

Abstract
Purpose of Review:
The aim of this review is to highlight the clinical presentation of a variety of inflammatory optic neuropathies through a case-based format. While emphasis will be placed on optic neuritis, which represents the most common acquired inflammatory optic nerve injury, the cardinal clinical features of other demyelinating forms of optic neuropathy and potential mimics for typical optic neuritis will be discussed.
Recent Findings:
Novel developments in the diagnostic evaluation, clinical associations, and treatment options for several inflammatory optic neuropathies will be described. Key points will emphasize clinical pearls and potential pitfalls that may thwart early diagnosis of underlying demyelinating, infectious, and inflammatory diseases presenting with optic nerve involvement.
Summary:
This review will aid the clinician in identifying potential red flags that should prompt consideration for causes of inflammatory optic nerve injury linked to neurologic and systemic disorders, and help guide safe and effective management for these conditions.
INTRODUCTION
Many potential causes of optic nerve inflammation exist, yet the vast majority of cases encountered in the clinical setting are optic neuritis. Idiopathic optic neuritis tends to affect young adults, is heralded by pain, and often has a self-limited course. While optic neuritis may be a sporadic event for some patients, one in every five individuals affected by multiple sclerosis (MS) will present with optic neuritis as the first manifestation of their disease. Therefore, recognizing the cardinal clinical features of optic neuritis is important, because the diagnosis may have potential long-term implications. By extension, it is also crucial that clinicians be aware of the red flags that may indicate the presence of an alternative diagnosis, including age of onset older than 45 years, absence of pain, atypical systemic symptoms and signs, and poor recovery. Optic neuritis associated with other demyelinating disorders, and conditions mimicking optic neuritis, may require a different course of management. Therefore, an in-depth understanding of inflammatory optic neuropathies is paramount, because it can help reduce the morbidity associated with these conditions and ensure the best possible patient care.
OPTIC NEURITIS ASSOCIATED WITH MULTIPLE SCLEROSIS
Typical optic neuritis is an inflammatory injury of the optic nerve, which can manifest as an isolated syndrome or, in some cases, represent a harbinger for the diagnosis of MS.1,2 With an incidence of 1 to 5 per 100,000 per year,2 optic neuritis is a common cause of acquired vision loss in young adults. Much of what is understood about the clinical presentation of optic neuritis has been derived from the initial experience and long-term follow-up from the Optic Neuritis Treatment Trial (ONTT).3,4,5 This randomized, multicenter study was initially designed to compare the benefits of treatment with IV methylprednisolone (250 mg administered every 6 hours for 3 days followed by oral prednisone [1 mg/kg/d] for 11 days), oral prednisone (1 mg/kg/d), or oral placebo in 457 patients with acute optic neuritis. Neuritis patients tend to be white (85%) women (77%) with a mean age of 32 years.3 In adults, the majority of optic neuritis cases are unilateral, but occasionally bilateral simultaneous vision loss is observed. Yet, in this setting, other less common demyelinating causes of optic neuropathy and potential optic neuritis mimics need to be considered, including neuromyelitis optica (NMO), toxic-metabolic optic neuropathies, and Leber hereditary optic neuropathy.2,6 Patients with typical optic neuritis often report subacute onset vision loss that worsens over hours to days. More than 90% of patients with optic neuritis experience pain within 1 to 2 weeks of symptom onset, which is frequently provoked by eye movements. One-third of patients may see intermittent sparkles of light in the affected eye, known as photopsia or phosphenes,2 although they may not divulge this information without prompting. Yet, a report of multiple “flashes or floaters” should trigger concern about a possible retinal mimic.
In the setting of suspected optic neuritis, several localizing features exist that, on initial examination, can be used to solidify the diagnosis. The severity of vision loss may range from mild (Snellen visual acuity equivalent of 20/20) to no light perception. In patients with unilateral optic nerve involvement, a relative afferent pupil defect will be apparent in the affected eye. In cases of bilateral asymmetric optic neuritis, a relative afferent pupillary defect will be found in the more severely affected eye. Alternatively, a relative afferent defect may not be detected if the severity of optic nerve injury is comparable between eyes in cases of bilateral sequential or simultaneous optic neuritis. Cecocentral (a defect extending from the blind spot to central fixation), altitudinal, and arcuate patterns of visual field loss may be observed. Dyschromatopsia, or decreased color vision, is also common. In cases of retrobulbar optic neuritis, the fundus examination is initially normal, whereas patients with anterior optic neuritis (sometimes referred to as papillitis)2,6 manifest mild to moderate optic disc swelling acutely, as illustrated in Case 2-1. The ONTT demonstrated that severe optic disc edema, vitreous cells, and hemorrhage are relatively uncommon findings in typical patients with optic neuritis and may herald a mimic, such as neuroretinitis.2,3,4,5,6 Not surprisingly, these atypical fundus features are associated with a reduced risk of developing MS, because the patient most likely does not have optic neuritis.
Case 2-1
A 28-year-old woman presented with subacute onset vision loss in the right eye and pain with eye movements. The examination showed a visual acuity of 20/100 in the right eye and 20/20 in the left eye, with a right relative afferent pupil defect. Visual field testing with automated perimetry showed a central defect in the right eye, whereas testing in the left eye was normal. The fundus examination revealed mild optic disc edema in the right eye and a normal-appearing optic nerve in the left eye (Figure 2-1). A baseline cranial MRI scan showed multifocal white matter lesions consistent with CNS demyelination.
Figure 2-1.
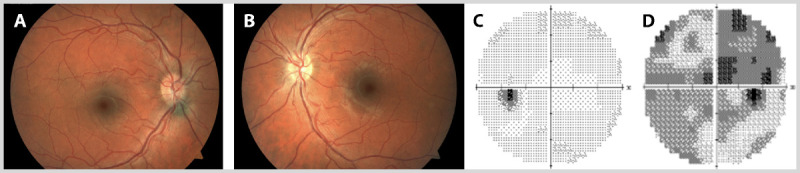
Right optic neuritis in a 28-year-old woman. Fundus examination revealed mild optic disc edema in the right eye (A) and a normal, although full-appearing, optic nerve in the left eye (B). Visual field testing with automated perimetry showed a normal left eye (C) and a central defect in the right eye (D).
Comment. This patient presented with typical signs and symptoms of demyelinating optic neuritis, including subacute vision loss (over 3 days) and pain with eye movements in the right eye. It is noteworthy that she also fits the typical demographic profile for this condition. In cases of typical optic neuritis, the optic nerve is normal in appearance, although in one-third of patients mild optic disc edema in the affected eye is observed. In most cases, visual recovery ensues within 3 to 6 weeks of symptom onset in typical optic neuritis. The Optic Neuritis Treatment Trial demonstrated that the baseline cranial MRI scan is the most useful predictor of this patient’s future risk of clinically definite multiple sclerosis, which given the presence of one or more white matter lesions, is 56% at 10 years, and 72% at 15 years.
With a classic history and a compatible examination, the diagnosis of optic neuritis can be made with a high degree of certainty on clinical grounds alone. Cranial MRI is performed because it is the best predictor of the patient’s future or current risk of MS, not because it is necessary to confirm the diagnosis of optic neuritis. In the original ONTT cohort, the 10-year risk of developing clinically definite MS was 22% in patients with no brain lesions on baseline MRI, as compared with 56% in patients with one or more lesions (Figure 2-24).5 After 15 years, 72% of patients with optic neuritis who had one or more white matter lesions on their initial MRI scan developed MS as compared with only 25% of patients with no baseline MRI lesions.6 With the benefit of 15 years of hindsight, the ONTT investigators concluded that the risk of developing MS was highest in the first 5 years after optic neuritis. Yet, a substantial risk persisted throughout the 15 years of follow-up in patients with optic neuritis who had initial brain MRI lesions.6,7 Among patients who were not diagnosed with MS at the 10-year examination, the probability of developing MS by the 15-year follow-up point was 32% when one or more baseline lesions were present versus 2% when there were no such lesions.6 It is noteworthy that, since the adoption of the revised McDonald criteria,8 the diagnosis of MS can be confirmed as early as the first clinical presentation (ie, at the time of the optic neuritis event) with cranial MRI.
Figure 2-2.
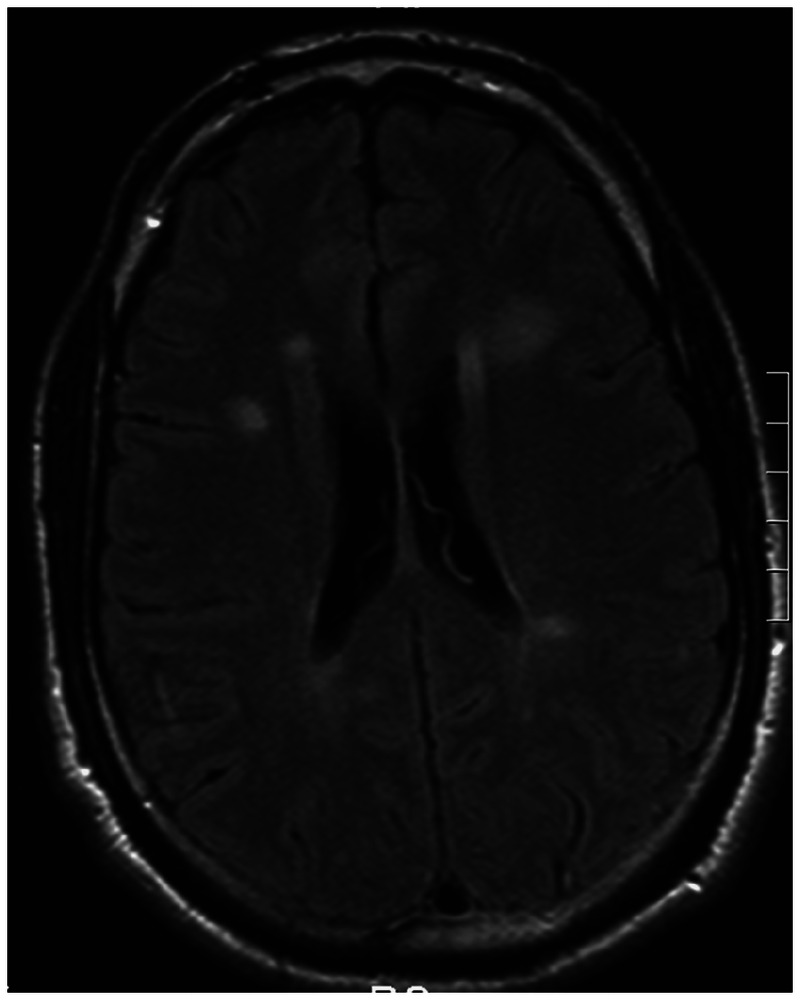
MRI of the brain showing characteristic white matter lesions in a patient with multiple sclerosis.
