

Abstract
Purpose of Review
The metabolic myopathies result from inborn errors of metabolism affecting intracellular energy production due to defects in glycogen, lipid, adenine nucleotides, and mitochondrial metabolism. This article provides an overview of the most common metabolic myopathies.
Recent Findings
Our knowledge of metabolic myopathies has expanded rapidly in recent years, providing us with major advances in the detection of genetic and biochemical defects. New and improved diagnostic tools are now available for some of these disorders, and targeted therapies for specific biochemical deficits have been developed (ie, enzyme replacement therapy for acid maltase deficiency).
Summary
The diagnostic approach for patients with suspected metabolic myopathy should start with the recognition of a static or dynamic pattern (fixed versus exercise-induced weakness). Individual presentations vary according to age of onset and the severity of each particular biochemical dysfunction. Additional clinical clues include the presence of multisystem disease, family history, and laboratory characteristics. Appropriate investigations, timely treatment, and genetic counseling are discussed for the most common conditions.
INTRODUCTION
The metabolic myopathies represent a heterogeneous group of disorders of cellular metabolism characterized by insufficient energy production as a result of specific defects of glycogen, lipid, or mitochondrial metabolism. Myoadenylate deaminase deficiency, a disorder of nucleotide metabolism, has traditionally been considered as a metabolic myopathy; however, the high prevalence of this disorder in the general population (2%) and the documentation of completely asymptomatic individuals render its pathogenicity into question.1
From a clinical standpoint, the metabolic myopathies can be viewed as static or dynamic disorders.2 The first group includes patients with fixed symptoms, such as weakness, often associated with systemic involvement (eg, cardiomyopathy, endocrinopathy, encephalopathy). Patients with dynamic disorders exhibit symptoms and signs related to exercise (cramps, myalgias, exercise intolerance, myoglobinuria). Metabolic myopathies should be considered in the differential diagnosis of patients with exercise-induced muscle symptoms, static or progressive myopathy, isolated neuromuscular respiratory weakness, and muscle disease associated with systemic conditions. Common presentations vary with age of onset, with older children and adults frequently having exercise intolerance, weakness, and myoglobinuria, whereas newborns and infants tend to present with hypotonia and severe multisystem disorders.
The diagnostic evaluation for a suspected metabolic myopathy varies according to age of onset, presentation (dynamic versus static symptoms), and comorbid conditions, and may include blood tests (serum creatine kinase [CK], lactate, acylcarnitine profile, and amino acids), urine testing (organic acids and myoglobin), EMG, forearm exercise testing, muscle biopsy, biochemical analysis, genetic testing, and muscle magnetic resonance spectroscopy (MRS). It is important to remember that patients with exercise intolerance may have normal laboratory and EMG testing during interictal periods. In these cases, a careful evaluation of the type of exercise that triggers symptoms (high intensity for less than 10 minutes versus low intensity for more than 10 minutes) and associated precipitating factors can help to narrow the differential diagnosis (Figure 3-1).
Figure 3-1.
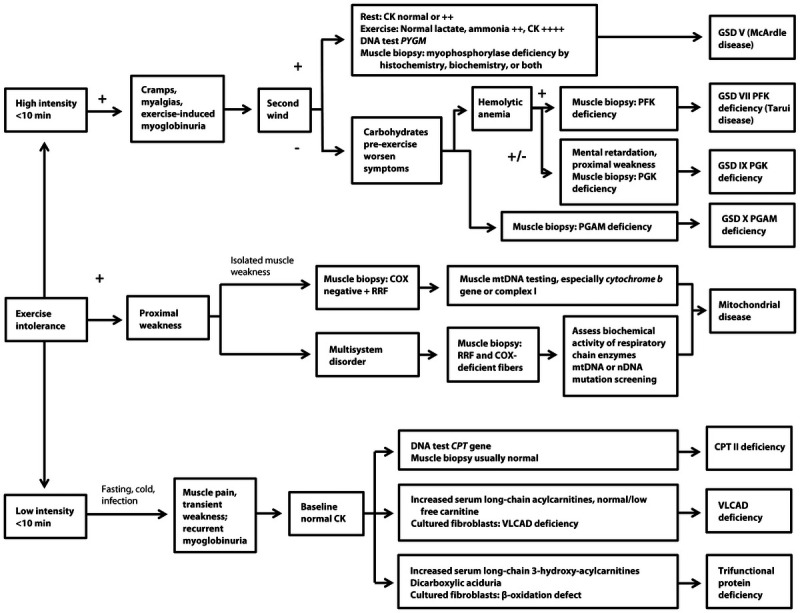
Clinical algorithm for patients with exercise intolerance in whom a metabolic myopathy is suspected. CK = creatine kinase; PYGM = muscle isoform of glycogen phosphorylase; PPL = myophosphorylase; GSD = glycogen storage disease; PFK = phosphofructokinase; PGK = phosphoglycerate kinase; PGAM = phosphoglycerate mutase; COX = cytochrome c oxidase; RRF = ragged red fibers; mtDNA = mitochondrial DNA; nDNA = nuclear DNA; CPT = carnitine palmitoyl transferase; VLCAD = very long chain acyl coenzyme A dehydrogenase. Modified from Berardo A, et al. Curr Neurol Neurosci Rep.2 © 2010, with permission from Springer Science + Business Media. link.springer.com/article/10.1007/s11910-010-0096-4.
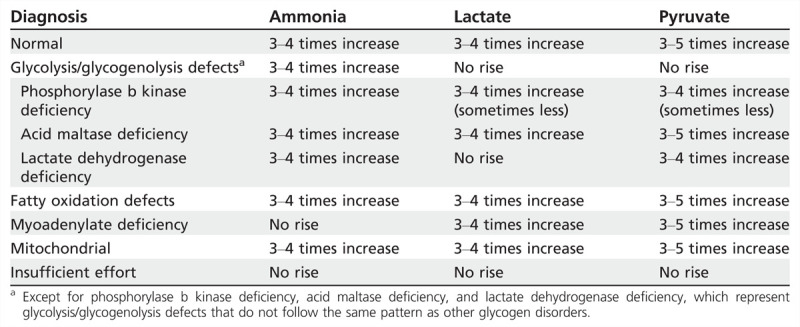
The forearm exercise test is a very helpful tool in the evaluation of patients with dynamic symptoms, particularly to differentiate those with defects of the glycolytic pathway. Several studies have shown that it can be performed in a safe and effective manner without need for ischemic conditions.3 Different protocols are available, but a common approach includes the insertion of a butterfly needle in the antecubital fossa. Baseline blood is drawn for ammonia, lactate, and pyruvate. The patient then opens and closes the hand rapidly and strenuously for 1 minute. Immediately after this and at 1, 2, 4, 6, and 10 minutes post exercise, further blood is drawn and levels of ammonia, lactate, and pyruvate are documented. The normal physiologic response is a threefold to fivefold rise above baseline in ammonia, lactate, and pyruvate. Common patterns seen in different metabolic disorders are shown in Table 3-1.
Table 3-1.
Forearm Exercise Test
Our knowledge of metabolic myopathies has expanded dramatically over the past decade, especially in the detection of specific genetic and biochemical defects of these disorders. New treatment options are now available for some of these conditions, making early recognition of paramount importance in order to reduce morbidity and mortality.
DISORDERS OF GLYCOGEN METABOLISM
Type II Glycogenosis
Glycogen storage disease (GSD) type II (acid maltase deficiency) is an autosomal recessive disorder caused by a deficiency of lysosomal α-glucosidase (Table 3-2). This enzyme cleaves 1,4 and 1,6 linkages in glycogen, and its deficiency results in glycogen accumulation (Figure 3-2). The threshold amount seems to vary depending on the organ, and the process by which skeletal muscle is eventually impaired is still not fully understood. Both atrophy and reduced performance by unit of muscle mass seem to have a role.4
Table 3-2.
Glycogen Storage Disorders Associated With Myopathies
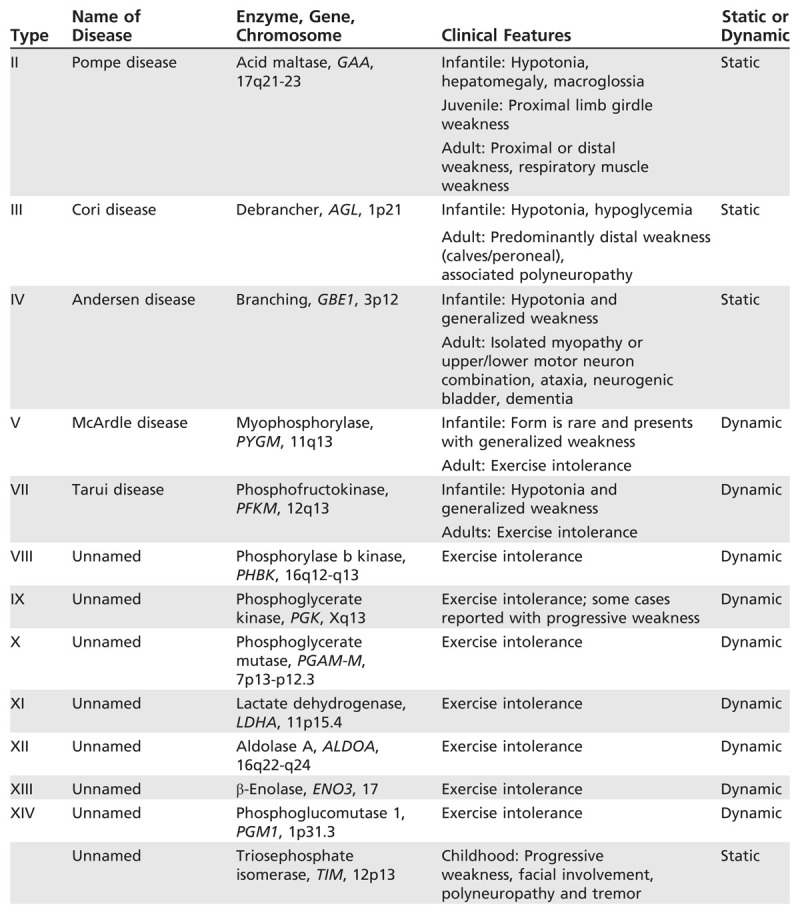
Figure 3-2.
