

Abstract
Purpose of Review:
This article reviews marketed pharmacologic treatments for Alzheimer disease as well as their efficacy, effectiveness, adverse effects, and issues involved in their use, including duration of treatment, adverse events, and controversies. Current experimental drug development, including challenges to developing successful drugs for Alzheimer disease, are also reviewed and assessed.
Recent Findings:
Cholinesterase inhibitors and memantine are the available pharmacologic treatment options. They show limited clinical effects over the shorter term for some patients, mild to moderate cholinergic adverse effects in a minority of patients, and potentially underappreciated toxicity over the longer term. No subsequent experimental drug in development has been successful thus far; there has not been a new drug marketed for Alzheimer disease since 2003.
Summary:
Cholinesterase inhibitors and memantine are marketed for the treatment of Alzheimer disease. Drug development programs aimed at new targets, including the amyloid-β cascade, have been unsuccessful thus far despite their designs to detect very small or minimal clinical effects from the experimental drugs. Marked advances in preclinical science nevertheless support a basis for considerable optimism that effective interventions will be found soon.
CURRENT MEDICATIONS APPROVED BY THE US FOOD AND DRUG ADMINISTRATION
The cholinesterase inhibitors tacrine, donepezil, rivastigmine, and galantamine were approved by the US Food and Drug Administration (FDA) for marketing in the United States for the treatment of Alzheimer disease in 1993, 1996, 2000, and 2001, respectively. Memantine was approved by the FDA in 2003 for the indication of moderately severe to severe Alzheimer disease.
The Cholinergic Hypothesis and Cholinesterase Inhibitors
The use of cholinesterase inhibitors for Alzheimer disease is based on the cholinergic hypothesis of memory impairment.1,2 The hypothesis implies that cholinergic deficits are responsible for cognitive and behavioral changes in patients with dementia and age-related memory impairment and, further, that pharmacologic augmentation of central cholinergic function will improve cognitive function. The cholinergic hypothesis is supported by observations of marked decline of the cholinergic corticobasal projections, loss of cholinergic cell bodies in the nucleus basalis, and reduced choline acetyltransferase activity, which is needed for acetylcholine synthesis. Further support for the hypothesis includes correlations between the above cholinergic deficits and neuritic, amyloid-β (Aβ) peptide–containing plaques and decline in cognitive test performance.
Historically, the targeted cholinergic treatment approaches have included (1) using acetylcholine precursors with the expectation that more acetylcholine will be produced; (2) using direct cholinergic agonists to mimic and replace the effects of acetylcholine; and (3) using cholinesterase inhibitors to inhibit the enzyme-induced metabolism of intrasynaptic acetylcholine. The first two approaches, using several different drugs, have shown no significant or meaningful clinical effects.3 In addition, barriers to the successful development of muscarinic agonists include the difficulty in finding a drug with clear M1 subtype agonism—not affecting other muscarinic receptor subtypes—and with few adverse effects.4
Tacrine. Tacrine is very rarely—if at all—used and is not actively marketed as it requires administration 4 times per day, a complicated four-step dose titration, and is associated with reversible direct hepatotoxicity requiring regular monitoring of serum transaminases. It is historically important as the first drug approved for Alzheimer disease, setting the roadmap for Alzheimer drug development, but will not be discussed further.
Donepezil. Donepezil is a long-acting reversible acetylcholinesterase inhibitor. Two phase 3 clinical trials showed evidence of efficacy for FDA approval. Additional randomized clinical trials were completed and include trials of 6 and 12 months’ duration and in severely impaired and nursing home patients, as reviewed in a Cochrane review.5 One non–industry-sponsored, randomized, placebo-controlled trial followed patients over several years and reported modest cognitive effects over 2 years but no significant effects on loss of function, nursing home placement, or health economic measures.6
Pharmacokinetics and drug interactions. Oral bioavailability approaches 100%, with peak concentration occurring in 3 to 4 hours (Tmax). It is both metabolized extensively in the liver and excreted unchanged in the urine. Donepezil has a long elimination half-life of 70 hours, and steady state occurs in approximately 2 weeks. A 23-mg extended-release formulation has been marketed, indicated for patients with moderate to severe Alzheimer disease who have been maintained on 10 mg/d and who might benefit from an increased dose. This formulation’s Tmax is approximately 8 hours, with peak plasma concentrations about twice as high compared to the 10 mg/d dose.
Rivastigmine. Rivastigmine is a pseudoirreversible cholinesterase inhibitor that is selective for acetylcholinesterase and butyrylcholinesterase. In the two published trials showing efficacy, doses were titrated weekly over 7 weeks to one of two dosage ranges, 1 mg/d to 4 mg/d or 6 mg/d to 12 mg/d, and dose decreases were not permitted, possibly contributing to less tolerability and seemingly more side effects.7,8
A transdermal patch formulation has been marketed based on a placebo-controlled study comparing a 17.4-mg patch, 9.5-mg patch, and 6 mg of orally administered rivastigmine twice per day in 1195 patients with moderately severe Alzheimer disease (ie, Mini-Mental State Examination [MMSE] scores of 10 to 20) over 6 months. All formulations showed efficacy, but fewer adverse events occurred with the patch formulations.9
Pharmacokinetics and drug interactions. Rivastigmine has little protein binding and is well absorbed. Although the elimination half-life is less than 2 hours, enzyme inhibition lasts about 9 hours. The drug is not metabolized by the liver. It is slowly hydrolyzed and then excreted by the kidneys. The pharmacokinetics of the transdermal patch shows maximum concentration in 8 to 16 hours and a 3-hour elimination half-life after the patch is removed. Its extrahepatic metabolism makes it unlikely to have significant pharmacokinetic interactions.
Galantamine. Galantamine is a reversible competitive acetylcholinesterase inhibitor with relatively less butyrylcholinesterase inhibition compared to rivastigmine. Competitive inhibitors potentially are less active in brain areas that have remaining high acetylcholine levels and more active in other areas. Galantamine also functions as an allosteric modulator of nicotinic receptors, possibly enhancing cholinergic transmission by presynaptic nicotinic stimulation.
Efficacy of galantamine has been demonstrated at doses of 8 mg twice a day and 16 mg twice a day, with fewer adverse effects at the lower dose, in at least four randomized trials of 3 and 6 months’ duration. A Cochrane review concluded that galantamine shows consistent positive effects of 3 to 6 months’ duration—with no additional improvement with doses over 16 mg/d—and that the frequency of gastrointestinal adverse events is similar to other cholinesterase inhibitors.10
Pharmacokinetics and drug interactions. Galantamine is well absorbed with approximately 90% bioavailability. Peak concentrations occur in approximately 1 hour. The half-life of the compound is approximately 7 hours. A sustained-release form is also available. It is metabolized by the liver and excreted in the urine. Galantamine does not inhibit CYP 2D6 or 3A4 and has little effect on the metabolism of other drugs; however, inhibitors of CYP 2D6 or 3A4, such as paroxetine and ketoconazole, respectively, may decrease clearance and increase the bioavailability and plasma levels of galantamine. Some other CYP 2D6 inhibitors include amitriptyline, fluoxetine, and quinidine.
Adverse effects of cholinesterase inhibitors. The most common adverse events due to cholinesterase inhibitors are cholinergically mediated and include nausea, diarrhea, vomiting, anorexia, and weight loss (Table 2-1). Muscle cramps are common with donepezil. Early cholinergic effects are frequently related to the initial dosing and titration of the medications. Reducing the dose temporarily and retitrating may reduce the reemergence of acute cholinergic adverse events. Many patients tend to become tolerant to the adverse events. Weight loss, fatigue, and anorexia may occur over time and be tolerated by patients and family and, hence, may not be recognized as adverse events induced by cholinesterase inhibitors. Few trials have directly compared cholinesterase inhibitors with respect to adverse events.3 More adverse events are observed, however, with 23 mg/d donepezil than with 10 mg/d of donepezil in a direct comparison, with many patients experiencing nausea and vomiting,11 and fewer adverse events are seen with transdermal rivastigmine compared to orally administered rivastigmine.
Table 2-1.
