

Abstract
Purpose of Review:
This article provides an overview of the neurologic complications found in the various endocrine disorders affecting adult patients. Specifically, disorders in pituitary hormones (prolactin, growth hormone, vasopressin, and oxytocin), thyroid hormones, adrenal hormones (glucocorticoids), and sex hormones (estrogen and testosterone) will be covered, with an emphasis on identifying the signs and symptoms in addition to diagnosing and managing these disorders.
Recent Findings:
Hyperthyroidism in the young was found to increase the risk for ischemic stroke in a recent prospective case-cohort study. The cognitive effects of hormonal therapy in postmenopausal women remain controversial, but a recent study found no benefit or risk in cognitive function when treating younger (50 to 55 years of age) postmenopausal women with hormonal therapy.
Summary:
Endocrine disorders can cause various neurologic complications, from insidious myopathy to acute encephalopathy. Diagnosing the endocrine disorder as the cause of the neurologic impairment is essential, as treating the underlying hormonal dysfunction will often rapidly reverse the neurologic symptoms. Ongoing research is needed to further clarify the role of hormonal dysfunction in neurologic disorders.
INTRODUCTION
Endocrine disorders, whether by excess or deficiency of any given hormone, cause widespread abnormal effects on numerous organs, often including the central and peripheral nervous systems. The neurologic complications of endocrine disorders have disparate manifestations, from subtle cognitive decline in a hypothyroid patient to motor abnormalities such as proximal muscle weakness in a patient with Cushing disease. The symptoms can range from indolent and chronic in nature to acute emergencies, such as myxedema coma. A neurologist may see a patient with primary endocrine disorders after a referral from an internist or endocrinologist to assist in the management of neurologic complications of his or her primary endocrine disease; however, since the neurologic complication may be the primary or initial symptom, the neurologist may be the first physician seeing the patient (Case 3-1). Because many of the neurologic complications from endocrine disorders are reversible, it is important for neurologists to be able to identify and manage neurologic complications of endocrine disorders. This review focuses primarily on the neurologic complications of nondiabetic endocrine disorders in adult patients. (For diabetes-related neuromuscular complications, see the article “Neuromuscular Complications of Diabetes Mellitus” by Dr Vera Bril in this issue of CONTINUUM.) This article does not address specific treatment and management of hypothalamic and pituitary tumors.
Case 3-1
A 24-year-old woman was brought to the hospital by her husband for evaluation of her anxiety, abnormal movements, and bizarre behavior. Several weeks before presentation, she developed progressively increasing anxiety, nervousness that had progressed recently to include wild mood swings, and bizarre behavior including inappropriate laughter. She had involuntary weight loss despite increased appetite. In the past 6 days, she began to have uncontrollable movements of her arms, feet, and head, including her tongue. These movements appeared to largely subside when she was sleeping. She took no medications, and her medical history was unremarkable, with no known neurologic diseases in her family.
On physical examination, the patient was thin, diaphoretic, and noticeably writhing her limbs and head. She was noted by the emergency department physician to have a diffusely enlarged thyroid gland on palpation. Her vital signs were remarkable for sinus tachycardia to 120 beats/min and an oral temperature of 39°C (102.2°F). Her speech was severely impaired by her continuous tongue movements, but she appeared to be able to follow commands, although at times she inappropriately burst out laughing or crying. The remainder of her neurologic examination was notable for choreiform movements of her bilateral limbs, head, and tongue, which severely impaired her gait as well as any coordinated movements. On command, she could temporarily suppress the choreiform movements of her arms and legs, but they would resume soon after.
Comment. This patient showed gradually worsening signs and symptoms of thyrotoxicosis. Chorea may be less appreciated as a symptom of thyrotoxicosis and therefore may lead to a mistaken initial diagnosis of Huntington disease. This would be a serious mistake, because thyrotoxicosis can be medically treated and may lead to devastating consequences if left untreated. This patient’s blood work was consistent with thyrotoxicosis, with an undetectable thyroid-stimulating hormone and abnormally high free thyroxine (T4) level. An endocrinologist started her on propranolol and propylthiouracil, which led to gradual improvement and resolution of her symptoms. Further endocrinologic diagnostic testing and management of her thyrotoxicosis was conducted once she was stable.
ENDOCRINE SYSTEMS
The endocrine system encompasses hormones released by cells that affect downstream target cells, usually after traveling through the circulatory system. The classic endocrine system can be conceptualized as a three-tiered pathway comprising the hypothalamus–pituitary–target organ (ie, thyroid gland, adrenal glands, and sex organs) as illustrated in Figure 3-1.1 Tight feedback regulation at each tier ensures that a homeostatic balance is achieved under various physiologic conditions. Endocrine diseases can be broadly categorized into three categories: (1) hormone deficiency; (2) hormone excess; and (3) altered tissue response to the hormone. Any of these mechanisms can lead to disruption of the homeostatic pathway, resulting in a myriad of clinical complications.
Figure 3-1.
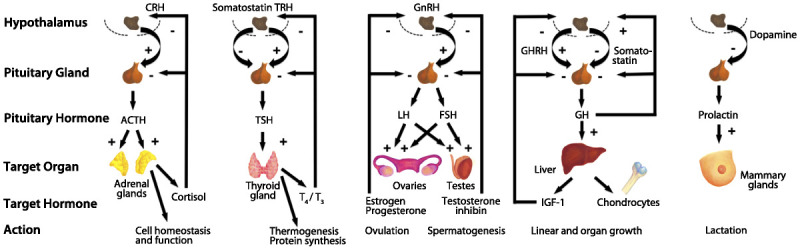
Control of hypothalamic–pituitary–target organ axes. CRH = corticotropin-releasing hormone; TRH = thyrotropin-releasing hormone; GnRH = gonadotropin-releasing hormone; GHRH = growth hormone–releasing hormone; ACTH = adrenocorticotropic hormone; TSH = thyroid-stimulating hormone; LH = luteinizing hormone; FSH = follicle-stimulating hormone; GH = growth hormone; T4 = thyroxine; T3 = triiodothyronine; IGF-1 = insulinlike growth factor 1. Modified with permission from Melmed S, J Clin Invest.1 © 2003, American Society for Clinical Investigation. www.jci.org/articles/view/20401. Figure redrawn by Chihiro Saka.
Pituitary Hormone Disorders
The pituitary gland has been called the “master gland” because it releases many of the regulatory hormones that affect downstream organs and their hormones (Figure 3-11). Pituitary disorders can arise from only a single hormonal pathway, such as a pituitary adenoma that secretes excess growth hormone (acromegaly), or can affect multiple hormonal pathways, such as due to hypopituitarism after pituitary apoplexy. Pituitary diseases often affect more than one hormonal pathway; therefore, a comprehensive endocrine evaluation is needed for any patient suspected to have a pituitary hormone disorder.
Prolactin. Prolactin is best known for its role in lactation and mammary gland development but can also affect sexual behavior and possibly other physiologic functions, such as the immune system.2 One of the major negative regulators for prolactin is dopamine released from the hypothalamus. Therefore, any alterations in dopaminergic signaling can affect prolactin levels, including those due to antipsychotic medications, especially the older “typical” agents, such as haloperidol, as well as some “atypical” ones, including risperidone.3
Generalized tonic-clonic and complex partial seizures have been associated with an acute and temporary rise in blood prolactin levels.4 Screening for elevated prolactin levels has been described as “probably a useful adjunct” in the appropriate clinical setting in distinguishing between epileptic and nonepileptic events (class B evidence) if a blood sample can be drawn within 10 to 20 minutes of the event.4 However, a normal serum prolactin level by itself is insufficient to make a diagnosis of a nonepileptic event because of its low sensitivity and low negative predictive value. Furthermore, other conditions, including syncope, may also cause hyperprolactinemia, thereby limiting the utility of screening for elevated prolactin levels.
Persistently elevated prolactin levels are most commonly associated with pituitary adenomas secreting prolactin (prolactinomas). The major symptoms from hyperprolactinemia are largely non-neurologic and include menstrual disorders, galactorrhea, osteoporosis, sexual dysfunction, and breast enlargement. Prolactinomas, like all pituitary masses, can cause headaches if large enough and may also lead to compression of cranial nerves in the cavernous sinuses when extending laterally, and in the optic chiasm when extending superiorly.5 Persistently elevated prolactin levels alone have not been reported to cause any significant neurologic complication. Multiple sclerosis has been recently associated with hyperprolactinemia, but the clinical significance remains unclear.6 No clinically significant neurologic complication from low prolactin levels has been reported.