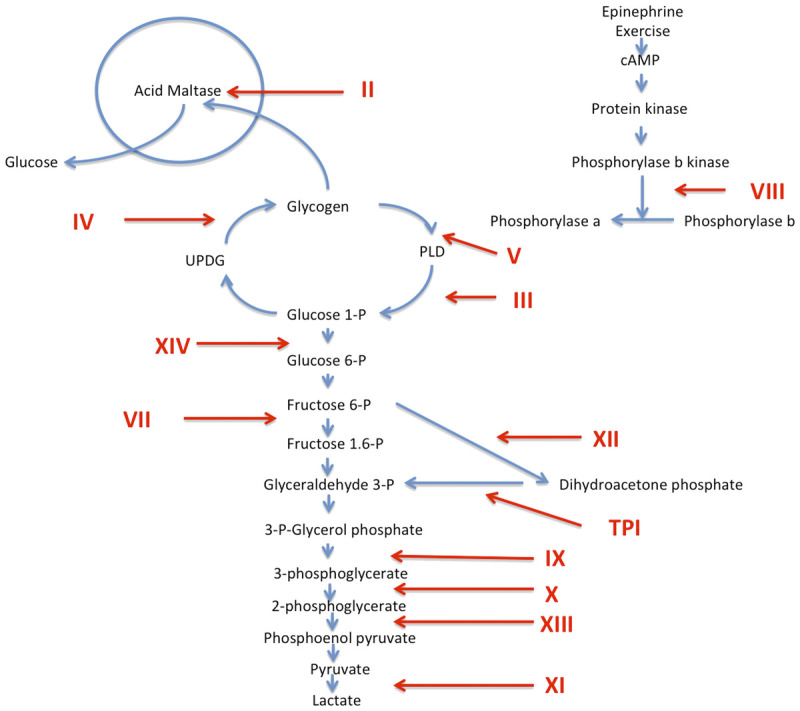
Glycolytic pathways. Glycogen storage diseases (GSDs) are designated with Roman numerals. GSD I and VI are not included since they do not cause muscle disease. The numerals denote defects in the following enzymes: II, acid maltase; III, debrancher; IV, brancher; V, myophosphorylase; VII, phosphofructokinase; VIII, phosphorylase b kinase; IX, phosphoglycerate kinase; X, phosphoglycerate mutase; XI, lactate dehydrogenase; XII, aldolase A; XIII, beta enolase, XIV, phosphoglucomutase 1. cAMP = cyclic adenosine monophosphate; UDPG = uridine diphosphoglucose; PLD = phosphorylase-limit dextrin; TPI = triose phosphate isomerase.
Clinical features. The disease has three phenotypical presentations: severe infantile form (Pompe disease), juvenile-onset type, and an adult-onset variant. The infantile form presents with cardiac symptoms, hypotonia, hepatomegaly, macroglossia, and failure to thrive. Respiratory muscle weakness and feeding difficulties are common. The disease is generally fatal by 2 years of age due to cardiorespiratory failure. The juvenile-onset form of GSD-II usually manifests during the first decade of life. Affected children have delayed gross motor development with proximal greater than distal limb-girdle weakness. Calf hypertrophy, waddling gait, and a positive Gower sign are commonly present. Cardiac and liver involvement is less common. Respiratory muscle weakness eventually develops, leading to death in the second or third decade of life.
Adult-onset acid maltase deficiency typically begins in the third or fourth decade of life (age range 18 to 65 years). Predominant proximal more than distal weakness is the main manifestation of the disease, similar to cases of polymyositis or limb-girdle muscular dystrophy (Case 3-1). Other presentations include scapuloperoneal weakness and rarely macroglossia. Hepatomegaly and cardiomegaly are usually not present; however, cardiac arrhythmias can develop. Involvement of the respiratory muscles is common and in some patients may represent the initial manifestation of the disease.
Case 3-1
A 50-year-old man presented to the clinic with progressive weakness affecting his legs more than his arms. He had always had difficulty going up and down the stairs and was never able to perform push-ups or pull-ups. He described a Gower sign when getting off the floor as a child. Weakness became more noticeable in his twenties, and by 40 years of age, he noticed difficulty breathing. He had no relevant personal or family medical history and was not taking any medications.
On examination, he displayed bilateral scapular winging with mild biceps and quadriceps atrophy. Mild calf hypertrophy was noted, and no clinical myotonia was seen on examination. He had proximal weakness (3/5) in the arms and legs as well as a mild waddling gait. Deep tendon reflexes were 1+ throughout, including biceps, triceps, patellar, and Achilles jerks. Plantar responses were flexor, and sensory examination was normal.
The serum creatine kinase was 750 U/L. Electrodiagnostic testing revealed normal nerve conduction studies with small-amplitude, brief-duration, polyphasic motor unit potentials and occasional myotonic discharges. α-Glucosidase activity (measured by the dried blood spot test) was 1.2 pmol/punch/hour (with normal being more than 10 pmol/punch/hour), and sequencing of the gene encoding α-glucosidase activity (GAA) confirmed a C.-32-13T>G mutation on chromosome 17.
Comment. This case illustrates that acid maltase deficiency can present with gradually progressive muscle weakness, often resembling a limb-girdle muscular dystrophy. The presence of myotonic discharges is not uncommon, especially over the paraspinal muscles. A dried blood spot measuring α-glucosidase activity can be obtained easily as a screening test, in some cases helping to reach a final diagnosis before more invasive procedures like muscle biopsy are performed. Acid maltase deficiency is one of the few metabolic myopathies with specific medical treatment and should not be missed.
Laboratory. Moderate elevations of the serum CK are common in all types of GSD-II, but occasionally adult patients can have normal levels. Findings from the forearm exercise test are normal. Motor and sensory nerve conduction study results are normal, and EMG often reveals increased insertional activity with fibrillations, positive sharp waves, complex repetitive discharges, and, not infrequently, myotonic discharges, especially in the paraspinal muscles. Motor unit potentials often display myopathic features with early recruitment. α-Glucosidase activity may be assayed in muscle fibers, fibroblasts, lymphocytes, and urine. A dried blood spot that measures α-glucosidase activity is the recommended screening test, followed by genetic testing if findings from the dried blood spot test are abnormal. ECG abnormalities can include left axis deviation, short PR interval, large QRS complexes, inverted T waves, and persistent sinus tachycardia. Wolff-Parkinson-White syndrome and hypertrophic cardiomyopathy have been reported. Pulmonary function tests frequently show reductions in forced vital capacity and maximal inspiratory and expiratory pressures.
Muscle biopsies have a pronounced vacuolar appearance, and periodic acid-Schiff (PAS) staining shows large deposits of glycogen in most fibers. The glycogen is digested by diastase, but some resistant material remains. The vacuoles also stain intensely to acid phosphatase, confirming that the vacuoles are secondary lysosomes (Figure 3-3). These vacuoles are very prominent in the infantile form, but in the childhood and adult forms they may be present in only 25% to 75% of fibers and could be absent in unaffected muscles. Free glycogen can be seen in the cytoplasm by electron microscopy; however, some of it may be lost during processing and the excessive amount is not always apparent.
Figure 3-3.
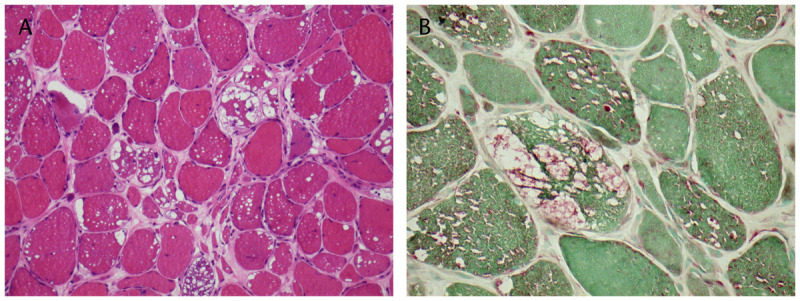
Acid maltase deficiency. Nonrimmed vacuoles in muscle fibers due to glycogen deposition. A, Hematoxylin and eosin stain; B, Acid phosphatase stain. Courtesy of Derek A. Mathis, MD.
Molecular genetics. GSD-II is inherited in an autosomal recessive manner. The gene encoding acid α-glucosidase (GAA) is localized to chromosome 17q25.2-q25.3 and contains 19 coding exons. The locus is very heterogeneous. More than 250 different missense, nonsense, and frame-shift mutations have been reported,5 and about 75% of these are considered pathogenic. C.-32-13T->G is the most common mutation in children and adults with a slowly progressive course of disease,6 while c.2560C>T is the most common mutation in the African American population. In general, α-glucosidase activity and clinical severity have an inverse correlation, but it is not considered 100% accurate.
Treatment. Alglucosidase alfa was approved in 2006 by the US Food and Drug Administration and the European Medicines Agency and became the first disease-specific treatment for childhood-onset Pompe disease. The Late-Onset Treatment Study in 20107 led to the approval of alglucosidase alfa for the treatment of late-onset Pompe disease in the United States. In this study, 90 patients ranging from 10 to 70 years old were randomized to receive biweekly infusions of alglucosidase alfa (20 mg/kg, based on body weight) or placebo. The two primary end points were distance traveled during a 6-minute walk test and the forced vital capacity percentage predicted in the upright position. At week 78, a modest but statistically significant improvement in favor of alglucosidase alfa was noticed in both parameters. Patients in the treatment and placebo group had similar adverse effects except for anaphylactic reactions that occurred only in patients treated with the medication. All alglucosidase alfa recipients tested negative for IgG anti–GAA antibodies at baseline and seroconverted by week 12. Patients treated with alglucosidase alfa who have persistently elevated titers should be closely monitored until the effect of the antibodies is more fully understood.
