

Abstract
Purpose of Review:
Toxic, nutritional, hereditary, traumatic, and neoplastic optic neuropathies result in significant disability due to visual dysfunction. Many of these conditions are treatable. Early diagnosis may allow for intervention to stabilize or improve vision and prevent unnecessary testing. These conditions have overlapping clinical features, and careful assessment of the visual system allows for accurate diagnosis and management.
Recent Findings:
Newer treatment strategies are available for optic neuropathies previously thought untreatable, such as some hereditary optic neuropathies. Many conditions that previously were felt to represent distinct diseases can be linked by a common pattern of mitochondrial dysfunction.
Summary:
The optic nerve is susceptible to a wide variety of pathologic processes. The correct diagnosis depends on a thorough history and detailed evaluation of the visual system. Certain optic neuropathies selectively affect the papillomacular bundle, and particular attention to this location can considerably narrow the differential diagnosis and subsequent workup.
INTRODUCTION
The optic nerve is susceptible to a number of pathologic processes, which reflect standard mechanisms of disease, eg, vascular, inflammatory, toxic, nutritional, compressive, infiltrative, hereditary, and traumatic. The term optic neuropathy denotes damage to the optic nerve from any etiology. The optic nerve has a relatively limited repertoire of responses to injury; therefore, many causes of optic nerve dysfunction may cause similar clinical manifestations. For example, optic nerve swelling can represent elevated intracranial pressure, inflammation from demyelinating optic neuritis, ischemic optic neuropathy, or an optic nerve sheath meningioma, with an identical appearance to the optic nerve for all of these diverse etiologies. The history and key elements of the examination (eg, visual field, visual acuity, color vision) help to distinguish among the various causes and guide workup. This article focuses on toxic, nutritional, hereditary, traumatic, and neoplastic optic neuropathies.
A metabolic optic neuropathy is defined as optic nerve dysfunction caused by an exogenous substance that causes relatively selective damage to the optic nerve (toxic optic neuropathy) or a nutritional deficiency that impairs optic nerve function (nutritional deficiency optic neuropathy). A hereditary optic neuropathy implies optic nerve damage secondary to an underlying genetic disease that includes optic neuropathy as a major component of its phenotype. Many of these conditions can be subsumed under a broader category of mitochondrial optic neuropathies. Emerging evidence suggests that a large number of optic neuropathies involve primary or secondary mitochondrial dysfunction.1
With some important exceptions, noted below, all metabolic (mitochondrial) optic neuropathies have similar clinical features and present in a similar manner. These conditions selectively affect the papillomacular bundle, the collection of retinal ganglion cells and axons that subserve central vision. Impairment of the papillomacular bundle causes characteristic clinical findings, including (1) reduced visual acuity, (2) impaired color perception, (3) central (involving fixation only) or cecocentral (involving fixation and the blind spot) visual field defects (Figure 5-1), and (4) temporal optic disc pallor.
Figure 5-1.
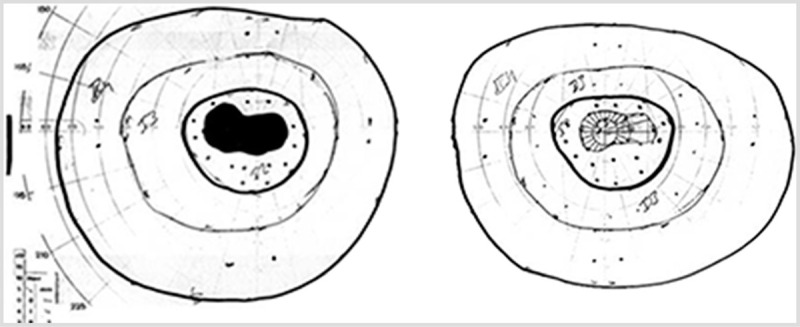
Goldmann perimetry visual fields in a patient with bilateral vitamin B12–deficiency optic neuropathy. Visual acuity was 20/200 in both eyes. Both eyes demonstrate cecocentral scotomas, defined by involvement of both fixation and the blind spot. The left eye is shown on the left and the right eye is shown on the right.
With some exceptions, all of the nutritional, toxic, and hereditary optic neuropathies described in this section have similar clinical presentations (Table 5-1). Onset is painless and in most cases bilateral and symmetric. A relative afferent defect is usually absent, given the symmetry of visual loss. The optic disc appearance early in the course often is normal, as it takes up to 8 weeks for visible pallor to develop after the optic nerve has been injured. True optic disc swelling and hyperemia are less common and when present are usually mild. Severe optic disc swelling with hemorrhages is rare. Bitemporal scotomas (Figure 5-2) can occasionally mimic the cecocentral scotomas seen with most of these disorders. Bitemporal scotomas usually indicate a lesion affecting the posterior aspect of the optic chiasm (eg, a suprasellar meningioma or craniopharyngioma) in which the macular fibers cross. Ethambutol optic neuropathy can cause a bitemporal scotoma,2 but a true bitemporal scotoma, with respect of the vertical meridian, is rare with most toxic and nutritional optic neuropathies. Therefore, unless compelling clinical evidence supports a metabolic or hereditary cause for optic neuropathy, neuroimaging is warranted to prevent delay of diagnosis.
Table 5-1.
Expected Clinical Characteristics of Most Toxic, Nutritional, and Hereditary Optic Neuropathies

Figure 5-2.
A bitemporal scotoma in a patient with ethambutol optic neuropathy. Note that the bitemporal defect involves only the central field and spares the periphery. The left eye is shown on the left and the right eye is shown on the right.
The papillomacular bundle is anatomically and physiologically distinct from other CNS axons and even from retinal ganglion cell neurons and axons (Figure 5-3). As part of the retinal nerve fiber layer, papillomacular bundle axons (along with other retinal ganglion cell axons) are unmyelinated and consequently cannot rely on saltatory conduction, resulting in higher energy demands for axonal transport.1,3 They are also narrower in caliber than other retinal ganglion cell axons, rendering them more vulnerable to energy depletion and the accumulation of reactive oxygen species.4 Mitochondria selectively accumulate at the optic nerve head, as this forms an anatomic “chokepoint,” where 1.2 million axons make a relatively acute turn to pass through an anatomically confined space. The optic nerve head is therefore a location with increased susceptibility to energy failure and oxidative stress. This helps explain why mitochondrial dysfunction (due to mutation, toxin, or nutritional deficiency), which should be present in every cell and tissue in the body, preferentially causes optic neuropathy.
Figure 5-3.
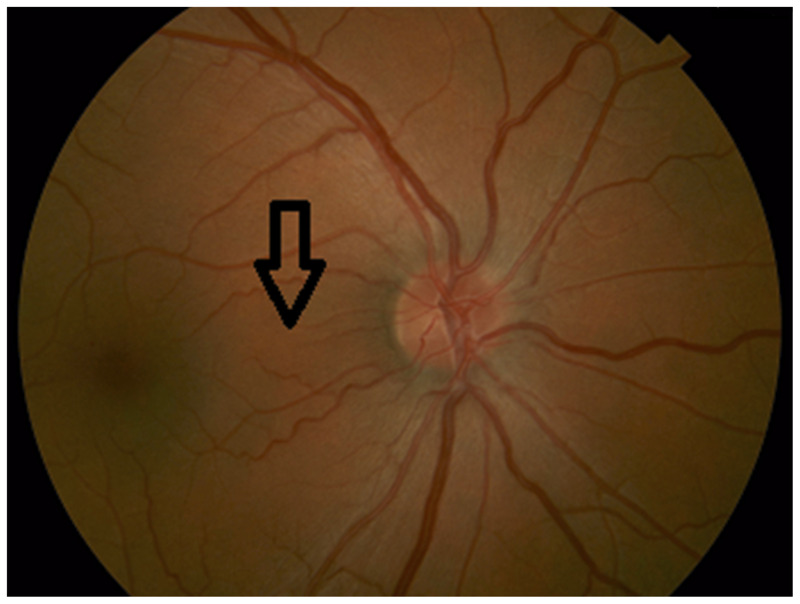
Color fundus photograph demonstrating papillomacular bundle fibers running from the temporal optic disc to the fovea (arrow).
Mitochondrial optic neuropathies (toxic, nutritional, and hereditary) can occasionally be mistaken for primary macular disease, or vice versa. Maculopathies can involve central vision but less frequently impair color vision, and usually cause central or paracentral, rather than cecocentral, visual field defects. Hydroxychloroquine-related maculopathy in particular can present with central visual loss and can mimic a primary optic neuropathy (Figure 5-4). The workup and management for a patient with maculopathy versus optic neuropathy differs considerably, but careful history taking and examination allow accurate localization (Table 5-2).
Table 5-2.
Clinical Features Differentiating Maculopathy From Optic Neuropathy
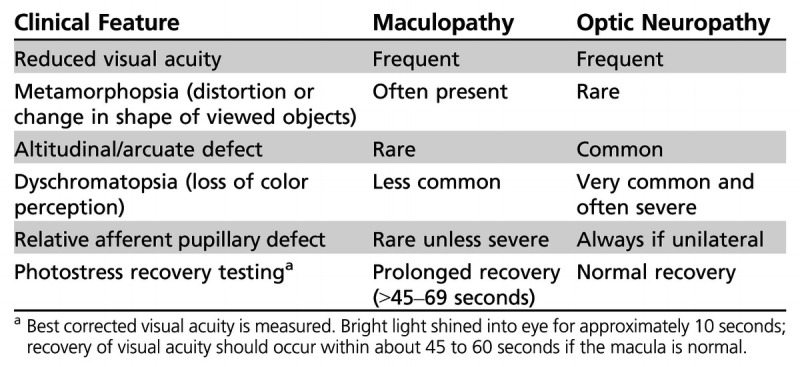
Figure 5-4.
Color fundus photographs demonstrate normal-appearing optic discs but pigmentary changes in both maculae (arrows) in a 72-year-old woman who was referred to the neuro-ophthalmology clinic with presumed bilateral optic neuropathy. She had a history of Sjögren syndrome and had been treated with hydroxychloroquine for the previous 20 years. Visual acuity was 20/40 in both eyes. The right eye is shown on the left, and the left eye is shown on the right.
NUTRITIONAL OPTIC NEUROPATHIES
Although a variety of nutritional deficiencies have been implicated in optic nerve dysfunction, many of these associations are anecdotal and a causal relationship is uncertain. Multiple nutritional deficiencies are often present, and identifying the specific vitamin responsible for the optic neuropathy can be challenging. With the exception of vitamin B12 deficiency, no nutritional deficiency has been proven unequivocally to cause optic neuropathy, and few good animal models are available for this condition.5
The prevalence of nutritional deficiency optic neuropathy is generally low in developed countries, although its incidence may be increasing with the rise in gastric bypass procedures and secondary reduction in vitamin absorption.6
Some general rules are applicable to all optic neuropathies causally related to nutritional deficiency. (1) The patient should be nutritionally deficient for a long-enough period, usually several months, to deplete nutrient stores. (2) Evidence of malnutrition (eg, weight loss, wasting) should be available, or a defined cause for the reduced vitamin level (eg, vitamin B12 deficiency in Crohn disease) should be determined. Other clinical findings may support a diagnosis of nutritional deficiency, including peripheral neuropathy, keratitis, or cutaneous or mucous membrane changes, although these are not always present.5
The clinical findings are remarkably similar for nearly all forms of nutritional deficiency optic neuropathy (Table 5-1). Rapid progression can occur in some cases. Visual loss in one eye and no findings in the other eye would argue strongly against nutritional deficiency optic neuropathy. The extent of visual acuity loss varies considerably, but visual acuity worse than hand motion is rare, as the non–papillomacular bundle fibers are spared. Color perception is affected early. Fundus examination may show temporal optic disc pallor, again reflecting papillomacular bundle loss; however, the fundus may be completely normal early on. All of the vitamins associated with nutritional deficiency optic neuropathy are involved in mitochondrial oxidative phosphorylation, and their deficiency would be expected to result in the depletion of energy and the accumulation of free radicals.7
The diagnosis of nutritional deficiency optic neuropathy can be supported by laboratory evidence, including serum vitamin levels, serum protein concentration, or routine laboratory studies such as blood count (eg, showing a macrocytic anemia with vitamin B12 deficiency).