Growth hormone. Growth hormone (GH), also known as somatotropin, is a polypeptide hormone released from the anterior pituitary gland, and its principle role is to mediate growth and metabolic functions.7 GH is regulated by hypothalamic signals, including GH-releasing hormone, which activates GH, and somatostatin, which inhibits GH release (Figure 3-11). Various other neuropeptides, neurotransmitters, hormones, and physiologic states, such as deep sleep, can also affect GH levels. One of the primary peripheral targets for GH is the liver, where GH activates its receptor to release insulinlike growth factor 1 (IGF-1), a major mediator of the growth-promoting effects of GH.
Excess GH in adults causes acromegaly and is most commonly caused by GH-secreting pituitary adenomas (or, rarely, by ectopic GH-releasing hormone secretion leading to pituitary hyperplasia).7 When this occurs in childhood, gigantism with accompanying acral and musculoskeletal changes is the most common manifestation. Other, more subtle changes can take years to manifest, however, and can occur in adult-onset acromegaly. These include reproductive and sexual abnormalities; visceromegaly (eg, tongue and thyroid) cardiac hypertrophy; hypertension; worsening carbohydrate and lipid metabolism leading to increased cardiovascular risk; and an increased incidence of colorectal neoplasms. The most common neurologic symptoms in acromegaly are usually neuromuscular in nature. Median neuropathy is common, with up to 64% of patients with acromegaly experiencing clinical symptoms attributable to median neuropathy and an even higher percentage of patients showing subclinical findings on nerve conduction studies.8 Treatment for median neuropathy in patients with acromegaly includes treating the underlying disease (ie, pituitary adenoma) as well as conservative and surgical management, just as in patients without acromegaly. In addition to peripheral neuropathies, proximal myopathy of unclear etiology can also be seen in some patients with acromegaly.9 Finally, sleep apnea (predominantly obstructive but also central) is common in acromegaly.7 Because acromegaly is an insidious disease, with diagnosis delayed 4 to 10 years after initial symptom onset, neurologists should be familiar with the overall clinical constellation associated with GH excess because the initial visit may be for the neurologic complications of the disease. Diagnosis for acromegaly can be made by measuring serum IGF-1 levels; if these are elevated, this is followed by measuring GH levels after an oral glucose load (GH suppression test).7 Abnormal results should trigger complete pituitary hormone testing and contrast-enhanced MRI of the sella turcica to localize and delineate the tumor.
GH deficiency during infancy or childhood results primarily in growth retardation and short stature. GH deficiency during adulthood can result in a different clinical picture, often presenting with seemingly nonspecific symptoms of fatigue, lack of energy, and reduced physical capacity.10 The signs of GH deficiency are also nonspecific and may include increased adiposity (mostly central) and decreased lean mass and bone mineral density, as well as hyperlipidemia and glucose intolerance or diabetes mellitus. In addition to nonspecific symptoms of low energy and mood, neurologic complications of GH deficiency in adult patients may also include poor concentration and memory. These symptoms generally improve with GH replacement therapy.
Vasopressin (antidiuretic hormone). Vasopressin, also known as arginine vasopressin or antidiuretic hormone (ADH), is a nonapeptide produced in the hypothalamus and stored in the posterior pituitary gland until it is secreted.11 The two classic functions of vasopressin are regulation of blood pressure and blood volume and regulation of osmolality.11,12 Vasopressin abnormalities are diagnosed by analyzing serum electrolytes (in particular, serum sodium) and serum osmolality, as well as urine electrolytes and urine osmolality. Analysis of serum vasopressin levels is often not readily available and not usually needed for diagnosis.
Excessive vasopressin leads to the appropriately labeled condition known as the syndrome of inappropriate ADH secretion (SIADH), leading to serum hypo-osmolality and its clinical consequences.13 Common etiologies of SIADH include tumors (especially pulmonary in origin leading to ectopic ADH production); CNS disorders such as mass lesions and trauma as well as inflammatory (eg, meningitis) and demyelinating disorders (eg, Guillain-Barré syndrome, multiple sclerosis, neuromyelitis optica); postsurgical complications; and drug-induced disorders. The clinical manifestations of SIADH are largely neurologic and may depend on the rate and severity of the hyponatremia associated with the condition.14 The neurologic symptoms range from mild headache and nausea to severe confusion, focal neurologic findings, seizures, and even death. Although a detailed discussion of treatment of SIADH is outside of the scope of this article, treatment of acute (ie, less than 48 hours in duration) SIADH involves reversing the electrolyte and osmolality deficits relatively quickly by hypertonic saline with or without a loop diuretic and more recently with vasopressin receptor antagonists (vaptans).11 Treatment of symptomatic chronic SIADH involves fluid restriction, especially for mild to moderate cases, and vaptans as needed. For chronic SIADH, rapid correction of the hyponatremia should be avoided because it may lead to central pontine and extrapontine myelinolysis (osmotic demyelination syndrome). To avoid this complication, maximal correction rate for the hyponatremia has been suggested to not exceed 0.5 mEq/L per hour, 12 mEq/L in the first 24 hours, and 18 mEq/L in the first 48 hours.
Vasopressin deficiency leads to diabetes insipidus (DI), which is characterized by excessive thirst, excretion of large amounts of diluted urine, and no change in urine concentration despite fluid restriction.15 DI has various causes (ie, neurohypophyseal, nephrogenic, gestational, primary, and psychogenic); however, DI caused by vasopressin deficiency is seen as a result of hypothalamic lesions (ie, neurohypophyseal DI and primary polydipsia), such as from neurosarcoidosis or neuromyelitis optica.15 Neurologic complications of vasopressin deficiency are largely a consequence of the DI and the resulting hypernatremia, often leading to encephalopathy,14 but other neurologic deficits may be associated with lack of vasopressin. In a study of 23 members of a large family carrying a heterozygous mutation in the vasopressin gene, extensive neuropsychological testing found modest but statistically significant deficits in specific attention and memory tasks independent from the symptoms of mild thirst and polyuria.16 Treatment of symptomatic neurohypophyseal DI in awake patients with intact thirst response often may be as simple as allowing the patient to freely drink and urinate as needed to maintain homeostasis, with the use of medications such as desmopressin (a synthetic analog to vasopressin) to help decrease the overall severity of the polydipsia and polyuria.15 Nonawake neurohypophyseal DI patients (such as postoperative patients or those who have experienced acute brain trauma) cannot self-regulate water intake; therefore, parenteral repletion with concomitant continuous infusion of low-dose vasopressin analogs may be used in concert with careful monitoring of serum sodium levels to avoid hyponatremia.
Oxytocin. Similar to vasopressin, oxytocin is a nonapeptide produced in the hypothalamus and stored in the posterior pituitary until its release into the circulatory system or to other regions of the brain.17 Its main known clinical function is in lactation and uterine contractions during pregnancy.
Excessive oxytocin can be caused by iatrogenic administration of oxytocin when trying to induce uterine contractions. Because oxytocin is an antidiuretic, continuous administration can lead to water intoxication and in some cases severe hyponatremia, resulting in seizures, coma, and rarely death.18 Other US Food and Drug Administration (FDA)–reported neurologic complications associated with pharmacologic use of oxytocin include cerebrovascular events such as subarachnoid hemorrhage.19
Oxytocin deficiency may be found when tumors compress the posterior pituitary or during conditions that lead to hypopituitarism. The clinical significance of oxytocin deficiency outside of its role in lactation and uterine contraction is unclear; however, oxytocin has been recently linked to improving neuropsychiatric and social behaviors.12 For example, intranasal administration of oxytocin has been found to increase trust, empathy, and generosity and to improve social anxiety.12 Preliminary small clinical trials using intranasal oxytocin on autism spectrum disorders appear promising, but more research is clearly needed.20
Thyroid Hormone Disorders
The thyroid gland produces two thyroid hormones, triiodothyronine (T3) and thyroxine (T4), which circulate in the blood and affect numerous organs. Thyroid hormone disorders can occur from any dysfunction in the hypothalamus-pituitary-thyroid axis (Figure 3-11). Although thyroid regulation is quite complex, thyroid abnormalities can be largely identified and classified by simple blood tests (Table 3-1).21,22 In clinical practice, the thyroid-stimulating hormone (TSH) level measured in the blood serves as the primary screening assay.23 TSH is secreted by the pituitary and regulated directly by thyroid hormone levels as well as by the thyrotropin-releasing hormone from the hypothalamus. If the TSH level is abnormal, further tests are needed; most commonly, free (unbound or active-form) T4 level is measured. Free T3 levels can also be useful, especially for hyperthyroidism, where in some cases free T4 can be normal but free T3 abnormally high. If hypothyroidism is suspected, it is generally not recommended to measure free T3 levels because these are often normal until the disease is severe. Caution must be used in interpreting these laboratory values in critically ill or pregnant patients and in those taking thyroid or antithyroid medications. In addition to blood tests, two radiologic tests, the radioactive iodine uptake test and thyroid scintigraphy, may be used to aid diagnosis and treatment of thyroid disorders.
