

Summary
Genetic association studies have demonstrated the critical involvement of the microglial immune response in Alzheimer’s disease (AD) pathogenesis. Phospholipase C-gamma-2 (PLCG2) is selectively expressed by microglia and functions in many immune receptor signaling pathways. In AD, PLCG2 is induced uniquely in plaque-associated microglia. A genetic variant of PLCG2, PLCG2P522R, is a mild hypermorph that attenuates AD risk. Here, we identified a loss-of-function PLCG2 variant, PLCG2M28L, that confers an increased AD risk. PLCG2P522R attenuated disease in an amyloidogenic murine AD model, whereas PLCG2M28L exacerbated the plaque burden associated with altered phagocytosis and Aβ clearance. The variants bidirectionally modulated disease pathology by inducing distinct transcriptional programs that identified microglial subpopulations associated with protective or detrimental phenotypes. These findings identify PLCG2M28L as a potential AD risk variant and demonstrate that PLCG2 variants can differentially orchestrate microglial responses in AD pathogenesis that can be therapeutically targeted.
Keywords: Microglia, Alzheimer’s disease, Phospholipase C-gamma-2, P522R variant, M28L variant, Amyloid pathology, Microglia-plaque interactions, Microglial uptake capacity, Neuroinflammation, Synaptic function, transcriptional programs
Etoc blurb
Human genetic studies implicate PLCG2 variants in Alzheimer’s Disease risk, but the mechanism is unclear. Tsai et.al. identify a PLCG2 loss-of-function variant that confers increased AD risk and demonstrate that gain- and loss-of-function PLCG2 AD variants uniquely alter microglial transcriptomes and direct plaque-responsive phenotypes, which may inform strategies to induce neuroprotective microglial responses to attenuate AD pathology.
Graphical Abstract
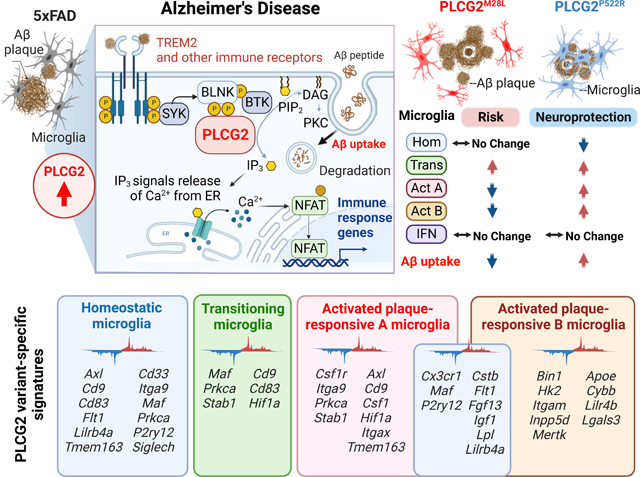
Introduction
A concerted effort to identify genes that confer altered risk for Alzheimer’s disease (AD) is leading to the recognition that many disease-risk genes are associated with the microglia-mediated immune response in the AD brain 1–3. These studies have provoked a renewed interest in immune mechanisms as therapeutic targets in AD, specifically genes encoding proteins involved in immune receptor signal transduction pathways 4. Phospholipase C-gamma-2 (PLCG2), a crucial signaling element employed by various immune receptors and expressed principally by microglia in the brain, is a key regulatory hub gene for immune signaling 5. The PLCG2 gain-of-function variant PLCG2P522R confers reduced AD risk 6. However, the role of PLCG2 in AD pathogenesis remains unclear.
PLCG2 is activated by tyrosine phosphorylation, principally by Bruton’s tyrosine kinase (BTK), following ligand binding to cell surface immune receptors, including triggering receptor expressed on myeloid cells 2 (TREM2). Alternatively, PLCG2 can be activated by association with activated forms of Rac GTPases 7,8. Tyrosine phosphorylation of PLCG2 induces a conformational change that displaces its autoinhibitory domain, activating its enzymatic activity. PLCG2 is recruited to the plasma membrane through pleckstrin homology (PH) domains and incorporated into a receptor-associated signaling complex 9. This translocation of PLCG2 to the plasma membrane allows its interaction with its membrane-associated substrate, 1-phosphatidyl-1D-myo-inositol 4,5-bisphosphate (PIP2). PIP2 hydrolysis yields the cytoplasmic secondary messengers 1D-myo-inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 release elevates intracellular calcium concentrations, promoting calcium-regulated transcription factor activation, including nuclear factor kappa B (NFKB) and nuclear factor of activated T-cells (NFAT) 10. In parallel, DAG promotes the activation of numerous downstream signaling cascades, ultimately regulating the cellular immune response 11. PLCG2 expression is increased in several brain regions in AD patients and the well-studied 5xFAD mouse model of amyloid pathology 12. A PLCG2 co-expression network analysis using microglial single-cell RNA-seq data previously revealed that PLCG2 is associated with inflammatory response-related pathways, consistent with studies documenting its critical involvement in the microglial immune response 12.