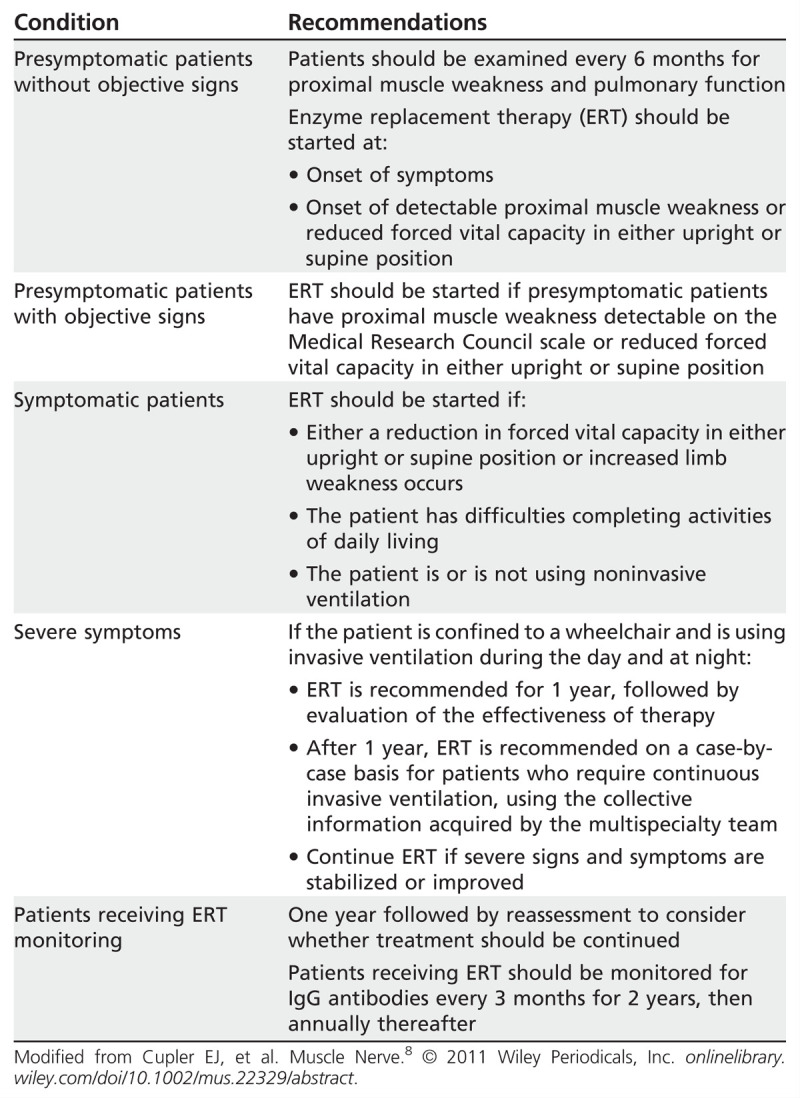
Consensus treatment recommendations based on stage and severity of Pompe disease have been recently published (Table 3-3).8 Overall, the clinical response observed in this trial and in subsequent studies9 suggests that prevention of further loss of muscle function is the target treatment goal for enzyme replacement therapy. Management of GSD-II patients should not only focus on enzyme replacement therapy but also include a multidisciplinary approach with emphasis on rehabilitation services, prevention and management of contractures and osteoporosis, and early recognition and treatment of cardiac, pulmonary, and gastrointestinal disease.
Table 3-3.
Treatment Recommendations Based on the Stage and Severity of Pompe Disease
Type III Glycogenosis
Debranching enzyme deficiency, also known as Cori disease or GSD-III, represents approximately 25% of all GSD. It is an autosomal recessive disorder due to a deficiency of amylo-1,6-glucosidase and 4-α-glucanotransferase enzymes, which degrade glycogen branches, releasing glucose in a two-step reaction catalyzed by its two distinct activities.
Clinical features. GSD-III has four clinical forms: GSD-IIIa, with lack of both glucosidase and transferase in liver and muscle; GSD-IIIb, with lack of both activities in liver only; GSD-IIIc, with selective loss of glucosidase activity; and GSD-IIId, with selective loss of transferase activity. Children with GSD-IIIa present with recurrent fasting hypoglycemia, seizures, hepatomegaly, decreased muscle tone, and growth retardation. Episodic hypoglycemia seems to resolve after puberty, and most patients have only minimal signs of liver disease afterward except for a small subgroup that may develop cirrhosis and hepatocellular carcinoma later in life. Adult-onset GSD-IIIa usually starts in the third or fourth decade with distal weakness, affecting calves and peroneal muscles predominantly, and variable proximal weakness with slow disease progression. A superimposed neuropathy can occur due to glycogen deposition in Schwann cells and axons. Rare cases of exercise intolerance have been reported, associated with cardiomyopathy.10 Respiratory weakness has been described, which sometimes can be very rapidly progressive.
Laboratory. CK elevations 2 to 20 times above normal levels are common. The forearm exercise test shows a normal increase in serum ammonia but no rise in lactate levels. ECG may reveal conduction defects, and echocardiogram may show findings suggestive of hypertrophic cardiomyopathy. Nerve conduction studies can be normal or show changes consistent with a mild sensorimotor polyneuropathy. Concentric needle EMG can demonstrate similar findings to those seen in acid maltase deficiency. Muscle biopsy reveals glycogen particles, which are PAS positive and digestible by diastase. Nonetheless, vacuoles do not stain with acid phosphatase, suggesting that glycogen does not primarily accumulate in the lysosome. Deficiency of debranching enzyme can be demonstrated with biochemical assay of muscle, fibroblasts, or lymphocytes.
Molecular genetics. The gene for debranching enzyme (AGL) is localized to chromosome 1p21, and six transcript isoforms have been isolated. Most GSD-IIIb patients have mutations in exon 3,11 whereas GSD-IIIa arises from downstream mutations.
Treatment. Specific therapy is not available for debranching enzyme deficiency. For management of hypoglycemic episodes during childhood, dietary measures have been recommended, with frequent daytime high-protein feedings and supplementation of uncooked cornstarch before sleep. Anecdotal cases of improved cardiac function after initiation of diet changes have been reported. Improvement of muscle symptoms with dietary changes has not been consistently proven.
Type IV Glycogenosis
GSD-IV, also known as Andersen disease, is an autosomal recessive disorder caused by branching enzyme deficiency. This enzyme catalyzes the last step in glycogen biosynthesis by transferring short glucosyl chains in α,1-6 glycosidic links to naked peripheral chains of nascent glycogen.
Clinical features. GSD-IV most commonly presents in children with failure to thrive, hepatosplenomegaly, and liver cirrhosis, leading to death at a young age. Neuromuscular symptoms vary according to the age of presentation with the following patterns: congenital, juvenile, and adult. The congenital form presents as fetal akinesia deformation sequence with multiple contractures (FADS) or as a severe congenital myopathy inconsistently associated with cardiomyopathy, often resembling Werdnig-Hoffman disease.12 The juvenile phenotype is dominated by myopathy or cardiomyopathy. The adult form can present as isolated myopathy or adult polyglucosan body disease characterized by a combination of upper and lower motor neuron syndromes, sensory nerve involvement, cerebellar ataxia, neurogenic bladder, and dementia.13
Laboratory. Serum CK can be normal or mildly elevated. ECG may show conduction defects, and echocardiogram may reveal a dilated cardiomyopathy. Nerve conduction studies are usually normal. Needle EMG demonstrates myopathic motor unit action potentials, occasionally with abnormal insertional activity. In adult polyglucosan body disease, an associated sensorimotor axonal polyneuropathy can be present, and EMG may reveal active denervation and reinnervation. Diagnosis is usually confirmed by determination of the branching enzyme activity in affected tissues. Muscle biopsy reveals deposition of PAS-positive, diastase-resistant filamentous polysaccharides (polyglucosan bodies). They are also found in the CNS, axons, Schwann cells, skin, liver, and cardiac muscle.
Molecular genetics. GSD-IV is an autosomal recessive disorder associated with mutations (deletion, nonsense, and missense) within the GBE1 gene localized to chromosome 3p12. More than 21 mutations have been identified, at least a quarter of which are localized to exon 12.14 There is significant phenotypic variability with differential expression of enzyme activity. The exact mechanism by which the abnormal accumulation of polysaccharides results in muscle damage remains unclear.
Treatment. Liver transplantation has been performed in children with both the classic and progressive hepatic form, with good results.15 Prevention of nutritional deficiencies and early recognition and management of cardiomyopathy are very important. Heart transplantation can be an option for severe cases of cardiomyopathy.
Type V Glycogenosis
GSD-V, also known as McArdle disease, is the most common disorder of carbohydrate metabolism. It is an autosomal recessive disorder caused by defects in the gene (PYGM) encoding for myophosphorylase enzyme. This enzyme is involved in the breakdown of glycogen to glucose for use in muscle. Myophosphorylase removes 1,4 glycosyl residues from outer branches of glycogen and adds inorganic phosphate to form glucose-1 phosphate.
Clinical features. GSD-V affects only skeletal muscle, and symptoms usually start during childhood with exercise intolerance, fatigue, myalgias, cramps, poor endurance, muscle swelling, and later fixed weakness. Symptoms are triggered by brief, intense activity but can also occur after prolonged low-intensity exercise. If patients continue to exercise, painful muscle cramping and contractures followed by myoglobinuria occurs (rare in children, more common after the second or third decade of life). A second-wind phenomenon is very characteristic of the disease, in which the muscle pain may dissipate after a brief rest period and allow the patient to resume exercise at the previous or a slightly reduced level (Case 3-2). It occurs due to a switch to alternative fuel substrates required for aerobic metabolism.16 Some patients with McArdle disease do not present with classic symptoms and may develop progressive muscle weakness.17
Case 3-2
A 17-year-old boy presented to the clinic 6 weeks after an episode of rhabdomyolysis. He had recently joined a soccer team and, during his first practice, noticed moderate to severe myalgia, cramping, and dark urine. He was seen at a local hospital, where his creatine kinase (CK) was found to be 8560 U/L. He denied any toxic exposure, and his personal and family history were unremarkable apart from two previous episodes of myalgia induced by brief exercise during his last summer camp. In the hospital, he was treated with aggressive IV hydration and a repeat CK 2 weeks later was 750 U/L. Findings from the general physical and neurologic examinations were completely normal. EMG and nerve conduction studies performed during the visit were normal. Forearm exercise test showed a normal increase in ammonia with no rise in lactate. Muscle biopsy showed abnormal glycogen accumulation in the subsarcolemmal and intermyofibrillar areas. Immunohistochemical staining for myophosphorylase was absent (Figure 3-4).
Figure 3-4.

Myophosphorylase deficiency. Hematoxylin eosin stain demonstrates subsarcolemmal vacuoles (A), immunohistochemical analysis for myophosphorylase confirms its absence (B), compared with normal control (C). Courtesy of Derek A. Mathis, MD.
Comment. This is a classic presentation of myophosphorylase deficiency. A negative family history is not uncommon for autosomal recessive disorders. Physical and electrophysiologic examinations frequently have normal results. A forearm exercise test with normal elevation of ammonia but no rise in lactate is consistent with a glycolysis/glycogenolysis disorder. Muscle biopsy shows abnormal glycogen accumulation, and immunohistochemical staining confirms the diagnosis.
Laboratory. Serum CK is usually elevated even between episodes of myoglobinuria (in contrast to another common cause of episodic myoglobinuria, carnitine palmitoyltransferase II [CPT II] deficiency, where interictal CK is usually normal). Forearm exercise test reveals a normal rise in ammonia but no significant lactate elevation. Results of routine nerve conduction studies are usually normal. Repetitive nerve stimulation after a short period of maximal exercise may show a decremental response. Needle EMG is usually normal but may reveal occasional myotonic discharges, fibrillations, and positive sharp waves in some patients. Early recruitment and myopathic potentials can also be seen.