Reprinted with permission from Costello FE, Goyal M, Neurol Clin.4 © 2010 Elsevier. www.neurologic.theclinics.com/article/S0733-8619(10)00049-6/abstract.
In the ONTT, the predictive value of other baseline factors for the development of MS varied depending on presence or absence of brain lesions on MRI at presentation. When MRI showed one or more lesions were present at study entry, no demographic or clinical characteristics were predictive of MS development. In contrast, among patients without baseline lesions on MRI, the risk of MS was higher for women when there was a history of a viral syndrome preceding the optic neuritis onset, and when the optic disc appeared normal at the time of visual loss.6 MS was more than twice as likely to develop in patients with monofocal optic neuritis when the retrobulbar part of the optic nerve was affected (31%) as opposed to the anterior optic nerve (14%), consistent with the belief that retrobulbar neuritis is the typical form of optic neuritis in MS.6,7 In the final 15-year ONTT follow-up, among patients with monofocal optic neuritis at study entry (no baseline brain lesions on MRI, no prior contralateral eye optic neuritis, and no prior neurologic symptoms or signs), the diagnosis of MS was not observed in any patient when initial ophthalmoscopy showed severe optic disc swelling (n = 21), disc or peripapillary hemorrhages (n = 16), or retinal macular exudates (n = 8); when no eye pain was present (n = 18); or when presenting visual acuity was reduced to no light perception (n = 6).6 Thus, in addition to highlighting the predictive value of the baseline MRI scan, the ONTT also demonstrated the importance of the initial fundus findings in determining the future risk of MS in patients with optic neuritis.
When the diagnosis of optic neuritis is not clear, other ancillary tests can be useful in the investigative process, although the role of these investigations has been diminished since the introduction of the revised McDonald criteria.8 CSF analysis can be used to detect evidence of CNS inflammation, characterized by hypercellularity and a raised protein concentration.2 Finding oligoclonal bands of immunoglobulins in the CSF can help differentiate between demyelinating optic neuritis and other causes of inflammatory optic neuritis.2 Patients with optic neuritis may have no oligoclonal bands or local intrathecal synthesis of immunoglobulins (unmatched bands compared with serum).2 In other demyelinating optic neuropathies, sarcoidosis, or vasculitis, there may be no oligoclonal bands or systemic production of immunoglobulins (matched oligoclonal bands in both CSF and serum).2 CSF analysis can also play a role in establishing a link with MS in patients with optic neuritis who have atypical features at presentation (eg, older age), or who have abnormal cranial MRI findings that are not classic for demyelination, including nonspecific punctate white matter lesions.2,7,9 In some cases, visual evoked potential (VEP) testing may also aid in establishing the mechanism of optic nerve injury. Patients with optic neuritis frequently (65% of cases) manifest increased VEP latencies and reduced amplitudes consistent with demyelination.2,7,9 Previously, VEP findings have been used to show dissemination of lesions in the CNS of patients deemed to be at risk of developing MS.2 Yet, VEP testing is not routinely indicated in the evaluation of optic neuritis because abnormal findings may also occur in patients with other conditions, such as optic nerve compression, infiltration, and nondemyelinating inflammation.7,9
While the diagnosis of typical optic neuritis is often apparent, misdiagnoses in clinical trials have occurred. Of 457 patients enrolled in the ONTT, three patients were later diagnosed with anterior ischemic optic neuropathy, two had compressive lesions, and two had optic neuritis associated with connective tissue diseases.2,3 The realization of an alternate diagnosis may arise after the initial cranial imaging study reveals an unexpected finding, such as a mass lesion, or when clinical recovery is worse than expected over the course of follow-up. Hence, persistent visual impairment should raise clinical suspicion for possible non-MS causes of optic neuritis and other potential mimics. High-dose IV methylprednisolone initiated within 8 days of initial presentation has been shown to hasten the clinical recovery but not change the long-term visual outcomes in patients with optic neuritis.2,3,7,10 In the original ONTT study, IV methylprednisolone was administered in divided doses, but for practical reasons 1000 mg IV methylprednisolone is often given as a single dose, or as 500 mg twice daily (with or without an oral taper) in clinical practice.7 In some centers, the oral equivalent (prednisone or methylprednisolone) of the original ONTT IV methylprednisolone dose is used in the outpatient setting.7 In 2000, the Quality Standards Subcommittee of the American Academy of Neurology reviewed the role of high-dose corticosteroids in the treatment of acute optic neuritis and concluded that:
Higher dose oral or parenteral methylprednisolone or ACTH [adrenocorticotropic hormone] may hasten the speed and degree of recovery of visual function in persons with acute monosymptomatic ON [optic neuritis]. There is, however, no evidence of long-term benefit for visual function. The decision to use these medications to speed recovery but not to improve ultimate visual outcome should therefore be based on other non–evidence-based factors such as quality of life, risk to the patient, visual function in the fellow eye, or other factors that the clinician deems appropriate.10
OPTIC NEURITIS NOT ASSOCIATED WITH MULTIPLE SCLEROSIS
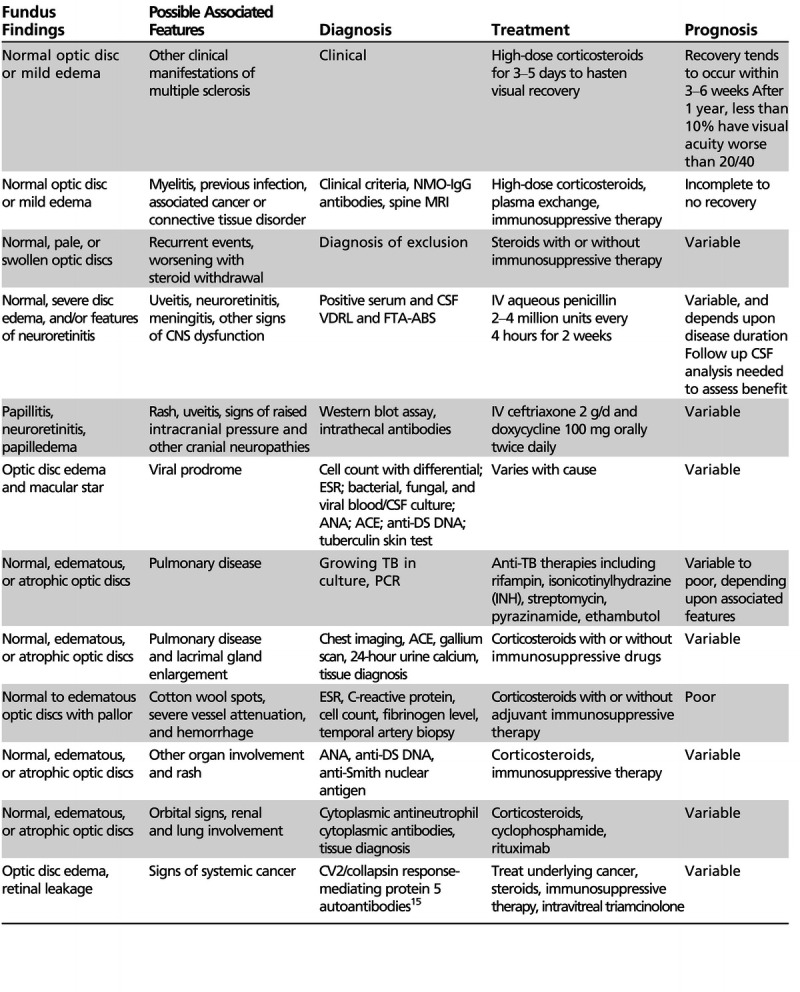
Several inflammatory forms of optic neuritis are not associated with MS but manifest as part of another demyelinating condition, including NMO,11,12 acute disseminated encephalomyelitis,11 chronic relapsing inflammatory optic neuropathy,11,12 antimyelin oligodendrocyte glycoprotein associated optic neuritis,13 and postvaccination-associated optic neuritis.14 These entities should be considered in patients who manifest atypical features, including pediatric or older age at onset, the presence of specific clinical signs (recurrent myelitis in NMO or encephalopathy in acute disseminated encephalomyelitis), or poor clinical recovery (Table 2-1).
Table 2-1.