Numerous genome-wide association studies (GWAS) revealed that an exonic variant of PLCG2, PLCG2P522R, is associated with reduced AD risk (OR=0.68, p=5.38E-10) 6,13, Lewy body dementia (LBD) and frontotemporal dementia (FTD); this variant is also associated with increased longevity 14. Among patients with mild cognitive impairment (MCI), PLCG2P522R carriers exhibit a slower cognitive decline rate and reduced cerebrospinal fluid (CSF) total tau and phospho-tau concentrations 15. Knock-in PLCG2P522R mice show modestly increased basal phospholipase activity 16 and macrophages with improved survival and viability, increased basal phagocytic activity, and, unexpectedly, elevated cytokine secretion 16. PLCG2P522R mice also exhibit an altered microglial gene expression profile involving genes linked to PLC signaling and pathways related to survival, proliferation, and inflammatory responses 16. Paradoxically, reduced expression of genes associated with the phagocytosis of fungal and bacterial particles and enhanced endocytosis of β-amyloid (Aβ) oligomers and dextran is observed in these mice 17. Andreone et al. knocked out PLCG2 from iPSC-derived microglia, revealing that PLCG2 is required for downstream signaling from TREM2, increasing microglial viability, phagocytosis, and cholesterol metabolism 5. The PLCG2P522R variant promotes cholesterol metabolism more effectively than the wild-type enzyme, consistent with the view that this variant is hypermorphic 5,16. Furthermore, PLCG2 acts broadly to transduce signals from many immune receptors independent of TREM2 5,18,19.
Mutant forms of PLCG2 in diverse immune cell populations are associated with peripheral immune disorders, including leukemias and lymphomas. Specifically, genomic deletions (exon 19 or exons 20 through 22) or somatic mutations (R665W or S707Y) within the regulatory domain of PLCG2 constitutively activate PLCG2 and lead to BTK inhibitor resistance in leukemia patients 20,21. Our genetic data analysis 22 revealed that the missense PLCG2M28L variant (rs61749044) previously found in patients with BTK inhibitor-resistant forms of chronic lymphocytic leukemia is associated with elevated AD risk 23. BTK inhibition in microglia arrests PLCG2 activation and inhibits phagocytosis 24, suggesting a critical role for PLCG2 in Aβ clearance and AD pathogenesis. Thus, understanding how PLCG2 variants affect the immune system and the brain has broad implications for the aging brain, AD, and neurodegenerative disorders.
Results
The PLCG2M28L variant is associated with elevated AD risk and reduced PLCG2 expression
We analyzed data from a large-scale GWAS of >94,000 individuals with late-onset AD 22 and identified a PLCG2 missense mutation (rs61749044) associated with elevated AD risk (OR=1.164; p=0.047) encoding the PLCG2M28L variant. This variant was also identified in a reanalysis of a European-American cohort database (OR=1.25 p=0.054) 25. The odds ratio, p-value, and dataset information of other large-scale GWAS studies are shown in Figures S1A and S1B. In contrast, the PLCG2 missense exonic variant encoding PLCG2P522R (rs72824905) protects against AD (OR=0.76, p=0.007) 22. This finding was validated in a European-American cohort (OR=0.63, p=0.002) 25 and was congruent with previous reports 6,26, verifying that PLCG2P522R is protective in AD (Figure S1A–C).
