

Abstract
Purpose of Review:
This article provides an update on the evaluation and treatment of neurosarcoidosis.
Recent Findings:
The broad range of clinical manifestations of neurosarcoidosis has recently expanded to include painful small fiber neuropathy. Although definitive diagnosis remains a challenge, fluorodeoxyglucose positron emission tomographic (FDG-PET) scan and high-resolution CT allow for improved detection of systemic sarcoidosis. In addition, endobronchial ultrasound-guided transbronchial needle aspiration provides a less invasive means of tissue confirmation of systemic sarcoidosis than mediastinoscopy. Although not standardized, treatment strategies for neurosarcoidosis now commonly include tumor necrosis factor-α antagonists in combination with corticosteroids and other cytotoxic agents for patients with severe disease.
Summary:
Advances in the diagnosis and management of neurosarcoidosis may benefit the patient and clinician faced with this multifaceted disease.
INTRODUCTION
Originally termed sarcoid in the late 19th century because the skin manifestations resembled sarcoma, sarcoidosis is now known as an immune-mediated multiorgan disorder of unknown cause that can affect any portion of the nervous system.1 Although the disease typically involves the lungs, skin, and lymph nodes, the most common neurologic manifestations are cranial neuropathies (especially involving the facial nerve) and aseptic meningitis.
Neurosarcoidosis is pathologically characterized by the presence of non-necrotizing granulomas composed of compact clusters of activated macrophages and other epithelioid cells surrounded by T lymphocytes (Figure 2-1). Granulomatous inflammation of the leptomeninges extends to the brain and spinal cord via Virchow-Robin spaces, which communicate with cervical lymph nodes that are thought to provide lymphatic drainage for the brain.2,3 In combination with CSF dynamics, the prominent size of these perivascular spaces at the base of the brain may partially account for the preferential involvement of the basal meninges, cranial nerves, and hypothalamic-pituitary structures in CNS sarcoidosis.4 The release of proinflammatory cytokines, such as tumor necrosis factor-α (TNFα), also plays a role in the pathogenesis of inflammation within the nervous system.5
Figure 2-1.
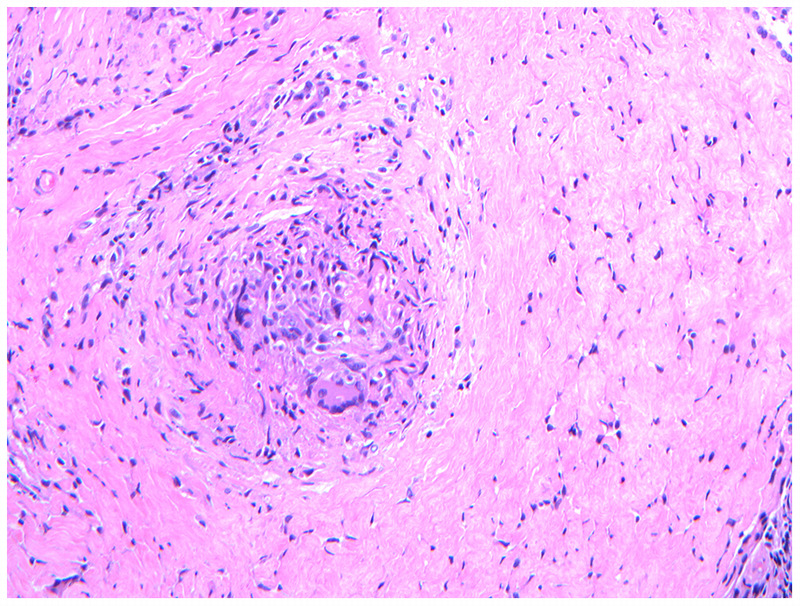
Noncaseating granuloma. Biopsy obtained from a dural-based frontal mass in a patient with neurosarcoidosis demonstrates a noncaseating granuloma with macrophages and multinucleated giant cells in the center.
Epidemiology
As reported by several retrospective case series, neurologic complications are seen in 5% to 10% of patients with sarcoidosis and are the presenting symptom in 50% to 70% of these cases.6,7,8,9,10 Rates of asymptomatic involvement may be higher; evidence of neurosarcoidosis has been found at autopsy in up to 15% of previously undiagnosed patients with systemic disease.11 A recent prospective study of 123 patients with sarcoidosis identified neurologic manifestations in 32 cases (26%).11 However, this may represent an overestimate, as peripheral nerve involvement in the form of a stocking-glove polyneuropathy or mononeuropathy (which included ulnar neuropathy and radiculopathy) in 23 cases was attributed to sarcoidosis in the absence of peripheral nerve histology. Isolated neurologic involvement with no evidence of systemic disease has been reported in 10% to 17% of neurosarcoidosis cases.8,12,13,14
Although the disease is more common in African Americans and people of Scandinavian descent, sarcoidosis may affect all ethnic groups with an estimated prevalence of 1 to 40 per 100,000 worldwide.13,15,16 Peak incidence is in the third to fifth decades for most individuals, although African American patients tend to present late in the fourth decade.16 Women also appear to have a later age of onset and are more frequently affected by neurosarcoidosis and systemic disease than men.15,16 Various infections (eg, mycobacteria and Propionibacterium acnes), genetic factors (eg, annexin A11 gene polymorphism involved in apoptosis), and environmental exposures (eg, pesticides, mold, and metallic particles) have been associated with the development of sarcoidosis, but no definitive causal link has been established.17,18,19
CLINICAL MANIFESTATIONS
The deposition of granulomatous inflammation throughout various areas of the central and peripheral nervous systems results in a vast array of clinical presentations.
Cranial Neuropathy
Facial nerve palsy is the most common neurologic manifestation reported in most case series and is bilateral in about 30% to 40% of patients.6,7,8,12 Symptoms are typically self-limited and resolve within a few weeks with little to no residual deficit, although weakness may recur. Optic neuropathy, which may also present bilaterally, is also common and was the most frequent neurologic finding in at least two case series; however, this may reflect referral bias because one series was derived from a multiple sclerosis population.9,20 Outcomes are mixed, with some series reporting improvement with treatment,6,11 while patients in others were left with marked visual impairment.13,20 Vestibulocochlear nerve involvement results in severe and often acute hearing loss that again may be unilateral or bilateral. Patients typically recover at least partially with corticosteroid treatment, although significant chronic hearing loss is not unusual with bilateral involvement. Less commonly, the trigeminal nerve may be affected, resulting in facial numbness or trigeminal neuralgia–like symptoms (Figure 2-2). Sarcoidosis may affect cranial nerves at the level of the nucleus or anywhere along the nerve tract. Inflammation of the leptomeninges or subarachnoid space may also result in isolated or multiple cranial nerve involvement.
Figure 2-2.