Histopathology reveals fiber-size variability with some regenerating muscle fibers, and excessive accumulation of glycogen in the subsarcolemmal and intermyofibrillar regions. Staining for myophosphorylase is absent; however, it may be falsely positive after an episode of myoglobinuria because of the presence of fetal isozyme that is immunohistochemically indistinct from the adult form. For this reason, some experts advocate waiting at least 1 month after a severe episode before performing a muscle biopsy. Biochemical assay for myophosphorylase activity reveals absent or severely reduced activity.
Molecular genetics. There are three isozymes of glycogen phosphorylase: one for muscle, one for brain, and one for cardiac muscle, encoded in different genes (chromosomes 11, 14, and 20, respectively).18 GSD-V results from homozygous or compound heterozygous mutations in the PYGM gene on chromosome 11q13. More than 100 mutations have been identified, the most common in the North American population involving pArg50X in exon 1 and pGly205Ser. Significant phenotypical variability has been documented among patients with the same type of mutation, and these differences seem to be at least partially explained by the presence of several phenotype modulators, such as the angiotensin-converting enzyme (ACE) I/D, I/I, or D/D polymorphisms (with I being the longer allele with lower ACE activity and D being the shorter allele with higher ACE activity).19
Treatment. A Cochrane review published in 2010 found minimal benefit with low-dose creatine (60 mg/kg/d), but higher doses actually resulted in worsening of clinical symptoms. Ramipril (2.5 mg/d), an ACE inhibitor, was effective only for patients with a polymorphism known as the D/D ACE phenotype.20 A carbohydrate-rich diet resulted in better exercise performance compared with a protein-rich diet. Two studies of oral sucrose given at different times and in different amounts before exercise showed an improvement in exercise performance. The benefits of 75 g of oral sucrose are usually short-lasting, and repeated use may lead to weight gain. Vitamin B6 supplementation (50 mg/d to 90 mg/d) also seems to reduce exercise intolerance and enhance performance, according to small clinical reports.21 Patients should avoid intense isometric exercises (eg, weight lifting) and maximum aerobic exercises (eg, sprinting). They should also take advantage of the second-wind phenomenon with a low-intensity, 5- to 15-minute warm-up period.22
Type VII Glycogenosis
GSD-VII, also known as Tarui disease, is caused by deficiency of phosphofructokinase (PFK), the rate-limiting enzyme in the glycolytic pathway that catalyzes the adenosine triphosphatase–dependent phosphorylation of fructose-6 phosphate to fructose 1,6-biphosphate.
Clinical features. Tarui disease most commonly manifests as exercise intolerance with myalgias, cramps, and recurrent myoglobinuria. The warm-up phenomenon is absent, and the incidence of myoglobinuria is less common compared with McArdle disease. Other important differences compared with myophosphorylase deficiency include worsening of muscle symptoms with glucose administration (the out-of-wind phenomenon) due to a reduction in availability of free fatty acids and ketones, and the presence in some patients of jaundice (hemolytic anemia) and gouty arthritis due to PFK deficiency in erythrocytes. Four clinical presentations have been described: classic form with exercise intolerance; severe infantile form with hypotonia, progressive myopathy, cardiomyopathy, and respiratory failure; a late-onset form with permanent myopathy appearing around the fifth decade; and a hemolytic form characterized by nonspherocytic anemia without muscle symptoms.
Laboratory. Serum CK is usually elevated, and mild anemia and reticulocytosis can be present. The nonischemic forearm exercise test reveals normal elevation in ammonia with no increase in lactate levels. Hyperbilirubinemia and hyperuricemia can also be seen. EMG may be normal or show myopathic changes (small, short-duration motor unit potentials). Muscle MR spectroscopy shows the accumulation, even with mild exercise, of glycolytic intermediates in the form of phosphorylated monoesters. Muscle biopsy shows glycogen accumulation in the periphery of the fibers. Biopsies of older patients tend to show abnormal polysaccharide accumulation that stains intensely with PAS but is diastase resistant. Immunohistochemistry documents deficiency of PFK.
Molecular genetics. PFK is composed of three subunits-liver (PFKL), muscle (PFKM), and platelet (PFKP)-encoded by different genes and expressed in a tissue-specific manner. Skeletal muscle expresses only the PFKM subunit, whereas, in erythrocytes, five different heterotetramers of PFKM and PFKL are present. The PFKM gene is located on chromosome 12, and more than 16 different mutations have been identified so far.23
Treatment. Unlike in GSD-V, the administration of glucose or fructose before physical activity is not beneficial in GSD-VII and in some patients can cause worsening of muscle symptoms. An aerobic conditioning program is often recommended.
Type VIII Glycogenosis
GSD-VIII is caused by deficiency of the phosphorylase b kinase (PBK) enzyme that catalyzes the conversion of inactive phosphorylase to the active form. In addition, the enzyme converts glycogen synthetase to a less active form.
Clinical features. PBK deficiency is classified in five clinical subtypes distinguished by inheritance and tissue involvement: liver disease, the most frequent, with X-linked inheritance pattern; liver disease with autosomal recessive inheritance; liver and muscle disease transmitted in an autosomal recessive trait characterized by hepatomegaly and nonprogressive myopathy in childhood; muscle disease with exercise intolerance and myoglobinuria in an autosomal recessive pattern; and a fatal infantile form, also autosomal recessive, with severe cardiomyopathy but no liver or skeletal muscle involvement.24
Laboratory features. Serum CK is normal or mildly elevated. EMG and nerve conduction studies are usually normal. An abnormal forearm exercise test with a flat lactic acid curve can be seen. Muscle biopsy results are normal or may show increased fiber-size variability, scattered necrotic fibers, and slight subsarcolemmal accumulation of glycogen. Biochemical analysis reveals decreased PBK activity.
Molecular genetics. The functional muscle PBK molecule is a polymer with four subunits: alpha, beta, gamma, and delta.25 The alpha subunit is encoded on chromosome X. The subunit responsible for muscle disease is located on chromosome 16q12-q13.
Treatment. No specific treatment is available for this condition.
Type IX Glycogenosis
GSD-IX results from a deficiency of phosphoglycerate kinase (PGK), the enzyme that catalyzes the transfer of the acyl phosphate group of 1,3-diphosphoglycerate to adenosine phosphate (ADP) with formation of 3-phosphoglycerate and adenosine triphosphate (ATP) in the terminal stage of the glycolytic pathway.
Clinical features. PGK deficiency was initially identified in 1968 in the erythrocytes of a homozygous female patient with hemolytic anemia.26 Additional case reports described other symptoms, including mental retardation, seizures, and stroke. Myopathy as the predominant manifestation was reported in 1981 in a teenager with exercise intolerance, cramps, and recurrent hemoglobinuria. Slowly progressive fixed weakness has also been reported.
Laboratory. Serum CK is usually mildly elevated. Most patients with myopathy do not have hemolytic anemia. The forearm exercise test shows a normal increase in ammonia without elevation in lactic acid levels. EMG and nerve conduction studies are usually normal. Muscle biopsy results may be normal or demonstrate mild diffuse glycogen accumulation that is more noticeable with electron microscopy.
Molecular genetics. The human PGK gene is located on Xq13. More than 20 mutations have been identified so far.
Treatment. No specific medical therapy is available for the myopathy.
Type X Glycogenosis
Phosphoglycerate mutase (PGAM) deficiency is a muscle glycogen storage disease that results in a terminal block in glycogenolysis. PGAM catalyzes the interconversion of 2-phosphoglycerate and 3-phosphoglycerate. Two subunits are present: muscle (PGAM-M) and brain (PGAM-B).
Clinical features. Initially described in 1981, PGAM deficiency leads to myopathy characterized by exercise intolerance, cramps and recurrent myoglobinuria. Initial symptoms usually develop in late childhood or adolescence. The disease is more common in African Americans.
Laboratory. Serum CK is usually mildly elevated between attacks. The exercise forearm test fails to show a normal rise in lactate. EMG is usually normal. Muscle biopsy reveals increased glycogen deposits by PAS or electron microscopy. Tubular aggregates have been reported in six out of 15 cases.27 Biochemical assay demonstrates markedly diminished activity of PGAM.
Molecular genetics. The PGAM-M gene is located on chromosome 7p12-7p13. The most common gene defect is a nonsense mutation at codon 78, but different mutations have been described in Italians, Japanese, and Pakistani patients.
Treatment. No specific medical treatment is available.
Type XI Glycogenosis
Lactate dehydrogenase catalyzes the interconversion of pyruvate and lactate with concomitant interconversion of NADH and the oxidized form of nicotinamide adenine dinucleotide (NAD+).
Clinical features. This is a rare autosomal recessive disorder characterized by exercise intolerance. Most cases have been described in families of Japanese origin. An associated generalized rash has been reported in some cases. Obstetric complications are common as a result of uterine stiffness, often requiring a cesarean section. Chronic renal failure may develop secondary to recurrent myoglobinuria.
Laboratory. Serum CK is elevated during episodes, with normal lactate dehydrogenase (LDH). Forearm exercise test reveals normal increase in pyruvate level without a rise in lactate. EMG and nerve conduction studies are normal. Results of muscle biopsy are usually normal, but biochemical assay reveals reduced LDH activity. On gel electrophoresis, less than 5% of the LDH-M isoform is present in muscle and blood.
Molecular genetics. LDH is a tetrameric enzyme composed of two subunits, M and H. The gene encoding the M subunit consists of seven exons and is located on chromosome 11p15.4.
Treatment. No specific medical treatment is available. Potential obstetric complications should be discussed with affected female patients.
Type XII Glycogenosis
GSD-XII is caused by deficiency of aldolase A. This enzyme catalyzes the conversion of fructose 1,6-phosphate to dihydroxyacetone phosphate and glyceraldehyde 3-phosphate.
Clinical features. GSD-XII presents during childhood with hemolytic anemia, jaundice, proximal weakness, and episodic rhabdomyolysis triggered by febrile illness.
Laboratory. Serum CK is elevated after exercise, febrile events, or anesthesia. Muscle pathology reveals variable fiber size, and electron microscopy shows distorted mitochondria and occasional lipid accumulations.
Molecular genetics. GSD-XII is an autosomal recessive disorder secondary to mutations localized to chromosome 16q22-q24.
Treatment. No specific medical treatment is available.
Type XIII Glycogenosis
β-Enolase is a metalloenzyme responsible for the catalysis of the conversion of 2-phosphoglycerate to phosphoenolpyruvate, the ninth and penultimate step of glycolysis.
Clinical features. A single case report is available of a middle-aged man with exercise intolerance, myalgias, and no associated systemic symptoms.
Laboratory. Serum CK is elevated during episodes but otherwise normal. Muscle biopsy reveals abnormal glycogen deposition in the cytoplasm. Selective β-enolase deficiency is demonstrated by immunohistochemistry.
Molecular genetics. Missense mutations (Gly156Asp and Gly374Glut) located on chromosome 17 have been identified.