Structurally, the P522 amino acid residue is positioned between an atypical split PH domain and the c-SH2 domain. The M28 mutation is positioned in the N-terminal PH domain, required for membrane localization (Figures 1A and 1B). We generated mice with the risk-conferring PLCG2M28L variant or the protective PLCG2P522R variant and crossed them with the 5xFAD amyloidogenic murine model of AD expressing five familial AD-associated mutations 27 to evaluate the impact of PLCG2 variants on AD pathogenesis. First, we characterized the effect of the variants on PLCG2 expression in the cortex of 5xFAD mice expressing PLCG2M28L (5xFADM28L) or PLCG2P522R (5xFADP522R). The PLCG2 variants did not alter brain mRNA expression versus those in control mice. However, Plcg2 gene expression was altered between 5xFADM28L and 5xFADP522R mice (Figure 1C). PLCG2 protein expression was lower in the brains of 5xFADM28L mice (0.47-fold) than in those in 5xFAD mice. A significant difference in PLCG2 protein expression was observed between 5xFADM28L and 5xFADP522R mice (Figure 1D). A reduction in PLCG2M28L protein expression was also observed in 5xFADM28L mouse spleens (Figure 1E). The basis of the reduced PLCG2M28L protein expression is presently unknown.
Figure 1. The PLCG2M28L variant is associated with AD risk and downregulates PLCG2 expression.
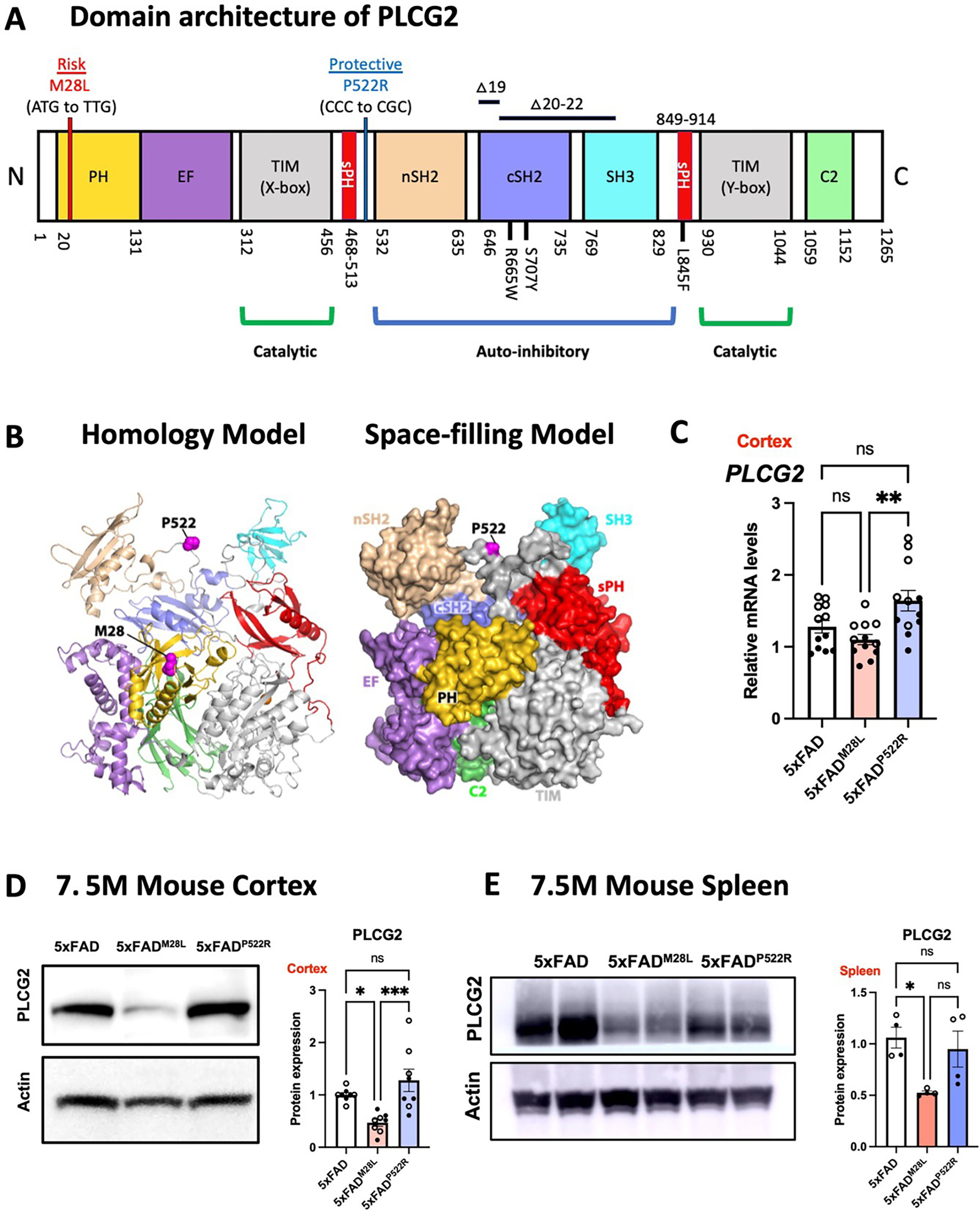
(A) Genetic linkage data of PLCG2M28L and PLCG2P522R with respect to AD risk are shown with the domain architecture of PLCG2 (to scale). Somatic mutations (R665W and S707Y) in PLCG2 are shown in the domain architecture. (B) PLCG2M28L (risk) and PLCG2P522R (protective) variants are mapped onto the structure of PLCG2 (magenta spheres) in both the homology model (left) and the space-filling model (right). (C). Gene expression of Plcg2 were assessed in cortical samples from 7.5-month-old 5xFAD, 5xFADM28L, and 5xFADP522R mice (n=12 per group; 6 male and 6 female mice). (D) Representative immunoblots and quantifications of PLCG2 protein expression in cortical lysates show reduced PLCG2 expression in 5xFADM28L mice (n=8 per group; 4 male and 4 female mice; 4 experiments). (E) Representative immunoblots and quantification of PLCG2 protein expression from the spleen show reduced PLCG2 expression in 5xFADM28L mice (n=4 per group; 2 male and 2 female mice; 2 experiments). All data are presented as the mean ± SEM, analyzed by an ordinary one-way ANOVA and Tukey’s multiple comparisons test. * P <0.05; ** P< 0.01; *** P< 0.001; ns: not significant. Male mice are marked with a solid circle (•), and the female mice are marked with a hollow circle (∘). See also Figure S1.
