

Abstract
Purpose of Review
The purpose of this review is to provide information regarding the diagnosis and natural history of some very rare disorders: congenital muscular dystrophies and congenital myopathies. Patients with these conditions share characteristics such as early onset of weakness and severe hypotonia. Other organs such as the brain, eyes, and skin may be involved. Diagnosis depends largely on recognition of phenotype, muscle biopsy, and mutation analysis.
Recent Findings
More than 30 genes have been associated with these diseases, most of which have only been recognized in the past decade. Increasing availability of DNA analysis has been important in decreasing delay in diagnosis.
Summary
Patients with congenital muscular dystrophy or congenital myopathy are at high risk of complications including restrictive lung disease, orthopedic deformities, seizures, cardiomyopathy, and malignant hyperthermia. Life expectancy varies with the severity of complications. Having an accurate and specific diagnosis allows the neurologist to carry out anticipatory guidance and appropriate monitoring. New hope exists for experimental treatments for congenital muscular dystrophy and congenital myopathy as our understanding of pathogenesis evolves.
CONGENITAL MUSCULAR DYSTROPHIES
Congenital muscular dystrophies are extremely rare and greatly heterogeneous neuromuscular disorders with onset at birth or early infancy, characterized by hypotonia, delayed motor development, and progressive weakness. The clinical presentation is variable and can affect other organs, including the eyes, brain, lungs, and heart. Serum creatine kinase (CK) is elevated in several but not all. Appropriate muscle biopsy studies are crucial for accurate diagnosis.
Since the discovery of primary merosin deficiency in 1994, our understanding of the molecular basis of these disorders has significantly increased.1 The prevalence and incidence of the congenital muscular dystrophies varies in different regions of the world. For example, in a recent study of 116 patients in the United Kingdom, the most common congenital muscular dystrophies were collagen VI–related disorders (19%), with α-dystroglycanopathy congenital muscular dystrophy (12%) and merosin-deficient congenital muscular dystrophy (MDC1A) (10%) being next in frequency.2 The Australian study by Peat and colleagues in 20083 showed dystroglycanopathy as the most common congenital muscular dystrophy (25%) on that continent, followed by collagen VI–related disorders (12%). Fukuyama congenital muscular dystrophy is the most prevalent form (49.2%) in Japan, followed by collagen VI deficiency at 7.2%.4
Clinical Aspects
Congenital muscular dystrophy manifests at birth or within the first 2 years of life. The typical presentation is congenital hypotonia, delayed motor skills, and slowly progressive muscle weakness. Phenotypic expression varies greatly among patients, such as with the distribution of hypotonia and weakness. Some patients have predominant axial hypotonia with head lag and later spine rigidity, as in selenoprotein 1 (SEPN1)–related and lamin A/C (LMNA)–related congenital muscular dystrophies, while patients with generalized hypotonia/weakness and contractures, with or without joint laxity, are likely to have collagen-related congenital muscular dystrophies.
CNS involvement and MRI findings are key in the differential diagnosis of congenital muscular dystrophy. Patients can present with mild to severe cognitive impairment and learning disabilities. Seizures occur in patients with MDC1A at a frequency of 8% to 20%.5 Brain MRI findings include white matter changes (T2 hyperintensity) and cortical dysplasia in α-dystroglycanopathy congenital muscular dystrophy. Ophthalmic abnormalities, including visual impairment and retinal abnormalities, are often present in α-dystroglycanopathy congenital muscular dystrophy. Cardiomyopathy can be seen in late stages but is usually limited to a few types of congenital muscular dystrophy including fukutin, fukutin-related protein (FKRP), protein-O-mannosyltransferase 1 (POMT1)–related congenital muscular dystrophies or limb-girdle muscular dystrophy, and LMNA-related congenital muscular dystrophy. In a recent study of 115 patients with α-dystroglycanopathy congenital muscular dystrophy in Italy,6 only seven were found to have abnormal cardiac function: five with dilated cardiomyopathy, one with a cardiac conduction defect, and one with mitral regurgitation. Sudden cardiac death was reported almost exclusively in LMNA-related congenital muscular dystrophy.
Respiratory failure can be an early symptom after birth, requiring ventilation. Otherwise, restrictive lung disease, nocturnal hypoventilation, and respiratory failure may not be evident until more advanced stages of disease. In the same Italian study, 14 patients out of 115 with α-dystroglycanopathy congenital muscular dystrophy had abnormal respiratory function. Ten of the 14 required nocturnal noninvasive ventilatory support (NIV), while the others required invasive ventilation.
In a case series of patients with SEPN1-related congenital muscular dystrophy, respiratory function data were collected from 41 patients between 1 and 60 years old. The need for nocturnal NIV increased with age. At the age of 15 years, 50% of the patients required a ventilator, with an increase to 75% at the age of 20 years. Sleep studies were found to be abnormal at a mean age of 13.2 years, anticipating the need for nocturnal NIV, which became necessary in 66% of patients during the second decade of life.7 Table 1-18,9 summarizes the clinical signs, imaging, and natural course of congenital muscular dystrophy.
Table 1-1.
Clinical Signs, Imaging, and Disease Course of Congenital Muscular Dystrophiesa
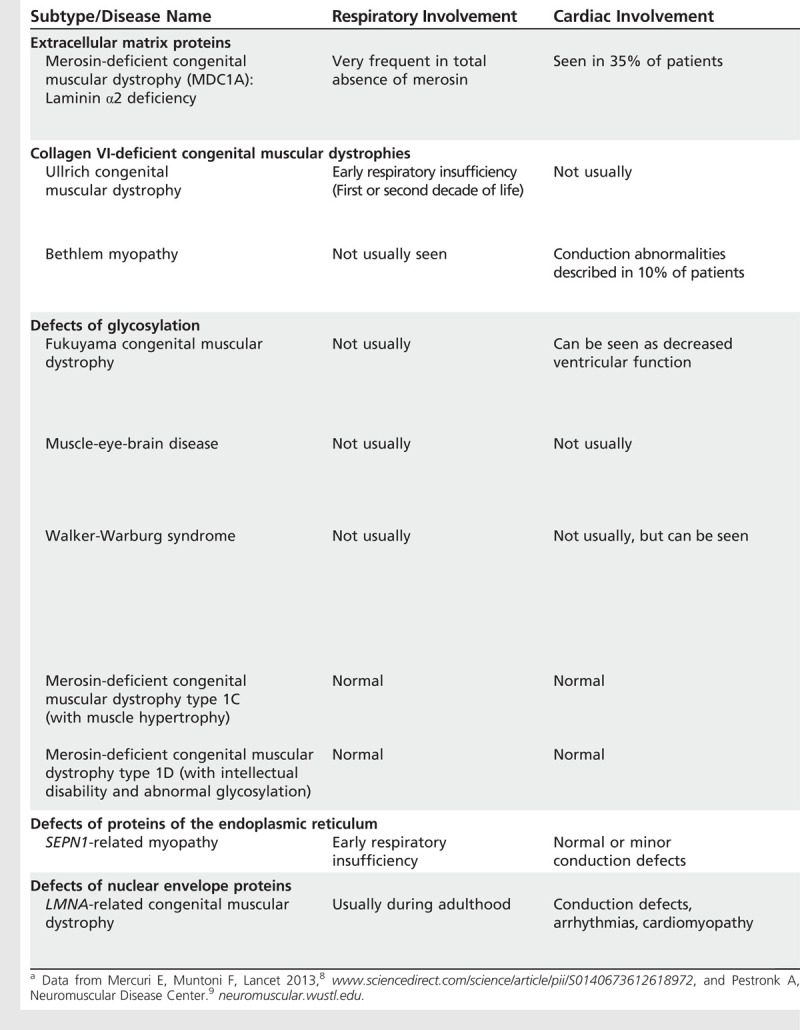
Table 1-1.
Continued
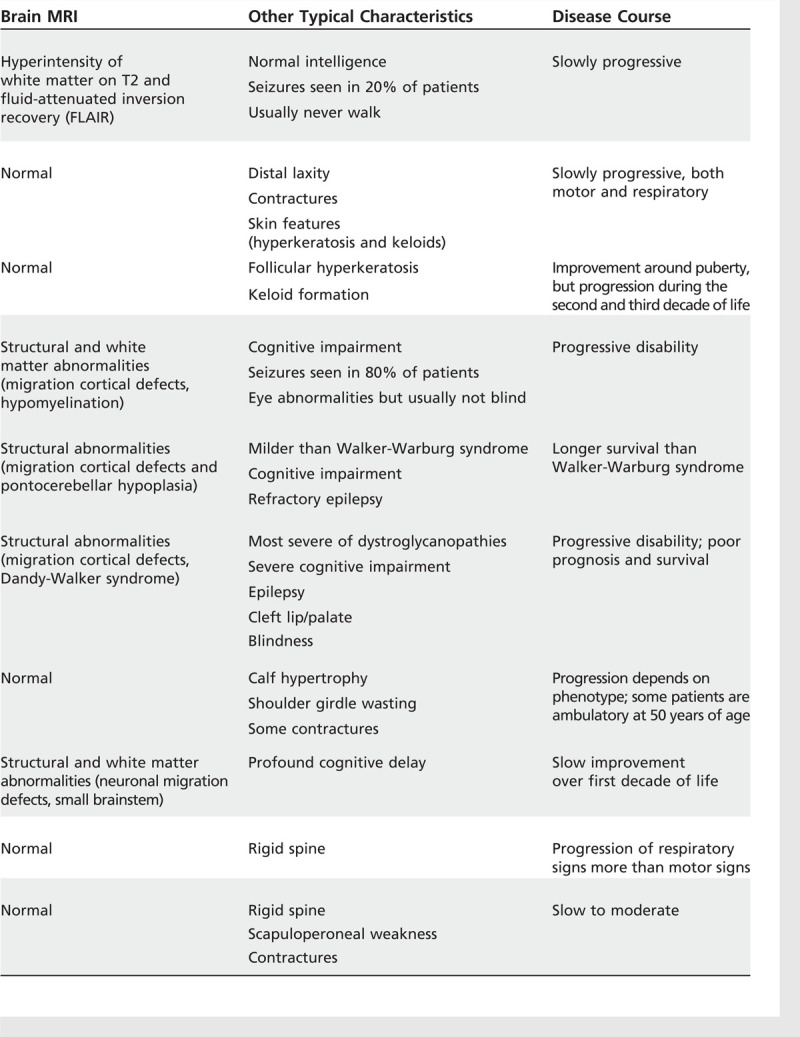
Classification
With advancing knowledge about the genetic defects causing congenital muscular dystrophy, the classificationfrequently requires updating. In 2004, Muntoni and colleagues10 suggested a classification according to the gene locus of the protein affected. A recent review in Lancet by Mercuri and Muntoni8 gave an updated classification based on clinical findings, informationabout the primary protein defect, and gene localization and function. The gene table of neuromuscular disorders is a comprehensive resource created by the World Muscle Society beginning in 1991.11 Data for Table 1-2 were taken from these sources.
Table 1-2.
