

Abstract
Purpose of Review
To discuss the clinical, laboratory, and histopathologic features and presumed pathogenic mechanisms of the four major categories of idiopathic inflammatory myopathy, namely dermatomyositis, polymyositis, immune-mediated necrotizing myopathy, and inclusion body myositis.
Recent Findings
Dermatomyositis, polymyositis, necrotizing myopathy, and inclusion body myositis are clinically, histologically, and pathogenically distinct. Polymyositis is a T cell–mediated disorder directed against muscle fibers. The pathogenesis of dermatomyositis, necrotizing myopathy, and inclusion body myositis are unknown. Dermatomyositis, polymyositis, and necrotizing myopathy are generally, but not always, responsive to immunosuppressive therapy, in contrast to inclusion body myositis, which is generally refractory to therapy.
Summary
The pattern of muscle weakness, other clinical features (eg, rash, concurrent interstitial lung disease), laboratory features (creatine kinase, autoantibodies), and muscle biopsies are useful in distinguishing subtypes of inflammatory myopathy and in guiding treatment. More research is necessary to unravel the exact pathogenic bases of these myopathies and identify better treatments.
INTRODUCTION
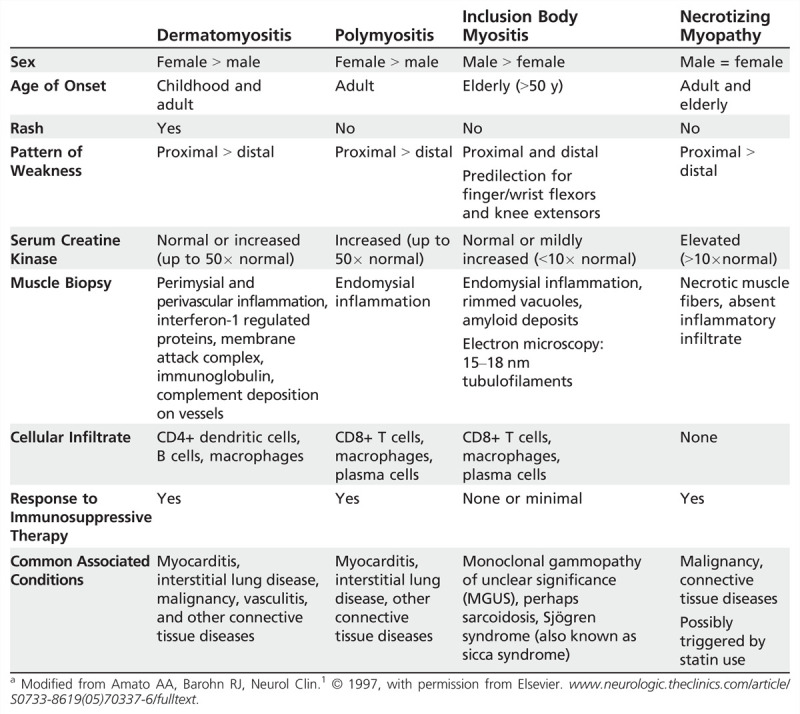
Idiopathic inflammatory myopathy can be broken into four major categories: dermatomyositis, polymyositis, immune-mediated necrotizing myopathy, and inclusion body myositis, which are clinically, histologically, and pathogenically distinct (Table 5-11).2,3,4,5,6,7,8,9,10 These disorders may occur in isolation or in association with cancer or with various connective tissue diseases (overlap syndromes).
Table 5-1.
Idiopathic Inflammatory Myopathies: Clinical and Laboratory Featuresa
DERMATOMYOSITIS
Clinical Features
The incidence of dermatomyositis is higher in women compared to men and can present at any age. Weakness can develop rather acutely (over several weeks) or insidiously (over months) and tends to affect the proximal greater than distal muscles in the legs more than the arms. Difficulties swallowing, chewing, and speaking occur in up to a third of patients secondary to masticatory, oropharyngeal, and esophageal muscle involvement. A characteristic rash typically accompanies or precedes the onset of muscle weakness. However, the rash can develop years after the onset of weakness, which could lead to an erroneous diagnosis of polymyositis. Additionally, some patients have a characteristic rash but never develop weakness (so-called amyopathic dermatomyositis or dermatomyositis sine myositis). Most patients with dermatomyositis have both skin and muscle involvement (Case 5-1), but on either end of the spectrum are rare patients who have only muscle or skin involvement.
Case 5-1
A 74-year-old woman began to notice a rash on her shins 3 months ago that gradually spread to cover the extensor surfaces of her upper limbs, then onto her face and trunk and the back of her neck. About 2 months ago, she started to notice weakness in her legs. She initially reported difficulty raising herself up off the toilet or chairs and walking up stairs. The weakness more recently spread to her arms. She denied shortness of breath at rest but had some dyspnea upon exertion. She had no myalgia, arthralgia, fever, chills, weight loss, or bowel or bladder problems.
Her medical history was remarkable for type 2 diabetes mellitus, for which she took glyburide. Family history was unrevealing.
Clinical examination was remarkable for a moderate-severe erythema on the patient’s face and scalp, neck and trunk (front and back), arms, knuckles (Gottron sign and papules), and periungual telangiectasia. She had mild alopecia as well.
Manual muscle testing revealed the following Medical Research Council scores: neck flexion 4−, neck extension 4+, shoulder abduction 4−, elbow extension and flexion 4, wrist flexion and extension 4+, hip flexion/abduction/extension 3, knee extension 5, knee flexion 4, ankle dorsiflexion 4+, and plantar flexion 5/5.
Her creatine kinase (CK) level was normal at 52 U/L. Her EMG revealed fibrillation potentials in proximal muscles as well as many small-amplitude, short-duration, polyphasic motor unit action potentials (MUAPs) that recruited very early. A biceps muscle biopsy was performed and revealed perivascular, perimysial inflammatory cell infiltration along with perifascicular atrophy. Jo-1 antibodies were not evident in her serum. Pulmonary function tests and ECG were normal. A malignancy workup was unrevealing. Dual-energy x-ray absorptiometry (DEXA) revealed osteoporosis.
Comment. This patient had classic clinical and histopathologic features of dermatomyositis, in which normal muscle enzymes (eg, CK) can be seen. She was started on prednisone 60 mg/d. Because of her diabetes and osteoporosis, she was also started on methotrexate 7.5 mg weekly at the same time. She gradually improved, and the treating physicians were able to slowly taper her off prednisone and maintain her on methotrexate 7.5 mg weekly.
The classical skin manifestations include a purplish discoloration of the eyelids (heliotrope rash), and papular, erythematous lesions over the knuckles (Gottron papules) (Figure 5-1). In addition, an erythematous, macular, sun-sensitive rash may appear on the face, neck, and anterior chest (V-sign); on the shoulders and upper back (shawl sign); and on the extensor surfaces of the elbows, knuckles, hips, knees, and malleoli (Gottron sign). The nail beds often have dilated capillary loops, occasionally with thrombi or hemorrhage. Subcutaneous calcifications may appear over pressure points (buttocks, knees, elbows), which can be complicated by ulceration of the overlying skin. Calcifications are more common in juvenile dermatomyositis, but some do develop in adult-onset cases.
Figure 5-1.
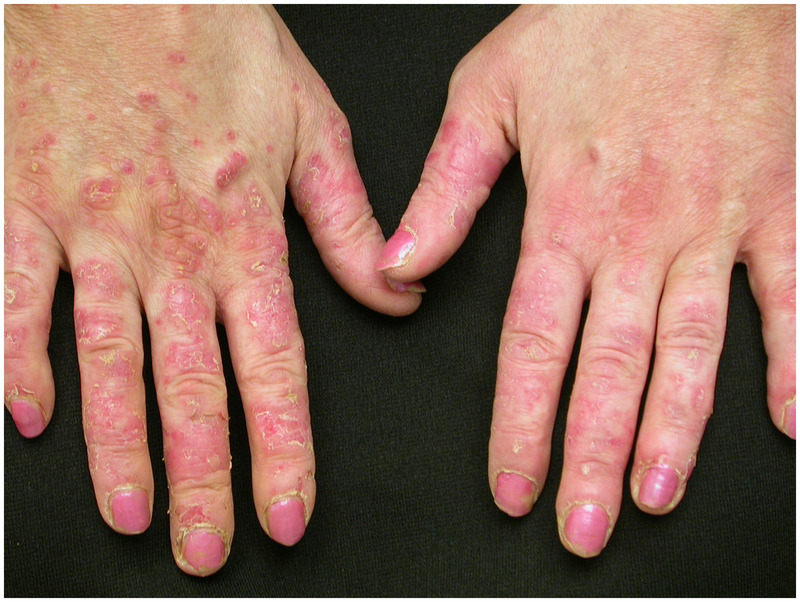
Dermatomyositis. Macular erythematous rash is seen over the extensor surface of the fingers along with cracked skin (mechanic hands) and nail bed changes.