OR odds ratio, N amino-terminus, C carboxyl-terminus, PH pleckstrin homology domain, EF EF hand motif, TIM TIM barrel, sPH split PH domain, nSH2 n-terminus, Src Homology 2 domain, cSH2 c-terminus Src Homology 2 domain, SH3 SRC Homology 3 domain, C2 C2 domain, WT wild-type,
PLCG2 variants affect plaque pathology and microglial uptake of Aβ aggregates
High-resolution T2*-weighted magnetic resonance imaging (MRI) was conducted using a 9.4T/30 MRI scanner to assess amyloid deposition in the brain (Figure 2A). Diffuse plaques were detected by immunofluorescence using an anti- Aβ antibody, 6E10, and compact plaques were visualized by X34 staining (Figure 2B). We observed exacerbated plaque deposition in the brain of 5xFADM28L mice compared to that in 5xFAD mice; 5xFADM28L mice demonstrated elevated hypointense MRI signals in the cortex (1.31-fold) and increased diffuse 6E10-positive and compact X34-stained amyloid deposits in the subiculum. In contrast, 5xFADP522R mice showed reduced hypointense MRI signals in the cortex (0.77-fold) and 6E10-positive amyloid deposits in the subiculum, demonstrating amyloid pathology attenuation. Notably, significant differences in diffuse, 6E10-positive and compact, X34-positive amyloid deposits were observed between 5xFADM28L and 5xFADP522R mice (Figure 2C). All genotypes had more diffuse 6E10-positive plaques than compact, strongly X34-positive plaques (Figure 2C). Moreover, compared to 5xFADM28L mice, 5xFADP522R mice demonstrated reduced 6E10-positive plaque numbers and size (Figure S2A) and reduced X34-positive plaque size (Figure S2B). The distribution of Aβ plaques was shifted, with a lower overall plaque burden (diffuse and compact) in 5xFADP522R mice than in control and 5xFADM28L mice (Figure 2D). Moreover, in comparison with 5xFAD mice, in 5xFADP522R mice, Aβ plaque formation shifted toward compact (strong X34-positive) plaques, and 5xFADM28L mice demonstrated more diffuse (Figure 2E). Collectively, these results establish a functional link between PLCG2 variants that confer elevated or reduced disease risk and amyloid pathology in a murine AD model.
Figure 2. PLCG2 variants affect plaque pathology and microglial uptake of Aβ aggregates.