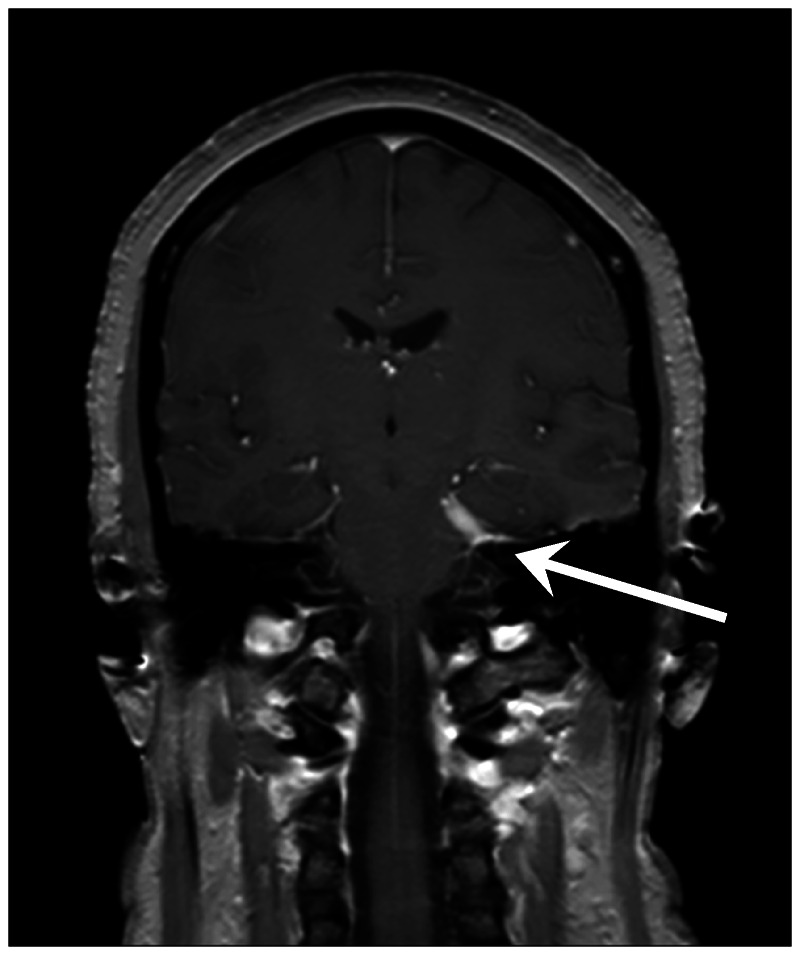
Trigeminal neuropathy in neurosarcoidosis. Dural-based mass along the left cavernous sinus is seen effacing the trigeminal cave (arrow), resulting in facial numbness and pain in a patient with neurosarcoidosis.
Meningitis
Leptomeningeal inflammation is frequently seen in neurosarcoidosis and was the most common neurologic finding in one series.21 Patients tend to present with a subacute or chronic meningitis predominantly involving the basal regions. Headache is a frequent concern and may be accompanied by other neurologic symptoms. In severe cases, hydrocephalus (both communicating and noncommunicating) may develop as a result of impaired arachnoid villi absorption or ependymal inflammation, as illustrated by Case 2-1. Cranial nerve palsies and hypothalamic-pituitary dysfunction may also occur with severe basal inflammation.
Case 2-1
A 32-year-old man presented with a 2-month history of progressively worsening headaches, double vision, nausea, and vomiting. Initial unenhanced MRI of the brain showed no abnormalities (Figure 2-3A). He subsequently developed generalized weakness and fell several times, prompting a gadolinium-enhanced MRI, which demonstrated leptomeningeal enhancement throughout the infratentorium and along the cranial nerves as well as ependymal enhancement of the third and fourth ventricles (Figure 2-3B). Serum angiotensin-converting enzyme (ACE) level was elevated, and CSF analysis showed a lymphocytic pleocytosis, reduced glucose of 23 mg/dL, and an elevated protein of 245 mg/dL. Although chest CT showed no abnormalities, follow-up fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed diffuse uptake in the hilar and mediastinal lymph nodes consistent with lymphadenopathy. The patient was diagnosed with probable neurosarcoidosis (he refused lymph node biopsy) and was started on prednisone and methotrexate. He improved almost immediately, but during the prednisone taper 6 months later, the patient developed acute confusion, difficulty walking, and polyuria. MRI showed a communicating hydrocephalus with basal meningeal inflammation (Figure 2-3C and D). A water deprivation test was positive for diabetes insipidus. With shunt placement and the addition of infliximab and desmopressin (methotrexate was discontinued), the patient returned to baseline mental status with residual gait imbalance and polyuria.
Figure 2-3.
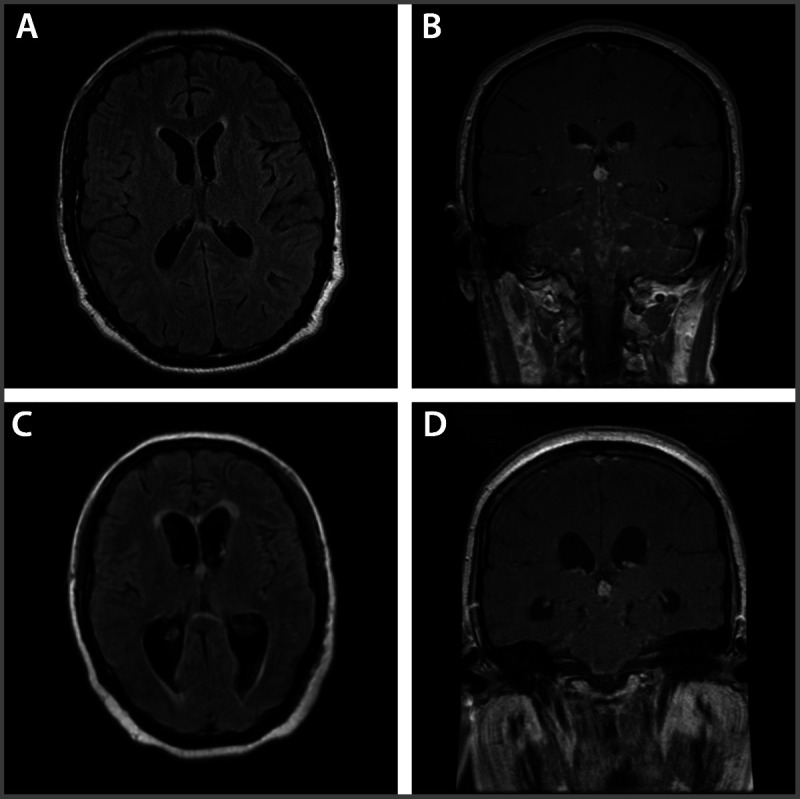
Progression of neurosarcoidosis in a patient with severe CNS involvement. A, Initial unenhanced MRI of the brain shows no abnormalities. B, Gadolinium-enhanced MRI demonstrating leptomeningeal enhancement throughout the infratentorium and along the cranial nerves, as well as ependymal enhancement of the third and fourth ventricles. C, D, MRI shows a communicating hydrocephalus with basal meningeal inflammation after the patient was tapered down on the steroid dose.
Comment. Given the nonspecific clinical presentation, as is often the case with neurosarcoidosis, optimized imaging studies are crucial for diagnosis. The initial normal MRI in the setting of a rapidly progressive neurologic decline underscores the importance of a gadolinium-enhanced study, especially for those with meningeal involvement. Similarly, the FDG-PET scan was able to detect evidence of systemic disease in the absence of CT abnormalities.
In this patient, like many others with moderate to severe CNS sarcoidosis, corticosteroids and methotrexate were insufficient for immunosuppression, which was evident during the prednisone taper.
The uncontrolled basal inflammation involved the hypothalamus and third and fourth ventricles, leading to diabetes insipidus and communicating hydrocephalus, respectively. Fortunately, the patient responded to more aggressive treatment with a tumor necrosis factor-α (TNFα) antagonist and shunt placement, although he may be left with some degree of diabetes insipidus and gait dysfunction indefinitely. The patient will need to be followed carefully as the underlying ependymal and CSF inflammation can lead to shunt obstruction. In addition, he will be at higher risk of infection given the presence of a foreign object (shunt) in the CNS, especially in the setting of chronic immunosuppression.