An Overview of the Inflammatory Optic Neuropathies
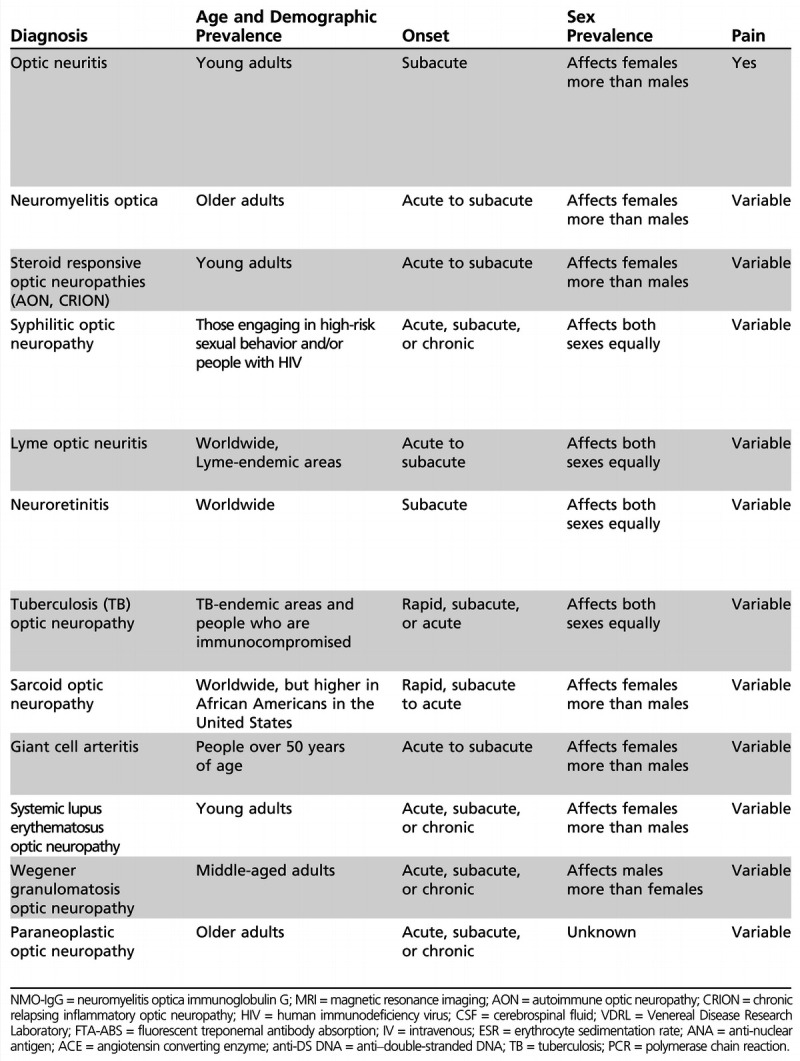
Table 2-1.
No caption available.
Neuromyelitis Optica
NMO spectrum disorders are typically characterized by a combination of optic neuritis and transverse myelitis. In NMO, severe inflammation and necrosis of the optic nerves and spinal cord often lead to marked disability and a decreased life expectancy, especially if untreated.7,16,17,18 Within 5 years of onset, approximately 50% of patients with NMO are blind in one or both eyes or require a walking aid, in sharp contrast to the relatively milder impact of MS on affected individuals with the same disease duration.17 Predictors of a worse prognosis in NMO include the number of relapses in the first 2 years of symptom onset, the severity of the first attack, and having an associated autoimmune disorder such as systemic lupus erythematosus (SLE).17 While other potential mimics may manifest with atypical signs and symptoms at initial presentation, optic neuritis associated with NMO can easily be overlooked if a high degree of suspicion is not present, since these patients often present with monocular involvement.16 Patients with poor visual recovery a month after symptom onset, a history of recurrent optic neuritis, bilateral optic neuritis, or symptoms of transverse myelitis should be investigated for NMO. Previously, the diagnosis of NMO was made predominantly on clinical grounds and with spinal MRI evidence showing extensive longitudinal cord lesions (Figure 2-3). Currently, 80% of NMO cases are associated with serum antibodies to aquaporin-4, the most abundant water channel protein in the CNS.16,17,18 Yet, anti-NMO-IgG antibodies are absent in 10% to 20% of patients with NMO, even with the most up-to-date assays.16 Thus, while this antibody has been incorporated into the diagnostic criteria for the disease, it is neither a necessary nor sufficient feature. In 2007, Wingerchuk and colleagues characterized the clinical spectrum of NMO, which includes simultaneous optic neuritis and myelitis, cases of myelitis and optic neuritis (in which events occur independently), and limited forms such as longitudinally extensive myelitis or recurrent optic neuritis.17 Notably, patients with NMO are on average older than their MS counterparts (the median age of onset is 40 years in NMO), with a strong female preponderance (female-to-male ratio is 9:1).16,17 In a recent retrospective study of 175 patients with NMO, Jarius and colleagues reported that, among patients with both optic neuritis and myelitis, isolated optic neuritis was the presenting event in 58% of cases. Interestingly, bilateral optic neuritis was more likely to occur in seronegative NMO cases.16 Furthermore, this study showed that there was a marked delay to diagnosis in patients with NMO, which ranged from 16 months if the disease started with myelitis and 55 months if optic neuritis was the initial clinical event. Unfortunately, a high proportion (43%) of patients with NMO in this study were initially incorrectly diagnosed with MS and managed inappropriately as a result. In fact, 53% of NMO patients in this study were treated at least once with interferon-β, a drug that is safe and effective in MS but considered to be harmful in NMO.16
Figure 2-3.
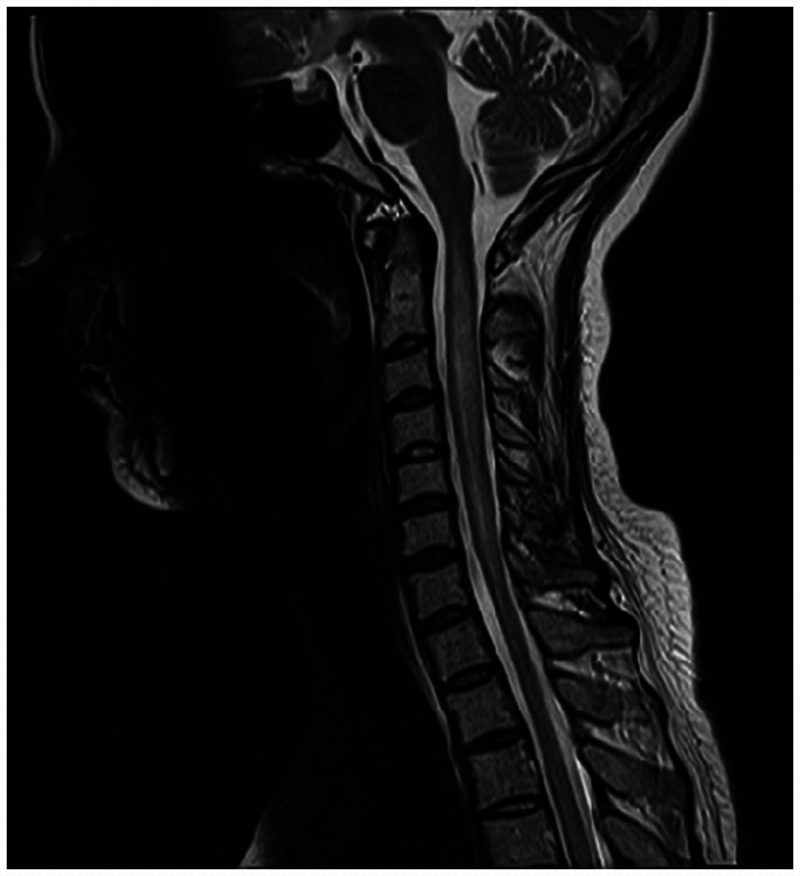
MRI of the cervical spinal cord showing a longitudinal lesion characteristic of neuromyelitis optica.
Courtesy of Jodie Burton, MD.
Recent reports have indicated that NMO can be associated with systemic infections (eg, tuberculosis, hepatitis, Lyme borreliosis, or syphilis) in up to one-third of seropositive patients.16 Patients with NMO spectrum disorders may also harbor other autoimmune conditions including SLE, Sjögren syndrome, thyroid disease, myasthenia gravis, and celiac disease.16,17,19 Classically, NMO attacks are treated with high-dose steroid therapy or plasma exchange treatments. Maintenance therapy may begin with prednisone and azathioprine, although mycophenolate mofetil can also be used.7 In recent years, rituximab has been shown to be a moderately effective agent,7 and tocilizumab, an interleukin-6 blocker, has been studied in patients with NMO with encouraging, albeit preliminary results.7 Eculizumab has demonstrated good success in a small open-label phase 2 trial, but the risk of meningitis and prohibitive cost of this therapy may limit its use.7 Despite emerging therapies for NMO, the prognosis for recovery after relapses remains guarded. Jarius and colleagues reported complete remission with therapy in 83 of 256 patients (32%), partial remission in 126 patients (49%), and no remission in 47 patients (18%). Furthermore, no differences in visual outcome were noted between seropositive (no or partial recovery in 67%) versus seronegative patients (71%).16
Autoimmune Optic Neuropathy
Autoimmune optic neuropathy was first described by Dutton and colleagues in 1982.20,21 Patients with autoimmune optic neuropathy are prone to recurrent optic neuritis events that respond to corticosteroid therapy. Vision loss may manifest during a steroid taper, which may be the first clue that these patients do not have typical optic neuritis. Autoimmune optic neuropathy tends to be more frequent in women. Pain is often not a prominent feature, and presenting visual acuity may range from normal to no light perception. Any pattern of visual field loss may manifest, and the optic nerve may appear mildly edematous during acute events.20 Because this diagnosis is rarely recognized initially, optic atrophy and severe vision loss may evolve over time. Moreover, the cause and pathogenesis of this condition are not known, therefore variable CSF, MRI, and serologic markers of disease have been reported. In one series, 79% of patients with autoimmune optic neuropathy had abnormal antinuclear antibody (ANA) results, and, in another, 82% of patients had elevated anticardiolipin antibody titers. In some cases, skin biopsy specimens from non–sun-exposed areas have shown leukocytoclastic or lymphohistiocytic vasculitis (or both) and immune-reactant deposition.20,22
Recurrent Demyelinating Optic Neuropathy
While optic neuritis is frequently limited to a single episode, 3% to 5% of patients experience recurrent episodes (affecting either or both eyes, sequentially or simultaneously) with negative preliminary investigations for MS, NMO, or other causes.11,12 A clinical entity referred to as recurrent (or relapsing) optic neuritis or (alternatively, neuropathy)11,12 has been described, particularly in the MS literature, and may overlap with autoimmune optic neuropathy. Although the lexicon used to describe these entities varies slightly, reports to date reveal two main forms of recurrent demyelinating optic nerve injury. The first, termed chronic relapsing inflammatory optic neuritis, is a painful, progressive condition that relapses after steroid withdrawal.11 The second, commonly referred to as relapsing idiopathic optic neuritis, is a nonprogressive condition that is not associated with steroid dependence.11,12 Similar to typical optic neuritis, these recurrent demyelinating optic neuropathy subtypes have occasionally been linked to CNS diseases. In one study, the combined conversion rate to MS or NMO was 27% at 5 years and 42% at 10 years.11 Approximately 20% to 25% of patients with recurrent demyelinating optic neuropathy have been shown to convert to NMO within 5 years, with a higher rate (50%) in the NMO-IgG seropositive group than in the seronegative group (10%).11 Therefore, it is important to remember that the numerous acronyms proposed to define forms of recurrent demyelinating optic nerve injury are merely clinical descriptors and do not tell us about the pathobiology of the conditions they are meant to represent. It is this author’s opinion that the longer the name of a medical syndrome is, and accordingly the acronym used to describe it, the less we understand it. Many of these recurrent optic neuritis subtypes fall into the broad category of steroid-responsive optic neuropathies, which over time can be linked with other systemic diseases such as NMO, SLE, sarcoidosis, and Wegener granulomatosis, to name a few (Table 2-1). Therefore, rigorous and repeated efforts should be made to investigate for these conditions over time in cases of relapsing demyelinating optic nerve injury, particularly when symptoms recur during a steroid taper. Patients with steroid-responsive optic neuropathies, regardless of etiology, are often treated with long-term, tapering corticosteroid regimens, with or without adjuvant immunosuppressive therapy.11,12,20
INFECTIOUS OPTIC NEUROPATHIES
Optic neuritis is rarely infectious in nature. More commonly, optic nerve involvement can occur in the clinical setting of neuroretinitis, which is characterized by vision loss, optic disc swelling, and exudative maculopathy (commonly referred to as a “macular star”) (Figure 2-4). Despite the ever-growing list of infectious, neoplastic, and inflammatory conditions linked with neuroretinitis, approximately 50% of cases are idiopathic.23 A complete workup including cranial imaging, lumbar puncture, and serologic evaluation in patients presenting with acute neuroretinitis is often necessary because treatments are directed toward the underlying mechanism or pathogen. The investigations involve specific testing for potential sources of infection, including syphilis, Lyme disease, histoplasmosis, brucellosis, chlamydia, HIV, West Nile virus, toxoplasmosis, Epstein-Barr virus, viral hepatitis B and C, and tuberculosis.23
Figure 2-4.