Interstitial lung disease is a complication occurring in approximately 10% to 20% of patients with dermatomyositis and manifests as dyspnea and nonproductive cough. Pulmonary function tests reveal reduced forced vital capacity (FVC) and decreased diffusing capacity of lungs for carbon monoxide (DLCO). Antibodies directed against T-histidyl transfer RNA synthetase-so-called anti–Jo-1 antibodies-are present in at least 50% of interstitial lung disease cases associated with inflammatory myopathies. Cardiomyopathy manifesting as arrhythmias or congestive heart failure is a less frequent complication but one of which clinicians need to be aware. Involvement of the skeletal and smooth muscles of the gastrointestinal tract can lead to dysphagia, aspiration, and delayed gastric emptying. Vasculopathy of the gut can result in gastrointestinal hemorrhage; this appears to be more common in juvenile dermatomyositis.
Incidence of cancer is increased, ranging from 6% to 45%, in adult dermatomyositis (usually over the age of 40 years), with most cases occurring within the first 2 years of diagnosis of dermatomyositis. Risk of cancer is not increased in juvenile dermatomyositis, and no correlation has been shown between the severity of weakness, rash, or creatine kinase levels and malignancy in adult dermatomyositis.
Because of the increased risk of cancer, a comprehensive history and annual physical examinations are recommended with breast and pelvic examinations for women and testicular and prostate examinations for men to search for an underlying malignancy. In addition, laboratory studies should be obtained, including a complete blood count (CBC),routine blood chemistries, serum immunofixation/immunoelectrophoresis, serum free light chains, urinalysis, and stool specimens for occult blood. Chest, abdominal, and pelvic CT scans are recommended as well as pelvic ultrasound and mammogram for women. Colonoscopy should be done on all patients over the age of 50 years or in those who have attributable gastrointestinal symptoms (eg, abdominal pain, constipation, blood in the stool).
Laboratory Features
Blood work. Necrosis of muscle fibers usually leads to increases in serum creatine kinase (CK), aldolase, myoglobin, lactate dehydrogenase, aspartate aminotransferase (AST), and alanine aminotransferase (ALT) levels. Serum CK is elevated in approximately 70% of dermatomyositis patients. However, serum CK levels do not correlate with the severity of weakness and can be normal even in markedly weak individuals, particularly in childhood dermatomyositis patients and in those with slow, insidious disease. In approximately 10% of cases, an aldolase level is elevated while the serum CK is still within normal limits. Erythrocyte sedimentation rate (ESR) is usually normal or only mildly elevated and is not a reliable indicator of disease severity.
Antinuclear antibodies (ANAs) have been detected in 24% to 60% of patients with dermatomyositis and are much more common in patients with overlap syndromes. Some patients have so-called myositis-specific antibodies, which may be useful in predicting response to therapy and prognosis but have never been studied prospectively regarding their predictive value. These antibodies have not been demonstrated to be pathogenic and may just represent an epiphenomenon. Nevertheless, they may be helpful in categorizing syndromes. For example, the antisynthetases such as anti–Jo-1 antibodies are associated with interstitial lung disease and Raynaud phenomena. Anti–Mi-2 antibodies have been reported in patients with acute onset, severe rash, and a good response to therapy. Antibodies directed against melanoma differentiation-associated 5 (anti-MDA5), also known as anti–CADM-140 antibodies, are associated with aggressive interstitial lung disease in Asians. Autoantibodies to p155/140 targeting transcriptional intermediary factor 1-γ (TIF1-γ) are found in adult cancer-associated dermatomyositis with an 89% specificity and 70% sensitivity.
Imaging. MRI may demonstrate signal abnormalities in affected muscles secondary to inflammation, replacement by fibrotic tissue, or atrophy. Although MRI has been advocated as a tool to indicate which muscle to biopsy, the authors have found that it adds little to a good clinical examination and EMG in defining the pattern of muscle involvement and determining which muscle to biopsy.
EMG. In patients with myositis, the characteristic EMG features observed include (1) increased insertional and spontaneous activity with fibrillation potentials, positive sharp waves, and occasionally pseudomyotonic or complex repetitive discharges; (2) short-duration, low-amplitude, polyphasic motor unit action potentials (MUAPs); and (3) early recruitment. Decreased recruitment (fast-firing MUAPs) may occur in the presence of marked loss of muscle fibers from advanced disease. Decreased insertional activity may be seen in long-standing cases if fibrofatty replacement of muscles has occurred. In addition, large-duration, polyphasic MUAPs may also be evident later in long-standing disease due to muscle fiber splitting and regeneration rather than a superimposed neurogenic process.
The authors have found EMG helpful in determining which muscle to biopsy in patients with only mild weakness. Furthermore, EMG may also be useful in the assessment of previously responsive myositis patients who become weaker, by differentiating an increase in disease activity from weakness secondary to type 2 muscle fiber atrophy from disuse or chronic steroid administration. In active myositis, abnormal insertional and spontaneous activity is expected, while isolated type 2 muscle fiber atrophy is not associated with such abnormal activity on EMG. However, EMG may remain abnormal, reflecting previous muscle damage. In such cases, skeletal muscle MRI may be of use because increased signal abnormalities reflective of active inflammation would be expected in a relapse but not in cases of type 2 muscle fiber atrophy.
Histopathology
The classic histology of dermatomyositis is perifascicular atrophy (Figure 5-2), although this is typically a late finding and, in the authors’ experience, is found in less than 50% of patients. Inflammatory cells are located around blood vessels (perivascular) in the perimysium and composed primarily of macrophages, B cells, and CD4+ plasmacytoid dendritic cells. Unlike that seen in polymyositis and inclusion body myositis (discussed later), invasion of non-necrotic muscle fibers is not prominent in dermatomyositis.
Figure 5-2.
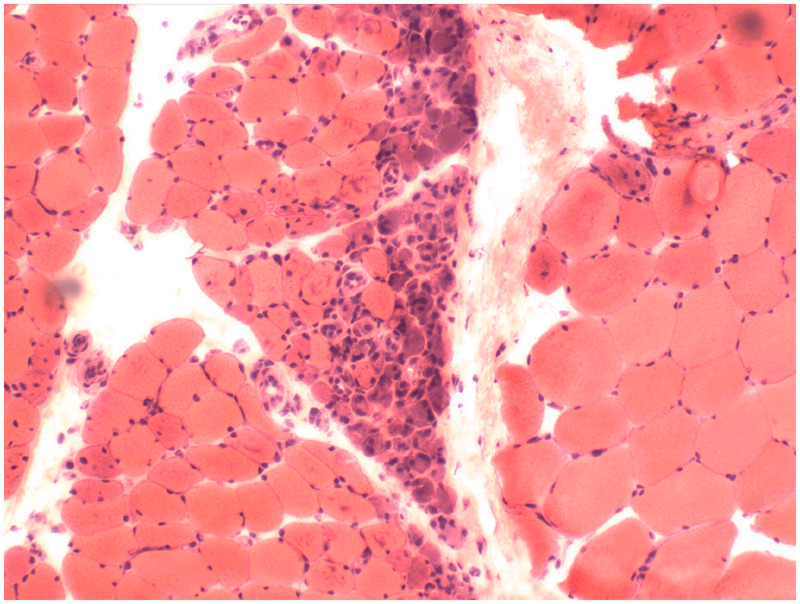
Dermatomyositis. Muscle biopsy demonstrates classic perifascicular atrophy of muscle fibers (hematoxylin and eosin stain).