Table 3-1.
Diagnosis of Major Thyroid Disorders by Routine Blood Testsa
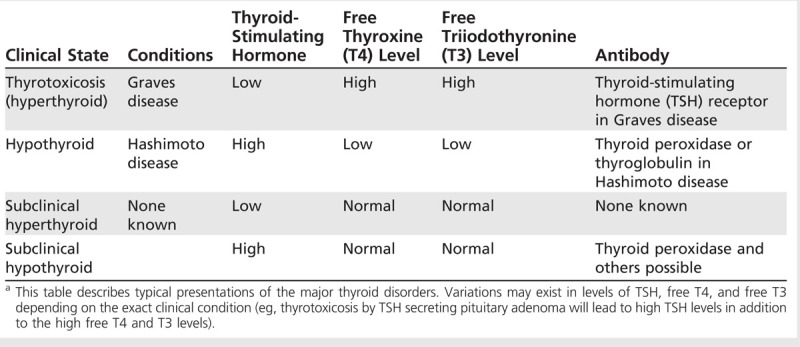
Thyroid hormone disorders are of particular importance to neurologists because both hypothyroidism and hyperthyroidism (thyrotoxicosis) can cause significant and widespread clinical problems, from cognitive dysfunction to muscle weakness.24 The neurologic complications can be insidious in nature (progressing over weeks to months) or acute, such as during a thyroid storm or myxedema coma, necessitating immediate intensive care.
Thyrotoxicosis and hyperthyroidism. Sustained hyperthyroidism or thyrotoxicosis can be seen in many conditions, including Graves disease (antibody against TSH receptor), toxic multinodular goiter, and, rarely, TSH-secreting pituitary adenoma.23 Acute thyrotoxicosis is also commonly seen in thyroiditis that can be associated with autoimmune conditions, including Hashimoto disease, viral or postviral, as well as drug-induced conditions, such as from amiodarone or lithium. Iatrogenic causes of thyrotoxicosis should always be considered in patients receiving exogenous thyroid replacement.
Systemic complications from thyrotoxicosis include goiter, proptosis, heat intolerance, weight loss, palpitations including atrial fibrillation, and irregular menses, but they can vary widely among patients and affect nearly every system in the body (Table 3-2).22 Neurologic complications due to thyrotoxicosis can also be quite varied and widespread, but they can be categorized mainly into the areas of neuropsychiatric and cognitive impairment; movement disorders; neuromuscular impairment; and rarely, seizures and other disorders.24,25
Table 3-2.
Non-neurologic Complications of Thyroid Disordersa
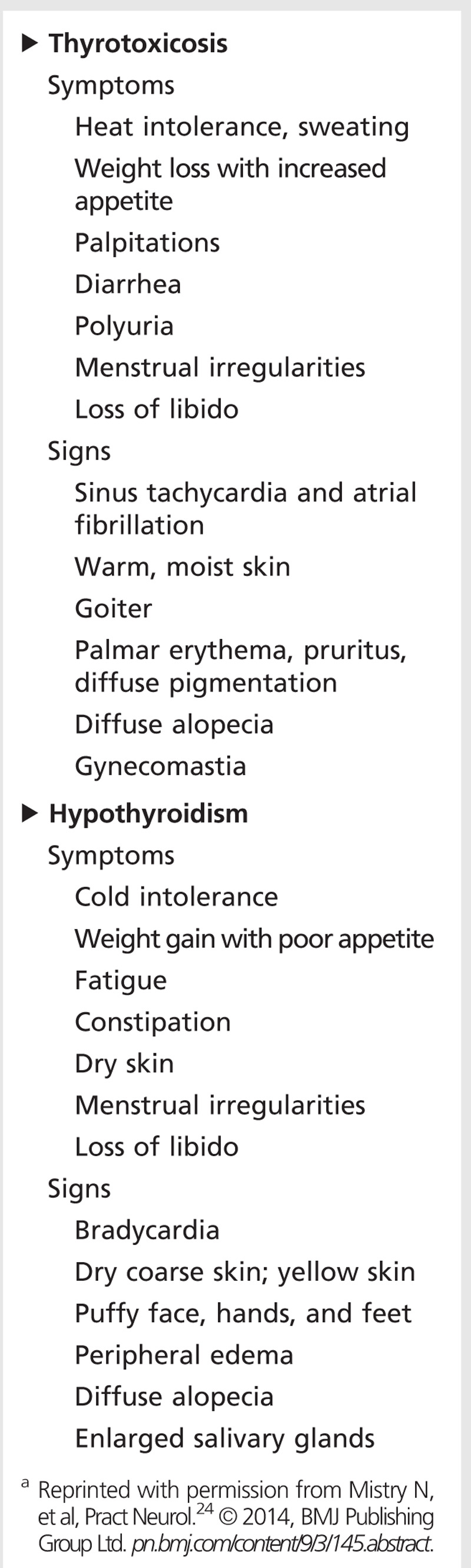
Typical neuropsychiatric impairments associated with thyrotoxicosis include anxiety, restlessness, emotional lability, difficulty concentrating, insomnia, depression, and even psychosis.24 Patients may be misdiagnosed as having primary mood or anxiety disorders, especially during the acute thyroid storm, when the predominant presentation may be severe agitation or psychosis. Affective impairments such as anxiety and depression are commonly found on neuropsychological testing in thyrotoxic patients; however, formal cognitive testing for memory and concentration on thyrotoxic patients is often inconclusive or conflicting, with some reports showing no significant impairment despite subjective concerns.26,27 The neuropsychiatric and cognitive presentation in thyrotoxic patients may also vary with age—younger patients tend to present with anxiety and hyperactivity, while elderly patients may present with cognitive symptoms and apathy, depression, or lethargy.
Movement disorders are often seen in thyrotoxic patients. The most common manifestation is tremor, which in one study occurred in approximately 60% to 80% of all thyrotoxic patients.28 The tremor is typically a postural tremor that is most commonly seen in the hands but can also affect various other body parts, such as the head, facial structures, trunk, and legs.25 β-Adrenergic overstimulation is believed to be a cause of the tremor; therefore, beta-blockers can often lead to dramatic improvement. Choreoathetosis is a much rarer manifestation and is most often seen in patients less than 40 years of age (Case 3-1).29 As with tremor, beta-blockers may be effective in treating the choreoathetosis.25 Both tremor and choreoathetosis resolve when patients become euthyroid.
Neuromuscular disorders that can occur in association with thyrotoxicity include myopathy (typically proximal), periodic paralysis, myasthenia gravis, and neuropathy.24,25 In one series of 21 patients with hyperthyroidism, 67% (14 of 21) of patients had neuromuscular symptoms, with 62% (13 of 21) having clinical weakness that correlated with free T4 levels but not with serum creatine kinase (CK) levels.30 The muscle weakness is primarily proximal, with extent and severity related to duration of hyperthyroidism; in long-standing hyperthyroid patients an insidious but significant loss in muscle mass may be present.25 Distal muscles are less commonly affected but may be involved later in the disease. Bulbar and oropharyngeal muscles are rarely affected, but cases of rapidly progressive and severe bulbar myopathy in chronic thyrotoxic patients experiencing an acute thyroid storm have been reported.31,32 In these cases, identification of the thyroid storm as the cause for the bulbar symptoms is critical because treatment with beta-blockers and antithyroid medication can often be lifesaving. Laboratory evaluation for thyrotoxic myopathy usually reveals normal serum CK levels unless rhabdomyolysis or inflammatory myopathy is also present.24,25 Muscle biopsy and neurophysiologic studies can show nonspecific changes. Acute treatment of thyrotoxic myopathy involves beta-blockers and antithyroid medications, with the ultimate goal of restoration to a euthyroid state.