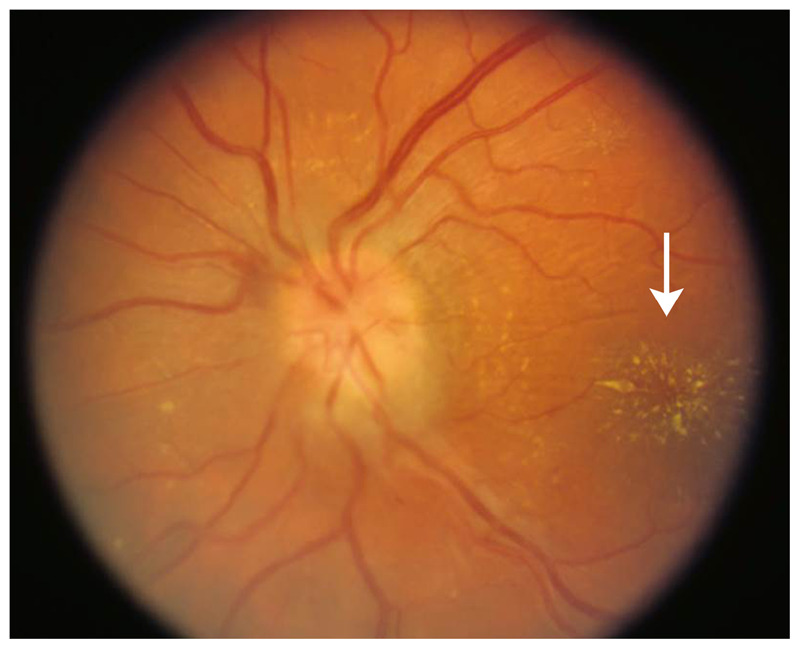
Fundus photo of a macular star (arrow) in a patient with neuroretinitis.
Courtesy of Julie Falardeau, MD.
Neuroretinitis
Neuroretinitis typically affects young, healthy adults, and no sex predilection exists.20,23 In approximately two-thirds of cases, patients present with an antecedent viral prodrome. The presenting visual acuity deficit can be mild to severe, and usually a relative afferent pupillary defect in the affected eye is present. The most common visual field defects noted are of the cecocentral and central types.20 While the severity of the clinical presentation can vary, patients generally present with stellate maculopathy and optic disc edema, which may be associated with exudative detachment in the peripapillary region. Ophthalmoscopy reveals vitreous cells in 90% of cases, and multiple focal yellow-white retinal lesions may appear, which can induce further retinal vascular injury in the form of branch retinal artery or vein occlusions.20,23 Optic disc edema associated with neuroretinitis typically begins to abate in 2 weeks, and to resolve within 3 months. In contrast, the macular star may be present for up to a year.20,23 Visual recovery usually starts within several weeks of symptom onset. In cases of recurrent idiopathic neuroretinitis, however, visual recovery is limited and vision loss is cumulative with repeated attacks, often resulting in permanent impairment.24 In this setting, immunosuppressive treatment may lessen the attack frequency.24
Catscratch disease, caused by Bartonella henselae, is a relatively common cause of neuroretinitis and represents one of several diseases caused by Bartonella species. It is a worldwide condition and typically affects children more often than adults, with a slight male preponderance.20 The majority of cases are caused by a cat scratch or bite, but the disease can also be transmitted to humans by dogs.20 Pain in or around the eye is a variable symptom, and vision loss may be mild to severe at onset.20 It is noteworthy that, in cases of catscratch disease, a macular star may not be initially evident, but may evolve over days to weeks. MRI of the optic nerve is usually normal, but CSF analysis may show lymphocytic pleocytosis.20 The diagnosis of catscratch disease is confirmed by the detection of serum antibodies to B. henselae. Medical treatments that may be used include 100 mg of oral doxycycline twice daily and up to 300 mg of rifampin twice daily for 4 to 6 weeks, or 250 mg of oral azithromycin daily for 5 days. In many cases, this condition is self-limited and treatment may not be needed.
Syphilis
Also known as the “great imitator,” syphilis is a bacterial infection caused by the spirochete Treponema pallidum.20 It is primarily a sexually transmitted disease, although vertical transmission can occur from mother to child.20 Primary, secondary, and tertiary stages of the disease exist; CNS involvement termed “neurosyphilis” may occur during any of these phases, with or without clinical manifestations. Optic neuropathy indicates evidence of active or clinically evident neurosyphilis, and usually manifests in later (secondary or tertiary) stages of the disease.20,25,26 The importance of syphilis as a cause of ocular disease has been recognized since 1858, but, over the years, the spectrum of ocular manifestations of this disease has changed, possibly due to the advent of antibiotic therapy.25 Approximately 18% of secondary syphilis cases develop neurologic or ophthalmic manifestations.20,25,26 The type of vision loss in patients affected by syphilis largely depends on the mechanism of injury. Visual symptoms can manifest as part of a meningeal process and may be accompanied by features of raised intracranial pressure, including papilledema. Patients with syphilitic perineuritis present with inflammation of the optic nerve sheaths, which is associated with optic disc swelling in the absence of increased CSF pressure, and visual impairment.20,25,26,27,28 In cases of optic neuropathy associated with syphilis, vision loss is often central and associated with dyschromatopsia. Pain may or may not be reported, and a variety of visual field defects may be seen.20 The optic disc appearance may be normal (in cases of retrobulbar optic nerve involvement), atrophic, or edematous, as illustrated in Case 2-2. Accompanying features of neuroretinitis may also be seen, including the manifestation of a macular star. The neuroimaging findings of syphilis are not particularly distinguishing, although meningeal enhancement may be observed. Similarly, CSF findings may include a normal cell count with differential normal protein level and normal Venereal Disease Research Laboratory (VDRL) results (diagnostic sensitivity is 27%). Specific testing with fluorescent treponemal antibody absorption (FTA-ABS) may be abnormal.20,25 Patients infected with HIV may have syphilis, even in the absence of positive VDRL or FTA-ABS serology. False negatives (32%) can occur in early primary, latent, or late syphilis and with concomitant HIV infection.20,25 Moreover, patients with concurrent HIV have a higher incidence of neurosyphilis, including ocular complications, which are more likely to be bilateral.25 Benzathine penicillin is the mainstay of therapy for primary and secondary syphilis but its CNS penetration is poor; therefore, in syphilitic optic neuropathy, long-term therapy with IV aqueous penicillin at a dose of 2 million to 4 million units every 4 hours for 2 weeks is standard treatment.20,25 Corticosteroid therapy may also be used.25
Case 2-2
A 58-year-old man with HIV presented with an 8-week history of progressive painless vision loss in the left eye. He denied any awareness of vision loss in the right eye. On examination, visual acuity was 20/80 in the left eye and 20/25 in the right eye with no relative afferent pupil defect. Fundus examination showed bilateral optic disc edema with surrounding choroidal infiltrate that was evident on fluorescein angiography (Figure 2-5A, B29). Visual field testing revealed a central visual field defect in the left eye and an enlarged blind spot in the right eye (Figure 2-5C, D29). Cranial, orbital, and spinal MRI results were normal. Syphilis serology (Venereal Disease Research Laboratory [VDRL] and fluorescent treponemal antibody absorption [FTA-ABS] results) were positive. Lumbar puncture showed a lymphocytic pleocytosis with an elevated protein count and an elevated opening pressure (27 cm of water).
Figure 2-5.