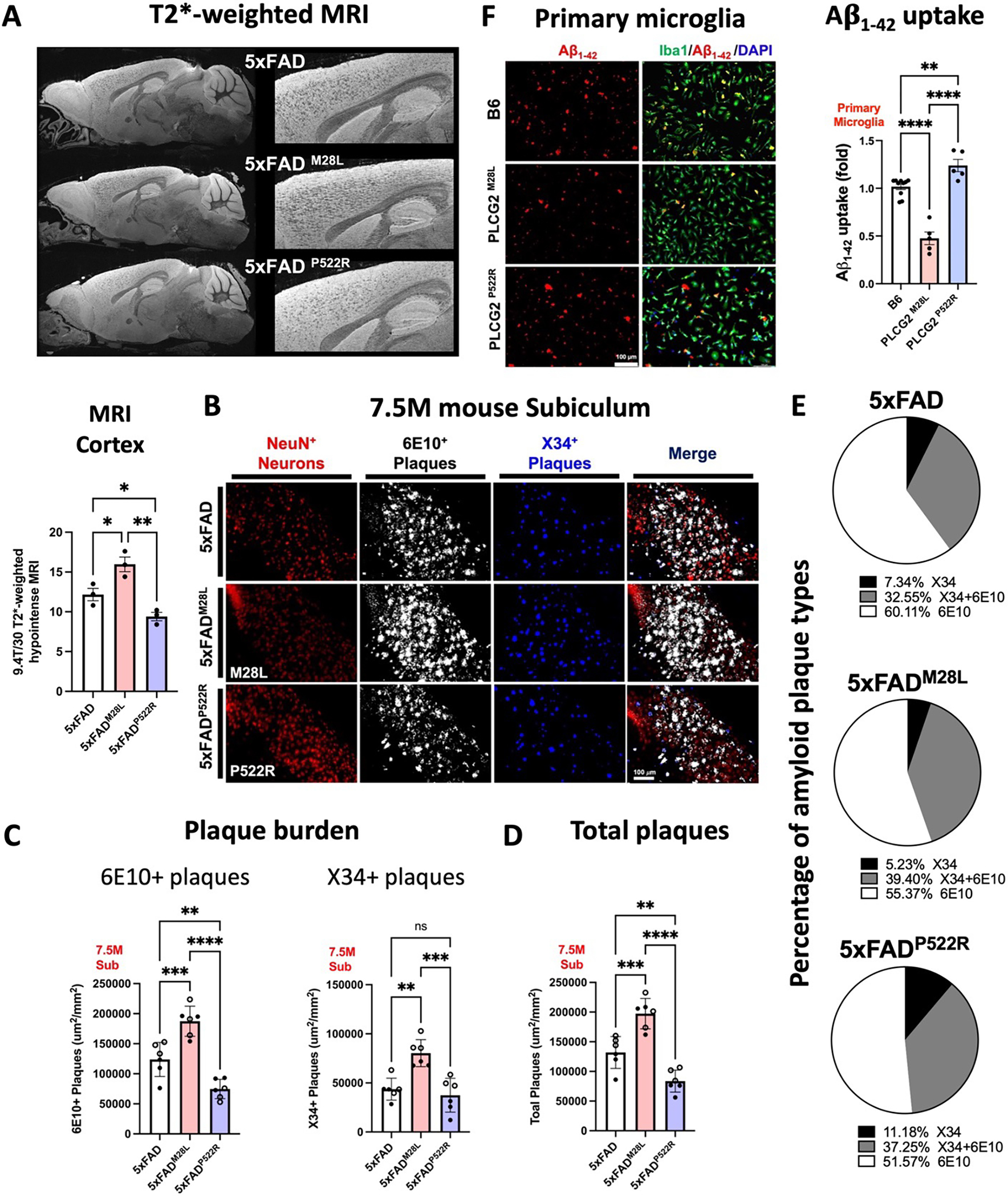
(A) Representative T2*-weighted images and quantitative hypointense signal results in the cortex of 7.5-month-old AD mice. (B) Representative images of amyloid plaques in the subiculum of 7.5-month-old AD mice (6 experiments). (C) Immunofluorescence analysis of diffuse 6E10 (white) and compact X34 (blue) positive plaque density in the subiculum. (D) Scatter plots show the quantification of the total plaque (6E10-positive and X34-positive) area in the subiculum. (E) Graphs denoting the percentage of plaques labeled with X34, 6E10, or their colocalized area. (F) Immunofluorescence analysis of primary murine microglia from B6, PLCG2M28L, and PLCG2P522R mice incubated with fluorescently labeled-Aβ1–42 aggregates (red). Cells were stained with Iba1 (microglia, green) and DAPI (nuclei, blue). Quantification results of Aβ uptake by fluorescence per cell are shown (5 experiments). All data are expressed as the mean values ± SEM and analyzed by an ordinary one-way ANOVA and Tukey’s multiple comparisons test (*P < 0.05, **P < 0.01, and ***P < 0.001; ns: not significant). Male mice: •; female mice: ∘. See also Figure S2.