Electron microscopy (EM) reveals small intramuscular blood vessels (arterioles and capillaries) with endothelial hyperplasia, microvacuoles, and tubuloreticular cytoplasmic inclusions; these abnormalities precede other structural abnormalities on EM. DNA microarray and immunohistochemistry studies of biopsied muscle tissue demonstrate an increased expression of genes induced by type 1 interferons.11 These interferons are synthesized by plasmacytoid dendritic cells in response to a serum factor or factors containing immune complexes of antibody, double-stranded DNA, or RNA viruses. Abundant plasmacytoid dendritic cells are evident in the dermatomyositis muscle biopsies. Increased expression of type 1 interferon-inducible proteins, such as myxovirus resistance 1 (MxA), are evident on blood vessels and muscle fibers (with a predilection for the perifascicular fibers). Interestingly, one postulated function of MxA is to form tubuloreticular inclusions around RNA viruses. These inclusions have the same morphology as the tubuloreticular inclusions seen on EM in blood vessels in dermatomyositis. Using immunoelectron microscopy, MxA was demonstrated within inclusions in vessels in dermatomyositis muscle biopsies.
Pathogenesis
Although capillary injury and perifascicular myofiber injury are key features of dermatomyositis muscle pathology, the central pathogenic disease question is how these two features relate-ie, whether the former causes the latter or some other mechanism causes them both. In this regard, the skin involvement of dermatomyositis has never been modeled as a result of ischemia. Dermatomyositis skin pathology is generally that of a cell-poor interface dermatitis in which loss of the basal layer of keratinocytes occurs in the absence or paucity of cellular inflammation. The topology of this cell loss is essentially the same as the perifascicular atrophy seen in muscle.
What dermatomyositis skin and muscle do have in common is the presence of molecular biomarkers of type 1 interferon (interferon-α, interferon-β, and others) exposure. The marked overproduction of type 1 interferon-inducible transcripts and proteins in muscle is remarkably unique to dermatomyositis in comparison to all other muscle diseases studied.11 Exposure of human skeletal muscle cell cultures to type 1 interferons produces a similar picture to that present in human dermatomyositis samples.
POLYMYOSITIS
Polymyositis is a heterogeneous group of disorders rather than a distinct entity. A major problem is the lack of universally accepted criteria for diagnosing polymyositis. The most commonly employed criteria were developed by Bohan and Peters in 1975,6,7 but these do not take into account advancements in our understanding of the immunopathogenesis of the various inflammatory myopathies or even the existence of inclusion body myositis and immune-mediated necrotizing myopathy. Several revised criteria for the various idiopathic inflammatory myopathies have been proposed. For definitive histopathologic diagnosis of polymyositis, many myopathologists want to see mononuclear inflammatory cells (CD8+ T cells) invading non-necrotic muscle fibers that express major histocompatibility 1 (MHC-1) antigen. However, this biopsy feature is also seen in inclusion body myositis and rarely in dystrophies. Further, mononuclear cell invasion of non-necrotic muscle fibers is uncommon in suspected cases of polymyositis, and some argue that it is not necessary for diagnosis of polymyositis. More frequently seen on biopsy are perivascular, perimysial inflammatory cell infiltrates, or endomysial inflammatory cells but no actual invasion of non-necrotic muscle fibers. Whether these cases represent polymyositis (with the absence of CD8+ T cells invading non-necrotic muscle fibers being due to sampling error), the same disorder as polymyositis, or a distinct type of inflammatory myopathy is unclear. Such perivascular, perimysial inflammation is common, particularly in patients with overlap myositis, but can be seen in dermatomyositis, inclusion body myositis, and occasionally in dystrophies.
For the various reasons listed above, it is impossible to derive from the literature the true incidence of polymyositis or its subtypes (eg, associated laboratory abnormalities or medical conditions-such as connective tissue disorders, interstitial lung disease, myocarditis, and cancer-that may accompany it) and prognosis. Prospective studies using more consensus-driven histopathologic criteria for polymyositis are needed to address these issues. Nevertheless, what is reported in the literature regarding polymyositis is summarized below.
Clinical Features
Polymyositis generally presents in patients over the age of 20 years and is more common in women. As with dermatomyositis, patients with polymyositis present with symmetric proximal arm and leg weakness that typically develops over several weeks or months. Approximately one-third of patients report swallowing difficulties. The cardiac and pulmonary complications of polymyositis are reportedly similar to those described in the dermatomyositis section. Myositis with secondary congestive heart failure or conduction abnormalities occurs in up to 1/3 of patients, but again, histopathologic confirmation of definite polymyositis using more up-to-date criteria is lacking in most of these studies.
Polyarthritis has been reported in as many as 45% of patients with polymyositis at the time of diagnosis. The risk of malignancy with polymyositis has been reported to be higher than the age-matched population but lower than that seen in dermatomyositis. However, it is likely that these epidemiologic studies included inclusion body myositis and dystrophy patients with inflammation, which are not associated with an increased risk of malignancy, so the true risk in polymyositis is probably higher than reported. Therefore, performance of a malignancy workup in patients with polymyositis is recommended, as discussed in the dermatomyositis section.
Laboratory Features
Blood work. Serum CK level is usually elevated fivefold or more in polymyositis cases. Unlike dermatomyositis and inclusion body myositis, in which the CK can be normal, the serum CK should never be normal in active polymyositis. Serum CK can be useful in monitoring response to therapy, but only in conjunction with the physical examination, as the CK level does not necessarily correlate with the degree of weakness.
Positive ANAs are reportedly present in 16% to 40% of patients with polymyositis. Again, however, the exact relationship of ANAs and connective tissue disorders in patients with histologically defined polymyositis is unclear. The questionable relationships of myositis-specific antibodies to polymyositis were previously addressed.
Imaging
MRI may demonstrate signal abnormalities in affected muscles secondary to inflammation and edema or replacement by fibrotic tissue (Figure 5-3).
Figure 5-3.
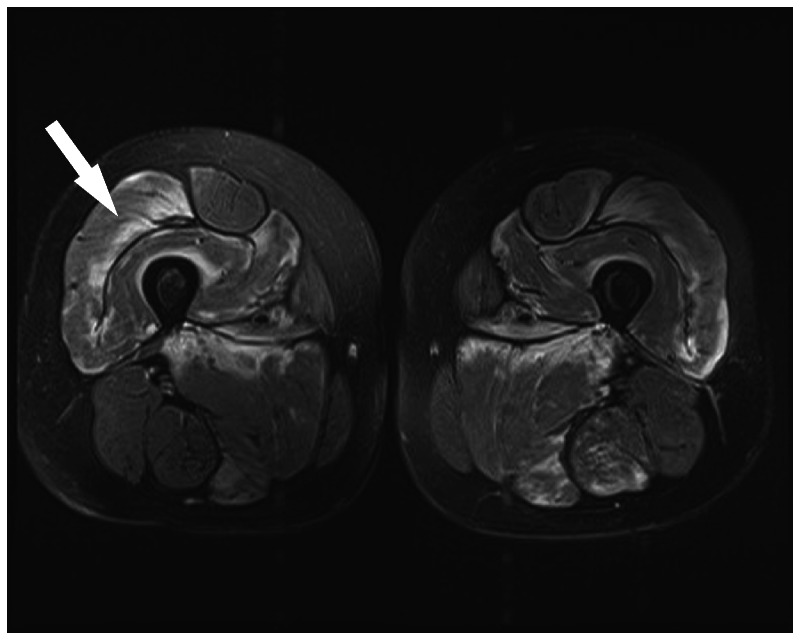
Skeletal muscle MRI scan. MRI (T2 with fat saturation) of thighs in a patient with polymyositis reveals increased signal in quadriceps (arrow) and, to a lesser extent, in the hamstrings.
EMG is usually abnormal in polymyositis, with increased insertional and spontaneous activity, small polyphasic MUAPs, and early recruitment. These abnormal features do not distinguish polymyositis from other inflammatory myopathies or myopathies with muscle membrane instability.
Histopathology
The predominant histologic features in polymyositis are variability in fiber size, scattered necrotic and regenerating fibers, and an inflammatory cell infiltrate (Figure 5-4). However, as mentioned previously, the specific characteristics of this inflammatory cell infiltrate have been a subject of recent debate. Small studies of polymyositis demonstrated that muscle biopsies demonstrate CD8+ T cells and macrophages invading non-necrotic muscle fibers expressing MHC-1 antigen. Subsequently, some authorities have argued that this histopathologic feature is required for the diagnosis of definite polymyositis. Other authorities feel that invasion of non-necrotic muscle fibers is not necessary and perivascular, perimysial, or endomysial inflammation without actual invasion of non-necrotic muscle fibers can suffice for diagnosis of polymyositis in the proper clinical context. In the authors’ opinion, however, demonstrating invasion of non-necrotic endomysial muscle fibers by T cells is required to make a definite diagnosis of polymyositis on histopathologic grounds, as perivascular, perimysial, and even endomysial inflammatory cell infiltrates can be seen in inclusion body myositis, dermatomyositis, and dystrophies.
