

Abstract
Purpose of Review
With transition to the genetic era, the number of muscular dystrophies has grown significantly, but so too has our understanding of their pathogenic underpinnings. Clinical features associated with each muscular dystrophy still guide us to the diagnosis. However, improved diagnostic abilities refine and expand phenotypic and genotypic correlates. This article discusses the epidemiology, clinical features, and diagnosis of these disorders.
Recent Findings
Some important recent advancements include (1) a much greater understanding of the pathogenetic pathways underlying facioscapulohumeral muscular dystrophy and myotonic dystrophy type 1; (2) the publication of diagnostic and treatment guidelines for Duchenne muscular dystrophy; and (3) further clarification of the many genetic muscle disorders presenting a limb-girdle pattern of weakness.
Summary
Muscular dystrophies are genetic, progressive, degenerative disorders with the primary symptom of muscle weakness. Duchenne, Becker, facioscapulohumeral, and myotonic muscular dystrophies are most prevalent and tend to have distinctive features helpful in diagnosis. The limb-girdle, Emery-Dreifuss, and oculopharyngeal muscular dystrophies are less common but often may also be diagnosed on the basis of phenotype. Researchers hope to help patients with future discoveries effective in slowing or halting disease progression, reversing or preventing underlying mechanisms, and repairing previously damaged muscle.
INTRODUCTION
Muscular dystrophies are genetic, progressive, degenerative disorders of muscle. Muscle weakness is the primary symptom. Clinical and pathologic criteria have been used in the past for classification, but now muscular dystrophies are mostly classified on a genetic basis.
DYSTROPHINOPATHIES
Dystrophinopathies include the spectrum of diseases associated with mutations in DMD, the gene for dystrophin. Most commonly, mutations in DMD present as Duchenne muscular dystrophy (DMD), but dystrophinopathies manifest a number of other phenotypes, including Becker muscular dystrophy (BMD); isolated quadriceps myopathy1; asymptomatic or minimally symptomatic hyperCKemia2; aches, pains, cramps, and rhabdomyolysis3; manifesting female carriers4; X-linked dilated cardiomyopathy5; and cognitive disorders such as developmental delay, autism spectrum disorders, attention deficit hyperactivity disorder, and impaired intelligence.6
Epidemiology
The incidence of DMD has been reported as 1:3500 live male births for many years. Newborn screening places the incidence closer to 1:5000 live male births. BMD has an incidence reported as approximately one-tenth to one-fifth that of DMD. However, ascertainment of milder phenotypes and a longer lifespan have yielded prevalence rates at 60% to 90% that of DMD. Female carriers should have an incidence comparable to DMD with a much higher prevalence due to a normal lifespan in most. Dystrophinopathies are X-linked disorders.
Clinical Features
Duchenne muscular dystrophy. Boys with DMD most often present between 3 and 5 years of age because of delayed motor milestones and falls, along with difficulty running and jumping. Typically, examination reveals calf hypertrophy, mildly lordotic posture, waddling of gait, and poor hip excursion during running (Figure 2-1). Boys with DMD also have head lag when pulled to sitting from supine and a partial Gower maneuver when rising from the floor. They tend to gain motor milestones through 6 to 7 years of age; however, progressive weakness ensues thereafter. Historically, boys with DMD transition to wheelchair use before their 12th birthdays.
Figure 2-1.
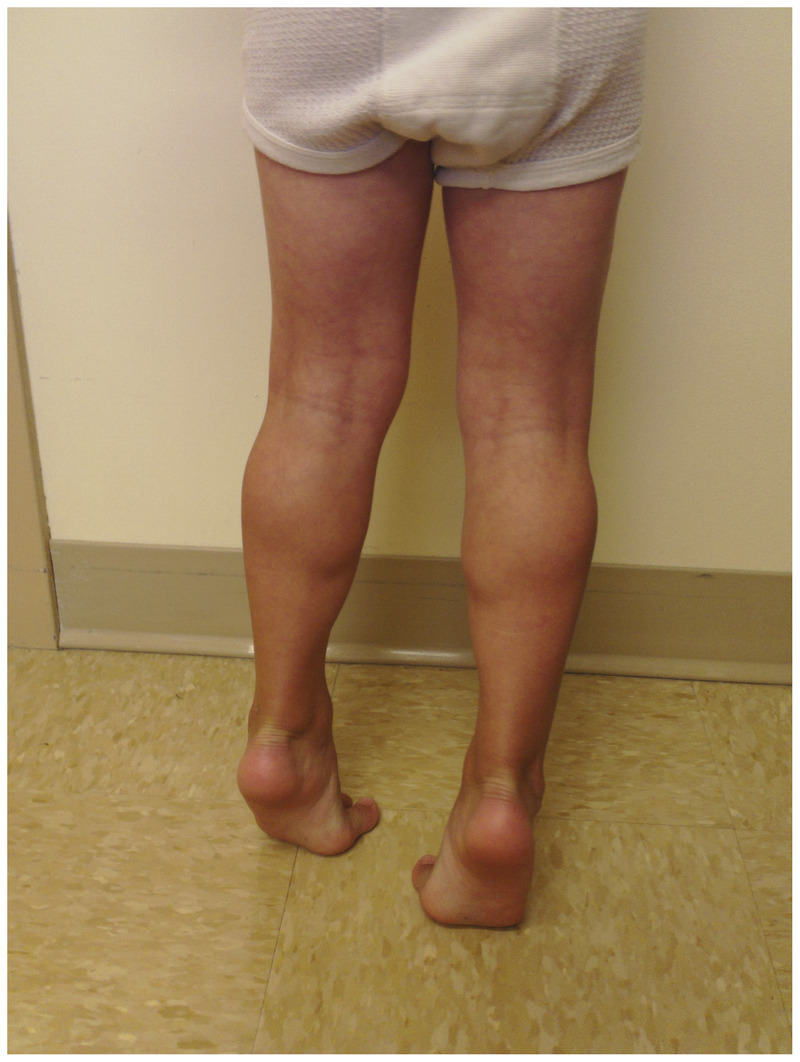
Calf hypertrophy in a 4-year-old boy with Duchenne muscular dystrophy.
By convention, if a boy with a dystrophinopathy stops walking before 12 years of age, he has DMD. Those who remain ambulatory beyond their 16th birthday are considered to have BMD. Finally, those who stop walking between their 12th and 16th birthdays have been designated as having an intermediate phenotype. Corticosteroids, which prolong ambulation by 1 to 3 years, are blurring the distinction of DMD versus BMD. Some investigators have transitioned to the more general term dystrophinopathies. This article will continue to follow the older nomenclature.
Joint contractures occur in the ankles even while still walking, and follow in the hips, knees, elbows, and wrists once power wheelchair use is full-time. Night splints, stretching, physical therapy, and standing boards forestall the development and decrease the severity of contractures. Kyphoscoliosis accelerates when ambulation ceases and sometimes requires spinal fusion for optimal respiratory function.
Pulmonary function declines continuously and should be monitored regularly by a pulmonologist with spirometry in the upright and supine positions. Noninvasive ventilation, especially at night, along with pulmonary hygiene (including mechanical insufflator-exsufflator devices) decrease infections and improve quality of life. Tracheostomy and mechanical ventilation is a choice made by some patients and their families later in disease.
Patients with DMD develop cardiac involvement and should be followed by a cardiologist via ECG, Holter monitor, and echocardiography. Sinus tachycardia is common. Atrial and ventricular arrhythmias may follow, associated with cardiac fibrosis and a dilated cardiomyopathy. Angiotensin-converting enzyme (ACE) inhibitors, beta-blockers, and other cardioprotective agents reduce progression and can improve ventricular function.
Cognitive involvement is common in DMD. Of patients with DMD, 30% have intellectual disability. The average patient’s IQ is 85, one standard deviation below the mean. Verbal IQ is more affected than performance IQ, and delayed language development is common. Attention deficit hyperactivity disorder (10% to 15%), autism spectrum disorder (3% to 6%), and obsessive compulsive disorder (5%) are also more prevalent in DMD.7
Management of DMD requires a multidisciplinary health care team.8,9,10 In addition to the cardiac, pulmonary, and orthopedic issues discussed above, the following specialties add to the quality of life in DMD: endocrinology to manage osteoporosis in immobile patients; ophthalmology to address cataract formation; nutritional medicine, speech therapy, and gastroenterology for optimal management of caloric intake, dysphagia, and possibly feeding-tube placement; developmental pediatrics and psychology for cognitive and behavioral issues; and rehabilitation medicine to optimize function and for provision of appropriate adaptive devices and wheelchairs.
Mean lifespan in DMD has increased from 19 years to more than 25 years. Some clinics have patients living into their thirties and even forties. Much of this increased longevity can be ascribed to improvements in care and corticosteroid use.
Corticosteroids should be offered to all boys with DMD. Corticosteroids prolong walking; transiently increase, and then slow the decline in, muscle strength; reduce falls; improve pulmonary measures; decrease scoliosis; and may improve cardiac and cognitive function. The literature does not allow consensus regarding when to start corticosteroids. Most often, boys are started on corticosteroids between 5 and 7 years of age, before they start to lose motor milestones. However, some specialists favor starting as early as 3 to 4 years of age. Optimal corticosteroid dosing for DMD is not defined, and a survey of Muscular Dystrophy Association clinics found more than 20 different corticosteroid regimens. Common corticosteroid regimens include (1) prednisone 0.75 mg/kg/d; (2) deflazacort 0.9 mg/kg/d; (3) prednisolone 0.75 mg/kg/d for 10 days alternating with 10 days off corticosteroids; and (4) prednisone 2.5 mg/kg to 5 mg/kg every Friday and Saturday. Before starting corticosteroids, patients should receive all of their immunizations, start on vitamin D, and have family dietary counseling. Clinics must monitor patients for corticosteroid side effects including weight gain, behavioral changes, growth retardation, hypertension, glucose intolerance, peptic ulcer disease, cataracts, acne, and fractures.8 Corticosteroid therapy continues to show benefit after loss of ambulation.
Becker muscular dystrophy. Severity of weakness varies substantially in patients with BMD. Weakness may begin as early as 5 to 6 years of age or may start in the fifth or sixth decade. Any male patient with a limb-girdle pattern of weakness at any age should be evaluated for a dystrophinopathy, irrespective of family history. On average, older onset of disease correlates with slower progression of weakness.
The extent of cardiac involvement does not necessarily correlate with skeletal muscle disease. Relatively preserved skeletal muscle strength and activity levels and retained ambulation place disproportional strain on a dysfunctional heart. As such, BMD may present with congestive heart failure in the face of minimal proximal weakness. Therefore, early referral with regular follow-up by a cardiologist is important, even without cardiac symptoms. Treatment with ACE inhibitors and/or beta-blockers forestalls failure, and left ventricular assist devices and cardiac transplantation are appropriate in selected individuals.
Respiratory dysfunction tends to be milder in BMD than DMD. However, referral for pulmonary evaluation with pulmonary function tests is indicated, and use of nocturnal noninvasive ventilation may be required by some.
In BMD, intellectual function is generally normal, although learning disabilities are slightly more common. Life expectancy is shortened for men with major cardiac and diaphragmatic involvement.
Dystrophinopathy gene carriers. Female gene carriers bear an underappreciated burden from mutations in DMD. Rarely, young girls present with a phenotype and disease course identical to DMD, usually associated with skewed X-chromosome inactivation. More often, symptomatic gene carriers manifest milder weakness with onset in the third decade of life or later. Creatine kinase (CK) levels may be elevated in 30% to 60% of carriers, whether they are symptomatic or not. This means that a normal CK level does not exclude carrier status. In a Dutch national survey of dystrophinopathy gene carriers from 18 to 60 years of age, 17% had muscle weakness varying from mild to severe, 8% had symptomatic dilated cardiomyopathy, and 18% had evidence for left ventricle dilation on echocardiography.11 Thus, nearly 40% had symptoms or signs that deserved further medical evaluation or observation. All female first-degree relatives of patients with DMD or BMD should undergo DNA analysis and, if they test positive, be evaluated for evidence of weakness or cardiac involvement.
Diagnosis
Dystrophinopathies are caused by mutations in DMD, one of the largest genes in humans with 2.3 megabases and inclusive of 0.1% of the entire human genome. Its 79 exons only make up 0.6% of the gene, with the remaining 99.4% dwelling in intronic sequences. DMD encodes the 3685–amino-acid, 427-kilodalton protein dystrophin, with eight promoters and seven different transcripts, which can be found in skeletal, cardiac, and smooth muscle fibers along with cortical, Purkinje, Schwann, glial, retinal, and kidney cells. Dystrophin has an N terminus anchored to actin, a long coiled-coil rod domain with 21 spectrin repeats, a cysteine-rich segment linked to β-dystroglycan at the sarcolemma, and a carboxy-terminal domain with many phosphorylation sites. Dystrophin, along with α-, β-, γ-, and δ-sarcoglycan, β-dystroglycan, sarcospan, syntrophins, dystrobrevin, and nitric oxide synthase, form the dystrophin-glycoprotein complex (Figure 2-212). This complex mechanically links the muscle fiber’s contractile apparatus, the sarcomere, to the extracellular matrix. Dystrophin also plays a role in communication across the sarcolemma.
A dystrophinopathy should be suspected in any male or female patient presenting with progressive limb-girdle weakness, especially if the patient has a positive family history, substantially elevated CK level, or cardiomyopathy.
The diagnosis of DMD should be the physician’s first thought when a 3- to 5-year-old boy who is physically slower than his peers presents with toe walking, large calves, neck weakness, a partial Gower’s sign, and a CK level greater than 3000 U/L. CK levels may be elevated 10- to 200-fold. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT), which are also muscle enzymes, are often elevated. This transaminitis reflects muscle involvement rather than liver disease, and this can be verified by checking the liver-specific transaminase γ-glutamyltransferase (GGT), which will be normal. In DMD, DNA analysis of the dystrophin gene is the first diagnostic procedure.
In patients with BMD or in manifesting female carriers, weakness may not be as pronounced, the CK level may be lower, and the initial procedure will often be electrodiagnostic testing. The EMG reveals myopathic motor units with or without muscle membrane instability. If the patient has no family history of a dystrophinopathy, then a muscle biopsy may be performed. Immunostaining for the N-terminal, rod, and C-terminal regions of dystrophin will usually reveal diffusely decreased, or patchy, staining of some but not all muscle fiber membranes. Confirmatory testing is through DNA analysis.
Mutation analysis consists of analysis for duplications and deletions via multiplex PCR or multiplex ligation-dependent probe amplification. If this is negative, then gene sequencing is undertaken. Mutations in DMD causative for DMD and BMD are fairly consistent when assessed across different countries,13,14,15,16 with the composition including 43% to 67% deletions and 9% to 11% duplications of exons, 16% to 26% point mutations, and 5% to 6% splice site mutations. Therapies allowing multiexon skipping from exons 45 through 55 would benefit more than 50% of patients with DMD.
Mutations disrupting the reading frame (out-of-frame mutations) in DMD create a truncated RNA transcript that is rapidly degraded. This leads to a virtual absence of dystrophin in muscle and a DMD phenotype. Mutations with maintenance of the reading frame (in-frame mutations) generate shorter or less stable dystrophin. Dystrophin with in-frame mutations retain their amino- and carboxy-terminus domains and thus still maintain the mechanical bridge between actin and β-dystroglycan. In-frame mutations more often lead to a BMD phenotype. In a large series, out-of-frame mutations led to a DMD phenotype in nearly 90% of cases; however, in-frame mutations led to a BMD phenotype in only approximately 60% of cases (Case 2-1).13
Case 2-1
A 4-year-old boy presented for neuromuscular evaluation. His birth was uncomplicated, but he did not sit until 12 months or walk until 20 months. At 2 years old, he only spoke two words and required speech therapy. At 4 years old, he fell more often than other children but continued to gain motor and language milestones. The family and the pediatrician were concerned about attention deficit hyperactivity disorder. On family history, a maternal second cousin carried the diagnosis of Becker muscular dystrophy but required full-time wheelchair use at 10 years of age.
On examination, the patient’s tongue and calves were large; he could not lift his head from the bed when supine; he could not arise from a squat and required the help of furniture to get up from the floor; and he had a slightly hyperlordotic posture when walking and diminished hip excursion when running.