Classification of Congenital Muscular Dystrophiesa,b
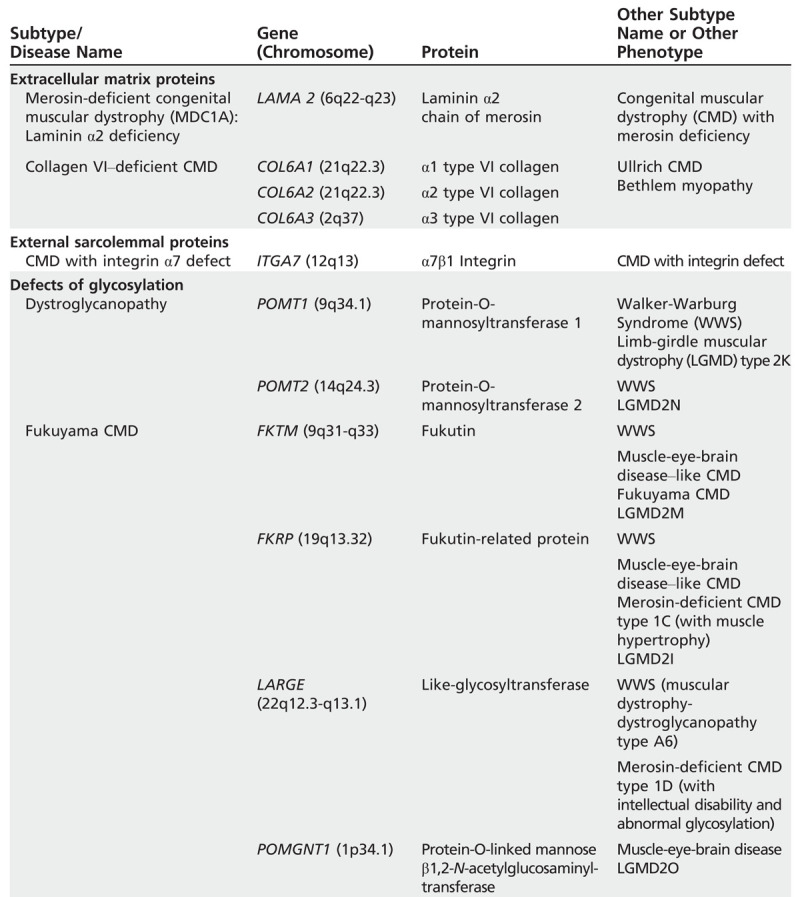
Table 1-1.
Continued
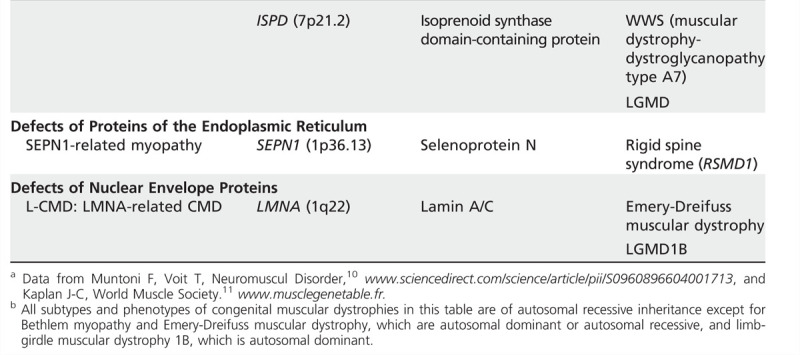
Diagnosis
The most important diagnostic tools are CK level, nerve conduction study and EMG with or without repetitive nerve stimulation, brain MRI, muscle biopsy, and specific genetic or metabolic testing. Most congenital muscular dystrophies show elevated CK values, with exceptions such as SEPN1-related congenital muscular dystrophy and collagen VI–deficient congenital muscular dystrophy, in which CK levels are often normal. MRI of the brain can be particularly helpful in the diagnosis of congenital muscular dystrophy with merosin deficiency and α-dystroglycanopathy congenital muscular dystrophy.
Muscle MRI may be useful to determine the specific or preferential involvement of certain muscle groups. In patients with Bethlem myopathy, the vasti muscles are the most frequently and most strikingly affected thigh muscles, with a rim of abnormal signal at the periphery of each muscle and relative sparing of the central part. The rectus femoris is affected, with a central area of abnormal signal within the muscle. Patients with Ullrich congenital muscular dystrophy have a more diffuse involvement of the thigh muscles, with relative sparing of sartorius, gracilis, and adductor longus. In SEPN1-related myopathy, involvement of sartorius and adductor longus muscles but sparing of the gracilis muscle is highly suggestive of the diagnosis.
In congenital muscular dystrophy, the muscle biopsy shows dystrophic changes with abnormal variation in fiber size (associated with whorled or split fibers) and rare hypercontracted fibers. An increase in internal nuclei is evident, with a variable increase in endomysial connective and adipose tissue. Prominent muscle necrosisis infrequent and may be absent in congenital muscular dystrophy (Figure 1-1).
Figure 1-1.
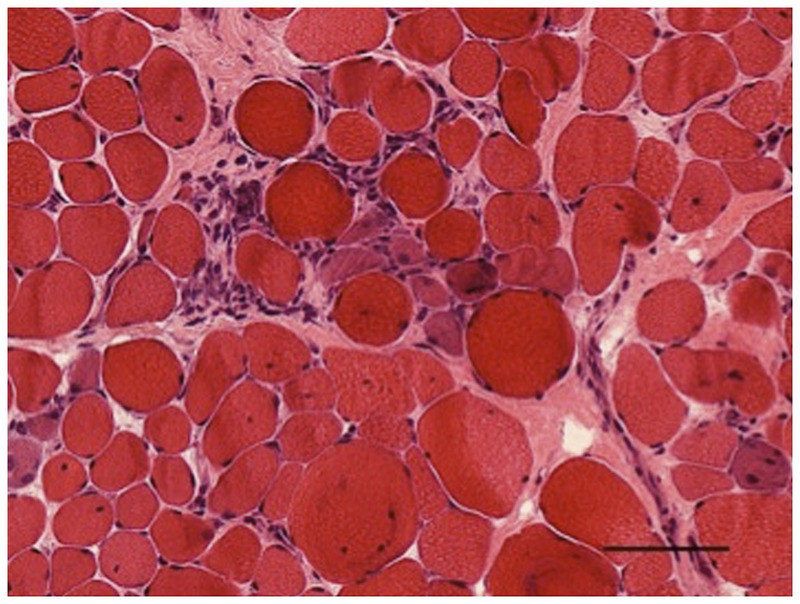
Hematoxylin and eosin–stained section of left quadriceps demonstrates hypertrophic fibers with sarcoplasmic disarray interspersed with atrophic fibers, some with sarcoplasmic basophilia consistent with regeneration. Occasional inflammatory cells are in the interstitial space (200×).
The immunohistochemical examination is extremely important in the differential diagnosis; specific antibodies for merosin, collagen VI, and glycosylated α-dystroglycan may identify specific protein deficiencies. Depending on the clinical findings, a muscle biopsy may be done early or late in the diagnostic process (Figure 1-2). Neurogenic changes may be prominent in MDC1A (merosin deficiency) while immunohistology shows complete or partial deficiency of laminin α2. In complete merosin deficiency, both the C-terminal and N-terminal antibodies to laminin α2 fail to stain muscle fibers. On the other hand, the light chains of laminin α2 (β1 and γ1) are preserved, and other laminin α chains (α4 and α5) are upregulated. Muscle biopsy in MDC1A may be neurogenic but is usually dystrophic. In collagen VI–deficient congenital muscular dystrophies, collagen IV shows normal expression, while collagen VI may or may not label normally. The muscle biopsy commonly shows a range from moderate myopathic changes to severe dystrophic features depending on the severity and duration of the disease. Collagen VI immunostaining is helpful if it shows anabsence or reduction in basement membrane (basal lamina) labeling, but normal labeling does not exclude Ullrich congenital muscular dystrophy or Bethlem myopathy. Immunohistochemistry in α-dystroglycanopathy congenital muscular dystrophy shows normal expression of β-dystroglycan in the sarcolemma, accompanied by absent or reduced α-dystroglycan.
Figure 1-2.
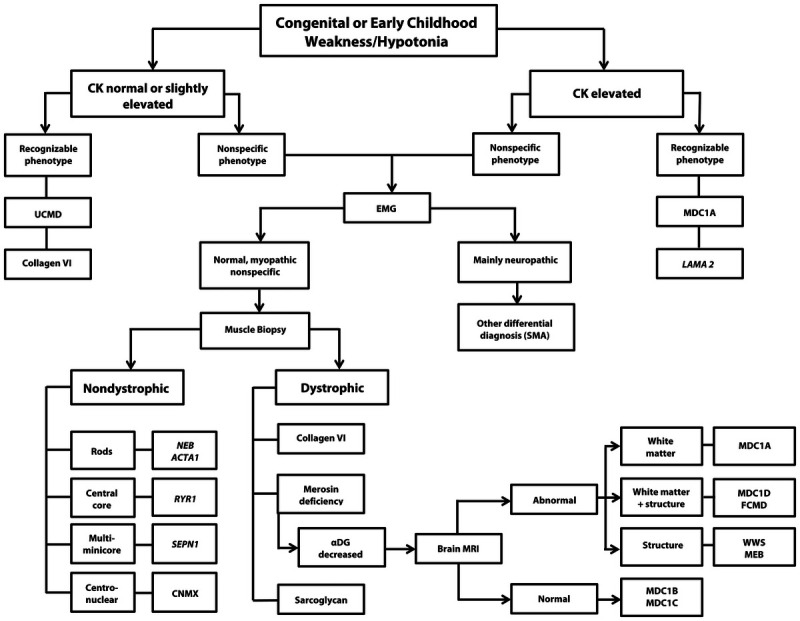
Algorithm for the diagnosis of congenital muscular dystrophy. CK = creatine kinase; UCMD = Ullrich congenital muscular dystrophy; EMG = electromyography; MDC1A = merosin-deficient congenital muscular dystrophy; LAMA 2 = laminin α2 gene; SMA = spinal muscular atrophy; NEB = nebulin gene; ACTA1 = gene encoding skeletal muscle actin; RYR1 = ryanodine receptor; MDC1D = congenital muscular dystrophy type 1D; FCMD = Fukuyama congenital muscular dystrophy; SEPN1 = selenoprotein 1; αDG = α-dystroglycanopathy; MRI = magnetic resonance imaging; WWS = Walker-Warburg syndrome; MEB = muscle-eye-brain disease; CNMX = X-linked centronuclear myopathy; MDC1B = congenital muscular dystrophy type 1B; MDC1C = congenital muscular dystrophy type 1C.
Congenital Muscular Dystrophy With Merosin Deficiency
MDC1A is caused by mutations in the laminin α2 gene (LAMA2) linked to chromosome 6q22-q23 and inherited as autosomal recessive. The prevalence varies in different areas of the world from 10% to 25% among patients with congenital muscular dystrophy. Laminins are extracellular glycoproteins that bind with other extracellular and transmembrane proteins to form the frame of the basal lamina that surrounds individual myofibers. Each laminin is a heterodimer composed of a heavy chain (α) and two light chains (β and γ). The major laminin of adult skeletal muscle is laminin-2 (also known as merosin), and only mutations of LAMA2 gene encoding laminin α2 cause muscular dystrophy.