Microglial phagocytosis is important for plaque remodeling and Aβ clearance 28. Therefore, we examined whether PLCG2 variants affect the intrinsic ability of microglia to take up Aβ. We cultured primary murine microglia from B6, PLCG2M28L, and PLCG2P522R mice and incubated the cells with 1.25 μM HiLyte Fluor 555-labeled Aβ1–42 aggregates for 30 min (Figure 2F). The basal uptake of Aβ1–42 aggregates was reduced in PLCG2M28L microglia (0.46-fold). Conversely, compared with B6 microglia, the hypermorphic P522R variant increased the microglial uptake of Aβ1–42 aggregates (1.22-fold). These results provide supportive evidence that the alteration of AD risk resulting from PLCG2 variants might be due to the microglial function of Aβ1–42 uptake in the context of amyloid pathology. These data demonstrate that plaque pathology is particularly sensitive to the PLCG2 variants, exhibiting large and bidirectional changes correlated with their ability to alter disease risk. The genotype-dependent alteration in plaque burden is consistent with the effect of the PLCG2 variants on the capacity of microglia to internalize aggregated forms of Aβ that affect plaque remodeling and overall burden.
PLCG2 variants differentially alter microglial phenotypes and responses to plaques in the 5xFAD mice.
Microglia–plaques interactions are prominent in AD pathology; thus, we investigated whether PLCG2 variants differentially affected microglial responses to plaques in 5xFAD mice. As expected, 7.5-month-old 5xFAD mice exhibited robust microglial clustering around plaques (Figure 3A), reflecting the migration of microglia to deposited Aβ, microglial process envelopment of the plaque and subsequent microgliosis. No significant changes in the area occupied by ionized calcium-binding adaptor molecule 1 (IBA1)-positive microglia between genotypes was observed; however, IBA1-positive microglial processes around the plaques were altered by the PLCG2 variants. Within X34-positive plaques, the percent area occupied by PLCG2M28L microglia was reduced compared to that in 5xFAD mice (Figure 3B–C). Moreover, we observed a similar percent area of C-type lectin domain containing 7A (CLEC7A)-positive microglia, a distinct amoeboid microglia subset 29, in both PLCG2M28L and PLCG2P522R mice. Compared to that observed in 5xFAD mice, the percent area of CLEC7A-positive microglia in X34-positive plaques was reduced in PLCG2M28L mice but not in 5xFADP522R mice. Thus, CLEC7A and PLCG2M28L microglia are less responsive to plaques than wild-type (WT) microglia or those expressing PLCG2P522R (Figure 3B and 3D). The numbers of ramified microglia expressing the homeostatic marker purinergic receptor P2Y12 were increased in 5xFADM28L versus 5xFAD and 5xFADP522R mice, suggesting an inability or impaired ability of the M28L variant-expressing microglia to transition to a disease-associated microglial phenotype. Although increased P2RY12-positive microglia were observed in 5xFADM28L mice, no genotype-dependent differences in P2RY12-positive microglia engaged with X34-positive plaques were observed (Figure 3B and 3E). Similar to our findings shown in Figure 2F, the percentage of colocalization of IBA1-positive and 6E10-positive stained regions was decreased by 0.62-fold in 5xFADM28L mice and increased by 1.57-fold in 5xFADP522R mice (Figure 3F).
Figure 3. PLCG2 variants differentially alter microglial phenotypes and responses to plaques in the 5xFAD mice.