His creatine kinase level was 31,188 U/L. His muscle biopsy was dystrophic (Figure 2-3), revealed areas of endomysial inflammation (Figure 2-4), and demonstrated diminished staining for the three epitopes of dystrophin (Figure 2-5). Western blot analysis revealed dystrophin quantity at 20% to 100% of normal with decreased dystrophin molecular size. This combination was interpreted to predict a mild or moderate Becker phenotype. Genetic testing of DMD revealed deletion of exons 58 through 67, an in-frame deletion interpreted to predict a Becker phenotype. However, his examination at 4 years of age was much more consistent with a Duchenne phenotype.
Comment. This case brings up two key points. (1) The clinical features of inability to lift the head from the bed or to arise from the floor at this age strongly suggest a Duchenne phenotype, and (2) the reading-frame rule for exon deletions and duplications holds true in most but not all cases. (Figure 2-6).
DMD and BMD are inherited in an X-linked recessive fashion, but in roughly 20% to 30% of cases, a mother does not test positive for the mutation in DMD. This is due to a high rate of new mutations, false maternity, and to germ-line mosaicism (in which the mutation is present in some tissues, such as ovaries, but not others, such as skeletal muscle). Because of the possibility of germ-line mosaicism, negative genetic testing in a mother does not preclude the possibility of an affected boy in a subsequent pregnancy. For this indication, preimplantation genetic testing is available.
FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY
As the name suggests, predominant involvement in facioscapulohumeral muscular dystrophy (FSHD) includes facial, periscapular, biceps, and triceps muscles. Pectoral muscles are distinctly weak, yet the deltoid muscles tend to be spared. Weakness spreads rostrocaudally with onset in the face, then the scapular region, followed by the proximal arms, then the legs. Different than in many muscular dystrophies, asymmetries are the norm in FSHD.
Epidemiology
Prevalence of FSHD is estimated at 7 per 100,000. However, with unrecognized mild cases, a normal lifespan, and improved genetic testing taken into consideration, actual prevalence may surpass 13 per 100,000, similar to the myotonic dystrophies and dystrophinopathies. FSHD is an autosomal dominant disorder. Although 70% to 90% of patients have a family history of FSHD, up to 30% of cases are sporadic.
Clinical Features
Onset of disease usually occurs in the patient’s teens, and 90% of patients show signs of disease by 20 years of age. Facial weakness most often is the earliest manifestation. Patients have difficulty pursing their lips and exhibit a transverse smile. Lips may have a pouty appearance. The palpebral fissures are widened, and patients have difficulty burying their eyelashes. Noteworthily, extraocular and bulbar muscles remain unaffected.
Since subtle perioral and periocular weakness is functionally nonlimiting early in disease, weakness in the arms, especially in overhead activities, often brings patients to medical attention. In the shoulder-girdle, scapular winging is prominent, (Figure 2-7) and pectoral muscle atrophy leads to reversal of the anterior axillary folds. Usually, the anterior axillary folds slant outward, toward the shoulder. In FSHD, due to pectoral atrophy and scapular laxity, the anterior axillary folds point inward, toward the neck, and the shoulders often slope downward. Additionally, patients may have the triple-hump sign, composed of the deltoid muscle, the bones of the shoulder, and the high-riding, winged scapula (when observing from the arm inward toward the neck) (Figure 2-8). Asymmetries in scapular winging, pectoral atrophy, or muscle weakness are common (Figure 2-9).
In the lower extremities, the ankle dorsiflexors weaken before proximal leg muscles in most patients. Sometimes this leads to foot drop with a slapped-foot gait. Weak abdominal muscles result in marked abdominal protuberance out of proportion to body habitus, and difficulties in performing sit-ups. Weak paraspinous muscles lead to an exaggerated lumbar lordosis.17 Pain is common in FSHD. Shoulder-girdle laxity leads to muscle and joint pain, but also to neurogenic pain from the weight of the arm tugging downward and stretching the brachial plexus and nerve roots. Neck and back pain due to paraspinous muscle weakness, with kyphosis and exaggerated lumbar lordosis, is also common.
Hearing loss is reported in 75% of patients with FSHD, and retinal vascular abnormalities in 60%. However, these hearing and retinal changes tend to be subclinical and asymptomatic in adult-onset disease, although more prevalent and problematic in infantile-onset FSHD. If the retinal vasculopathy progresses to an exudative retinopathy with visual loss unilaterally, this condition is known as Coat syndrome.
Cardiac arrhythmias are present in a slightly higher proportion of FSHD patients than controls but are generally asymptomatic. Because of the lack of significant bulbar, respiratory, and cardiac involvement in FSHD, life expectancy is normal; however, 20% of patients eventually require wheelchair use. Disease progression is steady in most, but some report stepwise, sudden declines in function. Earlier-onset disease correlates with greater functional disability.
Cases of FSHD sometimes present with atypical phenotypes, including minimal or no facial involvement in 5% to 10% of cases (therefore, the lack of obvious facial involvement should not dissuade the clinician from a diagnosis of FSHD); focal involvement of one limb or one side of the body; distal predominant disease; axial myopathy with camptocormia (forward flexion of the spine when standing or walking, but resolves when supine); and the more severe, infantile-onset disease.18 Infantile-onset disease afflicts approximately 4% of FSHD patients and may have associated dramatic facial weakness, early wheelchair need, mental retardation, severe hearing loss, and epilepsy.19
Diagnosis
In fully developed FSHD, the diagnosis is accomplished by simply asking the patient to remove his or her shirt. The distinctive reversal of the anterior axillary folds along with scapular winging, asymmetries, triple-hump sign, and facial weakness guide the diagnosis. Difficulty pursing the lips can be elicited by asking patients to whistle or demonstrate how to blow out a candle. Scapular winging stands out when patients raise their arms in a slow jumping-jack motion, bring their arms up in a forward motion, or attempt a push-up. In addition to visual inspection, it is useful to palpate the scapula with one hand during these maneuvers. This may detect subtle, early, asymmetric scapular winging. Pectoral muscle weakness can be evaluated by having patients attempt to hold their unclenched arms outstretched at the level of the navel (sternal head) and above eye level (clavicular head) while the examiner attempts to pull the arms apart.
CK levels range from normal to 1000 U/L. EMG reveals myopathic motor units with or without muscle membrane instability.
Definitive diagnosis of this autosomal dominant disorder is based on genetic analysis of a non–protein-coding region of chromosome 4q. The genetic basis of FSHD has been linked to a reduction in the number of 3.3 kilobase (kb) tandem repeats (termed D4Z4) on chromosome 4q35. Although a similar D4Z4 repeat sequence is found on chromosome 10q26, only patients with a reduction in the number of D4Z4 repeats on chromosome 4q35 develop FSHD. People with D4Z4 repeat sizes in excess of 50 kb (11 or more D4Z4 repeats) do not develop FSHD; those with repeats sizes smaller than 35 kb (8 or fewer D4Z4 repeats) develop FSHD; and those with 9 or 10 D4Z4 repeats tend to have mild or nonpenetrant disease. Interestingly, people with no D4Z4 repeats do not develop FSHD.
Due to cases that did not follow these rules, it was reported that a reduced number of D4Z4 repeats will manifest disease only when the following two permissive conditions coexist.20 Before the D4Z4 repeats on chromosome 4q35, one of three particular six-nucleotide simple sequence length polymorphisms (SSLPs) must be present (4A159, 4A161, or 4A168). In addition, following the D4Z4 repeats are two large sequence variations (4qA and 4qB), and only the 4qA variant is permissive of disease. So, for FSHD to manifest, the patient must have an allele meeting all of the following conditions: (1) on chromosome 4q35; (2) with a truncation of the D4Z4 repeat size to 8 to 10 or fewer; (3) in the presence of one of three permissive SSLPs (4A159, 4A161, or 4A168); and (4) with the 4qA variant at the terminus.
In patients with the above four genetic characteristics, the D4Z4 region is hypomethylated, leading to a more open chromatin structure. The final D4Z4 repeat sequence on chromosome 4q35 contains the gene DUX4. DUX4 transcripts are toxic in cell culture and may be responsible for abnormal myogenesis and myofiber apoptosis (Case 2-2).21
Case 2-2
A 37-year-old man who had recurrent right shoulder dislocations was referred by his orthopedist for evaluation for possible right brachial plexopathy. In high school, the patient had played volleyball and cricket in his native India. His dislocations occurred while spiking the ball during volleyball; although he was a bowler in cricket, he did not experience dislocations with that motion. He remembered long-standing pectoral muscle atrophy on his right only but had no family history of weakness, muscle atrophy, or asymmetric smile.
On examination, he could purse his lips and puff his cheeks without release of air. When smiling with lips closed, he displayed minimal elevation of the corners of the mouth. When smiling with teeth showing, he had a transverse smile. He showed minimal weakness of the muscles of the left upper lip. He had diminished bulk of the right pectoralis muscles. Mild scapular winging was evident bilaterally, more prominent on the left with shoulder extension and more prominent on the right with shoulder abduction. He had reversal of the anterior axillary folds on the right but not the left. A large, athletic man, he demonstrated full strength except as follows (Medical Research Council scale): shoulder abductors 4+/5, shoulder external rotators 4/5−, elbow flexors 4+/5, elbow extensors 5−/4+, finger extensors 5/4+; hip flexors 4+/4+, knee flexors 5/4+, ankle dorsiflexors 4+/5.
His EMG did not reveal a brachial plexopathy. Instead, myopathic motor units were seen in the deltoid, biceps, triceps, infraspinatus, serratus anterior, rhomboid major, and tibialis anterior muscles on the right. Facioscapulohumeral muscular dystrophy (FSHD) genetic testing revealed one normal allele of over 48 kb in size and one truncated allele of 33 kb. This confirmed the diagnosis of FSHD.
Comment. This case illustrates several important diagnostic features in FSHD. (1) Although facial weakness is nearly universally present, it may be subtle. (2) Asymmetries are the norm in this muscular dystrophy, sometimes strikingly so. (3) The finding of pectoral atrophy with reversal of the anterior axillary folds is highly predictive of the diagnosis.
Patients with the above genetics have been classified as having FSHD1, and make up approximately 90% to 95% of FSHD patients. Interestingly, approximately 5% of patients with FSHD do not have a truncated D4Z4 repeat size and yet have a classic FSHD phenotype. These patients also have hypomethylation of their D4Z4 region, and have been labeled as having FSHD2. FSHD2 is caused by autosomal dominant mutations in SMCHD1, the gene for structural maintenance of chromosomes flexible hinge domain-containing 1 protein.22
Of note, population-based genetic testing for FSHD has found a much higher prevalence of FSHD permissive alleles (perhaps 1.3% of people),23 which suggests that incomplete penetrance or further epigenetic factors may be involved in the manifestation of an FSHD phenotype.
MYOTONIC DYSTROPHIES
It is important to think of the two myotonic dystrophies not just as the most common muscular dystrophies in adults, but also as systemic disorders with effects on multiple organ systems. Initially described more than 100 years ago, myotonic dystrophy type 1 (DM1) affects strength, tone, the heart, breathing, swallowing, sleep, cognition, vision, and endocrine function. Myotonic dystrophy type 2 (DM2) may affect all these functions but overall tends toward onset later in life with less frequent and severe manifestations, except for pain and stiffness. Both are autosomal dominant disorders.
Epidemiology
The worldwide prevalence of myotonic dystrophy lies in the 5 to 20 per 100,000 range. The myotonic dystrophies afflict mostly people of European heritage, with lesser frequencies in Asia and a virtual absence of disease in sub-Saharan Africa. The minimum prevalence for the myotonic dystrophies in a northern England population exceeded 10 per 100,000 for type 1 and was only 0.2 per 100,000 for type 2.24 However, since pain and milder systemic symptoms in middle-aged and older patients with DM2 are often misdiagnosed or overlooked, the prevalence of DM2 may approach or surpass the more easily recognized DM1.25
Clinical Features
Myotonic dystrophy type 1. DM1 presents in four ways: classic adult-onset, congenital, childhood-onset, and late-onset oligosymptomatic.
The cardinal manifestations of adult-onset DM1 include weakness, cataracts, and myotonia (increased muscle irritability and contractility with impaired muscle relaxation) before age 50. A physician meeting a patient with DM1 for the first time often makes the diagnosis via the distinctive visible features, such as temporalis and masseter muscle wasting (“hatchet-face” look), male pattern baldness, and facial weakness (ptosis and a “fish-mouth” look) (Figure 2-10). Additionally, the patient’s inability to release a weak introductory handshake (reflecting grip myotonia and flexor digitorum profundus weakness) and a hypophonic, nasal dysarthria are other early clues.
Figure 2-10.
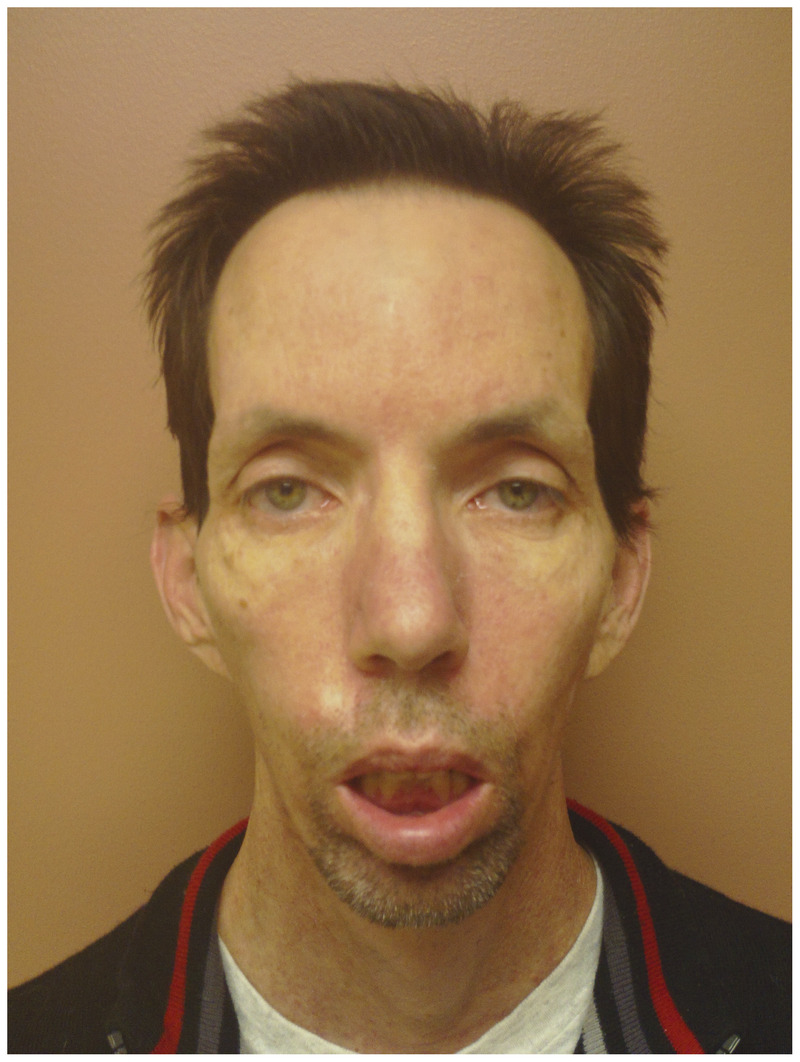
Man displaying the classic facial features of myotonic dystrophy type 1. He has male pattern baldness, temporalis muscle wasting, mild bilateral ptosis, and moderate perioral weakness with a “fish mouth” appearance.
Weakness in DM1 predominantly affects facial, oropharyngeal, long finger flexor, and foot/toe dorsiflexor muscles. Proximal weakness is not present early but may follow later in the course. Respiratory muscles can be affected early and disproportionately.
Grip myotonia reveals a 1-second to 5-second delay in fully extending the fingers after 10 seconds of forceful hand contraction. This grip myotonia shows a warm-up phenomenon, with faster relaxation on repeated contractions. Percussion myotonia may be elicited by tapping the thenar eminence or the forearm extensor muscles with a reflex hammer. Mexiletine (150 mg orally 3 times per day) significantly reduces relaxation times without serious adverse systemic or cardiac side effects, and some patients with DM1 obtain respite from stiffness and myotonia.26
Cataracts appear as multicolored, scintillating, punctate lens imperfections (“Christmas tree cataracts”) seen on slit lamp or ophthalmoscopic examination.