Vitamin B12–Deficiency Optic Neuropathy
Vitamin B12 deficiency in developed countries occurs most frequently in the setting of pernicious anemia. Other conditions that can result in vitamin B12 deficiency include damage to or surgical resection of the terminal ileum (in patients with Crohn disease) and previous bariatric surgery. Lack of vitamin B12 in the diet is the rarest cause, usually only seen in strict vegans. In addition to optic neuropathy, vitamin B12 deficiency can cause a megaloblastic anemia, as well as subacute combined degeneration of the spinal cord and peripheral neuropathy. A recent major review discusses current strategies for suspecting, identifying, and treating vitamin B12 deficiency.8
The optic neuropathy associated with vitamin B12 deficiency follows the standard rules outlined in this section and in Table 5-1. Vision loss may precede anemia or other neurologic symptoms. The diagnostic testing for vitamin B12 deficiency should include serum vitamin B12 levels; however, the diagnosis may not always be straightforward.8,9 Methylmalonic acid and homocysteine levels are both increased in vitamin B12 deficiency, and may suggest relative vitamin B12 deficiency even in the setting of low normal vitamin B12 levels. However, both methylmalonic acid and homocysteine levels can be elevated in patients with renal insufficiency, and homocysteine levels are influenced by a variety of factors, including tobacco and alcohol use.8 Misdiagnosis of vitamin B12–deficiency optic neuropathy may result in delay of diagnosis of the true underlying cause.
Vitamin B12–deficiency optic neuropathy is one of the few nutritional deficiency optic neuropathies with good experimental evidence for a causal relationship. Primate models for this condition show that the optic neuropathy is caused by demyelination of nerve fibers.10 Studies suggest that the mechanism of injury to the optic nerve may involve buildup of toxic levels of cyanide or improper fatty acid synthesis leading to dysfunction of myelin.10
Prompt recognition and treatment with parenteral hydroxycobalamin or high-dose oral vitamin B128 may reverse existing visual loss as well as prevent further visual deterioration.
Thiamine Deficiency
Thiamine (vitamin B1) is a cofactor for several mitochondrial enzymes, and in animal models thiamine deficiency results in impaired cellular production of energy. Visual loss in the setting of thiamine deficiency is rare. Optic neuropathy has been reported in association with Wernicke encephalopathy, traditionally in the context of alcohol abuse and malnutrition.11 The incidence of Wernicke encephalopathy may be increasing because of the popularity of bariatric surgery, particularly gastric bypass, the procedure with the greater risk of subsequent malabsorption. The optic neuropathy associated with Wernicke encephalopathy is to some degree distinct from other forms of nutritional deficiency optic neuropathy in that visual loss is often acute to subacute, and disc swelling (sometimes quite severe) is a prominent feature in most cases.12,13 The recovery of vision with appropriate thiamine supplementation is usually complete and often dramatic.
The diagnosis of Wernicke encephalopathy–associated optic neuropathy is in many cases presumptive, based on clinical features, neuroimaging findings, and response to thiamine treatment. Serum and plasma thiamine levels have poor sensitivity and specificity (less than 10% of blood thiamine is contained in plasma); and, since delayed treatment may lead to irreversible neurologic injury, supplemental thiamine is often given empirically before levels can be drawn.13 Whole blood thiamine testing is superior to serum or plasma levels, but results may take several days to obtain. An acute anterior optic neuropathy in the setting of malnutrition or previous gastric bypass procedure should raise suspicion for Wernicke encephalopathy and prompt expedited diagnostic testing and supplementation with thiamine.
Other Nutritional Deficiency Optic Neuropathies
Some reports suggest that optic neuropathy may be also associated with deficiencies in riboflavin, niacin, folic acid, and pyridoxine, but a causal relationship is uncertain, as these deficiencies are often found in severely malnourished patients, and identification of the specific vitamin deficiency leading to optic neuropathy is difficult.
Copper deficiency has been associated with optic neuropathy. Pineles and colleagues14 reported two patients with combined optic neuropathy and myelopathy secondary to copper deficiency. A confounding factor is that both patients also had vitamin B12 deficiency, but the authors argue that the visual loss progressed despite correction of vitamin B12 levels and only stabilized after copper supplementation. Neither patient recovered vision after supplementation. The mechanism is uncertain but may relate to impaired mitochondrial function secondary to hypocupremia.
A reasonable approach for patients with suspected nutritional deficiency optic neuropathy is to exclude alternate etiologies (with neuroimaging), and obtain vitamin B12 levels, complete blood cell count, and metabolic panel, and pursue less common nutritional deficiency optic neuropathies (eg, thiamine, copper) if the initial screen is negative and clinical suspicion remains high.
TOXIC OPTIC NEUROPATHIES
A toxic optic neuropathy refers to optic nerve dysfunction caused by exposure to an exogenous agent that is directly harmful to the optic nerve. As with nutritional deficiency optic neuropathy, a large number of drugs and toxins have been linked to optic neuropathy, but the strength of the relationship relies primarily on case reports and small case series, with no proven causal connection. Since in many cases the treatment of toxic optic neuropathy is discontinuation of the offending agent (which may be an essential treatment for the specific underlying disease), the clinician should establish as high a level of certainty as possible that the medication is causing the visual loss.
Sadun and colleagues propose five postulates, or criteria, for a toxic optic neuropathy, adapted to some degree from Hill’s causal criteria.1,15
Scientific plausibility for a causal relationship should be present: Why should retinal ganglion cells and their axons be susceptible?
A clinical dose-response curve (although this may not apply in some toxins that exhibit a threshold effect) should be present.
Longer duration of exposure is a risk factor.
Recovery to some degree should occur after removal of the offending toxin.
Asymmetry should be exceptional.
Along with these criteria, a perhaps more obvious criterion could be added: a reasonable temporal relationship, ie, the offending drug should be started before the onset of visual symptoms and be taken for a long-enough period of time to exert toxic effects. Such strict criteria may be difficult to satisfy in the clinical setting, since susceptibility varies from one individual to another, but the more of these criteria that are met, the greater the likelihood of causality. In this article, discussion will be limited to those toxic optic neuropathies that fulfill most of the postulates, and include several that are of historical or social or topical relevance. Table 5-3 provides a list of toxins and medications associated with optic neuropathy, highlighting those agents with the strongest associations.
Table 5-3.
Drugs and Toxins Associated With Toxic Optic Neuropathya

With some exceptions, toxic optic neuropathies obey the rules in Table 5-1 for mitochondrial and metabolic optic neuropathies. The diagnosis is usually based on a high index of clinical suspicion, excluding alternate causes (including compressive optic neuropathies), and the stabilization and improvement of vision after discontinuation of the toxin. In many (perhaps most) cases, the drug interferes with mitochondrial oxidative phosphorylation, triggering optic nerve dysfunction. Carelli and colleagues16 discuss the selective vulnerability of retinal ganglion cell neurons and axons to mitochondrial dysfunction, particularly in the context of toxic optic neuropathy.
Ethambutol
Ethambutol is the most common cause of toxic optic neuropathy, accounting for 100,000 new cases every year. The primary indication for ethambutol is the treatment of tuberculosis, and it is estimated that approximately 55% of patients treated for tuberculosis worldwide use ethambutol as part of their treatment regimen.17
Ethambutol inhibits arabinosyltransferase, an important enzyme in mycobacterial cell membrane synthesis. It also impairs oxidative phosphorylation and mitochondrial function by interfering with iron-containing complex I and copper-containing complex IV. It is a metal chelator and may reduce the level of copper, possibly contributing to the optic neuropathy; some evidence suggests that zinc may play a role in ethambutol toxicity.1,14,18 The mechanism of optic neuropathy is uncertain, but may be secondary to mitochondrial dysfunction at the level of retinal ganglion cell axons and neurons in the papillomacular bundle.
A clear dose-response effect exists. The incidence of visual dysfunction is as high as 50% at a dose of 60 mg/kg/d to 100 mg/kg/d, 5% to 6% at a dose of 25 mg/kg/d, and approximately 1% at a dose of less than 15 mg/kg/d. Since ethambutol is excreted renally, patients with renal insufficiency are at greater risk for visual loss, even at safe doses (Case 5-1). Other suggested risk factors include diabetes mellitus, alcoholism, advanced age, and preexisting visual loss. Optic neuropathy is rare with treatment duration of less than 2 months.1,16
Ethambutol optic neuropathy behaves as expected for a toxic optic neuropathy (Table 5-1). Patients usually report blurring of central vision, loss of contrast, and impaired color perception; dyschromatopsia (loss of color perception) may be the earliest symptom. The diagnosis is often straightforward, but exclusion of alternate causes (eg, compressive lesions, associated nutritional deficiency) may be necessary. The visual loss is often reversible with early discontinuation of the drug but may be permanent if treatment exceeds 6 months. It is common for the optic neuropathy to progress for several months after stopping the drug before stabilizing with some gradual improvement. Optic atrophy at presentation portends a worse prognosis, with lower likelihood of recovery.
Before starting ethambutol, all patients should have a baseline ophthalmic examination, including visual acuity, color vision, and formal visual fields. Patients using the recommend dosage should receive a follow-up examination, using the same metrics, every 1 to 3 months. Patients at higher risk may require more frequent monitoring. There is no consensus regarding treatment of ethambutol optic neuropathy, aside from discontinuing the medication. Routine screening for zinc and copper deficiency in patients with ethambutol optic neuropathy is unsupported by high-quality evidence but can be considered in individual patients, particularly those who fail to recover. The use of supplemental copper and zinc in a nondeficient patient, both to prevent and treat ethambutol optic neuropathy, is also unsupported by high-quality evidence but remains understudied.
Case 5-1
A 77-year-old woman presented to the neuro-ophthalmology clinic with an 8-month history of visual loss. She had been diagnosed with Mycobacterium avium-intracellulare complex over 1 year prior, and started on ethambutol approximately 7 to 8 months prior to onset of visual loss. During the next few months, her visual loss progressed. MRI of the brain was performed and was unremarkable.
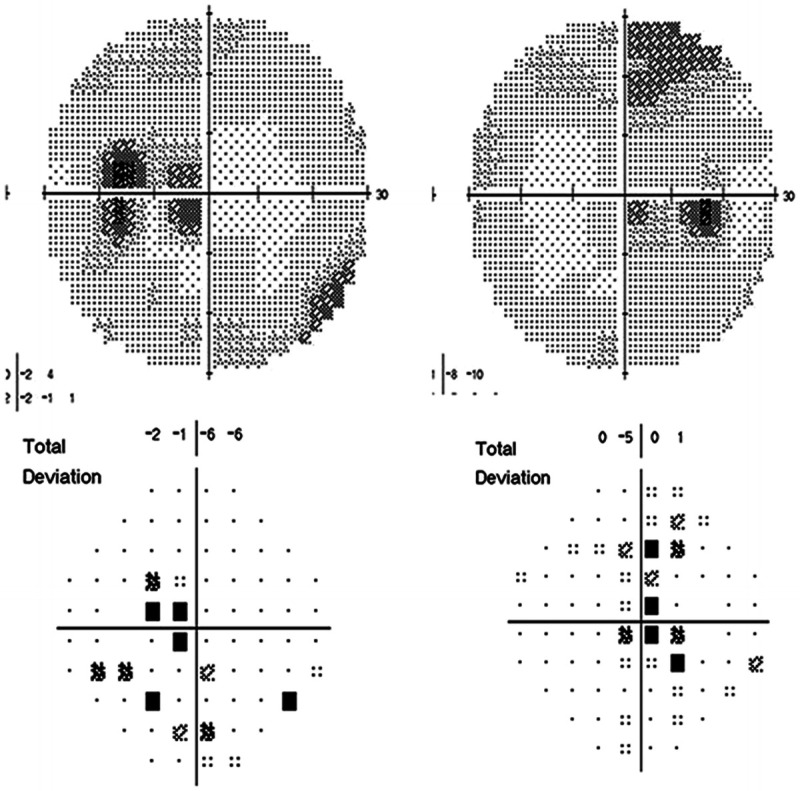
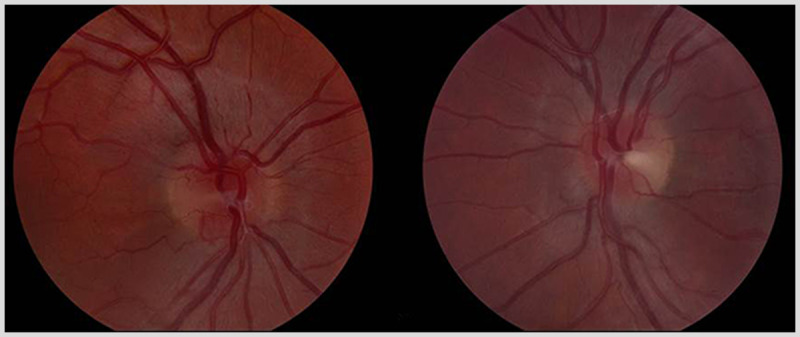
The patient was seen 16 months after initiation of ethambutol. Her dosage was 800 mg/d, which was 11 mg/kg/d for her weight. Visual acuity was 20/100 right eye, 20/70 left eye. Ishihara color plates were abnormal, 6/15 plates right eye, 5/15 plates left eye. No relative afferent pupillary defect was present. Fundus examination showed bilateral, temporal optic disc pallor Figure 5-5). Humphrey 24-4 automated visual fields were performed. The right eye showed an inferior central scotoma, while the left eye showed a cecocentral scotoma (Figure 5-6). The defect in the left eye resembled a temporal defect, but without respect of the vertical meridian.