Myelopathy
Sarcoidosis of the spinal cord most often affects the cervicothoracic regions and usually involves both the parenchyma and leptomeninges, although isolated cauda equina involvement can also occur.22 Patients typically present with difficulty walking and subjective leg weakness, but many may demonstrate normal strength on examination. Likewise, the reported sensory loss is often vague and diffuse, with inconsistent or normal sensory findings that may reflect the patchy and noncontiguous pattern of cord and leptomeningeal involvement seen on MRI. Bowel/bladder disturbance and sexual dysfunction are common. Although MRI lesions usually improve or resolve with treatment, most patients are left with significant disability, often with spinal cord atrophy.
Intracranial
Headache, seizures, and cognitive dysfunction are common manifestations of parenchymal lesions. Focal neurologic deficits may also arise depending on the location of the lesion. While the leptomeninges are often involved, parenchymal brain lesions may also occur in isolation.
Neuroendocrine
Hypothalamic-pituitary dysfunction occurs as a result of direct structural infiltration or basal meningeal involvement. Hypothalamic hypothyroidism, hypogonadotropism, and hyperprolactinemia are commonly seen endocrine abnormalities.23 Syndrome of inappropriate release of antidiuretic hormone secretion (SIADH), diabetes insipidus, and growth hormone deficiency have also been reported. Most patients are left with permanent endocrine dysfunction requiring lifelong hormonal therapy despite resolution of radiographic abnormalities.
Headache and Seizures
Acute or chronic meningitis, hydrocephalus, mass lesions, and opportunistic infections (due to chronic immunosuppression) may result in headaches or seizures, although both may also occur in the absence of a structural lesion. Other causes of headache in sarcoidosis include trigeminal neuropathy and exacerbation of coexisting chronic migraines.
Neurovascular
Stroke is a rare complication of neurosarcoidosis. Leptomeningeal inflammation, especially when severe, may penetrate the endothelial walls, resulting in large or small artery occlusion and infarct, as demonstrated in Figure 2-4.24 Other causes of stroke include compression of an intracranial artery by a sarcoidosis-associated mass lesion, CNS vasculitis, and cardiogenic emboli due to cardiac arrhythmias or a congestive cardiomyopathy due to cardiac granulomatous inflammation. Cerebral venous sinus thrombosis may also occur in association with the inflammatory process.
Figure 2-4.

Acute stroke due to neurosarcoidosis. A, Nodular leptomeningeal inflammation around the brainstem with patchy gadolinium enhancement is seen throughout the cervicothoracic cord (arrow) in a patient with severe neurosarcoidosis. B, Linear restricted diffusion is seen centrally within the cervicomedullary junction (arrow) consistent with acute infarct.
Neuromuscular
Large peripheral nerve fiber involvement is rare and mainly occurs via direct granulomatous infiltration or compression. Patients most commonly present with a painful non–length-dependent polyneuropathy or polyradiculoneuropathy. Other presentations include mononeuritis multiplex, entrapment syndromes, and a subacute demyelinating polyneuropathy similar to that seen with Guillain-Barré syndrome.25 Peripheral nerve vasculitis may also occur, resulting in local nerve ischemia and axon loss.26
More recently, small fiber neuropathy symptoms have been reported in up to 40% of patients with sarcoidosis.27 Sarcoidosis-associated small fiber neuropathy most often presents with somatic symptoms manifested as pain and paresthesia either in a distal stocking-glove distribution or a migratory non–length-dependent pattern involving the face, trunk, and proximal limbs. Autonomic manifestations include orthostatic intolerance, gastrointestinal dysmotility, and sweating abnormalities. Symptoms may be disabling even when the systemic disease is otherwise well controlled. Because the pathogenesis of sarcoidosis-associated small fiber neuropathy is thought to be systemic cytokine-mediated rather than a direct effect of granulomas, as it is with large fiber neuropathy, this manifestation cannot be classified as true neurosarcoidosis according to existing criteria. Rather, it is consistent with “paraneurosarcoidosis,” a term that was recently introduced in the updated version of A Case-Control Etiologic Sarcoidosis Study (ACCESS), which defines organ involvement in sarcoidosis.28 Of note, care must be taken to exclude all other potential etiologies of small fiber neuropathy (eg, diabetes mellitus, vitamin deficiency) before attributing it to sarcoidosis.
Granulomatous involvement of muscle has been found postmortem in up to 50% of patients with sarcoidosis, although findings are subclinical in most individuals.29 In symptomatic patients, the clinical presentation may range from acute myositis to gradually progressive weakness. Nodular myopathy characterized by palpable intramuscular nodules may also be seen and is more common in men.
Neuropsychiatric
While cognitive impairment may be seen with meningitis or an intracranial mass lesion, many patients report various neuropsychiatric symptoms, such as memory loss, fatigue, mood disturbances, and other behavioral changes, without evidence of a CNS lesion. These nonspecific symptoms, which are often referred to as “sarcoidosis fog,” tend to be multifactorial in nature as a result of the underlying systemic disease, medication side effects, and depression. Sleep disturbances, such as sleep apnea syndrome and primary hypersomnia, may also contribute to the sarcoidosis fog.
DIAGNOSIS
The challenge of treating patients with suspected neurosarcoidosis is to establish a firm diagnosis in those with no prior history of systemic disease; this requires histologic confirmation either directly from the affected nervous system tissue or preferably another more accessible organ that is also involved. Reliance on the neurologic presentation alone or corticosteroid responsiveness in the absence of neural tissue confirmation or supportive findings of systemic sarcoidosis may result in the misdiagnosis and delayed treatment of clinical or radiographic mimics, such as lymphoma, cryptococcosis, and tuberculosis.30
In patients with known systemic sarcoidosis and typical neurologic manifestations, neural tissue confirmation should still be considered if readily accessible (eg, muscle biopsy), but it is less critical for diagnosis and management. The focus of the evaluation in these patients is to confirm evidence of objective neurologic involvement and to exclude other potential etiologies (Table 2-1).
Table 2-1.
Differential Diagnosis of Neurosarcoidosis

The most widely accepted diagnostic criteria, by Zajicek and colleagues,10 classify neurosarcoidosis as definite when there is direct neural tissue confirmation, probable when neurologic inflammation is present with evidence of systemic sarcoidosis, and possible when the clinical presentation is typical but no other criteria have been met except for the exclusion of other potential etiologies (Table 2-2). Subsequent modifications by other authors include exclusion of the Kveim test (which is not approved by the US Food and Drug Administration and is no longer used in the United States because of the risk of infection) and inclusion of high-resolution CT and a bronchoalveolar lavage CD4/CD8 ratio greater than 3.5. The use of these diagnostic criteria has significant therapeutic implications because more aggressive treatment options may be considered for patients with definite and probable diagnoses.31,32
Table 2-2.
Diagnostic Criteria for Patients With a Clinical Presentation Suggestive of Neurosarcoidosis in Which All Other Diagnoses Have Been Excludeda

In patients with CNS findings, the initial diagnostic modality of choice is a gadolinium-enhanced MRI. Neurosarcoidosis is often referred to as “the great mimicker” on MRI because its radiographic appearance can range from an intraparenchymal tumorlike mass (Figure 2-5) to nonspecific periventricular white matter lesions (Figure 2-6). Meningeal involvement is common and may present in several forms, including focal dural thickening similar to a meningioma, diffuse pachymeningeal involvement as can be seen with intracranial hypotension, and leptomeningeal disease that may be basal or widespread (Figure 2-7). With severe meningeal inflammation, a communicating or noncommunicating hydrocephalus may develop, as demonstrated in Case 2-1.