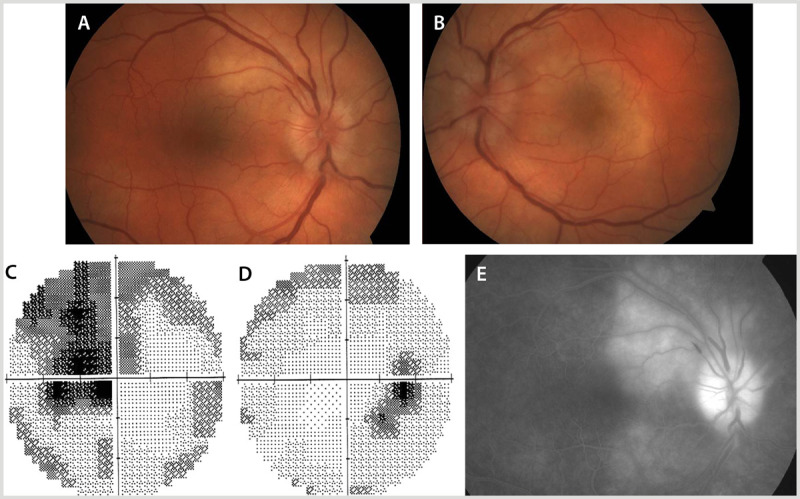
A 58-year-old man with syphilis presented with vision loss in the left eye. Fundus examination showed bilateral optic disc edema (A, B). Visual field testing revealed a central visual field defect in the left eye (C) and an enlarged blind spot with an inferior temporal arcuate area of vision loss in the right eye (D). Fluorescein angiography of the right eye showed choroidal infiltrate around the optic disc (E).
Panels A, B, and C reprinted with permission from Almekhlafi MA, et al, Neurology.29 © 2011 AAN Enterprises, Inc. www.neurology.org/content/77/5/e28.long.
Comment. The patient’s presentation was not typical for optic neuritis associated with multiple sclerosis because of his older age (older than 45 years) and lack of pain with symptom onset. Moreover, he reported progressive vision loss without significant improvement over an 8-week period, whereas typical optic neuritis tends to improve within 3 to 6 weeks. The presence of central vision loss in the absence of a relative afferent pupil defect suggested an alternate basis of vision loss involving the retina, choroid (or both), as opposed to the optic nerve, which was confirmed with fluorescein angiography. The patient was treated with IV aqueous penicillin at a dose of 2 million to 4 million units every 4 hours for 2 weeks, and his vision improved to 20/20 in each eye.
Lyme Optic Neuropathy
Lyme disease is caused by the spirochete Borrelia burgdorferi, which is a tick-borne pathogen.20 This condition is often characterized by a classic rash called erythema chronicum migrans. CNS involvement may occur days to weeks after the initial infection. Like syphilis, a tertiary stage of the disease may be delayed by months to years.20 Patients with Lyme disease may manifest inflammation of the ocular anterior segment, exudative retinal detachment, papilledema, and cranial neuropathies. Papilledema caused by raised intracranial pressure in Lyme meningitis occurs more frequently in children, although some adult cases have been reported. Occasionally, cases of retrobulbar optic neuritis, papillitis, neuroretinitis, and ischemic optic neuropathy have been associated with Lyme neuroborreliosis.20,30 In cases of Lyme disease–associated optic neuritis, vision loss is typically acute, and pain may occur at initial presentation. Visual acuity deficits range from mild to severe in cases of optic neuritis, whereas, for patients with perineuritis, visual acuity is generally well preserved. Optic disc edema can be observed in cases of papillitis and neuroretinitis. As in syphilis, MRI findings in Lyme disease are often not specific and may include meningeal involvement and cranial nerve enhancement.20 The CSF may show a lymphocytic pleocytosis with or without protein elevation.20 The standard screening test is an enzyme-linked immunosorbent assay (ELISA), whereas the Western blot assay is used to distinguish false-positive ELISA results from true infection. In cases of optic neuritis associated with CSF lymphocytic pleocytosis and cranial neuropathies, neuroborreliosis should be strongly considered, particularly in Lyme-endemic areas. Detection of intrathecal antibody against Lyme is diagnostic. Treatment for neuroborreliosis typically involves 2 g/d of IV ceftriaxone for 1 month.20,30
West Nile Virus
West Nile virus is maintained in an enzootic cycle of mosquitoes and birds and has evolved in its clinical expression since the 1950s. Previously, it was believed that the virus had very low affinity for the CNS and was more apt to cause fever, headache, lymphadenopathy, and skin rash.31 Recently, however, cases of meningoencephalitis have increased, for reasons unknown.31 West Nile virus can cause permanent visual loss and eye pain from retinitis, optic neuritis, and uveitis.31,32 In a retrospective review of ocular complications of West Nile virus, multifocal choroiditis was the most common ocular manifestation and typically had a benign clinical course.32 Less common ocular manifestations, including optic neuritis and occlusive vasculitis, may induce permanent visual deficits. Diabetic patients and those older than 50 years of age are believed to be more vulnerable to the severe effects of West Nile virus, including more deleterious ocular manifestations.32
Tuberculosis
Tuberculosis (TB) is bacterial infection caused by Mycobacterium tuberculosis, which on rare occasions causes optic neuritis. Since the emergence of AIDS, TB has made a resurgence in the clinical arena and is now considered a significant opportunistic infection in HIV patients.33 While TB typically presents with pulmonary involvement, extrapulmonary involvement is also relatively common in immunocompromised patients, particularly women and children.20,33 As with syphilis, manifestations of vision loss in TB patients vary with the mechanism of optic nerve involvement. Tuberculomas can cause mass effect on the anterior visual pathway, as demonstrated in Figure 2-6.4 Alternatively, there may be direct infiltration of the optic nerve or optic nerve inflammation caused by opticochiasmatic arachnoiditis. Pain is a variable finding, and vision loss may be mild to profound in TB-associated optic neuritis. The onset of visual symptoms may be rapid or gradual.20 The optic disc can be normal, edematous, or atrophic at presentation. In the setting of meningitis, multiple cranial neuropathies may be seen. MRI features may be nonspecific in TB, although skull-based dural disease is common, and optic nerve enhancement may or may not be evident. Similarly, the CSF findings may be difficult to distinguish from other entities, with elevated protein, mild pleocytosis, and low glucose being variably detected.20 The gold standard for diagnosis is the demonstration of tuberculous bacteria by smear or culture,20 yet recent studies have shown that PCR molecular assay testing of respiratory specimens can provide sensitive and rapid results.34,35 These techniques include commercially available nucleic acid amplification testing and other PCR-based methods. The sensitivity of PCR assay has been shown to vary (65% to 83%) between different testing techniques and across laboratories.35 For cases of TB with CNS involvement, the nested PCR assay has been reported to drastically increase the sensitivity and specificity of DNA amplification compared with conventional single-step PCR. However, the nested PCR assay of CSF is not widely used because it is a laborious and time-consuming procedure, and carries a high risk of cross-contamination.35 Treatment for TB-associated optic neuritis includes isonicotinic acid hydrazide (INH), ethambutol, rifampin, streptomycin, and pyrazinamide, and may vary between the immune-competent versus immune-compromised hosts. Of note, INH36 and ethambutol37 have been associated with vision loss, and ocular complications of TB need to be distinguished from iatrogenic vision loss caused by anti-TB therapy. Furthermore, HIV patients may require concomitant anti-TB treatment and antiretroviral therapy, and can be vulnerable to TB-associated immune reconstitution inflammatory syndrome (TB-IRIS).38 This entity is characterized by either deterioration of existing lesions (paradoxical TB-IRIS) or the appearance of new disease associated with previously subclinical infection.38 Manifestations of TB-IRIS need to be differentiated from other complications of antiretroviral therapy and HIV infection, including drug toxicity, opportunistic infections, treatment failure, and possible nonadherence to therapy. Notably, paradoxical worsening of lesions after initiating anti-TB treatment has been described in both immunocompetent and immunocompromised hosts. In a retrospective study of 659 patients with pulmonary TB who were HIV-negative, 16 developed paradoxical lesions, with an incidence of 2.4%. The risk of paradoxical worsening of TB lesions with therapy increased if patients had significantly decreased hemoglobin, albumin, body mass index, and baseline lymphocyte counts.39 Because TB is associated with different mechanisms of vision loss and potential complications arising from treatment, visual recovery may be poor.
Figure 2-6.
Suprasellar tuberculomas causing chiasmal compression. These lesions paradoxically enlarged after initiation of antituberculosis therapy in an immunocompetent 19-year-old man with tuberculosis meningitis and severe vision loss in both eyes.
Reprinted with permission from Costello FE, Goyal M, Neurol Clin.4 © 2010 Elsevier. www.neurologic.theclinics.com/article/S0733-8619(10)00049-6/abstract.
INFLAMMATORY OPTIC NEUROPATHIES ASSOCIATED WITH SYSTEMIC DISEASE
Rarely, optic neuritis may occur as part of a systemic disease such as SLE, sarcoidosis, or Sjögren syndrome. Given the multisystem nature of these conditions, early diagnosis and initiation of appropriate treatment can be paramount to reducing visual morbidity. To further complicate matters, medications, including some of the anti–tumor necrosis factor-α agents40 and hydroxychloroquine,41 can cause a demyelinating optic neuropathy and retinopathy, respectively. Thus, in patients presenting with vision loss secondary to systemic inflammatory conditions, it is important to distinguish the manifestations of the underlying disease from the effects of the therapies used to treat them.