Figure 5-5.
Fundus examination shows bilateral, temporal optic disc pallor in both eyes. The right eye is shown on the left and the left eye is shown on the right.
Figure 5-6.

Humphrey 24-2 automated visual fields show a cecocentral scotoma in the left eye (on left) and an inferior central scotoma in the right eye (on right).
Ethambutol optic neuropathy was suspected. Her infectious disease specialist was consulted, and he agreed to discontinue ethambutol. A follow-up MRI of the brain was unremarkable. Complete blood cell count, vitamin B12, and copper levels were normal.
She returned to clinic 10 weeks later. Visual acuity had improved to 20/50 right eye, 20/30 left eye. Color vision had also improved, to 8/11 plates right eye, 9/11 plates left eye. Fundus examination showed stable temporal optic disc pallor in both eyes. Humphrey 24-2 visual fields showed inferior central scotomas (Figure 5-7). She continued follow-up with a local ophthalmologist and had stabilized with no progression or further improvement 1 year after discontinuing the medication.
Figure 5-7.

Humphrey 24-2 visual fields show inferior central scotomas in both eyes. The left eye is shown on the left and the right eye is shown on the right.
Comment. Ethambutol optic neuropathy can occur even with safe dosages, and the presence of visible optic disc pallor and retinal nerve fiber layer thinning suggests a lower likelihood for complete recovery.
Methanol
Methanol is a colorless and odorless liquid present in antifreeze, paint remover, and windshield-washer fluid. It is most frequently ingested when added to ethyl alcohol. Methanol optic neuropathy is the best characterized and one of the best-proven toxic optic neuropathies.19
Patients ingesting methanol present with nausea, abdominal pain, metabolic acidosis, and visual loss, usually profound. The diagnosis is made by clinical findings and serum toxicology. Brain MRI may show bilateral putamenal hyperintensities.20 The visual loss is typically severe and bilateral. The optic disc is initially hyperemic with blurred margins. Some patients may regain vision after several weeks or longer. Lack of pupillary reactivity to light (implying severe bilateral optic nerve dysfunction) suggests a poor prognosis.
Methanol is ultimately metabolized to formic acid, which is a mitochondrial toxin known to impair cytochrome c oxidase. Humans and other primates are uniquely susceptible to methanol toxicity as they have a limited capacity to oxidize and thus detoxify formic acid.7
Treatment of methanol toxicity includes ethanol, dialysis, and 4-methylpyrazole, along with supportive care. Studies looking at high-dose corticosteroids for treatment of methanol optic neuropathy, however, lack control groups and suffer from retrospective analysis.21
Linezolid
Linezolid is an antibiotic designed to treat methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococcus. It acts by inhibiting protein synthesis via binding ribosomal RNA of the bacterial ribosomal subunit. Protein synthesis in mitochondria is also disrupted, since mitochondrial ribosomes are structurally similar to bacterial ribosomes. In a rat model, linezolid includes a dose-dependent decrease in the activity of mitochondrial complex I and complex IV.22
Linezolid has been associated with both optic and peripheral neuropathies.1 The duration of treatment is a key factor: neurotoxicity has been reported with prolonged treatment of 5 to 50 months.
The clinical presentation is nearly identical to other toxic and nutritional optic neuropathies. The optic discs are initially hyperemic and develop pallor over time. The diagnosis is usually predicated on the clinical features, appropriate temporal relationship and duration of treatment, and, in some cases, exclusion of alternate causes. Some patients recover vision after discontinuing the drug, but the peripheral neuropathy is usually permanent.
Amiodarone
Amiodarone is a cardiac antiarrhythmic medication that may produce an optic neuropathy characterized by insidious, slowly progressive visual loss, with prolonged disc swelling. After discontinuation of the medication, the disc swelling resolves, and visual acuity and visual fields stabilize. Although amiodarone optic neuropathy and nonarteritic anterior ischemic optic neuropathy share many similarities, occasionally several features (eg, rate of onset, duration of disc swelling) distinguish amiodarone optic neuropathy from (perhaps coincidental) anterior ischemic optic neuropathy in these patients.23 A recent critical review using the US Federal Drug Administration’s Adverse Event Reporting System and published case reports identified 296 reports of presumed amiodarone optic neuropathy.24 A true causal relationship is unproven, and the incidence is likely quite low. However, optic neuropathy developing in a patient using amiodarone should raise suspicion for this condition. If it is unilateral and behaves clinically like nonarteritic anterior ischemic optic neuropathy, observation is reasonable. If the optic disc swelling persists longer than 6 to 8 weeks, amiodarone optic neuropathy should be suspected. The patient’s cardiologist should be involved in the discussion of whether to discontinue the medication, since in some cases no viable alternatives to amiodarone may be available for cardiac rhythm control.
The mechanism of amiodarone optic neuropathy remains uncertain. In many ways, it breaks the rules of toxic optic neuropathies. The optic disc swelling is usually prominent, often with hemorrhages; it is unilateral rather than bilateral; and cecocentral scotomas are less common. One hypothesis is that amiodarone optic neuropathy results from a drug-induced lipidosis.25
Cyclosporine and Tacrolimus
Cyclosporine and tacrolimus are potent immunosuppressants, frequently used to prevent rejection in patients who have undergone organ transplantation. Both can cause posterior reversible encephalopathy syndrome (PRES) and cerebral visual loss, but case reports of optic neuropathy associated with both agents have been published. Optic disc edema was originally reported in a patient status post–bone marrow transplant treated with cyclosporine, and similar cases can be found in scattered reports in the literature.26 In some cases, the mechanism appears to be elevated intracranial pressure and papilledema, while in others a primary optic neuropathy is present. Recently, Venneti and colleagues27 described a case of asymmetric bilateral, demyelinating optic neuropathy presumed due to tacrolimus toxicity. The patient experienced rapidly progressive visual loss and underwent optic nerve biopsy in the blind eye for a definitive diagnosis. The biopsy showed prominent loss of myelin but was otherwise nondiagnostic. Several similar cases can be found in the literature. Some investigators have hypothesized that polymorphisms in the ATP-binding cassette, subfamily B (MDR/TAP), member 1 (ABCB1) gene protein, a P-glycoprotein that may influence elimination of tacrolimus from the CNS, may play a role in pathogenesis. Most of the reported cases showed no improvement after cessation of the drug, but visual loss stabilized.
Tobacco-Alcohol Amblyopia
Tobacco-alcohol amblyopia is a rare optic neuropathy related to a constituent of tobacco. It is often misdiagnosed and more commonly seen in tobacco users who do not smoke cigarettes (often pipe smokers). The literature regarding tobacco-alcohol amblyopia is confusing, likely because of its confusing name. This condition is not amblyopia in the current clinical meaning of the term (loss of vision in an otherwise normal eye that is caused by strabismus, uncorrected refractive error, or sensory deprivation at an early age), and no evidence suggests that alcohol itself is directly toxic to the optic nerve; indeed, the visual symptoms of so-called tobacco-alcohol amblyopia improve and resolve with adequate nutritional supplementation.28 Many experts feel that most patients diagnosed with tobacco-alcohol amblyopia have nutritional optic neuropathies, and that tobacco (particularly cigarette smoking) and alcohol play no causal role in visual loss.29 Clinicians should be reluctant to accept the diagnosis of tobacco-alcohol amblyopia without compelling evidence and exclusion of other disorders, particularly nutritional deficiency optic neuropathy.
HEREDITARY OPTIC NEUROPATHIES
Hereditary optic neuropathies share several clinical features with toxic and nutritional optic neuropathies (Table 5-1). As noted earlier in this article, with some exceptions, they fall within the broader category of mitochondrial optic neuropathies. The hereditary optic neuropathies may be grouped in several ways, including mode of inheritance, age at presentation, pattern of onset, and the presence of other neurologic and systemic symptoms. They are more easily divided in the following manner.
Optic neuropathy occurring primarily without any associated neurologic symptoms (monosymptomatic)
Optic neuropathy commonly with associated neurologic deficits
Optic neuropathy secondary to overall disease process (eg, Charcot-Marie-Tooth disease, spinocerebellar ataxias).
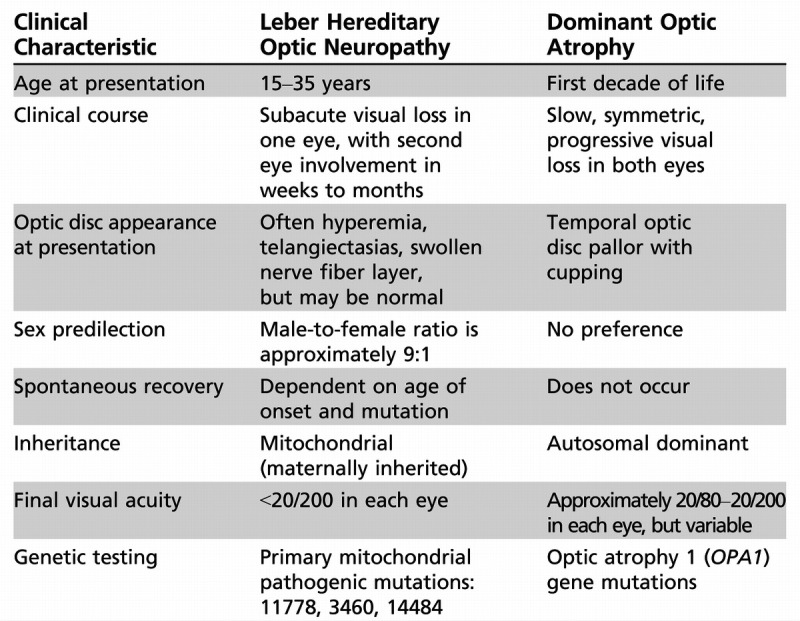
The latter two categories are beyond the scope of this review. The two most common primary, monosymptomatic hereditary optic neuropathies are Leber hereditary optic neuropathy and dominant optic atrophy. Table 5-4 summarizes the clinical features of both.
Table 5-4.