Figure 2-5.
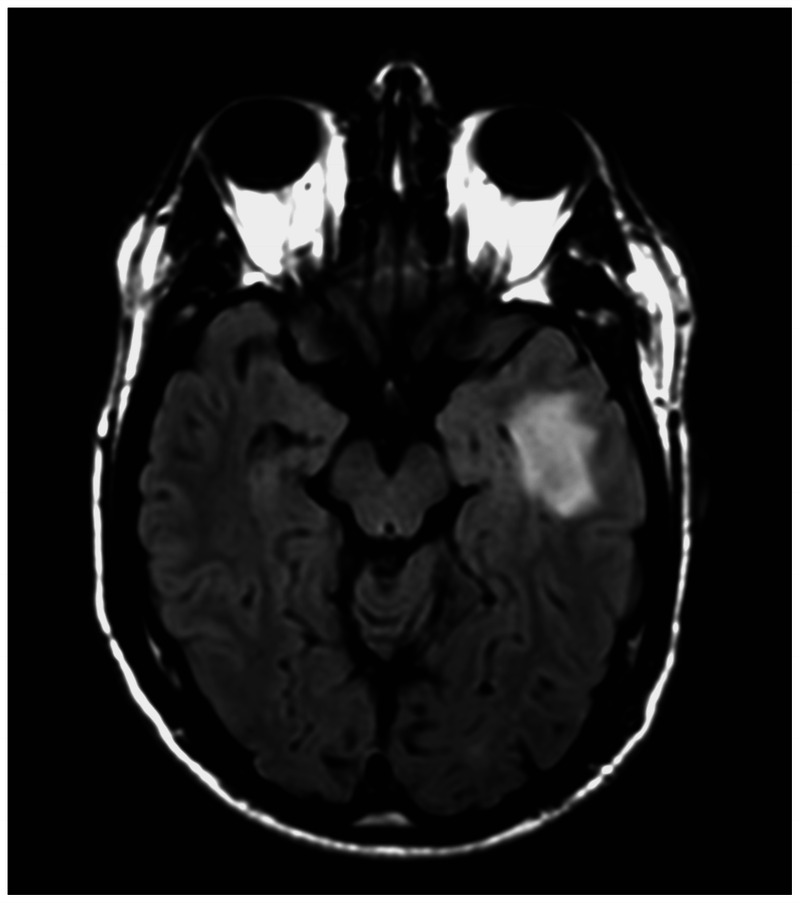
Heterogeneous left temporal mass with marked surrounding edema in a patient with multisystemic sarcoidosis.
Figure 2-6.
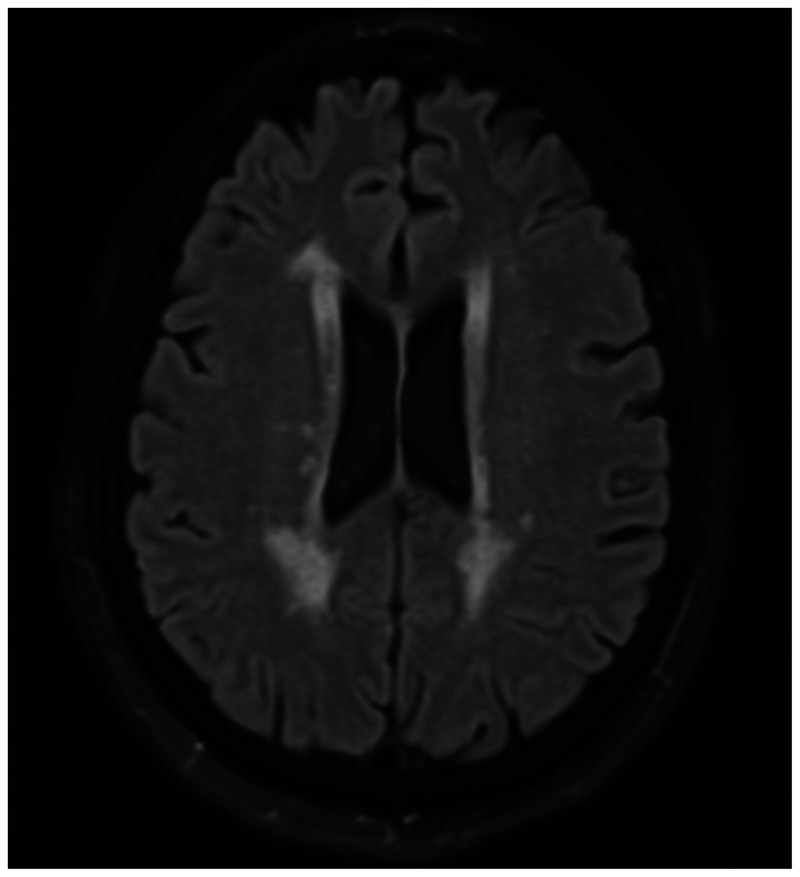
Nonspecific periventricular white matter changes are seen in a patient with systemic sarcoidosis and no risk factors for small vessel ischemic disease.
Figure 2-7.
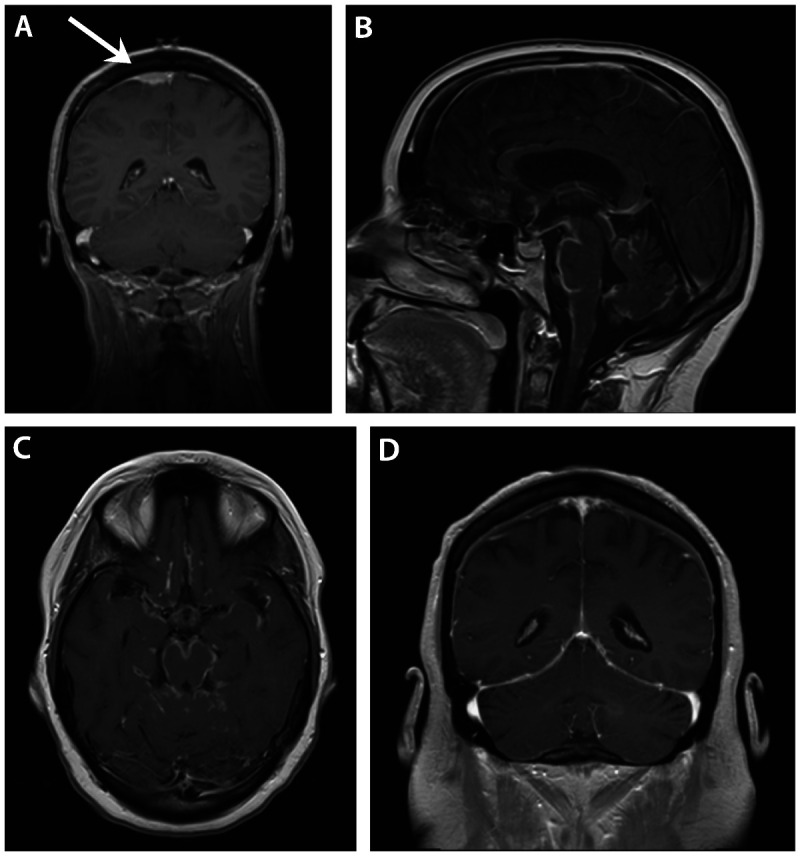
Meningeal involvement in neurosarcoidosis. A, Dural-based extraaxial mass overlying the dorsal right frontal lobe (arrow) with multiple nonnecrotizing granulomas on biopsy in a patient with neurosarcoidosis. B, Sagittal gadolinium-enhanced image of severe neurosarcoidosis affecting the basal meninges. C, Axial view of the same patient in Panel B. D, Diffuse pachymeningeal enhancement in a patient with neurosarcoidosis presenting with confusion and hearing loss.