Sarcoidosis
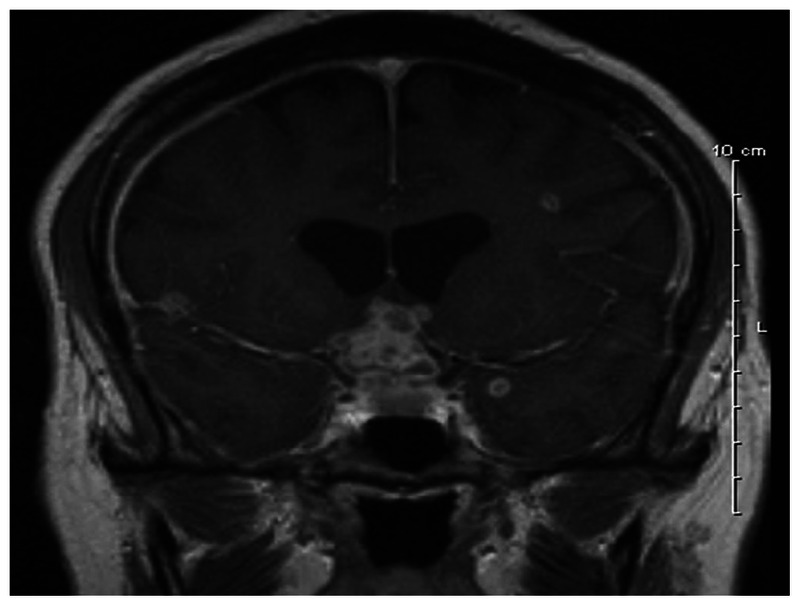
Sarcoidosis is a multisystem granulomatosis inflammatory condition that typically affects the lymphatic system, lungs, skin, and eyes. Like syphilis, sarcoidosis is a master mimicker, and sarcoid-associated optic neuropathy can present exactly like typical optic neuritis. Hence, the diagnosis can be missed unless there is an aberrant feature of the patient’s clinical presentation or course (including recurrent vision loss with steroid withdrawal) that prompts suspicion. Sarcoidosis affects all ethnic groups, although in the Unites States it is more prevalent in African Americans.20 Females are more commonly affected than males, and neurologic involvement occurs in 5% to 16% of cases.20 Ophthalmic involvement occurs in approximately 25% of patients, with a higher incidence noted in those with pulmonary disease. As Frohman has reported, the optic nerve can be affected via several mechanisms in sarcoidosis, including nonspecific inflammation of the optic nerve similar to typical optic neuritis (Figure 2-7); infiltration of the optic nerve or sheaths mimicking mass lesions, such as optic nerve glioma or meningioma; mass effect due to direct compression of the nerve; and parachiasmal involvement.20 Patients may or may not present with pain, and the extent of vision loss can be variable. Visual field defects tend to reflect the region of involvement in the anterior visual pathway affected, and central scotomas, altitudinal defects, or homonymous field loss may be seen.20 The optic nerve may be normal, atrophic, or edematous in appearance. Other findings that may provide a clue to the diagnosis include the presence of conjunctival nodules, keratic precipitates, iris nodules, uveitis, and lacrimal gland enlargement.20 In many cases of neurosarcoidosis, the patient may not have pulmonary features or any other associated findings; therefore, establishing the diagnosis can be challenging. In these cases, recognizing the cardinal ocular symptoms and signs can be helpful. The cranial and orbital MRI features include intense optic nerve and meningeal enhancement, particularly at the base of the skull in close proximity to the chiasm.20 The CSF analysis may show lymphocytic pleocytosis and an elevated protein level. If clinical suspicion exists, additional investigations to consider include chest imaging, total body gallium scan, pulmonary function tests, an anergy panel, and 24-hour urine calcium levels. To diagnose sarcoidosis, “tissue is the issue,” and potential sites to biopsy include the lacrimal glands, conjunctival lesions, or accessible lesions implicated on the gallium scan. Treatment regimens usually involve corticosteroids, sometimes in conjunction with other immunosuppressive agents.
Figure 2-7.
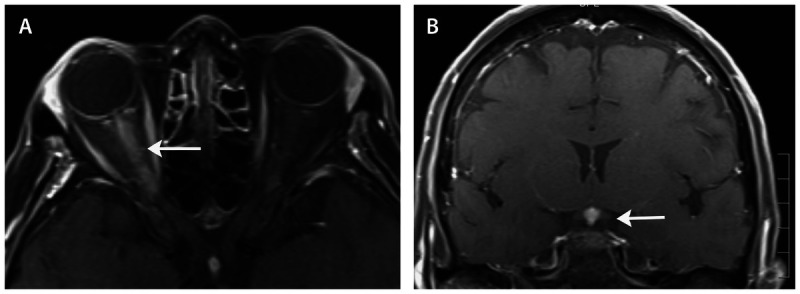
MRI (axial and coronal views) showing gadolinium enhancement of the right optic nerve (A, arrow) and pituitary stalk (B, arrow) in a patient with neurosarcoidosis.
Giant Cell Arteritis
Giant cell arteritis is covered in the article titled “Retinal and Optic Nerve Ischemia” by Valérie Biousse, MD, and Nancy Newman, MD, FAAN, in this issue of CONTINUUM.
Wegener Granulomatosis
Wegener granulomatosis is characterized by necrotizing granulomatous vasculitis affecting vessels in the upper and lower respiratory tracts, paranasal sinuses, kidneys, and lungs.20,42,43 It can occur in a limited ocular form, namely without pulmonary or renal involvement, which can thwart early diagnosis. Ocular involvement is seen in approximately 50% of patients and is usually bilateral. A classic nonocular feature is saddle-nose deformity. Orbital involvement can arise secondary to extension from the sinuses and may manifest with proptosis (45%), ocular motility deficits, and pain.20,42 Wegener granulomatosis generally affects patients 40 to 50 years of age, but has also been reported in children and younger adults. There is a predilection for male involvement, with a sex ratio (male to female) of 2:1.20,42 However, limited Wegener granulomatosis is more common in women. Investigations include cranial and orbital imaging to look for evidence of sinus inflammation, lacrimal gland enlargement as shown in Case 2-3, mastoid inflammation, and orbital masses (with infiltration and obliteration of the fat planes, midline involvement, and bone erosion).20,42 Serologic testing includes cytoplasmic antineutrophilic cytoplasmic antibodies (c-ANCA), which have good sensitivity and specificity for Wegener granulomatosis, resulting in positive results in 60% to 95% of patients with disseminated disease.20,42 The yield of testing decreases in cases of limited disease, however, and false-negative results can occur. Tissue diagnosis is ideal and may be obtained from the nasal sinuses or orbits. Treatment consists of high-dose corticosteroids, often in combination with cyclophosphamide or rituximab.20,42,43,44
Case 2-3
A 40-year-old woman presented with a 2-month history of pain, progressive proptosis, and vision loss in the left eye. Visual acuity was 20/20 in the right eye and 20/100 in the left eye, with a left relative afferent pupil defect. Color vision was normal in the right eye, but the patient could not read any Hardy-Rand-Ritter pseudoisochromatic plates with the left eye. Visual field testing was normal in the right eye and showed a cecocentral scotoma in the left eye. Fundus examination revealed a normal optic nerve appearance in the right eye (Figure 2-8A). In the left eye, the optic nerve was severely edematous and associated with venous stasis and hemorrhage (Figure 2-8B). Cranial and orbital imaging was performed to investigate for the presence of a mass lesion. A CT scan of the orbits showed a heterogenous enhancing mass in the apex of the orbit (Figure 2-8C, D), causing displacement of the left optic nerve with enhancement seen along the course of the optic nerve sheath. An orbital MRI scan (Figure 2-8E, F) showed a soft tissue mass around the left optic nerve that infiltrated the extraocular muscles and lacrimal gland. Orbital biopsy confirmed the diagnosis of Wegener granulomatosis.
Figure 2-8.
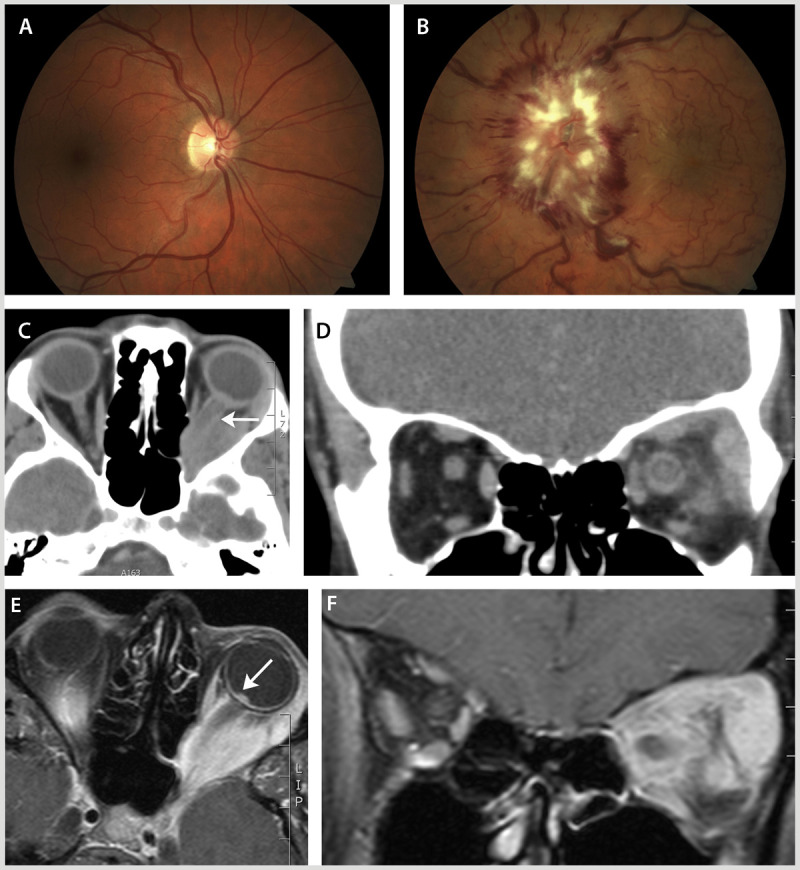
In this case of Wegener granulomatosis, fundus examination revealed a normal optic nerve appearance in the right eye (A) and severe optic disc edema extending to the macula, venous stasis, and hemorrhage in the left eye (B). CT scan of the orbits showed a heterogenous enhancing mass in the apex of the orbit (C, axial; D, coronal) causing displacement and enhancement of the left optic nerve (C, arrow). An orbital MRI (E, axial; F, coronal) showed a soft tissue mass around the left optic nerve that infiltrated the extraocular muscles and lacrimal gland. The edematous left optic nerve was evident relative to the surrounding vitreous fluid of the eye (E, arrow).
Comment. The presence of orbital signs (including proptosis) indicated a potential infiltrative or inflammatory basis of vision loss in this case, because orbital signs are not common in typical optic neuritis. Similarly, the history of persistent pain and progressive vision loss over a 2-month period argued against optic neuritis as the diagnosis in this case. This necessitated urgent orbital and cranial imaging that in Wegener granulomatosis may demonstrate evidence of sinus inflammation, lacrimal gland enlargement, mastoid inflammation, and orbital masses (with infiltration and obliteration of the fat planes, midline involvement, and bone erosion). After the initiation of corticosteroid treatment, this patient’s proptosis resolved, but she was left with a central visual defect and decreased visual acuity (20/200) in the left eye.