Clinical Characteristics of Leber Hereditary Optic Neuropathy and Dominant Optic Atrophy
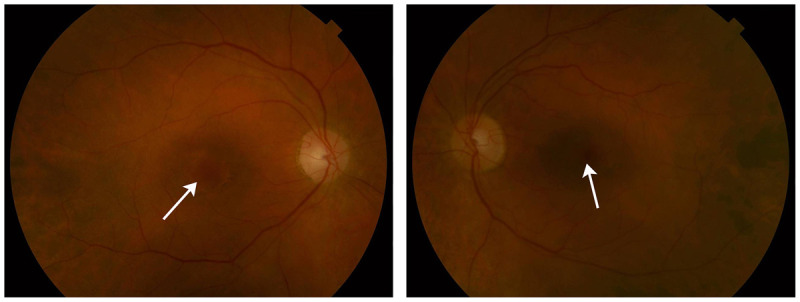
Leber Hereditary Optic Neuropathy
Leber hereditary optic neuropathy is the first human disease linked to a mutation in mitochondrial DNA.30,31 Originally thought to nearly exclusively affect young men, the advent of molecular genetic testing has expanded the spectrum of Leber hereditary optic neuropathy. Men between the ages of 15 and 35 remain the mostly frequently affected group. They may or may not have a relevant family history; in some series, up to 50% of patients have no family history of visual loss. Because mitochondria are transmitted by only the mother, offspring of male carriers are not at risk for visual loss. Three primary, pathogenic mitochondrial DNA point mutations have been identified: 11778, 3460, and 14484.
The classic presentation is painless, subacute visual loss in one eye, followed by visual loss in the contralateral eye within the next several weeks or months (Case 5-2). Visual loss usually progresses over several weeks to months, with final visual acuity of less than 20/200 in both eyes. On ophthalmoscopy, the classic, acute disc appearance is hyperemia, circumpapillary telangiectasia, and lack of leakage on retinal angiography (indicating pseudoedema rather than true disc swelling). Not all patients have the classic optic disc appearance at presentation; this varies depending on the specific mutation and the duration of visual loss. Indeed, the characteristic optic disc appearance may be seen in asymptomatic maternal relatives. Visual fields show characteristic central and cecocentral deficits. Leber hereditary optic neuropathy is exceedingly unlikely in a patient with a unilateral optic neuropathy that has been present for years. Over time, disc pallor develops as the hyperemia recedes. The visual prognosis depends on the specific mutation and age of onset. Patients with the 14484 mutation who lose vision early in life have the highest probability of recovering vision, while those harboring the 11778 mutation have the lowest likelihood of spontaneous recovery.32
Leber hereditary optic neuropathy and other diseases. Although most patients with Leber hereditary optic neuropathy mutations have isolated optic neuropathy and vision loss, a small subset of patients may have associated neurologic symptoms, including a multiple sclerosis–like illness, ataxia, and dystonia. These are referred to as Leber-plus syndromes. Multiple sclerosis and Leber hereditary optic neuropathy co-occur more frequently than might be expected from chance alone, and it remains unclear whether one condition can worsen or trigger the other, or whether this represents a distinct mitochondrial phenotype.33 Patients with Leber hereditary optic neuropathy may have cardiac arrhythmias, as seen with many mitochondrial myopathies.
Nutritional deficiency and toxic optic neuropathies and Leber hereditary optic neuropathy may coexist, and, in some cases, the nutritional deficiency or exogenous agent may trigger expression of the Leber hereditary optic neuropathy phenotype in patients harboring one of the pathogenic mutations.34 This phenomenon has been described most frequently in the setting of vitamin B12 deficiency31 but has also been reported in association with ethambutol optic neuropathy. The diagnosis of co-occurring Leber hereditary optic neuropathy should be considered in any patient with presumed nutritional deficiency optic neuropathy or toxic optic neuropathy if the visual loss fails to recover as expected, the disc appearance at presentation is suggestive of Leber hereditary optic neuropathy, or a suggestive maternal family history is present.
Pathogenesis. The exact mechanism of injury in Leber hereditary optic neuropathy is slowly being elaborated. The three primary mutations in mitochondrial DNA are located in genes encoding subunits of complex I of the electron transport chain. Although the pathogenic mutation is present in all somatic cells (in different concentrations, as a result of replicative segregation of mitochondrial into daughter cells), the optic nerve is selectively affected. This selective vulnerability is likely related to the unique anatomic and physiologic features of retinal ganglion cell axons described earlier, all of which contribute to lower the threshold for retinal ganglion cell loss, possibly by induced apoptosis.7,16,31
No treatment has yet been proven effective for Leber hereditary optic neuropathy, although several studies show promising results. In general, treatments for mitochondrial disorders are limited. General treatments include vitamins and cofactors (eg, coenzyme Q10) that might stabilize mitochondrial function and reduce oxidative stress. These therapies are harmless but expensive.
Brimonidine, a topical α2-agonist used in the treatment of glaucoma, may have neuroprotective effects on retinal ganglion cells. Since the putative antiapoptotic effects are mediated through complex I, this was thought to be a promising treatment for Leber hereditary optic neuropathy. However, brimonidine did not prevent second-eye vision loss in an open-label, nonrandomized multicenter study of nine patients with Leber hereditary optic neuropathy with first eye involvement.35
Idebenone is a short-chain benzoquinone. Although it is structurally similar to coenzyme Q10, idebenone crosses the blood-brain barrier and localizes in the mitochondria. A multicenter, double-blind, randomized controlled trial of idebenone versus placebo in the treatment of symptomatic patients with Leber hereditary optic neuropathy was conducted by Klopstock and colleagues.36 The study included 55 patients with Leber hereditary optic neuropathy treated with 900 mg idebenone for 24 weeks. Neither the primary nor secondary end points reached statistical significance. However, a post hoc analysis of those subjects with discordant visual acuity at baseline found a significant difference in secondary end points favoring the treatment group, corresponding to a difference of four to five lines of Snellen visual acuity. Although this study had many limitations, the results could suggest a role for idebenone in patients with Leber hereditary optic neuropathy in the early stages of visual loss.
The diagnosis of Leber hereditary optic neuropathy should be considered in any patient, regardless of age or sex, with severe, rapidly sequential or simultaneous bilateral optic neuropathy, particularly if the optic disc has a classic appearance. Although no proven treatments are available, early treatment with idebenone may be beneficial, and a specific genetic diagnosis allows for a more accurate prognosis and genetic counseling. The three pathogenic mutations can be screened for with commercially available genetic testing.
Case 5-2
A 17-year-old boy presented to the neuro-ophthalmology clinic with a 2-month history of painless visual loss in both eyes. He had initially noticed visual blurring from the left eye 2 months prior that progressed during a 2- to 3-week period. He was seen by a local ophthalmologist, who documented visual acuity of 20/20 right eye, counting fingers left eye. The left optic disc was felt to be swollen, and a diagnosis of optic neuritis was made. MRI of brain and orbit with and without gadolinium was unremarkable. He was treated with 3 days of IV methylprednisolone, 1 g/d, with no improvement.
One month after onset, he noted blurring of vision in the right eye, which worsened during the next several weeks. He was referred to the neuro-ophthalmology clinic and seen 2 months after onset of visual loss.
Medical history was unremarkable. He was a senior in high school and doing well academically. He denied alcohol and tobacco use. He had no family history of visual loss or neurologic disorders. An extensive systemic and neurologic review of systems was unremarkable. Visual acuity with best correction was 20/200 right eye, 20/400 left eye. He was unable to identify the Ishihara control plate with either eye. Pupils were equal, round, and reactive to light with a small left relative afferent pupillary defect. Ocular motility examination was normal. Fundus examination (Figure 5-8) showed mild hyperemia of the right optic disc, with swelling of the peripapillary nerve fiber layer and a few telangiectasias. The left optic disc showed minimal residual hyperemia, with developing temporal pallor and perhaps a few subtle peripapillary telangiectasias. Humphrey 24-2 visual fields demonstrated central scotomata in both eyes (Figure 5-9).
Figure 5-8.
Fundus examination shows mild hyperemia of the right optic disc (on left), with swelling of the peripapillary nerve fiber layer and a few telangiectasias. The left optic disc (on right) shows minimal residual hyperemia, with developing temporal pallor and possibly a few subtle peripapillary telangiectasias.
Figure 5-9.
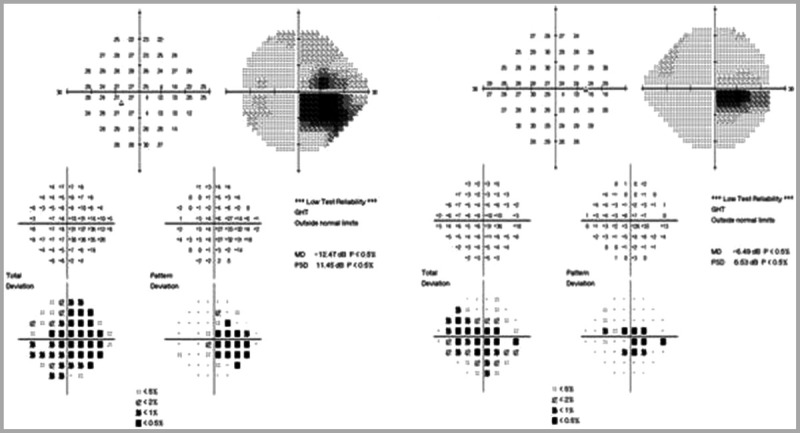
Humphrey 24-2 visual fields demonstrate central scotoma in both eyes. The left eye is shown on the left and the right eye is shown on the right.
Comment. Based on the patient’s age, clinical presentation, and fundus appearance, Leber hereditary optic neuropathy was suspected. Blood screening for the primary pathogenic mutations was positive for the 11778 mutation. Formal genetic counseling was requested for the maternal family relatives. The visual acuity continued to decline during the next 6 weeks, stabilizing at approximately 1/200 in both eyes. There had been no change at his last follow-up visit, several years after onset.
Leber hereditary optic neuropathy should always be considered in any patient presenting with severe, sequential, painless optic neuropathy. Family history is often negative, and the classic fundus findings may be absent. Accurate diagnosis allows for better prognosis and genetic counseling, and avoids unnecessary testing.
Dominant Optic Atrophy
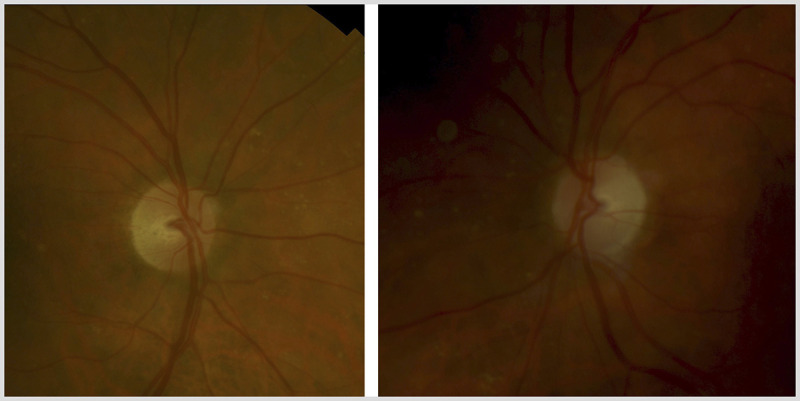
Dominant optic atrophy (also known as Kjer disease) is an autosomal dominantly inherited condition. It typically presents during childhood, often after a child has failed a school vision screening. Both optic nerves are involved symmetrically. Visual acuity is variable, but most patients lose vision slowly throughout life and retain visual acuity of 20/40 to 20/200.32 As a general rule, visual loss in dominant optic atrophy is less severe than in Leber hereditary optic neuropathy, with a mean visual acuity of 20/80 to 20/200. The clinical features are similar to other mitochondrial optic neuropathies (Table 5-1). Many (although not all) patients have prominent temporal optic disc pallor with a wedgelike excavation of the disc (Figure 5-10).
Figure 5-10.
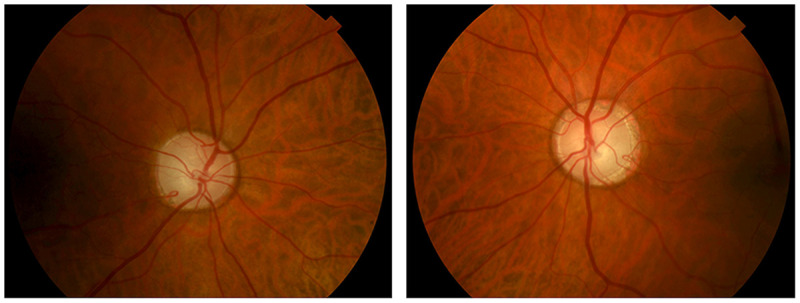
Color fundus photographs of a patient with genetically confirmed dominant optic atrophy. Note the prominent temporal optic disc pallor, with extensive optic disc cupping (cup-disc ratio of ~0.8 in both eyes). The right eye is shown on the left and the left eye is shown on the right.