Patients with complete absence of merosin expression present with early hypotonia, weakness (facial, proximal, and distal limb muscles), and delayed milestones. They never walk but have normal cognition despite white matter abnormalities. Contractures in elbows, hands, knees, and ankles are progressive (Figure 1-3). The phenotype of patients with residual (partial) merosin expression can range from mild to severe. Geranmayeh and colleagues5 presented the clinical and genetic features of 33 cases of complete merosin deficiency and 13 with residual merosin expression, and confirmed the correlation between levels of merosin expression and clinical severity. Patients with total absence of merosin had an earlier presentation, were more likely to lack independent ambulation, and required enteral feedings and ventilator support. Seizures (simple partial, complex partial, absence, or generalized) were reported in 23% (3/13) of patients with residual merosin, and 15% (5/33) from the absent merosin group. Previous studies reported seizures in 8% to 20% of thepatients. The EEG findings were nonspecific with focal or generalized spike-and-wave discharges.
Brain MRI usually shows T2 hyperintensity and fluid-attenuated inversion recovery (FLAIR) involvement of the white matter, mainly in the periventricular and frontal U fibers (Figure 1-4). The internal capsule, corpus callosum, cerebellum, and other dense fiber tracts are usually spared. Previous studies reported neuronal migration defects such as agyria or posterior fossa abnormalities (cerebellar hypoplasia) in around 5% of patients.12
Peripheral nerve may be affected, causing a motor and sensory neuropathy. The sural nerve shows a reduction in the number of myelinated nerve fibers, short internodal segments, excessively wide nodes of Ranvier, and variability in myelin thickness with redundant folds and tomacula.
Respiratory function is progressively reduced, and NIV in the form of bilevel positive airway pressure (BiPAP) is often required in the second or third decade of life. Patients with total absence of merosin expression are more likely to require NIV, but many patients require tracheostomy and mechanical ventilation. This is because weakness of facial and bulbar muscles make use of facial masks and control of oral secretions problematic.
Cardiac abnormalities are found in 35% of patients in the form of ventricular dysfunction, mitral regurgitation, and pulmonary hypertension. Thus, primary cardiomyopathy is rare in MDC1A.
Feeding difficulties occur secondary to facial weakness and oropharyngeal dysphagia. Enteral feeding is required in approximately half of patients with total absence of merosin. Enteral feeding is usually not required in the residual merosin expression group.
Diagnosis should be suspected in the presence of an elevated CK level (more than 5 times normal, especially in the first 2 years of life), abnormal white matter, progressive restrictive lung disease, and muscle pathology (Case 1-1). Diagnosis should be confirmed by muscle biopsy and mutation analysis (Figure 1-5).
Case 1-1
An 8-month-old boy was born after full-term pregnancy and normal delivery. His parents were nonconsanguineous and had no family history of neuromuscular diseases. The pregnancy was uneventful, but he was noted to be hypotonic at birth. He had recurrent episodes of pneumonia since the first month of life. Physical examination revealed a thin, alert infant with body weight in the third percentile. He was hypotonic with decreased spontaneous movements. He did not have antigravity movements and showed evidence of generalized muscle wasting. He had severe head lag with facial weakness, weak cry, and ineffective sucking. Deep tendon reflexes were diminished.
Serum creatine kinase (CK) was 1500 IU/L. Findings from both the metabolic screening and cardiac assessment were normal. Brain MRI demonstrated patchy T2 hyperintensity within the white matter, mainly in the periventricular area.
Electrophysiologic testing showed myopathic and neurogenic changes. Muscle biopsy showed fatty replacement of muscle fibers, and the preserved fibers exhibited variation in diameter. Immunohistochemical studies on the muscle biopsy showed a completely negative reaction for merosin.
The patient developed progressive respiratory failure and required noninvasive ventilation in the form of bilevel positive airway pressure (BiPAP). He was never able to walk and required gastrostomy tube feedings and full assistance for daily living activities. At 18 years, he could sit with full support; he had no spontaneous movements, multiple contractures, and severe levoscoliosis. His speech was hypernasal and his cognition was intact. He developed complex partial seizures at 15 years of age and was started on levetiracetam, with good control.
Comment. This patient had typical features of congenital muscular dystrophy with merosin deficiency (MDC1A). He presented with early hypotonia, weakness, delayed milestones, and inability to walk. The clues for his diagnosis were his elevated CK (5 times normal), normal cognition, and brain MRI findings showing typical T2 hyperintensity in white matter.
The differential diagnosis included spinal muscular atrophy and congenital myopathies. The elevated CK and leukodystrophy are both against those diagnoses. With this typical phenotype, mutation analysis of the merosin gene should be considered before proceeding with more invasive testing.
Diagnosis was supported by electrophysiologic testing showing myopathic and neurogenic changes and a muscle biopsy that showed typical dystrophic features, some neurogenic changes, and complete absence of merosin expression.
Patients with complete absence of merosin have severe disease and usually require early gastrostomy feedings, ventilatory support, and full assistance for activities of daily living. Death occurs between 15 and 30 years of age, usually secondary to respiratory failure.
Collagen VI–Deficient Congenital Muscular Dystrophy
Collagen VI–deficient congenital muscular dystrophy is the second most common variant of congenital muscular dystrophy worldwide and was originally described by Ullrich in 1930. Mutations in collagen VI genes (COL6) cause different phenotypes, including Ullrich congenital muscular dystrophy, Bethlem myopathy, and autosomal recessive myosclerosis myopathy. Ullrich congenital muscular dystrophy is usually an autosomal recessive disorder, but the disease has occasionally been reported to be caused by heterozygous mutations in the COL6A1 and COL6A2 genes.
Collagen VI is an extracellular matrix protein, which forms a microfibrillar network in close association with several other matrix constituents. It is composed of three peptide chains: α1(VI), α2(VI), and α3(VI). The α1(VI) and α2(VI) chains are encoded by COL6A1 and COL6A2, respectively, positioned head to tail on chromosome 21q22.3. The COL6A3 locus is on chromosome 2q37.
The classical clinical features of Ullrich congenital muscular dystrophy are congenital hypotonia, scoliosis or kyphosis of the spine, proximal joint contractures, torticollis, and distal joint hyperlaxity. Congenital hip dislocation may be present. The skin typically shows follicular hyperkeratosis (keratosis pilaris) over the extensor surfaces of the arms and legs, and abnormal keloid formation. This “rash” begins subtly in early childhood and becomes very prominent in adolescence (Figure 1-6).
Some patients never walk, whereas others achieve ambulation with delay up to the fourth year of life. Progressive functional difficulties secondary to increased contractures lead to loss of ambulation after a period of independence. Patients have normal cognitive function and brain MRI. Respiratory function progressively declines over time, leading to ventilatory insufficiency and often death in the first or second decade of life. Patients with Ullrich congenital muscular dystrophy have no cardiac involvement.
Bethlem myopathy is a mild disorder that is allelic to Ullrich congenital muscular dystrophy and inherited as autosomal dominant. Bethlem myopathy is characterized by slowly progressive muscle weakness and wasting with distal joint contractures affecting the long finger flexors, elbows, and ankles. The disorder shows a wide range of clinical symptoms, from mild myopathy to more severe cases with features of progressive muscular dystrophy.
Diagnosis can be suspected in the presence of a typical clinical phenotype, normal or slightly elevated CK level, muscle biopsy findings, and genetic analysis. The main differential diagnosis for COL6-related disorders includes Emery-Dreifuss muscular dystrophy, given the presence of prominent elbow contractures. However, the COL6-related disorders do not have any cardiac involvement. Molecular genetic confirmation is recommended because of the clinical similarities between Ullrich congenital muscular dystrophy and Emery-Dreifuss muscular dystrophy. Identifying the mutation is important in order to determine whether the inheritance is dominant, when family members would be at risk.
α-Dystroglycanopathies
The dystroglycanopathies are a group of disorders caused by abnormal glycosylation of α-dystroglycan. Dystroglycan is encoded by DAG1 and is composed of α-dystroglycan that lies outside the muscle cell and of β-dystroglycan, which is a transmembrane protein. This complex is a vital component of the dystrophin glycoprotein complex that links the extracellular matrix to the actin cytoskeleton. α-Dystroglycan serves as a receptor for several proteins in the extracellular matrix, including laminin, neurexin, agrin, biglycan, and perlecan. β-Dystroglycan associates with intracellular proteins, including dystrophin.
α-Dystroglycanopathies include Fukuyama congenital muscular dystrophy, muscle-eye-brain (MEB) disease, Walker-Warburg syndrome (WWS), congenital muscular dystrophy type 1C (FKRP-related or MDC1C), and congenital muscular dystrophy type 1D (LARGE-related or MDC1D), as well as the allelic limb-girdle muscular dystrophy presentations for these genes. These congenital muscular dystrophies are associated with CNS pathology and structural eye abnormalities. Diagnosis depends on recognition of the clinical phenotype, brain MRI, muscle biopsy (Figure 1-7), and DNA analysis.
Fukuyama congenital muscular dystrophy
Fukuyama congenital muscular dystrophy is an autosomal recessive disorder and the most common type of muscular dystrophy in Japan,13 where the incidence is 0.7 to 1.2 per 10,000 births. It is caused by a founder mutation of the fukutin gene on chromosome 9q31.14 Most of the patients described are from Japan, but mutations have been described in other ethnic groups.
Patients with Fukuyama congenital muscular dystrophy manifest with decreased fetal movements, congenital hypotonia, weakness, poor suck, weak cry, and motor delay. Muscle weakness is most evident in the proximal muscles of the upper body and distal muscles of the lower body (especially the gastrocnemius muscles). Facial weakness may cause mouth-breathing with inability to close the lips. Joint contractures are not marked at birth, but hip, knee, and ankle contractures appear before the first birthday. Muscle hypertrophy of the tongue and calf may be seen. Functional disability is severe, and most of the patients are never able to walk; they usually become bedridden before 10 years of age. In all cases, severe mental retardation is evident, with IQ scores from 30 to 50. Seizures occur in 80% of patients, often with onset at 3 years of age.
Eye abnormalities occur in more than 50% of patients and include myopia, cataracts, abnormal eye movements, pale optic discs, and retinal detachment. Patients are not blind, in contrast to WWS.15
The most common and characteristic changes in the CNS are brain malformations, which include polymicrogyria, pachygyria, and agyria of cerebrum and cerebellum (type II lissencephaly). Other findings include delayed myelination, fibroglial proliferation of the leptomeninges, focal interhemispheric fusion, fusion of cerebellar folia, hydrocephalus, and hypoplasia of the corticospinal tracts.16
Cardiac involvement occurs in patients with Fukuyama congenital muscular dystrophy, often becoming evident in late childhood in the second decade. Nakanishi and colleagues17 analyzed the cardiac function in 34 patients with Fukuyama congenital muscular dystrophy. Abnormally increased left ventricular end diastolic diameter was observed in two patients (6%), and decreased left ventricular systolic function (decreased left ventricular shortening fraction and/or mean velocity of circumferential fiber shortening) in 16 patients (47%).
Muscle-eye-brain disease
MEB disease is an autosomal recessivedisorder caused by homozygous loss-of-function mutations of POMGnT1 on chromosome 1p34-p32. Most of the patients with MEB disease have been from a small, isolated population in Finland, but patients in China, Korea, and Japan have been reported.
Clinical features include decreased fetal movements in utero, severe congenital hypotonia, progressive weakness, and moderate to severe global developmental delay. Patients have profound facial and neck weakness, and many are unable to roll over or sit; some achieve minimal ambulation, but they become rapidly bedridden. Refractory epilepsy, severe intellectual disability, and behavioral disorders are common in MEB disease. Brain malformations include frontoparietal pachygyria, polymicrogyria, lissencephaly type II, hydrocephalus, cerebellar vermis hypoplasia, and brainstem hypoplasia. Structural eye abnormalities are very common, including microphthalmia, retinal detachment, retinal hypoplasia, anterior chamber malformation, and cataracts.