Sjögren Syndrome
Sjögren syndrome is a systemic autoimmune disease characterized by dry eyes (keratoconjunctivitis) and dry mouth (xerostomia).20 Primary Sjögren tends to be limited to the oral and lacrimal glands, causing sicca syndrome. Patients who manifest features of other conditions, including rheumatoid arthritis, SLE, vasculitis, scleroderma, polymyositis, primary biliary cirrhosis, or chronic active hepatitis, are characterized as having secondary Sjögren syndrome.20 This condition is usually seen in middle-aged adults, with a female-to-male sex ratio of 9:1.20 Thirty percent of patients with rheumatoid arthritis have associated Sjögren syndrome, and may carry the human leukocyte antigens (HLA) DQ*0501 allele, HLA-B8, HLA-DR3, and/or HLA-DRw52. CNS involvement occurs in 2% to 25% of people with Sjögren syndrome and is much less common than peripheral nervous system involvement in affected individuals.20 Sjögren syndrome has been associated with NMO spectrum disorders and, accordingly, severe vision loss and optic atrophy can manifest in affected patients, as demonstrated in Figure 2-9.16,45,46 Present studies indicate that optic neuritis in patients with Sjögren syndrome who are seropositive for NMO-IgG have two coexisting autoimmune diseases rather than a secondary vasculitic complication of the systemic disease.13 The diagnosis of Sjögren syndrome can be determined by testing for rheumatoid factor (50% of patients) and antibodies against antigens known as Ro (SSA) and La (SSB).18 Treatment commonly involves corticosteroids and immunosuppressive therapies.18
Figure 2-9.
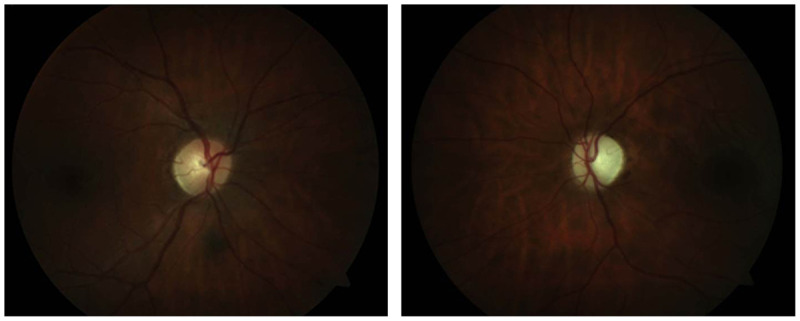
Left optic atrophy in a 35-year-old woman with Sjögren syndrome and neuromyelitis optica spectrum disorder. The right optic nerve was normal in appearance. The right eye is shown on the left and the left eye is shown on the right.
Systemic Lupus Erythematosus
As in sarcoidosis, SLE can present with a myriad of ocular manifestations that may be a harbinger of the disease or a marker of disease activity in those with an established diagnosis. From a clinical perspective, patients with SLE and optic nerve involvement may be distinguished from patients presenting with typical optic neuritis because the former often have coexisting features of systemic disease, including rash, fever, weight loss, and other organ involvement. While relatively rare (affecting 1% of patients),20,47,48 optic nerve involvement in SLE may manifest as optic neuritis or ischemic optic neuropathy. The onset of vision loss is usually painless, subacute, progressive, and severe. Visual acuity in SLE-associated optic nerve injury is usually worse than 20/200, whereas in the ONTT only 36% of typical optic neuritis patients had similar visual dysfunction. In SLE, visual recovery is not as robust as typical optic neuritis, with only 50% of patients recovering better than 20/25, and 38% of patients having a postacute visual acuity worse than 20/200.20,47,48 The increased severity of disease in SLE-associated optic injury is believed to occur because the primary process is considered an ischemic injury to the nerve as opposed to a primary demyelinating insult.20,47,48 When SLE is suspected, the ANA level may be abnormal, with a speckled pattern most commonly seen.20 While ANA titers can be sensitive, this test is not specific for SLE, and serologic markers including anti–double-stranded DNA and anti-Smith antibodies can help confirm the diagnosis.20 Antiphospholipid antibodies are also seen with increased frequency in patients with SLE who have manifestations of ophthalmic or neurologic disease.20,47,48 The inflammatory optic neuropathy associated with SLE can respond dramatically to corticosteroid therapy, and early treatment is associated with better visual outcomes. Standard therapy includes high-dose corticosteroids followed by an extended oral taper. Therapeutic benefits have also been shown with immunosuppressive agents such as cyclophosphamide, cyclosporine, methotrexate, and azathioprine.20,47,48
Paraneoplastic Optic Neuropathy
Paraneoplastic optic neuropathy is covered in the article titled “Metabolic, Hereditary, Traumatic, and Neoplastic Optic Neuropathies” by Gregory Van Stavern, MD, in this issue of CONTINUUM.
CONCLUSION
The inflammatory optic neuropathies represent a group of conditions often characterized by subacute onset vision loss, pain, and variable visual recovery. Optic neuritis associated with MS is the most common inflammatory optic neuropathy encountered in clinical practice, and typically affects young white women. This syndrome is generally self-limited and carries a favorable prognosis for clinical recovery. Optic neuritis is a cause for concern as it may be the first clinical manifestation of MS. In cases of typical optic neuritis, the baseline MRI scan is the most potent predictor for the current or future risk of MS. The revised McDonald criteria have arguably reduced the role of CSF analysis or VEP testing in the evaluation of optic neuritis, yet, for patients with atypical features, including age older than 45 years, persistent pain, systemic symptoms and signs, abnormal fundus findings, and poor visual recovery, additional investigations need to be considered. Optic nerve involvement associated with other demyelinating conditions (including NMO) and other mimics must be distinguished from typical optic neuritis, because they can implicate underlying and potentially life-threatening systemic conditions. For this reason, an in-depth understanding of the presenting features, diagnostic evaluation, and treatment of inflammatory optic neuropathies is important in day-to-day clinical practice.
KEY POINTS
One in every five individuals affected by multiple sclerosis will present with optic neuritis as the first manifestation of their disease. Therefore, recognizing the cardinal clinical features of optic neuritis is important, because the diagnosis has potential long-term implications.
In patients with unilateral optic nerve involvement, a relative afferent pupil defect will be apparent in the affected eye. In cases of bilateral asymmetric optic neuritis, a relative afferent pupillary defect will be found in the more severely affected eye. Alternatively, a relative afferent defect may not be detected if the severity of optic nerve injury is comparable between eyes in cases of bilateral sequential or simultaneous optic neuritis.
Severe optic disc edema, vitreous cells, and hemorrhage are relatively uncommon findings in typical optic neuritis and may herald an alternate diagnosis.
The baseline cranial MRI scan is used to predict the current or future risk of multiple sclerosis in patients presenting with acute optic neuritis.
The decision to use high-dose corticosteroids for the treatment of typical optic neuritis should be done with the understanding that this treatment approach may hasten recovery but does not change patients’ long-term visual outcomes or future risk of multiple sclerosis.
Neuromyelitis optica-associated optic neuritis is most often unilateral at presentation and can closely mimic typical optic neuritis. A high degree of suspicion must be maintained for neuromyelitis optica, because early diagnosis and treatment can prevent disability in this condition.
Currently, 80% of cases of neuromyelitis optica are associated with serum antibodies to aquaporin-4, the most abundant water channel protein in the CNS. Yet, anti-NMO-IgG antibodies are absent in 10% to 20% of patients with neuromyelitis optica, even with the most up-to-date assays.
There can be a marked delay to diagnose patients with neuromyelitis optica (NMO), which ranges from 16 months if the disease starts with myelitis to 55 months if optic neuritis is the initial clinical event. Unfortunately, a high proportion (43%) of patients with NMO are initially incorrectly diagnosed with multiple sclerosis and managed inappropriately as a result. In a recent study, 53% of patients with NMO were treated at least once with interferon-β, a drug that is safe and effective in multiple sclerosis but considered to be harmful in NMO.
Steroid-responsive optic neuropathies can be linked with other systemic diseases such as neuromyelitis optica, systemic lupus erythematosus, sarcoidosis, and Wegener granulomatosis, to name a few; therefore, rigorous and repeated efforts should be made to investigate for these conditions over time in cases of relapsing demyelinating optic nerve injury, particularly when symptoms recur during a steroid taper.
Optic nerve edema may manifest with a macular star in cases of neuroretinitis, which can arise from a wide variety of potential infections.
Lyme disease should be considered for patients presenting with optic neuritis, CSF lymphocytic pleocytosis, and associated cranial neuropathies.
Sjögren syndrome has been shown to coassociate with neuromyelitis optica spectrum disorders, and affected individuals may manifest severe vision loss.
Optic neuropathy associated with systemic lupus erythematosus can be quite severe, and early recognition and treatment can improve clinical outcomes.
Footnotes
Relationship Disclosure: Dr Costello has served on the steering committee of Novartis Corporation, has received a grant from the Multiple Sclerosis Society of Canada, has received personal compensation for advisory board participation from Allergan, Inc, for speaking engagements with EMD Serono, Inc, and for a commissioned article from International Scholarly Research Notices. Dr Costello has received paid travel accommodations/meeting expenses as a speaker from the European Committee for Treatment and Research in Multiple Sclerosis and the Americas Committee for Treatment and Research in Multiple Sclerosis.
Unlabeled Use of Products/Investigational Use Disclosure: Dr Costello reports no disclosure.