The specific phenotype varies considerably within and among families, because of incomplete penetrance and variable expressivity. Progressive visual loss occurs in up to 67% of patients, again with a large degree of variability. A clinical rule of thumb is that visual loss occurs at a rate of one line of Snellen visual acuity per decade of life. Spontaneous recovery of vision is not a feature of dominant optic atrophy.
Similar to Leber hereditary optic neuropathy and Leber plus, up to 20% of patients with dominant optic atrophy develop a syndromal form of the disorder, most commonly with sensorineural hearing loss, but also myopathy, peripheral neuropathy, ataxia, and ophthalmoplegia.37
Approximately 60% of cases of dominant optic atrophy result from mutations in the optic atrophy 1 (OPA1) gene located on chromosome 3q28-29. This gene encodes OPA1, a dynaminlike guanosine triphosphatase (GTPase) attached to the inner membrane of the mitochondrial cristae. A variety of impairments in mitochondrial function have been implicated with OPA1 protein dysfunction, including mitochondrial fusion, membrane stabilization, oxidative phosphorylation, and apoptosis.1,32 Oxidative stress likely has a key role in dominant optic atrophy pathogenesis, as in Leber hereditary optic neuropathy. Genetic testing for OPA1 mutations is commercially available. The sensitivity is high in large pedigrees but lower in patients with no relevant family history.
No proven treatments are available for dominant optic atrophy, although ongoing studies are using idebenone and similar mitochondrial cofactors.
TRAUMATIC OPTIC NEUROPATHIES
Traumatic optic neuropathy can occur through a direct or indirect mechanism. Direct mechanisms disrupt optic nerve fibers and include penetrating orbital trauma, nerve avulsion from the globe, optic nerve crush injuries, or laceration by bone fragments. More commonly, traumatic optic neuropathy results from an indirect injury in which the forces (often acceleration or deceleration) produced by an orbital or cranial impact lead to traction, elevated intraorbital pressure, compression by orbital hemorrhage or optic nerve sheath hematoma, or axonal shearing.38
Most cases of traumatic optic neuropathy involve localized damage to the intracanalicular segment of the nerve. This is thought to occur by a shearing injury analogous to diffuse axonal injury, possibly followed by ischemic injury as reactive edema generates compression within the optic canal, and further inflammatory injury as the tissues respond to traumatic insult.39
Traumatic optic neuropathy is uncommon but not rare. In one review of 326 patients seen in a tertiary care neuro-ophthalmology service after head injury, deficits consistent with traumatic optic neuropathy were seen in 12%, with 7% diagnosed as indirect traumatic optic neuropathy.40
Clinical examination and appropriate neuroimaging play a vital role in identifying the location and nature of the injury. However, the expected findings of an optic neuropathy (eg, reduced visual acuity, visual field loss, relative afferent pupillary defect) may be difficult to detect in a patient with an acute head injury who is not fully cooperative, delaying the diagnosis until the patient has retained consciousness and can participate in the examination. Orbital CT can assess for retrobulbar hemorrhage, optic nerve sheath hematoma, and bone fragments compressing or lacerating the optic nerve. MRI is of less utility in traumatic optic neuropathy, often not showing any signal changes. However, diffusion tensor imaging, an MRI technique that tracks motion of water molecules along nerve fiber tracts, demonstrates lower axial and mean diffusivity in optic nerves with traumatic optic neuropathy, acting as a biomarker of injury and potentially predicting the potential for visual recovery.41
Treatment
No single treatment protocol has proven effective for traumatic optic neuropathy. Management options include observation, systemic corticosteroids, surgical decompression of the optic canal, or a combination of corticosteroids and surgical decompression. Several of the existing strategies draw inferences from treatment trials involving other forms of CNS injury.
Steroid treatment for brain edema was introduced in the early 1960s, and became a common treatment for patients with severe traumatic brain injury by the 1970s, after a randomized trial showed improved mortality rates. Several subsequent trials, however, have failed to show appreciable benefit.
The Corticosteroid Randomization After Significant Head Injury (CRASH) trial of 10,008 patients from 239 hospitals in 49 countries showed a detrimental effect of methylprednisolone therapy, with increased 2-week mortality (21% versus 18%) and increased relative risk of death by 6 months (1.15) in the steroid group. Furthermore, no significant difference in death or disability occurred between the steroid and nonsteroid groups when stratified by injury severity, leading the authors to conclude that corticosteroid treatment provides no benefit after head injury.42
No high-quality evidence is available to guide the management of traumatic optic neuropathy. Two Cochrane reviews searching for randomized controlled trials comparing steroids, surgery, or observation alone found only one article meeting inclusion criteria.43,44 Steroids have been used to treat traumatic optic neuropathy, based on extrapolation from previous studies evaluating spinal cord trauma,45 but their use has declined given the results of the CRASH study. Surgical optic canal decompression (by orbitotomy or endoscopic approach) is also performed at some centers, citing the potential to relieve pressure in the optic canal and remove bony fragments that threaten the nerve. Yang and colleagues reported visual acuity improvement in 40.6% of traumatic optic neuropathy patients treated with endoscopic decompression surgery after a trial of megadose steroids had failed to produce any effect.39
For patients presenting with acute traumatic optic neuropathy, clinical decisions should be made on a case-by-case basis, keeping in mind the possibility of spontaneous recovery in some patients.
NEOPLASTIC OPTIC NEUROPATHIES
Neoplasms can affect the optic nerve directly or indirectly. Benign or malignant neoplasms can cause compression of the optic nerve. The neoplasms can develop intrinsically from nerve tissue itself or grow adjacent to the nerve. Less commonly, infiltration or metastasis from a remote site may occur. Table 5-5 summarizes neoplastic lesions of the optic nerve, based on location.
Table 5-5.
Neoplastic Lesions of the Optic Nerve
A neoplastic optic neuropathy may occur in the setting of a known, primary tumor or be the presenting manifestation of cancer. The neurologist should have a high index of suspicion for a neoplastic, compressive optic neuropathy for the following reasons.
Early detection of the tumor may result in improved survival.
In many cases, the tumor may pose a risk to the unaffected eye.
Visual loss may recover once the tumor is treated.
Compressive, neoplastic optic neuropathies are among the most treatable forms of optic nerve dysfunction, and dramatic recovery may occur with decompression of the visual pathways.
Neoplastic lesions may affect the orbital, intracranial, and prechiasmal optic nerve. The visual symptoms typically develop slowly, over weeks and months, and it is common for patients to have seen several eye care providers with no diagnosis. Although uncommon, some patients harboring compressive lesions may experience sudden vision loss, due to intratumoral hemorrhage and/or necrosis. Perhaps more commonly, the onset may appear sudden because the patient happens to cover the sound eye and becomes aware of long-standing visual loss.
The earliest abnormalities present include an ipsilateral relative afferent pupillary defect and dyschromatopsia, as expected for any optic neuropathy. Visual field defects are almost always present and may be of any type. However, temporal hemianopic defects, a central scotoma that extends into the periphery temporally, and a “junctional” scotoma strongly suggest a compressive lesion. The fundus appearance may differ somewhat depending on the location of the tumor. Intracranial, intracanalicular, and posterior orbital compressive lesions typically produce no disc swelling. Optic disc swelling is more commonly found with intraorbital tumors near the optic nerve head and may be accompanied by shunt vessels that bypass the central retinal vein to drain into the vortex veins and ciliary circulation (Figure 5-11). These shunt vessels are common with optic nerve sheath meningiomas but may also occur with gliomas.
Figure 5-11.
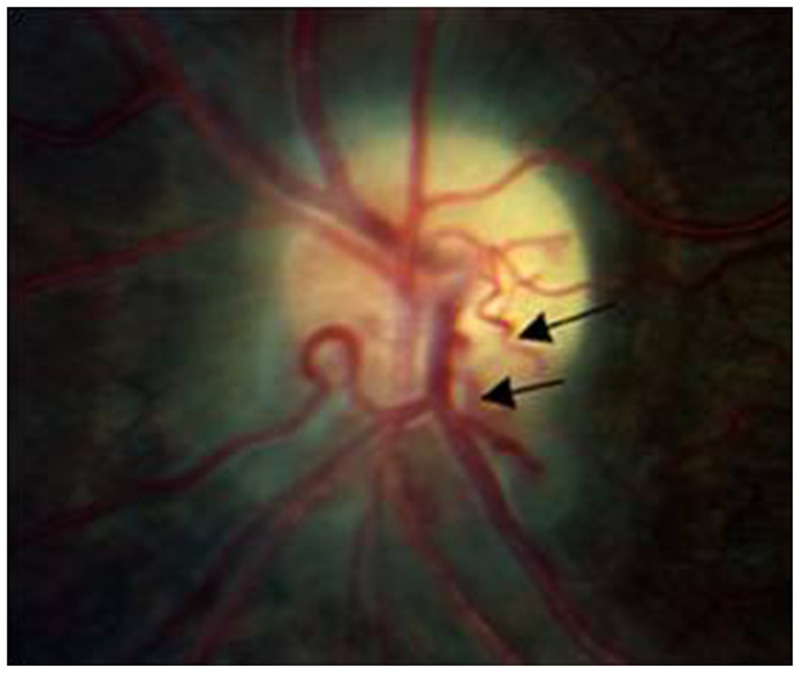
Color disc photograph of a patient with optic nerve sheath meningioma, demonstrating severe optic disc pallor, peripapillary gliotic changes suggesting previous disc swelling, and shunt vessels (arrows) on the surface of the disc. These vessels reflect diversion of retinal venous drainage from the central retinal veins to the vortex veins draining the choroid and outer retina.
Pituitary tumors and other parasellar lesions may cause intracranial optic nerve as well as chiasmal compression. Lesions that affect the anterior angle of the chiasm and the distal optic nerve produce a distinct syndrome characterized by a temporal defect and decreased visual acuity in the ipsilateral eye, and a superotemporal defect with preserved visual acuity in the contralateral eye (Figure 5-12). The ipsilateral temporal visual field defect may be hemianopic if all crossing fibers are involved or scotomatous if only macular crossing fibers are involved. The origin of the superotemporal, contralateral visual field defect is of both historical and clinical interest. Wilbrand proposed that crossed fibers originating from ganglion cells inferior and nasal to the fovea in the contralateral eye extend anteriorly (less than 2 mm) into the involved optic nerve and are thus subject to compression. This anatomic configuration is known as the Wilbrand knee.46 Although some believe that Wilbrand knee is simply a pathologic artifact rather than a true anatomic structure, its clinical relevance remains unquestioned, and the finding of this junctional visual field defect provides strong evidence of a lesion at the anterior angle of the chiasm, mandating neuroimaging. Pituitary tumors can rarely present with a unilateral optic neuropathy, particularly in patients in whom the optic chiasm is postfixed (located posterior to the pituitary gland, placing the intracranial nerves in a position more vulnerable to compression).
Figure 5-12.
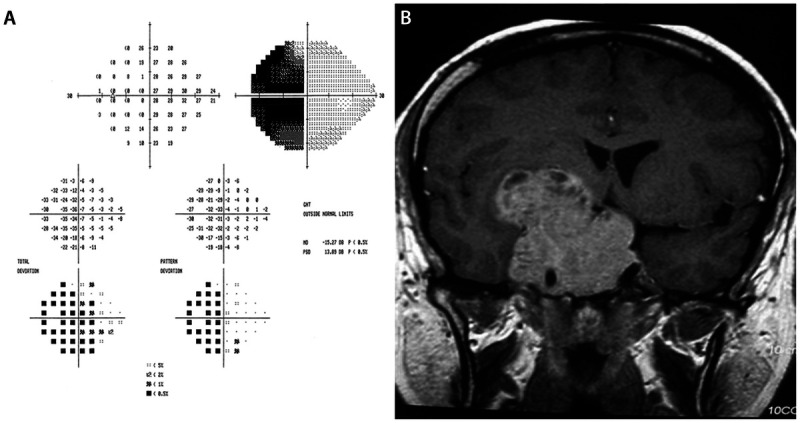
Pituitary macroadenoma in a 34-year-old patient who presented with a 1-month history of progressive visual loss in the right eye. The left eye was asymptomatic. Visual acuity was light perception only in the right eye, and 20/20 in the left eye. A dense right relative afferent pupillary defect was present. Fundus examination of each eye was normal. Humphrey 24-2 automated visual field was performed in the left eye and demonstrated a complete temporal hemianopic defect (A). The combination of a right optic neuropathy and temporal defect in the left eye was compatible with a junctional scotoma, indicating a lesion compressing the right posterior optic nerve and the anterior right optic chiasm. This was confirmed with MRI showing a massive pituitary macroadenoma (B).