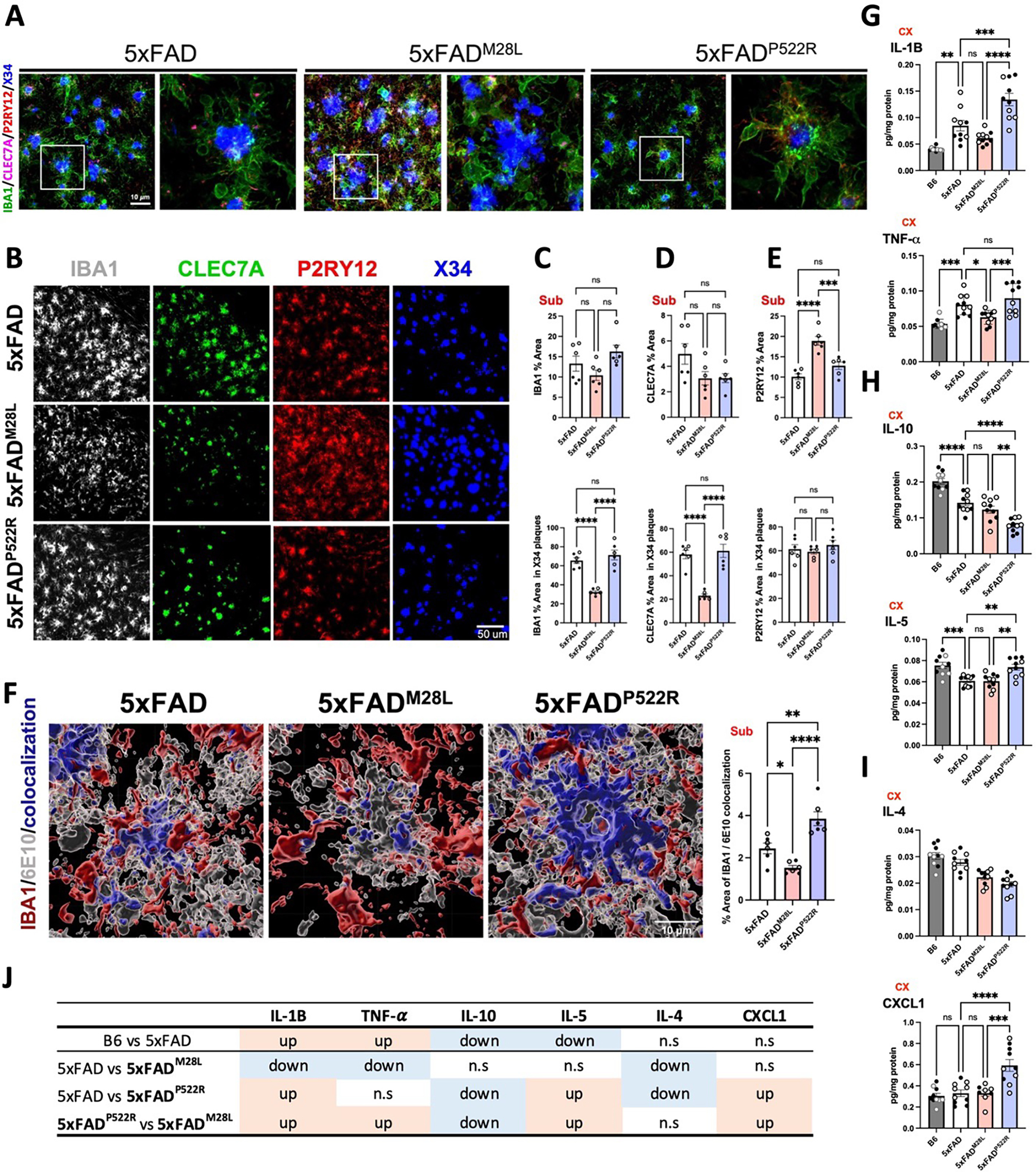
Confocal images (A) and representative images (B) of 7.5-month-old 5xFAD, 5xFADM28L, and 5xFADP522R mouse subiculum stained with IBA1, CLEC7A, and P2RY12 to label microglia and X34 to label amyloid plaques (n=6 per group; 3 male and 3 female mice; 6 experiments). Bar, 10 μm. (C-E) Scatter plots show quantification of IBA1 (C), CLEC7A (D), and P2RY12 (E) staining in the subicula of 7.5- month-old mice (n=6,). The upper graphs show the total percentage of the area stained. The bottom graphs show the quantification of the percentage volume within individual plaque areas. (F) Representative images and quantitative results of 6E10-positive Aβ internalization by IBA1-positive microglia in subicula using Imaris software (n=6 per group; 3 male and 3 female mice; 6 experiments). (G-I) Protein concentrations of cytokines were measured from the cortex of 7.5-month-old mouse brains (n=10). (J) Table summarizing the results from G to I. All data were normalized by total protein. All data are expressed as the mean values ± SEM and analyzed by an ordinary one-way ANOVA and Tukey’s multiple comparisons test (*P < 0.05, **P < 0.01, and ***P < 0.001; ns: not significant). Male mice: •; female mice: ∘.