Figure 5-4.

Polymyositis. Muscle biopsy demonstrates endomysial mononuclear inflammatory cell infiltrate surrounding and invading non-necrotic muscle fibers (modified Gomori one-step trichrome stain).
The endomysial inflammatory cells consist primarily of activated CD8+ (cytotoxic) alpha-beta T cells and macrophages. Although B cells are rare, plasma cells are common in the endomysium and likely account for the increased expression of immunoglobulin genes on microarray experiments.
Pathogenesis
Polymyositis is likely caused by a human leukocyte antigen–restricted, antigen-specific, cell-mediated immune response directed against muscle fibers. The trigger of this autoimmune attack is not known, but viral infections have been speculated. However, no conclusive evidence supports this hypothesis.
OVERLAP SYNDROMES
The term “overlap syndrome” is applied when dermatomyositis or polymyositis is associated with another well-defined connective tissue disorder such as scleroderma, mixed connective tissue disease, Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis. Retrospective series of patients suggest that the myositis associated with overlap syndromes is more responsive to immunosuppressive treatment than isolated dermatomyositis and polymyositis, but again, prospective studies are lacking.
INCLUSION BODY MYOSITIS
Clinical Features
Inclusion body myositis is characterized by slowly progressive proximal and distal weakness in the arms and legs that usually develops after the age of 50 years (see Table 5-11). Many experts consider it to be the most common myopathy (apart from sarcopenia of aging) in patients over the age of 50 years. The slow, progressive nature of the myopathy probably accounts in part for the delay in diagnosis that averages 6 to 7 years after the onset of symptoms. Unlike dermatomyositis and polymyositis, inclusion body myositis is more common in men than in women.
The clinical hallmark of inclusion body myositis is early weakness and atrophy of the quadriceps, flexor forearm muscles (ie, wrist and finger flexors), and the ankle dorsiflexors, that is often asymmetric. Many patients develop dysphagia than can be severe enough to require esophageal dilation or cricopharyngeal myotomy. Facial weakness can also be demonstrated. No association with myocarditis, lung disease, or malignancy is evident. The course of the disease is a slow progression, and it does not typically respond to immunotherapies.
Laboratory Features
Blood work. Serum CK is normal or only mildly elevated (usually less than 10-fold above normal). Some clinicians have reported positive ANAs in approximately 20% of their patients with inclusion body myositis. Antibodies directed against cytosolic 5′-nucleotidase 1A (cN1A) have been detected in as many as two-thirds of inclusion body myositis patients, whereas their prevalence in dermatomyositis, polymyositis, and other neuromuscular disorders is much lower.12,13
Imaging. Skeletal muscle MRI scans demonstrate atrophy and signal abnormalities in affected muscle groups. In the thighs, there appears to be a predilection for the vastus lateralis and medialis with sparing of the rectus femoris (Figure 5-5).
Figure 5-5.
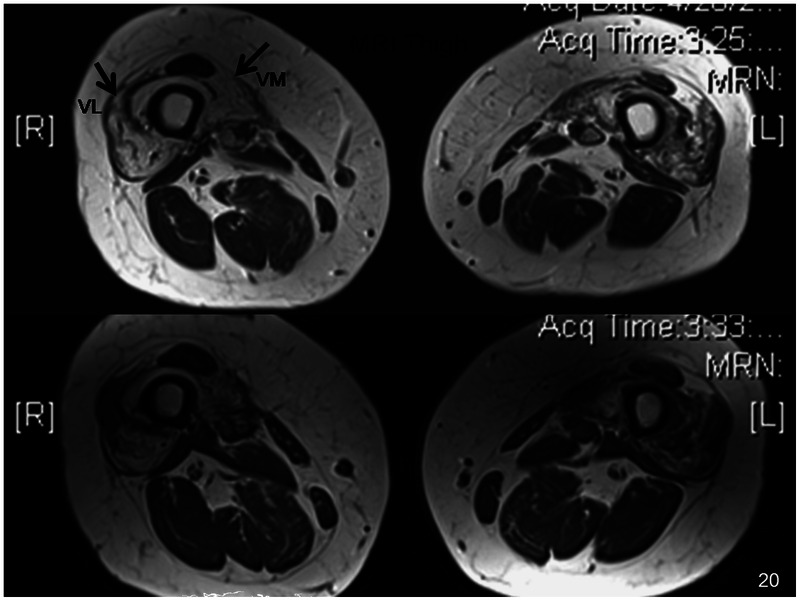
Inclusion body myositis. Skeletal muscle MRI scan of thighs reveals atrophy of muscles and fibrofatty replacement, particularly affecting the vastus lateralis (VL) and vastus medialis (VM) muscles (arrows) with relative sparing of the rectus femoris muscle in between.
EMG. Nerve conduction studies reveal evidence of a mild axonal sensory neuropathy in up to 30% of patients. EMG demonstrates increased spontaneous and insertional activity, small polyphasic MUAPs, and early recruitment. In addition, large polyphasic MUAPs can also be demonstrated in one-third of patients. This finding may lead to the misinterpretation of a neurogenic process and misdiagnosis in some patients as having ALS. However, large polyphasic MUAPs can also be seen in myopathies (ie, polymyositis, dermatomyositis, muscular dystrophies), which probably reflects the chronicity of the disease process rather than a neurogenic etiology.
Histopathology
Muscle biopsy characteristically reveals endomysial inflammation, small groups of atrophic fibers, eosinophilic cytoplasmic inclusions, and muscle fibers with one or more rimmed vacuoles lined with granular material (Figure 5-6). Endomysial inflammatory cells (macrophages and CD8+ lymphocytes) appear to surround and invade non-necrotic fibers. MHC-1 antigens are expressed on necrotic and non-necrotic muscle fibers. The T-cell receptor repertoire of the inflammatory cells have an oligoclonal pattern of gene rearrangement, although there is heterogeneity in the CDR3 domain. These findings suggest that the T-cell response is not directed against a muscle-specific antigen, although a superantigen could trigger the response. Oligoclonal plasma cells are also quite prominent in the endomysium, but their pathogenic role is unclear. Amyloid deposition in vacuolated muscle fibers and, to a lesser extent, within nuclei can be demonstrated on Congo red staining using polarized light or fluorescence techniques. The numbers of ragged red fibers and cyclooxygenase (COX)–negative fibers are also increased. On EM, 15-nm to 21-nm cytoplasmic and intranuclear tubulofilaments are found in vacuolated muscle fibers, although they can be difficult to see. Not all of the features of inclusion body myositis are evident on any given muscle biopsy, which probably accounts for many cases of inclusion body myositis being misdiagnosed as polymyositis (Case 5-2).
Figure 5-6.
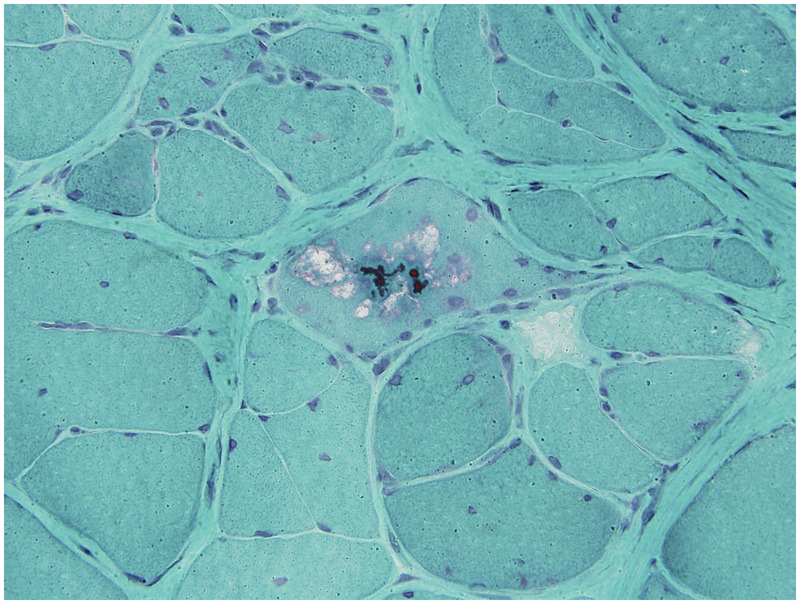
Inclusion body myositis. Muscle biopsy reveals muscle fiber with rimmed vacuole and cytoplasmic body inclusions (modified Gomori one-step trichrome stain).
