

Abstract
Like all sick children, children with CKD need access to safe and effective medicines that have been formulated and examined specifically for them. Despite legislation in the United States and the European Union that either mandates or incentivizes programs for children, conducting trials to advance the treatment of children continues to prove to be a challenge for drug developers. This is also the case for drug development in children with CKD, where trials face challenges in recruitment and completion and where there remains a substantial time lag between initial approval of a medicinal product for use in adults and completion of studies that result in the addition of pediatric-specific labeling for the same indication. The Kidney Health Initiative commissioned a workgroup of diverse stakeholders (https://khi.asn-online.org/projects/project.aspx?ID=61), including participants from the Food and Drug Administration and the European Medicines Agency, to think carefully through the challenges in drug development for children with CKD and how to overcome them. This article provides an overview of the regulatory frameworks in the United States and the European Union that govern pediatric drug development, the current landscape of drug development and approval for children with CKD, the challenges in conduct and execution of these drug trials, and the progress that has been made to facilitate drug development for children with CKD.
Keywords: chronic kidney disease, clinical trial, ESKD, kidney disease, pediatric nephrology, pediatrics
Introduction
As with all sick children, children with kidney diseases need access to safe and effective medicines that have been formulated and examined specifically for them. Despite legislation in the United States and the European Union (EU) that either mandates or incentivizes drug development programs for children, conducting trials to advance the treatment of children continues to prove to be a challenge for drug developers. These challenges require close collaboration with all stakeholders to think carefully about what data are needed to support pediatric approval of drugs and how to optimally design trials to collect those data.
One specific example of children faced with limited approved therapeutic options is children with CKD. Pediatric CKD is relatively uncommon compared with adult CKD, and development of products for use in small populations presents unique challenges requiring thoughtful planning early in the product development process. There are also ethical considerations that must be addressed when conducting clinical research in pediatric populations. Furthermore, age-appropriate formulations must be developed for many drugs to treat the youngest patients with CKD. Other operational and feasibility issues that hamper successful trial completion include the lack of coordination across pediatric clinical trial networks, the need to collect appropriate safety data that sometimes require longer-term follow-up, and the development of pediatric-specific clinical outcome measures and end points.
The Kidney Health Initiative (KHI), a public-private partnership between the American Society of Nephrology, the US Food and Drug Administration (FDA), and over 100 member companies and organizations, commissioned a workgroup to take on an inventory of challenges, share insights and lessons learned, and develop an approach for overcoming barriers to drug development for children with kidney disease. Through this partnership with the nephrology community and FDA, KHI seeks to foster the development of patient-centered therapies for kidney diseases by advancing the scientific understanding of the kidney health and patient safety implications of new and existing medical products.1
The workgroup took advantage of the diversity of constituent members within KHI and convened a group that included representatives for patients and caregivers, health care providers, researchers, professional organizations and research networks, industry partners, and regulators. By convening these diverse stakeholders, the workgroup was able to develop resources for the community to facilitate a streamlined approach to pediatric drug development among stakeholders. This article provides an overview of the regulatory frameworks in the United States and the European Union that govern pediatric drug development, the current landscape of drug development for children with CKD, the challenges in development and execution of pediatric drug trials, and the progress that has been made to facilitate drug development for children with kidney diseases. The article focuses on the regulations by the FDA and European Medicines Agency (EMA) because they have had pediatric legislation in place longer than other regions. However, several regions are developing or have recently developed legislation to support pediatric drug development. In addition, there are robust mechanisms for global regulators to discuss pediatric drug development, including the Pediatric Cluster (further defined below) and the International Council for Harmonisation (ICH), a joint regulatory-industry initiative that works on international harmonization of drug development including in pediatrics to reduce the likelihood of substantial differences between regions.
Legal Regulatory Framework for Pediatric Drug Development in the United States and the European Union
The US- and EU-specific legislation that govern the development of medicines for pediatric use aim to produce high-quality evidence to support the authorization of medicines for children by providing a system of obligations/requirements and incentives to be followed by the applicants.2 Incentives for industry sponsors include extension of patent life or marketing exclusivity, while obligations include the requirement to conduct studies in pediatric patients under certain circumstances. These laws are intended to promote plans for studies in pediatric populations early during overall product development. A comparison of the regulatory processes, which are similar but not identical in the two regions, was recently published as a joint effort by the regulatory agencies,3 and an overview is provided in Table 1.