Specific involvement of the neuroendocrine structures may be seen as thickening and enhancement of the pituitary stalk and/or hypothalamus (Figure 2-8).23 In addition to masslike presentations, parenchymal brain and spinal cord lesions may appear patchy or densely infiltrative, often with nodular meningeal thickening (Figure 2-9). In one spinal cord sarcoidosis series, intramedullary lesions were found to extend three or more vertebral levels in 17 of 22 patients (78%), similar to that seen in neuromyelitis optica.22 Gadolinium enhancement of CNS lesions is often seen with active disease, although acute lesions may sometimes be nonenhancing. Finally, gadolinium-enhanced MRI of the musculoskeletal system has also been shown to help detect subclinical muscle involvement, which may be nodular in appearance.33
Figure 2-8.
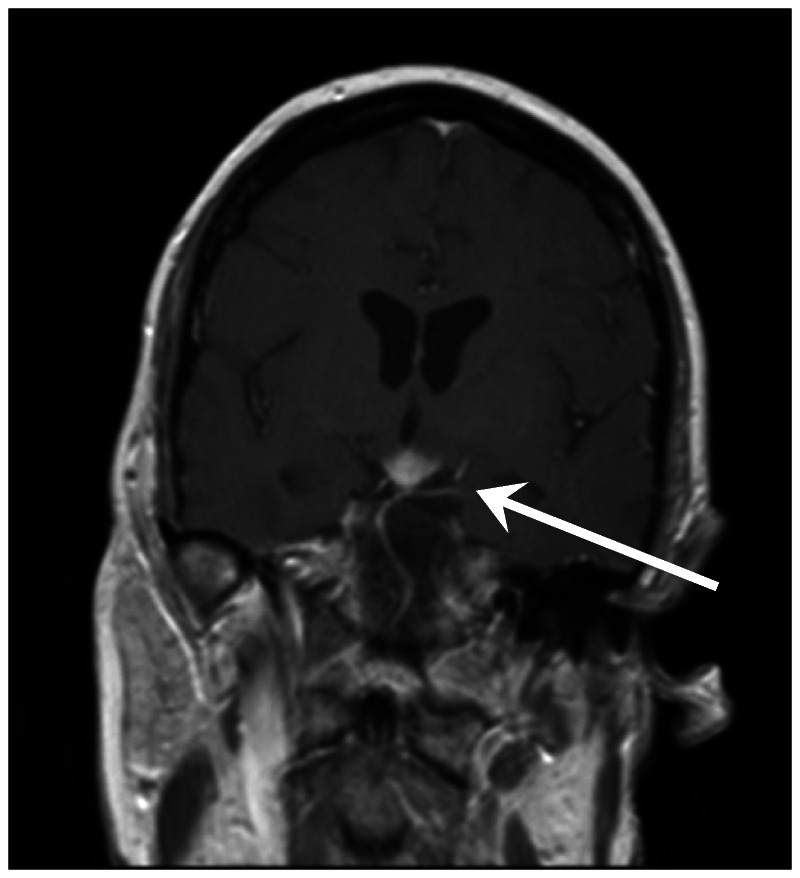
Gadolinium enhancement of the hypothalamus in a patient with diabetes insipidus due to neurosarcoidosis.
Figure 2-9.
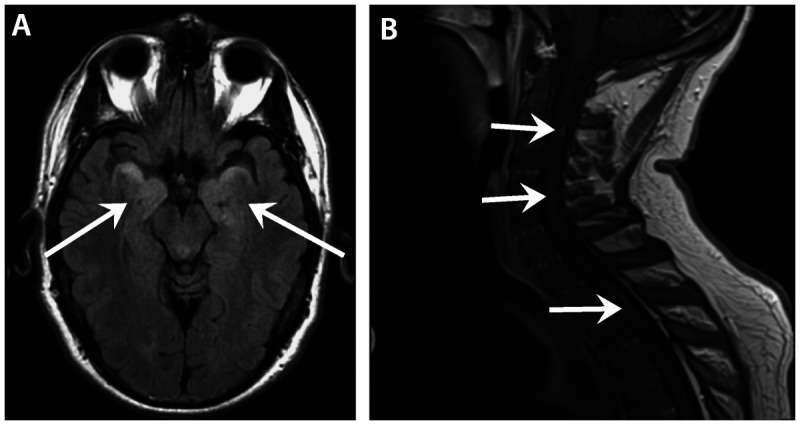
Intraparenchymal brain and spinal cord manifestations of neurosarcoidosis. A, Patchy increased signal intensity is seen on fluid-attenuated inversion recovery (FLAIR) imaging of bilateral mesial temporal lobes (arrows) in a patient with neurosarcoidosis presenting with memory loss and confusion. B, Noncontiguous, patchy gadolinium enhancement is seen throughout the cervicothoracic cord (arrows), presenting as diffuse sensory loss and subjective weakness in a patient with neurosarcoidosis.