REFERENCES
- 1.Miller D,, Barkhof F,, Montalban X, et al. Clinically isolated syndromes suggestive of multiple sclerosis, part 1: natural history, pathogenesis, diagnosis and prognosis. Lancet Neurol 2005; 4 (5): 281–288. [DOI] [PubMed] [Google Scholar]
- 2.Hickman SJ,, Dalton CM,, Miller DH,, Plant GT. Management of acute optic neuritis. Lancet 2002; 360 (9349): 1953–1962. [DOI] [PubMed] [Google Scholar]
- 3.Beck RW,, Cleary PA,, Anderson MM, Jr, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med 1992; 326 (9): 581–588. [DOI] [PubMed] [Google Scholar]
- 4.Costello FE,, Goyal M. Neuroimaging in neuro-ophthalmology. Neurol Clin 2010; 28 (3): 757–787. [DOI] [PubMed] [Google Scholar]
- 5.Beck RW,, Trobe JD,, Moke PS, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol 2003; 121 (7): 944–949. [DOI] [PubMed] [Google Scholar]
- 6.Optic Neuritis Study Group. Visual function 15 years after optic neuritis: a final follow-up report from the Optic Neuritis Treatment Trial. Ophthalmology 2008; 115 (6): 1079–1082. [DOI] [PubMed] [Google Scholar]
- 7.Costello F,, Burton JM. An approach to optic neuritis: the initial presentation. Expert Rev Ophthalmol 2013; 8 (6): 539–551. [Google Scholar]
- 8.Polman CH,, Reingold SC,, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011; 69 (2): 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balcer LJ. Clinical practice. Optic neuritis. N Engl J Med 2006; 354 (12): 1273–1280. [DOI] [PubMed] [Google Scholar]
- 10.Kaufman DI,, Trobe JD,, Eggenberger ER,, Whitaker JN. Practice parameter: the role of corticosteroids in the management of acute monosymptomatic optic neuritis. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000; 54 (11): 2039–2044. [DOI] [PubMed] [Google Scholar]
- 11.de Seze J. Atypical forms of optic neuritis. Rev Neurol (Paris) 2012; 168 (10): 697–701. [DOI] [PubMed] [Google Scholar]
- 12.Petzold A,, Pittock S,, Lennon V, et al. NMO-IgG (Aquaporin-4) autoantibodies in immune-mediated optic neuritis. J Neurol Neurosurg Psychiatry 2010; 81 (1): 109–111. [DOI] [PubMed] [Google Scholar]
- 13.Kezuka T,, Usui Y,, Yamakawa N, et al. Relationship between NMO-antibody and anti-MOG antibody in optic neuritis. J Neuroophthalmol 2012; 32 (2): 107–110. [DOI] [PubMed] [Google Scholar]
- 14.Stubgen JP. A literature review on optic neuritis following vaccination against virus infections. Autoimmun Rev 2013; 12 (10): 990–997. [DOI] [PubMed] [Google Scholar]
- 15.Jarius S,, Wandinger KP,, Borowski K, et al. Antibodies to CV2/CRMP5 in neuromyelitis optica-like disease: case report and review of the literature. Clin Neurol Neurosurg 2012; 114 (4): 331–335. [DOI] [PubMed] [Google Scholar]
- 16.Jarius S,, Ruprecht K,, Wildemann T, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optical: a multicenter study of 175 patients. J Neuroinflammation 2012; 9: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wingerchuk DM,, Lennon VA,, Lucchinetti CF, et al. The spectrum of neuromyelitis optica. Lancet Neurol 2007; 6 (9): 805–815. [DOI] [PubMed] [Google Scholar]
- 18.Lennon VA,, Wingerchuk DM,, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364 (9451): 2106–2112. [DOI] [PubMed] [Google Scholar]
- 19.Matijaca M,, Pavelin S,, Kaliterna DM, et al. Pathogenic role of aquaporin antibodies in the development of neuromyelitis optica in a woman with celiac disease. Isr Med Assoc J 2011; 13 (3): 182–184. [PubMed] [Google Scholar]
- 20.Frohman LP. Other inflammatory optic neuropathies. Neuro-ophthalmology the practical guide. New York: Thieme, 2005: 202–216. [Google Scholar]
- 21.Dutton JJ,, Burde RM,, Warren FA,, Klingele TG. Autoimmune retrobulbar optic neuritis. Am J Ophthalmol 1982; 94 (1): 11–17. [DOI] [PubMed] [Google Scholar]
- 22.Bielory L,, Kupersmith M,, Warren F, et al. Skin biopsies in the evaluation of atypical optic neuropathies. Ocul Immunol Inflamm 1993; 1 (3): 231–242. [DOI] [PubMed] [Google Scholar]
- 23.Ray S,, Gragoudas E. Neuroretinitis. Int Ophthalmol Clin 2001; 41 (1): 83–102. [DOI] [PubMed] [Google Scholar]
- 24.Sundaram SV,, Purvin VA,, Kawasaki A. Recurrent idiopathic neuroretinitis: natural history and effect of treatment. Clin Experiment Ophthalmol 2010; 38 (6): 591–596. [DOI] [PubMed] [Google Scholar]
- 25.Reed JB,, Scales DK,, Wong MT, et al. Bartonella henselae neuroretinitis in cat scratch disease. Diagnosis, management, and sequelae. Ophthalmology 1999; 106 (1): 1–2. [DOI] [PubMed] [Google Scholar]
- 26.Syphilis and the eye: 30573-SD/102386. Optocase: Optometry Continuing Education (CE). www.optocase.com/archives/Syphilis_and_the_Eye.aspx. Accessed April 21, 2014.
- 27.Smith GT,, Goldmeier D,, Migdal C. Neurosyphilis with optic neuritis: an update. Postgrad Med J 2006; 82 (963): 36–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gartaganis S,, Georgiou S,, Monastirli A, et al. Asymptomatic bilateral optic perineuritis in secondary syphilis. Acta Derm Venereol 2000; 80 (1): 75–76. [DOI] [PubMed] [Google Scholar]
- 29.Almekhlafi MA,, Williams G,, Costello F. Clinical reasoning: optic disc swelling in a patient with AIDS. Neurology 2011; 77 (5): e28–e32. [DOI] [PubMed] [Google Scholar]
- 30.Träisk F,, Lindquist L. Optic nerve involvement in Lyme disease. Curr Opin Ophthalmol 2012; 23 (6): 485–490. [DOI] [PubMed] [Google Scholar]
- 31.Anninger W,, Lubow M. Visual loss with West Nile virus infection: a wider spectrum of a “new” disease. Clin Infect Dis 2004; 38 (7): e55–e56. [DOI] [PubMed] [Google Scholar]
- 32.Chan CK,, Limstrom SA,, Tarasewicz DG,, Lin SG. Ocular features of west nile virus infection in North America: a study of 14 eyes. Ophthalmology 2006; 113 (9): 1539–1546. [DOI] [PubMed] [Google Scholar]
- 33.Jaafar J,, Hitam WH,, Noor RA. Bilateral atypical optic neuritis associated with tuberculosis in an immunocompromised patient. Asian Pac J Trop Biomed 2012; 2 (7): 586–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rozales FP,, Machado AB,, DE Paris F, et al. PCR to detect Mycobacterium tuberculosis in respiratory tract samples: evaluation of clinical data. Epidemiol Infect 2013; 10: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi T,, Tamura M,, Takasu T. The PCR-based diagnosis of central nervous system tuberculosis. Up to date. Tuberc Res Treat 2012; 2012: 831292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kulkarni HS,, Keskar VS,, Bavdekar SB,, Gabhale Y. Bilateral optic neuritis due to isoniazid (INH). Indian Pediatr 2010; 47 (6): 533–535. [DOI] [PubMed] [Google Scholar]
- 37.Ezer N,, Benedetti A,, Darvish-Zargar M,, Menzies D. Incidence of ethambutol-related visual impairment during treatment of active tuberculosis. Int J Tuberc Lung Dis 2013; 17 (4): 447–455. [DOI] [PubMed] [Google Scholar]
- 38.Agarwal U,, Kumar A,, Behera D, et al. Tuberculosis associated immune reconstitution inflammatory syndrome in patients infected with HIV: meningitis a potentially life threatening manifestation. AIDS Res Ther 2012; 9 (1): 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng SL,, Wang HC,, Yang PC. Paradoxical response during anti-tuberculosis treatment in HIV-negative patients with pulmonary tuberculosis. Int J Tuberc Lung Dis 2007; 11 (12): 1290–1295. [PubMed] [Google Scholar]
- 40.Faillace C,, de Almeida JR,, de Carvalho JF. Optic neuritis after infliximab therapy. Rheumatol Int 2013; 33 (4): 1101–1103. [DOI] [PubMed] [Google Scholar]
- 41.Mititelu M,, Wong BJ,, Brenner M, et al. Progression of hydroxychloroquine toxic effects after drug therapy cessation: new evidence from multimodal imaging. JAMA Ophthalmol 2013; 131 (9): 1187–1197. [DOI] [PubMed] [Google Scholar]
- 42.Hogan RN. Orbital inflammation and infection. Neuro-ophthalmology the practical guide. New York: Thieme, 2005: 356–385. [Google Scholar]
- 43.Huchzermeyer C,, Mardin C,, Holbach L, et al. Successful remission induction with a combination therapy of rituximab, cyclophosphamide, and steroids in a patient with refractory optic neuritis in Wegener’s granulomatosis. Clin Rheumatol 2013; 32 (suppl 1): S97–101. [DOI] [PubMed] [Google Scholar]
- 44.Taylor SR,, Salama AD,, Joshi L, et al. Rituximab is effective in the treatment of refractory ophthalmic Wegener’s granulomatosis. Arthritis Rheum 2009; 60 (5): 1540–1547. [DOI] [PubMed] [Google Scholar]
- 45.Tan P,, Yu WY,, Umapathi T,, Lim SA. Severe optic neuritis in a patient with combined neuromyelitis optica spectrum disease and primary Sjögren’s syndrome: a case report. J Med Case Rep 2012; 6 (1): 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurne A,, Isikay IC,, Karlioguz K, et al. A clinically isolated syndrome: a challenging entity multiple sclerosis or collagen tissue disorders: clues for differentiation. J Neurol 2008; 255 (11): 1625–1635. [DOI] [PubMed] [Google Scholar]
- 47.Palejwala NV,, Walia HS,, Yeh S. Ocular manifestations of systemic lupus erythematosus: a review of the literature. Autoimmune Dis 2012; 2012: 290898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Belmont HM. Treatment of systemic lupus erythematosus—2013 update. Bull Hosp Jt Dis 2013; 71 (3): 208–213. [PubMed] [Google Scholar]