Case 5-2
A 71-year-old right-handed man was incidentally noted to have slightly elevated serum creatine kinase (CK) levels for the past 10 years during his routine cardiology checkups, in the 400 U/L to 600 U/L range. Over the past 6 years, he had noticed increasing difficulty with walking, particularly involving climbing stairs. He also noted difficulty grasping with his hands and mild difficulty swallowing.
He was seen by an outside provider, who performed a biceps muscle biopsy that was interpreted as polymyositis. He was treated with high-dose prednisone, and then methotrexate was added. His serum CK decreased to the normal range, but he did not feel his strength or function had improved. Therefore, the prednisone and methotrexate were discontinued, and the patient was referred for a second opinion.
His medical history was remarkable for hypertension and coronary artery disease. He took hydrochlorothiazide and aspirin. Family history was unremarkable.
Physical examination was remarkable for moderate atrophy of his thighs and forearms bilaterally. Manual muscle testing revealed the following Medical Research Council scores: neck flexors 4, neck extension 5, shoulder abduction 5, elbow flexion and extension 4, wrist extension 4+, wrist flexion 4−, finger extension 4 on the right and 4− on the left, deep finger flexors 4−, flexor pollicis longus 4−, hip flexion/abduction/extension 4, knee extension 3−, knee flexion 4−, foot dorsiflexion 0, and ankle plantar flexion 5. He had a steppage gate that was stable with a cane. The patient was unable to get out of a chair without using his arms. Deep tendon reflexes were 3 at the biceps, triceps, brachioradialis, and knees, and 2 at the ankles. Plantar responses were flexor bilaterally.
His outside muscle biopsy slides revealed moderate variation in myofiber size, with many atrophic fibers (some in small groups) and rare slightly hypertrophic fibers measuring up to 120 μm. Mononuclear inflammatory cell infiltrates were present in the endomysium and perimysium, with inflammatory cells surrounding and invading non-necrotic muscle fibers. Scattered muscle fibers contained rimmed vacuoles and eosinophilic inclusions.
Comment. This patient had a classic clinical pattern of weakness seen in inclusion body myositis. His muscle biopsy also had typical features, but these were missed by the outside pathologist. At any rate, as many as 30% of muscle biopsies in such patients do not show all of the typical features (eg, rimmed vacuoles or inclusions), which is why it is imperative to do a good clinical examination. Not recognizing that he had a typical pattern of weakness seen in inclusion body myositis led to a misdiagnosis of polymyositis and treatment with prednisone and methotrexate, which were not effective. He was referred for physical, occupational, and speech therapy and given ankle-foot orthoses, adaptive equipment, and home exercises, which improved his function and quality of life.
Pathogenesis
Inclusion body myositis is a poorly understood disease. Although various pathophysiologic aspects have been reported, understanding of any underlying unifying mechanism is lacking. This topic has been recently reviewed,14 and that review is paraphrased here. Two aspects that have been studied are myonuclear degeneration and autoimmunity.
Degeneration of myonuclei and their association with rimmed vacuoles were noted in histochemical studies. Most rimmed vacuoles contain nuclear proteins and are derived from nuclei. Immunohistochemical evidence shows that rimmed vacuoles are lined with the nuclear membrane proteins lamin A/C and emerin as well as other nuclear proteins (histone H1, histone 2AX, DNA-PK, Hu70, and Hu80). An accumulation of mislocalized nucleic acid–binding proteins (including TDP-43, a predominantly nuclear heterogeneous nuclear ribonucleoprotein [hnRNP] that undergoes nucleocytoplasmic shuttling and associates with translation machinery in the cytoplasm) was identified in inclusion body myositis nonnuclear sarcoplasm.
Of the four largest categories of inflammatory myopathy, inclusion body myositis muscle has the greatest degree of adaptive immune response. Highly refined T-cell and B-cell responses are present in inclusion body myositis muscle, and the disease has commonly been categorized as having prominent cytotoxic T-cell destruction of myofibers. Extensive research on inclusion body myositis muscle T cells has failed to define their autoantigen, but recent studies have demonstrated a prominent B-cell response against a 43-kDa muscle protein identified as cN1A (also known as NT5C1A).12,13 The detection of serum and plasma antibodies against this muscle protein is highly sensitive and specific for inclusion body myositis among muscle diseases. Furthermore, the immunohistochemical localization of cN1A protein in areas of rimmed vacuole formation suggest that inclusion body myositis myonuclear degeneration and autoimmunity are mechanistically linked, but does not indicate that these anti-cN1A antibodies are themselves pathogenic.
IMMUNE-MEDIATED NECROTIZING MYOPATHY
Clinical Features
This category of idiopathic myopathy has only been recently identified as a probable distinct autoimmune myositis. In the authors’ experience, nearly 20% of inflammatory myopathy patients have immune-mediated necrotizing myopathy. Patients present with proximal weakness and often myalgia that may begin acutely or more insidiously. Patients may have an underlying connective tissue disease (usually scleroderma or mixed connective tissue disease) or cancer (paraneoplastic necrotizing myopathy) or be idiopathic. The most common associated malignancies are gastrointestinal tract adenocarcinomas and small cell and non–small cell carcinomas of the lung. Again, as in polymyositis and dermatomyositis, a malignancy workup should be performed on all patients. In the authors’ experience, most patients have immune-mediated necrotizing myopathy that was likely triggered by statin use (discussed in greater detail in “Toxic Myopathies,” by Dr Andrew L. Mammen in this issue of CONTINUUM). Patients generally improve with immunosuppressive and immunomodulating therapies but, in the authors’ experience, are more difficult to treat than those with dermatomyositis or polymyositis. Most patients require corticosteroids plus another immunosuppressive agent and occasionally IV immunoglobulin (IVIg) or rituximab (Case 5-3).
Case 5-3
A 61-year-old woman with a history of diabetes and hyperlipidemia presented with a 3-month history of soreness and weakness in her arms and legs. Six months earlier she was noted to have creatine kinase (CK) levels over 3400 U/L. She had been on simvastatin at that time but had no muscle weakness. The statin was stopped, but the CK continued to rise, and she had become weak. Her medical history was otherwise unremarkable.
Physical examination was remarkable for the absence of any rash. Manual muscle testing revealed the following Medical Research Council scores: neck flexion 4−, neck extension 4+, shoulder abduction 4, elbow flexion/extension 4+, wrist extension/flexion 5, finger extension/finger flexion 5, hip flexion/extension/abduction 4−, knee flexion 4+, knee extension 5, ankle dorsiflexion 4+, and ankle plantar flexion 5.
Her serum CK was over 10,000 U/L. An EMG was not done as it was obvious that with a CK of that magnitude she had a myopathy. A biceps muscle biopsy revealed many scattered regenerating and necrotic fibers undergoing myophagocytosis. Aside from the inflammatory cells invading necrotic muscle fibers, inflammation was minimal. Anti–signal recognition particle (SRP) antibodies were tested for but absent. A malignancy workup was negative.
Comment. The presumptive diagnosis was immune-mediated necrotizing myopathy that was most likely triggered by statin use. She was started on both prednisone and methotrexate because this disorder can be difficult to treat and because she also had diabetes. She slowly improved but flared when prednisone was tapered below 20 mg/d. Therefore, IV immunoglobulin (IVIg) was added, and the treating physicians subsequently were able to taper her off prednisone.
Laboratory Features
Blood work. Serum CK is usually markedly elevated. Antinuclear antibodies suggestive of an underlying connective tissue disorder may be found. Patients with immune-mediated necrotizing myopathy may have anti–signal recognition particle (SRP) antibody, which is associated with an acute-onset necrotizing myopathy that may be associated with a dilated cardiomyopathy and often is poorly responsive to standard immunosuppression. Recent studies have demonstrated that patients with statin-associated immune-mediated necrotizing myopathy, particularly if over 50 years of age, often have autoantibodies directed, interestingly enough, against hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase.15,16 These antibodies are not typically found in patients who take statins but have no symptoms or in those who have myopathic symptoms/signs that reverse upon discontinuation of statins.