Table 1.
EU and US legislation on pediatric drug development
| Attribute | US FDA BPCA/FDASIA 2012 |
US FDA PREA/FDASIA 2012 |
EU-EMA (Paediatric Regulation 1901/2006) |
|---|---|---|---|
| Pediatric plan | WR | PSP | PIP |
| Scope of pediatric development | Any indication in the pediatric population (not limited to adult indication) | Same as adult indication | Derived from adult indication, within the same condition, voluntary PIPs for other indications possible |
| Types of products | All medicinal products | New medicinal products and biosimilars | New medicinal products; authorized products under patent/SPC if applying for new indication/route of administration/pharmaceutical form |
| Orphan-designated products | Included | Excluded | Included |
| Timing of submission | Anytime adequate data are available | End of phase 2 in adults | End of phase 1 in adults |
| Pediatric development | Optional | Mandatory unless waived | Mandatory unless waived |
| Applies to | Incentive | Requirement | Incentive and requirement (obligations) |
| Main reward | 6-mo patent extension on the moiety | None | 6-mo SPC extension (patent) for the product |
EU, European Union; US, United States; FDA, Food and Drug Administration; BPCA, Best Pharmaceuticals for Children Act; FDASIA, FDA Safety and Innovation Act; PREA, Pediatric Research Equity Act; EMA, European Medicines Agency; WR, written request; PSP, pediatric study plan; PIP, Paediatric Investigation Plan; SPC, Supplementary Protection Certificate.
The United States
Over the past 20 years, there has been considerable advancement in pediatric drug development under the US legislative framework, resulting in more consistent inclusion of children in drug development programs and a higher proportion of FDA-approved drugs containing pediatric use information in labeling than that before the laws were passed.4 This progress has been largely because of the FDA's authority, under the 2002 Best Pharmaceuticals for Children Act (BPCA)5 and the 2003 Pediatric Research Equity Act (PREA),6 to incentivize and require, respectively, the development of drugs and biological products in children. Since the passage of BPCA and PREA, the FDA has approved over 1000 new labeling changes with pediatric-specific use information (Figure 1A).7,8
Figure 1.
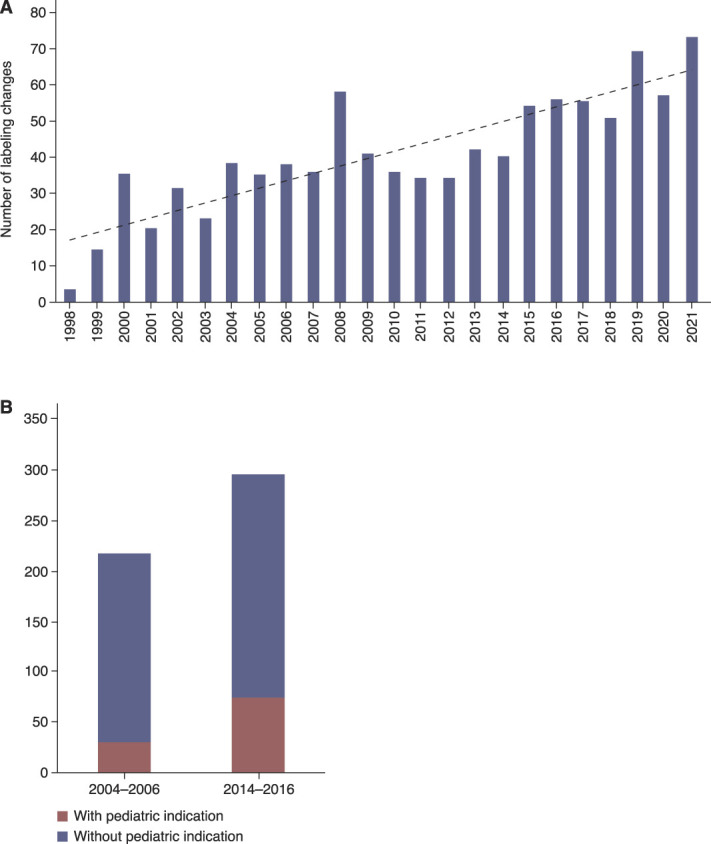
Increase of pediatric label changes in the United States and the European Union. (A) Number of pediatric labeling changes that occurred pursuant to PREA, BPCA, and the Pediatric Rule from 1998 to 2021. Figure reprinted from ref. 7. (B) EMA marketing authorizations of medicines with pediatric indication. After the EU Paediatric Regulation9 entered into force in 2007, the number of new medicines with a pediatric indication at the time of first marketing authorization plus the number of new pediatric indications added to authorized medicines more than doubled from 30 of 217 approvals (14%) in 2004–2006 to 74 of 295 approvals (25%) in 2014–2016.11 BPCA, Best Pharmaceuticals for Children Act; EMA, European Medicines Agency; EU, European Union; PREA Pediatric Research Equity Act.