Walker-Warburg syndrome
WWS is an autosomal recessive disorder caused by mutations in the POMT1 (chromosomal locus 9q34.1), POMT2 (chromosomal locus 14q24.3), fukutin (FKTM), and FKRP genes. WWS is the most severe of the dystroglycanopathies. Patients present with paralysis at birth, severe hypotonia, weak cry, weak suck, and profound global developmental delay. With rare exceptions, patients never reach any psychomotor milestones; they usually have severe cognitive impairment and epilepsy. Brain abnormalities include microcephaly, hydrocephalus, cobblestone lissencephaly, severely hypoplastic cerebellum and brainstem, with a Dandy-Walker malformation in 15% to 20%. Ocular abnormalities include severe congenital myopia, glaucoma, pallor of the optic discs, retinal hypoplasia, microphthalmia, cataracts, and iris malformations. Cleft lip and palate and occipital encephalocele may distinguish WWS from other dystroglycanopathies.
CONGENITAL MYOPATHIES
Congenital myopathies are rare inherited neuromuscular disorders with early-onset hypotonia and weakness, normal to slightly elevated CK levels, and specific histopathologic characteristics on muscle biopsy. Congenital myopathies were named for histologic abnormalities seen in muscle described after the use of cryostat sections and histochemistry became known. They are usually nonprogressive with stable hypotonia and weakness.
The first description of a nonprogressive myopathy was by Magee and Shy in 1956.18 Since then, several other congenital myopathies have been described with different molecular backgrounds and phenotypic expressions. Identification of mutations and expansion of genotype-phenotype correlation has informed current classification.
Epidemiologic details of congenital myopathies are limited. Congenital myopathies are very rare, although they are more common than the congenital muscular dystrophies. The prevalence of specific congenital myopathies has only been estimated in some regions of the world. Recent studies have shown that core myopathy is more prevalent than nemaline myopathy.
In a recent study by Maggie and colleagues19 in the United Kingdom, 54 biopsies from patients with congenital myopathies were analyzed. More than 50% of the subjects had core myopathy (central core disease and multiminicore disease), and only 17% had nemaline myopathy. Another important finding was that more thanhalf of the genetically confirmed cases were due to ryanodine receptor (RYR1) mutations.
Other studies were from Amburgey and colleagues,20 who showed the prevalence of congenital myopathies in a US cohort, and Norwood and colleagues,21 who analyzed the prevalence of genetic muscle disease in northern England. In the latter study, the prevalence of congenital myopathy (adult and pediatric) was 1:135,000, with central core disease at 1:249,000.21,22,23
Clinical Aspects
Congenital myopathies have similar clinical and histologic phenotypes, so specific diagnosis may depend on mutation analysis. The typical clinical presentation may be indistinguishable from congenital muscular dystrophies. Congenital hypotonia, early muscle weakness, facial muscle involvement, and delayed acquisition of motor milestones are frequent manifestations. Facial weakness is often pronounced and predominately affects the lower face and mouth. In contrast to the congenital muscular dystrophies, congenital myopathies are nonprogressive or very slowly progressive. Some patients present late in childhood or adulthood with minimal symptoms.
Cognitive impairment is rare but may be seen in congenital fiber-type disproportion. Severe facial weakness with bulbar and respiratory muscle involvement in the newborn period is typically seen in nemaline myopathy and severe forms of centronuclear myopathy. Involvement of the extraocular muscles is a feature of centronuclear myopathies. Predominantly axial weakness with marked head lag, early-onset scoliosis, and respiratory muscle involvement out of proportion to skeletal muscle weakness are commonly seen in myopathies caused by SEPN1 mutations. Cardiac involvement is rare in congenital myopathies but may be seen with mutations in the titin (TTN) and cardiac myosin heavy chain 7 (MYH7) genes.
Orthopedic complications such as kyphoscoliosis, contractures, and fetal akinesia with arthrogryposis are particularly common in nemaline myopathy. Hip dislocation at birth is suggestive of RYR1 mutations but can be seen with all congenital myopathies. Distal muscle involvement is observed in patients with mutations in the nebulin gene (NEB), TPM3, MYH7, and DNM2, with foot drop and pes cavus.
Diagnosis
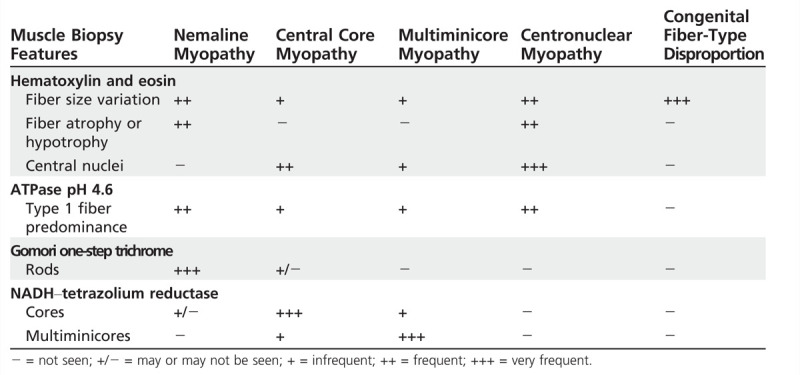
CK levels are either normal or mildly elevated. Electrophysiologic studies may rule out neuropathy, neuromuscular junction defects, and myotonia. In nemaline myopathy, 90% of the cases have normal CK, and the EMG shows myopathic features. Neuropathic changes can be seen in severe cases. Diagnosis depends on muscle biopsy with appropriate histochemical studies and electron microscopy. Biopsy findings will guide genetic testing and confirmation of the type of congenital myopathy (Table 1-3).
Table 1-3.
Muscle Biopsy Features of Congenital Myopathies
Classification
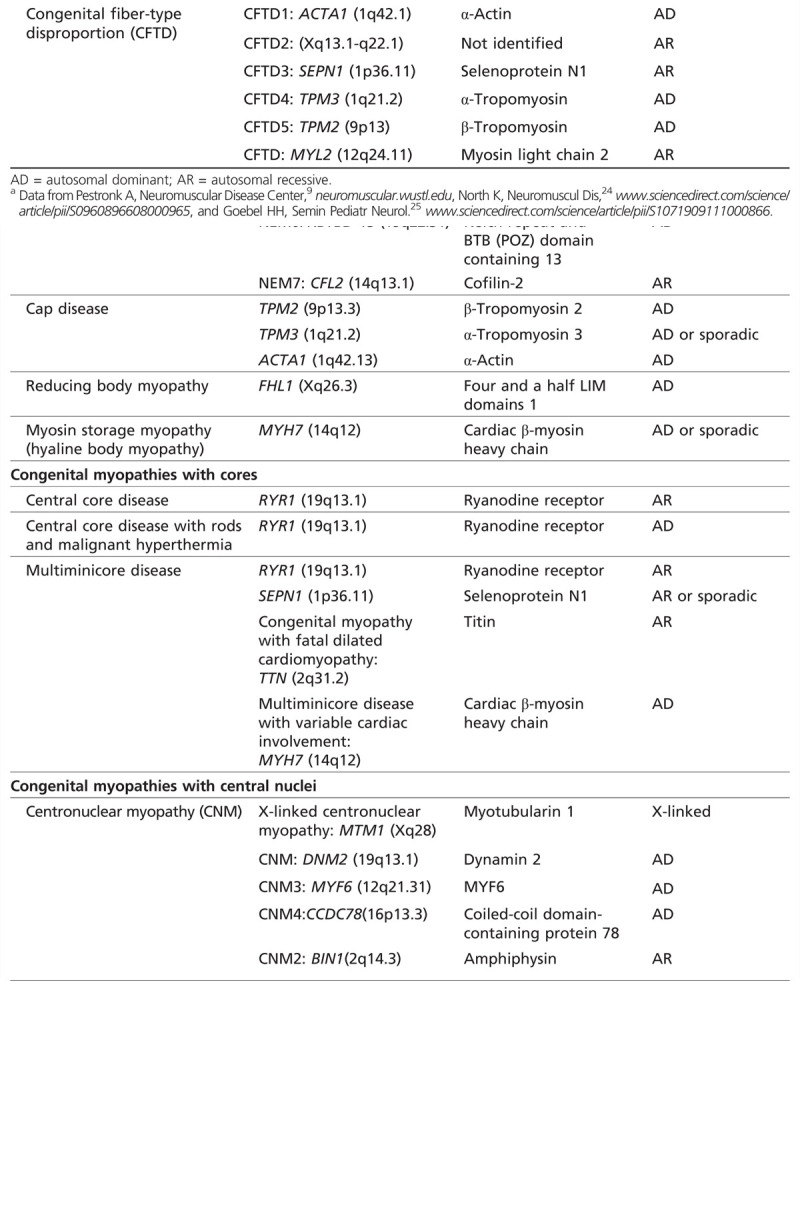
The congenital myopathies are named on the basis of the predominant histopathologic, immunohistochemical, and electron microscopic features (Table 1-424,25). The main categories are congenital myopathies with protein accumulation, congenital myopathies with cores, congenital myopathies with central nuclei, and congenital myopathies with fiber-type disproportion.
Table 1-4.
Classification of Congenital Myopathiesa
Congenital Myopathies With Protein Accumulation
The first clinical description of nemaline myopathy was by Shy and Engel in 1963.26 In 1995, Laing and colleagues27 identified the first genetic cause of nemaline myopathy: heterozygous mutation in the TPM3 gene, affecting members of a large family with autosomal dominantly inherited disease. Since then, mutations in seven other genes have been associated with nemaline myopathy. Fifty percent of nemaline myopathy cases are associated with mutations in NEB (nemaline myopathy type 2 [NEM2]) and 20% to 25% with ACTA1 mutations (nemaline myopathy type 3 [NEM3]).
Nemaline myopathies occur with several clinical presentations: severe congenital, intermediate congenital, typical congenital, childhood-onset, and adult-onset forms.28 Typical nemaline myopathy with infantile onset is characterized by generalized hypotonia, with particular weakness of the bulbar, neck flexor, and respiratory muscles. Weakness is usually proximal, but distal limb (knee flexors and ankle dorsiflexors) weakness eventually occurs. Facial weakness, nasal voice, high palate, and micrognathia are common. Extraocular muscles are usually spared as well as the heart, but not always. Respiratory involvement is frequently seen in the severe forms of nemaline myopathy.
Joint hypermobility, finger contractures, and chest deformities are common. The spine is hyperlordotic, sometimes rigid, and scoliosis may develop before or after puberty. However, weakness shows little or no progression (Figure 1-8).
Muscle biopsy. All of the known nemaline myopathy genes so far identified encode structural components of the sarcomeric thin filaments or proteins that help regulate turnover of the thin filament. Alteration causes formation of abnormal rodlike structures (nemaline bodies) that may disrupt the ability of the myofiber to generate adequate force during contraction. Alpha-actinin makes up most of the rod protein and stains like Z bands on electron microscopy. Rods typically tend to cluster under the sarcolemma but may be found diffusely scattered throughout the sarcoplasm or in the nucleus (intranuclear rods). Not all of the muscle fibers necessarily contain nemaline rods. Rods tend to occur in type 1 fibers and are visible on Gomori one-step trichrome stain as dark red-blue structures,29 between 1 and 7μm in length. Rods may increase in number with age. A commonly associated histopathologic feature is the predominance of type 1 fibers that are significantly smaller than type 2 fibers. These characteristics are shared by all of the nemaline myopathies (Figure 1-9, Figure 1-10).