Cardiac evaluation and management is of utmost importance for reducing morbidity and mortality in DM1. Cardiac involvement correlates with cytosine, thymine, guanine (CTG) trinucleotide repeat size and severity of weakness. A meta-analysis found that arrhythmias and conduction blocks occur frequently in patients (first-degree atrioventricular block in 28.2%, QTCc greater than 440 ms in 22%, QRS greater than 120 ms in 19.9%, frequent premature ventricular contractions in 14.6%, atrial fibrillation/flutter in 5%, right/left bundle branch block in 4.4/5.7%, and nonsustained ventricular tachycardia in 4%). Left ventricular dysfunction (defined as left ventricular ejection fraction less than 50%) is uncommon at only 7.2% of patients, and does not occur before age 40. The mean age of death in DM1 was reported to be 53.2 years in the 1990s, with nearly a third of deaths due to cardiovascular diseases or sudden death.27,28 Sudden cardiac death risk is 0.56% per year,27 and the majority of cases of sudden death are due to ventricular tachyarrhythmias.29 Annual monitoring by a cardiologist and consideration for early pacemaker/defibrillator placement is recommended.
Respiratory muscle weakness predisposes a patient to pneumonia and nocturnal hypoxemia and should be surveilled via seated and supine pulmonary function testing. More than 30% of patients with DM1 experience fatigue, excessive daytime somnolence, or postprandial sleepiness, which are likely underappreciated in this population. Evaluations with sleep studies and treatment with modafinil or positive airway pressure are warranted. In DM1, one-third of deaths stem from respiratory failure and pneumonia.
Endocrine dysfunction is common, and patients should be monitored for diabetes mellitus, thyroid deficiency, hyperlipidemia, and testosterone insufficiency. Male hypogonadism, male infertility, and miscarriages are common.
In the classic form of the disease, impaired executive function, avoidant personality, depression, apathy, and mild cognitive impairment are reported. Brain MRI demonstrates white matter changes, more so than atrophy, especially involving the temporal tips (Figure 2-11).
Figure 2-11.
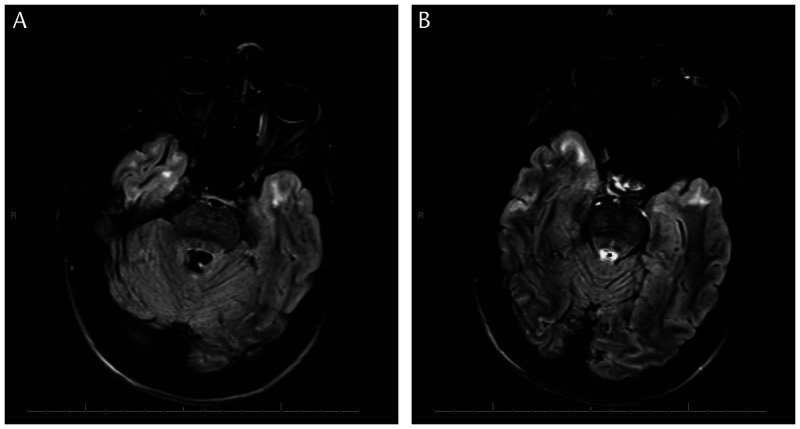
Brain MRI of a 36-year-old woman with myotonic dystrophy type 1 showing increased signal intensity on axial fluid-attenuated inversion recovery (FLAIR) sequences in the temporal tips, seen on two slices (A and B).
Dysphagia, constipation, diarrhea, bloating, and abdominal pain are frequent gastrointestinal concerns. Interestingly, antimyotonic therapy improves these symptoms in some patients. Dysphagia can become problematic and require attention later in the course. Cholelithiasis is increased.
Anesthesia and sedation pose a greater risk of arrhythmias and can result in prolonged respiratory depression. A patient’s initial diagnosis may be made after postoperative failure to wean off the ventilator.
Congenital myotonic dystrophy occurs in 25% of children of mothers with DM1. Some of these mothers with DM1 are undiagnosed and manifest few or no manifestations of disease. For this reason, it is imperative to evaluate women of reproductive age with myotonia on EMG for DM1. Clinically, congenital myotonic dystrophy presents with a much more severe phenotype: hypotonia, respiratory distress, failure to thrive due to weak suck, delayed motor and language milestones, and mild-severe cognitive dysfunction with IQ levels from 40 to 80 in most patients. Examination usually reveals prominent facial weakness with a fish-mouth appearance, high arched palate, arthrogryposis, hypotonia, and weakness. As infants and young children, no clinical or EMG myotonia is evident, and muscle biopsy results are normal or nonspecific. Brain MRI often has evidence for global atrophy, white matter changes, and callosal thinning.
Childhood-onset DM1 may present with learning difficulties in the early school years before development of prominent weakness. The extent of weakness parallels the length of disease. Thus, a 30-year-old patient with childhood-onset DM1 will be weaker than a 30-year-old patient with adult-onset DM1, and the same will hold true at age 50.
At the opposite end of the spectrum, the oligosymptomatic DM1 patient with a smaller than 150 CTG repeat expansion may only develop cataracts or male pattern baldness. Other oligosymptomatic people with DM1 might have mild weakness, ptosis, or dysphagia late in life, which may be difficult to discern from normal aging.30
Myotonic dystrophy type 2. DM2 most often presents in middle age or later and may vary significantly within families. Muscle pain and stiffness often predominate, although clinical and EMG myotonia may be minimal or even absent. This myalgic pain may limit activities and even lead to disability. In contrast to DM1, weakness is predominant proximally, although often the deep finger flexors are also substantially affected. Often, proximal lower extremity weakness with greater difficulty ascending stairs is the first sign in an elderly individual. Calf hypertrophy often occurs, but facial muscles are relatively uninvolved.
In most patients, the systemic manifestations in DM2 are less frequent and milder than in DM1. One in five patients with DM2 experience cardiac arrhythmia or conduction block, and endocrinopathies arise in roughly the same proportion of patients. Cataracts occur in nearly all patients, although generally later in disease. Increased signal intensity lesions can be seen in the white matter on MRI in more than half of DM2 patients, but cognitive and behavioral dysfunction is uncommon. Respiratory insufficiency is not a feature of this disorder, and with medical management, patients with DM2 have a normal lifespan. Childhood cases are very uncommon, and congenital onset remains the subject of publishable case reports.
Diagnosis
Clinical features usually are sufficient to make the diagnosis of DM1. In mild or early cases of DM1, and in DM2, further evaluation will be necessary.
CK levels in both disorders tend to be normal to mildly elevated (most often less than 1000 U/L). Needle EMG reveals overlapping decrescendo-crescendo myotonia in adults with DM1. Myotonia is absent in congenital DM1 and usually not present in the first decade of life. In DM2, the same prominent EMG myotonia may be present. However, in some patients with DM2, short decrescendo myotonic discharges may be infrequently seen or absent. No myotonia may be found on the initial EMG in 10% to 15% of DM2 cases.31
Genetic testing usually obviates the need for muscle biopsy in classic DM1 cases and in DM2 cases with prominent EMG myotonia. The muscle pathology in DM2 demonstrates pyknotic nuclear clumps, small type 2a fibers, and myofibers with increased internal nuclei (often more than 5 per fiber) (Figure 2-12).
Figure 2-12.
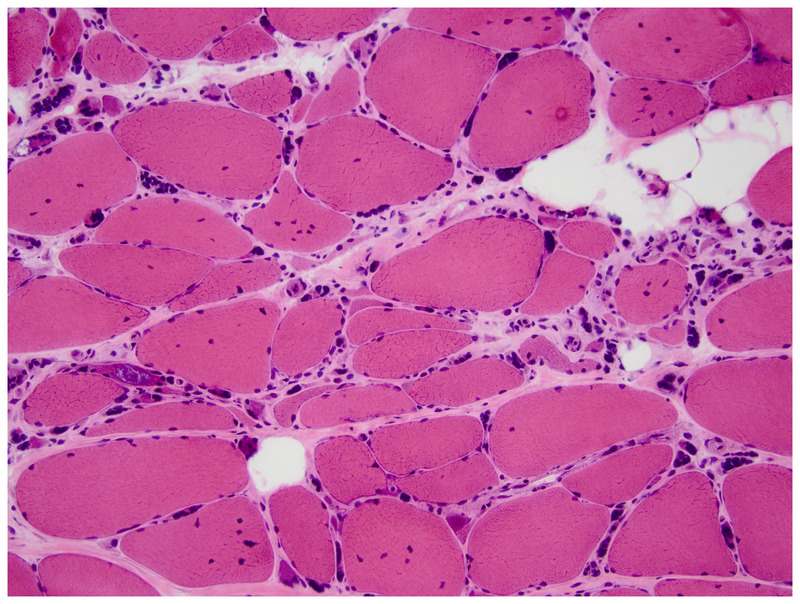
Muscle biopsy of a patient with myotonic dystrophy type 2. Note several muscle fibers with >5 internal nuclei and multiple pyknotic nuclear clumps superimposed on dystrophic appearing muscle (hematoxylin and eosin). Courtesy of Jennifer W. Baccon, MD, PhD.
Repeat expansion mutations are the underlying genetic basis for both myotonic dystrophies. DM1 is caused by a (CTG)n trinucleotide repeat expansion in the 3′-untranslated region of the dystrophia myotonica protein kinase gene (DMPK) on chromosome 19q13.3. DM2 is caused by a (CCTG)n quadruple nucleotide repeat expansion in intron 1 of the zinc finger protein 9 gene (ZNF9) on chromosome 3q21.3. In both DM1 and DM2, these repeat expansions produce aberrant RNA transcripts that collect into nuclear aggregates. Downstream effects of these aggregates include (1) interaction with proteins involved in RNA metabolism, and (2) mis-splicing of genes responsible for the multiple organ system involvement.
Individuals unaffected with DM1 have 4 to 37 CTG trinucleotide repeats. These repeat lengths tend to remain stable. People with (CTG)38-50 are considered to have premutations, and those with (CTG)51-100 have protomutations. These patients may not have evident disease or only minimal manifestations, such as late-onset cataracts. However, their CTG repeats are unstable and may expand in subsequent generations. (CTG)100-1000 repeat lengths yield classic disease, while very large repeat expansions, (CTG)1000-4000+, are associated with severe, often congenital, disease. In DM1, CTG repeat expansion can occur with either maternal or paternal transmission, but extreme expansions with congenital-onset disease occur almost always with maternal transmission. Trinucleotide repeat expansion size correlates inversely with age of onset and parallels severity of disease. CTG repeat length varies in different organs and tends to increase in size through life (Case 2-3).
Case 2-3
At 38 years of age, a father of three young children first noticed worsening mild dysarthria, dysphagia, fatigue, and excessive daytime sleepiness. He had no known family history of muscle disease, although his mother died of a cardiac arrest in her fifties, his 40-year-old sister had similar symptoms, and his 52-year-old brother had cataracts. His children were asymptomatic.
Examination revealed temporalis muscle wasting, mild orbicularis oculi weakness, slight finger flexor weakness with obvious grip myotonia, and mild ankle and toe extensor weakness. His creatine kinase level was 84 U/L. EMG revealed distal predominant myotonia and myopathic motor units. Genetic testing revealed a 168 CTG repeat expansion in the 3′-untranslated region of DMPK, confirming the diagnosis of myotonic dystrophy type 1 (DM1).
Over the ensuing decade, he had a cardiac ablation and pacemaker/defibrillator placed for arrhythmia; pulmonary function testing, which revealed a forced vital capacity of 1.84 L (43% predicted); bilevel positive airway pressure (BiPAP) initiated for sleep apnea; treatment for mild diabetes mellitus, thyroid disease, and vitamin B12 deficiency; cataracts removed; a swallowing study documenting mild-moderate aspiration; and botulinum toxin started for sialorrhea. He continued to work. His children, now 21, 19, and 15 years of age, had 530, 515, and 253 CTG repeat expansions, respectively. The oldest two children were symptomatic by that time.
Comment. This case illustrates two key features in DM1. (1) DM1 is not just a muscular dystrophy but, more importantly, a multisystem disorder. (2) Anticipation often occurs from generation to generation, with larger CTG repeat expansions and younger onset in each subsequent generation. Anticipation occurs with maternal or paternal transmission. However, very large CTG repeat expansions (1500 to 4000 or more), which result in severe, congenital myotonic dystrophy, nearly always emerge only with maternal transmission.
In DM2, age of onset and disease severity do not correlate well with CCTG repeat size. Although age of onset may vary somewhat within a family, overall, anticipation does not occur. This may be due to the fact that CCTG repeats frequently undergo both expansions and contractions.
The underlying pathogenesis of the myotonic dystrophies is thought to involve one or all three of the following mechanisms.
1. Transcription of expanded DNA nucleotide repeat sequences into mutant RNA with expanded cytosine, uracil, guanine (CUG) repeats interferes with trans-acting RNA-binding proteins with resultant abnormal RNA splicing for key proteins in muscle, brain, and endocrine glands.
2. Haplo-insufficiency of DMPK occurs in tissues of DM1 patients, and reduced DMPK levels in mice replicate the features of DM1, such as muscle weakness, cardiac conduction defects, and sodium channel abnormalities.
3. Expanded nucleotide repeats exert modifying effects on neighboring genes, possibly through chromatin condensation, hypermethylation, or disruption. As an exciting model for patient therapeutics, researchers showed that silencing expression of pathogenic RNA through antisense oligonucleotides reduced the levels of toxic RNA in mouse and human DM1 cells and a DM1 mouse model.32
LIMB-GIRDLE MUSCULAR DYSTROPHIES
Before the 1950s, specific muscular dystrophies were confined to Duchenne, facioscapulohumeral, and myotonic. All other patients were lumped into the limb-girdle muscular dystrophies (LGMDs). As currently defined, LGMDs are genetic muscle diseases with postnatal onset of progressive weakness and muscle atrophy affecting proximal upper and lower extremity muscles. Since the early 1990s, more than 25 genetic muscle disorders have fallen under this rubric. Despite a uniform underlying phenotype, LGMDs possess not only substantial genetic variability, but also phenotypic, pathogenic, and regional variability.
Many LGMDs display distinctive features that are valuable to clinicians in the diagnostic process. Age of onset, mode of inheritance, pattern of muscle weakness, extent of contractures, cardiopulmonary involvement, CK level, and muscle biopsy features all help distinguish one LGMD from another (Table 2-1).33 Patient phenotypes may even vary within a family, with the same mutation generating a proximal predominant pattern in one member and distal involvement in another.
Table 2-1.
Limb-Girdle Muscular Dystrophies
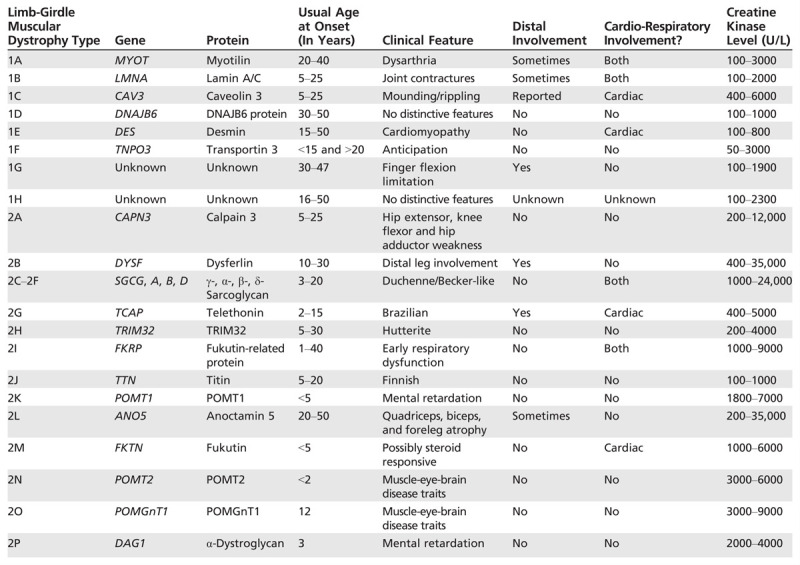
LGMDs stem from dysfunctional proteins at all levels of the muscle fiber: nucleus; intermediate filaments; sarcomere; sarcoplasm; sarcolemma; sarcolemmal repair, maintenance, trafficking, and signal transduction; and extracellular matrix (Figure 2-1334). Nomenclature for the LGMDs relies on a number for mode of inheritance (1 for autosomal dominant and 2 for autosomal recessive) and a letter for order of discovery (the chromosomal locus for LGMD1A was delineated before LGMD1B). However, LGMDs currently are more often named after the protein involved (eg, LGMD2A = calpainopathy).