The European Union
The EU Paediatric Regulation9 entered into force in 2007 and combines incentives and requirements for pediatric drug development into a single law. It requires that each application for marketing authorization of a new medicinal product and certain applications for variations of already authorized medicines include a Paediatric Investigation Plan agreed upon with the Paediatric Committee of the EMA unless an exemption (waiver) is granted. The Regulation has also led to the creation of a network of pediatric research at the EMA (Enpr-EMA), which facilitates research to increase the availability of medicines authorized for use in the pediatric population.10
The Regulation has had a substantial positive effect on pediatric medicine development in Europe. An analysis of two 3-year periods before and a few years after the implementation of the Regulation showed that the number of new medicines with a pediatric indication at the time of first marketing authorization plus the number of new pediatric indications added to authorized medicines more than doubled (Figure 1B).11 In the first 10 years since the entry into force of the Regulation, between 2007 and 2016, a total of 273 new medicines and 43 additional pharmaceutical forms appropriate for the use in children were authorized in the European Union.11
International Regulatory Concordance
The publication of the 2000 ICH E11 guidance, “Clinical Investigation of Medicinal Products in the Pediatric Population,” was an important milestone for global pediatric drug development because it marked the first time that regulatory bodies from around the world collaborated to establish general principles relating to the development of drugs in children.12 The guidance emphasized that the best therapy for pediatric patients is an approved therapy and that the safety and efficacy of drugs in development should be studied and understood in children if those drugs are anticipated to be used in the pediatric population. An addendum to this guidance, ICH E11(R1), published in 2017, provided additional regulatory guidance on current and evolving topics in global pediatric drug development, including a discussion of the use of pediatric extrapolation.12
The FDA and EMA, together with regulators from other regions, strive to enable timely, ethical, and sound global development programs through regular interactions. The Pediatric Cluster was set up by the FDA and EMA in 2007 as a forum for discussion of pediatric plans across regions through monthly teleconferences with the aim to harmonize requirements on scientific issues.13 The Pediatric Cluster has since expanded to include other regulatory agencies, i.e., Pharmaceuticals and Medical Devices Agency Japan, Health Canada, and Therapeutic Goods Administration Australia (as an observer). Since 2013, the FDA and EMA have also issued common commentaries, which are informal, nonbinding comments to sponsors regarding pediatric development plans that have been submitted to both the FDA and EMA. A drug developer may request that a common commentary be issued.
One area that has evolved significantly during recent years and has had a substantial effect on the development of pediatric therapeutics both in the United States and the European Union is the use of pediatric extrapolation. Pediatric extrapolation is defined in the ICH E11(R1) guideline as “an approach to providing evidence in support of effective and safe use of drugs in the pediatric population when it can be assumed that the course of the disease and the expected response to a medicinal product would be sufficiently similar in the pediatric [target] and reference (adult or other pediatric) population.”12 Pediatric extrapolation can extend what is known (e.g., information on efficacy, safety, and/or dosing) in the reference population (adult or other pediatric) to the target population (pediatric) on the basis of an assessment of the relevant similarities of disease and response to therapy in the two populations.14 In scenarios where the disease and treatment response are sufficiently similar in adults and pediatric patients, efficacy can be leveraged from adults, obviating the need to collect pediatric efficacy data. In these cases, collection of pharmacokinetic data to confirm that the pediatric doses achieve the same exposures found to be efficacious in adults along with pediatric safety data may be sufficient to support product approval in a pediatric population. Depending on the uncertainties that exist about disease and/or treatment response similarity, additional clinical investigations may be needed in pediatric patients to bridge the knowledge gaps to support product approval in a pediatric population. Regulatory guidances detailing the approaches to pediatric extrapolation continue to evolve.15–17 Clearly, the appropriate use of pediatric extrapolation can increase the efficiency of pediatric therapeutic development by optimizing the use of existing data.