Figure 1-10.
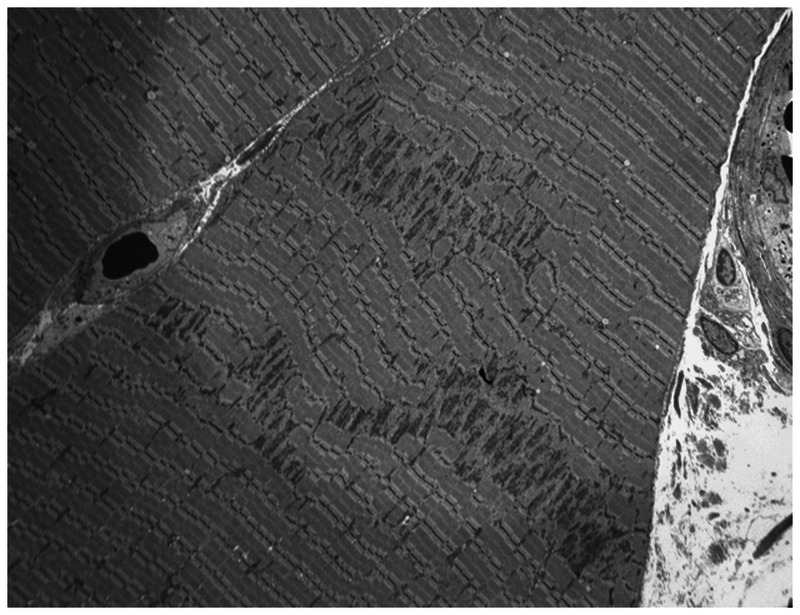
Electron microscopy showing minicores affecting focal areas of the fiber. A myofibrillary disruption and paucity of mitochondria is shown, as well as degeneration of the sarcomere and transverse tubules.
NEB-associated myopathies. Mutations in NEB located on chromosome 2q23.3 cause recessively inherited nemaline myopathy30 and account for 50% of all cases of nemaline myopathy. Wallgren-Pettersson and colleagues in 1999 reported 41 unrelated families with NEM2 suggested by linkage analysis. In the same year, Pelin and colleagues31 demonstrated mutations in the NEB gene in autosomal recessive typical nemaline myopathy. Since then more than 60 mutations have been described, with point mutations being most common. Mutations in NEB can also cause congenital fiber-type disproportion type 1, core-rod, cap, and zebra body myopathies.
ACTA1-related myopathies. Mutations in the gene encoding skeletal muscle actin (ACTA1), located on chromosome 1q42.13, cause sporadic or autosomal dominant nemaline myopathy and occasionally autosomal recessive nemaline myopathy. These mutations account for 20% to 25% of all cases of nemaline myopathy.32 ACTA1 mutations were first described by Nowak and colleagues.33 Fifteen mutations were identified in 14 families, 11 of which were associated with nemaline myopathy. Since then, at least four more mutations have been identified; most of these are heterozygous missense mutations.
Variable onset and clinical presentation within families is frequently observed.Mild phenotypes have symptoms after the first decade of life, with facial, axial, and proximal weakness. Progression may be very slow over decades, and respiratory failure can occur as late as the fifth decade of life.
De novo dominantly inherited mutations in ACTA1 account for 50% of patients with the severe congenital form of nemaline myopathy. Severe phenotypes present with marked congenital weakness, mainly axial and proximal but sparing the extraocular muscles. Bulbar symptoms and early respiratory failure are commonly seen. Rarely reported are frontal lobe hypoplasia and lateral ventricle dilatations, as well as skeletal dysplasia, hepatomegaly, and urinary tract stenosis. Cardiac function is usually normal. Death from respiratory failure may occur soon after birth.
Congenital Myopathies With Cores
Core myopathies, specifically central core disease and multiminicore disease, are characterized by the histopathologic finding of focally reduced oxidative and glycolytic enzymatic activity.34 Central cores extend along the longitudinal axis of the myofiber. Minicores are small multifocal areas, each affecting only a few sarcomeres. Core myopathies are the most common type of congenital myopathies in all ethnic groups. Mutations in skeletal muscle ryanodine receptor 1 (RYR1) gene are the most common cause of core myopathies, followed by SEPN1 mutations. All patients with core myopathies, regardless of mutation, are at high risk for malignant hyperthermia.
RYR1-related core myopathies. RyR1 is a ligand-gated calcium channel receptor, which serves as a calcium release channel of the sarcoplasmic reticulum as well as a bridging structure connecting the sarcoplasmic reticulum and transverse tubule. RYR1 gene mutations (located in chromosome 19q13.1) cause central core myopathy, multiminicore disease, congenital fiber-type disproportion, and malignant hyperthermia.
RYR1-related central core myopathy is caused by dominant (some de novo) mutations, usually missense. Patients present clinically with infantile onset of static or slowly progressive proximal weakness involving hip girdle and axial muscles or, less frequently, during adulthood with exclusive axial weakness. Almost all patients with central core disease achieve the ability to walk independently (Case 1-2). Extraocular muscles are spared, facial weakness is mild, and no cardiac involvement occurs. Bulbar and respiratory involvement are uncommon but have been observed in severe neonatal cases. Decreased fetal movements and breech presentation are seen, with or without hip dislocation and foot deformities. Exercise-induced myalgia and cramps are common.35,36 RYR1 mutations can cause malignant hyperthermia without myopathy.
Case 1-2
A 7-year-old girl had weakness, motor delay, and leg cramps. She had been born at full term with no complications during pregnancy or delivery but with congenital hip dislocation and clubfoot. She started walking at 19 months old and was a toe walker until 3 years of age. She had difficulties running and keeping up with her peers as well as climbing stairs and getting up from a squatting position. She had leg cramping almost every day. Cramps were partially relieved with warm baths, walking, or pain medication. She had one episode of rigidity during tonsillectomy. Her family history was remarkable for multiple family members with similar symptoms, including her maternal great-grandmother, mother, older sister, and younger twin brothers.
Physical examination revealed a narrow face, high palate, mild facial weakness, mild ptosis, generalized hypotonia, and mild proximal weakness. Shoulder abduction and hip flexion were 4+/5 bilaterally. Deep tendon reflexes were absent. She had mild thoracolumbar scoliosis. No ophthalmoplegia was evident, and forced vital capacity was 82% predicted. Echocardiogram was normal, as was CK, and EMG showed myopathic units. Muscle biopsy showed variable fiber size and central nuclei. Oxidative enzyme staining showed one or more large cores in most fibers and marked type 1 fiber predominance. RYR1 genetic analysis revealed a heterozygous deletion in exon 101 of the RYR1 gene.
Comment. This case is typical for a patient with central core myopathy. She had proximal weakness that was mild but with severe hypotonia. Her reports of exercise-induced myalgia and cramps are important clues to diagnosis, as is the history suggestive of an episode of malignant hyperthermia. Her diagnosis depended on muscle biopsy that showed central cores by histochemical stains, and DNA analysis confirmed the RYR1 mutation.
Her disease showed slow progression, and she never required ventilatory or nutritional support. She and her relatives remained at risk for malignant hyperthermia, and if she had children in the future, each would have a 50% risk of inheriting the disease.
Malignant hyperthermia is a hypermetabolic response to potent inhalation agents (such as halothane, sevoflurane, and desflurane), the depolarizing muscle relaxant succinylcholine, and rarely to stresses such as vigorous exercise and heat. Clinical signs are elevation of expired carbon dioxide, muscle rigidity, rhabdomyolysis, hyperthermia, acidosis, and hyperkalemia. Treatment must be emergent and include IV fluids, rapid cooling, and dantrolene.
Muscle biopsy. Central core myopathy muscle histology is dramatic and shows well-demarcated cores, located centrally, peripherally, or more than one per fiber and of variable size extending along the fiber length on longitudinal sections. Histochemical staining-specifically, NADH–tetrazolium reductase (TR)-shows absence of oxidative and glycolytic enzymatic activity from the central core region (Figure 1-11). These findings are also evident in other stains, such as succinate dehydrogenase and cytochrome c oxidase. Sometimes the cores are surrounded by a thin rim of high-oxidative enzyme activity, giving the appearance of “rimmed cores.” Central cores are typically found in type 1 fibers; marked type 1 predominance or uniformity on myosin ATPase staining and type 1 fiber hypotrophy are typical and may be the only abnormal features. An increase in internal nucleation and other myopathic features are often seen. The degree of histopathologic changes may be variable depending on sampling site and the age of the patient.
Figure 1-11.
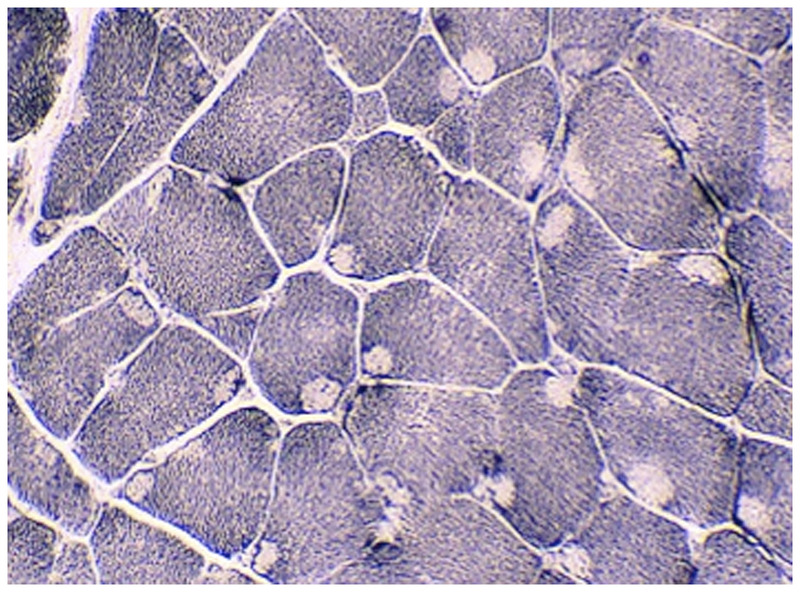
Central core myopathy. NADH–tetrazolium reductase–stained section of left quadriceps showing predominant type 1 fibers with cores affecting the majority of fibers. Cores extend along the fiber length on longitudinal sections and are typically well demarcated. Courtesy of Emily Herndon, MD.
Typical findings on electron microscopy include focal reduction or absence of mitochondria, variable degrees of myofibrillar disorganization, and accumulation of abnormal Z-band material within the core area. Some (“structured”) cores preserve a degree of myofibrillar organization and therefore retain some myosin ATPase activity; other (“unstructured”) cores do not and can be found in the same muscle biopsy. The sarcoplasmic reticulum and T tubule structures may be increased within the core area.