Maintenance Dosages and Adverse Events of Marketed Cholinesterase Inhibitorsa
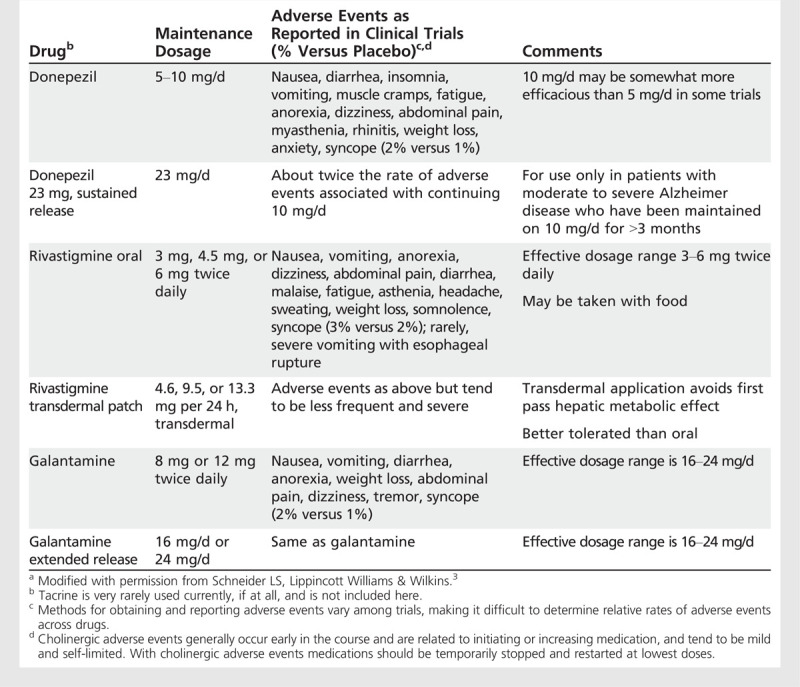
Anorexia varies in incidence from 8% to 25% at higher doses of cholinesterase inhibitors compared with 3% to 10% in patients on placebo and may be dose related. Similarly, the proportion of patients with weight loss in clinical trials (ie, losing greater than 7% of their weight) ranges from 10% to 24% in patients taking higher doses compared to 2% to 10% of patients treated with placebo.
General precautions, listed in the prescribing information for the drugs, should be considered when using cholinesterase inhibitors. These include increased gastric acid secretion; increased risk for gastrointestinal bleeding, especially in patients concurrently using anti-inflammatories; sinus bradycardia, especially in patients with sick sinus and other supraventricular conduction delays; exacerbation of obstructive pulmonary disease and asthma; urinary outflow obstruction; and risk of seizures. Bradycardia may lead to syncope, falls, and injury. Finally, cholinesterase inhibitors may prolong the effects of succinylcholine-type muscle relaxants.
Long-Term Safety of Cholinesterase Inhibitors
The long-term safety of cholinesterase inhibitors has not been systematically studied. An analysis of Canadian medical and prescription records, however, indicated that patients on cholinesterase inhibitors (mainly donepezil) were hospitalized for syncope nearly twice as often as people with dementia who did not receive these drugs. Moreover, they showed a 69% increased risk for bradycardia, a 49% increased risk for having a pacemaker implanted, and an 18% increased risk for hip fractures.12 The absolute incidence of these events is about 2% of treated patients per year, implying that one patient will be hospitalized for syncope for every 50 to 100 patients treated for 1 year.
Effectiveness of Cholinesterase Inhibitors
Most investigations of drugs that demonstrate efficacy for Alzheimer disease have been done in 3- or 6-month trials in patients with mild to moderate Alzheimer disease. In addition, studies with donepezil have demonstrated efficacy for patients with severe Alzheimer disease. Despite differences in mechanism of action and dosing levels, no evidence exists for efficacy differences between the three cholinesterase inhibitors. The relevant clinical question, however, is to what degree these consistent but modest effects translate into meaningful clinical improvement.
In a Cochrane review,13 the drugs are associated with an overall mean 2.4 points effect (range 1.1 to 3.9) over placebo on the Alzheimer Disease Assessment Scale—Cognitive Subscale (ADAS-Cog) over 6 months, and absolute mean improvement over baseline with treatment ranges from worsening or no change to improvement of about 1.9 ADAS-Cog points (Figure 2-1). This difference represents 10% or less change on the ADAS-Cog, which is within the bounds of the scale’s test-retest error. Although clearly some patients improved substantially with cholinesterase inhibitors, some also worsened to a greater extent than those treated with placebo. Withdrawals due to adverse events, particularly gastrointestinal adverse effects, were higher for all three cholinesterase inhibitors compared with placebo. Intolerability may have more to do with initial titration than with longer-term treatment, at least with respect to nausea and vomiting.
Figure 2-1.
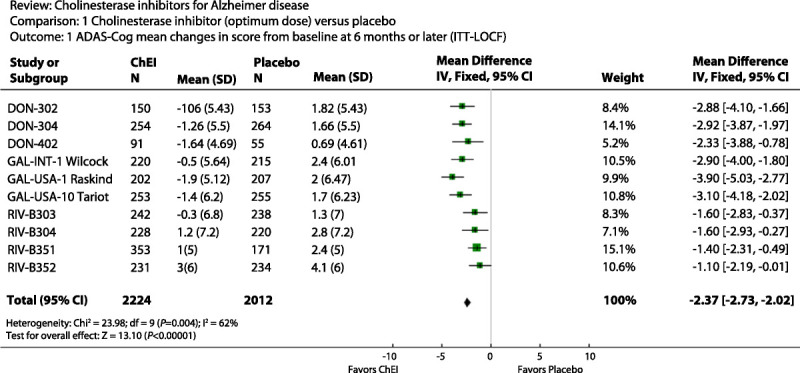
Cholinesterase inhibitors, optimum dose versus placebo. The figure shows the mean drug-placebo difference on the ADAS-Cog from several clinical trials along with 95% confidence interval widths displayed as horizontal lines. The overall mean effect is −2.37 points with 95% confidence intervals of −2.73 to −2.03.
ADAS-Cog = Alzheimer Disease Assessment Scale—Cognitive Subscale; ITT-LOCF = intention to treat last observation carried forward; ChEI = cholinesterase inhibitors; SD = standard deviation; IV, Fixed = inverse variance, fixed effect; CI = confidence interval; Chi2 = Chi squared; df = degrees of freedom; Z = Z score.
Reprinted with permission from Birks J, Cochrane Database Syst Rev.13 © 2006, John Wiley & Sons, Inc. onlinelibrary.wiley.com/doi/10.1002/14651858.CD005593/abstract.
Because of the design of the trials and the modest therapeutic effect, it is difficult to identify individual treatment responders, especially those who may be benefiting by a couple of points. Overall, the use of cholinesterase inhibitors involves balancing the modest expectations for benefit with the potential for adverse effects due to the drugs, and considerable clinical judgment.
Cholinesterase Inhibitors for Severe Alzheimer Disease
Donepezil is the only cholinesterase inhibitor specifically labeled for patients with severe Alzheimer disease (ie, with MMSE scores of 10 or less). The efficacy evidence is based on three 6-month, randomized, placebo-controlled clinical trials.4 Effects were modest, on the order of several points on the Severe Impairment Battery (SIB), a scale ranging from 100 to 0.14 In addition, the 23-mg extended-release formulation of donepezil is intended to be used in more moderate to severe cases after a patient has been treated with 10 mg/d for at least 3 months and when the clinician is uncertain whether the patient is benefiting from the 10-mg dose. In the only clinical trial to document its efficacy, 1467 patients with Alzheimer disease with MMSE scores from 20 to 0 who had been treated with donepezil 10 mg/d were randomized to 23 mg/d or to continue their 10 mg/d dose for 6 months.11 A significant 2.2 mean drug-placebo difference on the SIB was not supported by efficacy on the clinician’s global assessments, and dropouts were substantially greater with the higher dose compared to continuing the 10 mg dose, 30% versus 18%. A post hoc analysis indicated that the more severe patients showed a somewhat greater 3.1 effect on the SIB and significance on the global assessment of 0.09 points on a scale from 1 to 7. The 23-mg formulation was approved for marketing by the FDA despite recommendations for nonapproval by two FDA officers. A citizen watchdog group has expressed concern about the added risks of the higher dose and has sued the FDA for its removal. As donepezil is now generically manufactured, the branded 23-mg extended-release formulation sells at a premium over the generics and is heavily promoted. Clinicians should be cautious, however, not to use 20 mg of generic donepezil in place of the 23-mg branded dose, as the formulations are different.
Dosage and Use of Cholinesterase Inhibitors
Donepezil is started at 5 mg/d and can be increased to 10 mg/d after 4 weeks. Both 5 mg/d and 10 mg/d are effective doses, but the 10-mg/d dose is somewhat more so when the dosing groups are directly compared. The sustained-release 23-mg/d donepezil formulation is indicated for moderate to severe Alzheimer disease but only for patients who have been treated with 10 mg/d for at least 3 months.
The starting dose of rivastigmine is 1.5 mg twice a day with meals, increased to 3 mg twice per day after 2 weeks. Subsequent increases to 4.5 mg and 6 mg twice a day are determined by tolerability and can be considered after 2 weeks of treatment. Higher daily doses are associated with better efficacy than are lower doses. The transdermal patch is started at 4.6 mg (5 cm2) per day and increased to one 9.5-mg (10 cm2) patch per day after 4 weeks, if tolerated. A 13.3-mg (15 cm2) patch was recently approved, allowing for a further increase after 4 weeks at the 9.5-mg dose. The maintenance doses are 4.6 mg/d, 9.5 mg/d, or 13.3 mg/d.