In addition to advances in the appropriate application of pediatric extrapolation, increases in the understanding of the use of drug development strategies and tools (e.g., state-of-the art experimental systems, such as organoids and cells on a chip, modeling and simulation techniques, novel statistical strategies, innovative study designs, and pediatric-specific biomarkers and clinical end points) have all increased the efficiency and feasibility of pediatric clinical therapeutic development. When pediatric extrapolation is not acceptable, independent substantiation of efficacy and safety may be needed in the target pediatric population.
Current Landscape of Drug Development in Children with CKD
FDA and EMA approvals of drugs and therapeutic biologics for complications associated with pediatric CKD are provided in Table 2. Supplemental Table 1 contains more details on a list of some FDA-approved drugs and therapeutic biologics for the treatment of complications associated with adult and pediatric CKD. There are currently approved treatments of anemia (erythropoiesis-stimulating agents and iron agents approved for iron deficiency anemia), genetic diseases with kidney manifestations, growth failure, hyperkalemia, hyperphosphatemia, hypertension, and secondary hyperparathyroidism. This list is not exhaustive, aiming instead to highlight the effect of BPCA and PREA on key areas of drug approval in children with CKD. A notable effect of the legislation was a surge in the number of antihypertensive medications that were approved for children. These have greatly expanded the therapeutic armamentarium for pediatric nephrologists who care for children with hypertension, which is often a consequence of CKD.
Table 2.
US Food and Drug Administration and European Medicines Agency approvals of drugs and biologics for complications associated with pediatric CKD
| Indication | FDA | EMA |
|---|---|---|
| All | 35 | 16 |
| Anemia: Erythropoiesis-simulating agents | 3 | 4 |
| Anemia: Iron agents specifically approved for iron deficiency anemia in CKD | 2 | 0 |
| Genetic diseases with kidney manifestations | 8 | 6 |
| Growth failure | 2 | 2 |
| Hyperkalemia | 1 | 0 |
| Hyperphosphatemia | 1 | 1 |
| Hypertension | 16 | 2a |
| Secondary hyperparathyroidism | 2 | 1 |
Approvals of drugs and biologics listed in Supplemental Tables 1 and 2. FDA, Food and Drug Administration; EMA, European Medicines Agency.
In the European Economic Area, antihypertensive drugs are authorized mainly on a national level and these are not included.
The EMA has also approved several pediatric investigation plans for the treatment of CKD and the complications associated with it as presented in Supplemental Table 2. These products have been authorized by the centralized procedure, i.e., assessed by the EMA's committees and authorized in all EU member states as well as in the European Economic Area. However, it needs to be taken into account that in the European Economic Area, antihypertensive drugs are authorized mainly on a national level, and these are not included in the tables. Among the 35 approvals by the FDA and the 16 by the EMA in the tables, there is an overlap of eight approvals for the same drug/formulation(s) approved by the FDA and EMA (see annotation in Supplemental Tables 1 and 2).
Current Barriers and Operational Challenges
Despite the successes as evidenced by the list of approved drugs to treat complications associated with CKD in children, development of pediatric therapeutics for kidney disease continues to face challenges and, as with other therapeutic areas, there remains a substantial time lag between initial adult approval and completion of studies that results in the addition of pediatric-specific labeling for the same indication. One important reason for delay in the completion of pediatric studies is that once a drug is approved in adults, off-label use can occur in children. It should be noted that the FDA and EMA do not regulate the practice of medicine or the off-label use of drugs.