Neuroimaging is indicated for all patients suspected of having a compressive, neoplastic optic neuropathy. The ideal modality is MRI of the brain, with and without gadolinium, with fat-saturated orbital views. This offers superb views of the globe, optic nerve head, optic nerve sheath, extraocular muscles, and orbital apex. It is also superior to CT for the evaluation of the intracanalicular and intracranial optic nerve, the pituitary fossa, and the cavernous sinuses.
Treatment depends on the specific type of tumor, location, size, and degree of optic nerve dysfunction. The prognosis for visual outcome is related in part to duration of compression, as well as retinal ganglion cell and retinal nerve fiber layer integrity. Patients with severe optic atrophy at presentation (indicating loss of neurons and axons) have a poorer prognosis for recovery, although some of these patients may have partial improvement after decompression.
A comprehensive review of all tumors that may compress the optic nerve is beyond the scope of this article. However, any patient presenting with an unexplained optic neuropathy should be suspected of harboring a compressive lesion, and neuroimaging is indicated unless a well-defined cause is known. Close communication between the neuro-ophthalmologist and a neurosurgeon well versed in treating these tumors is essential for optimal management.
Primary Optic Nerve Neoplasms
Optic glioma. Optic gliomas are the most common intrinsic tumors of the visual pathway and occur in two forms. The most common is the juvenile, benign, pilocytic astrocytoma; the much rarer form is malignant glioblastoma of the optic nerve. There is some variability in the nomenclature; optic glioma is often used to refer to gliomas involving the orbital optic nerve, while optic pathway glioma refers to tumors involving the optic chiasm and/or hypothalamus.
Optic pathway glioma usually presents in the first decade of life, and 90% present within the first 2 decades of life. They can occur in the setting of neurofibromatosis type 1 (NF 1), and approximately one-fourth of patients with optic nerve and chiasmal glioma have NF 1, while about 15% of patients with NF 1 develop these lesions. Two-thirds of patients with optic pathway gliomas have a normal neuro-ophthalmic examination, with no evidence of visual loss.47
When present, visual loss is slowly progressive, and the examination shows the expected features of optic nerve dysfunction. If the orbital optic nerve is affected, optic disc swelling, proptosis, and occasionally ocular motor deficits may be present. Chiasmal and hypothalamic gliomas often present as large masses even though they are typically well differentiated and low grade (most often pilocytic) and may be associated with hydrocephalus and the diencephalic syndrome.
The diagnosis is made by neuroimaging, which typically shows kinking, fusiform enlargement, and enhancement of the optic nerve (Figure 5-13). In general, MRI is superior to CT, since it will better evaluate intracranial extension and chiasmal or hypothalamic involvement.
Figure 5-13.
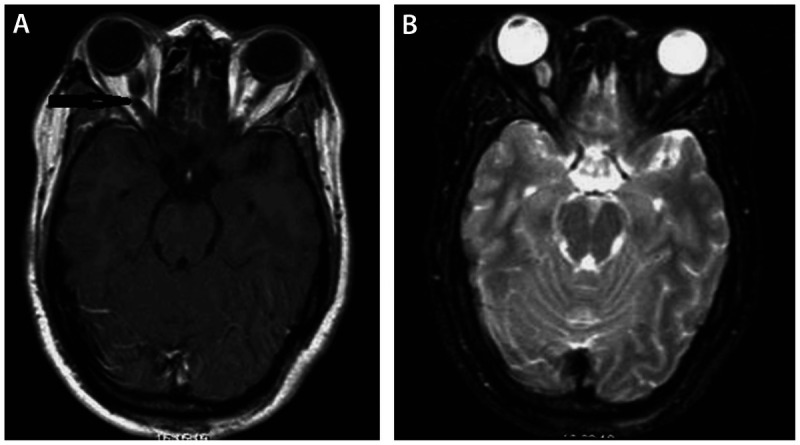
Brain MRI of a 26-year-old woman who presented with incidentally discovered orbital mass and headache. T1 axial gadolinium enhanced image (A) showed enlargement with kinking of the right optic nerve (arrow), demonstrated also on an axial T2-weighted fat-saturated image (B). These features are compatible with orbital optic glioma. The patient had a completely normal neuro-ophthalmic examination, and observation was planned.
The clinical course of juvenile optic pathway glioma is extremely variable, complicated to some degree by the fact that, in many case series, progression is defined radiologically, with no careful documentation of visual function. Monitoring optic glioma in very young children is challenging, as visual metrics are more difficult to obtain.
Treatment of optic pathway glioma is highly individualized and depends in part on the location of the tumor, the size, associated neurologic deficits and clinical findings (eg, hydrocephalus and pituitary/hypothalamic dysfunction), and the degree of visual loss. Surgical resection of an orbital optic glioma is impossible without sacrificing the nerve; for chiasmal and hypothalamic gliomas, surgical treatment is a viable option, particularly if associated hydrocephalus is present. Orbital optic gliomas rarely progress to involve the optic chiasm, so surgical resection to prevent intracranial extension is almost never indicated. Radiation therapy carries the risk of developmental delay, delayed radionecrosis, and future malignancies, and no clear evidence suggests that treatment alters long-term prognosis. However, external beam radiation (less than 45 Gy) has been used in patients with progressive tumor to diminish tumor bulk, decrease recurrence rates, prolong recurrence-free survival, and improve vision. Chemotherapy rather than radiation is often preferred as initial treatment, especially in children younger than 5 years old, for whom there is a high risk of adverse effects on cognitive function with radiation.
It is currently recommended that patients with optic pathway gliomas confined to the orbit be monitored for sustained progression of visual loss before considering treatment. Visual acuity remains the best clinical measure available to monitor disease, although accuracy and reliability of testing highly depend on the child’s cooperation. Surveillance MRI scans and careful neuro-ophthalmic examinations should be performed periodically. Treatment is indicated when evidence shows sustained progression of visual loss or the development of disfiguring proptosis.47,48
Optic nerve sheath meningioma. Optic nerve sheath meningiomas are the second most common primary optic nerve tumor (Case 5-3). They account for 2% of all orbital tumors and 1% to 2% of all meningiomas. It is important to distinguish these tumors from external (eg, paraclinoid, sphenoid wing) meningiomas that compress the optic nerve because the treatment differs considerably. Optic nerve sheath meningiomas are benign neoplasms that arise from the meningoepithelial cells along the optic nerve sheath. They most frequently occur in middle-aged adults, with a mean age of 41 years and a female predominance. Optic nerve sheath meningiomas and neurofibromatosis type 2 are loosely associated.49
The clinical presentation of optic nerve sheath meningioma is a slowly progressive visual loss with associated findings of optic nerve dysfunction, which is similar to other compressive optic neuropathies. Proptosis occurs in up to 60% of patients and may be the presenting sign. If the lesion involves the orbital optic nerve, optic disc swelling may be present and demonstrate shunt vessels. Most optic nerve sheath meningiomas (approximately 95%) are unilateral, but when bilateral, 65% are located in the optic canals. Proptosis is common but is rarely the presenting symptom.
Neuroimaging using MRI of brain and orbits, with and without gadolinium, is the diagnostic modality of choice. Optic nerve sheath meningioma may radiologically mimic some infiltrative and inflammatory disorders (eg, lymphoma, sarcoidosis, and idiopathic optic perineuritis), but the clinical history is usually sufficient to allow accurate diagnosis. Biopsy is rarely indicated, as it carries the risk of inducing further damage to the optic nerve.
The prognosis is variable, although most series suggest that progressive visual loss occurs without treatment in most patients. Recent literature suggests that patients with visual acuity of 20/50 or better may retain this level of visual function for at least 5 years, but at least 85% of patients with optic nerve sheath meningioma lose vision over time.50,51
Treatment for most patients is limited to observation and radiation. Observation is usually the first option in patients at either extreme, those with good visual function or very poor visual function at baseline (with the latter group having minimal potential for functional recovery). Careful follow-up is crucial, with repeat examinations every 3 to 6 months and follow-up MRI every 6 to 12 months. Involvement of the fellow eye due to intracranial extension may be more of a theoretic than actual risk, as bilateral cases are more likely related to intracranial meningiomas spreading into the orbits. Most neuro-ophthalmologists recommend that treatment be offered when visual acuity drops below 20/40 or with sustained progression of visual field loss.
Surgical resection plays a limited role in the management of optic nerve sheath meningioma, and is employed only when the patient has disfiguring proptosis and no useful vision, or with aggressive tumors that have intracranial extension. Even in these cases, surgery is associated with a high rate of local recurrence and orbital invasion and is typically expected to sacrifice the affected optic nerve.
Case 5-3
A 54-year-old woman presented to the neuro-ophthalmology clinic with a 3- to 4-month history of progressive visual loss in the left eye. She had been evaluated by an outside ophthalmologist and found to have visual acuity of 20/40 in both eyes, with a swollen left optic nerve. Brain MRI at an outside hospital showed possible enhancement of the left optic nerve, suggestive of demyelinating optic neuritis. The remainder of the study was normal, with no demyelinating lesions. The patient reported generalized clouding and blurring of vision in the left eye and no visual symptoms from the right eye. She denied pain on eye movements, transient visual loss, double vision, or other focal neurologic symptoms.
Medical history was significant for hypertension and hyperlipidemia. She denied tobacco and alcohol use. An extensive systemic review of systems was unremarkable.
Visual acuity was 20/20 right eye, 20/30 left eye. Ishihara color testing showed 11/11 plates right eye, 3/11 plates left eye. Pupils were equal and reactive, with a moderate left relative afferent pupillary defect. External examination showed no ptosis or proptosis. The slit-lamp and ocular motility examinations were unremarkable. Dilated fundus examination showed a normal right optic disc and severe, diffuse optic disc swelling in left eye, with congestion (Figure 5-14). Humphrey 24-2 visual fields were performed and showed mild, nonspecific generalized depression in the right eye and severe generalized depression in the left eye (Figure 5-15).
Figure 5-14.
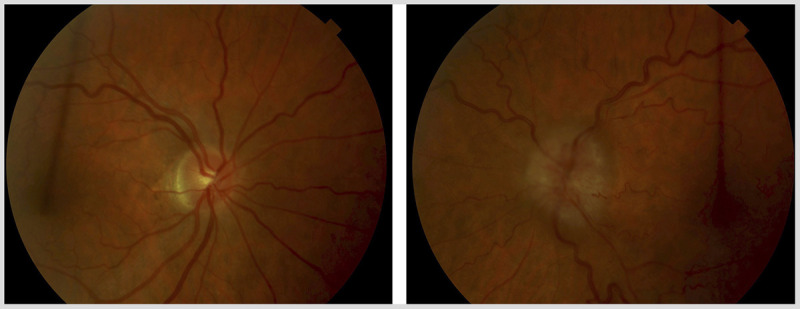
Dilated fundus examination shows severe, diffuse optic disc swelling in the left eye (on right) with congestion and a normal right optic disc (on left).
Figure 5-15.