Treatment. No specific medical treatment is available.
Type XIV Glycogenosis
Phosphoglucomutase 1 catalyzes the transfer of phosphate between positions 1 and 6 of glucose.
Clinical features. Affected individuals present during the first or second decade of life with exercise intolerance and episodic rhabdomyolysis. A second-wind phenomenon has been described, and patients may later develop fixed weakness.
Laboratory. Serum CK is mildly elevated at rest and increases during attacks. The forearm exercise test shows normal elevation of lactate with up to 4 times increase in ammonia. Muscle biopsy may reveal abnormal glycogen deposition.
Molecular genetics. Mutations in PGM1 located on chromosome 1p31.3 are responsible for this disease.
Treatment. No specific medical therapy is available for this disease.
Triosephosphate Isomerase Deficiency
Triosephosphate isomerase (TPI) enzyme catalyzes the reversible interconversion of the triose phosphate isomers dihydroxyacetone phosphate and D-glyceraldehyde 3-phosphate.
Clinical features. TPI deficiency is the most severe glycolytic enzyme defect associated with progressive neurologic dysfunction. Onset is usually before 2 years of age with hemolytic anemia, recurrent infections, slowly progressive proximal and facial weakness, and CNS dysfunction (including mental retardation, dystonia, cerebellar symptoms, and tremor). Some patients develop an associated sensorimotor polyneuropathy. Survival is limited, with most children dying before 6 years of age.
Laboratory. Serum CK is normal or mildly elevated. Nerve conduction studies can be normal or show changes consistent with an axonal sensorimotor polyneuropathy. EMG shows myopathic potentials. Muscle pathology reveals fiber degeneration and regeneration with increased glycogen in some muscle fibers. Biochemical analysis reveals reduced TPI activity in different tissues.
Molecular genetics. The TIM gene is located on chromosome 12p13, and the most common mutation is Glu104Asp.28
Treatment. No specific medical therapy is available.
DISORDERS OF LIPID METABOLISM
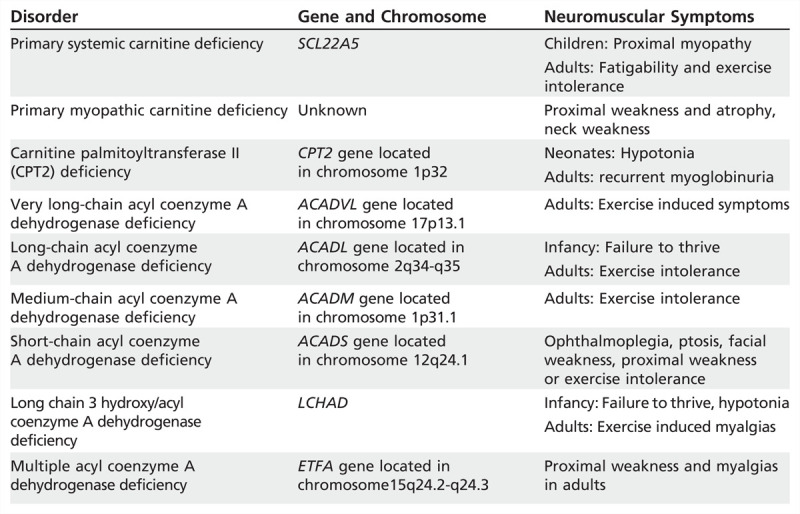
Carnitine Transporter Deficiency
Carnitine transporter deficiency is the most common disorder of lipid metabolism (Table 3-4). It can be primary-as a result of mutations in the gene codifying for the sodium-dependent carnitine transporter (OCTN2), a protein responsible for the transport of carnitine across cell membranes-or secondary to systemic conditions (including renal Fanconi syndrome, organic acidurias, respiratory chain defects, chronic hemodialysis, and malnutrition) and with certain medications (valproate, zidovudine).
Table 3-4.
Disorders of Lipid Metabolism
Clinical features. Systemic primary carnitine deficiency is an autosomal recessive disorder of carnitine transportation characterized by episodes of hypoketotic hypoglycemia, hepatomegaly, elevated transaminases, and hyperammonemia in infants. Children present with myopathy, elevated CK, and cardiomyopathy, while adults present with fatigability. Pregnancy may unmask symptoms such as decreased stamina and cardiac arrhythmias.
Laboratory. Serum CK can be normal in almost half of affected patients, but elevations up to 15 times normal can also be seen. EMG may be normal or reveal myopathic motor unit potentials with increased insertional activity in the form of fibrillations, positive sharp waves, or complex repetitive discharges, especially in severe cases. Liver enzymes are also elevated in the primary systemic form. Echocardiogram may reveal a dilated or hypertrophic cardiomyopathy. Reduced carnitine levels in plasma, liver, heart, and muscle are present. Muscle biopsy demonstrates vacuoles as well as abnormal accumulation of lipid in the subsarcolemmal and intermyofibrillar regions. Electron microscopy also reveals lipid deposition. Type I fibers are preferentially affected. Muscle carnitine levels are severely decreased (less than 2% to 4% of normal levels) in patients with the primary form, compared with secondary causes in which free levels are moderately reduced (25% to 50%). Diagnosis can be confirmed through genetic testing or skin biopsy with assessment of carnitine transport in skin fibroblasts (less than 10% of normal levels).
Molecular genetics. This disorder is transmitted in an autosomal recessive pattern, and is due to mutations of the SLC22A5 gene located on chromosome 5q23. More than 100 mutations have been reported so far.
Treatment. Patients may benefit from oral carnitine 50 mg/kg/d to 400 mg/kg/d divided in three doses.29 Common side effects include diarrhea and abdominal discomfort. During acute episodes, IV glucose may be helpful. Patients should follow a low-fat diet and avoid fasting.
Carnitine Palmitoyltransferase II Deficiency
CPT II is a peripheral inner mitochondrial membrane protein that catalyzes the transesterification of palmitoylcarnitine back into palmitoyl–coenzyme A (CoA), which is now an activated substrate for beta-oxidation inside the matrix (Figure 3-5).
Figure 3-5.
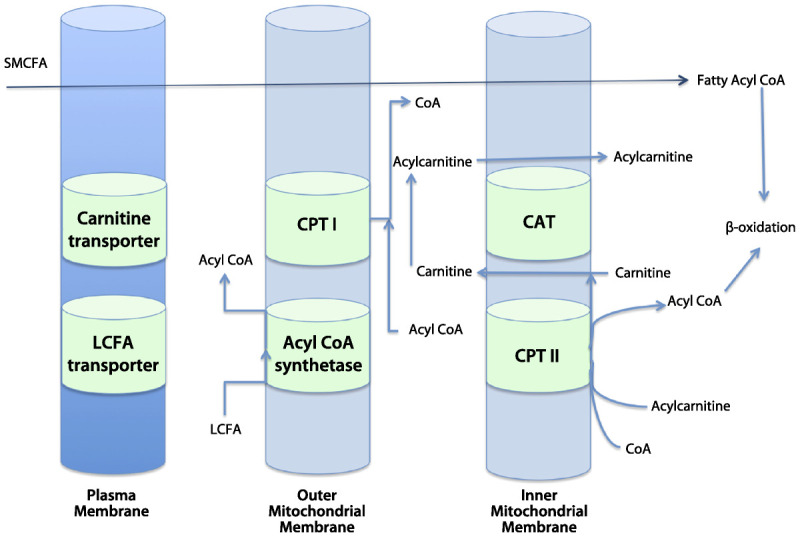
Schematic illustration of the oxidation of fatty acids in mitochondria. Short- and medium-chain fatty acids (SMCFA) freely cross the mitochondrial membrane, whereas long-chain fatty acids (LCFA) require a specific transporter system. Acyl coenzyme A (acyl-CoA) synthetase transforms the LCFA into acyl-CoA, and the coenzyme A (CoA) is exchanged for carnitine by carnitine palmitoyltransferase I (CPT I). Acylcarnitine is then transported across the inner mitochondrial membrane by carnitine acylcarnitine translocase (CAT). Carnitine is removed from acylcarnitine by carnitine palmitoyltransferase II (CPT II) to form acyl-CoA, which can undergo beta oxidation. Modified with permission from van Adel BA, Tarnopolsky MA, J Clin Neuromuscul Dis.1 © 2009, Lippincott Williams & Wilkins. journals.lww.com/jcnmd/Abstract/2009/03000/Metabolic_Myopathies__Update_2009.3.aspx.
Clinical features. Phenotypical expression of CPT II deficiency is dependent on the severity of enzyme deficiency and has been described in a severe neonatal form, severe infantile form, and, more commonly, an adult myopathic form.
The lethal neonatal form begins during the first few days of life with encephalopathy, cardiomyopathy, hypoglycemia, hepatomegaly, and seizures. Death usually occurs during the first week. Fasting or infection usually triggers the severe infantile form; common manifestations include hypoglycemia, hepatomegaly, cardiac arrhythmias and cardiomyopathy, hypotonia, and weakness. In adults, CPT II deficiency is the most common cause of recurrent myoglobinuria. Usual manifestations include myalgias and cramps after intense or prolonged exercise, febrile illness, or fasting (Case 3-3). Medications like valproate sometimes can trigger these symptoms. Physical examination usually shows normal findings between attacks.
Case 3-3
A 20-year-old college student presented to the emergency department with progressive, severe incapacitating pain in both thighs, inability to walk, and dark urine. The day before, she had been asymptomatic and participated in a strenuous 3-hour soccer practice. She had a history of occasional muscle cramps during exercise but never as severe. She denied any drug use, and her personal and family history were unremarkable. Examination revealed severe tenderness to palpation of both quadriceps muscles, and mild tenderness to palpation of the gastrocnemius muscles. Strength was 5/5 in upper extremities, 2/5 hip flexion and knee extension with significant limitations due to pain, and 5/5 strength distally. Deep tendon reflexes, sensory examination findings, and coordination were normal. Initial laboratory tests were remarkable for mild leukocytosis, with 12,500 white blood cells/μL; creatine kinase (CK) 210,000 U/L; aspartate aminotransferase (AST) 9600 IU/L; creatinine 2.4 mg/dL; and blood urea nitrogen (BUN) 32 mg/dL. MRI showed diffuse swelling of the quadriceps muscles with blurred margins and hyperintense signal on short T1 inversion recovery (STIR) and T2-weighted images, and hypointense to isointense signal on T1-weighted image. After hydration and rest, her kidney function and serum CK gradually normalized. Later, a forearm exercise test was performed, with normal results. Serum carnitine levels were normal, and acylcarnitine profile showed a high ratio of palmitoyl carnitine (C16:0) and oleoyl carnitine (C18:1) to acetylcarnitine (C2). Muscle biopsy performed 6 weeks after the episode showed mild fiber type 2 predominance, and carnitine palmitoyltransferase II (CPT II) activity was very reduced (less than 10% of normal levels).