Initial dosing for galantamine is 4 mg twice a day, raised to 8 mg twice a day after 4 weeks. The dose can be raised to 12 mg twice a day after another 4 weeks if patients are tolerating the medication but do not appear to be benefiting from it. The initial dose of the extended-release formulation is 8 mg/d, raised to 16 mg/d after 4 weeks, and can be increased to 24 mg/d based on tolerability and benefit. With all cholinesterase inhibitors, raising the dose too soon increases risks for cholinergic adverse events.
Cholinesterase Inhibitors for Mild Cognitive Impairment
Cholinesterase inhibitors are not indicated for mild cognitive impairment (MCI), yet their use may be common practice, and nearly half of patients so diagnosed in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) were receiving them.15 The several randomized, placebo-controlled trials of cholinesterase inhibitors for MCI used the typical doses for Alzheimer disease, tended to be long-term (from 2 to 4 years), and the onset of Alzheimer dementia as main end points. Nevertheless, the two 2-year galantamine trials,16 one 4-year rivastigmine trial,17 and one over-3-year donepezil trial18 were not positive on their primary outcomes and showed an excess in adverse events (summarized in at least three systematic reviews10,19,20). Uncertainty regarding definitions of MCI limit inferences that can be drawn from the outcomes of these trials as there were broad variations in cognitive impairment and progression to Alzheimer disease. The two galantamine trials, when combined, were particularly concerning for an excess in deaths associated with the drug. In some trials, nominally significant drug-placebo differences on the ADAS-Cog were observed at earlier time points during the first year. The effects were not sustained, however, and appeared more evident in the patients with more severe cognitive impairment and in APOE*E4 carriers.
MEMANTINE
Memantine was approved by the FDA in late 2003 for moderate to severe Alzheimer disease. It is characterized as a moderate-affinity, uncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist; a rationale for its use is that it may protect against overstimulation of NMDA receptors that may occur in Alzheimer disease as well as consequent glutamate- and calcium-mediated neurotoxicity.
The basis for approval was positive outcomes on two 6-month-long placebo-controlled clinical trials. In one trial cholinesterase inhibitors were not allowed,21 and in the other trial, all patients had been taking donepezil for at least 6 months (over 2 years on average) before being randomized to memantine or placebo.22 A third moderate to severe Alzheimer disease trial did not show significant effects for memantine, however.23
The trials were similar to the design of cholinesterase inhibitors trials but included only patients with MMSE scores less than 15—hence the moderate to severe Alzheimer disease indication—whereas mild to moderate Alzheimer disease trials are usually operationalized as MMSE 10 to 26, and for memantine as 10 to 22.
The SIB, a global assessment, and an activities of daily living (ADLs) inventory were primary outcome measures. The SIB is used as the cognitive outcome measure instead of the ADAS-Cog because the patients are too severely impaired to perform on the latter. Although the outcomes are different, the standardized statistical magnitudes of benefit from memantine in two of the three moderate to severe Alzheimer disease trials is similar to modest effect sizes seen in cholinesterase inhibitors trials with patients with mild to moderate Alzheimer disease.
It is important to note that only one of three trials of memantine in mild to moderate Alzheimer disease that were of similar design to cholinesterase inhibitor trials showed statistically significant improvement on the ADAS-Cog and global assessment.24 Two others did not show significant drug-placebo differences,25,26,27 and in pooled analyses did not show efficacy for mild Alzheimer disease.28 Hence, memantine has not been approved by the FDA for patients with mild Alzheimer disease.28
A once-per-day 28-mg sustained-release formulation was approved by the FDA in 2010 but has not yet been marketed. The basis for FDA approval was a 6-month, placebo-controlled, Alzheimer disease trial involving 677 patients with MMSE scores between 3 and 12 (completed in 2010 but unpublished), in which there were statistically significant effects favoring sustained-release memantine on the SIB and global assessment but not on ADLs. Reported adverse events were similar to placebo.
Controlled clinical trials of memantine are summarized in a Cochrane review that concluded that memantine had a small beneficial effect in moderate to severe Alzheimer disease and was well tolerated.27
Mechanism of Action
The therapeutic mechanism of action of memantine is unknown, but it may act as an open-channel NMDA receptor antagonist that does not have apparent pharmacologic activity until higher glutamate levels trigger the receptor and cause the ion channel to open. It is speculated that the drug then enters the channel, blocking it and preventing calcium influx, depolarization, and hyperactivation of the neuron. Memantine could be viewed as a modulator of glutamatergic activity. As this hypothesized mechanism is apparently neuroprotective, it is not clear what exactly is involved in memantine’s short-term and symptomatic effect in 6-month trials. Memantine may have effects on long-term potentiation that may correlate with a short-term effect on memory.29,30
The above is speculative, however, and potential long-term efficacy has not been tested in the long-term and large clinical trials that would be required.
Pharmacokinetics
Memantine is well absorbed, not affected by food; bioavailability approaches 100%, and it is widely distributed throughout the body. Plasma protein binding is about 45%; the time to maximum plasma concentration is between 3 to 7 hours and elimination half-life is 60 to 80 hours. Memantine undergoes minimal hepatic metabolism, and it is mostly excreted unchanged in the urine.
Adverse Effects
Adverse events are infrequent but can include headache, dizziness, confusion, somnolence, and infrequent hallucinations. In clinical trials, the frequency of gastrointestinal symptoms is less than placebo, and, for example, diarrhea occurred half as often.
The actions of memantine may reduce the cholinergic effects of donepezil (although this has not been formally studied). There appear to be no adverse drug interactions with cholinesterase inhibitors.
Dosage and Use
Per prescribing information, memantine is started at 5 mg/d for 1 week and then increased weekly by 5 mg/d until 10 mg twice daily is reached. The reason for this titration regimen—considering memantine’s long half-life and high degree of tolerability—is because it was used in the trials that supported its marketing approval, and other regimens have not been tested.
In clinical practice, memantine is either prescribed alone or added to a cholinesterase inhibitor, often after the latter has been used for a time. Some clinicians, however, start memantine along with or very soon after a cholinesterase inhibitor, an off-label use. Its duration of effectiveness is not known beyond the 6-month length of the clinical trials; however, it tends to be prescribed for indefinite periods. Some open-label observations from clinic cohorts have suggested that therapy with a cholinesterase inhibitor and memantine together may ameliorate the course of Alzheimer disease,31,32 while observations from clinical trial extensions and ADNI suggest that any effects may wane over 6 to 12 months of treatment.15,33,34
The practical issue of how long to treat is particularly challenging in patients with severe dementia and poor quality of life, especially when the clinician is uncertain as to whether or not an individual patient is benefiting.
CEREBROLYSIN AND GINKGO BILOBA
Cerebrolysin and Ginkgo biloba standardized extract are mentioned because they are licensed or on medication formularies of many countries (not the United States). Cerebrolysin is a peptide and amino acid preparation from porcine brain that may have neurotrophic actions in preclinical models. It is administered intravenously or intramuscularly 5 days a week for 4-week periods. Cognitive effects are considered to last up to 3 months. Several 6-month placebo-controlled trials in patients with Alzheimer disease showed equivocal outcomes.35
Extracts from leaves of the G biloba, or maidenhair, tree are widely sold in the United States as food supplements for which health claims are not permitted and most insurance plans will not reimburse.36 A specific standardized extract, EGb 761 (ie, standardized to a certain level of flavonoids and ginkgolides), is approved by the formularies in some countries, most notably Germany and France.
Rationales for G biloba extract as an Alzheimer disease treatment are that in preclinical models the flavonoids and ginkgolides are antioxidants, appear neuroprotective, may inhibit Aβ42-induced neuron death, enhance neurogenesis, and inhibit Aβ aggregation. None of these properties has been demonstrated in humans.
G biloba extract is also used as a memory enhancer in people without Alzheimer disease; however, clinical trials in older and younger adults who do not have cognitive impairment show mixed results at best.36
A Cochrane systematic review that included 35 clinical trials reported inconsistent evidence that G biloba had clinically significant benefits for dementia or cognitive impairment.37 One FDA regulatory–quality 6-month trial in mild to moderate Alzheimer disease was conducted in the United States with the expectation for gaining FDA approval but failed to demonstrate efficacy.38 A 6-month trial conducted at British primary care sites also failed to show efficacy.39
The controversies about EGb 761 include its promotion in the United States for Alzheimer disease and a tepid endorsement from the German Institute for Quality and Efficiency in Health Care that it might improve ADLs, show cognitive benefit, and improve behavioral symptoms, but only in dementia patients with “psychopathologic” symptoms, conclusions, however, that relied on only two studies conducted in Eastern Europe.40
G biloba EGb 761 extract is also notable because three prevention trials to delay the onset of Alzheimer disease or MCI41,42,43 did not yield significant results. Two trials included 3069 and 2854 nonimpaired or MCI patients followed over 5 years. In sum, little, if any, evidence exists for G biloba extract either improving symptoms or preventing Alzheimer disease.