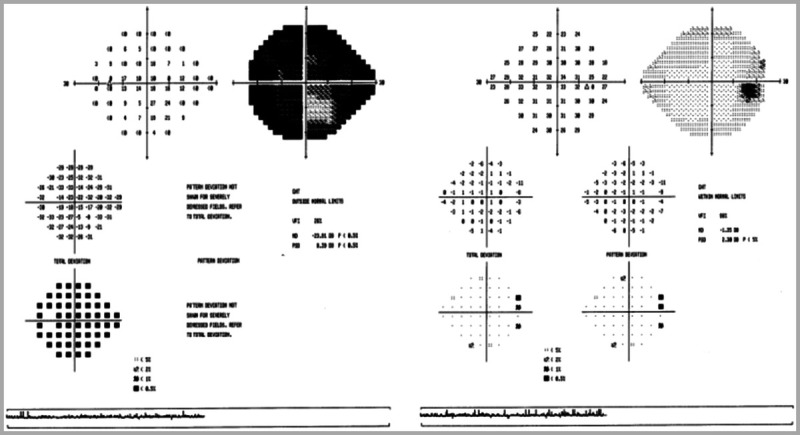
Humphrey 24-2 visual fields show severe generalized depression in the left eye (on left) and mild, nonspecific generalized depression in the right eye (on right).
Comment. The slowly progressive, painless visual loss and the severe optic disc swelling argued strongly against demyelinating optic neuritis and favored a compressive lesion. The original MRI was unavailable and had not included fat-saturated orbital images. Repeat brain MRI with dedicated, fat-saturated orbital images, with and without gadolinium, demonstrated intense perineural enhancement of the intraorbital and intracanalicular left optic nerve and was otherwise normal (Figure 5-16). Although optic nerve sheath meningioma was the likely diagnosis, the rapid progression of visual loss raised concern for alternate etiologies, including idiopathic optic perineuritis, sarcoidosis, syphilis, and lymphoma. Laboratory studies and CT of the chest, abdomen, and pelvis were unremarkable. The patient received a short course of oral corticosteroids, with no improvement. She was seen 3 months later, and visual acuity in the left eye had declined to 20/60. Repeat MRI showed no change, and review by neuroradiology staff agreed that optic nerve sheath meningioma was the likely diagnosis. She was referred to radiation oncology. She received fractionated radiation treatment using the intensity-modulated radiation therapy protocol, with a total dosage of 50 Gy in 28 fractions.
Figure 5-16.
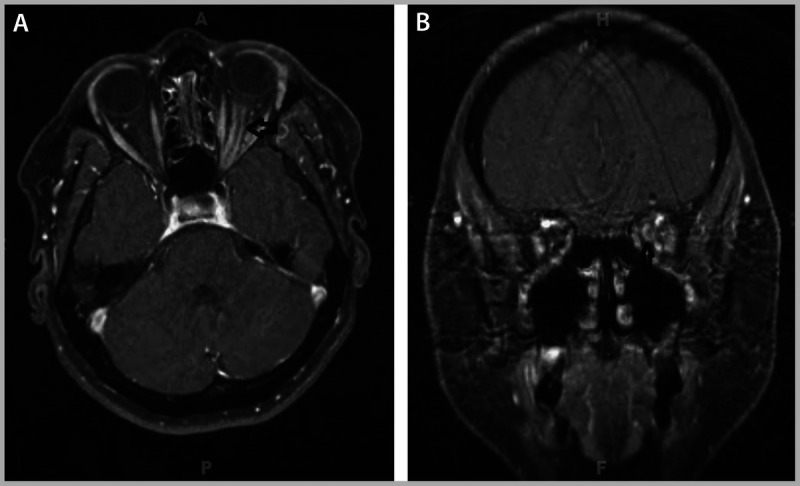
Repeat brain MRI (A, axial view; B, coronal view) with dedicated, fat-saturated orbital images, with and without gadolinium, demonstrated intense perineural enhancement of the intraorbital and intracanalicular left optic nerve and was otherwise normal.
She developed radiation keratopathy and later developed superimposed functional overlay, with nonphysiologic visual field changes. The optic disc swelling gradually resolved with supervening pallor, and she remained stable at her last visit, nearly 3 years after treatment.
Optic nerve sheath meningioma is an uncommon tumor, and the radiologic appearance can mimic other disorders, which may need to be excluded through ancillary testing. The prognosis is variable, but most patients progress over time. The tumor is highly radiosensitive, and radiation therapy remains the primary treatment modality.
PARANEOPLASTIC OPTIC NEUROPATHIES
Although still relatively rare, paraneoplastic visual loss is most commonly caused by carcinoma-associated retinopathy, an immune-mediated condition involving destruction of photoreceptors in the outer retina. Even more rarely, the optic nerve itself may be the primary target of a paraneoplastic disorder. Malik and colleagues52 in 1992 were the first to suggest an immunologic basis for optic neuropathy in the setting of a primary malignancy. At least 56 cases of paraneoplastic optic neuropathy have since been identified.53 Most cases are associated with small cell lung cancer, but other tumors also have been implicated, including B-cell lymphoma, uterine sarcoma, nasopharyngeal carcinoma, prostate cancer, and renal cell carcinoma.
Patients with paraneoplastic optic neuropathy typically present with subacute, painless visual loss. Although presentation varies, Cross and colleagues54 described a classic triad of optic disc edema, leakage from the optic disc vasculature on retinal angiography, and vitreous cells. Visual fields are abnormal, showing any type of optic nerve–related defect. Neuroimaging may be normal or show patchy T2-signal changes and enhancement of the optic nerves. The optic neuropathy is nearly always associated with multifocal neurologic deficits, including brainstem, spinal cord, or cerebellar dysfunction. Some patients have presented with optic nerve and spinal cord dysfunction simulating neuromyelitis optica.53
Most patients are seropositive for a serum antibody to a 62-kD neuronal antigen, collapsin response mediator protein-5 (CRMP-5).54 This protein is found in normal adult retina, optic nerve, and central and peripheral nervous system neurons. Aberrant expression of this protein has been found in small cell lung cancer tumor cells, and the host immune response invoked by neoplastic CRMP-5 is thought to cross-react with native tissues, resulting in the visual and neurologic manifestations of paraneoplastic optic neuropathy.
Paraneoplastic optic neuropathy should be suspected in any patient with a known primary malignancy and optic nerve dysfunction, particularly in patients with small cell lung cancer. Metastatic infiltration, carcinomatous meningitis, or treatment-related injury (eg, chemotherapy or radiation optic neuropathy) should also be in the differential and excluded through appropriate neuroimaging and laboratory studies. An isolated optic neuropathy, particularly with no disc edema or vitreous cells, is highly unlikely to represent paraneoplastic optic neuropathy, and this condition should be low in the differential in these cases. A CRMP-5 assay is commercially available, and the presence of CRMP-5 in the serum or CSF should mandate an expedited search for a primary malignancy.
Treatment of the primary tumor may stabilize or improve visual function in some patients. Adjunctive immunosuppressive therapies may be an additional treatment option, although the treatment results from individual case reports have been mixed.
KEY POINTS
A wide variety of optic neuropathies can manifest with similar clinical findings. The management and prognosis for each optic neuropathy may differ considerably. Historical clues and key elements of the examination, coupled with selective ancillary tests, are usually sufficient to achieve an accurate diagnosis.
With a few exceptions, all metabolic and hereditary optic neuropathies selectively affect the papillomacular bundle, a collection of retinal ganglion cells and their axons that subserves central vision, contrast sensitivity, and color vision. The resulting defects (reduced visual acuity, dyschromatopsia, cecocentral visual field defects, temporal optic disc pallor) reflect preferential papillomacular bundle dysfunction, and allow for a narrowed differential diagnosis.
The papillomacular bundle axons are smaller in caliber than other retinal ganglion cell fibers and are more vulnerable to energy depletion and reactive oxygen species. They are therefore uniquely susceptible to mitochondrial dysfunction from any cause.
Macular disease can mimic optic neuropathy, and failure to accurately localize may result in unnecessary neurodiagnostic testing and delay in diagnosis. Features favoring maculopathy over optic neuropathy include metamorphopsia, sparing of color vision, presence of pain, and lack of a relative afferent pupillary defect.
Very few nutritional deficiencies have been conclusively proven to cause optic neuropathy. Many patients harbor more than one nutritional deficiency, so clinicians should exercise caution when attributing optic neuropathy to a nutritional deficiency without good evidence.
The vitamins implicated in nutritional deficiency optic neuropathy are involved in mitochondrial energy production, and deficiency of any of these can result in bioenergetic failure and increased oxidative stress.
Optic neuropathy resulting from B12 deficiency should follow the “rules” of nutritional deficiency optic neuropathy. Laboratory confirmation of B12 deficiency is sometimes challenging, and incorrect diagnosis of B12 deficiency optic neuropathy can result in the delay of diagnosis and treatment.
An acute or subacute bilateral optic neuropathy in the setting of suspected nutritional deficiency should prompt expedited testing for and treatment of thiamine deficiency, even if more classic features of Wernicke encephalopathy are absent.
Most of the reported associations between specific drugs/toxins and optic neuropathy are based on anecdotal reports, and a true causal relationship is uncertain. Before recommending discontinuation of a specific agent, the physician should establish with as much certainty as possible that the medication is responsible for the visual loss, particularly if it is a medication vital to the treatment of another disease.
Ethambutol optic neuropathy is the best described and most common toxic optic neuropathy. This condition can occur even at “safe” doses and may cause irreversible visual loss if not detected early. All patients on ethambutol should receive periodic screening, with special attention paid to papillomacular bundle function (eg, visual acuity, color vision, visual field).
Linezolid has been associated with both optic neuropathy and peripheral neuropathy. The risk of toxicity increases with the duration of treatment. Discontinuation of the medication may result in some recovery of vision, but the peripheral neuropathy is typically permanent.
Tobacco-alcohol amblyopia is a rare and poorly characterized disorder. In many cases, the visual loss is secondary to nutritional deficiency. Neurologists should be reluctant to accept the diagnosis of tobacco-alcohol amblyopia without excluding other conditions, particularly nutritional deficiency.
Leber hereditary optic neuropathy typically presents with subacute, painless visual loss in one eye, with fellow eye involvement within weeks or months, usually progressing to severe visual loss over several months. Such a presentation should prompt suspicion of and genetic testing for Leber hereditary optic neuropathy, even if there is no relevant family history.
Hereditary optic neuropathy and nutritional deficiency or toxic optic neuropathy may coexist in the same patient, and nutritional deficiency may trigger visual loss in patients harboring one of the primary mutations for Leber hereditary optic neuropathy. The possibility of coexisting Leber hereditary optic neuropathy should be considered in any patient with nutritional deficiency or toxic optic neuropathy who fails to recover after repletion of the vitamin or removal of the offending agent, particularly if the visual loss is severe (worse than 20/200).
Dominant optic atrophy is the most common hereditary optic neuropathy worldwide. It causes slowly progressive visual loss throughout life. Since the degree of visual loss is variable, there may be no relevant family history, as affected family members may be unaware of their mild form of the disease. Genetic testing is commercially available.
MRI is of limited utility in the diagnosis and management of indirect traumatic optic neuropathy. The neuroimaging modality of choice is high-resolution axial and coronal orbital CT, screening for canalicular fracture and orbital hematoma.
There is no treatment proven effective for indirect traumatic optic neuropathy. The diagnosis can be challenging, particularly in an acutely head injured patient who cannot cooperate with the examination. Any interventions should be decided on a patient-by-patient basis.
Compressive, neoplastic optic neuropathies are often very treatable, and even profound visual loss may be reversible with decompression of the optic pathways. Clinicians should maintain a high index of suspicion for a compressive lesion in any patient with optic neuropathy.
A temporal hemianopic defect in one eye combined with an optic nerve-related defect in the fellow eye (junctional scotoma) on visual field testing strongly suggests a compressive lesion at the optic chiasm, and urgent neuroimaging is mandated.
Most patients with an orbital optic glioma have an excellent prognosis. Observation should be the first-line management option for most cases.
Optic nerve sheath meningioma is typically a slow-growing tumor and is not amenable to surgical resection. Treatment is usually reserved for patients with sustained progression of visual loss and involves some form of radiation therapy.
Paraneoplastic optic neuropathy is rare, and usually associated with other neurologic deficits, such as ataxia. The optic discs are commonly swollen, with inflammatory cells present in the posterior vitreous. Most patients are seropositive for collapsin response mediator protein-5 (CRMP-5).
Footnotes
Relationship Disclosure: Dr Van Stavern reports no disclosure.
Unlabeled Use of Products/Investigational Use Disclosure: Dr Van Stavern reports no disclosure.