Comment. CPT II is the most common cause of myoglobinuria in adults. Usual trigger factors include prolonged strenuous exercise or fasting. The forearm exercise test helps to exclude certain glycogen storage disorders such as McArdle disease. Serum carnitine is normal or mildly decreased, and the acylcarnitine profile helps to narrow the differential diagnosis. CPT II enzyme activity or genetic testing are confirmatory.
Laboratory. Serum CK is normal or mildly elevated between attacks and increases significantly during episodes of rhabdomyolysis. Serum carnitine levels are normal or mildly decreased. Forearm exercise test findings are normal, and EMG can be normal or show myopathic units. Muscle pathology can be normal or reveal increased lipid content, especially with electron microscopy. Tandem mass spectrometry of serum/plasma acylcarnitines (ie, the acylcarnitine profile) helps to reach a diagnosis. The finding suggestive of a defect in mitochondrial beta-oxidation (and therefore suspect for CPT II deficiency) is an elevation of C12 to C18 acylcarnitines (in which C stands for the number of carbon atoms), notably of C16 and C18:1. The differential diagnosis for an elevation of C12 to C18 acylcarnitines includes glutaric acidemia type II and carnitine-acylcarnitine translocase deficiency, which can be excluded by additional screening of urinary metabolites such as glutaric and 3-OH-glutaric acids. Diagnosis is confirmed by detection of reduced CPT II enzyme activity in skeletal muscle homogenates or cultured skin fibroblasts.
Molecular genetics. CPT II deficiency is caused by mutations on chromosome 1p32 (most commonly p.Ser113Leu, p.Pro50His, and p.Lys414ThrfsX7). If none or only one of these mutations is identified, sequence analysis of the entire coding region can be performed.
Treatment. Patients are advised to follow a low-fat diet with frequent meals and to avoid prolonged exercise and fasting. A small case study with bezafibrate 200 mg 3 times a day showed significant increase in palmitoyl L-carnitine levels and reduced frequency of attacks.30 Another small study with triheptanoin diet (30% of daily calories) demonstrated increased exercise tolerance.31
Very-Long-Chain Acyl–Coenzyme A Dehydrogenase Deficiency
Very-long-chain acyl-CoA dehydrogenase (VLCAD) is involved in the initial step of mitochondrial beta-oxidation, and is active toward CoA esters of longer-chain substrates: arachidoyl-CoA, behenoyl-CoA, and lignoceroyl-CoA.
Clinical features. Three phenotypes have been identified: an infantile form with recurrent hypoketotic hypoglycemia, hepatomegaly, cardiomyopathy, hypotonia, and very high mortality; a hepatic form with episodic hypoglycemia, hepatomegaly but no systemic involvement, and low mortality rate; and a rare myopathic form in children or adults with episodic myalgias, rhabdomyolysis, and variable systemic features.
Laboratory. Serum CK is normal except in cases of myoglobinuria. Free plasma carnitine level is normal or mildly decreased. Acylcarnitine profile shows elevated tetradecanoic acid (C14:1). Muscle biopsy results are normal or may show increased lipid deposition. VLCAD immunostain is reduced. In addition, VLCAD protein activity in fibroblasts is decreased.
Molecular genetics. VLCAD deficiency is an autosomal recessive disorder secondary to mutations in the gene ACADVL located on chromosome 17p13.1.
Treatment. Infants with the more severe forms are typically placed on a low-fat formula, with supplemental calories provided through medium-chain triglycerides (MCT). A variety of strategies for the low-fat diet are used, ranging from 13% to 39% of calories as total fat, with an additional 15% to 18% of calories supplied as MCT oil for patients most strictly restricted from long-chain fats. Extra MCT have demonstrated benefit in older patients with long-chain defects who have exercise intolerance. Triheptanoin has been used in a few individuals with the goal of providing calories as well as anaplerotic (ie, alternative substrate) carbons; however, its efficacy remains controversial.
Long-Chain Acyl–Coenzyme A Dehydrogenase Deficiency
Long-chain acyl-CoA dehydrogenase (LCAD) acts on fatty acyl-CoA derivatives whose acyl residues contain more than 12 carbon atoms; patients with LCAD deficiency, therefore, have impaired ability to metabolize long-chain fatty acids.
Clinical features. Phenotypical variation is significant, but most patients present during infancy with failure to thrive, hepatomegaly, cardiomegaly, nonketotic hypoglycemia, and encephalopathy. Other presentations include exercise-induced rhabdomyolysis in adults or progressive limb-girdle weakness.
Laboratory. Serum CK is normal between attacks. Total and free carnitine levels are reduced, but long-chain acylcarnitine esters are increased. Oil Red O staining of muscle biopsies demonstrates increased lipid deposition. Diagnosis can be confirmed with reduced LCAD enzyme activity in cultured fibroblasts.
Molecular genetics. LCAD deficiency is an autosomal recessive disorder caused by mutations of the ACADL gene located on chromosome 2q34-q35. Some patients previously diagnosed with LCAD deficiency may actually have VLCAD deficiency, a much more common disorder.
Treatment. MCT supplementation during periods of increased activity may improve the metabolic control of children with LCAD deficiency after exercise.32
Medium-Chain Acyl–Coenzyme A Dehydrogenase Deficiency
Medium-chain acyl-CoA dehydrogenase (MCAD) works in the degradation of fatty acyl-CoA derivatives whose acyl residues contain four to 14 carbon atoms.
Clinical features. MCAD is the most common form of acyl-CoA deficiency, but it is rarely associated with skeletal muscle or cardiac disease. Typical presentation is very similar to LCAD, presenting before 2 years of age with lethargy, hypoketotic hypoglycemia, hepatomegaly, and encephalopathy. Older children and adults can have exercise-induced symptoms or mild myopathy.
Laboratory. CK level is usually normal. Carnitine levels are low. Dicarboxylic, adipic, and sebacic acids are increased in urine. MCAD activity is diminished to less than 10% in muscle, fibroblasts, and liver.
Molecular genetics. Mutations in the gene ACADM located on chromosome 1p31.1 are responsible for this disorder (K304E is the most common).
Treatment. Patients should avoid fasting for more than 12 hours, and infants affected should have frequent feedings and receive 2 g/kg of uncooked cornstarch at bedtime. A relatively low-fat diet may also be beneficial.
Short-Chain Acyl–Coenzyme A Dehydrogenase Deficiency
Short-chain acyl-CoA dehydrogenase (SCAD) enzyme acts on fatty acyl-CoA derivatives whose acyl residues contain four to six carbon atoms.
Clinical features. Affected infants present with failure to thrive and nonketotic hypoglycemia. Older patients may present with ophthalmoplegia, ptosis, facial weakness, and cardiomyopathy, or exercise-induced symptoms and progressive weakness.
Laboratory. Serum CK is usually normal apart from episodes of rhabdomyolysis. Urinary excretion of ethylmalonate and methylsuccinate is increased.
Molecular genetics. Mutations in the ACADS gene located on chromosome 12q24.31 are responsible for this disorder.
Treatment. Patients should avoid fasting for long periods of time.
Multiple Acyl–Coenzyme A Dehydrogenase Deficiency (MADD)
Electron-transfer-flavoprotein alpha-polypeptide (ETFA) participates in catalyzing the initial step of the mitochondrial fatty acid beta-oxidation. It shuttles electrons between primary flavoprotein dehydrogenases and the membrane-bound electron transfer flavoprotein ubiquinone oxidoreductase.
Clinical features. MADD most commonly affects adults in the third or fourth decade of life, with proximal weakness and myalgias. A severe neonatal form is characterized by acidosis, hypoglycemia, and high mortality rate.
Laboratory. Serum CK is usually elevated 2 to 20 times above normal. EMG shows myopathic changes. Carnitine levels are reduced while urine glutaric and ethylmalonic acids are elevated.
Molecular genetics. MADD is caused by mutations in the gene codifying ETFA located on chromosome 15q24.2-q24.3.
Treatment. Riboflavin (100 mg/d) seems to be beneficial in some patients. The effect of carnitine supplementation and coenzyme Q10 is controversial.
MITOCHONDRIAL MYOPATHIES
The mitochondrial myopathies are a complex group of diseases related to structural, biochemical, or genetic defects in the mitochondria. The most common neuromuscular feature of mitochondrial disease is myopathy, causing proximal weakness often accompanied by exercise intolerance out of proportion to the weakness. Ptosis and chronic progressive external ophthalmoparesis are also common manifestations, often associated with dysphagia.33 Peripheral neuropathies are common. Table 3-5 shows the most frequent mitochondrial disorders associated with myopathy. Given the considerable heterogeneity of these disorders, a detailed description of each mitochondrial disorder is beyond the scope of this article, and only some of the most common entities are reviewed.
Table 3-5.
Most Common Mitochondrial Disorders Associated With Myopathies
Myoclonic Epilepsy With Ragged Red Fibers
Myoclonic epilepsy with ragged red fibers (MERRF) is a multisystem disorder characterized by myoclonus followed by generalized epilepsy, ataxia, weakness, and dementia.34
Clinical features. Myoclonus is usually the first symptom, most commonly emerging during late adolescence or young adulthood. Myoclonus can be stimulus-sensitive or present at rest, and generalized seizures may frequently occur.
Other common manifestations include ataxia, sensorineural hearing loss, short stature, dementia, optic atrophy, muscular weakness, and atrophy. An associated sensorimotor polyneuropathy with pes cavus can be seen. Clinical features and age of onset are variable, and not all patients present with the full syndrome. Other, less common systemic features include cardiac arrhythmias with conduction block, pigmentary retinopathy, pyramidal signs, and multiple lipomatosis. This condition is inherited in a mitochondrial pattern, which is also known as maternal inheritance.
Laboratory. Serum CK is normal or mildly elevated. Lactate and pyruvate are commonly elevated at rest and increase with physical activity. Brain-imaging studies may reveal cerebral and cerebellar atrophy. Nerve conduction studies may demonstrate an axonal polyneuropathy, and EMG often shows myopathic potentials. Muscle biopsy reveals subsarcolemmal accumulation of abnormal mitochondria, best seen on the modified trichrome stain, giving the characteristic appearance of “ragged red” fibers (Figure 3-6). These fibers may be more basophilic and granular with hematoxylin and eosin, and they react intensely with succinic dehydrogenase and NADH–tetrazolium reductase (TR) and may lack cytochrome oxidase activity. On electron microscopy, an increased number of enlarged mitochondria with abnormal cristae and occasional inclusions can be seen.
Figure 3-6.