Imaging. MRI may demonstrate signal abnormalities in affected muscles secondary to inflammation and edema. EMG demonstrates increased insertional and spontaneous activity, myopathic MUAPs, and early recruitment similar to the other described inflammatory myopathies.
Histopathology
The most prominent feature on muscle biopsy is scattered necrotic muscle fibers (Figure 5-7). By Bohan and Peters criteria,6,7 patients with these necrotizing myopathies might be misdiagnosed as polymyositis. Inflammatory cell infiltration is sparse and confined mainly to necrotic muscle fibers. The sarcolemma of non-necrotic muscle fibers usually express MHC-1, and membrane attack complex deposition may occur as well. Thickened, pipestem capillaries may be evident on routine histochemistry and EM.
Figure 5-7.
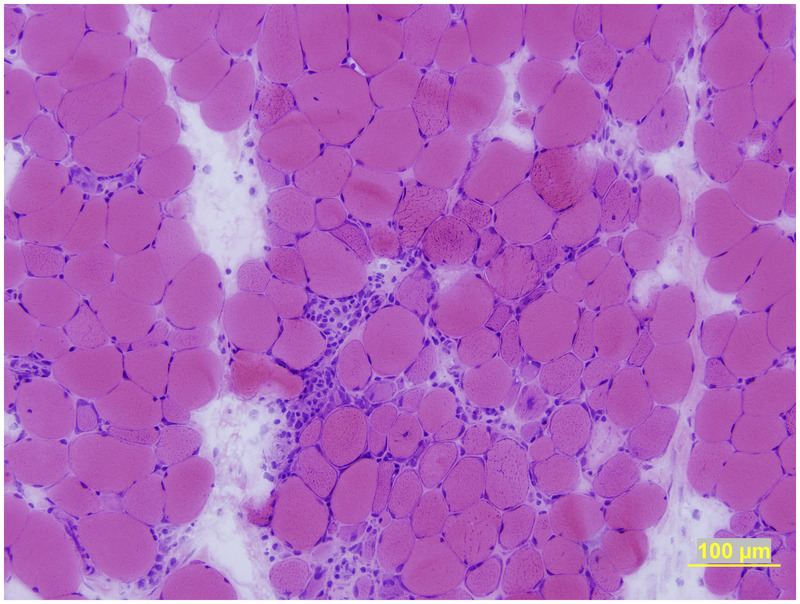
Necrotizing myositis. Muscle biopsy reveals scattered necrotic fibers, some in the process of undergoing phagocytosis. Unlike polymyositis, there is scant, if any, inflammatory cell infiltrate, except in fibers undergoing phagocytosis (hematoxylin and eosin stain).
Pathogenesis
The pathogenesis of this necrotizing myopathy is unknown; however, the deposition of membrane attack complex on small arterioles and capillaries with thickened endothelial walls suggests a humorally mediated microangiopathy.
TREATMENT OF INFLAMMATORY MYOPATHIES
Dermatomyositis, polymyositis, and necrotizing myopathy typically respond to immunotherapy, while inclusion body myositis usually does not. Although class 1 evidence of any specific therapy is lacking, the authors have used several standard treatments based on experience. There is no one correct way to treat myositis, and the authors refer readers to several reviews.2,5 That said, the standard of care is high-dose corticosteroids. Equipoise exists regarding when to start second-line agents (see below). Concurrently, the authors recommend physical, occupational, and speech/swallowing therapy as warranted.
Corticosteroids. Corticosteroids are considered the first-line treatment of choice for dermatomyositis, polymyositis, and necrotizing myopathy. Approaches to dosing steroids vary; the most common regimen is to start patients on high-dose daily prednisone (eg, 60 mg/d or 0.75 mg/kg/d to 1.5 mg/kg/d). In patients with severe weakness, a short course of IV methylprednisolone (1 g/d for 3 days) may be given, continuing with high-dose prednisone until muscle strength normalizes, improvement in strength has reached a plateau, or at least normalization of the serum CK occurs (this typically takes 3 to 6 months). Thereafter, the authors decrease the prednisone by 10 mg/d every 4 weeks until the patient is on 20 mg/d; at this dose, prednisone is decreased by 5 mg/d every 4 weeks until the patient is on 10 mg/d, then decreased by 2.5 mg/d every 4 weeks. Although most patients improve, the response may not be complete, and most will require at least a small dose of prednisone or a second-line agent to have a sustained remission. In those patients who do not respond at all to high-dose prednisone, the clinician needs to consider alternative disorders (eg, inclusion body myositis or an inflammatory muscular dystrophy).
As noted above, equipoise also exists regarding when to start second-line agents (eg, methotrexate, azathioprine, mycophenolate, or immunoglobulin). The clinician must review with the patient the increased risks of immunosuppression versus possible benefits (eg, faster improvement, steroid-sparing effect). The authors usually start a second-line agent along with corticosteroids in patients with severe weakness or other organ system involvement (eg, myocarditis, interstitial lung disease), those with increased risk of steroid complications (eg, diabetics, patients with osteoporosis, or postmenopausal women), and those known to have difficult-to-treat myositis (eg, immune-mediated necrotizing myopathy). A second-line agent should also be strongly considered in patients who fail to significantly improve after 2 to 4 months of treatment or who experience an exacerbation during treatment with prednisone. The authors follow the serum CK levels; however, adjustments of immunotherapies are primarily based on the objective clinical examination. While an increasing serum CK may precede a relapse, the authors would not increase dosages unless it were accompanied by increased weakness; in such cases, it is advisable to hold the dosages and follow the patient more closely.
Concurrent Management. In patients who have interstitial lung disease or are on prednisone plus another immunosuppressive agent, the authors start a prophylactic antibiotic for pneumocystis (eg, sulfamethoxazole and trimethoprim). Because of the potential for osteoporosis with chronic steroid administration, the authors obtain a baseline dual-energy x-ray absorptiometry (DEXA) and follow yearly while the patient is on corticosteroids. The authors also start calcium supplementation (1 g/d) and vitamin D (800 IU/d). Depending on results of the DEXA and other risk factors (eg, postmenopausal status), the authors may also treat patients with a bisphosphonate. In addition, patients are counseled on a low-sodium, low-carbohydrate, high-protein diet, and treating physicians should monitor fasting blood glucose and serum potassium levels, blood pressure, and eyes (for cataracts) while patients are on high doses of prednisone.
Second-Line Therapies
Methotrexate. Methotrexate is often regarded as the second-line treatment of choice in dermatomyositis, polymyositis, and immune-mediated necrotizing myopathy. The authors usually initiate methotrexate orally at 7.5 mg/wk, gradually increased by 2.5mg each week or two up to 25mg/wk as appropriate and tolerated. The dosage needs to be adjusted in patients with renal insufficiency. All patients are concomitantly given folate or folinic acid. Methotrexate has many potential side effects (Table 5-21). Because one rare side effect of methotrexate is pulmonary fibrosis, the authors typically avoid using it in patients with interstitial lung disease or Jo-1 antibodies. In patients treated with methotrexate, pulmonary function tests (FVC and DLCO) should be periodically repeated. The authors monitor the pulmonary function tests (FVC and DLCO), CBC, renal and liver function tests (ALT, AST, bilirubin), and gamma-glutamyl transpeptidase (GGT). GGT is the most reliable indicator of hepatic dysfunction, because AST and ALT can be elevated from muscle involvement alone.
Table 5-2.