Figure 2-13.
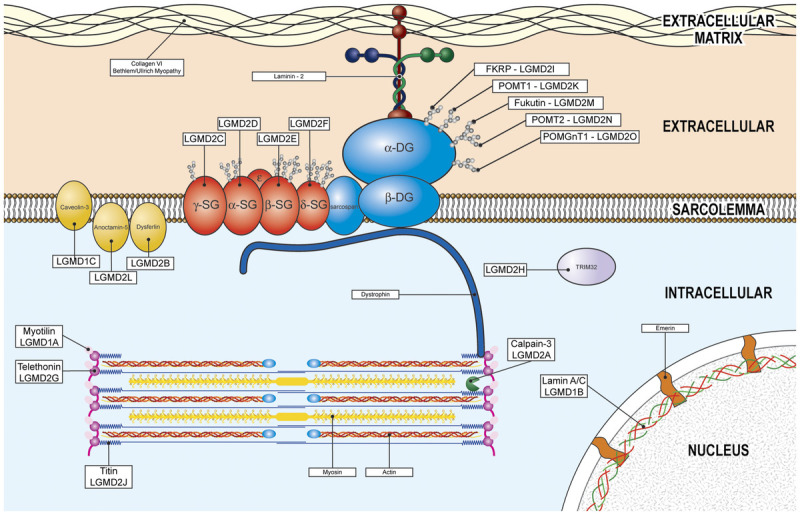
Subcellular localization of the limb-girdle muscular dystrophies. FKRP = fukutin-related protein; LGMD = limb-girdle muscular dystrophy; POMT = protein-O-mannosyltransferase; DG = dystroglycan; POMGnT1 = protein-O-linked mannose β1, 2-N-acetylglucosaminyltransferase; SG = sarcoglycan; TRIM32 = tripartite motif containing protein 32. Reprinted from Aminoff MJ, Daroff RB, Academic Press.34 © 2014, with permission from Elsevier.
Epidemiology
Some LGMDs appear to have regional prominence. While calpainopathies tend to be the most common LGMD in most populations sampled, the genes for Fukutin-related protein (FKRP) and anoctaminopathies have high prevalence rates in Northern Europe. Most reports of titinopathies emanate from Finland, while telethoninopathies are rare outside of the Canadian Hutterite population.
The United States is a melting pot of immigrants from around the world. In an evaluation of 488 genetically confirmed LGMD cases in the US population, calpainopathies comprised approximately 30% of the cases, while sarcoglycanopathies, dysferlinopathies, and FKRP accounted for approximately 15% to 20% of cases each (Figure 2-14).35 While clarity regarding the relative prevalence of each LGMD is improving, the prevalence in the population is not well defined. In the north of England, the minimum prevalence of LGMDs was 2.3 per 100,000, with individual LGMDs having minimum prevalence rates of less than 1 per 1,000,000 up to 6 per 1,000,000.24 Other estimates have placed LGMD prevalence higher. Regional, isolated populations may have much higher prevalence rates, as much as 5 to 15 per 100,000, similar to dystrophinopathies, myotonic dystrophy, and FSHD. Until the advent of population-based genome sequencing, reports likely will underestimate true prevalence.
Figure 2-14.
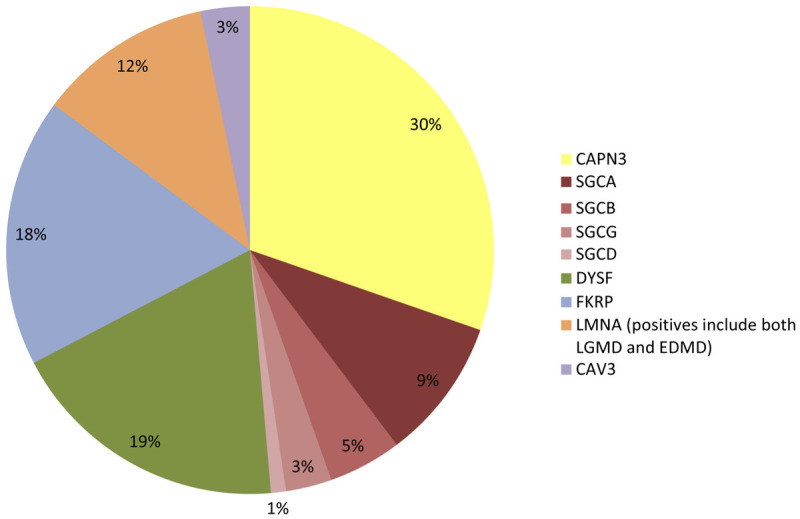
Relative prevalence of the limb-girdle muscular dystrophy subtypes in the United States. CAPN3 = Calpain 3 (limb-girdle muscular dystrophy [LGMD] type 2A); SGCA = α-sarcoglycan (LGMD2D); SGCB = β-sarcoglycan (LGMD2E); SGCG = γ-sarcoglycan (LGMD2C); SGCD = δ-sarcoglycan (LGMD2F); DYSF = dysferlin (LGMD2B); FKRP = Fukutin-related protein (LGMD2I); LMNA = Lamin A/C (LGMD1B); EDMD = Emery-Dreifuss muscular dystrophy; CAV3 = Caveolin 3 (LGMD1C).
Clinical and Laboratory Features
Some generalities can be made about the autosomal dominant and autosomal recessive LGMDs. Autosomal dominant LGMDs occur less frequently, have lower CK rates, and account for only around 10% to 15% of cases. Autosomal recessive LGMDs tend to have much higher CK levels and comprise the majority of cases.
Autosomal dominant limb-girdle muscular dystrophies. Limb-girdle muscular dystrophy type 1A. Myotilinopathies account for less than 2% of LGMD, and more often present as distal myopathies. Patients often present with both proximal and distal leg weakness in their twenties. A distinctive, nasal dysarthria and hypophonia may accompany the weakness or may be the sole manifestation in some family members.36
Limb-girdle muscular dystrophy type 1B. Mutations in the nuclear envelope proteins lamin A and lamin C underlie a vast array of disorders in addition to LGMD1B, including autosomal dominant Emery-Dreifuss muscular dystrophy, a dilated cardiomyopathy, a heart-hand syndrome, Dunnegan lipodystrophy, a mandibuloacral dysplasia, a restrictive dermopathy, an axonal neuropathy, and metabolic syndrome. Sometimes, these clinical presentations overlap like a Venn diagram in patients. Laminopathies make up 5% to 10% of LGMDs and generally present in the first few decades of life. Patients may have early joint contractures (especially of the elbows, Achilles tendon, or neck). Cardiac involvement relatively uniformly starts in the second and third decades of life regardless of skeletal muscle involvement. Dysrhythmias and conduction block may occur early in disease, with dilated cardiomyopathy later in the course. Pacemaker/defibrillator placement is common by the patient’s twenties.
Limb-girdle muscular dystrophy type 1C. Caveolinopathies make up less than 3% of LGMD but may also present as rippling muscle disease, distal myopathy, or asymptomatic hyperCKemia. Caveolinopathies can present in the first decade of life, and generally have the highest CK levels of the autosomal dominant LGMDs-as high as 3000 U/L to 5000 U/L. Some patientshave an electrically silent, self-propagating, rolling or rippling movement across their muscle with passive stretch or tapping of muscles. Others may note focal, rapid contraction, and localized mounding of muscles with a knock or blow to the muscle.
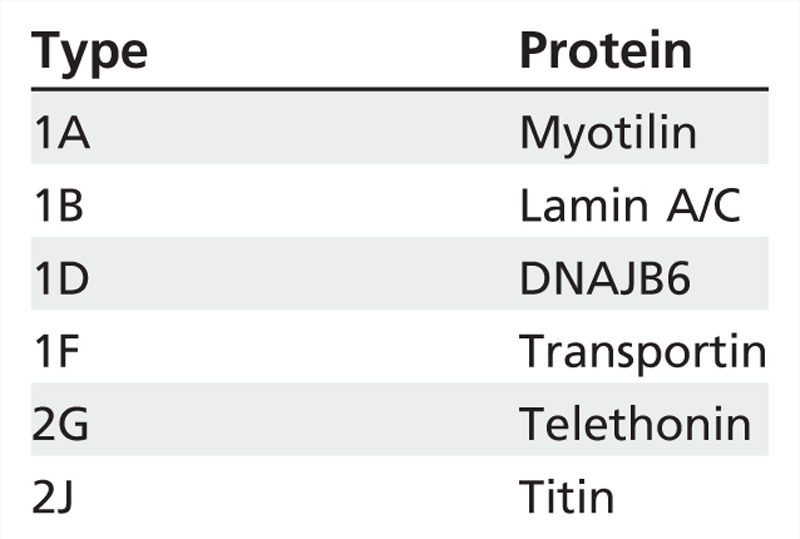
Limb-girdle muscular dystrophy type 1D. Mutations in DNAJB6 are a rare cause of LGMD that presents in the teens through upper middle age with proximal weakness and no distinctive features. Most patients remain ambulatory into their sixties. Muscle biopsies may have rimmed vacuoles (Table 2-2).37
Table 2-2.
Limb-Girdle Muscular Dystrophies With Rimmed Vacuoles
Limb-girdle muscular dystrophy type 1E. Desmin is an intermediate filament spanning from the sarcolemma to the sarcomere and through to the nucleus. Patients with desminopathy have onset of weakness after puberty, mildly elevated CK levels, and develop a dilated cardiomyopathy with conduction defects.38
Limb-girdle muscular dystrophy type 1F. Transportinopathies have scapulopelvic weakness with symptomonset in subsequent generations commencing at earlier ages, which perhaps suggests genetic anticipation.39
Limb-girdle muscular dystrophy type 1G. Patients have onset of proximal weakness between 30 and 47 years of age, progress slowly, and have progressive limitation of finger and toe flexion.
Limb-girdle muscular dystrophy type 1H. A single report has been published of a 12-member extended family with onset of slowly progressive, lower extremity weakness in the second through fifth decades, reduced reflexes, proximal muscle atrophy, and calf hypertrophy.
Inclusion body myopathy with Paget disease and frontotemporal dementia. Patients with this autosomal dominant muscle disease-due to mutations in VCP, the valosin-containing protein gene-have midlife onset of proximal predominant weakness mimicking an autosomal dominant LGMD, with or without Paget disease. Frontotemporal dementia may follow a decade or so later. Extended family members may have any combination of weakness, Paget disease, and dementia. A thorough family history will help make this diagnosis.
The clinical phenotype is useful in ferreting out the correct autosomal dominant LGMD roughly 50% of the time (Figure 2-15).
Figure 2-15.
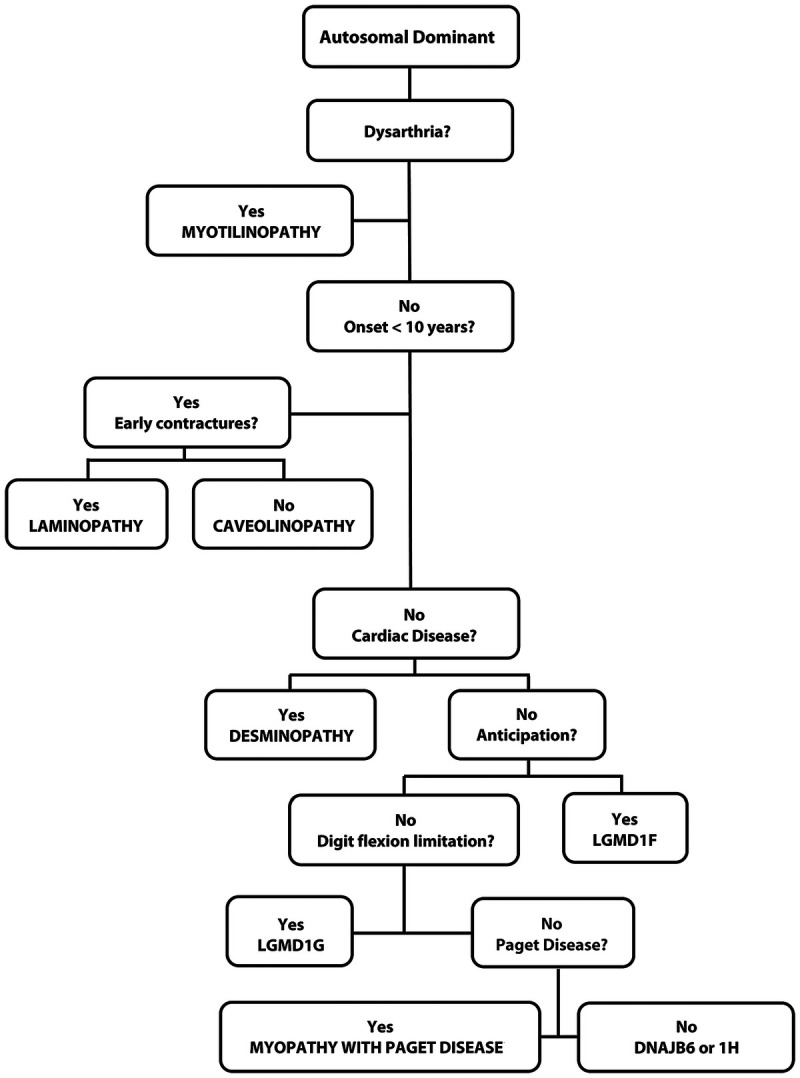
Algorithm for the diagnosis of the autosomal dominant limb-girdle muscular dystrophies.
Autosomal recessive limb-girdle muscular dystrophies. Limb-girdle muscular dystrophy type 2A. The most common LGMD in most geographic regions, calpainopathies are estimated to represent anywhere from 9% to 40% of cases. CAPN3 encodes a muscle-specific, calcium-activated neutral protease that binds to titin. Clinical onset varies widely (2 to 53 years of age), but 75% of cases have onset before 20 years of age. Proximal lower extremity muscles are weaker than shoulder-girdle muscles from the outset, and hip extensor, hip adductor, and knee flexor weakness and atrophy is often disproportionately severe. Scapular winging and abdominal laxity are common, but clinical respiratory and cardiac dysfunction seldom affect patients. Around 50% of patients no longer ambulate independently within 2 decades. CK levels average approximately 4000 U/L but may surpass 20,000 U/L. Muscle biopsies often reveal lobulated fibers on oxidative enzyme stains (Figure 2-16).40
Figure 2-16.
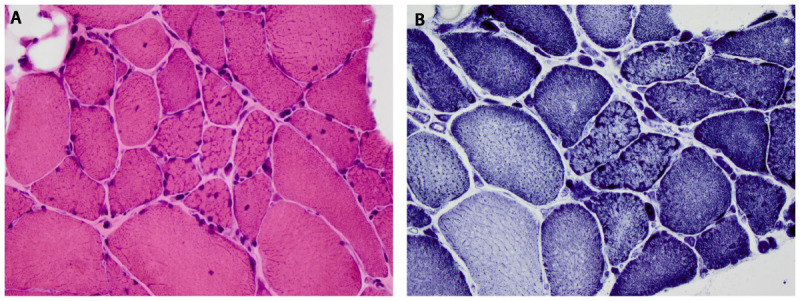
Muscle biopsy in limb-girdle muscular dystrophy type 2A, calpainopathy, demonstrating lobulated fibers as seen on hematoxylin and eosin (A) and nicotinamide adenine dinucleotide (NADH) (B) stains. Courtesy of Jennifer W. Baccon, MD, PhD.