Fortunately, the number of children with CKD in the United States and the European Union is limited. Most of the available epidemiological data are collected through kidney failure registries, which reflect the geographic availability of health services that cover KRT. According to the United States Renal Data System annual data report, there were 5405 children aged 0 to 17 years with prevalent kidney failure as of 2018. Kidney transplantation was the most common kidney replacement modality. Of the 838 children aged 0–17 years entering kidney failure in 2018, over 600 initiated dialysis and the remaining children received a kidney transplant.18 The number of children with CKD not receiving KRT is more uncertain because earlier stages of CKD are frequently asymptomatic and underreported, and there is a lack of population-based ascertainment.19 The estimated number of children with nondialysis-dependent CKD and kidney failure is similar in the United States and in Europe. The number of children on dialysis in the United States and the European Union is between 1000 and 2000 in each area, with approximately 5000–10,000 children with earlier stages of CKD in each area. Globally, these numbers underestimate the incidence and prevalence of CKD and kidney failure in the pediatric population because epidemiology data are not as readily available in developing economies. The number of children with CKD decreases from older to younger age,20 making the study of the youngest population especially challenging. Furthermore, kidney diseases in children are heterogeneous and vary in their age at onset and their clinical presentations, including complications such as hypertension, anemia, electrolyte disorders, recurrent urinary infections, and bone disease. This heterogeneity adds to the challenge of drug development in this population.21
Clinical trials in pediatric CKD are operationally challenging because children with earlier stages of CKD are dispersed geographically and may receive care jointly by their primary care physician and a pediatric nephrology specialist. Once on dialysis, many children receive their care in specialized dialysis centers affiliated with tertiary academic centers, but some do receive care in dialysis units operated by independent dialysis organizations. The largest pediatric dialysis centers may care for as many as 70 children, whereas for most centers, the pediatric dialysis census is only 10–15 patients. The preferred modality of KRT is often peritoneal dialysis, especially in younger children. In addition, approximately one third of children receive a kidney transplant within the first year after initiating dialysis, representing a common reason for early discontinuation in drug studies. Of the small population of children with CKD, only a fraction can be expected to meet customary eligibility criteria for a trial.
Owing to the pediatric regulatory requirements, there is now active competition across different trials to enroll children with CKD who are clustered at a limited number of pediatric nephrology research sites and academic centers. As a result, clinical trial programs in children with CKD face significant recruitment and completion issues.
One approach to address these operational challenges is the development of large research networks for children with CKD, on dialysis or with a kidney transplant. The North American Pediatric Renal Trials and Collaborative Studies and the Pediatric Nephrology Research Consortium (formerly known as the Midwest Pediatric Nephrology Consortium) in the United States as well as the European Study Consortium for Chronic Kidney Disorders Affecting Pediatric Patients in Europe are three important examples. These networks have built registries and trial consortia that facilitate investigator or drug company–sponsored research. Yet each network may not be large enough to provide complete enrollment numbers for a particular study. In addition, there are also several disease-specific networks, such as the Nephrotic Syndrome Study Network and the Cure Glomerulonephropathy consortium, which can provide access to specific disease groups, and grant-supported academic prospective registry studies of children with CKD, such as the Cardiovascular Comorbidity in Children with Chronic Kidney Disease Study (4C) in the European Union and the Chronic Kidney Disease in Children Cohort Study in the United States.22,23 These cohorts can be queried to understand distributions of clinical characteristics and eligibility criteria.