MEDICAL FOODS
A medical food is a food formulated for the dietary management of an illness that has distinctive nutritional requirements, and is intended to be used under medical supervision.44 Thus physicians may write prescriptions for medical foods.
A formulation of medium-chain triglycerides is marketed as a medical food for Alzheimer disease. The rationale for this formulation proposes that Alzheimer disease may result in part from mitochondria dysfunction and impaired glucose metabolism; therefore, enriching a diet with a food that is converted to ketones that would presumably enhance electron transport in mitochondria would be therapeutic.45 A randomized, placebo-controlled, 12-week trial in Alzheimer disease showed an improvement in cognitive function after 6 weeks that was lost at 12 weeks; another trial in MCI was also negative. The main adverse events reported by the manufacturer are gastrointestinal symptoms; cautions include risk for ketoacidosis in patients at risk, including those with alcohol abuse history and poorly controlled diabetes.
Another medical food, marketed in late 2012 in Europe and Brazil, is a combination of compounds including uridine, choline, omega-3 fatty acids, phospholipids, B vitamins, and antioxidants.46 The rationale is that the combination enhances dendritic spine growth, synapse formation, and neurotransmitter precursors, ultimately improving cognitive function. Thus far, results of a 12-week, placebo-controlled trial in 225 patients with Alzheimer disease were not significant on most outcomes; and the primary outcome, a neuropsychological memory composite score, from a 24-week trial with 259 patients with mild Alzheimer disease, showed a statistical trend in favor of the formulation. No increase in adverse events over placebo was reported in the later trial.
CONTROVERSIES ON THE EFFECTIVENESS OF CURRENT TREATMENTS
Considerable controversy surrounds the use of currently available drugs for treating Alzheimer disease, involving not only the limited efficacy and adverse effects but also the desperation and unmet needs of many patients and their families, as well as the frustration of clinicians. This controversy is played out between two positions: either that the drugs are effective and should be considered the standard of care for Alzheimer disease, or that they are ineffective and not worthwhile in terms of cost and adversity. Hence, the overarching controversy is whether or not their use yields clinically meaningful or therapeutically useful outcomes, and for whom. As examples, the Agency for Healthcare Research and Quality (AHRQ) stated that “treatment of dementia with cholinesterase inhibitors and memantine can result in statistically significant but clinically marginal improvement in measures of cognition and global assessment of dementia.”47 The United Kingdom’s National Institute for Health and Clinical Excellence (NICE) and a health assessment group initially concluded in 2006 that the cholinesterase inhibitors can delay cognitive impairment for 6 months,48 but that they are not cost-effective.49 Additional concerns raised included the fact that the real impact of treatment is difficult to assess because of the limited methodologic quality of the trials, a lack of generalizability, the fact that patients with medical comorbidities were not included in the trials, and that the mean scores are difficult to interpret.48 NICE is less restrictive, however, in their revised opinion.
Pharmaceutical advertising campaigns, some of which have resulted in FDA warnings for overstating efficacy, have contributed to the perception of effectiveness that influences both physicians and caregivers.50,51
Efficacy Versus Effectiveness
The issue of efficacy versus effectiveness of cholinesterase inhibitors is illustrated and discussed in Figure 2-2.52 Despite trends showing clear statistical significance, it can be seen that there is substantial overlap in outcomes between drug and placebo patients. Even the patients who improve to the greatest extent on the ADAS-Cog are still in the moderately severe dementia range. This illustrates the meaning of the “small” or “modest” cognitive effects of cholinesterase inhibitors and makes inferences about this effect, indicating improved health outcomes, difficult.
Figure 2-2.
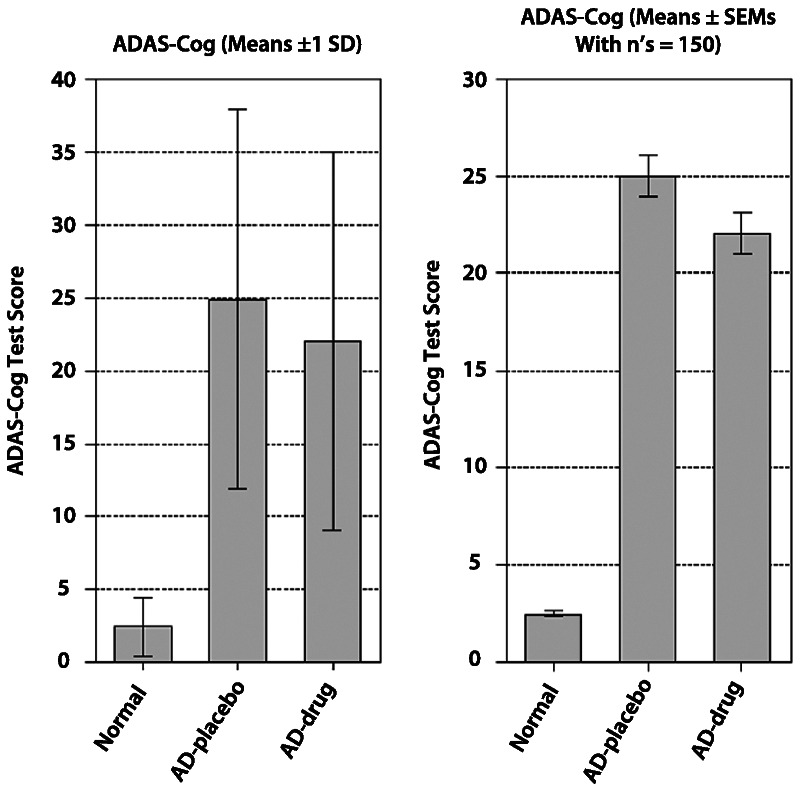
Comparison of the identical effect of donepezil from a clinical trial compared to placebo, with standard deviation (SD) bars (left) reflecting the distribution of outcomes and standard error of the mean (SEM) limits (right) reflecting the precision of the outcomes. The narrow standard error bar indicates the relatively high precision and strong statistically significant drug-placebo estimate of the difference. The SD limits show how, despite the statistical significance, substantial overlap occurs in outcomes between patients treated with donepezil and patients treated with placebo, such that very few individuals can be seen to benefit on ADAS-Cog scores. Even the patients who improve to the greatest extent (eg, those outside the 1 SD limit) are still in the moderately severe dementia range.
ADAS-Cog = Alzheimer Disease Assessment Scale—Cognitive Subscale; AD = Alzheimer disease.
Reprinted from Lindner MD, et al, Academic Press.52 © 2008, with permission from Elsevier.
It is difficult to identify the individual patient who benefits from cholinesterase inhibitors or memantine because the outcome measures and mean changes on scale scores do not identify responders. Further, most trials have not taken caregiver views into account, although one that did shows that important aspects of the treatment response are missed by current measures.53
Duration of Treatment
Placebo-controlled clinical trials with marketed cholinesterase inhibitors generally have lasted 6 months, with a few exceptions lasting up to 12 months or longer. Inferences are made that if the drugs are effective over this period then they will continue to be beneficial far longer, perhaps indefinitely. Over the long term, however, as patients inevitably worsen, it becomes even more difficult to determine whether any given individual is benefiting from the drugs.
In some 3-month-long and 6-month-long trials, after medication has been discontinued patients on average return to the cognitive level of the patients contemporaneously treated with placebo within 6 weeks. Such findings are taken to indicate that the drugs have overall symptomatic effects and that continuous use is required to maintain benefits.
Some observational studies using clinic databases or open-label extensions of clinical trials suggest that patients who continue cholinesterase inhibitors over at least 1 year have a delay in nursing-home placement compared to those who cannot tolerate or do not take them, and that the addition of memantine could further contribute to the delay.33,32,54,55 These observations, however, are not controlled and are subject to the potential bias that patients who experience a less-progressive course continue their medications, while patients who are destined to progress more quickly do not continue, resulting in apparent therapeutic effects that are illusory. Moreover, comparisons are made between cohorts from time periods both before and after the ready availability of the cholinesterase inhibitors.56 These observational studies, however, contrast with the long-term controlled trials in MCI and with observations from the ADNI15 and Australian Imaging Biomarkers and Lifestyle datasets,57 where the use of cholinesterase inhibitors over the long term is associated with faster decline. Thus, duration of treatment remains an unresolved issue.