Limb-girdle muscular dystrophy type 2B. Mutations in DYSF, the gene for dysferlin, underlie both LGMD and Miyoshi myopathy (distal myopathy with predominant involvement of the calves), and identical mutations can account for either phenotype. Several cases of symptomatic carriers have been reported. Dysferlinopathies account for 5% to 35% of LGMD diagnoses and tend to be more prevalent in Asian populations and regions surrounding the Mediterranean Sea. Onset occurs between 15 and 30 years of age in 75% of patients. When young, patients develop normally in cognitive and motor spheres, some actually being quite athletically gifted. Weakness begins in the legs in nearly all cases, but many have a component of distal leg weakness. The extent of lower leg weakness delineates whether the limb-girdle or Miyoshi moniker fits most appropriately (Figure 2-17). Cardiac dysfunction and respiratory compromise are rare and reportable. Serum CK levels may be substantially elevated, as great as 40,000 U/L. Muscle biopsies are notable in that up to 50% may show a component of endomysial or perimysial infiltrate, sometimes leading to the misdiagnosis of polymyositis. A very large protein, dysferlin likely participates in membrane trafficking, fusion, and repair, along with roles in mitochondrial function.41
Figure 2-17.
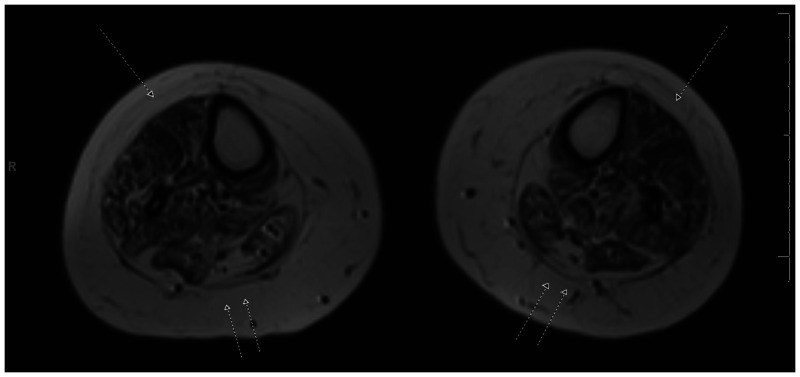
MRI of the calves in a patient with limb-girdle muscular dystrophy type 2B dysferlinopathy. Note the relative preservation of muscle in the anterior compartment (single arrows) of the forelegs compared to the marked fatty and fibrous replacement in the posterior compartment (double arrows).
Limb-girdle muscular dystrophy types 2C through F. The sarcoglycans form a heterotetrameric complex spanning the sarcolemma in association with dystrophin, sarcospan, and the dystroglycans. This dystrophin-glycoprotein complex provides a mechanical bridge between the extracellular basement membrane, the cytoskeleton, and the intracellular contractile proteins of the sarcomere. This complex also facilitates cell signaling and trafficking in concert with neuronal nitric oxide synthase, dystrobrevin, and caveolin 3. LGMDs due to the different sarcoglycans yield similar clinical and laboratory characteristics. Sarcoglycanopathies comprise 10% to 20% of LGMD cases with higher prevalence rates in some North African and Brazilian populations. Symptoms most often begin between 1 and 15 years of age with pelvic-girdle weakness. Common examination features include calf hypertrophy, scapular winging, macroglossia, and lumbar hyperlordosis (Table 2-3). The breadth of severity mirrors dystrophinopathies and includes a severe, Duchenne-like progression, but milder cases include a Becker-like phenotype, exercise intolerance, or even asymptomatic hyperCKemia. Cardiac and respiratory impairment affect patients as early as 10 years into disease and may be severe. CK values range from 1000 U/L to 25,000 U/L. Muscle biopsies usually reveal diminished immunostaining for all four sarcoglycans, regardless of which sarcoglycan carries the mutation (Case 2-4).
Table 2-3.
Muscular Dystrophies With Calf Hypertrophy

Case 2-4A
A 21-year-old white woman of Brazilian heritage was the product of a normal birth, had normal motor and cognitive developmental milestones, and participated in competitive volleyball and basketball through the ninth grade. From 15 years of age onward, weakness progressed to the point of wheelchair use. Early in her course, she could no longer stand on her tiptoes. Her examination revealed normal cardiopulmonary function along with symmetrical proximal predominant weakness (shoulders 4+, distal upper extremity 5, hip girdle 2, distal lower extremity 4−); no scapular winging; and bilateral calf atrophy. Her creatine kinase (CK) levels were 17,730 U/L and 9499 U/L. A muscle biopsy demonstrated a severe dystrophy with absent immunostaining for dysferlin (Figure 2-18). Genetic testing revealed homozygous DYSF missense mutations (c.5429G>A; p.Arg1810Lys), which confirmed the diagnosis of dysferlinopathy.
Figure 2-18.
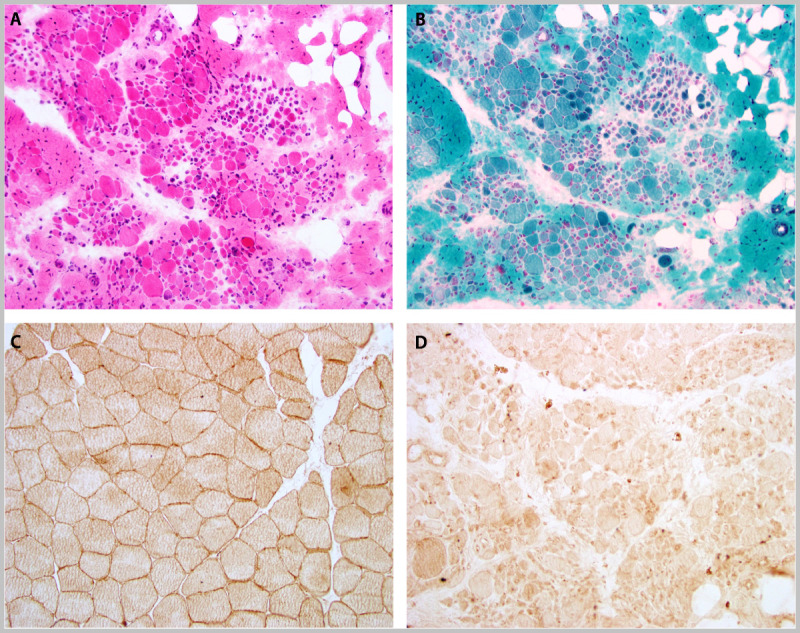
Muscle biopsy in a patient with limb-girdle muscular dystrophy type 2B, dysferlinopathy, and absent dysferlin immunostaining. A, hematoxylin and eosin; B, Gomori one-step trichrome; C, dysferlin immunostain-control; and D, dysferlin immunostain-patient. Courtesy of Charles S. Specht, MD.
Case 2-4B
A 19-year-old white woman first noted exercise intolerance while riding her bicycle at 4 years of age. Living in Brazil from 2 to 7 years of age, she became a very talented soccer player but could only play for 10 to 20 minutes before muscle soreness, aching, and pain set in. She had several episodes of dark urine. She reported no immediate or remote family history of muscle disease. The results of her neurologic examination (including strength) were normal, including the ability to run, do three deep knee bends, and jump effortlessly. CK levels were 8990 U/L, 9725 U/L, and 18,567 U/L were in her medical record. Her muscle biopsy revealed minimal variability in fiber size, approximately 12% of fibers with internal nuclei (Figure 2-19), and diminished immunostaining for the four sarcoglycans (Figure 2-20). Genetic testing revealed two heterozygous, disease-associated, missense mutations in SGCA, the gene for α-sarcoglycan (c.229C>T; p.Arg77Cys and c.850C>T; p.Arg284Cys).
Figure 2-19.
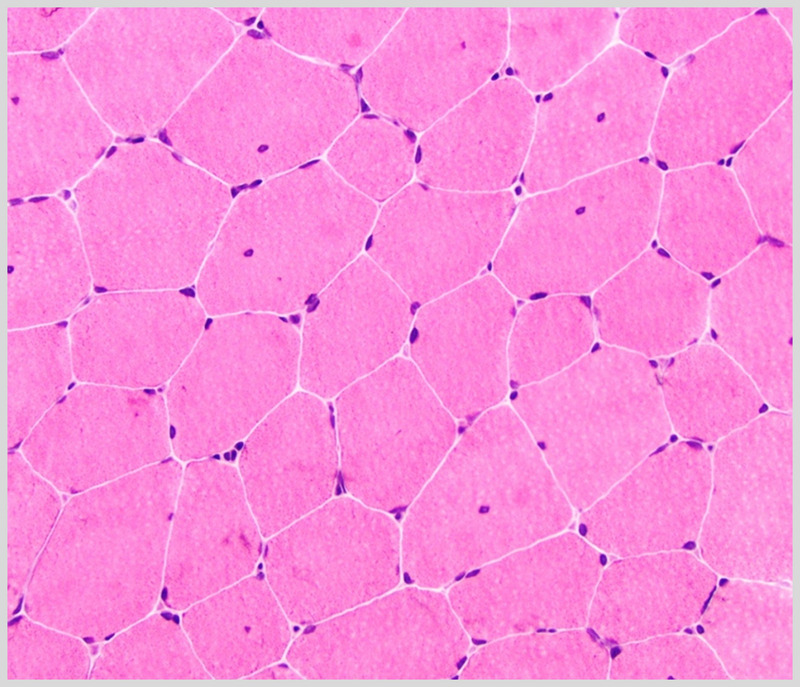
Muscle biopsy with minor abnormalities, including minimal variability in fiber size and approximately 12% of fibers with internal nuclei, in a woman with limb-girdle muscular dystrophy type 2D α-sarcoglycanopathy who presented with exercise intolerance and creatine kinase levels >10,000 U/L (hematoxylin and eosin). Courtesy of Charles S. Specht, MD.
Figure 2-2.
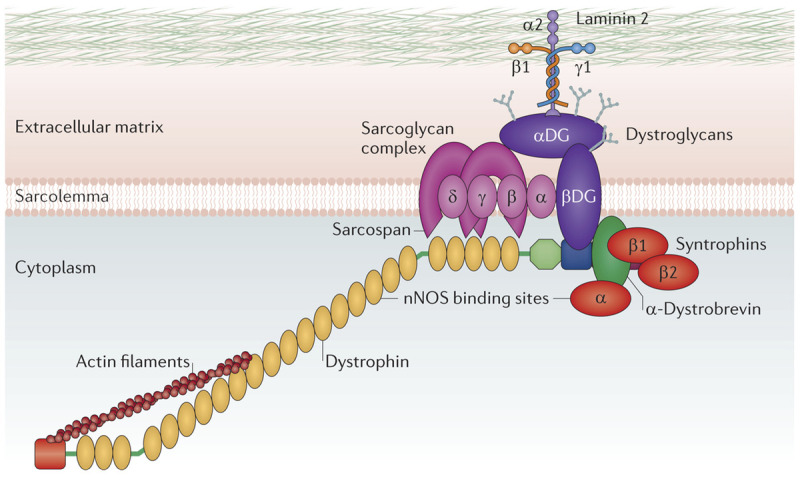
Dystrophin glycoprotein complex. Dystrophin, along with α-, β-, γ-, and δ-sarcoglycan, β-dystroglycan, sarcospan, syntrophins, dystrobrevin, and nitric oxide synthase form the dystrophin-glycoprotein complex. Reprinted with permission from Fairclough RJ, et al, Nat Rev Genet.12 © 2013, Nature Publishing Group. http://www.nature.com/nrg/journal/v14/n6/abs/nrg3460.html.
Figure 2-20.
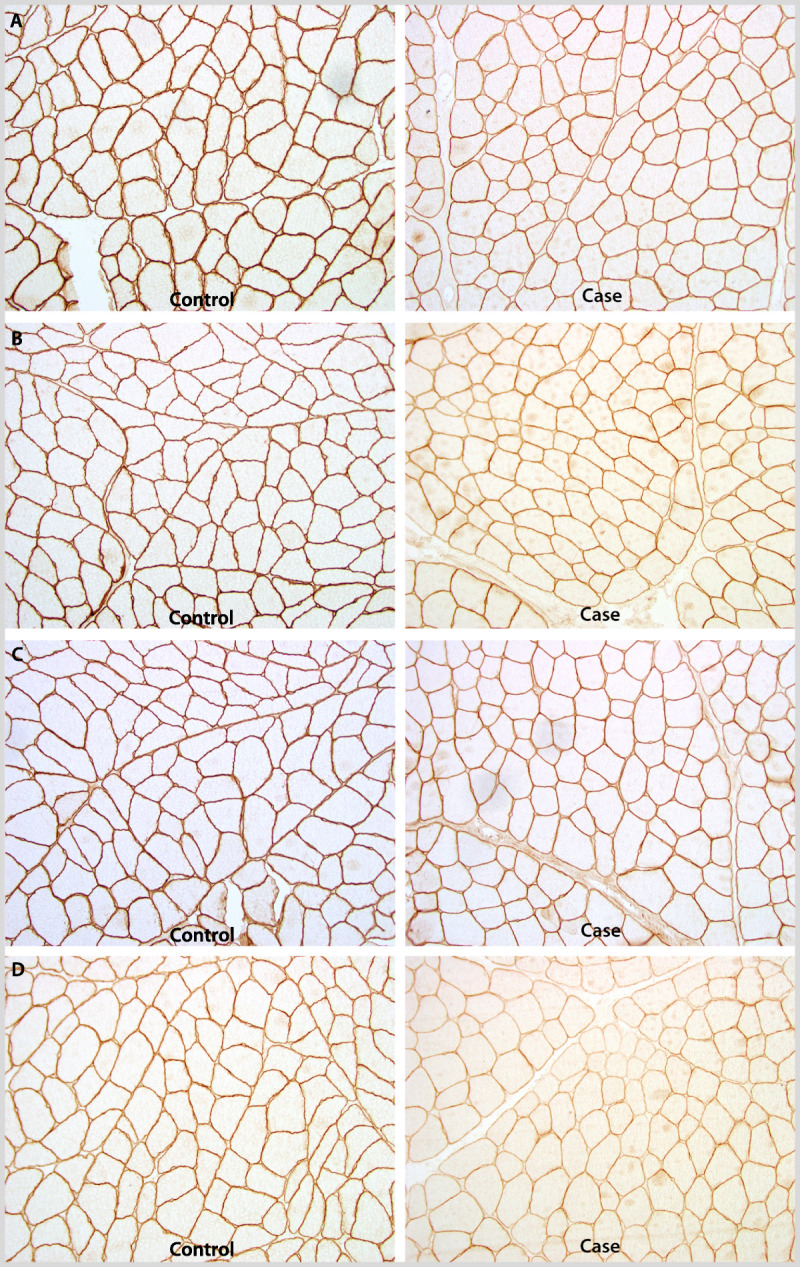
Muscle biopsy immunostained for A, α-sarcoglycan; B, β-sarcoglycan; C, γ-sarcoglycan; and D, δ-sarcoglycan demonstrating diminished staining for all four in a woman with limb-girdle muscular dystrophy type 2D, α-sarcoglycanopathy. Courtesy of Charles S. Specht, MD.
Comment. Dysferlinopathies are somewhat overly represented in Brazil. In addition, sarcoglycanopathies are more common. These two cases illustrate several important features about LGMDs in general and these common LGMD subtypes. (1) LGMDs have significant variability in their clinical severity, from asymptomatic hyperCKemia to rapidly progressive, Duchenne-like muscular dystrophies. (2) Cardiopulmonary involvement varies among the LGMDs, with little cardiopulmonary dysfunction in dysferlinopathies and the potential for significant cardiac involvement in sarcoglycanopathies. Ironically, despite no clinical skeletal muscle weakness in the 19-year-old athlete with a sarcoglycanopathy, the concern for cardiac dysfunction remains much greater in her than in the 21-year-old with dysferlinopathy in a wheelchair. (3) Dysferlinopathies often have distal weakness or atrophy, even relatively early in disease. (4) In patients with CK levels persistently at or above 10,000 U/L, the differential diagnosis is relatively limited (Table 2-4).
Table 2-4.
Muscular Dystrophies With Very High Creatine Kinase Levels (>10,000 U/L)

Limb-girdle muscular dystrophy type 2G. Telethoninopathy is a rare disorder reported mostly in Brazil. Disease begins before 15 years of age and involves the proximal and distal lower extremity and proximal upper extremity. The need for wheelchair use ensues by the patient’s forties. Generally, CK levels seldom exceed 2000 U/L. Muscle biopsy features include prominent rimmed vacuoles.