CSF analysis is another diagnostic tool that can provide evidence of inflammation and exclude other potential etiologies. While the CSF abnormalities are nonspecific for CNS sarcoidosis, most affected patients will have at least one of the following: elevated protein (most common), mononuclear pleocytosis, or the presence of oligoclonal bands or increased IgG indices. Hypoglycorrhachia (reduced CSF glucose) can also be seen. In one-third of patients, the CSF may be normal even in the presence of gadolinium-enhancing MRI lesions or a diagnostic brain biopsy.7,9 Other important CSF studies include cytology with flow cytometry to evaluate for malignancy, and cultures, polymerase chain reaction assays, and antibody titers to exclude a variety of infections. Despite being a product of granulomas, CSF angiotensin-converting enzyme (ACE) is an insensitive marker of neurosarcoidosis that is elevated in only 25% to 35% of patients and does not appear to correlate with the degree of clinical activity.12,14,34,35
EMG or nerve conduction studies may be useful in detecting muscle or large fiber nerve involvement in patients with sensorimotor changes. If neurosarcoidosis is highly suspected and no other tissue is accessible for confirmation, biopsy of the nerve or muscle may be considered. Although this was previously performed in patients with any kind of neurologic involvement, including isolated CNS disease, as a “blind biopsy,” the yield in patients without peripheral nervous system symptoms or signs is low. In those with prominent small fiber nerve involvement, skin biopsy with intraepidermal nerve fiber density evaluation, quantitative sudomotor axon reflex testing, tilt table testing, and other cardiovascular autonomic studies can be used for diagnosis. Confocal corneal microscopy is a new noninvasive means of detecting small fiber neuropathy that may also be helpful.36
Systemic Evaluation
For patients with suspected neurosarcoidosis and no prior history of systemic involvement, a careful investigation in pursuit of an extraneural biopsy target or other findings supportive of systemic disease is appropriate. As with CSF findings, no single laboratory or imaging study has definitively indicated sarcoidosis. While serum angiotensin-converting enzyme levels are still widely used and are part of the original diagnostic criteria, in isolation the test is neither specific nor sufficiently sensitive for diagnosing sarcoidosis and may be affected by genetic polymorphisms.17,37 Soluble interleukin-2 receptor levels have emerged as a potential serologic marker that has been found to correlate with disease activity.38 However, its value in the initial diagnostic evaluation is still unknown, because elevated levels may also be seen in leukemia, lymphoma, and other immune-mediated disorders.39
In addition to the studies listed under the diagnostic criteria in Table 2-2, routine laboratory tests to look for liver enzyme abnormalities, hypercalcemia/hypercalcuria, purified protein derivative (PPD) or interferon-γ release assay for tuberculosis, CT of the abdomen and pelvis, pulmonary function tests with diffusion study, and ophthalmic evaluation with slit-lamp examination should be performed. Conjunctival biopsy may also be considered, even if no pathology is apparent (eg, conjunctival nodules); in one series, 10 of 26 patients with neurosarcoidosis (38%) and no specific ocular abnormalities were found to have a positive diagnostic biopsy.40 In patients with characteristic pulmonary lesions or intrathoracic lymphadenopathy, endobronchial ultrasound-guided transbronchial needle aspiration is a promising new procedure that has not only been found to have a high diagnostic yield (84% to 93% sensitivity) for sarcoidosis, but also has reduced costs by 40% compared with mediastinoscopy.17,41
If initial screening tests fail to disclose evidence of systemic disease, further evaluation with a whole body fluorodeoxyglucose positron emission tomography (FDG-PET) scan should be considered. Although tissue biopsy is still often needed, given the lack of adequate specificity for sarcoidosis, FDG-PET scan is superior to a gallium 67 scan in detecting evidence of occult systemic disease (up to 97% sensitivity in one prospective series compared with 88% with gallium 67 scans) and is also occasionally helpful in demonstrating neurologic involvement not visualized on MRI.33,42,43 It can also be used to follow disease activity, especially in combination with CT.
For most patients with documented systemic disease, CNS biopsy is not necessary. However, in those with isolated CNS involvement, tissue biopsy of the affected region is usually the final and most definitive step in diagnosing neurosarcoidosis and should be considered only after significant effort has been made to look for systemic disease. A CNS biopsy may also be indicated to rule out alternative diagnoses when a patient does not respond to immunosuppressive therapy or has worsening lesions.
TREATMENT
While no definitive cure currently exists for neurosarcoidosis, the goals of management are to stop the ongoing inflammation, prevent worsening of disease, and restore neurologic function if possible with the use of immunosuppressive therapy. Because of the infrequent occurrence of the disease, there is a lack of data derived from rigorously designed clinical trials to establish specific treatment guidelines. Current strategies are largely based on anecdotal reports and empiric observations. With optimal immunosuppressive therapy, clinical remission occurs in up to 70% of patients.13
Corticosteroids are the first line of treatment. For most patients, the initial starting dose of oral prednisone is 0.5 mg/kg/d to 1 mg/kg/d or about 60 mg/d for at least 3 months, depending on the severity of disease. Pulse dose IV methylprednisolone at 1000 mg/d for 5 days followed by maintenance oral corticosteroids should be considered for patients with potentially disabling neurologic disease, such as spinal cord lesions, severe meningitis with or without hydrocephalus, and intracranial disease with edema. Over the next several months, patients should be carefully monitored with close clinical follow-up and imaging studies, especially while tapering the dose. Although corticosteroids are effective in the treatment of neurosarcoidosis, both clinicians and patients should be aware of the numerous complications that may arise with long-term treatment (eg, diabetes mellitus, mood changes, steroid-induced myopathy, weight gain, cataracts, and epidural lipomatosis) so that the appropriate symptomatic management or a more rapid tapering schedule may be considered if warranted.
Based on anecdotal experience, combination therapy with aggressive corticosteroid-sparing cytotoxic agents should be considered early on for patients with significant CNS involvement—especially those with spinal cord disease, diencephalic lesions, and severe meningitis—for two main reasons. The first is that corticosteroid monotherapy at moderate doses (less than 0.5 mg/kg/d) is often insufficient in controlling active inflammation despite the appearance of stabilization or slight improvement on MRI. As soon as the corticosteroids are tapered down from the initial aggressive burst dose, many patients experience a relapse or—worse yet—further extension of the existing lesions. The second is to reduce the incidence of corticosteroid complications as most of these patients will require ongoing immunosuppression for at least 6 months and in some cases several years. This may allow for a more expedited transition to a minimal daily dose (typically prednisone 10 mg/d) or, less commonly, discontinuation of the corticosteroids once the disease is stable.
Of the cytotoxic agents used for neurosarcoidosis, TNFα antagonists are the newest and most promising. Infliximab and adalimumab are monoclonal antibodies directed against TNFα, a proinflammatory cytokine secreted by macrophages that contributes to granulomatous inflammation. Infliximab, the more studied of the two, is now widely used in systemic sarcoidosis and, in combination with corticosteroids, appears effective in treating refractory neurosarcoidosis, including severe cases affecting the meninges, brain, and spinal cord.44,45 Serious complications include progressive multifocal leukoencephalopathy, demyelinating CNS lesions, malignancy, and tuberculosis infection or reactivation.
Mycophenolate mofetil is an oral agent that has been beneficial in treating moderate to severe disease, allowing for tapering or discontinuation of corticosteroids in patients with CNS involvement, although it was not helpful in patients with sarcoidosis-related myopathy.44,46,47 Methotrexate and azathioprine are other conventional agents that may be considered. Their effects in more severe cases, however, may not be as robust in comparison with other agents previously discussed, although in combination with corticosteroids and infliximab they may be helpful for refractory disease. Radiation therapy has also been reported to be helpful in refractory cases of neurosarcoidosis, particularly in those with pituitary dysfunction.48 See Table 2-3 for other treatment options.
Table 2-3.
Treatment Options for Neurosarcoidosis

The treatment of small fiber neuropathy represents a new therapeutic paradigm for sarcoidosis-associated disease that is currently being explored. Although symptoms are often refractory to corticosteroids, IV immunoglobulin and TNFα antagonists have been used successfully in a small number of patients to treat both the somatic and autonomic manifestations of small fiber neuropathy.49,50 ARA-290, an experimental erythropoietin agonist with activity against receptors involved in cytokine production, was recently shown to be of benefit in reducing pain and autonomic symptoms in a pilot trial.51
Supportive therapy with a multidisciplinary team approach is key. The patient should be referred to the appropriate specialists for treatment of neuroendocrine dysfunction, respiratory disease, ophthalmic lesions, or other complications. Pain management and counseling are often helpful for those with chronic pain, depression, and other neuropsychiatric complications. Physical therapy and exercise may benefit those with deconditioning and fatigue. Dietary consultation may help to control corticosteroid side effects of weight gain and hyperglycemia. Finally, referral to a sarcoidosis center with a team of specialists who regularly treat sarcoidosis should also be considered.
CONCLUSION
Neurosarcoidosis is a heterogeneous disorder whose clinical course is dependent on the area of the nervous system affected. Ideally, the diagnosis should be based on histologic confirmation either from the affected tissue or other involved organs. Treatment with aggressive immunosuppression, especially in patients with severe CNS involvement, should be instituted early in the course of the disease in an effort to reduce long-term complications.
KEY POINTS
Neurosarcoidosis is characterized by granulomatous inflammation that preferentially involves the basal meninges and cranial nerves but can affect any portion of the nervous system.