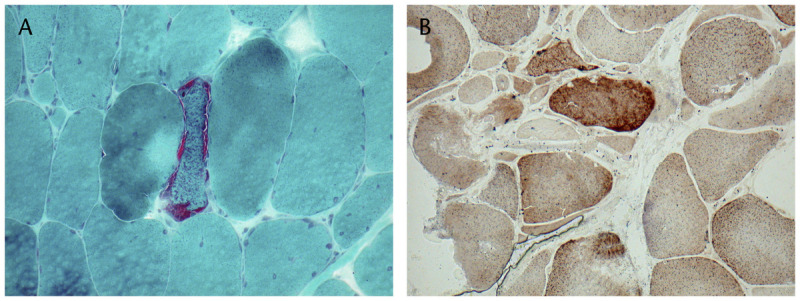
Ragged red fibers revealed by the modified Gomori one-step trichrome stain (A) and combined cytochrome oxidase/succinate dehydrogenase stain (B). They are formed by the subsarcolemmal accumulation of abnormal mitochondria. Courtesy of Derek A. Mathis, MD.
Molecular genetics. The mitochondrial DNA (mtDNA) gene MT-TK encoding transfer RNA (tRNA) lysine is the gene most commonly associated with MERRF. Four MT-TK mutations (m.8344A>G, m.8356T>C, m.8363G>A, and m.8361G>A) account for approximately 90% of cases.35
Treatment. No specific treatments are available. Seizures should be managed with appropriate anticonvulsants. Aerobic exercise is helpful, and physical therapy might improve some impaired motor abilities.
Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes Syndrome
Mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) syndrome is a metabolic disorder that usually presents with seizures and strokelike events in young people. It is a prototypical mitochondrial disorder in that it exhibits a maternal pattern of inheritance and features a variable proportion of mutated mitochondria in different tissues over time (heteroplasmy). It affects tissues of high metabolic demand (brain, muscle), and a given tissue appears to require a certain levelof affected mitochondria before clinical symptoms occur (threshold effect).36
Clinical features. Common clinical features of MELAS syndrome include headaches, seizures, hearing loss, muscle weakness with exercise intolerance, and strokelike episodes, which typically improve or fluctuate (usually cortical blindness, hemianopia, hemiparesis). Attacks may be precipitated by exercise or infection.
Laboratory. Serum CK may be elevated or normal. The majority of patients have raised CSF and serum lactate levels. Serial brain MRI shows lesions that are not restricted to arterial territories and migrate over time. They often affect the occipital/parietal regions. Deep gray matter structures like the thalami can also be affected. Brain lesions tend to affect the cortical ribbon with relative sparing of the deep white matter. EMG may be normal, although, in patients with severe weakness, it can show myopathic potentials. Muscle biopsy findings in MELAS syndrome are similar to those described in MERRF.
Molecular genetics. MELAS syndrome is caused in approximately 80% of cases by a mitochondrial RNA (mtRNA) Leu (MTTL1) mutation (A3243G). Other, less common mutation foci include T3271C and A3252G. Mutations in MTTV, MTTK, MTTF, MTTS1, MTND5, MTND4, and MTCYB have all been described with MELAS syndrome.
Treatment. Different treatment approaches have been tried in patients with MELAS syndrome, with mixed results. A 3-year randomized study with dichloroacetate was stopped early because of a very high incidence or worsening of polyneuropathy. Coenzyme Q10 is considered safe but has poor CNS penetration and has been tried in several small studies with variable success. L-Arginine showed promise in the acute treatment of strokelike episodes, but additional data are required.37
Kearns-Sayre Syndrome
The diagnosis of Kearns-Sayre syndrome (KSS) is based on three classic features: progressive external ophthalmoplegia, pigmentary retinopathy, and heart block.
Clinical features. Symptoms usually develop before the second decade of life. Accompanying features may include proximal myopathy, short stature, dementia, ataxia, sensorineural hearing loss, and endocrinopathies. This condition is generally not inherited but arises from mutations in the body’s cells that occur after conception. Rarely, it can be transmitted with a maternal inheritance pattern.
Laboratory. Serum CK is usually normal. Serum lactate and pyruvate are commonly elevated, and CSF reveals high protein and homovanillic acid levels with very low 5-methyltetrahydrofolate. Conduction defects can be seen on ECG. EMG may be normal or demonstrate myopathic potentials. Nerve conduction studies are normal unless an associated axonal sensory or sensorimotor polyneuropathy is present. Muscle biopsy shows ragged red fibers, with most fibers being cytochrome oxidase (COX)–negative.
Molecular genetics. More than 150 different mtDNA deletions have been associated with KSS. A deletion of 4977 base pairs known as m.8470_13446del4977 is encountered most frequently. The same mutation and numerous other types of deletions of varying length have been identified in Pearson syndrome (sideroblastic anemia and exocrine pancreas dysfunction) and progressive external ophthalmoplegia (PEO).
Treatment. Recommended strategies for treatment include the placement of cardiac pacemakers in individuals with cardiac conduction blocks, cochlear implants and hearing aids for sensorineural hearing loss, hormone replacement for endocrinopathies, folic acid supplementation in individuals with KSS with low CSF folic acid, administration of coenzyme Q10 and L-carnitine, and rehabilitation services.
Mitochondrial Neurogastrointestinal Encephalopathy
The main clinical components of mitochondrial neurogastrointestinal encephalopathy (MNGIE) include severe gastrointestinal dysmotility, cachexia, ptosis, external ophthalmoplegia, sensorimotor polyneuropathy, and leukoencephalopathy.
Clinical features. Patients develop muscle weakness and atrophy, more prominent distally. Sensation is commonly lost in a stocking and glove pattern, and patients are hyporeflexic or areflexic. Ptosis and ophthalmoparesis are present in most patients. Other accompanying symptoms may include pigmentary retinopathy, sensorineural hearing loss, facial weakness, and dysarthria (Case 3-4). The earliest and most common symptoms are related to gastrointestinal dysmotility: dyspepsia, bloating, cramps, intolerance of large meals, nausea, vomiting, and diarrhea.38 This condition is transmitted in an autosomal recessive pattern.
Case 3-4
A 26-year-old woman was admitted to the hospital for evaluation of nausea, vomiting, and progressive weight loss. Symptoms had been present intermittently over the past 9 months, and she had lost at least 20.4 kg (45 lbs). A neurologic consult was requested as she also reported proximal and distal weakness affecting the lower extremities, mild distal paresthesia, and increased difficulty walking. Her medical and family history were unremarkable, and she had no history of drug or toxic exposure. Neurologic examination was positive for mild bilateral ptosis with restriction of horizontal eye movements. No muscle atrophy was evident, and strength examination showed Medical Research Council (MRC) grade 4/5 bilaterally in the hip flexors, abductors, and knee extensors, with grade 4+/5 in the tibialis anterior and gastrocnemius. Deep tendon reflexes were mildly decreased (1+) symmetrically in both lower extremities. Sensory examination showed mildly decreased vibratory sensation in both feet. Serum creatine kinase was normal, and both serum lactate and pyruvate were mildly elevated. Nerve conduction studies revealed a mild mixed axonomyelinic polyneuropathy. Needle EMG showed some chronic reinnervation changes in distal muscles but also myopathic motor unit potentials without fibrillations or positive sharp waves in the iliopsoas, quadriceps, and gluteus medius. Muscle biopsy of the right vastus lateralis showed ragged red fibers (Figure 3-6A) with cyclooxygenase-negative fibers and succinic dehydrogenase-positive fibers. Thymidine phosphorylase enzyme activity in leukocytes was less than 10%.
Comment. The combination of gastrointestinal dysmotility, polyneuropathy, myopathy, ptosis, and external ophthalmoplegia is consistent with mitochondrial neurogastrointestinal encephalopathy (MNGIE). A mitochondrial myopathy is confirmed with muscle biopsy findings, and a definite diagnosis could be reached by measuring thymidine phosphorylase activity or plasma thymidine and deoxyuridine levels, or with molecular testing.
Laboratory. Brain MRI reveals prominent white matter changes even in the absence of significant hemispheric symptoms. Serum CK is normal or mildly elevated. Plasma thymidine levels greater than 3 μmol/L and deoxyuridine concentration greater than 5 μmol/L can be sufficient to confirm the diagnosis. Thymidine phosphorylase enzyme activity in leukocytes less than 10% can also be confirmatory. Nerve conduction studies show demyelinating or mixed axonomyelinic changes. EMG varies according to muscle selection with possible denervation and chronic reinnervation changes, while in proximal muscles a myopathic pattern can be seen. Muscle biopsy demonstrates ragged red fibers, increased NADH and succinic dehydrogenase staining, and COX-negative staining. Reduced COX and other respiratory complex activities are demonstrable on biochemical assays of muscle tissue.
Molecular genetics. Mutations are detected in genomic DNA by sequencing the TYMP exons and flanking regions in 100% of individuals with enzymatically proven MNGIE disease. POLG1 mutations may cause MNGIE-like syndrome without leukoencephalopathy and normal thymidine plasma levels.39
Treatment. Early attention to swallowing difficulties and airway protection are very important, and nutritional support should be provided. No specific medical therapy is available for this disease. Liver transplantation remains controversial, and stem cell transplantation shows some promising results.
CONCLUSIONS
The discovery of the basic genetic and biochemical changes responsible for the development of metabolic myopathies should help us expand the diagnostic tools and treatment options for these disorders in the near future.
KEY POINTS
From a clinical standpoint, the metabolic myopathies can be viewed as static or dynamic disorders.
Metabolic myopathies should be considered in the differential diagnosis of patients with exercise-induced muscle symptoms, static or progressive myopathy, isolated neuromuscular respiratory weakness, and muscle disease associated with systemic conditions.
The forearm exercise test is a very helpful tool in the evaluation of patients with dynamic symptoms, particularly to differentiate those with defects of the glycolytic pathway.
Glycogen storage disease (GSD) type II has three phenotypical presentations: severe infantile form (Pompe disease), juvenile-onset type, and an adult-onset variant.
A dried blood spot that measures α-glucosidase activity is the recommended screening test for glycogen storage disease type II, followed by genetic testing if results are abnormal.
Prevention of further loss of muscle function is the target treatment goal for enzyme replacement therapy in patients with acid maltase deficiency.
Deficiency of debranching enzyme can be demonstrated with biochemical assay of muscle, fibroblasts, or lymphocytes.
The adult form of glycogen storage disease type IV can present as isolated myopathy or adult polyglucosan body disease characterized by a combination of upper and lower motor neuron syndromes, sensory nerve involvement, cerebellar ataxia, neurogenic bladder, and dementia.
A second-wind phenomenon is very characteristic of glycogen storage disease type V, in which the muscle pain may dissipate after a brief rest period and allow the patient to resume exercise at the previous or slightly reduced level.