Limb-girdle muscular dystrophy type 2H. A mild, autosomal recessive LGMD affects Hutterites in Manitoba. In the first decade, slowly progressive weakness ensues with prominent initial quadriceps and pelvic-girdle involvement, followed by trapezius and deltoid weakness. CK levels tend to be less than 2000 U/L.
Limb-girdle muscular dystrophy type 2I. Fukutin-related protein is one of a number of proteins involved in glycosylation of α-dystroglycan, a member of the dystrophin-glycoprotein complex. Defects in this glycosylation process lead to muscle dysfunction (Figure 2-13). Mutations in FKRP likely account for approximately 15% of LGMD patients, commonly those of Northern European heritage. Symptoms develop over a broad age range, from 1 to 50 years, most often before the third decade. Initial pelvifemoral weakness later extends into the distal legs and proximal arms. Calf hypertrophy, tongue hypertrophy, and lumbar lordosis are quite common (Table 2-5). Prominent respiratory and cardiac dysfunction may occur early and may not parallel skeletal muscle involvement. Forced vital capacity may shrink below 50%, especially when measured in the supine position.CK levels range from 500 U/L toover 10,000 U/L. Muscle biopsies have dystrophic features with diminished immunostaining for glycosylated α-dystroglycan.42
Table 2-5.
Distinctive Clinical Characteristics of Some Muscular Dystrophies

Limb-girdle muscular dystrophy type 2J. The enormous, filamentous protein titin spans half the sarcomere from Z disc to M line and contains binding sites for calpain 3 (see the section on LGMD2A). Single mutations in TTN, the gene for titin, lead to tibial muscular dystrophy, a late-onset distal disorder common in Finland. Occasional patients have been reported to have mutations in both TTN alleles. They develop a typical LGMD phenotype with onset in the first to third decades. CK levels are high (2000 U/L to 10,000 U/L). Western blot analysis reveals loss of not only titin, but also calpain 3, suggesting an indirect effect of calpain 3 deficiency on the LGMD phenotype.
Limb-girdle muscular dystrophy type 2L. Discovered in 2009, anoctaminopathies have been found to be a quite common cause of LGMD in Northern European populations. Mutations in ANO5 also yield a distal leg phenotype. Onset of LGMD occurs after the first decade, generally in adulthood, and some require wheelchair use after 10 to 20 years. Patients may have prominent, asymmetric thigh atrophy. CK levels are often very high at 500 U/L to 35,000 U/L, mirroring dysferlinopathies. Cardiac and respiratory dysfunction are not reported.43
Limb-girdle muscular dystrophy types 2K, M, N, O, and P. All of these LGMDs manifest disease through hypoglycosylation of α-dystroglycan. They are quite rare, with a paucity of published cases. Most often, these genes present as congenital muscular dystrophies or muscle-eye-brain disease.
Figure 2-21 walks through the biopsy evaluation of autosomal recessive LGMDs.
Figure 2-21.
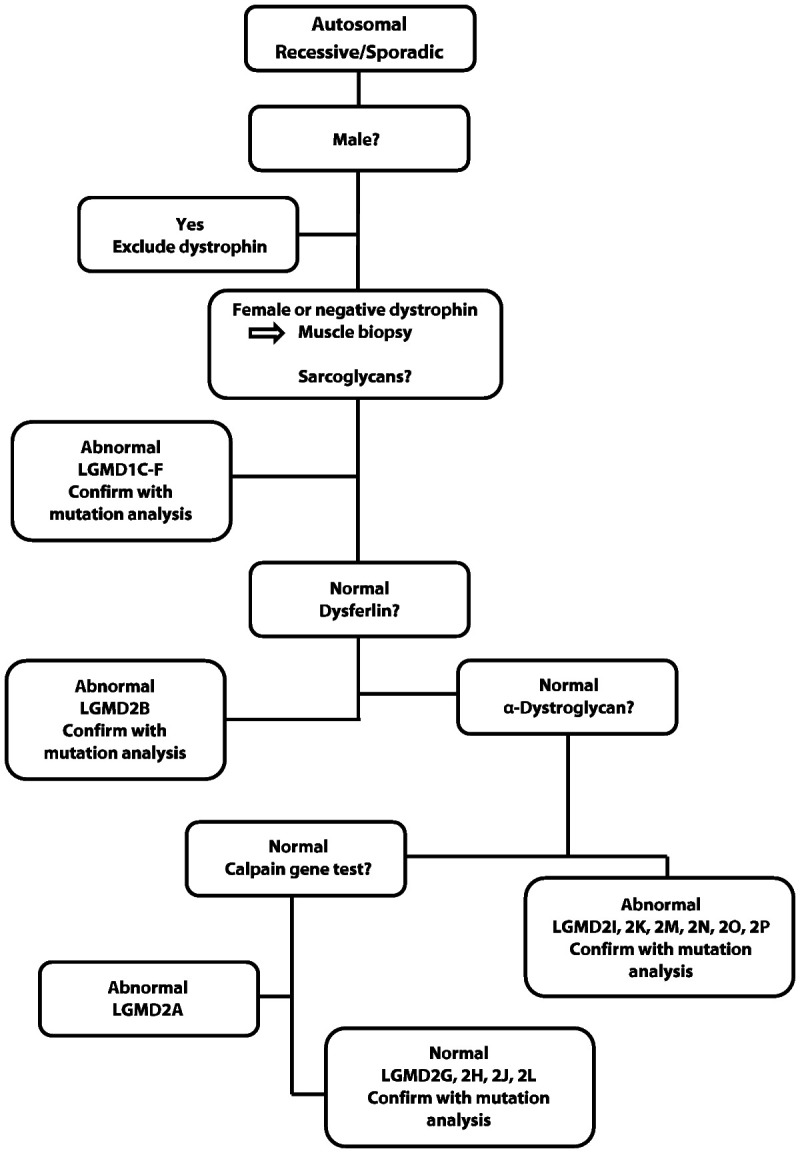
Algorithm for muscle biopsy diagnosis of autosomal recessive limb-girdle muscular dystrophies.
Diagnosis
The differential diagnosis of proximal predominant weakness is broad. Acquired muscle disorders, such as toxic, endocrine, and autoimmune myopathies, and nonmuscle disorders, such as myasthenia gravis and spinal muscular atrophy, all occur at much higher rates than the LGMDs. These should be considered and excluded early in the evaluation. Dystrophinopathies (in patients with Duchenne or Becker muscular dystrophies and in manifesting carriers) are more common than LGMDs. Therefore, dystrophin mutation analysis should come early in the evaluation of male patients and be considered in female patients. FSHD without prominent facial involvement can masquerade as LGMD and actually made up 3% of cases referred for a LGMD clinical trial.44 Even without facial involvement, FSHD generally can be excluded by the absence of its cardinal clinical features of reversed anterior axillary folds and triple-hump sign. Testing for Pompe disease should also come early in the diagnostic evaluation now that effective treatment exists with enzyme replacement therapy. Metabolic, mitochondrial, and myofibrillar myopathies should also be considered. Guidelines for the diagnosis and management of the LGMDs have been published.45
EMERY-DREIFUSS MUSCULAR DYSTROPHY
Epidemiology
Emery-Dreifuss muscular dystrophy (EDMD) is uncommon but has a distinctive phenotype (Figure 2-22). The prevalence of genetically confirmed cases in Northern England was 0.33 per 100,000.24 However, the true prevalence of all genetic subtypes probably lies near 1 per 100,000.
Figure 2-22.
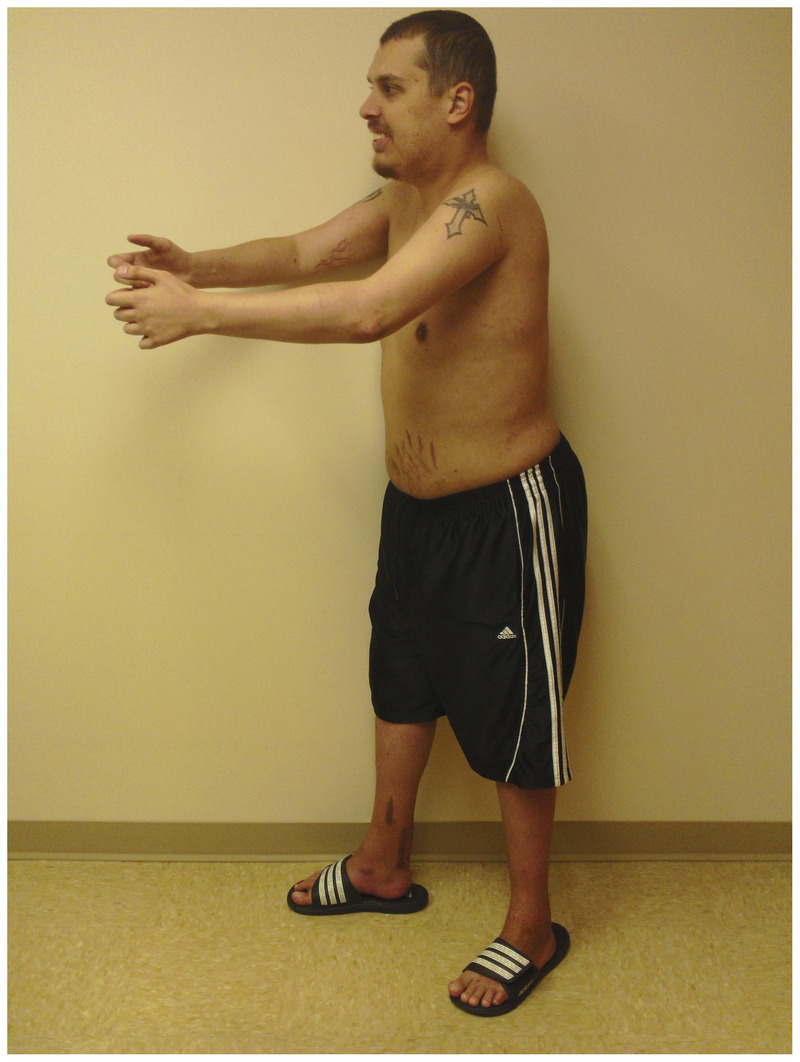
A 24-year-old man with Emery-Dreifuss muscular dystrophy. He has had prominent contractures of his elbows, spine, and ankles from an early age. He is maximally extending his elbows. The scars around his ankles stem from Achilles tendon lengthening surgeries at 12 years of age for ankle plantar flexor contractures.
Clinical Features
The cardinal triad associated with EDMD includes (1) contractures of the elbow flexors, ankle plantar flexors, and spine early in disease; (2) childhood onset of humeroperoneal weakness; and (3) cardiac disease with arrhythmias, conduction block, and cardiomyopathy.46 Patients’ health is normal at birth and in the first few years. Disease generally begins in the first or second decade of life with weakness or contractures. Biceps and triceps weakness and wasting, relative preservation of deltoid muscle strength, and ankle dorsiflexion weakness comprise the typical early pattern. Most patients remain ambulatory for 20 years or more after diagnosis.
A key early diagnostic feature may be a child’s inability to fully extend the elbows-evidence for subtle contractures. Likewise, toe walking may reflect ankle contractures. Progressive contractures lead to the typical phenotype, including a rigid spine.
Cardiac conduction system defects usually follow the muscle disease but may trigger the initial evaluation. First-degree atrioventricular block, right bundle branch block, and atrial fibrillation/flutter are common. The major risk for mortality is sudden cardiac death, most commonly related to severe bradyarrhythmias or ventricular tachyarrhythmias. Pacemakers are insufficient in EDMD to prevent mortality; therefore, patients require primary placement of combined pacemakers with internal cardiac defibrillators. While ECG abnormalities are uncommon in the first decade of life, the vast majority of patients have an irregular heart rhythm by the fourth decade.47 Sudden death in the second decade is documented but most commonly occurs after the third decade.48
Very prominent interfamilial and intrafamilial phenotypic variability may be seen for both skeletal and cardiac muscle involvement, even with the same mutation.
Diagnosis
Later in the disease course, the phenotype, with prominent contractures easily allows the diagnosis. CK levels are usually normal or mildly elevated, and virtually always less than 2000 U/L. The EMG reveals a mostly nonirritable myopathy. An ECG and cardiologic evaluation should always be part of the EDMD diagnostic evaluation.
An EDMD phenotype may stem from mutations in six different genes, some X-linked recessive and others autosomal dominant in inheritance pattern (Table 2-6). All of these proteins act and interact at the inner nuclear membrane. The exact mechanism of disease is not known but may relate to fragility of the nuclear envelope under stress forces of muscle contraction and/or to the roles of these proteins in chromatin scaffolding with effects on gene expression.
Table 2-6.
Genetic Causes of Emery-Dreifuss Muscular Dystrophy
Similar to dystrophinopathies, it is important to remember that female carriers of the X-linked forms of EDMD (mutations in EMD and FHL1) may develop weakness or cardiac disease. They should be referred for neuromuscular and cardiac evaluation.
OCULOPHARYNGEAL MUSCULAR DYSTROPHY
Epidemiology
Found clustered in certain geographic regions, oculopharyngeal muscular dystrophy (OPMD) remains an otherwise fairly uncommon disorder. In the province of Quebec, Canada, a founder mutation has led to a carrier rate of nearly 1 per 1000. A high concentration also exists among the Bukhara Jews in Israel and Hispanics in New Mexico. Elsewhere, the prevalence is less than 1 per 100,000.
Clinical Features
Clinically, the cardinal features of OPMD include ptosis and dysphagia (Figure 2-23). Onset most often occurs in the fifth or sixth decade of life with ptosis. Before diagnosis, patients often unconsciously contract their frontalis muscles to raise their eyebrows. They also gradually develop a backward head tilt to increase their field of vision. Blepharoplasty readily corrects the ptosis, although repeat procedures are sometimes necessary. Extraocular muscle dysfunction ensues in two-thirds of patients, with predominant involvement of upgaze. With slow progression of eye muscle weakness, patients rarely experience diplopia. Dysphagia usually follows ptosis by several years, but occasionally is the first symptom. The major cause for morbidity and mortality in OPMD, dysphagia may lead to coughing, choking, laryngeal penetration, and aspiration pneumonia. Without interventions later in disease (such as cricopharyngeal myotomy, upper esophageal sphincter dilatation, or gastrostomy/jejunostomy tube placement), phagophobia, anorexia, and cachexia supervene. Although lifespan is normal in OPMD, lack of treatment of dysphagia is the most common cause for premature death in patients with the disease. Of patients with OPMD, 71% eventually develop lower extremity weakness, and 38% develop upper extremity weakness.49 The weakness is slowly progressive and rarely disabling. A proportion of patients also develop tongue atrophy (82%), facial muscle weakness (43%), or change in voice-hypophonia, hoarseness, and hypernasality (67%) (see Table 2-5).49 Cognitive dysfunction has been described in homozygous patients, and, recently, subtle executive dysfunction has been seen in heterozygotes.50
Figure 2-23.

Patient with oculopharyngeal muscular dystrophy showing bilateral ptosis and facial weakness. Note the use of his frontalis muscles to help hold his eyes open. Courtesy of Sarah Youssof, MD.
Figure 2-3.
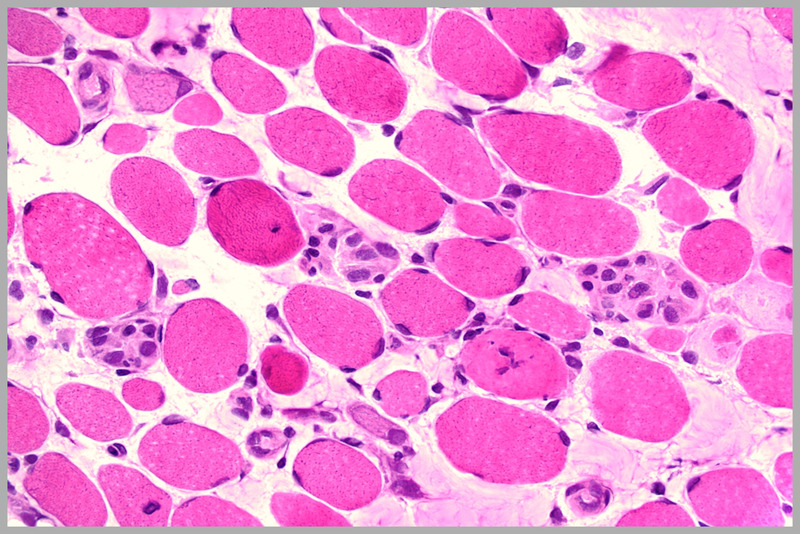
Dystrophic muscle in Duchenne muscular dystrophy with marked variability in fiber size, increased endomysial connective tissue, regenerating fibers, and hypercontracted fibers (hematoxylin and eosin). Courtesy of Charles S. Specht, MD.