Effects from Withdrawal of Cholinesterase Inhibitors or Memantine
Discontinuation of cholinesterase inhibitors has been associated with worsening of cognition and confusion in some patients. This effect was evident in a clinical trial in which donepezil was stopped after a fixed period of 12 weeks and patients were then randomized to continuing drug or to placebo58 as well as when patients were discontinued from some 6-month trials. Yet worsening of behavior and confusion do not appear common when the drugs are stopped in clinical practice, as is frequently done. In clinical practice only 19% to 23% of patients continued to take donepezil or rivastigmine for more than 1 year, and about one-third discontinued the drugs within 2 months.59
Tapering and withdrawal of donepezil after maintenance treatment for an average of 2 to 3 years was formally tested in a randomized controlled trial of severely impaired patients with Alzheimer disease; continuing donepezil was compared with discontinuing it, and, simultaneously, starting memantine was compared with not starting it.60 Over the 1-year follow-up period, continuing donepezil was associated with better cognitive scores and ADLs, and adding memantine when donepezil was discontinued was better than not adding it. Many patients, however, discontinued donepezil without difficulty; notably, only half of the patients who were assigned to continue donepezil actually continued treatment beyond the 1-year follow-up, suggesting that many patients perceived that continuing donepezil, at least under double-blinded conditions, was not effective. Thus, the outcomes support decisions either to continue medication or to taper and discontinue it when physicians are uncertain of continuing benefit.61 This trial also did not support the typical use for memantine as an add-on to donepezil, showing that the add-on was not better than continuing donepezil alone, a finding that adds to the controversy of whether the drugs taken together are better than either alone. It is generally good practice to taper these medications before discontinuing, even though both donepezil and memantine have long terminal half-lives.
Effectiveness for Treatment of Disruptive Behaviors
The evidence base for advocating cholinesterase inhibitors and memantine for treating disruptive behaviors in Alzheimer disease consists of post hoc analyses of individual items on behavior rating scales from randomized trials. In a summary analysis of 14 cholinesterase inhibitor trials that assessed effects on behavior in post hoc analyses, only three showed significant effects for improving behavior62; none of these effects was large.13 Another meta-analysis reported significant but trivial effects on behavior in the more mildly cognitively impaired patients, but no effect in the more severely impaired.63 The fact that patients were not chosen for having behavior problems and their symptom ratings were rather low at baseline limits reasonable inferences about clinical efficacy.
One randomized placebo-controlled trial prospectively assessed donepezil’s efficacy for improving behavior in patients who had clinically important agitation and did not find an effect for donepezil over placebo, which itself had a moderate effect.64 Of considerable importance was that donepezil did improve cognitive function in this trial, suggesting that the effects on cognition do not necessarily affect behavior. A similarly designed randomized controlled trial, but with memantine, did not demonstrate its efficacy at improving behavior, either.65
As many patients with disruptive behaviors are treated with both cholinesterase inhibitors and memantine, the opportunities to use the drugs specifically to treat agitation and disruptive behavior are limited, however. Finally, a clinician would want to be sure that the cholinesterase inhibitor is not exacerbating restlessness, agitation, or sleeplessness.
OVERVIEW OF ALZHEIMER DISEASE DRUGS IN DEVELOPMENT
Not for the want of trying over the past 2 decades, and despite substantial progress in understanding pathogenetic processes associated with Alzheimer disease, no new practical treatments have been developed. A 2008 estimate identified 172 experimental drugs for Alzheimer disease that had been in phase 1 to phase 3 development, half of which had already failed by that time, and nearly all of those have now been discontinued.52 This enormously high failure rate is due almost entirely to efficacy and safety issues.
The phenotypic heterogeneity and pleomorphic expression of the illness implies that many potential therapeutic targets may exist. The many drugs under development can be grouped into approaches directed at Aβ and tau proteins; at neurotrophic, neuroprotective, and anti-inflammatory mechanisms; and at various neuroreceptors. The dominant target areas currently are drugs targeting Aβ, neuronal nicotinic receptors, and 5-HT subtype receptors.
Regulatory Requirements
Regulatory criteria for marketing symptomatic and disease-modifying therapies require demonstrating improvements in cognition and ADLs, overall improvements compared to placebo, and adequate safety.66 For drug development programs, pharmaceutical companies plan fairly standard protocols, usually including patients with mild or mild to moderate Alzheimer disease and using standard outcome measures (such as the ADAS-Cog),67 standard ADLs, and global change and severity scales, regardless of whether efficacy is being tested over the short or long term.
Cholinesterase inhibitors, memantine, and small molecules that are considered to have symptomatic effects are tested over 6 months. Drugs in development that are considered as modifiers of illness progression have been tested generally in more mildly impaired patients and over 18 months.
The Amyloid Cascade Hypothesis
The amyloid cascade hypothesis is the most-researched conceptual framework for Alzheimer disease, and it markedly influences drug development.68 The gist of the amyloid cascade hypothesis is that Aβ deposition drives tau phosphorylation and tangle formation and neuron death.69 The pathologic and clinical expression of Alzheimer disease results from the increased production and/or impaired clearance of various Aβ peptides produced by variations in the processing of the neuronal membrane protein amyloid precursor protein (APP) and that one or several forms of Aβ drive pathogenesis.68,70 Although Aβ40 is the most prevalent Aβ peptide, Aβ42 and perhaps others have greater propensity to aggregate into oligomers, fibrils, and amyloid-containing deposits that are thought to be toxic. Amyloid plaques, protofibrils, oligomeric and monomeric forms of Aβ may each be responsible for the pathogenic expression of the illness. For example, Aβ oligomers may show synaptic toxicity effects, and plaque-derived Aβ fibrils may be proinflammatory and neurotoxic.
Anti-Amyloid Approaches
There are several Aβ-targeted experimental approaches, including modulation of Aβ production, inhibition of Aβ aggregation, enhancement of Aβ degradation, and use of passive and active immunization to raise antibodies that target and remove Aβ. Unfortunately, although several drugs have been demonstrated to be active at their intended targets, none has had significant clinical effects. In 2011 and 2012, two negative trials of γ-secretase inhibitors, semagacestat and avagacestat, and several negative trials of monoclonal antibodies, bapineuzumab and solanezumab were reported. Other Aβ antibodies, Aβ vaccines, γ-secretase modulators, and β-secretase inhibitors continue to be tested, as well as methods to modify or enhance the function of apolipoprotein E4 to increase brain clearance of Aβ.71 There are now several clinical examples demonstrating that reducing Aβ in the brain is possible but that decreasing production or reducing fibrils or plaques is not clearly associated with clinical improvement and could be associated with harm in some circumstances.72,73,74,75,76
Tau in Alzheimer Disease and Anti-Tau Approaches to Therapy
The neurofibrillary tangles that define and are characteristic of Alzheimer disease correlate with the clinical severity of dementia.77 The tangles represent the aggregation and accumulation of hyperphosphorylated forms of the microtubule-associated protein tau. Microtubule-bound soluble tau supports axonal transport. Hyperphosphorylation of tau might disrupt microtubules and axonal transport and lead to the formation of soluble tau aggregates and insoluble paired helical filaments, and could contribute to neurodegeneration.
A current hypothesis is that Alzheimer pathology starts as pretangles in proximal axons of the noradrenergic locus ceruleus that spreads by neuron-to-neuron and trans-synaptic transport of tau aggregates to the entorhinal cortex, hippocampus, and neocortex.78 This prionlike, protein-templating cross-neuronal propagation hypothesis suggests several interventions aimed at tau and also small-molecule interventions targeted toward midbrain monoaminergic systems.
Anti-tau therapeutic strategies. The range of therapeutic approaches under development include inhibiting tau kinases; enhancing phosphatase activity in an effort to enhance microtubule stability; blocking or inhibiting tau hyperphosphorylation, tau aggregates, and filament formation; and enhancing clearance of aggregates with drugs or antibodies. A few of these approaches have progressed from preclinical to phase 2 clinical trials.79,80
Tau pathology is not specific to Alzheimer disease, however, and occurs in several other disorders, including frontotemporal dementia, corticobasal degeneration, and progressive supranuclear palsy; any “anti-tau” approach could be considered for any of the tauopathies.
Other Approaches and Small Molecules
Most of the “small molecules” approaches target neurotransmitter receptors. They include nicotinic neuronal receptor agonists or modulators, drugs that are active at serotonin (5-hydroxytryptophan subtype receptors), histamine (H3 subtype receptor) antagonists, metabolic enhancers, phosphodiesterase type 4 (PDE 4) enzyme inhibitors, γ-aminobutyric acid A receptor modulators, monoamine oxidase type B enzyme inhibitors, and group 2 metabotropic glutamate receptor inhibitors. Some of these drugs have effects on Aβ generation or secretase activity in preclinical models. For example, alpha-7 neuronal nicotinic modulators may enhance cholinergic function and alter Aβ. H3 antagonists may act on H3 presynaptic autoreceptors to increase cholinergic and monoaminergic neurotransmitter release and are associated with enhanced cognitive function in preclinical models. Group 2 metabotropic glutamate inhibitors might combine cognitive effects, neurogenesis effects, and reduction of Aβ42.