RYR1-related multiminicore disease. RYR1-related multiminicore disease is generally associated with recessive mutations. Patients may present during infancy or childhood with hypotonia, variable degrees of weakness (mostly axial muscles, pelvic girdle, and moderate hand weakness), occasional facial weakness, and extraocular muscle weakness (decreased horizontal and vertical gaze). Patients may report muscle pain after exertion. Hyperlaxity, heel cord contractures, and arthrogryposis at birth can be seen. The course is usually nonprogressive.
Bulbar involvement is less common than in nemaline myopathy, and respiratory insufficiency is rare. Right ventricular cardiac impairment secondary to respiratory failure was reported in severe cases. Other congenital cardiac defects, such as mitral valve prolapse, have been reported.
Muscle biopsy. Multiminicore disease must be diagnosed by muscle histology that demonstrates multiple “minicores” with sarcomeric disorganization and diminished oxidative activity that correlate with focal lack of mitochondria in muscle fibers (Figure 1-12). As with central cores, minicores may only be evident with use of certain histochemical stains. They appear as clear areas that do not stain with oxidativeenzymes like NADH-TR, succinate dehydrogenase, and cytochrome c oxidase. Minicores affect both type 1 and type 2 fibers and are short in length, spanning only a few sarcomeres in the muscle fiber longitudinal axis. Myosin ATPase may be normal, but frequently shows type 1 fiber predominance. Relative hypotrophy of type 1 fibers may be seen wherein the mean diameter of type 1 fibers is smaller than that of type 2 fibers.
Figure 1-12.
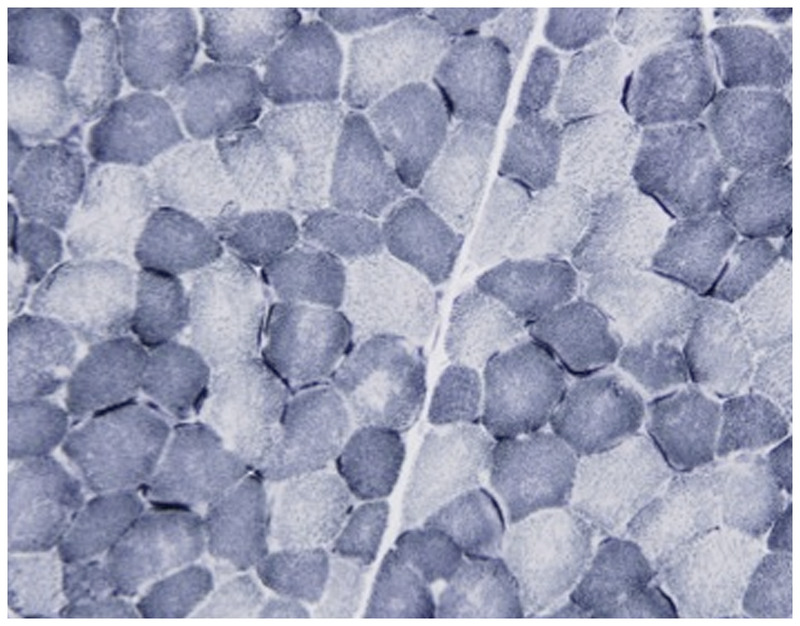
Multiminicore disease. NADH–tetrazolium reductase–stained section of left quadriceps demonstrating multiple areas in both fiber types of varying size and number devoid of oxidative enzyme stain compatible with multiminicore structures. Minicores are usually perpendicular to the longitudinal axis. Courtesy of Emily Herndon, MD.
Figure 1-3.
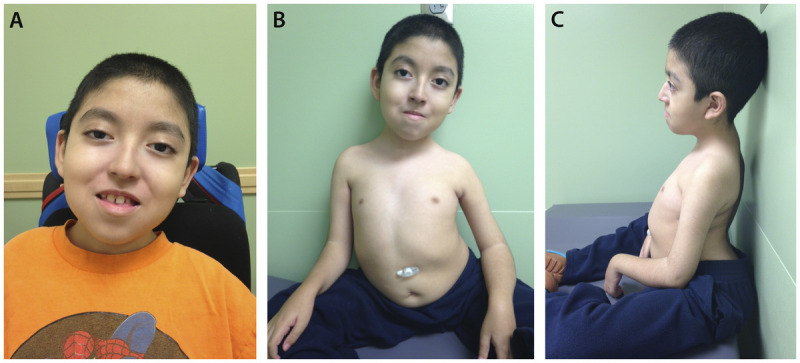
An 8-year-old boy with merosin-deficient congenital muscular dystrophy confirmed by DNA mutation analysis, which revealed a mutation of the LAMA 2 gene. A, Evidence of bilateral ptosis and facial weakness; B, severe dextroscoliosis and the presence of a gastrostomy tube; C, hyperlordosis and contractures at the elbow and wrist.
Figure 1-4.
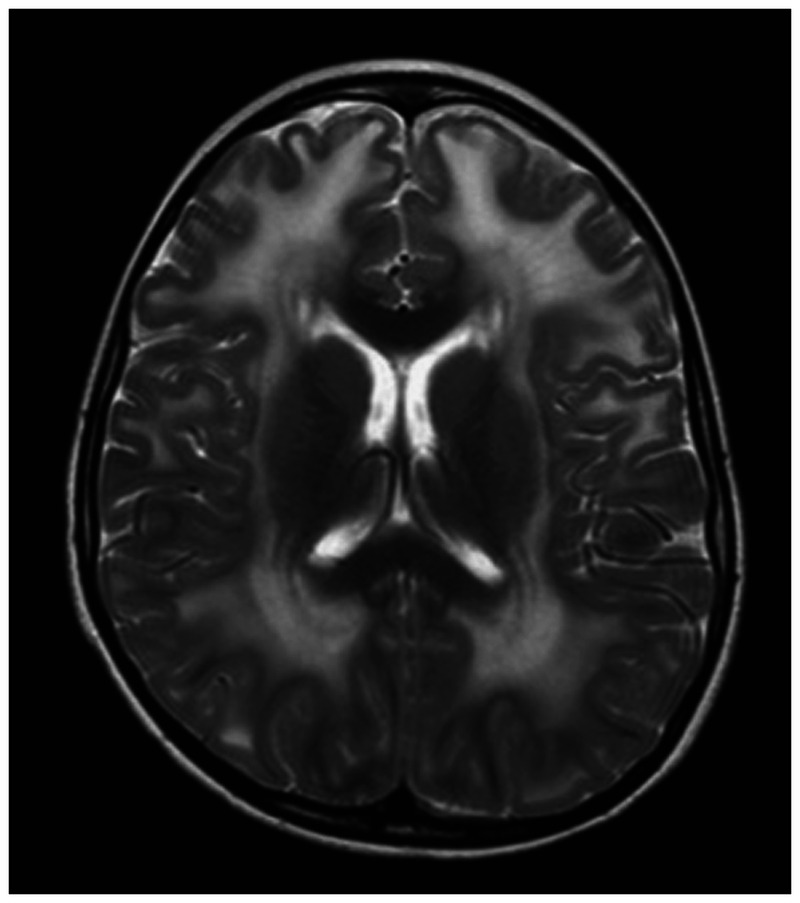
Brain MRI with diffuse prolonged T2 signal seen in the deep and subcortical white matter through both cerebral hemispheres in a 10-year-old boy with merosin-deficient congenital muscular dystrophy.
Figure 1-5.
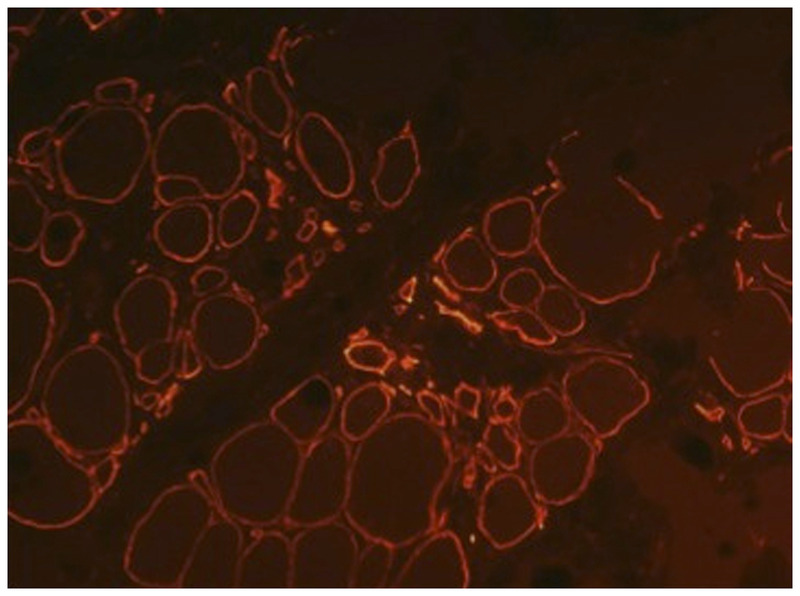
Merosin-deficient congenital muscular dystrophy (MDC1A). Immunohistochemical stain with an antibody to laminin α2 showing partial expression of merosin (200×).
Figure 1-6.
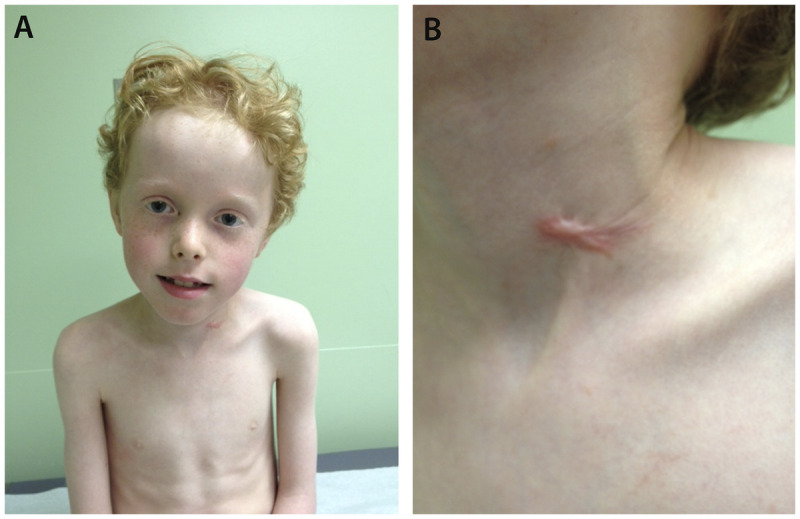
A 9-year-old boy with Ullrich congenital muscular dystrophy. A, Patient shows muscle wasting and bilateral elbow contractures and, B, abnormal keloid formation.
Figure 1-7.
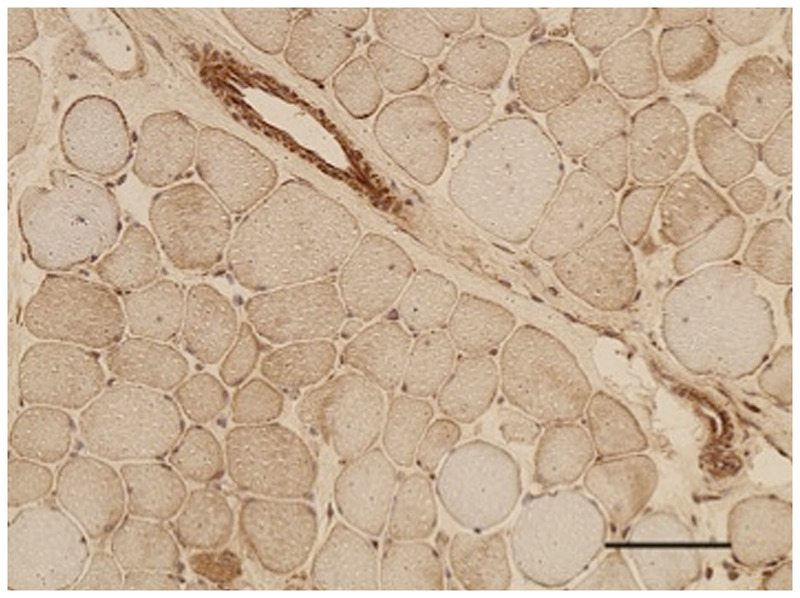
Dystroglycanopathy. Immunohistochemical stain with an antibody to α-dystroglycan reveals attenuated sarcolemmal reactivity in most fibers (200×). Courtesy of Emily Herndon, MD.