Figure 2-4.
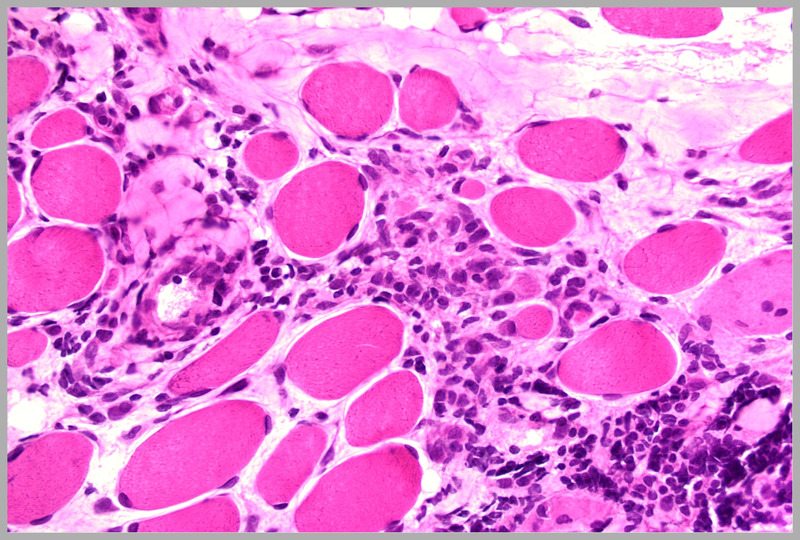
Inflammation in a muscle biopsy of Duchenne muscular dystrophy. Note the significant inflammatory infiltrate in the right lower quadrant of the photo (hematoxylin and eosin). Courtesy of Charles S. Specht, MD.
Figure 2-5.
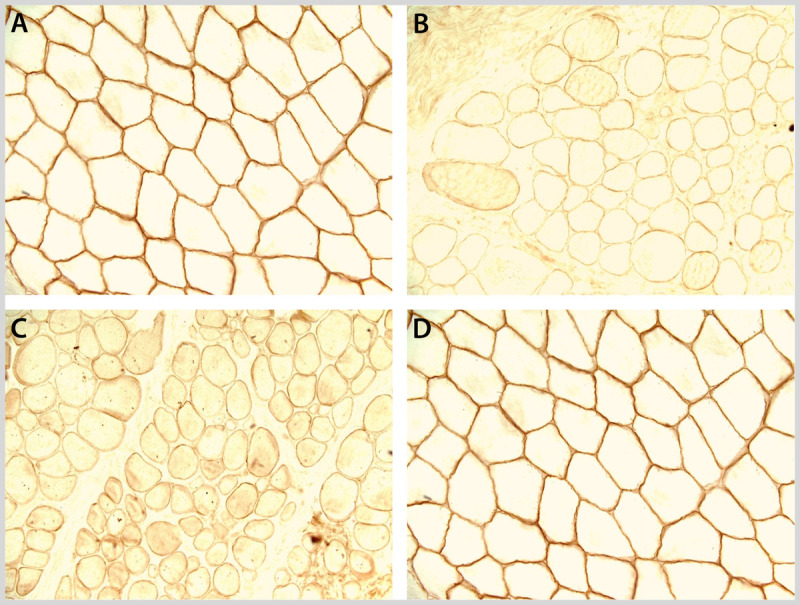
Dystrophin immunostaining (400X). Normal control (A) and decreased immunostaining to the N terminus (B), rod domain (C), and C terminus (D). Courtesy of Charles S. Specht, MD.
Figure 2-6.

Reading frame rule for dystrophinopathies. Each exon must connect with the preceding and following exon, but not all exons can interact with each other. For example, deletion of exon 39 would require exon 38 and exon 40 to reconnect, and they can do so. This would lead to a slightly shorter protein of fairly good function, and the reading frame would be maintained. This would yield a Becker phenotype. However, with deletion of exon 50, exon 49 and exon 51 cannot reconnect. This leads to a dystrophin RNA transcript that stops at exon 49. These truncated RNA transcripts rapidly undergo RNA decay leading to a near total lack of dystrophin and a Duchenne phenotype. In this case, the reading frame was broken. Deletion of exons 58 to 67 in the 4-year-old boy in Case 2-1 allows exon 57 to anneal to exon 68, retaining the reading frame. However, dystrophin’s attachment site to β-dystroglycan at the sarcolemma falls within exons 58 to 67. Thus, the patient’s dystrophin cannot anchor to the sarcolemma, leading to significant sheer forces and subsequent muscle fiber damage. Patients whose mutations cannot anchor to the sarcomere or the sarcolemma manifest the more severe Duchenne phenotype. Courtesy of Annemieke Aartsma-Rus, PhD.
Figure 2-7.
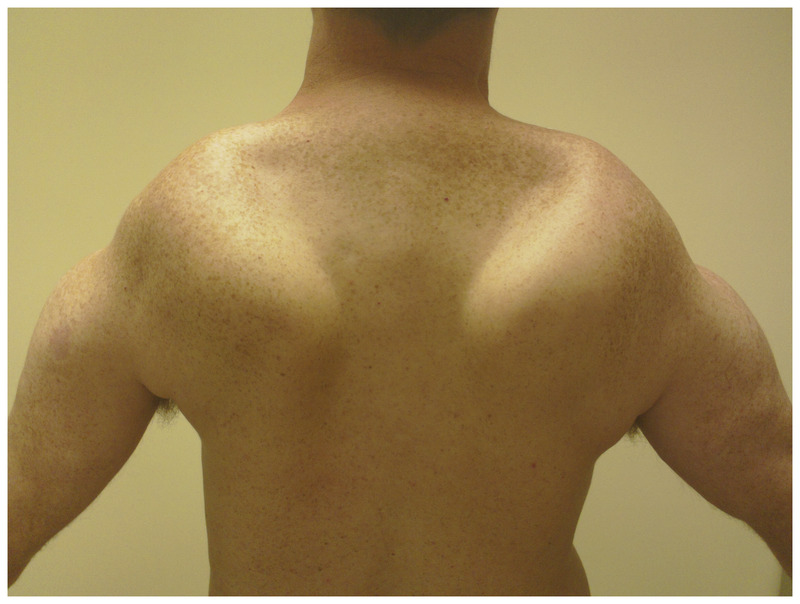
Man with facioscapulohumeral muscular dystrophy and bilateral scapular winging.
Figure 2-8.
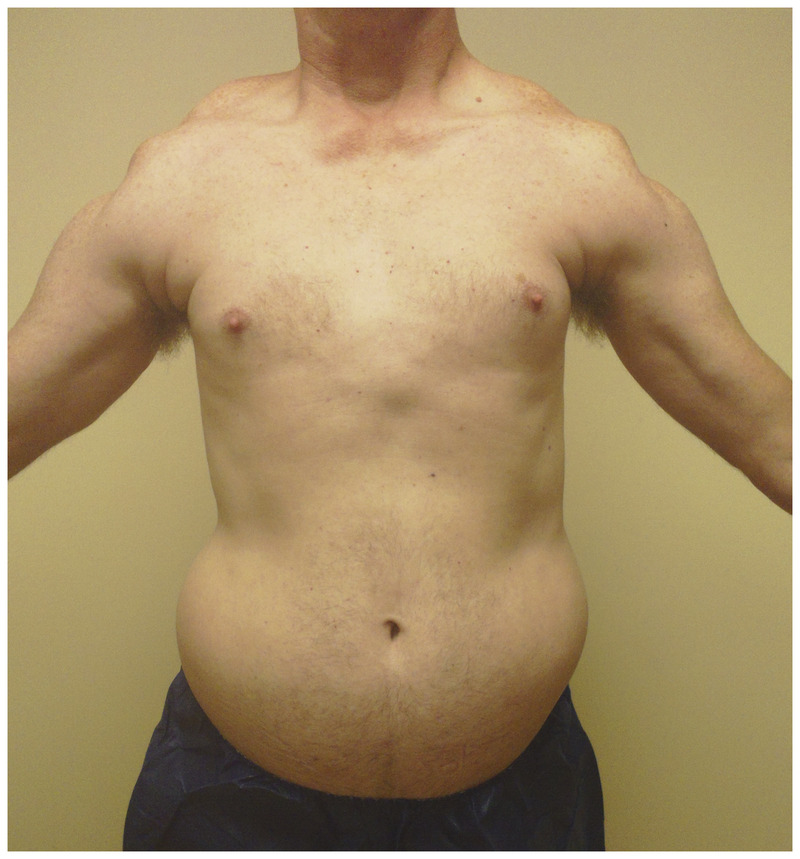
Prominent reversal of the anterior axillary folds, abdominal laxity, and the “triple hump” sign (protuberant deltoid muscle, acromioclavicular junction, and overriding scapula) in facioscapulohumeral muscular dystrophy.
Figure 2-9.
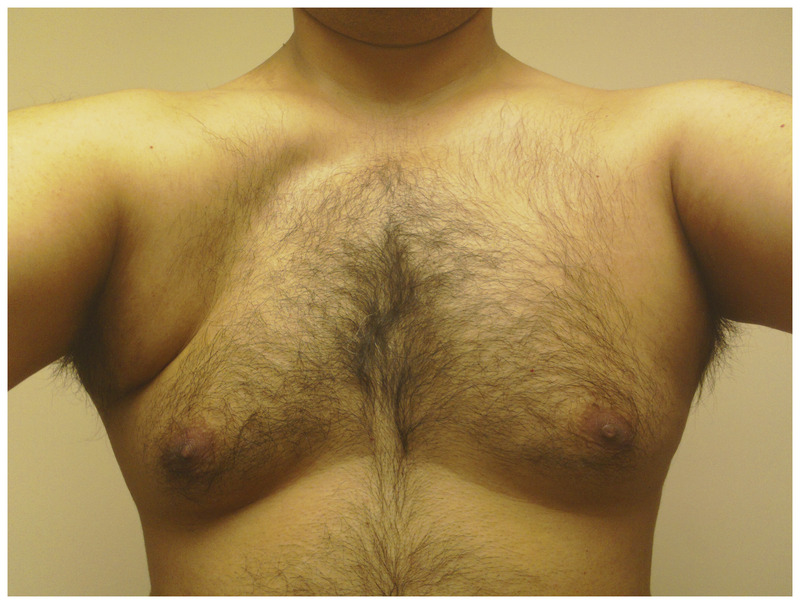
Asymmetric involvement in facioscapulohumeral muscular dystrophy. Note the marked pectoral atrophy and reversal of the anterior axillary fold on the right with relative preservation of pectoral muscle bulk on the left.
Diagnosis
Diagnosis can be made on clinical grounds with genetic confirmation. The differential diagnosis includes myasthenia gravis, mitochondrial myopathy, and myotonic dystrophy. In myasthenia gravis, the ptosis is usually variable and fatigable. Mitochondrial myopathies are maternally transmitted, have less prominent dysphagia, and often have other organ system involvement, such as neuropathy, deafness, cardiomyopathy, or seizures. Myotonic dystrophy has distinctive features such as temporal wasting, “Christmas tree” cataracts, distal weakness, clinical myotonia, arrhythmogenic cardiac disease, and endocrinopathies. In OPMD, the CK level most often is normal and is always less than 1000 U/L. Muscle biopsy is not needed but may show rimmed vacuoles on light microscopy and 8.5 nm intranuclear tubular filaments on electron microscopy. Genetic testing reveals (GCN)12-17 trinucleotide repeat expansions in exon 1 of PABPN1, the polyadenylation-binding protein nuclear 1 gene. OPMD was initially described as a (GCG)8-13/polyalanine disease. It is now known that the polyalanine repeat sequence contains not only GCG/alanine trinucleotide repeats, but also GCA/alanine trinucleotide repeats. The normal number of GCN repeats is 10. Across multiple ethnic groups, the total number of GCN trinucleotide repeats in OPMD ranges from 12 to 17. Therefore, the nomenclature for the most common repeat length in French Canadians with OPMD, previously (GCG)9, is now called (GCN)13. Because (GCN)12-17 trinucleotide repeat sequences are mitotically and meiotically stable, anticipation does not occur inOPMD. Patients homozygous for PABPN1 (GCN)n repeat expansions develop disease 18 years earlier, have greater limb weakness, and show evidence of cognitive dysfunction on neuropsychological testing. Two percent of the French Canadian population carry a (GCN)11 allele instead of the normal (GCN)10 allele and do not manifest disease in the heterozygous state. However, patients inheriting a (GCN)11 allele from each parent will develop recessive OPMD. These patients manifest a phenotype that may be milder or more severe than typical OPMD patients heterozygous for the PABPN1 (GCN)13 allele. The mechanism underlying OPMD is not fully understood but likely involves retention of misfolded proteins in the nucleus.
FINAL COMMENTS
During the transition into the genetic era, evaluation of muscular dystrophies is changing. In the early 1990s, nearly all patients with DMD underwent muscle biopsy as the initial diagnostic procedure. By the late 1990s, initial DMD genetic testing became the norm (with muscle biopsy performed only when no duplication or deletion was found). This diagnostic paradigm shift is happening now on a much larger scale. With the advent of commercially available next-generation (massively parallel sequencing) panels with additional duplication/deletion analysis, exome sequencing and genome sequencing, medical evaluation stands at the cusp of transformation to a genetics-first strategy. Substantial expansion in our understanding of genetic muscle disease will ensue as the genotypic spectrum of most syndromes and phenotypic variability of individual genes will surely broaden. However, significant challenges (false positive/negative results, ethnic variability, epigenetic factors, environmental influences) in interpretation of the data lie ahead as we embrace these new technologies.
USEFUL WEBSITES
Neuromuscular Disease Center
http://www.neuromuscular.wustl.edu
Exceptional website with bulleted format, easy navigation, significant breadth, and frequent updates.
Jain Foundation LGMD subtyping tool
http://www.jain-foundation.org/lgmd-subtyping-diagnosis-tool
A helpful computer-aided diagnostic tool for the LGMDs.
Genetic Testing Registry
http://www.ncbi.nlm.nih.gov/sites/GeneTests/?db=GeneTests
Excellent source to find genetic testing laboratories, especially for uncommon muscular dystrophies. Gene Reviews provide useful overviews of muscular dystrophies and are updated regularly.
ClinicalTrials.gov
clinicaltrials.gov/
A ready resource to direct patients toresearch opportunities for their condition.
Muscular Dystrophy Association
mdausa.org/
Provides overviews of muscular dystrophies for clinicians, descriptions of current research, and patient-centered information.
FSH Society
Myotonic Dystrophy Foundation
Parent Project Muscular Dystrophy
http://www.parentprojectmd.org/
Jain Foundation
KEY POINTS
Muscular dystrophies are genetic, progressive, degenerative disorders of muscle, of which muscle weakness is the primary symptom.
Most commonly, mutations in the gene for dystrophin present as Duchenne muscular dystrophy, but dystrophinopathies manifest a number of other phenotypes, including Becker muscular dystrophy; isolated quadriceps myopathy; asymptomatic or minimally symptomatic hyperCKemia; aches, pains, cramps, and rhabdomyolysis; manifesting female carriers; X-linked dilated cardiomyopathy; and cognitive disorders.
The incidence of Duchenne muscular dystrophy has been reported as 1:3500 live male births for many years. Newborn screening places the incidence closer to 1:5000 live male births.
Boys with Duchenne muscular dystrophy most often present between 3 and 5 years of age because of delayed motor milestones and falls, along with difficulty running and jumping.
Management of Duchenne muscular dystrophy requires a multidisciplinary health care team.