Thyrotoxic periodic paralysis is a relatively uncommon but well-recognized complication of thyrotoxicosis affecting mainly male patients of East Asian descent but also rarely white patients.33,34 These patients have increased cellular sodium/potassium–adenosine triphosphatase pump activity leading to a net increase in transport of potassium to the intracellular space leading to hypokalemia and weakness. A typical presentation involves a 20- to 40-year-old man of East Asian descent with no family history and often no other signs of thyrotoxicity presenting with frequent attacks of transient (ie, lasting a few hours to up to 72 hours) weakness ranging from mild proximal weakness to complete flaccid paralysis. The attacks usually occur during the early morning hours and may be preceded by a large carbohydrate meal, heavy alcohol intake, or strenuous exercise (attacks occur during the rest period).33 Bulbar, respiratory, and ocular muscles are generally spared, and mentation and sensations are intact. Electrocardiac changes are extremely common and include sinus tachycardia, high QRS voltage, atrioventricular blocks, and electrical changes associated with hypokalemia such as prominent U waves.33 During an attack, laboratory blood analysis typically finds hypokalemia with serum potassium levels ranging from 1.1 mmol/L to 3.4 mmol/L, and hypophosphatemia. In contrast to nonthyrotoxic hypokalemic periodic paralysis, thyrotoxic periodic paralysis usually shows a marked decline in urine phosphate excretion, and one report suggests using spot urine calcium/phosphate ratio of greater than 1.4 mmol/L, as a diagnostic tool to help distinguish between the two.33,35 The diagnosis remains largely clinical, supported by laboratory and other findings. Treatment of an acute attack of thyrotoxic periodic paralysis includes careful potassium supplementation (intravenous if severe weakness or respiratory compromise is present, with extreme caution for possible severe rebound hyperkalemia that can lead to fatal cardiac arrhythmias) and nonselective beta-blockers.33 Definitive treatment is curative therapy of the underlying thyrotoxic state; patients who achieve a euthyroid state rarely have a recurrence.
The association between myasthenia gravis and autoimmune thyroid disease has long been recognized, with an estimated 7% prevalence of Graves disease and 3% prevalence of Hashimoto thyroiditis in patients who have myasthenia gravis.25,36 The reverse appears to be less common, with studies finding less than 1% of thyrotoxic patients with myasthenia gravis.25 In one study, patients with both myasthenia gravis and Graves disease were found to have a younger age of onset of their myasthenia, lower frequency of acetylcholine receptor antibodies, and higher frequency of thymic hyperplasia.37 Both thyrotoxicosis and hypothyroidism may exacerbate symptoms of myasthenia, which suggests that controlling thyroid disease in these patients may improve their overall neuromuscular status.25
Compared with myopathic symptoms, neuropathy is less commonly recognized in thyrotoxic patients. However, in one series of 21 thyrotoxic patients, 14% reported neuropathic symptoms such as numbness, and 24% had neuropathic changes on neurophysiologic studies.31 Furthermore, an acute to subacute neuropathy with paraplegia or quadriplegia, originally referred to as Basedow paraplegia by Charcot in 1889, has been rarely reported in the literature.38,39 This rare thyrotoxic neuropathy is classically described as symmetric weakness (involving both proximal and distal muscles, usually with lower extremities affected more than upper extremities), decreased tendon reflexes, minimal or no sensory disturbances, and sparing of the sphincter. Electrophysiologic studies show a mixed axonal and demyelinating sensorimotor neuropathy. Restoration to a euthyroid condition usually, but not always, resolves the neuropathic condition in these patients.
Other rare neurologic manifestations of thyrotoxicosis have been reported in the literature, including seizures and corticospinal tract disease.24,40,41 Since restoration to a euthyroid state reverses many of the neurologic deficits, including those presenting with corticospinal tract disease, accurate diagnosis of thyrotoxicosis in these patients is critical because they may be mistaken for having a progressive and irreversible condition such as motor neuron disease. Finally, thyrotoxicosis can lead to an increased risk of developing atrial fibrillation or hypercoagulability, which are both risk factors for stroke.42 Perhaps as a result of the increase in these stroke risk factors, a recent prospective case-cohort study of young patients with hyperthyroidism found an overall 1.44 times greater (95% CI, 1.02 to 2.12; P=.038) risk of having an ischemic stroke compared with a control group.43
Hypothyroidism. The most common form of primary hypothyroidism in adults is autoimmune thyroiditis (Hashimoto disease).21 Iatrogenic causes include postablative thyroiditis in hyperthyroid patients, previous neck irradiation, as well as medications including lithium and sulfonamides. Iodine deficiency leading to endemic goiter may be a common cause of hypothyroidism in areas where people do not have access to iodine in the diet.
As in thyrotoxicosis, hypothyroidism can affect all organ systems. Although symptoms of hypothyroidism may vary depending on the patient’s age and sex, some of the most common systemic manifestations of hypothyroidism include weight gain, fatigue, constipation, cold intolerance, dry skin, myxedema, hair thinning or loss, and menstrual irregularities (Table 3-2).21 Neurologic complications of hypothyroidism can be classified as neuropsychiatric and cognitive impairment; movement disorders; neuromuscular impairment; or headaches.
The association between cognitive and neuropsychiatric impairment and hypothyroidism has been long recognized, and the evaluation of a patient’s thyroid status by blood TSH levels is commonly incorporated in most dementia screens to assess for potentially reversible causes of dementia.26,44 Overt clinical hypothyroidism leads to various types of cognitive impairment, including minimal to severe deficits in general intelligence, psychomotor speed, visual-spatial skills, and memory, as well as neuropsychiatric changes such as depression and paranoia.44 Thyroxine replacement therapy typically rapidly reverses these deficits. However, formal testing in euthyroid patients on thyroxine replacement therapy has been reported to show poor performance in some neurocognitive functions, such as complex attention tasks and verbal memory tests.45 Subclinical hypothyroidism (elevated TSH but normal free T4 levels) has also been linked by some studies to poor cognitive and neuropsychiatric function; however, because the results are often conflicting from one study to another, it remains unclear whether subclinical hypothyroidism leads to significant cognitive or neuropsychiatric impairments.46
Myxedema coma caused by severe untreated hypothyroidism is a true medical emergency, and, in addition to coma, these patients may have seizures, including status epilepticus.47 Therefore, all coma patients with unclear etiology—especially those with hypothermia, hyponatremia, or hypercapnia—should be evaluated for thyroid dysfunction and, if confirmed, should receive parental T3 or T4 treatment in addition to supportive care in an intensive care unit setting.24,47
Cerebellar (predominantly gait) ataxia may be a presenting clinical feature, particularly in autoimmune-mediated hypothyroid (Hashimoto disease) patients.24 One study of 320 patients diagnosed with progressive nonfamilial ataxia found 22 patients with hypothyroidism and positive thyroid antibodies.48 While cerebellar ataxia may be reversible in many forms of hypothyroidism, some patients with autoimmune hypothyroid disease have been reported to have irreversible cerebellar degeneration that does not improve despite thyroxine replacement therapy.24,49 Brain MRI in these cases may show cerebellar degeneration with atrophy of the vermis and other structures.
Neuromuscular complications due to hypothyroidism are quite common, with up to 79% of patients in one series reporting symptoms consistent with neuromuscular dysfunction; in this series, formal testing revealed 38% of the patients to have clinical signs of muscle weakness, 42% to have signs of sensorimotor axonal neuropathy, and 29% to have carpal tunnel syndrome (often bilateral).30 On physical examination, proximal muscle weakness may be seen, in addition to the characteristic “hung up” or decreased or absent deep tendon reflexes.30,50 If extensor plantar responses or decreased vibratory sensation is found on examination, it should lead to evaluation for vitamin B12 deficiency because of the association of pernicious anemia with autoimmune hypothyroidism.24 Thyroxine replacement therapy improves the neuromuscular symptoms in a majority of patients; however, persistent clinical weakness can occasionally be seen even a year after treatment initiation.30 In addition, a prospective cohort study on neurologically asymptomatic primary hypothyroid patients found abnormal nerve conduction studies or EMG in 83% of the patients, including 74% with myopathic changes (predominantly proximal upper extremities) and 52% with neuropathic changes (most commonly median motor neuropathy).51
Headache is a common symptom, affecting as many as 30% of hypothyroid patients, and is typically characterized as bilateral and nonpulsatile (without nausea and vomiting), usually remitting after achieving a euthyroid state.5,52 The mechanism for primary hypothyroidism causing headaches, if any, is unknown, but careful evaluation is needed because headaches can also be a manifestation of pituitary adenoma in patients with hypothyroidism.5
Hashimoto encephalopathy is a clinical syndrome characterized by the association of antithyroid antibodies with encephalopathy and other signs and symptoms of CNS dysfunction (including seizures, ataxia, myoclonus, and strokelike symptoms).53 The relationship between the thyroid antibodies and the encephalopathy remains unclear, and patients diagnosed with this syndrome may be euthyroid. It is of particular importance to identify Hashimoto encephalopathy as a cause of “reversible” dementia as these patients respond to treatment with steroids or other immunosuppressive treatment.
Adrenal Hormone Disorders
The adrenal gland produces three main types of hormones: glucocorticoids (eg, cortisol, corticosterone), mineralocorticoids (eg, aldosterone, deoxycorticosterone), and gonadal hormones (mainly androgens). Mineralocorticoid disorders may affect neurologic function secondarily (through regulation of sodium, water balance, and blood pressure) and will not be addressed further here.