Although neurologic complications of sarcoidosis are seen in only 5% to 10% of cases, they are often the presenting symptom in these patients.
Facial nerve palsy is one of the most common manifestations of neurosarcoidosis and may be bilateral in about a third of cases. Symptoms are usually self-limited.
Subacute or chronic meningitis is frequently seen in neurosarcoidosis and in severe cases may result in hydrocephalus. Hypothalamic-pituitary dysfunction and cranial neuropathy may also occur with prominent basal inflammation.
Spinal cord sarcoidosis most often involves the cervicothoracic region and may be patchy and noncontiguous.
Symptoms of small fiber neuropathy, which include both somatic (painful paresthesia) and autonomic manifestations, are seen in up to 40% of patients with sarcoidosis.
Many patients with neurosarcoidosis report neuropsychiatric symptoms, such as mood disturbances, fatigue, and memory loss, which are often multifactorial and referred to as “sarcoidosis fog.”
Although a definite diagnosis of neurosarcoidosis requires histologic confirmation of the affected tissue, the presence of CNS inflammation compatible with neurosarcoidosis in patients with evidence of systemic involvement is often sufficient for diagnosis and initiation of treatment.
Neurosarcoidosis is known as “the great mimicker” because of its vast range of MRI appearances, which include a tumorlike mass, periventricular white matter lesions, and focal or diffuse leptomeningeal disease.
While no specific CSF abnormality is diagnostic of neurosarcoidosis, an elevated CSF protein is most commonly seen in patients with active disease, although up to one-third of patients may have normal CSF findings.
The soluble interleukin-2 receptor level is a potential serologic marker of disease activity that is increasingly being used in sarcoidosis.
Fluorodeoxyglucose–positron emission tomographic imaging is superior to gallium scans in demonstrating evidence of systemic disease in patients with suspected neurosarcoidosis.
Corticosteroids in combination with tumor necrosis factor-α antagonists or other cytotoxic agents should be considered in the treatment of moderate to severe neurosarcoidosis, which often requires aggressive immunosuppression for at least 6 months to a year, if not longer.
A multidisciplinary team approach is key in the management of neurosarcoidosis. Supportive therapy with counseling, physical therapy, pain management, and nutritional consultation should be considered for patients with acute and chronic disease.
Footnotes
Relationship Disclosure: Dr Tavee is involved in a clinical trial funded by Araim Pharmaceuticals, Inc, evaluating the use of ARA290 in patients with small fiber neuropathy related to sarcoidosis. Dr Stern has served as an expert witness, as editor of The Neurologist, and serves on the Data and Safety Monitoring Board and as a medical safety monitor at the National Institute of Neurological Disorders and Stroke. Dr Stern receives research support from the National Institute of Neurological Disorders and Stroke and Remedy Pharmaceuticals.
Unlabeled Use of Products/Investigational Use Disclosure: Drs Tavee and Stern report no disclosures.
REFERENCES
- 1.Sharma OP. Definition and history of sarcoidosis. European Respiratory Society Monograph 2005; 32: 1–12. [Google Scholar]
- 2.Mirfakhraee M,, Crofford MJ,, Guinto FC, Jr, et al. Virchow-Robin space: a path of spread in neurosarcoidosis. Radiology 1986; 158 (3): 715–720. [DOI] [PubMed] [Google Scholar]
- 3.Schley D,, Carare-Nadi R,, Please CP, et al. Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J Theor Biol 2006; 238 (4): 962–974. [DOI] [PubMed] [Google Scholar]
- 4.Nowak DA,, Widenka DC. Neurosarcoidosis: a review of its intracranial manifestation. J Neurol 2001; 248 (5): 363–372. [DOI] [PubMed] [Google Scholar]
- 5.Heij L,, Dahan A,, Hoitsma E. Sarcoidosis and pain caused by small-fiber neuropathy. Pain Res Treat 2012. Epub 2012 Dec 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stern BJ,, Krumholz A,, Johns C, et al. Sarcoidosis and its neurological manifestations. Arch Neurol 1985; 42 (9): 909–917. [DOI] [PubMed] [Google Scholar]
- 7.Delaney P. Neurologic manifestations in sarcoidosis review of the literature, with a report of 23 cases. Ann Intern Med 1977; 87 (3): 336–345. [DOI] [PubMed] [Google Scholar]
- 8.Chapelon C,, Ziza JM,, Piette JC, et al. Neurosarcoidosis: signs, course and treatment in 35 confirmed cases. Medicine (Baltimore) 1990; 69 (5): 261–276. [PubMed] [Google Scholar]
- 9.Joseph FG,, Scolding NJ. Neurosarcoidosis: a study of 30 new cases. J Neurol Neurosurg Psychiatry 2009; 80 (3): 297–304. [DOI] [PubMed] [Google Scholar]
- 10.Zajicek JP,, Scolding NJ,, Foster O, et al. Central nervous system sarcoidosis—diagnosis and management. QJM 1999; 92 (2): 103–117. [DOI] [PubMed] [Google Scholar]
- 11.Ricker W,, Clark M. Sarcoidosis; a clinicopathologic review of 300 cases, including 22 autopsies. Am J Clin Pathol 1949; 19 (8): 725–749. [DOI] [PubMed] [Google Scholar]
- 12.Allen RK,, Sellars RE,, Sandstrom PA. A prospective study of 32 patients with neurosarcoidosis. Sarcoidosis Vasc Diff Lung Dis 2003; 20 (2): 118–125. [PubMed] [Google Scholar]
- 13.Stern BJ,, Aksamit A,, Clifford D,, Scott TF. Neurologic presentations of sarcoidosis. Neurol Clin 2010; 28 (1): 185–198. [DOI] [PubMed] [Google Scholar]
- 14.Scott TF,, Yandora K,, Valeri A, et al. Aggressive therapy for neurosarcoidosis: long-term follow-up of 48 treated patients. Arch Neurol 2007; 64 (5): 691–696. [DOI] [PubMed] [Google Scholar]
- 15.Iannuzzi MC,, Rybicki BA,, Teirstein AS. Sarcoidosis. N Engl J Med 2007; 357 (21): 2153–2165. [DOI] [PubMed] [Google Scholar]
- 16.Baughman RP,, Teirstein AS,, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001; 164 (10 pt 1): 1885–1889. [DOI] [PubMed] [Google Scholar]
- 17.Culver DA. Sarcoidosis. Immunol Allergy Clin North Am 2012; 32 (4): 487–511. [DOI] [PubMed] [Google Scholar]
- 18.Ishige I,, Usui Y,, Takemura T,, Eishi Y. Quantitative PCR of mycobacterial and propionibacterial DNA in lymph nodes of Japanese patients with sarcoidosis. Lancet 1999; 354 (9173): 120–123. [DOI] [PubMed] [Google Scholar]
- 19.Hofmann S,, Franke A,, Fischer A, et al. Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat Gener 2008; 40 (9): 1103–1106. [DOI] [PubMed] [Google Scholar]