Mean lifespan in Duchenne muscular dystrophy has increased from 19 years to more than 25 years. Much of this increased longevity can be ascribed to improvements in care and corticosteroid use.
Severity of weakness varies substantially in patients with Becker muscular dystrophy. Weakness may begin as early as 5 to 6 years of age or may start in the fifth or sixth decade.
In a Dutch national survey of dystrophinopathy gene carriers from 18 to 60 years of age, 17% had muscle weakness varying from mild to severe, 8% had symptomatic dilated cardiomyopathy, and 18% had evidence for left ventricle dilation on echocardiography.
A dystrophinopathy should be suspected in any male or female patient presenting with progressive limb-girdle weakness, especially if the patient has a positive family history, substantially elevated creatine kinase level, or cardiomyopathy.
Prevalence of FSHD is estimated at 7 per 100,000. However, with unrecognized mild cases, a normal lifespan, and improved genetic testing taken into consideration, actual prevalence may surpass 13 per 100,000, similar to the myotonic dystrophies and dystrophinopathies.
In facioscapulohumeral muscular dystrophy, scapular winging is prominent, and pectoral muscle atrophy leads to reversal of the anterior axillary folds. Additionally, patients may have the triple-hump sign.
Because of the lack of significant bulbar, respiratory, and cardiac involvement in facioscapulohumeral muscular dystrophy, life expectancy is normal; however, 20% of patients eventually require wheelchair use.
For facioscapulohumeral muscular dystrophy to manifest, the patient must have an allele meeting all of the following conditions: (1) on chromosome 4q35; (2) with a truncation of the D4Z4 repeat size to 8 to 10 or fewer (3) in the presence of one of three permissive simple sequence length polymorphisms (4A159, 4A161, or 4A168); and (4) with the 4qA variant at the terminus.
It is important to think of the two myotonic dystrophies not just as the most common muscular dystrophies in adults, but also as systemic disorders with effects on multiple organ systems.
A physician meeting a patient with myotonic dystrophy type 1 for the first time often makes the diagnosis via the distinctive visible features, such as temporalis and masseter muscle wasting (“hatchet face” look), male pattern baldness, and facial weakness (ptosis and a “fish mouth” look).
Cardiac evaluation and management is of utmost importance for reducing morbidity and mortality in myotonic dystrophy type 1.
Congenital myotonic dystrophy occurs in 25% of children of mothers with myotonic dystrophy type 1. Some of these mothers with myotonic dystrophy type 1 are undiagnosed and manifest few or no manifestations of disease. For this reason, it is imperative to evaluate women of reproductive age with myotonia on EMG for myotonic dystrophy type 1.
Myotonic dystrophy type 2 most often presents in middle age or later and may vary significantly within families. Muscle pain and stiffness often predominate. In most patients, the systemic manifestations in myotonic dystrophy type 2 are less frequent and milder than in myotonic dystrophy type 1.
Repeat expansion mutations are the underlying genetic basis for both myotonic dystrophies. Myotonic dystrophy type 1 is caused by a (CTG)n trinucleotide repeat expansion in the 3′-untranslated region of the dystrophia myotonica protein kinase gene on chromosome 19q13.3. Myotonic dystrophy type 2 is caused by a (CCTG)n quadruple nucleotide repeat expansion in intron 1 of the zinc finger protein 9 gene on chromosome 3q21.3.
In myotonic dystrophy type 1, CTG repeat expansion can occur with either maternal or paternal transmission, but extreme expansions with congenital-onset disease occur almost always with maternal transmission.
Anticipation does not occur in myotonic dystrophy type 2.
Limb-girdle muscular dystrophies are genetic muscle diseases with postnatal onset of progressive weakness and muscle atrophy affecting proximal upper and lower extremity muscles.
In an evaluation of 488 genetically confirmed cases of limb-girdle muscular dystrophy in the US population, calpainopathies comprised approximately 30% of the cases, while sarcoglycanopathies, dysferlinopathies, and Fukutin-related protein accounted for approximately 15% to 20% of cases each.
Autosomal dominant limb-girdle muscular dystrophies occur less frequently, have lower creatine kinase rates, and account for only around 10% to 15% of cases. Autosomal recessive limb-girdle muscular dystrophies tend to have much higher creatine kinase levels and comprise the majority of cases.
Patients with limb-girdle muscular dystrophy type 1B may have early joint contractures. Those with limb-girdle muscular dystrophy type 1C may have electrically silent, self-propagating, rolling or rippling with passive stretch or tapping of muscles.
The most common limb-girdle muscular dystrophy in most geographic regions, calpainopathies are estimated to represent anywhere from 9% to 40% of cases; hip extensor, hip adductor, and knee flexor weakness and atrophy is often disproportionately severe.
Mutations in DYSF, the gene for dysferlin, underlie both limb-girdle muscular dystrophy and Miyoshi myopathy (distal myopathy with predominant involvement of the calves), and identical mutations can account for either phenotype.
Mutations in FKRP, the gene for Fukutin-related protein, likely account for approximately 15% of LGMD patients. Prominent respiratory and cardiac dysfunction may occur early and may not parallel skeletal muscle involvement.
Mutations in ANO5 also yield a distal leg phenotype. Creatine kinase levels are often very high at 500 U/L to 35,000 U/L, mirroring dysferlinopathies.
The cardinal triad associated with Emery-Dreifuss muscular dystrophy includes (1) contractures of the elbow flexors, ankle plantar flexors, and spine early in disease; (2) childhood onset of humeroperoneal weakness; and (3) cardiac disease with arrhythmias, conduction block, and cardiomyopathy.
An ECG and cardiologic referral should always be part of the Emery-Dreifuss muscular dystrophy diagnostic evaluation.
Similar to dystrophinopathies, it is important to remember that female carriers of the X-linked forms of Emery-Dreifuss muscular dystrophy (mutations in EMD and FHL1) may develop weakness or cardiac disease.
Clinically, the cardinal features of oculopharyngeal muscular dystrophy include ptosis and dysphagia.
Although lifespan is normal in oculopharyngeal muscular dystrophy, lack of treatment of dysphagia is the most common cause for premature death in patients with the disease.
With the advent of commercially available exome sequencing and genome sequencing, medical evaluation stands at the cusp of transformation to a genetics-first strategy. Significant challenges in interpretation of the data lie ahead as we embrace these new technologies.
Footnotes
Relationship Disclosure: Dr Wicklund has participated as a site investigator for clinical trials sponsored by Eli Lilly and Company and Genzyme Corporation.
Unlabeled Use of Products/Investigational Use Disclosure: Dr Wicklund discusses the unlabeled use of prednisone, prednisolone, and deflazacort for the treatment of Duchenne muscular dystrophy and of mexiletine for the treatment of myotonia in myotonic dystrophy.
REFERENCES
- 1.Sunohara N,, Arahata K,, Hoffman EP, et al. Quadriceps myopathy: forme frusta of Becker muscular dystrophy. Ann Neurol 1990; 28 (5): 634–639. [DOI] [PubMed] [Google Scholar]
- 2.Melis MA,, Cau M,, Muntoni F, et al. Elevation of serum creatine kinase as the only manifestation of an intragenic deletion of the dystrophin gene in three unrelated families. Eur J Paediatr Neurol 1998; 2 (5): 255–261. [DOI] [PubMed] [Google Scholar]
- 3.Veerapandiyan A,, Shashi V,, Jiang YH, et al. Pseudometabolic presentation of dystrophinopathy due to a missense mutation. Muscle Nerve 2010; 42 (6): 975–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soltanzadeh P,, Friez MJ,, Dunn D, et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul Disord 2010; 20 (8): 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferlini A,, Sewry C,, Melis MA, et al. X-linked dilated cardiomyopathy and the dystrophin gene. Neuromuscul Disord 1999; 9 (5): 339–346. [DOI] [PubMed] [Google Scholar]
- 6.North KN,, Miller G,, Iannaccone ST, et al. Cognitive dysfunction as the presenting feature of Becker’s muscular dystrophy. Neurology 1996; 46 (2): 461–465. [DOI] [PubMed] [Google Scholar]
- 7.Hendriksen JG,, Vles JS. Neuropsychiatric disorders in males with duchenne muscular dystrophy: frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive–compulsive disorder. J Child Neurol 2008; 23 (5): 477–481. [DOI] [PubMed] [Google Scholar]
- 8.Bushby K,, Finkel R,, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacologic and psychosocial management. Lancet Neurol 2010; 9 (1): 77–93. [DOI] [PubMed] [Google Scholar]
- 9.Bushby K,, Finkel R,, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 2010; 9 (2): 177–189. [DOI] [PubMed] [Google Scholar]
- 10.Kinnett K,, Cripe LH. Transforming Duchenne care. Fort Lauderdale, FL: Neuromuscul Disord 2013;23(8):690–695. [DOI] [PubMed] [Google Scholar]
- 11.Hoogerwaard EM,, Bakker E,, Ippel PF, et al. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in the Netherlands: a cohort study. Lancet 1999; 353 (9170): 2116–2119. [DOI] [PubMed] [Google Scholar]
- 12.Fairclough RJ,, Wood MJ,, Davies KE. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat Rev Gen 2013; 14 (6): 373–378. [DOI] [PubMed] [Google Scholar]
- 13.Flanigan KM,, Dunn DM,, von Niederhausern A, et al. Mutation spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 2009; 30 (12): 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tuffery-Giraud S,, Béroud C,, Leturcq F, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat 2009; 30 (6): 934–945. [DOI] [PubMed] [Google Scholar]
- 15.Takeshima Y,, Yagi M,, Okizuka Y, et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet 2010; 55 (6): 379–388. [DOI] [PubMed] [Google Scholar]
- 16.Mah JK,, Selby K,, Campbell C, et al. A population-based study of dystrophin mutations in Canada. Can J Neurol Sci 2011; 38 (3): 465–474. [DOI] [PubMed] [Google Scholar]
- 17.Orrell RW. Facioscapulohumeral dystrophy and scapuloperoneal syndromes. Handb Clin Neurol 2011; 101: 167–180. [DOI] [PubMed] [Google Scholar]
- 18.Hassan A,, Jones LK,, Milone M,, Kumar N. Focal and other unusual presentations of facioscapulohumeral muscular dystrophy. Muscle Nerve 2012; 46 (3): 421–425. [DOI] [PubMed] [Google Scholar]
- 19.Klinge L,, Eagle M,, Haggerty ID, et al. Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul Disord 2006; 16 (9–10): 553–558. [DOI] [PubMed] [Google Scholar]
- 20.Lemmers RJ,, van de Vliet PJ,, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010; 329 (5999): 1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Statland JM,, Tawil R. Facioscapulohumeral muscular dystrophy: molecular pathological advances and future directions. Curr Opin Neurol 2011; 24 (5): 423–428. [DOI] [PubMed] [Google Scholar]
- 22.Lemmers RJ,, Tawil R,, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet 2012; 44 (12): 1370–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scionti I,, Greco F,, Ricci G, et al. Large-scale population analysis challenges the current criteria for molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am J Hum Genet 2012; 90 (4): 628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Norwood FL,, Harling C,, Chinnery PF, et al. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 2009; 132 (pt 11): 3175–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suominen T,, Bachinski LL,, Auvinen S, et al. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet 2011; 19 (7): 776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Logigian EL,, Martens WB,, Moxley RT, 4th, et al. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology 2010; 74 (18): 1441–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petri H,, Vissing J,, Witting N, et al. Cardiac manifestations of myotonic dystrophy type 1. Int J Cardiol 2012; 160 (2): 82–88. [DOI] [PubMed] [Google Scholar]
- 28.Mathieu J,, Allard P,, Potvin L, et al. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology 1999; 52 (8): 1658–1662. [DOI] [PubMed] [Google Scholar]
- 29.Groh WJ,, Groh MR,, Saha C, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med 2008; 358 (25): 2688–2697. [DOI] [PubMed] [Google Scholar]
- 30.Udd B,, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 2012; 11 (10): 891–905. [DOI] [PubMed] [Google Scholar]
- 31.Young NP,, Daube JR,, Sorenson EJ,, Milone M. Absent, unrecognized, and minimal myotonic discharges in myotonic dystrophy type 2. Muscle Nerve 2010; 41 (6): 758–762. [DOI] [PubMed] [Google Scholar]
- 32.Mulders SA,, van den Broek WJ,, Wheeler TM, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci U S A 2009; 106 (33): 13915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosales XQ,, al-Dahhak R,, Tsao CY. Childhood onset of limb-girdle muscular dystrophy. Pediatr Neurol 2012; 46 (1): 13–23. [DOI] [PubMed] [Google Scholar]
- 34.Aminoff MJ,, Daroff RB, eds. Encyclopedia of the neurological sciences. 2nd edition. Academic Press, 2014. [Google Scholar]
- 35.Wicklund M,, DiVincenzo C,, Liaquat K,, Karbassi I. Relative prevalence of limb girdle muscular dystrophies in the United States population. Neurology 2013; 80 (1001): P07.030. [Google Scholar]
- 36.Mitsuhashi S,, Kang PB. Update on the genetics of limb girdle muscular dystrophy. Semin Pediatr Neurol 2012; 19 (4): 211–218. [DOI] [PubMed] [Google Scholar]
- 37.Harms MB,, Sommerville RB,, Allred P, et al. Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann Neurol 2012; 71 (3): 407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greenberg SA,, Salajegheh M,, Judge DP, et al. Etiology of limb girdle muscular dystrophy 1D/1E determined by laser capture microdissection proteomics. Ann Neurol 2012; 71 (1): 141–145. [DOI] [PubMed] [Google Scholar]
- 39.Melia MJ,, Kubota A,, Ortolano S, et al. Limb-girdle muscular dystrophy 1F is caused by a microdeletion in the transportin 3 gene. Brain 2013; 136 (pt 5): 1508–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fardeau M,, Hillaire D,, Mignard C, et al. Juvenile limb-girdle muscular dystrophy. Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain 1996; 119 (pt 1): 295–308. [DOI] [PubMed] [Google Scholar]
- 41.Amato AA,, Brown RH, Jr. Dysferlinopathies. Handb Clin Neurol 2011; 101: 111–118. [DOI] [PubMed] [Google Scholar]
- 42.Stensland E,, Lindal S,, Jonsrud C, et al. Prevalence, mutation spectrum and phenotypic variability in Norwegian patients with limb girdle muscular dystrophy 2I. Neuromuscul Disord 2011; 21 (1): 41–46. [DOI] [PubMed] [Google Scholar]
- 43.Magri F,, Del Bo R,, D’Angelo MG, et al. Frequency and characterization of anoctamin 5 mutations in a cohort of Italian limb-girdle muscular dystrophy patients. Neuromuscul Disord 2012; 22 (11): 934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore SA,, Shilling CJ,, Westra S, et al. Limb-girdle muscular dystrophy in the United States. J Neuropathol Exp Neurol 2006; 56 (10): 995–1003. [DOI] [PubMed] [Google Scholar]
- 45.Norwood F,, de Visser M,, Eymard B, et al.; EFNS Guideline Task Force. EFNS guideline on diagnosis and management of limb girdle muscular dystrophies. Eur J Neurol 2007; 14 (12): 1305–1312. [DOI] [PubMed] [Google Scholar]
- 46.Puckelwartz M,, McNally EM. Emery-Dreifuss muscular dystrophy. Handb Clin Neurol 2011; 101: 155–166. [DOI] [PubMed] [Google Scholar]
- 47.van Berlo JH,, de Voogt WG,, van der Kooi AJ, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl) 2005; 83 (1): 79–83. [DOI] [PubMed] [Google Scholar]
- 48.Meune C,, van Berlo JH,, Anselme F, et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med 2006; 354 (2): 209–210. [DOI] [PubMed] [Google Scholar]
- 49.Bouchard JP,, Brais B,, Brunet D, et al. Recent studies on oculopharyngeal dystrophy in Quebec. Neuromuscul Disord 1997; 7 (suppl 1): S22–S29. [DOI] [PubMed] [Google Scholar]
- 50.Dubbioso R,, Moretta P,, Manganelli F, et al. Executive functions are impaired in heterozygote patients with oculopharyngeal muscular dystrophy. J Neurol 2012; 259 (5): 833–837. [DOI] [PubMed] [Google Scholar]