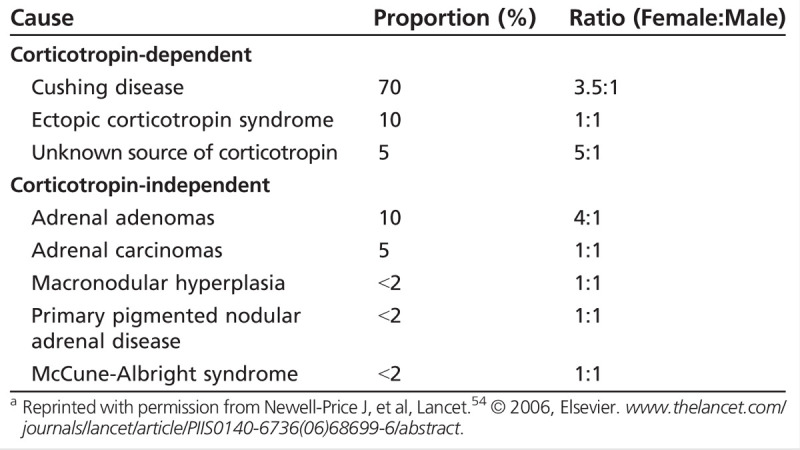
Glucocorticoid excess (Cushing syndrome) is most commonly seen when supraphysiologic amounts of exogenous glucocorticoids are administered to treat other medical conditions (Case 3-2).54 Endogenous glucocorticoid excess is most often (in 80% to 85% of cases) due to corticotropin-dependent causes, of which 80% are caused by pituitary adenomas (Cushing disease), with the remaining 20% occurring from ectopic corticotropin secretion (Table 3-3).54 Corticotropin-independent causes of endogenous glucocorticoid excess include adrenal adenomas and carcinomas.54 Once exogenous causes are ruled out, careful testing for endogenous glucocorticoid excess, which involves measuring urine cortisol, late-night salivary cortisol, or dexamethasone suppression test, supplemented with imaging studies, are needed for the diagnosis.56,57
Table 3-3.
Causes of Endogenous Glucocorticoid Excessa
Case 3-2
A 40-year-old man was self-referred for a neurologic evaluation of leg and arm weakness. For the past few months, he had progressively increasing difficulty getting up from a chair and more recently had trouble opening jars. He admitted that he had not been exercising and gained a lot of weight in the past 2 years. He noted that friends have commented on his appearance and how different he looks. His medical history was notable for a 10-year history of psoriasis, which had worsened over the past 2 years despite applying a lot of “cream” that a friend, who also had psoriasis, had been giving to him. He denied using any other medications and had no significant family history. His blood pressure was 146/90 mm Hg. On physical examination, he had a bloated appearance with mooning of his face and noticeable acne, central obesity, and prominent striae on his abdomen. His skin was very thin with bruise marks visible throughout. Neurologic examination was notable for marked bilateral proximal weakness more prominent in the lower than the upper extremities but was otherwise unremarkable.
Comment. The differential for progressive muscle weakness is wide; however, this patient has cardinal features of excess glucocorticoid (Cushing syndrome), including moon face, central obesity, acne, striae, and proximal weakness. Some of these features may be dismissed by the patient or be mild in nature, thus delaying the diagnosis. The cream the patient was using was found to be a high-potency steroid cream, which he was applying excessively. Cushing syndrome has been associated with all forms of steroid preparations including nasal, aerosol, oral, IM, epidural, and even topical steroids, especially if applied excessively at high doses.56 For this patient, laboratory values were unremarkable, including normal serum creatine kinase and thyroid-stimulating hormone levels. An abnormal 24-hour urinary cortisol level and failure to suppress cortisol levels after an overnight dexamethasone suppression test helped confirm the diagnosis. With assistance of a dermatologist, the high-potency steroid cream was switched to judicious use of a low-dose steroid cream to avoid a rebound phenomenon and led to a gradual improvement in the patient’s symptoms.
Glucocorticoids affect numerous organs (Figure 3-258). Systemic complications from glucocorticoid excess, most commonly described in Cushing disease, include central obesity, gonadal dysfunction, hirsutism, skin thinning, hypertension, glucose intolerance, osteopenia or skeletal fractures, and nephrolithiasis. Neurologic complications from glucocorticoid excess include cognitive and neuropsychiatric impairments, and myopathy. Glucocorticoid excess leads to impairments in several cognitive domains (including processing speed, auditory attention, working memory, and reading speed), which were found to be independent from fatigue and mood disorders that often coexist.59 Neuropsychiatric impairments from glucocorticoid excess include depression with fatigue and insomnia, but mania and anxiety may also be seen.54,56 While improvement in the cognitive and neuropsychiatric impairments can be seen after treatment and cure of the underlying cause of glucocorticoid excess, these impairments can persist long afterward.54,59 The myopathy from glucocorticoid excess can be a characteristic symptom of the disease, with proximal muscle weakness and normal serum CK levels.56
Figure 3-2.
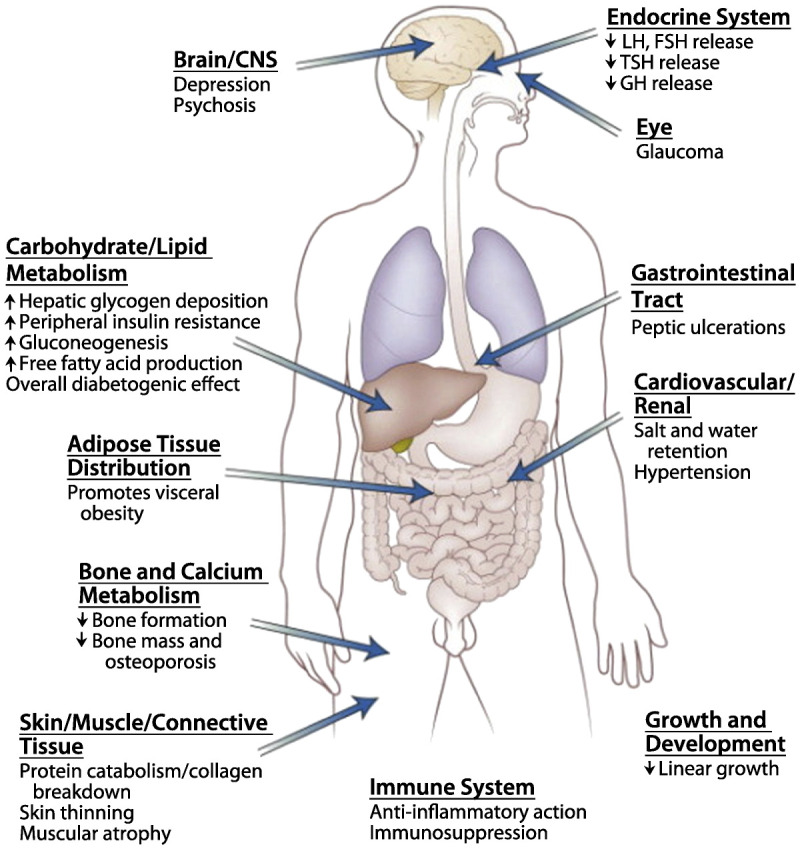
The principal sites of action of glucocorticoids in humans, highlighting some of the consequences of glucocorticoid excess. CNS = central nervous system; LH = luteinizing hormone; FSH = follicle-stimulating hormone; TSH = thyroid-stimulating hormone; GH = growth hormone. Modified with permission from Melmed S, et al, eds, Saunders Elsevier.58 © 2012, Elsevier.
Glucocorticoid deficiency is most commonly seen as part of adrenal insufficiency, either as a primary disorder (ie, Addison disease) or from secondary causes, which often have an intact mineralocorticoid system (Table 3-458). Of particular interest to neurologists is that both traumatic brain injury and acute cervical spinal cord injury may lead to adrenal insufficiency, with one study finding a 22% incidence in patients with cervical spinal cord injury and neurogenic shock, and other studies finding an incidence as high as 50% in intubated patients with traumatic brain injury.60,61 Systemic complications from adrenal insufficiency include weakness and fatigue, anorexia, hyperpigmentation, gastrointestinal symptoms, hypotension, and electrolyte disturbances. Neurologic complications from adrenal insufficiency are generally due to secondary effects, such as from the cardiovascular and metabolic problems. Primary neurologic complications from adrenal insufficiency are generally nonspecific but are commonly neuropsychiatric in nature, including depression or even psychosis, which may be the first presenting symptoms of the disease.62 During an Addisonian or adrenal crisis, loss of consciousness and seizures may develop. Adrenal insufficiency can be diagnosed by the cosyntropin test, and acute treatment involves careful replacement with glucocorticoids and, if needed, mineralocorticoids.63
Table 3-4.