In glycogen storage disease type V, serum creatine kinase is usually elevated even between episodes of myoglobinuria (in contrast to another common cause of episodic myoglobinuria, carnitine palmitoyltransferase II deficiency, where interictal creatine kinase is usually normal).
Ramipril (2.5 mg/d), an angiotensin-converting enzyme inhibitor, was effective only for patients with a polymorphism known as the deletion/deletion angiotensin-converting enzyme phenotype of McArdle disease.
Unlike glycogen storage disease type V, in glycogen storage disease type VII the administration of glucose or fructose before physical activity is not beneficial, and in some patients can cause worsening of muscle symptoms. An aerobic conditioning program is often recommended.
Potential obstetric complications of glycogen storage disease type XI should be discussed with affected female patients.
Glycogen storage disease type XII presents during childhood with hemolytic anemia, jaundice, proximal weakness, and episodic rhabdomyolysis triggered by febrile illness.
Triosephosphate isomerase deficiency is the most severe glycolytic enzyme defect associated with progressive neurologic dysfunction.
Children with carnitine deficiency present with myopathy, elevated creatine kinase, and cardiomyopathy, while adults present with fatigability. Pregnancy may unmask symptoms such as decreased stamina and cardiac arrhythmias.
In adults, carnitine palmitoyltransferase II deficiency is the most common cause of recurrent myoglobinuria.
Infants with the more severe forms of very-long-chain acyl–coenzyme A dehydrogenase are typically placed on a low-fat formula, with supplemental calories provided through medium-chain triglycerides.
The most common neuromuscular feature of mitochondrial disease is myopathy, causing proximal weakness often accompanied by exercise intolerance out of proportion to the weakness.
Common clinical features of mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) syndrome include headaches, seizures, hearing loss, muscle weakness with exercise intolerance, and strokelike episodes, which typically improve or fluctuate (usually cortical blindness, hemianopia, hemiparesis). Attacks may be precipitated by exercise or infection.
Recommended strategies for treatment of Kearns-Sayre syndrome include the placement of cardiac pacemakers in individuals with cardiac conduction blocks, cochlear implants and hearing aids for sensorineural hearing loss, hormone replacement for endocrinopathies, folic acid supplementation in individuals with low CSF folic acid, administration of coenzyme Q10 and L-carnitine, and rehabilitation services.
ACKNOWLEDGMENT
Thanks to Dr Derek A. Mathis, Lt Col, USAF, MC, Department of Pathology San Antonio Military Medical center for assistance with the pathology slides.
Footnotes
Relationship Disclosure: Dr Tobon reports no disclosure.
Unlabeled Use of Products/Investigational Use Disclosure: Dr Tobon reports no disclosure.
REFERENCES
- 1.van Adel BA,, Tarnopolsky MA. Metabolic myopathies: Update 2009. J Clin Neuromuscul Dis 2009; 10 (3): 97–121. [DOI] [PubMed] [Google Scholar]
- 2.Berardo A,, DiMauro S,, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep 2010; 10 (2): 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hogrel JY,, Laforet P,, Ben Yaou R, et al. A non-ischemic forearm exercise test for the screening of patients with exercise intolerance. Neurology 2001; 56 (12): 1733–1738. [DOI] [PubMed] [Google Scholar]
- 4.van der Ploeg AT,, Reuser AJ. Pompe’s disease. Lancet 2008; 372 (9646): 1342–1353. [DOI] [PubMed] [Google Scholar]
- 5.Kroos M,, Hoogeveen-Westerveld M,, van der Ploeg A,, Reuser AJ. The genotype-phenotype correlation in Pompe disease. Am J Med Genet C Semin Med Genet 2012; 160 (1): 59–68. [DOI] [PubMed] [Google Scholar]
- 6.Kroos M,, Hoogeveen-Westerveld M,, Michelakakis H, et al. Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Hum Mutat 2012; 33 (8): 1161–1165. [DOI] [PubMed] [Google Scholar]
- 7.van der Ploeg AT,, Clemens PR,, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 2010; 362 (15): 1396–1406. [DOI] [PubMed] [Google Scholar]
- 8.Cupler EJ,, Berger KI,, Leshner RT, et al. Consensus treatment recommendations for late-onset Pompe disease. Muscle Nerve 2012; 45 (3): 319–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toscano A,, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol 2013; 260 (4): 951–959. [DOI] [PubMed] [Google Scholar]
- 10.Lucchiari S,, Santoro D,, Pagliarani S,, Comi GP. Clinical, biochemical and genetic features of glycogen debranching enzyme deficiency. Acta Myol 2007; 26 (1): 72–74. [PMC free article] [PubMed] [Google Scholar]
- 11.Shen J,, Bao Y,, Liu HM, et al. Mutations in exon 3 of the glycogen debranching enzyme gene are associated with glycogen storage disease type III that is differentially expressed in liver and muscle. J Clin Invest 1996; 98 (2): 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruno C,, Cassandrini D,, Assereto S, et al. Neuromuscular forms of glycogen branching enzyme deficiency. Acta Myol 2007; 26 (1): 75–78. [PMC free article] [PubMed] [Google Scholar]
- 13.Mochel F,, Schiffmann R,, Steenweg ME, et al. Adult polyglucosan body disease: natural history and key magnetic resonance imaging findings. Ann Neurol 2012; 72 (3): 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Massa R,, Bruno C,, Martorana A, et al. Adult polyglucosan body disease: proton magnetic resonance spectroscopy of the brain and novel mutation in the GBE1 gene. Muscle Nerve 2008; 37 (4): 530–536. [DOI] [PubMed] [Google Scholar]
- 15.Davis MK,, Weinstein DA. Liver transplantation in children with glycogen storage disease: controversies and evaluation of the risk/benefit of this procedure. Pediatr Transplant 2008; 12 (2): 137–145. [DOI] [PubMed] [Google Scholar]
- 16.Vissing J,, Haller RG. Mechanisms of exertional fatigue in muscle glycogenoses. Neuromuscul Disord 2012; 22 (suppl 3): S168–S171. [DOI] [PubMed] [Google Scholar]
- 17.Nadaj-Pakleza AA,, Vincitorio CM,, Laforet P, et al. Permanent muscle weakness in McArdle disease. Muscle Nerve 2009; 40 (3): 350–357. [DOI] [PubMed] [Google Scholar]
- 18.Quinlivan R,, Buckley J,, James M, et al. McArdle disease: a clinical review. J Neurol Neurosurg Psychiatry 2010; 81 (11): 1182–1188. [DOI] [PubMed] [Google Scholar]
- 19.Martinuzzi A,, Sartori E,, Fanin M, et al. Phenotype modulators in myophosphorylase deficiency. Ann Neurol 2003; 53 (4): 497–502. [DOI] [PubMed] [Google Scholar]
- 20.Quinlivan R,, Martinuzzi A,, Schoser B. Pharmacological and nutritional treatment for McArdle disease (glycogen storage disease type V). Cochrane Database Syst Rev 2010; (12): CD003458. [DOI] [PubMed] [Google Scholar]
- 21.Sato S,, Ohi T,, Nishino I,, Sugie H. Confirmation of the efficacy of vitamin B6 supplementation for McArdle disease by follow-up muscle biopsy. Muscle Nerve 2012; 45 (3): 436–440. [DOI] [PubMed] [Google Scholar]
- 22.Quinlivan R,, Vissing J,, Hilton-Jones D,, Buckley J. Physical training for McArdle disease. Cochrane Database Syst Rev 2011; (12): CD007931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Musumeci O,, Bruno C,, Mongini T, et al. Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII). Neuromuscul Disord. 2012; 22 (4): 325–330. [DOI] [PubMed] [Google Scholar]
- 24.DiMauro S,, Spiegel R. Progress and problems in muscle glycogenoses. Acta Myol 2011; 30 (2): 96–102. [PMC free article] [PubMed] [Google Scholar]
- 25.Echaniz-Laguna A,, Akman HO,, Mohr M, et al. Muscle phosphorylase b kinase deficiency revisited. Neuromuscul Disord 2010; 20 (2): 125–127. [DOI] [PubMed] [Google Scholar]
- 26.Kraus AP,, Langston MF, Jr,, Lynch BL. Red cell phosphoglycerate kinase deficiency. A new cause of non-spherocytic hemolytic anemia. Biochem Biophys Res Commun 1968; 30 (2): 173–177. [DOI] [PubMed] [Google Scholar]
- 27.Salameh J,, Goyal N,, Choudry R, et al. Phosphoglycerate mutase deficiency with tubular aggregates in a patient from panama. Muscle Nerve 2013; 47 (1): 138–140. [DOI] [PubMed] [Google Scholar]
- 28.Serdaroglu G,, Aydinok Y,, Yilmaz S, et al. Triosephosphate isomerase deficiency: a patient with Val231Met mutation. Pediatr Neurol 2011; 44 (2): 139–142. [DOI] [PubMed] [Google Scholar]
- 29.Magoulas PL,, El-Hattab AW. Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J Rare Dis 2012; 7: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonnefont JP,, Bastin J,, Behin A,, Djouadi F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med 2009; 360 (8): 838–840. [DOI] [PubMed] [Google Scholar]
- 31.Roe CR,, Yang BZ,, Brunengraber H, et al. Carnitine palmitoyltransferase II deficiency: successful anaplerotic diet therapy. Neurology 2008; 71 (4): 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gillingham MB,, Scott B,, Elliott D,, Harding CO. Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab 2006; 89 (1–2): 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finsterer J. Inherited mitochondrial disorders. Adv Exp Med Biol 2012; 942: 187–213. [DOI] [PubMed] [Google Scholar]
- 34.Pfeffer G,, Chinnery PF. Diagnosis and treatment of mitochondrial myopathies. Ann Med 2013; 45 (1): 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis RL,, Sue CM. The genetics of mitochondrial disease. Semin Neurol 2011; 31 (5): 519–530. [DOI] [PubMed] [Google Scholar]
- 36.Goodfellow JA,, Dani K,, Stewart W, et al. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes: an important cause of stroke in young people. Postgrad Med J 2012; 88 (1040): 326–334. [DOI] [PubMed] [Google Scholar]
- 37.Kerr DS. Treatment of mitochondrial electron transport chain disorders: a review of clinical trials over the past decade. Mol Genet Metab 2010; 99 (3): 246–255. [DOI] [PubMed] [Google Scholar]
- 38.Garone C,, Tadesse S,, Hirano M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain 2011; 134 (pt 11): 3326–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang S,, Dimberg EL,, Milone M,, Wong LJ. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like phenotype: an expanded clinical spectrum of POLG1 mutations. J Neurol 2012; 259 (5): 862–868. [DOI] [PubMed] [Google Scholar]