Furthermore, there is a lack of coordination and prioritization of those drug development programs that may be deemed most necessary and impactful by all stakeholders, including patients and their caregivers. When setting priorities across and within studies, including the voice of patients or parents/caregivers is essential in ensuring proper investigational focus, balancing trade-offs between potential risks and benefits, and optimizing developmental plans and study design. The importance of listening to these vital stakeholders' perspectives from the early stages of clinical trial planning is increasingly recognized, e.g., in Enpr-EMA's recommendations on preparedness of pediatric clinical trials.24
Ongoing Efforts to Overcome Barriers to Drug Development in Children with CKD
The work of the KHI workgroup on overcoming barriers to drug development in children with CKD resulted in the launch of the Kidney Pediatric Accelerator Trial Clearing House program in 2018. The intent of this program was to enable feasibility assessment for the available patient populations through data sharing and access to CKD pediatric registries, to facilitate assessment of the capacity of various pediatric kidney clinical trial organizations, and to assist with the identification of expertise that can provide consultation on study planning. Additional information about the Kidney Pediatric Accelerator Trial Clearing House and the request form can be found on the KHI website: www.kidneyhealthinitiative.org. Pediatric drug development continues to be a priority of KHI with efforts focused on IgA nephropathy, C3 glomerulopathy, and a community-based prioritization effort all launching over the past year. KHI has also supported complementary programs such as the Pediatric Inclusion in the Evaluation of Novel Therapies (PIONEER) initiative stemming from NephCure Kidney International's Gateway Initiative. Additional activities in the community dedicated to children with CKD have been established, including those by the Kidney Research Network (https://www.kidneyresearchnetwork.org/) and the American Society of Pediatric Nephrology Therapeutics Development Committee, which has published on enhancing clinical trial development for pediatric kidney diseases and highlighted different approaches to trial design by academic and industry sponsors.25
Summary
Regulatory frameworks across regions have been successful in planning for necessary pediatric studies early during product development resulting in pediatric use information in labeling, but more still needs to be done to overcome the drug development challenges specific to pediatric CKD. Coordination of efforts in drug development is necessary to better serve the needs of these children and their families. Addressing the many challenges requires collaboration with industry sponsors, academic investigators, regulatory authorities, patients and caregivers, advocacy groups, and professional groups to develop and implement innovative approaches to pediatric therapeutics development. The KHI aims to foster collaboration across all stakeholders to address these challenges so that timely and efficacious and safe therapies are available for the benefit of all children with CKD.
Supplementary Material
Acknowledgments
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the US Food and Drug Administration, the European Medicines Agency, or one of its committees or working parties, nor do they necessarily reflect the official policies of any KHI member organization, the US Department of Veterans Affairs, the US Department of Health and Human Services, or of Agios Pharmaceuticals, nor does any mention of trade names, commercial practices, or organization imply endorsement by the US Government.
This work was supported by the Kidney Health Initiative (KHI), a public-private partnership between the American Society of Nephrology, the US Food and Drug Administration, and >100 member organizations and companies to enhance patient safety and foster innovation in kidney disease. We acknowledge the workgroup members for their support over the duration of this project, including Deepa Chand, Pamela Duquette, Debbie Gipson, Teresa Vu Lewis, Amy Mason, Alicia Neu, Jesse Roach, Franz Schaefer, Michelle Rheault, and Bill Schnaper. KHI funds were used to defray costs incurred during the conduct of the project, including project management support, which was expertly provided by American Society of Nephrology staff members Meaghan Malley, Elle Silverman, and Melissa West. There was no honorarium or other financial support provided to KHI workgroup members. The authors of this paper have final review authority and are fully responsible for its content. KHI makes every effort to avoid actual, potential, or perceived conflicts of interest that may arise as a result of industry relationships or personal interests among the members of the workgroup. More information on KHI, the workgroup, or the conflict of interest policy can be found at www.kidneyhealthinitiative.org.
Disclosures
G.F. Egger and C. Pallidis report employment with the European Medicines Agency. S.L. Goldstein reports consultancy for Akebia, Baxter Healthcare, Bayer, Bioporto, Inc., BioProducts Labs, Fresenius, Kaneka, Inc, Leadiant, MediBeacon, Medtronic, NuWellis, Otsuka, Reata, and Renibus; ownership interest in MediBeacon, Inc; research funding from Baxter Healthcare, Bioporto, ExThera, Medtronic, and NuWellis; honoraria from Baxter Healthcare and Fresenius; patents or royalties from Vigilanz; an advisory or leadership role for MediBeacon; and speakers bureau for Baxter Healthcare and Fresenius. B.L. Laskin reports ownership interest in Acorda Therapeutics, AT&T, Charter Communications, Comcast, DHT Holdings, Duke Energy, Electra Battery Materials, Ford Motor, Happiness Development, Johnson Controls, Medtronic, Nu Holdings, TE Connectivity, Verizon, and Warner Brothers Discovery; research funding from Viracor-Eurofins; research sample testing free of charge; and patent: Compositions and Methods for Treatment of HSCT-Associated Thrombotic Microangiopathy (US Patent Number PCT/US2014/055922, 2014). M.A. Malley reports employment with Travere Therapeutics and ownership interest in Travere Therapeutics and Altimmune. A. Thompson and L. Yao report employment with the US Food and Drug Administration. S. Tuchman reports ownership interest in Apple, American Electric Power, Boeing, BP, and Disney. S. Tuchman's spouse reports employment with the Eating Recovery Center. K. Uhlig reports employment with Agios Pharmaceuticals, ownership interest in Agios Pharmaceuticals, and an advisory or leadership role for Kidney Health Initiative Board of Directors. K. Uhlig's spouse reports consultancy for S2N Health. All remaining authors have nothing to disclose.