Etiology of Adrenal Insufficiencya

Gonadal Hormone Disorders
The two main gonadal hormones, estrogen and testosterone, are regulated by complex hypothalamic-pituitary-gonadal pathways (Figure 3-12) that both decrease with aging. Any disruption of the hypothalamic-pituitary-gonadal pathway can lead to disorders in gonadal hormones, but for brevity this review focuses mainly on aging-related changes in gonadal hormones.
Estrogen deficiency (menopause). Menopause in women leads to a complex and dramatic shift in the gonadal hormones, culminating in a low estrogen state.64 The systemic complications from menopause include irregular menses followed by amenorrhea; vasomotor symptoms, including hot flashes; urogenital atrophy; and osteoporosis. Based on early observational studies, hormonal replacement therapy (estrogen alone or in combination with progesterone) was commonly given to menopausal women for treatment of these symptoms and as primary prevention for osteoporosis and cardiovascular disease. Neurologic complications are less specific, but early observational studies in menopausal women found a decrease in cognitive ability (including an increased risk of developing Alzheimer disease) and increase in mood disorders such as depression that were improved with hormonal replacement therapy. The use of hormonal therapy in postmenopausal women sharply declined after the Women’s Health Initiative (WHI) studies were published in 2002.65 As part of the WHI, a large randomized trial of postmenopausal women receiving hormonal therapy (combination estrogen and progesterone) concluded that the risks of therapy (including small but increased risks of cardiovascular disease, stroke, thromboembolic events, and breast cancer) outweighed any benefits (ie, decreased risk of fractures caused by osteoporosis).65,66 Furthermore, studies of hormonal replacement not only failed to find any improvement in cognition and prevention of dementia but interestingly found a small but significant detrimental effect on cognition in postmenopausal women aged 65 years or older.67,68
Since the publication of WHI studies, the data have been reevaluated, with many experts suggesting that the results may not be applicable to all women, in particular younger postmenopausal women, because the women in the studies were generally older (average age 63 years) and more advanced into menopause.65 In addition, the absolute risks for developing the adverse risks reported in WHI were small (8 strokes per 10,000 person-years for combination and 12 events per 10,000 person-years for estrogen alone) and could have been labeled as rare occurrences.64,65 This culminated in a 2013 global consensus statement of several international societies stating that, for women younger than 60 years of age or within 10 years after menopause, hormonal therapy may be beneficial and effective for vasomotor symptoms and prevention of osteoporosis-related fracture and may decrease coronary heart disease risk.69 However, it was noted that therapy should be tailored to each individual’s potential benefits and risks, in particular of venous thrombosis, stroke, heart disease, and breast cancer. The consensus statement did not mention the effects of hormonal therapy on cognition or dementia. Some have argued that treating younger postmenopausal women during a “window of opportunity” may be beneficial for cognitive function and dementia prevention.65 However, a recent large randomized placebo-controlled trial found no overall sustained benefit or risk to cognitive function in younger postmenopausal women (aged 50 to 55 years) who received hormonal therapy.70 It is still unclear whether the risk for developing Alzheimer dementia is decreased in younger postmenopausal women receiving hormonal therapy. Therefore, further research is needed before hormonal therapy in postmenopausal women can be recommended for the prevention of cognitive impairment or dementia. In addition to cognition and dementia, menopause and levels of sex hormones (exogenous hormonal therapy or changes in endogenous levels) also affect other neurologic conditions, such as migraine and epilepsy, which have been recently reviewed.71,72,73
Testosterone deficiency. Adult androgen deficiency or hypogonadism in men can be from any disorder resulting in failure to produce physiologic levels of testosterone due to disruption of the hypothalamic-pituitary-testicular axis.74 Hypogonadism in men can be classified as primary if it is associated with disorders of the testes and secondary if it results from disorders of the hypothalamus or pituitary gland.75 Identifying the cause of hypogonadism is important because secondary hypogonadism may be due to a pituitary adenoma, which would lead to different clinical management. Morning serum testosterone levels should be measured to help confirm the diagnosis in patients with symptoms of androgen deficiency.74,75 In order to distinguish primary from secondary causes of hypogonadism, levels of serum-luteinizing hormone and follicle-stimulating hormone can be measured and will be low or inappropriately normal in secondary causes. Testosterone also gradually decreases with natural aging and is lower in older men than in younger men; however, it is unclear whether the age-related decline in testosterone levels is an adaptive or pathologic response.75 Systemic complications from androgen deficiency in men include decreases in sexual function and secondary male sexual characteristics as well as less specific signs and symptoms of decreased energy, decrease in muscle mass, increase in adiposity, and mild normocytic normochromic anemia.74 Neurologic complications of androgen deficiency are largely nonspecific, including neuropsychiatric symptoms of depressed mood and decrease in concentration and memory. Because several, albeit small, placebo-controlled randomized trials with testosterone therapy in androgen-deficient older men have failed to show significant improvements in cognition with normal aging or in mild Alzheimer disease, testosterone therapy for cognitive improvement remains unproven.76,77 Therefore, any potential benefits may be outweighed by the costs and burden of testosterone administration along with the unknown long-term risks, including the potentially increased risk of prostate cancer, cardiovascular diseases, and ischemic stroke.74,78
CONCLUSIONS
Neurologic complications from endocrine disorders are as varied as the endocrine hormones that cause the disorders. Identification of the endocrine disorder as the cause of the neurologic symptom is essential because effective treatment of the hormonal disorder often leads to resolution of the neurologic symptoms. Although many of the neurologic complications from endocrine disorders have been well classified, many areas remain controversial and require further research, including identification of the neurologic complications, if any, from subclinical thyroid diseases, and whether hormonal therapy in postmenopausal women is beneficial in reducing the risk of dementia from Alzheimer disease or other causes.
KEY POINTS
As many of the neurologic complications from endocrine disorders are reversible, it is important for neurologists to be able to identify and manage the neurologic complications of endocrine disorders.
The classic endocrine system can be conceptualized as a three-tiered pathway consisting of the hypothalamus–pituitary–target organ with tight feedback regulation at each tier.
Neurologic complications from growth hormone excess (ie, acromegaly) include peripheral neuropathies (most commonly median neuropathy), proximal myopathy, and sleep apnea (both obstructive and central).
The syndrome of inappropriate antidiuretic hormone secretion (SIADH) causes neurologic symptoms associated with the rate and severity of hyponatremia, ranging from mild nonspecific symptoms such as headache and nausea to critical symptoms of confusion, seizures, and even death.
In chronic SIADH rapid correction of hyponatremia should be avoided because of concern for osmotic demyelination syndrome.
Excess iatrogenic oxytocin administration may lead to water intoxication and severe hyponatremia leading to seizures, coma, and even death.
In clinical practice, blood thyroid-stimulating hormone (TSH) levels serve as the primary screening assay for thyroid hormone disorders; if abnormal TSH levels are found, free thyroxine (T4) and occasionally free triiodothyronine (T3) levels should be measured.
Both hypo- and hyperthyroidism (thyrotoxicosis) can cause a wide range of neurologic symptoms that are often reversible.
Neurologic complications that can occur in association with thyrotoxicosis can include neuropsychiatric and cognitive impairment; movement disorders, most commonly tremors and rarely choreoathetosis; neuromuscular impairment, including proximal myopathy, myasthenia gravis, periodic paralysis, and neuropathy; and seizures and corticospinal disorders (rarely).
Tremors in thyrotoxic patients typically present as persistent and fine; they are most commonly seen in the hands but can also affect other body parts, such as head, facial structures, trunk, and legs.
In cases of neuromuscular weakness of unclear etiology, a screen for thyroid disease may be useful to identify those patients with thyrotoxic neuromuscular complications, as treating the underlying thyroid disease often completely resolves the condition.
Acute attacks of thyrotoxic periodic paralysis are treated with careful potassium supplementation and nonselective beta-blockers.
Young patients who are hyperthyroid have an increased risk for ischemic stroke.
Neurologic complications from hypothyroidism include neuropsychiatric and cognitive impairment; movement disorders, in particular cerebellar ataxia; neuromuscular impairment; and headaches.
Myxedema coma is a true medical emergency and should be rapidly recognized in all severely cognitively impaired patients, especially those having features of hypothermia, hyponatremia, hemodynamic instability or hypercapnia, in addition to any other signs of hypothyroidism.
Cerebellar ataxia may be a presenting clinical feature in autoimmune-mediated hypothyroidism, and MRI may show cerebellar atrophy.
Glucocorticoid excess is most commonly seen when supraphysiologic amounts of exogenous glucocorticoids are administered to treat other medical conditions.
Traumatic brain injury and acute cervical spinal cord injury can lead to adrenal insufficiency.