Immunosuppressive/Immunomodulating Therapy for Inflammatory Myopathiesa
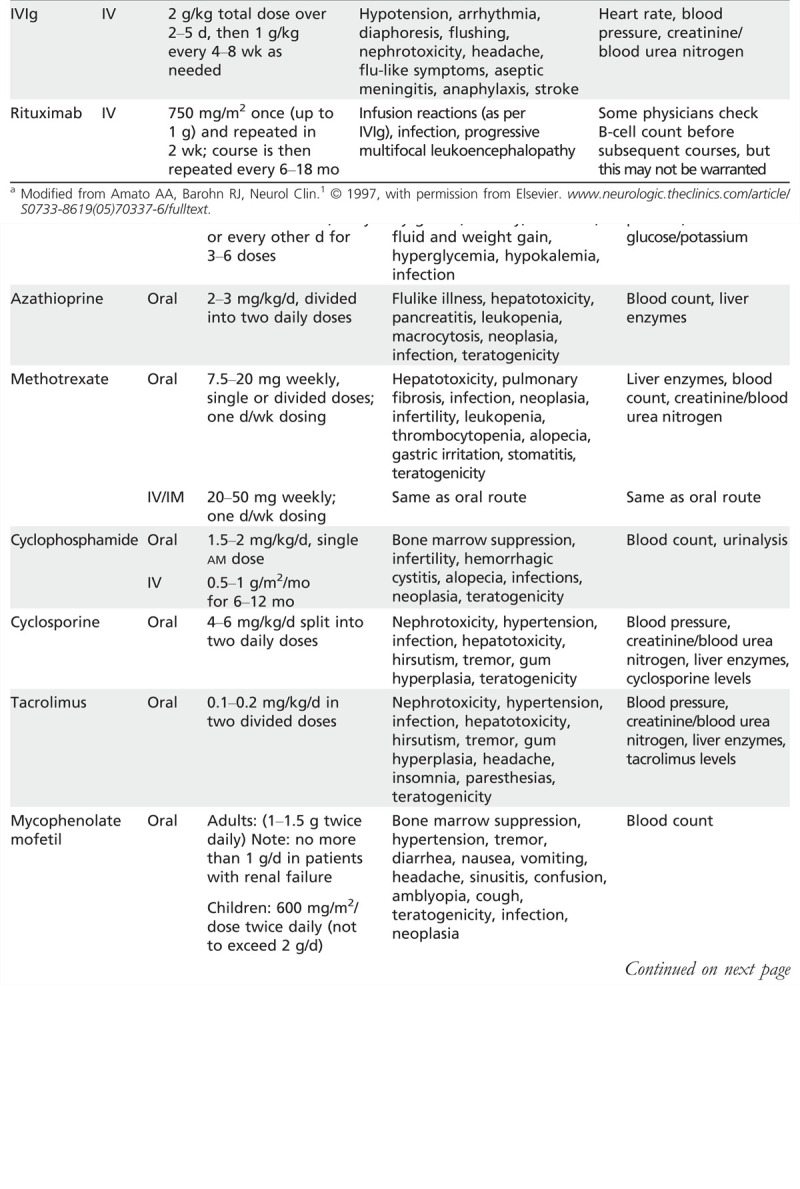
Azathioprine. Azathioprine is another frequently used second-line agent, but the authors prescribe it less often than methotrexate, as it tends to take a little longer to work. The authors usually screen patients for thiopurine methyltransferase (TPMT) deficiency; the authors avoid using azathioprine in those who are homozygous for TPMT mutations because of an increased risk for severe bone marrow toxicity. The authors begin azathioprine at 50 mg twice daily and then increase 50 mg every 2 weeks up to 2 mg/kg/d to 3 mg/kg/d. Approximately 12% of patients do not tolerate azathioprine and develop fever, abdominal pain, nausea, vomiting, and anorexia. Azathioprine has several other potential side effects (see Table 5-21). Allopurinol should be avoided, because combination with azathioprine increases the risk of bone marrow and liver toxicity.
Mycophenolate mofetil. The authors use mycophenolate mofetil in patients who do not tolerate methotrexate or azathioprine or in whom those treatments are contraindicated. The authors initiate treatment at 1.0 g twice daily; it can be increased to 3 g/d in divided doses as needed. In patients with renal insufficiency, the dosage is limited to 500 mg twice daily.
IV immunoglobulin. The authors typically reserve treatment with IVIg in polymyositis, dermatomyositis, and immune-mediated necrotizing myopathy patients who are refractory to prednisone and one of the other above second-line agents. However, in patients with severe myositis, the authors may start treatment with a triple cocktail (eg, prednisone, methotrexate, and IVIg). The authors give IVIg (2 g/kg total dose) over 2 to 5 days and repeat infusions performed at monthly intervals for at least 3 months, then decrease the dosage to 1 g/kg/month.
Rituximab. Rituximab is a monoclonal antibody that targets B cells. Studies involving small series of patients have suggested that rituximab may be an effective therapy in dermatomyositis and polymyositis. A large, prospective, double-blind NIH trial found no benefit, but the study design had significant flaws. In the authors’ experience, rituximab may be beneficial in patients with refractory dermatomyositis, polymyositis, and immune-mediated necrotizing myopathy. The authors use it in patients who are refractory to prednisone and at least one of the other second-line agents discussed above, at a dosage of 750 mg/m2 (up to 1 g) IV, with the infusion repeated in 2 weeks. A course of rituximab, as above, is usually repeated every 6 to 18 months. There is a risk of progressive multifocal leukoencephalopathy (PML), which, although low, should be discussed with patients before prescribing rituximab.
Cyclosporine and tacrolimus. Cyclosporine and tacrolimus may be beneficial, but their cost and potential side effects (see Table 5-21) have limited their use. Cyclosporine is started at a dose of 3.0 mg/kg/d to 4.0mg/kg/d in two divided doses and gradually increased to 6.0 mg/kg/d as necessary to achieve a trough serum cyclosporine level of 50 ng/mL to 200 ng/mL. The authors start tacrolimus at a dose of 0.1 mg/kg and increase it up to 0.2 mg/kg (in two divided doses daily) as needed to achieve a trough level of 5 ng/mL to 15 ng/mL. Blood pressure, electrolytes, renal function, CBC, and liver function tests are monitored closely, and the doses are adjusted if renal insufficiency develops.
Cyclophosphamide. Cyclophosphamide is rarely used because of its potential side effects, and the authors reserve it for patients refractory to other treatments discussed above. Typically, the authors treat patients with IV cyclophosphamide at 0.5 g/m2/mo to 1 g/m2/mo for 6 to 12 months. Cyclophosphamide can also be given orally (1.0 mg/kg/d to 2.0 mg/kg/d), although oral administration may present a greater risk of hemorrhagic cystitis. Before IV cyclophosphamide treatment, patients should be hydrated with IV fluids to help avoid hemorrhagic cystitis. Urinalysis and CBC should be monitored closely.
Tumor necrosis factor-α (TNF-α) blockers. The results of these agents in the myositides have been mixed, and therefore the authors tend to avoid them.
Other Therapies. Physical and occupational therapy are important and may help improve function and reduce type 2 fiber muscle atrophy associated with chronic steroids and inactivity. Speech/swallowing therapy is beneficial in patients with dysphagia, which is very common in inclusion body myositis patients and often requires esophageal dilatation or cricopharyngeal myotomy. Some patients need a feeding tube to prevent recurrent aspiration.
SUMMARY
Dermatomyositis, polymyositis, necrotizing myopathy, and inclusion body myositis are clinically, histologically, and pathogenically distinct. In particular, the pattern of muscle involvement, other organ system involvement, the presence of certain autoantibodies, and muscle biopsy findings are useful in distinguishing these different types of inflammatory myopathy. Polymyositis is a T cell–mediated disorder directed against muscle fibers. The pathogenesis of dermatomyositis, necrotizing myopathy, and inclusion body myositis are unknown. Dermatomyositis, polymyositis, and necrotizing myopathy are generally, but not always, responsive to immunosuppressive therapy, in contrast to inclusion body myositis, which is generally refractory to therapy.
KEY POINTS
Idiopathic inflammatory myopathy can be broken into four major categories: dermatomyositis, polymyositis, immune-mediated necrotizing myopathy, and inclusion body myositis, which are clinically, histologically, and pathogenically distinct.
Most patients with dermatomyositis have both skin and muscle involvement, but on either end of the spectrum are rare patients who have only muscle or skin involvement.
Interstitial lung disease is a complication occurring in approximately 10% to 20% of patients with dermatomyositis and manifests as dyspnea and nonproductive cough.
Incidence of cancer is increased, ranging from 6% to 45%, in adult dermatomyositis (usually over the age of 40 years), with most cases occurring within the first 2 years of diagnosis of dermatomyositis.
