,, Sugiura M, et al. Alcoholic neuropathy is clinicopathologically distinct from thiamine-deficiency neuropathy. Ann Neurol 2003; 54 (1): 19–29. [DOI] [PubMed] [Google Scholar]
- 25.Mellion M,, Gilchrist JM,, de la Monte S. Alcohol-related peripheral neuropathy: nutritional, toxic, or both? Muscle Nerve 2011; 43 (3): 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vittadini G,, Buonocore M,, Colli G, et al. Alcoholic polyneuropathy: a clinical and epidemiological study. Alcohol Alcohol 2001; 36 (5): 393–400. [DOI] [PubMed] [Google Scholar]
- 27.Monforte R,, Estruch R,, Valls-Sole J, et al. Autonomic and peripheral neuropathies in patients with chronic alcoholism. A dose-related toxic effect of alcohol. Arch Neurol 1995; 52 (1): 45–51. [DOI] [PubMed] [Google Scholar]
- 28.Singer W,, Spies JM,, McArthur J, et al. Prospective evaluation of somatic and autonomic small fibers in selected autonomic neuropathies. Neurology 2004; 62 (4): 612–618. [DOI] [PubMed] [Google Scholar]
- 29.Maisel NC,, Blodgett JC,, Wilbourne PL, et al. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction 2013; 108 (2): 275–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palliyath SK,, Schwartz BD,, Gant L. Peripheral nerve functions in chronic alcoholic patients on disulfiram: a six month follow up. J Neurol Neurosurg Psychiatry 1990; 53 (3): 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin F,, Peters TJ. Alcoholic muscle disease. Alcohol Alcohol 1985; 20 (2): 125–136. [PubMed] [Google Scholar]
- 32.Preedy VR,, Adachi J,, Ueno Y, et al. Alcoholic skeletal muscle myopathy: definitions, features, contribution of neuropathy, impact and diagnosis. Eur J Neurol 2001; 8 (6): 677–687. [DOI] [PubMed] [Google Scholar]
- 33.Urbano-Marquez A,, Estruch R,, Fernandez-Sola J, et al. The greater risk of alcoholic cardiomyopathy and myopathy in women compared with men. JAMA 1995; 274 (2): 149–154. [DOI] [PubMed] [Google Scholar]
- 34.Jung MK,, Callaci JJ,, Lauing KL, et al. Alcohol exposure and mechanisms of tissue injury and repair. Alcohol Clin Exp Res 2011; 35 (3): 392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez-Sola J,, Nicolas JM,, Fatjo F, et al. Evidence of apoptosis in chronic alcoholic skeletal myopathy. Hum Pathol 2003; 34 (12): 1247–1252. [DOI] [PubMed] [Google Scholar]
- 36.Fernandez-Sola J,, Nicolas JM,, Sacanella E, et al. Low-dose ethanol consumption allows strength recovery in chronic alcoholic myopathy. QJM 2000; 93 (1): 35–40. [DOI] [PubMed] [Google Scholar]
- 37.Rodrigues M,, Rocha S,, Machado A,, Guimaraes A. Is there really an acute alcohol-related axonal polyneuropathy? J Neuropsychiatry Clin Neurosci 2011; 23 (4): E31. [DOI] [PubMed] [Google Scholar]
- 38.Haller RG. Experimental acute alcoholic myopathy—a histochemical study. Muscle Nerve 1985; 8 (3): 195–203. [DOI] [PubMed] [Google Scholar]
- 39.Vella LD,, Cameron-Smith D. Alcohol, athletic performance and recovery. Nutrients 2010; 2 (8): 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Preedy VR,, Adachi J,, Peters TJ, et al. Recent advances in the pathology of alcoholic myopathy. Alcohol Clin Exp Res 2001; 25 (5 suppl ISBRA): 54S–59S. [DOI] [PubMed] [Google Scholar]