A large randomized placebo-controlled trial in younger (aged 50 to 55 years) postmenopausal women receiving hormonal therapy recently found no overall sustained benefit or risk in cognitive function after a 7-year follow-up.
Testosterone therapy for cognitive impairment or Alzheimer disease in androgen-deficient men remains unproven.
Footnotes
Relationship Disclosure: Dr Ishii has received an unrestricted research grant from the Leon Levy Foundation.
Unlabeled Use of Products/Investigational Use Disclosure: Dr Ishii reports no disclosure.
REFERENCES
- 1.Melmed S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest 2003; 112 (11): 1603–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ignacak A,, Kasztelnik M,, Sliwa T, et al. Prolactin—not only lactotrophin. A “new” view of the “old” hormone. J Physiol Pharmacol 2012; 63 (5): 435–443. [PubMed] [Google Scholar]
- 3.Haddad PM,, Wieck A. Antipsychotic-induced hyperprolactinaemia: mechanisms, clinical features and management. Drugs 2004; 64 (20): 2291–2314. [DOI] [PubMed] [Google Scholar]
- 4.Chen DK,, So YT,, Fisher RS; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Use of serum prolactin in diagnosing epileptic seizures: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2005; 65 (5): 668–675. [DOI] [PubMed] [Google Scholar]
- 5.Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013; 33 (9): 629–808. [DOI] [PubMed] [Google Scholar]
- 6.Zhornitsky S,, Yong VW,, Weiss S,, Metz LM. Prolactin in multiple sclerosis. Mult Scler 2013; 19 (1): 15–23. [DOI] [PubMed] [Google Scholar]
- 7.Melmed S. Medical progress: acromegaly. N Engl J Med 2006; 355 (24): 2558–2573. [DOI] [PubMed] [Google Scholar]
- 8.Baum H,, Ludecke DK,, Herrmann HD. Carpal tunnel syndrome and acromegaly. Acta Neurochir (Wien) 1986; 83 (1–2): 54–55. [DOI] [PubMed] [Google Scholar]
- 9.Pickett J,, Layzer RB,, Levin SR, et al. Neuromuscular complications of acromegaly. Neurology 1975; 25 (7): 638–645. [DOI] [PubMed] [Google Scholar]
- 10.Melmed S. Idiopathic adult growth hormone deficiency. J Clin Endocrinol Metab 2013; 98 (6): 2187–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peri A. Clinical review: the use of vaptans in clinical endocrinology. J Clin Endocrinol Metab 2013; 98 (4): 1321–1332. [DOI] [PubMed] [Google Scholar]
- 12.Insel TR. The challenge of translation in social neuroscience: a review of oxytocin, vasopressin, and affiliative behavior. Neuron 2010; 65 (6): 768–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellison DH,, Berl T. The syndrome of inappropriate antidiuresis. N Engl J Med 2007; 356 (20): 2064–2072. [DOI] [PubMed] [Google Scholar]
- 14.Samuels MA,, Seifter JL. Encephalopathies caused by electrolyte disorders. Semin Neurol 2011; 31 (2): 135–138. [DOI] [PubMed] [Google Scholar]
- 15.Oiso Y,, Robertson GL,, Nørgaard JP,, Juul KV. Clinical review: treatment of neurohypophyseal diabetes insipidus. J Clin Endocrinol Metab 2013; 98 (10): 3958–3967. [DOI] [PubMed] [Google Scholar]
- 16.Bruins J,, Kovács GL,, Abbes AP, et al. Minor disturbances in central nervous system function in familial neurohypophysial diabetes insipidus. Psychoneuroendocrinology 2006; 31 (1): 80–91. [DOI] [PubMed] [Google Scholar]
- 17.Viero C,, Shibuya I,, Kitamura N, et al. REVIEW: oxytocin: crossing the bridge between basic science and pharmacotherapy. CNS Neurosci Ther 2010; 16 (5): e138–e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergum D,, Lonnée H,, Hakli TF. Oxytocin infusion: acute hyponatraemia, seizures and coma. Acta Anaesthesiol Scand 2009; 53 (6): 826–827. [DOI] [PubMed] [Google Scholar]
- 19.US Food and Drug Administration label for Pitocin (oxytocin). FDA.gov. www.accessdata.fda.gov/drugsatfda_docs/label/2007/018261s028lbl.pdf. Published March 2007. Accessed February 18, 2014.
- 20.Guastella AJ,, Einfeld SL,, Gray KM, et al. Intranasal oxytocin improves emotion recognition for youth with autism spectrum disorders. Biol Psychiatry 2010; 67 (7): 692–694. [DOI] [PubMed] [Google Scholar]
- 21.Gaitonde DY,, Rowley KD,, Sweeney LB. Hypothyroidism: an update. Am Fam Physician 2012; 86 (3): 244–251. [PubMed] [Google Scholar]
- 22.Brent GA. Clinical practice. Graves’ disease. N Engl J Med 2008; 358 (24): 2594–2605. [DOI] [PubMed] [Google Scholar]
- 23.Bahn Chair RS,, Burch HB,, Cooper DS, et al. American Thyroid Association; American Association of Clinical Endocrinologists. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid 2011; 21 (6): 593–646. [DOI] [PubMed] [Google Scholar]
- 24.Mistry N,, Wass J,, Turner MR. When to consider thyroid dysfunction in the neurology clinic. Pract Neurol 2009; 9 (3): 145–156. [DOI] [PubMed] [Google Scholar]
- 25.Kung AW. Neuromuscular complications of thyrotoxicosis. Clin Endocrinol (Oxf) 2007; 67 (5): 645–650. [DOI] [PubMed] [Google Scholar]
- 26.Samuels MH. Cognitive function in untreated hypothyroidism and hyperthyroidism. Curr Opin Endocrinol Diabetes Obes 2008; 15 (5): 429–433. [DOI] [PubMed] [Google Scholar]
- 27.Vogel A,, Elberling TV,, Hørding M, et al. Affective symptoms and cognitive functions in the acute phase of Graves’ thyrotoxicosis. Psychoneuroendocrinology 2007; 32 (1): 36–43. [DOI] [PubMed] [Google Scholar]
- 28.Nordyke RA,, Gilbert FI, Jr,, Harada AS. Graves’ disease. Influence of age on clinical findings. Arch Intern Med 1988; 148 (3): 626–631. [DOI] [PubMed] [Google Scholar]
- 29.Heffron W,, Eaton RP. Thyrotoxicosis presenting as choreoathetosis. Ann Intern Med 1970; 73 (3): 425–428. [DOI] [PubMed] [Google Scholar]
- 30.Duyff RF,, Van den Bosch J,, Laman DM, et al. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68 (6): 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kammer GM,, Hamilton CR, Jr. Acute bulbar muscle dysfunction and hyperthyroidism. Am J Med 1974; 56 (4): 464–470. [DOI] [PubMed] [Google Scholar]
- 32.Boddu NJ,, Badireddi S,, Straub KD, et al. Acute thyrotoxic bulbar myopathy with encephalopathic behaviour: an uncommon complication of hyperthyroidism. Case Rep Endocrinol 2013; 2013: 369807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Falhammar H,, Thorén M,, Calissendorff J. Thyrotoxic periodic paralysis: clinical and molecular aspects. Endocrine 2013; 43 (2): 274–284. [DOI] [PubMed] [Google Scholar]
- 34.Lin SH. Thyrotoxic periodic paralysis. Mayo Clinic Proc 2005; 80 (1): 99–105. [DOI] [PubMed] [Google Scholar]
- 35.Lin SH,, Chu P,, Cheng CJ, et al. Early diagnosis of thyrotoxic periodic paralysis: spot urine calcium to phosphate ratio. Crit Care Med 2006; 34 (12): 2984–2989. [DOI] [PubMed] [Google Scholar]
- 36.Mao ZF,, Yang LX,, Mo XA, et al. Frequency of autoimmune diseases in myasthenia gravis: a systematic review. Int J Neurosci 2011; 121 (3): 121–129. [DOI] [PubMed] [Google Scholar]
- 37.Kanazawa M,, Shimohata T,, Tanaka K,, Nishizawa M. Clinical features of patients with myasthenia gravis associated with autoimmune diseases. Eur J Neurol 2007; 14 (12): 1403–1404. [DOI] [PubMed] [Google Scholar]
- 38.Feibel JH,, Campa JF. Thyrotoxic neuropathy (Basedow’s paraplegia). J Neurol Neurosurg Psychiatry 1976; 39 (5): 491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen YH,, Lin HJ,, Chen KT. Rare presentations of hyperthyroidism—Basedow’s paraplegia and pancytopenia. Am J Emerg Med 2009; 27 (2): 258.e1–258.e2. [DOI] [PubMed] [Google Scholar]