Serum creatine kinase levels do not correlate with the severity of weakness and can be normal even in markedly weak individuals, particularly in childhood dermatomyositis patients and in those with slow, insidious disease.
MRI in patients with dermatomyositis may demonstrate signal abnormalities in affected muscles secondary to inflammation, replacement by fibrotic tissue, or atrophy.
In dermatomyositis, decreased recruitment (fast-firing motor unit action potentials) may occur in the presence of marked loss of muscle fibers from advanced disease.
The classic histology of dermatomyositis is perifascicular atrophy, although this is typically a late finding and in the authors’ experience is found in less than 50% of patients.
For definitive histopathologic diagnosis of polymyositis, many myopathologists want to see mononuclear inflammatory cells (CD8+ T cells) invading non-necrotic muscle fibers that express major histocompatibility 1 antigen.
As with dermatomyositis, patients with polymyositis present with symmetric proximal arm and leg weakness that typically develops over several weeks or months.
Performance of a malignancy workup in patients with polymyositis is recommended.
Serum creatine kinase can be useful in monitoring response to therapy in dermatomyositis, polymyositis, and immune-mediated necrotizing myopathy, but only in conjunction with the physical examination, as the creatine kinase level does not necessarily correlate with the degree of weakness.
The predominant histologic features in polymyositis are variability in fiber size, scattered necrotic and regenerating fibers, and an inflammatory cell infiltrate.
Demonstrating invasion of non-necrotic endomysial muscle fibers by T cells is required to make a definite diagnosis of polymyositis on histopathologic grounds, as perivascular, perimysial, and even endomysial inflammatory cell infiltrates can be seen in inclusion body myositis, dermatomyositis, and dystrophies.
The endomysial inflammatory cells consist primarily of activated CD8+ (cytotoxic) alpha-beta T cells and macrophages.
Inclusion body myositis is characterized by slowly progressive proximal and distal weakness in the arms and legs that usually develops after the age of 50 years.
The clinical hallmark of inclusion body myositis is early weakness and atrophy of the quadriceps, flexor forearm muscles (ie, wrist and finger flexors), and the ankle dorsiflexors that is often asymmetric.
Antibodies directed against cytosolic 5′-nucleotidase 1A have been detected in as many as two-thirds of patients with inclusion body myositis, whereas their prevalence in dermatomyositis, polymyositis, and other neuromuscular disorders is much lower.
Muscle biopsy in patients with inclusion body myositis characteristically reveals endomysial inflammation, small groups of atrophic fibers, eosinophilic cytoplasmic inclusions, and muscle fibers with one or more rimmed vacuoles lined with granular material.
Not all of the features of inclusion body myositis are evident on any given muscle biopsy, which probably accounts for many cases of inclusion body myositis being misdiagnosed as polymyositis.
In most patients who have immune-mediated necrotizing myopathy, the disease was likely triggered by statin use.
Most patients with immune-mediated necrotizing myopathy require corticosteroids plus another immunosuppressive agent and occasionally IV immunoglobulin or rituximab.
Patients with immune-mediated necrotizing myopathy may have anti–signal recognition particle antibody, which is associated with an acute-onset necrotizing myopathy that may be associated with a dilated cardiomyopathy and often is poorly responsive to standard immunosuppression.
Patients with statin-associated immune-mediated necrotizing myopathy, particularly if over 50 years of age, often have autoantibodies directed, interestingly enough, against hydroxymethylglutaryl coenzyme A reductase.
The most prominent feature of immune-moderated necrotizing myopathy on muscle biopsy is scattered necrotic muscle fibers.
Corticosteroids are considered the first-line treatment of choice for dermatomyositis, polymyositis, and necrotizing myopathy.
In patients with severe weakness, a short course of IV methylprednisolone (1 g/d for 3 days) may be given, continuing with high-dose prednisone until muscle strength normalizes, improvement in strength has reached a plateau, or at least normalization of the serum creatine kinase occurs (this typically takes 3 to 6 months).
The authors usually start a second-line agent along with corticosteroids in patients with severe weakness or other organ system involvement (eg, myocarditis, interstitial lung disease), those with increased risk of steroid complications (eg, diabetics, patients with osteoporosis, or postmenopausal women), and those known to have difficult-to-treat myositis (eg, immune-mediated necrotizing myopathy).
The authors follow the serum creatine kinase levels when treating the inflammatory myopathies; however, adjustments of immunotherapies are primarily based on the objective clinical examination.
Gamma-glutamyl transpeptidase is the most reliable indicator of hepatic dysfunction, because aspartate aminotransferase and alanine aminotransferase can be elevated from muscle involvement alone.
A large, prospective, double-blind NIH trial of rituximab found no benefit, but the study design had significant flaws. In the authors’ experience, rituximab may be beneficial in patients with refractory dermatomyositis, polymyositis, and immune-mediated necrotizing myopathy.
Physical and occupational therapy are important and may help improve function and reduce type 2 fiber muscle atrophy associated with chronic steroids and inactivity.
Dysphagia is very common in patients with inclusion body myositis and often requires esophageal dilatation or cricopharyngeal myotomy.
Footnotes
Relationship Disclosure: Dr Amato has served on the medical advisory board of Biogen Idec, as a consultant for MedImmune, LLC, and provided expert testimony for litigation on a neuropathy case. Dr Amato serves as an associate editor of Neurology. Dr Greenberg has served as a consultant for aTyr Pharma, as an expert witness in litigation pertaining to copper deficiency, and is supported by a grant from the Muscular Dystrophy Association.
Unlabeled Use of Products/Investigational Use Disclosure: Drs Amato and Greenberg discuss therapies for the treatment of myositis, all of which are unlabeled.
REFERENCES
- 1.Amato AA,, Barohn RJ. Idiopathic inflammatory myopathies. Neurol Clin 1997; 15 (3): 615–648. [DOI] [PubMed] [Google Scholar]
- 2.Amato AA,, Barohn RJ. Evaluation and treatment of inflammatory myopathies. J Neurol Neurosurg Psychiatry 2009; 80 (10): 1060–1068. [DOI] [PubMed] [Google Scholar]
- 3.Amato AA,, Barohn RJ. Inclusion body myositis: old and new concepts. J Neurol Neurosurg Psychiatry 2009; 80 (11): 1186–1193. [DOI] [PubMed] [Google Scholar]
- 4.Amato AA,, Gronseth GS,, Jackson CE, et al. Inclusion body myositis: clinical and pathological boundaries. Ann Neurol 1996; 40 (4): 581–586. [DOI] [PubMed] [Google Scholar]
- 5.Amato AA,, Russell J. Neuromuscular disease. New York: McGraw-Hill, 2008. [Google Scholar]
- 6.Bohan A,, Peters JB. Polymyositis and dermatomyositis (first of two parts). N Eng J Med 1975; 292 (7): 344–347. [DOI] [PubMed] [Google Scholar]
- 7.Bohan A,, Peters JB. Polymyositis and dermatomyositis (second of two parts). N Eng J Med 1975; 292 (8): 403–407. [DOI] [PubMed] [Google Scholar]
- 8.Grable-Esposito P,, Katzberg HD,, Greenberg SA, et al. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010; 41 (2): 185–190. [DOI] [PubMed] [Google Scholar]
- 9.Griggs RC,, Askanas V,, DiMauro S, et al. Inclusion body myositis and myopathies. Ann Neurol 1995; 38 (5): 705–713. [DOI] [PubMed] [Google Scholar]
- 10.Lotz BP,, Engel AG,, Nishino H, et al. Inclusion body myositis. Observations in 40 patients. Brain 1989; 112 (pt 3): 727–747. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg SA,, Pinkus JL,, Pinkus GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 2005; 57 (5): 664–678. [DOI] [PubMed] [Google Scholar]
- 12.Pluk H,, van Hoeve BJ,, van Dooren SH, et al. Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann Neurol 2013; 73 (3): 397–407. [DOI] [PubMed] [Google Scholar]
- 13.Larman HB,, Salajegheh M,, Nazareno R, et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol 2013; 73 (3): 408–418. [DOI] [PubMed] [Google Scholar]
- 14.Greenberg SA. Pathogenesis and therapy of inclusion body myositis. Curr Opin Neurol 2012; 25 (5): 630–639. [DOI] [PubMed] [Google Scholar]
- 15.Mammen AL, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63 (3): 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mammen AL, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012; 64 (2): 269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]