CHALLENGES OF DEVELOPING EFFECTIVE TREATMENTS FOR ALZHEIMER DISEASE
The considerable challenges to developing effective treatments for Alzheimer disease include the uncertainty and lack of validated drug and molecular targets and the ability to conduct efficient clinical development programs. Establishing validated drug targets requires greater understanding of the pathogenic processes leading to illness. For example, the amyloid cascade may be well understood and potential drugs targeting various components are apparent. Yet it is possible that no intervention in this area will demonstrate efficacy because the amyloid cascade—as strongly associated with Alzheimer pathology as it is—may still not be a relevant therapeutic target.
Barriers to developing successful drugs for Alzheimer disease include current translational models from animal to man (ie, efficacy in mice does not predict efficacy in humans), the various clinical presentations of the illness, and the numerous potential pathologic targets for new drugs. It is possible that newer approaches to prevention trials, earlier interventions, interventions aimed at particular clinical and biological subtypes of Alzheimer disease, and smaller phase 2a trials to gain early signals of potential efficacy may be helpful. Although there are many explanations for the failures in trials, the most likely are that the experimental drugs do not work.
SUMMARY
The currently marketed medications for Alzheimer disease are the cholinesterase inhibitors and memantine. Vitamins, food supplements, and G biloba extract have not been shown to be effective. No pharmacologic approaches have been demonstrated to prevent or delay onset of MCI or Alzheimer dementia.
New drug development has mainly targeted the amyloid hypothesis and thus far has been unsuccessful. Other experimental approaches for which there are some indications for efficacy in early-phase development include small molecules that are active at nicotinic and serotoninergic receptor subsites. Several approaches to targeting tau protein are gaining momentum.
As there are many potential and no validated drug targets for Alzheimer disease, experimental drug development faces numerous challenges. Finally, drug development will be influenced by somewhat different diagnostic criteria for AD and for prodromal AD than used in the past; the use of diagnostic, prognostic, and predictive biomarkers; and the potential to make pharmacologic interventions before the onset of cognitive symptoms. Both substantial hope and challenges lie ahead.
KEY POINTS
The cholinergic hypothesis of memory impairment implies that cholinergic deficits are responsible for cognitive and behavioral changes in patients with dementia and age-related memory impairment, and that augmentation of central cholinergic function will improve cognitive function.
Historically, the targeted cholinergic treatment approaches have included using (1) acetylcholine precursors; (2) direct cholinergic agonists; and (3) cholinesterase inhibitors.
The most common adverse events due to cholinesterase inhibitors include nausea, diarrhea, vomiting, anorexia, and weight loss. Muscle cramps are common with donepezil.
Early cholinergic effects are frequently related to the initial dosing and titration of the medications.
Anorexia varies in incidence from 8% to 25% at higher doses of cholinesterase inhibitors compared with 3% to 10% in patients on placebo and may be dose related. The proportion of patients with weight loss in clinical trials ranges from 10% to 24% in patients taking higher doses compared to 2% to 10% of placebo-treated patients.
An analysis of Canadian medical and prescription records showed that patients on cholinesterase inhibitors were hospitalized for syncope nearly twice as often as people with dementia who did not receive these drugs.
Despite differences in mechanism of action and dosing levels, no evidence exists for efficacy differences between the three cholinesterase inhibitors. In a Cochrane review, the drugs are associated with an overall mean 2.4 points effect over placebo on the Alzheimer Disease Assessment Scale—Cognitive Subscale.
A 23-mg extended-release formulation of donepezil is intended to be used after a patient has been treated with 10 mg/d for at least 3 months and when the clinician is uncertain whether the patient is benefiting from the 10-mg dose.
Cholinesterase inhibitors are not indicated for mild cognitive impairment, yet their use may be common practice. Clinical trials of cholinesterase inhibitors in MCI were not positive on their primary outcomes and showed an excess in adverse events.
Memantine was approved by the US Food and Drug Administration in late 2003 for moderate to severe Alzheimer disease. The basis for approval was positive outcomes on two 6-month-long placebo-controlled clinical trials. In one trial cholinesterase inhibitors were not allowed, and in another, patients had been taking donepezil for at least 6 months (over 2 years on average). A third trial did not show significant effects.
Only one of three trials of memantine in mild to moderate Alzheimer disease showed significant improvement on the Alzheimer Disease Assessment Scale—Cognitive Subscale and global assessment. Memantine has not been approved by the US Food and Drug Administration for patients with mild Alzheimer disease.
A Cochrane review concluded that memantine had a small beneficial effect in moderate to severe Alzheimer disease and was well tolerated.
Adverse events with memantine are infrequent but can include headache, dizziness, confusion, somnolence, and infrequent hallucinations. In clinical trials, the frequency of gastrointestinal symptoms is less than placebo; diarrhea occurred half as often.
Extracts from leaves of the Ginkgo biloba, or maidenhair, tree are widely sold in the United States as food supplements for which health claims are not permitted. A specific standardized extract, EGb 761, is approved by the formularies of Germany and France.
G biloba extract is also used as a memory enhancer in people without Alzheimer disease; however, clinical trials in older and younger adults who do not have cognitive impairment show mixed results at best.
A Cochrane review that included 35 clinical trials reported inconsistent evidence that G biloba had clinically significant benefits for dementia or cognitive impairment.
A medical food is a food formulated for the dietary management of an illness that has distinctive nutritional requirements, and is intended to be used under medical supervision.
A formulation of medium-chain triglycerides is marketed as a medical food for Alzheimer disease in the United States. Another medical food, marketed in late 2012 in Europe and Brazil, is a combination of compounds including uridine, choline, omega-3 fatty acids, phospholipids, B vitamins, and antioxidants, intended to enhance synaptic function and neurotransmitters, presumably improving cognitive function. Controlled trials of these two medical foods have not been positive.
It is difficult to identify the individual patient who benefits from cholinesterase inhibitors or memantine because the outcome measures and mean changes on scale scores do not identify responders.
Discontinuation of cholinesterase inhibitors has been associated with worsening of cognition and confusion in some patients in trials. Yet worsening of behavior and confusion do not appear common when the drugs are stopped in clinical practice. In clinical practice, 19% to 23% of patients continued to take donepezil or rivastigmine for more than 1 year, and about one-third discontinued the drugs within 2 months.
In a withdrawal trial after maintenance treatment with donepezil for 2 to 3 years in severely impaired patients with Alzheimer disease, continuing donepezil was associated with better cognitive scores and activities of daily living. Many patients discontinued donepezil without difficulty, and only half of the patients assigned to continue donepezil actually continued treatment beyond the 1-year follow-up. Thus, the outcomes support decisions either to continue medication or to taper and discontinue it when physicians are uncertain of continuing benefit.
Only three of 14 trials showed significant effects for cholinesterase inhibitors improving behavior; none of these effects was large. Trivial effects were reported in the more mildly cognitively impaired patients, but no effect was reported in the more severely impaired.
Regulatory criteria for marketing symptomatic and disease-modifying therapies require demonstrating improvements in cognition and activities of daily living, overall improvements compared to placebo, and adequate safety.
The gist of the amyloid cascade hypothesis is that amyloid-β deposition drives tau phosphorylation, tangle formation, and neuron death.
There are several amyloid-β–targeted experimental approaches, including modulation of amyloid-β production, inhibition of amyloid-β aggregation, enhancement of amyloid-β degradation, and use of passive and active immunization to raise antibodies that target and remove amyloid-β.
The range of anti-tau therapeutic approaches under development include inhibiting tau kinases; enhancing phosphatase activity in an effort to enhance microtubule stability; blocking or inhibiting tau hyperphosphorylation, tau aggregates, and filament formation; and enhancing clearance of aggregates with drugs or antibodies.
Challenges to developing effective treatments for Alzheimer disease include the uncertainty and lack of validated drug and molecular targets and the ability to conduct efficient clinical development programs. Establishing validated drug targets requires greater understanding of the pathogenic processes leading to illness.
Footnotes
Relationship Disclosure: Dr Schneider has served on the scientific advisory boards of or has consulted for AC Immune; Accera, Inc; Allon Therapeutics, Inc; AstraZeneca; Baxter; Biogen Idec; Biotie Therapies; California Department of Justice; Elan Corporation; Eli Lilly Corporation; EnVivo Pharmaceuticals; Hoffmann-La Roche, Inc; Janssen Pharmaceuticals, Inc; Johnson & Johnson Services, Inc; Lundbeck, Merck & Co, Inc; Phloronol, Inc; Piramal Life Sciences; Takeda Pharmaceutical Company Limited; TauRx Ltd; Toyama Chemical Company, Ltd; and Zinfandel Pharmaceuticals, Inc. Dr Schneider and the University of Southern California have received research support from Baxter; Eli Lilly Corporation; Genentech, Inc; and TauRx Therapeutics. Dr Schneider has received grants from the NIH and the State of California.
Unlabeled Use of Products/Investigational Use Disclosure: Dr Schneider discusses the unlabeled use of donepezil, galantamine, rivastigmine, and memantine. Information on drugs is provided for general purposes only and not relied on for prescribing. Before prescribing any of the drugs discussed, the physician should be knowledgeable about the full prescribing information that can be obtained from the manufacturers.