Figure 1-8.
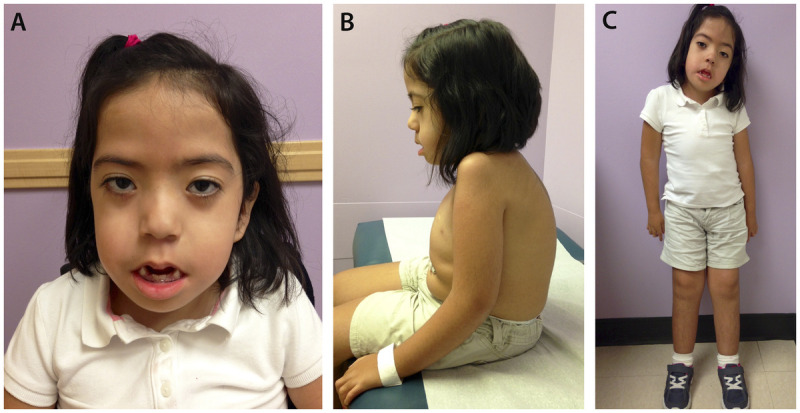
A 7-year-old girl with nemaline myopathy diagnosed by muscle biopsy. A, Myopathic face with severe facial weakness and micrognathia; B, chest wall deformity, kyphosis, and gastrostomy tube; C, dextroscoliosis and muscle atrophy more notable in the upper extremities.
Figure 1-9.
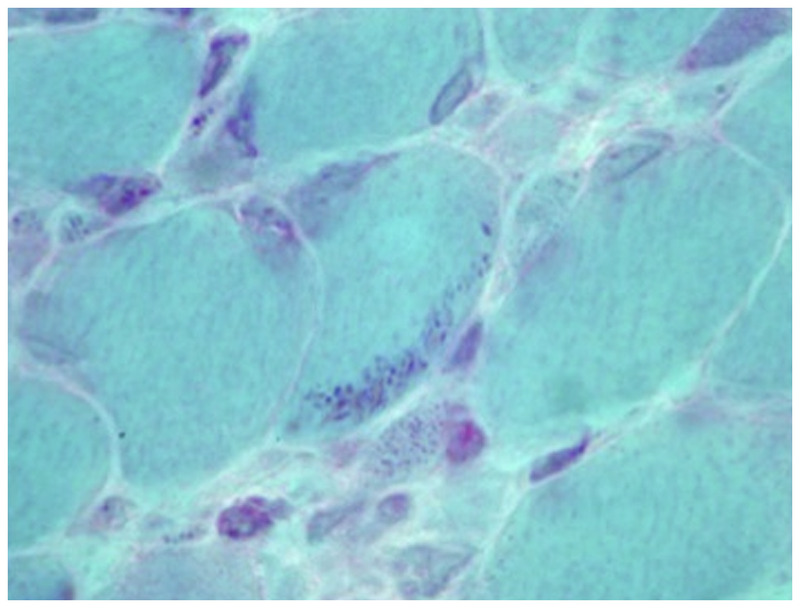
Nemaline myopathy. Gomori one-step trichrome-stained section of left quadriceps showing multiple clusters of red staining rods at the periphery of most fibers, which is compatible with nemaline rods (400×). Courtesy of Emily Herndon, MD.
For all RYR1-related core myopathies, the CK level is normal. Muscle MRI suggests that the most affected muscles are the quadriceps in the anterior thigh, in contrast to other congenital myopathies that usually affect the posterior thigh compartment. In the lower legs, abnormality of soleus and peroneal muscles is predominant.37
SEPN1-related core myopathies. The SEPN1 gene encodes selenoprotein N (SelN), which contains the amino acid selenocysteine (Sec). Mutations in this gene can prevent Sec incorporation and reduction of both SelN mitochondrial RNA and protein levels. Selenoprotein N expression occurs early in embryogenesis and appears to be necessary for intracellular calcium homeostasis and protection against redox-related cellular damage. SEPN1 gene (located on chromosome 1p36.11) mutations are associated with congenital muscular dystrophy with early spine rigidity, classical multiminicore disease, congenital fiber-type disproportion type 3, and desmin-related myopathy with Mallory body–like inclusions. SEPN1 mutations so far account for 50% of patients with the classic phenotype of multiminicore disease.38
Clinically, patients with SEPN1-related disease manifest with decreased fetal movements, hypotonia at birth, predominantly axial weakness, weak neck flexors, mild facial weakness, spinal rigidity, and scoliosis usually after the first decade of life. Patients have early motor developmental delay, but the majority of these patients achieve and maintain independent ambulation.7,39 Bulbar weakness is reported, but extraocular muscles are usually spared. Respiratory compromise is disproportionate to extremity weakness and progressive. Respiratory difficulties are more common after the first decade, associated with nocturnal hypoventilation.40 Cardiac involvement is rare, but septal defects and conduction block have been described.
Muscle biopsy. Muscle histology looks exactly like that of RYR1-related multiminicore disease. Some patients may show no cores or minicores but only mild myopathic changes with small, type 1 fibers.
Centronuclear (Myotubular) Myopathies
The centronuclear myopathies are characterized by abnormal muscle histology with internal nuclei, sometimes in central chains with longitudinal orientation as seen in embryonic myotubes. Centronuclear myopathy can be classified according to the mode of inheritance and clinical presentation. Centronuclear myopathies can be associated with X-linked, dominant, or recessive mutations. The most common and severe is the X-linked centronuclear myopathy. Less common and usually less severe are those inherited as autosomal dominant or recessive diseases.41
X-linked centronuclear myopathy is caused by mutations in the MTM1 gene located on chromosome Xq28, causing abnormalities in the myotubularin protein. Male infants have marked hypotonia and symmetric proximal and distal muscle weakness. Respiratory muscle impairment leads to early respiratory failure. Facial weakness, ptosis, and ophthalmoplegia are common, as well as impaired bulbar function with feeding difficulties. Skeletal features typically include macrocephaly, narrowface, and long digits. Systemic features may include pyloric stenosis, spherocytosis, gallstones, kidney stones, bleeding diathesis, and hepatic dysfunction.42 Heterozygous female carriers of mutations may present with limb-girdle and facial weakness. Weakness is nonprogressive, but most survivors (80%) remain ventilator dependent.
Muscle biopsy. Muscle histology shows a majority of muscle fibers with central nuclei and predominance of type 1 fibers. In transverse sections, the central areas of the fibers show increased periodic acid-Schiff staining with reduced myofibrillar ATPase reaction. The histologic findings are similar to those seen in congenital myotonic muscular dystrophy (DM1). In infancy, centronuclear myopathy must be distinguished from DM1, generally by mutation analysis.
Congenital Fiber-Type Size Disproportion
Congenital fiber-type size disproportion is defined by the muscle biopsy finding of small, type 1 fibers compared to type 2. Diagnosis is considered after other congenital myopathies have been excluded, because the finding of small, predominately type 1 fibers can be seen in any of the congenital myopathies. The absence of rods, cores, or centronuclear chains makes congenital fiber-type size disproportion a diagnosis of exclusion.
Congenital and childhood presentation with mild to severe generalized hypotonia and weakness of the extremities, face, and bulbar and respiratory muscles is similar to all other congenital myopathies. Ophthalmoplegia can be infrequently seen, as well as skeletal abnormalities such as congenital arthrogryposis and scoliosis.43,44 The clinical course can be static in half of the patients; otherwise, improvement of the symptoms or slow progression can be seen. CK levels are normal or less than 3 times the upper limit of normal. Diagnosis depends on the muscle biopsy findings, and testing for at least five different genes associated with congenital fiber type size disproportion is available.
USEFUL WEBSITES
CureCMD
curecmd.org
Joshua Frase Foundation
A Foundation Building Strength for Nemaline Myopathy
http://www.buildingstrength.org
Muscular Dystrophy Association
mda.org
KEY POINTS
Congenital muscular dystrophy manifests at birth or within the first 2 years of life.
CNS involvement and MRI findings are key in the differential diagnosis of congenital muscular dystrophy.
The most important diagnostic tools for congenital muscular dystrophy are creatine kinase level, nerve conduction study or EMG with or without repetitive nerve stimulation, brain MRI, muscle biopsy, and specific genetic or metabolic testing.
In congenital muscular dystrophy, the muscle biopsy shows dystrophic changes.
The major laminin of adult skeletal muscle is laminin-2 (also known as merosin), and only mutations of LAMA2 gene encoding laminin α2 cause muscular dystrophy.
Patients with complete absence of merosin expression present with early hypotonia, weakness (facial, proximal, and distal limb muscles), and delayed milestones. They never walk but have normal cognition despite white matter abnormalities.
Patients with total absence of merosin have an earlier presentation, are more likely to lack independent ambulation, and require enteral feedings and ventilator support. Seizures (simple partial, complex partial, absence, or generalized) are reported in around 23% of patients with residual merosin and in 15% of patients with absent merosin.
In merosin-deficient congenital muscular dystrophy, peripheral nerve may be affected, causing a motor and sensory neuropathy.
Collagen VI–deficient congenital muscular dystrophy is the second most common variant of congenital muscular dystrophy worldwide.
The classical clinical features of Ullrich congenital muscular dystrophy are congenital hypotonia, scoliosis or kyphosis of the spine, proximal joint contractures, torticollis, and distal joint hyperlaxity. Congenital hip dislocation may be present. The skin typically shows follicular hyperkeratosis (keratosis pilaris) over the extensor surfaces of the arms and legs, and abnormal keloid formation.
Bethlem myopathy is a mild disorder that is allelic to Ullrich congenital muscular dystrophy and inherited as autosomal dominant.
The dystroglycanopathies are a group of disorders caused by abnormal glycosylation of α-dystroglycan.
Fukuyama congenital muscular dystrophy is an autosomal recessive disorder and is the most common type of muscular dystrophy in Japan.
Functional disability in patients with Fukuyama congenital muscular dystrophy is severe, and most of the patients are never able to walk; they usually become bedridden before 10 years of age.
Seizures occur in 80% of patients with Fukuyama congenital muscular dystrophy, often with onset at 3 years of age.
Most of the patients with muscle-eye-brain disease have been from a small, isolated population in Finland, but patients in China, Korea, and Japan have been reported.
Refractory epilepsy, severe intellectual disability, and behavioral disorders are common in muscle-eye-brain disease.
Walker-Warburg syndrome is the most severe of the dystroglycanopathies.