Funding
This work was supported by R18FD005283 from the US Food and Drug Administration.
Author Contributions
Conceptualization: Stuart L. Goldstein, Katrin Uhlig.
Project administration: Meaghan A. Malley, Katrin Uhlig.
Supervision: Katrin Uhlig.
Writing – original draft: Gunter F. Egger, Stuart L. Goldstein, Mona Khurana, Benjamin L. Laskin, Meaghan A. Malley, Chrissi Pallidis, Aliza Thompson, Shamir Tuchman, Katrin Uhlig, Lynne Yao.
Writing – review & editing: Gunter F. Egger, Stuart L. Goldstein, Benjamin L. Laskin, Chrissi Pallidis, Aliza Thompson, Shamir Tuchman, Katrin Uhlig, Lynne Yao.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/CJN/B768.
Supplemental Table 1. FDA-approved drugs and biologics marketed for the treatment of complications associated with adult and pediatric CKD.
Supplemental Table 2. EMA-approved medicinal products and paediatric investigation plans (PIPs) for the treatment of complications associated with adult and pediatric CKD.
References
- 1.Archdeacon P, Shaffer RN, Winkelmayer WC, Falk RJ, Roy-Chaudhury P. Fostering innovation, advancing patient safety: the kidney health initiative. Clin J Am Soc Nephrol. 2013;8(9):1609–1617. doi: 10.2215/CJN.01140113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatti S, Sanders C. Paediatric Regulations: US and EU: Similar but Different. European Pharmaceutical Contractor, Spring 2011. Accessed February 20, 2023. http://www.samedanltd.com/magazine/11/issue/149/article/2893 [Google Scholar]
- 3.Penkov D, Tomasi P, Eichler I, Murphy D, Yao LP, Temeck J. Pediatric medicine development: an overview and comparison of regulatory processes in the European Union and United States. Ther Innov Regul Sci. 2017;51(3):360–371. doi: 10.1177/2168479017696265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bourgeois FT, Kesselheim AS. Promoting pediatric drug research and labeling—outcomes of legislation. N Engl J Med. 2019;381(9):875–881. doi: 10.1056/NEJMhle1901265 [DOI] [PubMed] [Google Scholar]
- 5.Best Pharmaceuticals for Children Act, Pub. L. 107-109. (2002). Reauthorized in 2007 by Pub. L. 110-85 (Food and Drug Administration Amendments Act), and Permanently Reauthorized in 2012 by Pub. L. 112-114 (Food and Drug Administration Safety and Innovation Act).
- 6.Pediatric Research Equity Act. Pub L. 108-155 (2003). Reauthorized in 2007 by Pub. L. 110-85 (Food and Drug Administration Amendments Act), and Permanently Reauthorized in 2012 by Pub. L. 112-114 (Food and Drug Administration Safety and Innovation Act).