REFERENCES
- 1.Bartus R,, Dean R,, Beer B, et al.. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982; 217 (4558): 408–414. [DOI] [PubMed] [Google Scholar]
- 2.Drachman DA,, Leavitt J. Human memory and the cholinergic system. A relationship to aging? Arch Neurol 1974;30(2):113–121. [DOI] [PubMed] [Google Scholar]
- 3.Schneider LS. Antidementia drugs. In: Sadock BJ,, Sadock MD,, Ruiz P, editors. Comprehensive textbook of psychiatry. 9th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2009: 4119–4130. [Google Scholar]
- 4.McArthur RA,, Gray J,, Schreiber R. Cognitive effects of muscarinic M1 functional agonists in non-human primates and clinical trials. Curr Opin Investig Drugs 2010; 11 (7): 740–760. [PubMed] [Google Scholar]
- 5.Birks J,, Harvey RJ. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev 2006; (1): CD001190. [DOI] [PubMed] [Google Scholar]
- 6.Courtney C,, Farrell D,, Gray R, et al.. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet 2004; 363 (9427): 2105–2115. [DOI] [PubMed] [Google Scholar]
- 7.Corey-Bloom J,, Anand R,, Veach J. A randomized trial evaluating the efficacy and safety of ENA 713 (rivastigmine tartrate), a new acetylcholinesterase inhibitor, in patients with mild to moderately severe Alzheimer’s disease. Int J Geriatr Psychopharmacol 1998; 1: 55–65. [Google Scholar]
- 8.Rosler M,, Anand R,, Cicin-Sain A, et al.. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ 1999; 318 (7184): 633–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winblad B,, Grossberg G,, Frölich L, et al.. IDEAL: a 6-month, double-blind, placebo-controlled study of the first skin patch for Alzheimer disease. Neurology 2007; 69 (4 suppl 1): S14–S22. [DOI] [PubMed] [Google Scholar]
- 10.Loy C,, Schneider L. Galantamine for Alzheimer’s disease and mild cognitive impairment. Cochrane Database Syst Rev 2006; (1): CD001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gill SS,, Anderson GM,, Fischer HD, et al.. Syncope and its consequences in patients with dementia receiving cholinesterase inhibitors: a population-based cohort study. Arch Intern Med 2009; 169 (9): 867–873. [DOI] [PubMed] [Google Scholar]
- 12.Farlow MR,, Salloway S,, Tariot PN, et al.. Effectiveness and tolerability of high-dose (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer’s disease: a 24-week, randomized, double-blind study. Clin Ther 2010; 32 (7): 1234–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev 2006; (1): CD005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitt FA,, Ashford W,, Ernesto C, et al.. The severe impairment battery: concurrent validity and the assessment of longitudinal change in Alzheimer’s disease. Alzheimer Dis Assoc Disord 1997; 11 (suppl 2): S51–S56. [PubMed] [Google Scholar]
- 15.Schneider LS,, Insel PS, Weiner MWAlzheimer’s Disease Neuroimaging Initiative. Treatment with cholinesterase inhibitors and memantine of patients in the Alzheimer’s Disease Neuroimaging Initiative. Arch Neurology 2011; 68 (1): 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winblad B,, Gauthier S,, Scinto L, et al.. Safety and efficacy of galantamine in subjects with mild cognitive impairment. Neurology 2008; 70 (22): 2024–2035. [DOI] [PubMed] [Google Scholar]
- 17.Feldman HH,, Ferris S,, Winblad B, et al.. Effect of rivastigmine on delay to diagnosis of Alzheimer’s disease from mild cognitive impairment: the InDDEx study. Lancet Neurol 2007; 6 (6): 501–512. [DOI] [PubMed] [Google Scholar]
- 18.Petersen RC,, Thomas RG,, Grundman M, et al.. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 2005; 352 (23): 2379–2388. [DOI] [PubMed] [Google Scholar]
- 19.Raschetti R,, Albanese E,, Vanacore N,, Maggini M. Cholinesterase inhibitors in mild cognitive impairment: a systematic review of randomised trials. PLoS Med 2007; 4 (11): e338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birks J,, Flicker L. Donepezil for mild cognitive impairment. Cochrane Database Syst Rev 2006; (3): CD006104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reisberg B,, Doody R,, Stoffler A, et al.. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 2003; 348 (14): 1333–1341. [DOI] [PubMed] [Google Scholar]
- 22.Tariot PN,, Farlow MR,, Grossberg GT, et al.. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 2004; 291 (3): 317–324. [DOI] [PubMed] [Google Scholar]
- 23.van Dyck CH,, Tariot PN,, Meyers B, et al.. A 24-week randomized, controlled trial of memantine in patients with moderate-to-severe Alzheimer disease. Alzheimer Dis Assoc Disord 2007; 21 (2): 136–143. [DOI] [PubMed] [Google Scholar]
- 24.Peskind ER,, Potkin SG,, Pomara N, et al.. Memantine treatment in mild to moderate Alzheimer disease: a 24-week randomized, controlled trial. Am J Geriatr Psychiatry 2006; 14 (8): 704–715. [DOI] [PubMed] [Google Scholar]
- 25.Bakchine S,, Loft H. Memantine treatment in patients with mild to moderate Alzheimer’s disease: results of a randomised, double-blind, placebo-controlled 6-month study. J Alzheimers Dis 2008; 13 (1): 97–107. [DOI] [PubMed] [Google Scholar]
- 26.Porsteinsson AP,, Grossberg GT,, Mintzer J, et al.. Memantine treatment in patients with mild to moderate Alzheimer’s disease already receiving a cholinesterase inhibitor: a randomized, double-blind, placebo-controlled trial. Curr Alzheimer Res 2008; 5 (1): 83–89. [DOI] [PubMed] [Google Scholar]
- 27.McShane R,, Areosa Sastre A,, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev 2006; (2): CD003154. [DOI] [PubMed] [Google Scholar]
- 28.Schneider LS,, Dagerman KS,, Higgins JPT,, McShane R. Lack of evidence for the efficacy of memantine in mild Alzheimer disease. Arch Neurol 2011; 68 (8): 991–998. [DOI] [PubMed] [Google Scholar]
- 29.Frankiewicz T,, Parsons CG. Memantine restores long term potentiation impaired by tonic N-methyl-D-aspartate (NMDA) receptor activation following reduction of Mg2+ in hippocampal slices. Neuropharmacology 1999; 38 (9): 1253–1259. [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Coria H,, Green KN,, Billings LM, et al.. Memantine improves cognition and reduces Alzheimer’s-like neuropathology in transgenic mice. Am J Pathol 2010; 176 (2): 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopez OL,, Becker JT,, Wahed AS, et al.. Long-term effects of the concomitant use of memantine with cholinesterase inhibition in Alzheimer disease. J Neurol Neurosurg Psychiatry 2009; 80 (6): 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rountree SD,, Chan W,, Pavlik VN, et al.. Persistent treatment with cholinesterase inhibitors and/or memantine slows clinical progression of Alzheimer disease. Alzheimers Res Ther 2009; 1 (2): 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reisberg B,, Doody R,, Stoffler A, et al.. A 24-week open-label extension study of memantine in moderate to severe Alzheimer disease. Arch Neurol 2006; 63 (1): 49–54. [DOI] [PubMed] [Google Scholar]
- 34.Schneider LS. Open-label extension studies and misinformation. Arch Neurol 2006; 63 (7): 1036; author reply 1036–1037. [DOI] [PubMed] [Google Scholar]
- 35.Alvarez XA,, Cacabelos R,, Sampedro C, et al.. Combination treatment in Alzheimer’s disease: results of a randomized, controlled trial with cerebrolysin and donepezil. Curr Alzheimer Res 2011; 8 (5): 583–591. [DOI] [PubMed] [Google Scholar]
- 36.Schneider LS. Ginkgo biloba extract and preventing Alzheimer disease. JAMA 2008; 300 (19): 2306–2308. [DOI] [PubMed] [Google Scholar]
- 37.Birks J,, Grimley Evans J. Ginkgo biloba for cognitive impairment and dementia. Cochrane Database Syst Rev 2009(1):CD003120. [DOI] [PubMed] [Google Scholar]
- 38.Schneider LS,, DeKosky ST,, Farlow MR, et al.. A randomized, double-blind, placebo-controlled trial of two doses of Ginkgo biloba extract in dementia of the Alzheimer’s type. Curr Alzheimer Res 2005; 2 (5): 541–551. [DOI] [PubMed] [Google Scholar]
- 39.McCarney R,, Fisher P,, Iliffe S, et al.. Ginkgo biloba for mild to moderate dementia in a community setting: a pragmatic, randomised, parallel-group, double-blind, placebo-controlled trial. Int J Geriatr Psychiatry 2008; 23 (12): 1222–1230. [DOI] [PubMed] [Google Scholar]
- 40.Institute for Quality and Efficiency in Health Care. Alzheimer’s disease: only a few therapies provide proof of benefit to patients. www.iqwig.de/index.948.en.html?random=667c6a. Updated April 30, 2010. Accessed January 3, 2013.