Ocular abnormalities in Walker-Warburg syndrome include severe congenital myopia, glaucoma, pallor of the optic discs, retinal hypoplasia, microphthalmia, cataracts, and iris malformations.
Congenital myopathies are rare inherited neuromuscular disorders with early-onset hypotonia and weakness, normal to slightly elevated creatine kinase levels, and specific histopathologic characteristics on muscle biopsy.
Congenital myopathies have similar clinical and histologic phenotypes, so specific diagnosis may depend on mutation analysis.
In contrast to the congenital muscular dystrophies, congenital myopathies are nonprogressive or very slowly progressive.
Diagnosis of congenital myopathies depends on muscle biopsy with appropriate histochemical studies and electron microscopy. Biopsy findings will guide genetic testing and confirmation of the type of congenital myopathy.
Fifty percent of nemaline myopathy cases are associated with mutations in NEB (nemaline myopathy type 2) and 20% to 25% with ACTA1 mutations (nemaline myopathy type 3).
Nemaline myopathies occur with several clinical presentations: severe congenital, intermediate congenital, typical congenital, childhood-onset, and adult-onset forms.
All of the known nemaline myopathy genes so far identified encode structural components of the sarcomeric thin filaments or proteins that help regulate turnover of the thin filament.
A commonly associated histopathologic feature of nemaline myopathy is the predominance of type 1 fibers that are significantly smaller than type 2 fibers.
De novo dominantly inherited mutations in ACTA1 account for 50% of patients with the severe congenital form of nemaline myopathy.
All patients with core myopathies, regardless of mutation, are at high risk for malignant hyperthermia.
Exercise-induced myalgia and cramps are common in patients with central core disease.
Malignant hyperthermia is a hypermetabolic response to potent inhalation agents (such as halothane, sevoflurane, and desflurane), the depolarizing muscle relaxant succinylcholine, and rarely to stresses such as vigorous exercise and heat.
SEPN1 mutations so far account for 50% of patients with the classic phenotype of multiminicore disease.
Centronuclear myopathies can be associated with X-linked, dominant, or recessive mutations.
The absence of rods, cores, or centronuclear chains makes congenital fiber-type size disproportion a diagnosis of exclusion.
Footnotes
Relationship Disclosure: Dr Iannaccone has received research support from GlaxoSmithKline; Isis Pharmaceuticals, Inc; the NIH; and Santhera Pharmaceuticals. Dr Castro reports no disclosure.
Unlabeled Use of Products/Investigational Use Disclosure: Drs Iannaccone and Castro report no disclosures.
REFERENCES
- 1.Tome FM,, Evangelista T,, Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency. CR Acad Sci III 1994; 317 (4): 351–357. [PubMed] [Google Scholar]
- 2.Clement EM,, Feng L,, Mein R, et al. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001-2008. Neuromuscul Disord 2012; 22 (6): 522–527. [DOI] [PubMed] [Google Scholar]
- 3.Peat RA,, Smith JM,, Compton AG, et al. Diagnosis and etiology of congenital muscular dystrophy. Neurology 2008; 71 (5): 312–321. [DOI] [PubMed] [Google Scholar]
- 4.Okada M,, Kawahara G,, Noguchi S, et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007; 69 (10): 1035–1042. [DOI] [PubMed] [Google Scholar]
- 5.Geranmayeh F,, Clement E,, Feng LH, et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 2010; 20 (4): 241–250. [DOI] [PubMed] [Google Scholar]
- 6.Pane M,, Messina S,, Vasco G, et al. Respiratory and cardiac function in congenital muscular dystrophies with alpha dystroglycan deficiency. Neuromuscul Disord 2012; 22 (8): 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scoto M,, Cirak S,, Mein R, et al. SEPN1-related myopathies: clinical course in a large cohort of patients. Neurology 2011; 76 (24): 2073–2078. [DOI] [PubMed] [Google Scholar]
- 8.Mercuri E,, Muntoni F. Muscular dystrophies. Lancet 2013; 381 (9869): 845–860. [DOI] [PubMed] [Google Scholar]
- 9.Pestronk A. Neuromuscular Disease Center. neuromuscular.wustl.edu. Accessed July 30, 2013.
- 10.Muntoni F,, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004; 14 (10): 635–649. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan J-C. GeneTable of neuromuscular disorders. World Muscle Society. http://www.musclegenetable.fr. Updated January 9, 2013. Accessed July 30, 2013.
- 12.Philpot J,, Cowan F,, Pennock J, et al. Merosin-deficient congenital muscular dystrophy: the spectrum of brain involvement on magnetic resonance imaging. Neuromuscul Disord 1999; 9 (2): 81–85. [DOI] [PubMed] [Google Scholar]
- 13.Fukuyama Y,, Osawa M. A genetic study of the Fukuyama type congenital muscular dystrophy. Brain Dev 1984; 6 (4): 373–390. [DOI] [PubMed] [Google Scholar]
- 14.Toda T,, Kobayashi K,, Kondo-Iida E, et al. The Fukuyama congenital muscular dystrophy story. Neuromuscul Disord 2000; 10 (3): 153–159. [DOI] [PubMed] [Google Scholar]
- 15.Chijiiwa T,, Nishimura M,, Inomata EI, et al. Ocular manifestations of congenital muscular dystrophy (Fukuyama type). Ann Ophthalmol 1983; 15 (10): 921–928. [PubMed] [Google Scholar]
- 16.Yoshioka M,, Saiwai S,, Kuroki S,, Nigami H. MR imaging of the brain in Fukuyama-type congenital muscular dystrophy. AJNR Am J Neuroradiol 1991; 12 (1): 63–65. [PMC free article] [PubMed] [Google Scholar]
- 17.Nakanishi T,, Sakauchi M,, Kaneda Y, et al. Cardiac involvement in Fukuyama-type congenital muscular dystrophy. Pediatrics 2006; 117 (6): e1187–e1192. [DOI] [PubMed] [Google Scholar]
- 18.Magee KR,, Shy GM. A new congenital non-progressive myopathy. Brain 1956; 79 (4): 610–621. [DOI] [PubMed] [Google Scholar]
- 19.Maggie L,, Scoto M,, Cirak S, et al. Congenital myopathies-clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul Disord 2013; 23 (3): 195–205. [DOI] [PubMed] [Google Scholar]
- 20.Amburgey K,, McNamara N,, Bennet LR, et al. Prevalence of congenital myopathies in a representative pediatric United States population. Ann Neurol 2011; 70 (4): 662–665. [DOI] [PubMed] [Google Scholar]
- 21.Norwood FL,, Harling C,, Chinnery PF, et al. Prevalence of genetic muscle disease on Northern England: in-depth analysis of a muscle clinic population. Brain 2009; 132 (pt 11): 3175–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallgren-Pettersson C,, Pelin K,, Hilpelä P, et al. Clinical and genetic heterogeneity in autosomal recessive nemaline myopathy. Neuromusc Disord 1999; 9 (8): 564–572. [DOI] [PubMed] [Google Scholar]
- 23.Feng JJ,, Marston S. Genotype-phenotype correlations in ACTA1 mutations that cause congenital myopathies. Neuromuscul Disord 2009; 19 (1): 6–16. [DOI] [PubMed] [Google Scholar]
- 24.North K. What’s new in congenital myopathies? Neuromuscul Disord 2008; 18 (6): 433–442. [DOI] [PubMed] [Google Scholar]
- 25.Goebel HH. Congenital myopathies. Introduction. Semin Pediatr Neurol 2011; 18 (4): 213–215. [DOI] [PubMed] [Google Scholar]
- 26.Shy GM,, Engel WK,, Somers JE,, Wan T. Nemaline myopathy: a new congenital myopathy. Brain 1963; 86: 793–810. [DOI] [PubMed] [Google Scholar]
- 27.Laing NG,, Wilton SD. A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy NEM1. Nat Genet 1995; 10 (2): 249. [DOI] [PubMed] [Google Scholar]
- 28.Ryan MM,, Schnell C,, Strickland CD, et al. Nemaline myopathy: a clinical study of 143 cases. Ann Neurol 2001; 50 (3): 312–320. [DOI] [PubMed] [Google Scholar]
- 29.Gonatas NK. The fine structure of the rod-like bodies in nemaline myopathy and their relation to the Z-discs. J Neuropathol Exp Neurol 1966; 25 (3): 409–421. [DOI] [PubMed] [Google Scholar]
- 30.Arts WF,, Bethlem J,, Dingemans KP,, Eriksson AW. Investigations on the inheritance of nemaline myopathy. Arch Neurol 1978; 35 (2): 72–77. [DOI] [PubMed] [Google Scholar]
- 31.Pelin K,, Hilpela P,, Donner K, et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci U S A 1999; 96 (5): 2305–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sparrow JC,, Nowak KJ,, Durling HJ, et al. Muscle disease caused by mutations in the skeletal muscle alpha-actin gene (ACTA1). Neuromuscul Disord 2003; 13 (7–8): 519–531. [DOI] [PubMed] [Google Scholar]
- 33.Nowak KJ,, Wattanasirichaigoon D,, Goebel HH, et al. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet 1999; 23 (2): 208–212. [DOI] [PubMed] [Google Scholar]
- 34.Dubowitz V,, Pearse AG. Oxidative enzymes and phosphorylase in central-core disease of muscle. Lancet 1960; 2 (7140): 23–24. [DOI] [PubMed] [Google Scholar]
- 35.Jungbluth H,, Lillis S,, Zhou H, et al. Late-onset axial myopathy with cores due to a novel heterozygous dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord 2009; 19 (5): 344–347. [DOI] [PubMed] [Google Scholar]
- 36.Patterson VH,, Hill TR,, Fletcher PJ,, Heron JR. Central core disease: clinical and pathological evidence of progression within a family. Brain 1979; 102 (3): 581–594. [DOI] [PubMed] [Google Scholar]
- 37.Jungbluth H,, Sewry C,, Brown SC, et al. Minicore myopathy in children: a clinical and histopathological study of 19 cases. Neuromuscul Disord 2000; 10 (4–5): 264–273. [DOI] [PubMed] [Google Scholar]
- 38.Ferreiro A,, Estournet B,, Chateau D, et al. Multi-minicore disease-searching for boundaries: phenotype analysis of 38 cases. Ann Neurol 2000; 48 (5): 745–757. [PubMed] [Google Scholar]
- 39.Ferreiro A,, Quijano-Roy S,, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 2002; 71 (4): 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rowe PW,, Eagle M,, Pollitt C, et al. Multicore myopathy: respiratory failure and paraspinal muscle contractures are important complications. Dev Med Child Neurol 2000; 42 (5): 340–343. [DOI] [PubMed] [Google Scholar]
- 41.Romero B. Centronuclear myopathies: a widening concept. Neuromuscul Disord 2010; 20 (4): 223–228. [DOI] [PubMed] [Google Scholar]
- 42.Herman GE,, Finegold M. Medical complications in long-term survivors with X-linked myotubular myopathy. J Pediatr 1999; 134 (2): 206. [DOI] [PubMed] [Google Scholar]
- 43.Clarke NF. Congenital fibre type disproportion-a syndrome at the crossroads of the congenital myopathies. Neuromuscul Disord 2011; 21 (4): 252–253. [DOI] [PubMed] [Google Scholar]
- 44.Nance J,, Dowling J,, Gibbs E,, Bonnemann C. Congenital myopathies: an update. Curr Neurol Neurosci Rep 2012; 12 (2): 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]