- 7.US Food and Drug Administration and American Academy of Pediatrics. Historic Milestone: 1,000 Drugs, Biologics Have New Pediatric Use Information in Labeling. AAP News FDA Updates. Accessed February 20, 2023. https://www.fda.gov/science-research/pediatrics/aap-news-fda-updates [Google Scholar]
- 8.US Food and Drug Administration. Pediatric Labeling Changes. Accessed March 3, 2022. https://www.fda.gov/science-research/pediatrics/pediatric-labeling-changes [Google Scholar]
- 9.European Commission. Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on Medicinal Products for Pediatric Use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. Accessed February 15, 2023. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32006R1901&from=EN [Google Scholar]
- 10.European Medicines Agency. European Network of Paediatric Research at the European Medicines Agency (Enpr-EMA). Accessed March 3, 2022. https://www.ema.europa.eu/en/partners-networks/networks/european-network-paediatric-research-european-medicines-agency-enpr-ema [DOI] [PubMed] [Google Scholar]
- 11.Tomasi PA, Egger GF, Pallidis C, Saint-Raymond A. Enabling development of paediatric medicines in Europe: 10 years of the EU paediatric regulation. Paediatr Drugs. 2017;19(6):505–513. doi: 10.1007/s40272-017-0261-1 [DOI] [PubMed] [Google Scholar]
- 12.International Council for Harmonisation. Addendum to ICH E11: Clinical Investigation of Medicinal Products in the Pediatric Population. Accessed March 3, 2022. https://database.ich.org/sites/default/files/E11_R1_Addendum.pdf [PubMed] [Google Scholar]
- 13.US Food and Drug Administration. International Collaboration/Pediatric Cluster. Accessed February 20, 2023. https://www.fda.gov/science-research/pediatrics/international-collaboration-pediatric-cluster [Google Scholar]
- 14.European Medicines Agency. ICH E11(R1) Guideline on Clinical Investigation of Medicinal Products in the Pediatric Population Step 5. Accessed March 3, 2022. https://www.ema.europa.eu/en/ich-e11r1-step-5-guideline-clinical-investigation-medicinal-products-pediatric-population [Google Scholar]
- 15.European Medicines Agency. Reflection Paper on the Use of Extrapolation in the Development of Medicines for Paediatrics. Accessed March 3, 2022. https://www.ema.europa.eu/en/documents/scientific-guideline/adopted-reflection-paper-use-extrapolation-development-medicines-paediatrics-revision-1_en.pdf [Google Scholar]
- 16.US Food and Drug Administration. General Clinical Pharmacology Considerations for Pediatric Studies for Drugs and Biological Products. Accessed March 3, 2022. https://www.fda.gov/media/90358/download [Google Scholar]
- 17.European Medicines Agency. ICH E11A Harmonized Guideline on Pediatric Extrapolation Step 2b. Accessed October 10, 2022. https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-guideline-e11a-pediatric-extrapolation-step-2b_en.pdf [Google Scholar]
- 18.United States Renal Data System. USRDS 2019 Annual Data Report: Epidemiology of Kidney Disease in the United States. Accessed March 3, 2022. https://www.usrds.org/media/2371/2019-executive-summary.pdf [Google Scholar]
- 19.Harambat J, Godron A, Ernould S, Rigothier C, Llanas B, Leroy S. Prediction of steroid-sparing agent use in childhood idiopathic nephrotic syndrome. Pediatr Nephrol. 2013;28(4):631–638. doi: 10.1007/s00467-012-2365-8 [DOI] [PubMed] [Google Scholar]
- 20.North American Pediatric Renal Trials and Collaborative Studies. NAPRTCS 2011 Annual Dialysis Report. Accessed March 3, 2022. https://naprtcs.org/system/files/2011_Annual_Dialysis_Report.pdf [Google Scholar]
- 21.Foster BJ, Warady BA. Clinical research in pediatric nephrology: challenges and strategies to address them. J Nephrol. 2009;22(6):685–693. PMID: 19967646 [PubMed] [Google Scholar]
- 22.Center for Pediatrics and Adolescent Medicine. The Cardiovascular Comorbidity in Children with Chronic Kidney Disease Study (4C Study). Accessed October 10, 2022. http://4c-study.eu/ [Google Scholar]
- 23.The Chronic Kidney Disease in Children Cohort Study (CKiD) Accessed October 10, 2022. https://repository.niddk.nih.gov/studies/ckid/ [Google Scholar]
- 24.Siapkara A, Fracasso C, Egger GF, et al. Recommendations by the European Network of Paediatric Research at the European Medicines Agency (Enpr-EMA) Working Group on preparedness of clinical trials about paediatric medicines process. Arch Dis Child. 2021;106(12):1149–1154. doi: 10.1136/archdischild-2020-321433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnaper H, Flynn J, Gross C, et al. Enhancing clinical trial development for pediatric kidney diseases. Pediatr Res. 2017;82(5):727–732. doi: 10.1038/pr.2017.180 [DOI] [PMC free article] [PubMed] [Google Scholar]