- 20.Pawate S,, Moses H,, Sriram S. Presentations and outcomes of neurosarcoidosis: a study of 54 cases. QJM 2009; 102 (7): 449–460. [DOI] [PubMed] [Google Scholar]
- 21.Aksamit A. Neurosarcoidosis. Continuum (Minneap Minn) 2008; 14 (1): 181–196. [Google Scholar]
- 22.Sohn M,, Culver DA,, Judson MA, et al. Spinal cord neurosarcoidosis. Am J Med Sci 2013. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 23.Langrand C,, Bihan H,, Raverot G, et al. Hypothalamo-pituitary sarcoidosis: a multicenter study of 24 patients. QJM 2012; 105 (10): 981–995. [DOI] [PubMed] [Google Scholar]
- 24.Navi BB,, DeAngelis LM. Sarcoidosis presenting as brainstem ischemic stroke. Neurology 2009; 72 (11): 1021–1022. [DOI] [PubMed] [Google Scholar]
- 25.Burns TM,, Dyck PJB,, Aksamit AJ,, Dyck PJ. The natural history and long-term outcome of 57 limb sarcoidosis neuropathy cases. J Neurol Sci 2006; 244 (1–2): 77–87. [DOI] [PubMed] [Google Scholar]
- 26.Said G,, Lacroix C,, Planté-Bordeneuve V, et al. Nerve granulomas and vasculitis in sarcoid peripheral neuropathy A clinicopathological study of 11 patients. Brain 2002; 125 (pt 2): 264–275. [DOI] [PubMed] [Google Scholar]
- 27.Hoitsma E,, Marziniak M,, Faber CG, et al. Small fibre neuropathy in sarcoidosis. Lancet 2002; 359 (9323): 2085–2086. [DOI] [PubMed] [Google Scholar]
- 28.Judson MA,, Costabel U,, Drent M, et al. The WASOG Sarcoidosis Organ Assessment Instrument Investigators The WASOG sarcoidosis organ assessment instrument: an update of a previous clinical tool. Sarcoidosis Vasc Diffuse Lung Dis 2014; 31 (1): 19–27. [PubMed] [Google Scholar]
- 29.Wallace SL,, Lattes R,, Malia JP,, Ragan C. Muscle involvement in Boeck’s sarcoid. Ann Intern Med 1958; 48 (3): 497–511. [DOI] [PubMed] [Google Scholar]
- 30.Marangoni S,, Argentiero V,, Tavolato B. Neurosarcoidosis. Clinical description of 7 cases with a proposal for a new diagnostic strategy. J Neurol 2006; 253 (4): 488–495. [DOI] [PubMed] [Google Scholar]
- 31.Costabel U,, Hunninghake GW. ATS/ERS/WASOG statement on sarcoidosis. Sarcoidosis Statement Committee. American Thoracic Society. European Respiratory Society. World Association for Sarcoidosis and Other Granulomatous Disorders. Eur Respir 1999; 14 (4): 735–737. [DOI] [PubMed] [Google Scholar]
- 32.Baughman RP,, Lower EE. Novel therapies for sarcoidosis. Semin Respir Crit Care Med 2007; 28 (1): 128–133. [DOI] [PubMed] [Google Scholar]
- 33.Mana J,, Gámez C. Molecular imaging in sarcoidosis. Curr Opin Pulm Med 2011; 17 (5): 325–331. [DOI] [PubMed] [Google Scholar]
- 34.Oksanen V,, Fyhrquist F,, Gronhagen-Riska C,, Somer H. CSF angiotensin converting enzyme in neurosarcoidosis. Lancet 1985; 1 (8436): 1050–1051. [DOI] [PubMed] [Google Scholar]
- 35.Dale JC,, O’Brien JF. Determination of angiotensin-converting enzyme levels in cerebrospinal fluid is not a useful test for the diagnosis of neurosarcoidosis. Mayo Clin Proc 1999; 74 (5): 535. [DOI] [PubMed] [Google Scholar]
- 36.Tavakoli M,, Quattrini C,, Abbott C, et al. Corneal confocal microscopy a novel noninvasive test to diagnose and stratify the severity of human diabetic neuropathy. Diabetes Care 2010; 33 (8): 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomita H,, Ina Y,, Sugiura Y, et al. Polymorphism in the angiotensin-converting enzyme (ACE) gene and sarcoidosis. Am J Respir Crit Care Med 1997; 156 (1): 255–259. [DOI] [PubMed] [Google Scholar]
- 38.Grutters JC,, Fellrath JM,, Mulder L, et al. Serum soluble interleukin-2 receptor measurement in patients with sarcoidosis: a clinical evaluation. Chest 2003; 124 (1): 186–195. [DOI] [PubMed] [Google Scholar]
- 39.Rubin LA,, Nelson DL. The soluble interleukin-2 receptor: biology, function, and clinical application. Ann Intern Med 1990; 113 (8): 619–627. [DOI] [PubMed] [Google Scholar]
- 40.Leavitt JA,, Campbell RJ. Cost-effectiveness in the diagnosis of sarcoidosis: the conjunctival biopsy. Eye 1998; 12 (pt 6): 959–962. [DOI] [PubMed] [Google Scholar]
- 41.Navani N,, Lawrence DR,, Kolvekar S, et al. Endobronchial ultrasound-guided transbronchial needle aspiration prevents mediastinocopies in the diagnosis of isolated mediastinal lymphadenopathy: a prospective trial. Am J Respir Crit Care Med 2012; 186 (3): 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keijsers RG,, Grutters JC,, Thomeer M, et al. Imaging the inflammatory activity of sarcoidosis: sensitivity and inter observer agreement of (67)Ga imaging and (18)F-FDG PET. Q J Nucl Med Mol Imaging 2011; 55 (1): 66–71. [PubMed] [Google Scholar]
- 43.Huang JF,, Aksamit AJ,, Staff NP. MRI and PET imaging discordance in neurosarcoidosis. Neurology 2012; 79 (10): 1070. [DOI] [PubMed] [Google Scholar]
- 44.Moravan M,, Segal BM. Treatment of CNS sarcoidosis with infliximab and mycophenolate mofetil. Neurology 2009; 72 (4): 337–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santos E,, Shaunak S,, Renowden SA,, Scolding NJ. Treatment of refractory neurosarcoidosis with infliximab. J Neurol Neurosurg Psychiatry 2010; 81 (3): 241–246. [DOI] [PubMed] [Google Scholar]
- 46.Androdias G,, Maillet D,, Marignier R, et al. Mycophenolate mofetil may be effective in CNS sarcoidosis but not in sarcoid myopathy. Neurology 2011; 76 (13): 1168–1172. [DOI] [PubMed] [Google Scholar]
- 47.Segal BM. Neurosarcoidosis: diagnostic approaches and therapeutic strategies. Curr Opin Neurol 2013; 26 (3): 307–313. [DOI] [PubMed] [Google Scholar]
- 48.Motta M,, Alongi F,, Bolognesi A, et al. Remission of refractory neurosarcoidosis treated with brain radiotherapy: a case report and a literature review. Neurologist 2008; 14 (2): 120–124. [DOI] [PubMed] [Google Scholar]
- 49.Parambil JG,, Tavee JO,, Zhou L, et al. Efficacy of intravenous immunoglobulin for small fiber neuropathy associated with sarcoidosis. Respir Med 2011; 105 (1): 101–105. [DOI] [PubMed] [Google Scholar]
- 50.Tavee J,, Culver D. Sarcoidosis and small-fiber neuropathy. Curr Pain Headache Rep 2011; 15 (3): 201–206. [DOI] [PubMed] [Google Scholar]
- 51.Heij L,, Niesters M,, Swartjes M, et al. Safety and efficacy of ARA 290 in sarcoidosis patients with symptoms of small fiber neuropathy: a randomized, double-blind pilot study. Mol Med 2012; 18: 1430–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]