REFERENCES
- 1.Wang MY,, Sadun AA. Drug-related optic neuropathies. J Neuro Ophthalmol 2013; 33 (2): 172–178. [DOI] [PubMed] [Google Scholar]
- 2.Kho RC,, Al-Obailan M,, Arnold AC. Bitemporal visual field defects in ethambutol-induced optic neuropathy. J Neuroophthalmol 2011; 31 (1): 121–126. [DOI] [PubMed] [Google Scholar]
- 3.Pan BX,, Ross-Cisneros FN,, Carelli V, et al. Mathematically modeling the involvement of axons in Leber’s hereditary optic neuropathy. Invest Ophthalmol Vis Sci 2012; 53 (12): 7608–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadun AA,, Win PH,, Ross-Cisneros FN, et al. Leber’s hereditary optic neuropathy differentially affects smaller axons in the optic nerve. Trans Am Ophthalmol Soc 2000; 98: 223–232; discussion 232–235. [PMC free article] [PubMed] [Google Scholar]
- 5.Phillips P. Toxic and deficiency optic neuropathies. In: Miller NR, ed. Walsh and Hoyt’s clinical neuro-ophthalmology. Newman, NJ: Lippincott Williams & Wilkins, 2005; 447–462. [Google Scholar]
- 6.Juhasz-Pocsine K,, Rudnicki SA,, Archer RL, et al. Neurologic complications of gastric bypass surgery for morbid obesity. Neurology 2007; 68 (21): 1843–1850. [DOI] [PubMed] [Google Scholar]
- 7.Sadun AA,, La Morgia C,, Carelli V. Mitochondrial optic neuropathies: our travels from bench to bedside and back again. Clin Experiment Ophthalmol 2013; 41 (7): 702–712. [DOI] [PubMed] [Google Scholar]
- 8.Stabler SP. Clinical practice. Vitamin B12 deficiency. N Engl J Med 2013; 368 (2): 149–160. [DOI] [PubMed] [Google Scholar]
- 9.Grzybowski A. Problems related to the diagnosis of B12 deficiency optic neuropathy. Acta Ophthalmol 2014; 92 (1): e74–e75. [DOI] [PubMed] [Google Scholar]
- 10.Chester EM,, Agamanolis DP,, Harris JW, et al. Optic atrophy in experimental vitamin B12 deficiency in monkeys. Acta Neurol Scand 1980; 61 (1): 9–26. [DOI] [PubMed] [Google Scholar]
- 11.Longmuir R,, Lee AG,, Rouleau J. Visual loss due to Wernicke syndrome following gastric bypass. Semin Ophthalmol 2007; 22 (1): 13–19. [DOI] [PubMed] [Google Scholar]
- 12.Surges R,, Beck S,, Niesen SA, et al. Sudden bilateral blindness in Wernicke’s encephalopathy: a case report and review of the literature. J Neurol Sci 2007; 260 (1–2): 261–264. [DOI] [PubMed] [Google Scholar]
- 13.Ball GFM. Vitamins: their role in the human body. Oxford, United Kingdom: Blackwell Publishing, 2004: 273–288. [Google Scholar]
- 14.Pineles SL,, Wilson CA,, Balcer LJ, et al. Combined optic neuropathy and myelopathy secondary to copper deficiency. Surv Ophthalmol 2010; 55 (4): 386–392. [DOI] [PubMed] [Google Scholar]
- 15.Hill AB. The environment and disease: association or causation? Proc R Soc Med 1965; 58: 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carelli V,, La Morgia C,, Sadun A. Mitochondrial dysfunction in optic neuropathies: animal models and therapeutic options. Curr Opin Neurol 2013; 26 (1): 52–58. [DOI] [PubMed] [Google Scholar]
- 17.World Health Organization. Global tuberculosis control: surveillance, planning, financing: WHO report 2008. WHO/HTM/TB/2008.393. Geneva, Switzerland: World Health Organization, 2008. [Google Scholar]
- 18.Chung H,, Yoon YH,, Hwang JJ, et al. Ethambutol-induced toxicity is mediated by zinc and lysosomal membrane permeabilization in cultured retinal cells. Toxicol Appl Pharmacol 2009; 235 (2): 163–170. [DOI] [PubMed] [Google Scholar]
- 19.Sharpe JA,, Hostovsky M,, Bilbao JM,, Rewcastle NB. Methanol optic neuropathy: a histopathological study. Neurology 1982; 32 (10): 1093–1100. [DOI] [PubMed] [Google Scholar]
- 20.Bhatia R,, Kumar M,, Garg A,, Nanda A. Putaminal necrosis due to methanol toxicity. Pract Neurol 2008; 8 (6): 386–387. [DOI] [PubMed] [Google Scholar]
- 21.Desai T,, Sudhalkar A,, Vyas U,, Khamar B. Methanol poisoning: predictors of visual outcome. JAMA Ophthalmol 2013; 131 (3): 358–364. [DOI] [PubMed] [Google Scholar]
- 22.De Vriess AS,, Coster RV,, Smet J, et al. Linezolid-induced inhibition of mitochondrial protein synthesis. Clin Infect Dis 2006; 42 (8): 1111–1117. [DOI] [PubMed] [Google Scholar]
- 23.Johnson LN,, Krohel GB,, Thomas ER. The clinical spectrum of amiodarone associated optic neuropathy. J Natl Med Assoc 2004; 96 (11): 1477–1491. [PMC free article] [PubMed] [Google Scholar]
- 24.Passman RS,, Bennett CL,, Pupura JM, et al. Amiodarone-associated optic neuropathy: a critical review. Am J Med 2012; 125 (5): 447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mansour AM,, Puklin JE,, O’Grady R. Optic nerve ultrastructure following amiodarone therapy. J Clin Neuroophthalmol 1988; 8 (4): 231–237. [PubMed] [Google Scholar]
- 26.Akagi T,, Manabe S,, Ishigooka H. A case of cyclosporine induced optic neuropathy with a normal therapeutic level of cyclosporine. Jap J Ophthalmol 2010; 54 (1): 102–112. [DOI] [PubMed] [Google Scholar]
- 27.Venneti S,, Moss HE,, Levin MH, et al. Asymmetric bilateral demyelinating optic neuropathy from tacrolimus toxicity. J Neurol Sci 2011; 301 (1–2): 112–115. [DOI] [PubMed] [Google Scholar]
- 28.Carroll FD. Nutritional amblyopia. Arch Ophthalmol 1966; 76 (3): 406–411. [DOI] [PubMed] [Google Scholar]
- 29.Grzybowski A,, Holder GE. Tobacco optic neuropathy (TON)—the historical and present concept of the disease. Acta Ophthalmol 2011; 89 (5): 495–499. [DOI] [PubMed] [Google Scholar]
- 30.Wallace DC,, Singh G,, Lott MT, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988; 242 (4884): 1427–1430. [DOI] [PubMed] [Google Scholar]
- 31.Sadun AA,, La Morgia C,, Carelli V. Leber’s hereditary optic neuropathy. Curr Treat Options Neurol 2011; 13 (1): 109–117. [DOI] [PubMed] [Google Scholar]
- 32.Newman NJ. Treatment of hereditary optic neuropathies. Nat Rev Neurol 2012; 8 (10): 545–556. [DOI] [PubMed] [Google Scholar]
- 33.Palace J. Multiple sclerosis associated with Leber’s hereditary optic neuropathy. J Neurol Sci 2009; 286 (1–2): 24–27. [DOI] [PubMed] [Google Scholar]
- 34.Li J,, Rucker JC. Irreversible optic neuropathy in wernicke encephalopathy and leber hereditary optic neuropathy. J Neuroophthalmol 2010; 30 (1): 49–53. [DOI] [PubMed] [Google Scholar]
- 35.Newman NJ,, Biousse V,, David R, et al. Prophylaxis for second eye involvement in leber hereditary optic neuropathy: an open-labeled, non-randomized multicenter trial of topical brimonidine purite. Am J Ophthalmol 2005; 140 (3): 407–415. [DOI] [PubMed] [Google Scholar]
- 36.Klopstock T,, Yu-Mai-Wan P,, Dimitriadis K, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 2011; 134 (pt 9): 2677–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu-Wai-Man P,, Griffiths PG,, Gorman GS, et al. Multi-system neurologic disease is common in patients with OPA1 mutations. Brain 2010; 133 (pt 3): 771–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacobs SM,, Van Stavern GP. Neuro-ophthalmic deficits after head trauma. Curr Neurol Neurosci Rep 2013; 13 (11): 389. [DOI] [PubMed] [Google Scholar]
- 39.Yang QT,, Zhang GH,, Liu X, et al. The therapeutic efficacy of endoscopic optic nerve decompression and its effects on the prognoses of 96 cases of traumatic optic neuropathy. J Trauma Acute Care Surg 2012; 72 (5): 1350–1355. [DOI] [PubMed] [Google Scholar]
- 40.Van Stavern GP,, Biousse V,, Lynn MJ, et al. Neuro-ophthalmic manifestations of head trauma. J Neuroophthalmol 2001; 21 (2): 112–117. [DOI] [PubMed] [Google Scholar]
- 41.Bodanapally UK,, Kthirkamanathan S,, Geraymovych E, et al. Diagnosis of traumatic optic neuropathy: application of diffusion tensor magnetic resonance imaging. J Neuroophthalmol 2013; 33 (2): 128–133. [DOI] [PubMed] [Google Scholar]
- 42.Edwards P,, Arango M,, Balica L, et al. Final results of MRC CRASH, a randomized placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet 2005; 365 (9475): 1957–1959. [DOI] [PubMed] [Google Scholar]
- 43.Yu-Wai-Man P,, Griffiths PG. Steroids for traumatic optic neuropathy. Cochrane Database Syst Rev 2013; 6: CD006032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu-Wai-Man P,, Griffiths PG. Surgery for traumatic optic neuropathy. Cochrane Database Syst Rev 2005; (4): CD005024. [DOI] [PubMed] [Google Scholar]
- 45.Bracken MB,, Shepard MJ,, Holford TR, et al. Methylprednisolone or tirilazad mesylate administration after acute spinal cord injury: 1-year follow up. Results of the third National Acute Spinal Cord Injury randomized controlled trial. J Neurosurg 1998; 89 (5): 699–706. [DOI] [PubMed] [Google Scholar]
- 46.Wilbrand H,, Saenger A. Die Neurologie des Auges. Wiesbaden. J Bergman 1904; 3: 98–120. [Google Scholar]
- 47.Avery RA,, Fisher MJ,, Liu GT. Optic pathway glioma. J Neuroophthalmol 2011; 31 (3): 269–278. [DOI] [PubMed] [Google Scholar]
- 48.Nicolin G,, Parkin P,, Mabbot D, et al. Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer 2009; 53 (7): 1231–1237. [DOI] [PubMed] [Google Scholar]
- 49.Shapey J,, Sabin HI,, Danesh-Meyer H,, Kaye AH. Diagnosis and management of optic nerve sheath meningiomas. J Clin Neurosci 2013; 20 (8): 1045–1056. [DOI] [PubMed] [Google Scholar]
- 50.Turbin RE,, Thompson CR,, Kennerdall JS, et al. A long term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology 2002; 109 (5): 890–899. [DOI] [PubMed] [Google Scholar]
- 51.Paulsen F,, Doerr S,, Wilhem H, et al. Fractionated stereotactic radiotherapy in patients with optic nerve sheath meningioma. Int J Radiat Onc Biol Phys 2012; 82 (2): 773–778. [DOI] [PubMed] [Google Scholar]
- 52.Malik S,, Furlan AJ,, Sweeney PJ, et al. Optic neuropathy: a rare paraneoplastic syndrome. J Clin Neuroophthalmol 1992; 12 (3): 137–141. [PubMed] [Google Scholar]
- 53.Rahimy E,, Sarraf D. Paraneoplastic and non-paraneoplastic retinopathy and optic neuropathy: evaluation and management. Surv Ophthalmol 2013; 58 (5): 430–458. [DOI] [PubMed] [Google Scholar]
- 54.Cross SA,, Salomao DR,, Parisi JE, et al. Paraneoplastic autoimmune optic neuritis with retinitis defined by CRMP-5 IgG. Ann Neurol 2003; 54 (1): 38–50. [DOI] [PubMed] [Google Scholar]