- 40.Garcia CA,, Fleming RH. Reversible corticospinal tract disease due to hyperthyroidism. Arch Neurol 1977; 34 (10): 647–648. [DOI] [PubMed] [Google Scholar]
- 41.Song TJ,, Kim SJ,, Kim GS, et al. The prevalence of thyrotoxicosis-related seizures. Thyroid 2010; 20 (9): 955–958. [DOI] [PubMed] [Google Scholar]
- 42.Squizzato A,, Gerdes VE,, Brandjes DP, et al. Thyroid diseases and cerebrovascular disease. Stroke 2005; 36 (10): 2302–2310. [DOI] [PubMed] [Google Scholar]
- 43.Sheu JJ,, Kang JH,, Lin HC,, Lin HC. Hyperthyroidism and risk of ischemic stroke in young adults: a 5-year follow-up study. Stroke 2010; 41 (5): 961–966. [DOI] [PubMed] [Google Scholar]
- 44.Bauer M,, Goetz T,, Glenn T,, Whybrow PC. The thyroid-brain interaction in thyroid disorders and mood disorders. J Neuroendocrinol 2008; 20 (10): 1101–1114. [DOI] [PubMed] [Google Scholar]
- 45.Wekking EM,, Appelhof BC,, Fliers E, et al. Cognitive functioning and well-being in euthyroid patients on thyroxine replacement therapy for primary hypothyroidism. Eur J Endocrinol 2005; 153 (6): 747–753. [DOI] [PubMed] [Google Scholar]
- 46.Joffe RT,, Pearce EN,, Hennessey JV, et al. Subclinical hypothyroidism, mood, and cognition in older adults: a review. Int J Geriatr Psychiatry 2013; 28 (2): 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klubo-Gwiezdzinska J,, Wartofsky L. Thyroid emergencies. Med Clin North Am 2012; 96 (2): 385–403. [DOI] [PubMed] [Google Scholar]
- 48.Hadjivassiliou M,, Boscolo S,, Tongiorgi E, et al. Cerebellar ataxia as a possible organ-specific autoimmune disease. Mov Disord 2008; 23 (10): 1370–1377. [DOI] [PubMed] [Google Scholar]
- 49.Selim M,, Drachman DA. Ataxia associated with Hashimoto’s disease: progressive non-familial adult onset cerebellar degeneration with autoimmune thyroiditis. J Neurol Neurosurg Psychiatry 2001; 71 (1): 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eslamian F,, Bahrami A,, Aghamohammadzadeh N, et al. Electrophysiologic changes in patients with untreated primary hypothyroidism. J Clin Neurophysiol 2011; 28 (3): 323–328. [DOI] [PubMed] [Google Scholar]
- 51.El-Salem K,, Ammari F. Neurophysiological changes in neurologically asymptomatic hypothyroid patients: a prospective cohort study. J Clin Neurophysiol 2006; 23 (6): 568–572. [DOI] [PubMed] [Google Scholar]
- 52.Moreau T,, Manceau E,, Giroud-Baleydier F, et al. Headache in hypothyroidism. Prevalence and outcome under thyroid hormone therapy. Cephalalgia 1998; 18 (10): 687–689. [DOI] [PubMed] [Google Scholar]
- 53.Olmez I,, Moses H,, Sriram S, et al. Diagnostic and therapeutic aspects of Hashimoto’s encephalopathy. J Neurol Sci 2013; 331 (1–2): 67–71. [DOI] [PubMed] [Google Scholar]
- 54.Newell-Price J,, Bertagna X,, Grossman AB,, Nieman LK. Cushing’s syndrome. Lancet 2006; 367 (9522): 1605–1617. [DOI] [PubMed] [Google Scholar]
- 55.Kelly A,, Nelson K,, Goodwin M,, McCluggage J. Iatrogenic Cushing’s syndrome. Br Med J 1972; 4 (5832): 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nieman LK,, Biller BM,, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008; 93 (5): 1526–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tritos NA,, Biller BM,, Swearingen B. Management of Cushing disease. Nat Rev Endocrinol 2011; 7 (5): 279–289. [DOI] [PubMed] [Google Scholar]
- 58.Melmed S,, Polonsky KS,, Larsen PR,, Kronenberg HM, eds. Williams textbook of endocrinology. 12th edition. Philadelphia, PA: Saunders Elsevier, 2012. [Google Scholar]
- 59.Ragnarsson O,, Berglund P,, Eder DN,, Johannsson G. Long-term cognitive impairments and attentional deficits in patients with Cushing’s disease and cortisol-producing adrenal adenoma in remission. J Clin Endocrinol Metab 2012; 97 (9): E1640–E1648. [DOI] [PubMed] [Google Scholar]
- 60.Dusick JR,, Wang C,, Cohan P, et al. Pathophysiology of hypopituitarism in the setting of brain injury. Pituitary 2012; 15 (1): 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pastrana EA,, Saavedra FM,, Murray G, et al. Acute adrenal insufficiency in cervical spinal cord injury. World Neurosurg 2012; 77 (3–4): 561–563. [DOI] [PubMed] [Google Scholar]
- 62.Anglin RE,, Rosebush PI,, Mazurek MF. The neuropsychiatric profile of Addison’s disease: revisiting a forgotten phenomenon. J Neuropsychiatry Clin Neurosci 2006; 18 (4): 450–459. [DOI] [PubMed] [Google Scholar]
- 63.Arlt W. The approach to the adult with newly diagnosed adrenal insufficiency. J Clin Endocrinol Metab 2009; 94 (4): 1059–1067. [DOI] [PubMed] [Google Scholar]
- 64.Henderson VW. The neurology of menopause. Neurologist 2006; 12 (3): 149–159. [DOI] [PubMed] [Google Scholar]
- 65.Lobo RA. Where are we 10 years after the Women’s Health Initiative? J Clin Endocrinol Metab 2013; 98 (5): 1771–1780. [DOI] [PubMed] [Google Scholar]
- 66.Rossouw JE,, Anderson GL,, Prentice RL, et al. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women principal results from the women’s health initiative randomized controlled trial. JAMA 2002; 288 (3): 321–333. [DOI] [PubMed] [Google Scholar]
- 67.Rapp SR,, Espeland MA,, Shumaker SA, et al. Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003; 289 (20): 2663–2672. [DOI] [PubMed] [Google Scholar]
- 68.Shumaker SA,, Legault C,, Rapp SR, et al. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003; 289 (20): 2651–2662. [DOI] [PubMed] [Google Scholar]
- 69.de Villiers TJ,, Gass ML,, Haines CJ, et al. Global consensus statement on menopausal hormone therapy. Climacteric 2013; 16 (2): 203–204. [DOI] [PubMed] [Google Scholar]
- 70.Espeland MA,, Shumaker SA,, Leng I, et al. Long-term effects on cognitive function of postmenopausal hormone therapy prescribed to women aged 50 to 55 years. JAMA Intern Med 2013; 73 (15): 1429–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brandes JL. Migraine in women. Continuum (Minneap Minn) 2012; 18 (4 Headache): 835–852. [DOI] [PubMed] [Google Scholar]
- 72.Pack AM,, Reddy DS,, Duncan S,, Herzog A. Neuroendocrinological aspects of epilepsy: important issues and trends in future research. Epilepsy Behav 2011; 22 (1): 94–102. [DOI] [PubMed] [Google Scholar]
- 73.Herzog AG. Epilepsy. Continuum (Minneap Minn). 2009; 15 (2 Neuroendocrinology): 37–66. [Google Scholar]
- 74.Bhasin S,, Cunningham GR,, Hayes FJ, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2010; 95 (6): 2536–2559. [DOI] [PubMed] [Google Scholar]
- 75.Bhasin S,, Basaria S. Diagnosis and treatment of hypogonadism in men. Best Pract Res Clin Endocrinol Metab 2011; 25 (2): 251–270. [DOI] [PubMed] [Google Scholar]
- 76.Emmelot-Vonk MH,, Verhaar HJ,, Nakhai Pour HR, et al. Effect of testosterone supplementation on functional mobility, cognition, and other parameters in older men: a randomized controlled trial. JAMA 2008; 299 (1): 39–52. [DOI] [PubMed] [Google Scholar]
- 77.Lu PH,, Masterman DA,, Mulnard R, et al. Effects of testosterone on cognition and mood in male patients with mild Alzheimer disease and healthy elderly men. Arch Neurol 2006; 63 (2): 177–185. [DOI] [PubMed] [Google Scholar]
- 78.Vigen R,, O’Donnell CI,, Baron AE, et al. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA 2013; 310 (17): 1829–1836. [DOI] [PubMed] [Google Scholar]