- 41.DeKosky ST,, Williamson JD,, Fitzpatrick AL, et al.. Ginkgo biloba for prevention of dementia: a randomized controlled trial. JAMA 2008; 300 (19): 2253–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vellas B,, Andrieu S,, Ousset PJ, et al.. The GuidAge study: Methodological issues. A 5-year double-blind randomized trial of the efficacy of EGb 761(R) for prevention of Alzheimer disease in patients over 70 with a memory complaint. Neurology 2006; 67 (9 suppl 3): S6–S11. [DOI] [PubMed] [Google Scholar]
- 43.Dodge HH,, Zitzelberger T,, Oken BS, et al.. A randomized placebo-controlled trial of Ginkgo biloba for the prevention of cognitive decline. Neurology 2008; 70 (19 pt 2): 1809–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.U.S. Food and Drug Administration. Guidance for industry: frequently asked questions about medical foods. www.fda.gov/Food/GuidanceComplianceRegulatoryInformation/GuidanceDocuments/MedicalFoods/ucm054048.htm. Updated July 27, 2011. Accessed February 8, 2013.
- 45.Henderson ST,, Vogel JL,, Barr LJ, et al.. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond) 2009; 6 (1): 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheltens P,, Twisk JWR,, Blesa R, et al.. Efficacy of Souvenaid in mild Alzheimer’s disease: results from a randomized, controlled trial. J Alzheimers Dis 2012; 31 (1): 225–236. [DOI] [PubMed] [Google Scholar]
- 47.Raina P,, Santaguida P,, Ismaila A, et al.. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med 2008; 148 (5): 379–397. [DOI] [PubMed] [Google Scholar]
- 48.Takeda A,, Loveman E,, Clegg A, et al.. A systematic review of the clinical effectiveness of donepezil, rivastigmine and galantamine on cognition, quality of life and adverse events in Alzheimer’s disease. Int J Geriatr Psychiatry 2006; 21 (1): 17–28. [DOI] [PubMed] [Google Scholar]
- 49.Loveman E,, Green C,, Kirby J, et al.. The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer’s disease. Health Technol Assess 2006; 10 (1): iii–iv, ix,–xi, 1–160. [DOI] [PubMed] [Google Scholar]
- 50.Watson SM. NDA # 20-690 Aricept (donepezil hydrochloride) tablets. 2010 warning letters and untitled letters to pharmaceutical companies. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM201238.pdf. Dated February 4, 2010. Accessed January 3, 2013.
- 51.Abrams T. NDA 20-823/21-025: Exelon (rivastigmine tartrate) capsules and oral solution: MACMIS ID #14943. Warning letter. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/ucm054180.pdf. Accessed February 12, 2012.
- 52.Lindner MD,, McArthur RA,, Deadwyler S, et al.. Development, optimization and use of preclinical behavioral models to maximize the productivity of drug discovery for Alzheimer’s disease. In: McArthur RA,, Borsini F, eds. Animal and translational models for CNS drug discovery. San Diego, CA: Academic Press, 2008: 93–157. [Google Scholar]
- 53.Rockwood K,, Fay S,, Song X, et al.. Attainment of treatment goals by people with Alzheimer’s disease receiving galantamine: a randomized controlled trial. CMAJ 2006; 174 (8): 1099–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Atri A,, Shaughnessy L,, Locascio JJ, et al.. Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis Assoc Disord 2008; 22 (3): 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geldmacher DS,, Provenzano G,, McRae T, et al.. Donepezil is associated with delayed nursing home placement in patients with Alzheimer’s disease. J Am Geriatr Soc 2003; 51 (7): 937–944. [DOI] [PubMed] [Google Scholar]
- 56.Schneider LS,, Qizilbash N. Delay in nursing home placement with donepezil. J Am Geriatr Soc 2004; 52 (6): 1024–1026. [DOI] [PubMed] [Google Scholar]
- 57.Sona A,, Zhang P,, Ames D, et al.. Predictors of rapid cognitive decline in Alzheimer’s disease: results from the Australian imaging, biomarkers and lifestyle (AIBL) study of ageing. Int Psychogeriatr 2011; 24 (2): 1–8. [DOI] [PubMed] [Google Scholar]
- 58.Holmes C,, Wilkinson D,, Dean C, et al.. The efficacy of donepezil in the treatment of neuropsychiatric symptoms in Alzheimer disease. Neurology 2004; 63 (2): 214–219. [DOI] [PubMed] [Google Scholar]
- 59.Mauskopf JA,, Paramore C,, Lee WC,, Snyder EH. Drug persistency patterns for patients treated with rivastigmine or donepezil in usual care settings. J Manag Care Pharm 2005; 11 (3): 231–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Howard R,, McShane R,, Lindesay J, et al.. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med 2012; 366 (10): 893–903. [DOI] [PubMed] [Google Scholar]
- 61.Schneider LS. Discontinuing donepezil or starting memantine for Alzheimer’s disease. N Engl J Med 2012; 366 (10): 957–959. [DOI] [PubMed] [Google Scholar]
- 62.Rodda J,, Morgan S,, Walker Z. Are cholinesterase inhibitors effective in the management of the behavioral and psychological symptoms of dementia in Alzheimer’s disease? A systematic review of randomized, placebo-controlled trials of donepezil, rivastigmine and galantamine. Int Psychogeriatr 2009; 21 (5): 813–824. [DOI] [PubMed] [Google Scholar]
- 63.Campbell N,, Ayub A,, Boustani MA, et al.. Impact of cholinesterase inhibitors on behavioral and psychological symptoms of Alzheimer’s disease: a meta-analysis. Clin Interv Aging 2008; 3 (4): 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Howard RJ,, Juszczak E,, Ballard CG, et al.. Donepezil for the treatment of agitation in Alzheimer’s disease. N Engl J Med 2007; 357 (14): 1382–1392. [DOI] [PubMed] [Google Scholar]
- 65.Fox C,, Crugel M,, Maidment I, et al.. Efficacy of memantine for agitation in Alzheimer’s dementia: a randomised double-blind placebo controlled trial. PLoS One 2012; 7 (5): e35185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneider LS. Issues in design and conduct of clinical trials for cognitive-enhancing drugs. In: McArthur RA,, Borsini F, eds. Animal and translational models for CNS drug discovery. San Diego, CA: Academic Press, 2008: 21–76. [Google Scholar]
- 67.Mohs RC,, Knopman D,, Petersen RC, et al.. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer’s Disease Assessment Scale that broaden its scope. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997; 11 (suppl 2): S13–S21. [PubMed] [Google Scholar]
- 68.Golde TE,, Schneider LS,, Koo EH. Anti-Aβ therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron 2011; 69 (2): 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hardy J,, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 1991;12(10):383–388. [DOI] [PubMed] [Google Scholar]
- 70.Hardy J,, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002; 297 (5580): 353–356. [DOI] [PubMed] [Google Scholar]
- 71.Castellano JM,, Kim J,, Stewart FR, et al.. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med 2011; 3 (89): 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Holmes C,, Boche D,, Wilkinson D, et al.. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008; 372 (9634): 216–223. [DOI] [PubMed] [Google Scholar]
- 73.Rinne JO,, Brooks DJ,, Rossor MN, et al.. 11C-PiB PET assessment of change in fibrillar amyloid-[beta] load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol 2010; 9 (4): 363–372. [DOI] [PubMed] [Google Scholar]
- 74.Gilman S,, Koller M,, Black RS, et al.. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005; 64 (9): 1553–1562. [DOI] [PubMed] [Google Scholar]
- 75.Salloway S,, Sperling R,, Gilman S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 2009;73(24):2061–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ostrowitzki S,, Deptula D,, Thurfjell L, et al.. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol 2011; 69 (2): 198–207. [DOI] [PubMed] [Google Scholar]
- 77.Blessed G,, Tomlinson BE,, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry 1968; 114 (512): 797–811. [DOI] [PubMed] [Google Scholar]
- 78.Braak H,, Del Tredici K. Alzheimer’s pathogenesis: is there neuron-to-neuron propagation? Acta Neuropathol 2011; 121 (5): 589–595. [DOI] [PubMed] [Google Scholar]
- 79.Boutajangout A,, Sigurdsson EM,, Krishnamurthy PK. Tau as a therapeutic target for Alzheimer’s disease. Curr Alzheimer Res 2011; 8 (6): 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fuentes P,, Catalan J. A clinical perspective: anti taus treatment in Alzheimer’s disease. Curr Alzheimer Res 2011; 8 (6): 686–688. [DOI] [PubMed] [Google Scholar]