IBA1 Ionized calcium binding adaptor molecule 1, CLEC7A C-type lectin domain family 7 member A, P2RY12 Purinergic Receptor P2Y12, TNF-a Tumor necrosis factor-alpha, IL interleukin, CXCL1 C-X-C motif chemokine ligand 1
We next investigated whether the PLCG2 variants resulted in different cytokine concentrations in the 5xFAD brain using a Meso Scale Discovery (MSD) cytokine panel. The concentrations of many cytokines were altered by PLCG2 variants (Figure 3G–J). In the comparison between B6 and 5xFAD mice, the concentrations of two proinflammatory cytokines, interleukin-1 β (IL-1B), tumor necrosis factor-alpha (TNF-α), were increased Figure 3G), and those of two other cytokines, IL-10, IL-5, were decreased (Figure 3H), consistent with previous findings 30. Moreover, the concentrations of IL-1β, TNF-α, IL-5, and the chemokine (C-X-C motif) ligand 1 (CXCL1) were increased, and IL-10 concentrations were reduced in 5xFADP522R versus 5xFADM28L mice (Figure 3I). These findings suggest that the perturbation of PLCG2-dependent signaling can positively and negatively alter cytokine expression, potentially contributing to the altered amyloid pathology.
The hypermorphic P522R variant ameliorates impaired synaptic function in 5xFAD mice
Cognitive decline correlates with synaptic dysfunction. Thus, we assessed the effect of PLCG2 genotypes on learning and memory in 5xFAD mice, which demonstrate a robust deficit in working memory starting at 4 to 5 months of age 27. At 6 months of age, compared to B6 control mice, 5xFAD and 5xFADM28L mice exhibited impaired performance on a Y-maze task; however, no significant genotype-related differences in working memory impairment were observed between these two strains. Notably, 5xFADP522R mice were behaviorally similar to B6 control mice in Y-maze task performance, reflecting normal cognition (Figure 4A). Long-term potentiation (LTP) involves the persistent strengthening of synapses and signal transmission between neurons and is the most widely proposed mechanism subserving memory in the hippocampus. Aβ exposure is associated with impaired synaptic function and blocks LTP 31. We assessed synaptic plasticity in our mouse models, which showed different working memory impairment severity resulting from the PLCG2 variants. Field recordings of LTP of excitatory postsynaptic potentials (fEPSPs) in hippocampal area CA1 revealed diminished LTP in the 5xFAD and 5xFADM28L mice relative to the B6 controls. The LTP was equivalent in the 5xFADP522R and B6 mice; however, 5xFADP522R demonstrated greater LTP than 5xFAD and 5xFADM28L mice, supporting a protective role for PLCG2P522R in preserving synaptic plasticity in 5xFAD mice (Figure 4B and 4C). Next, we measured fEPSP output responses to electrical stimulation. Compared to B6 control mice, output responses were reduced in 5xFAD and 5xFADM28L mice. 5xFADP522R mice did not differ from B6 mice and had greater responses than 5xFADM28L mice (Figure 4D and 4E). No genotype-related differences in paired-pulse ratio responses were observed, suggesting that presynaptic plasticity was similar among the mouse strains (Figure S3A). Furthermore, we performed whole-cell patch-clamp electrophysiological recordings of hippocampal area CA1 pyramidal neurons to ascertain synaptic differences that could explain the reduced LTP and output responses in 5xFAD and 5xFADM28L mice. Spontaneous excitatory postsynaptic current (sEPSC) analyses revealed significant effects on sEPSC frequency and sEPSC amplitudes in 5xFADP522R versus 5xFAD or 5xFADM28L mice. There were no differences in these measures between 5xFADP522R and B6 control mice (Figure S3B). We further evaluated differences in excitatory transmission by measuring EPSCs mediated by AMPA and NMDA glutamate receptors. There were smaller AMPA/NMDA receptor current ratios in the 5xFAD and 5xFADM28L mice relative to the B6 mice, but no difference between the 5xFADP522R mice and the B6 mice (Figure S3C). In measures of inhibitory transmission, the spontaneous inhibitory postsynaptic current (sIPSC) frequency and amplitudes were lower in 5xFAD and 5xFADM28L mice relative to B6 mice. Similarly, 5xFADP522R mice showed a similar pattern to the B6 mice (Figure S3D). The reduced LTP and input-output response in 5xFAD and 5xFADM28L mice likely results from predominating reduced postsynaptic glutamate receptor responses rather than the expected increased GABA transmission. These findings document a functional role of PLCG2 in governing synaptic functionality, as evidenced by the effect of its hypermorphic variant PLCG2P522R. However, we did not detect differences between 5xFAD mice and 5xFADM28L mice.
Figure 4. The hypermorphic P522R variant ameliorates impaired synaptic function in 5xFAD mice.
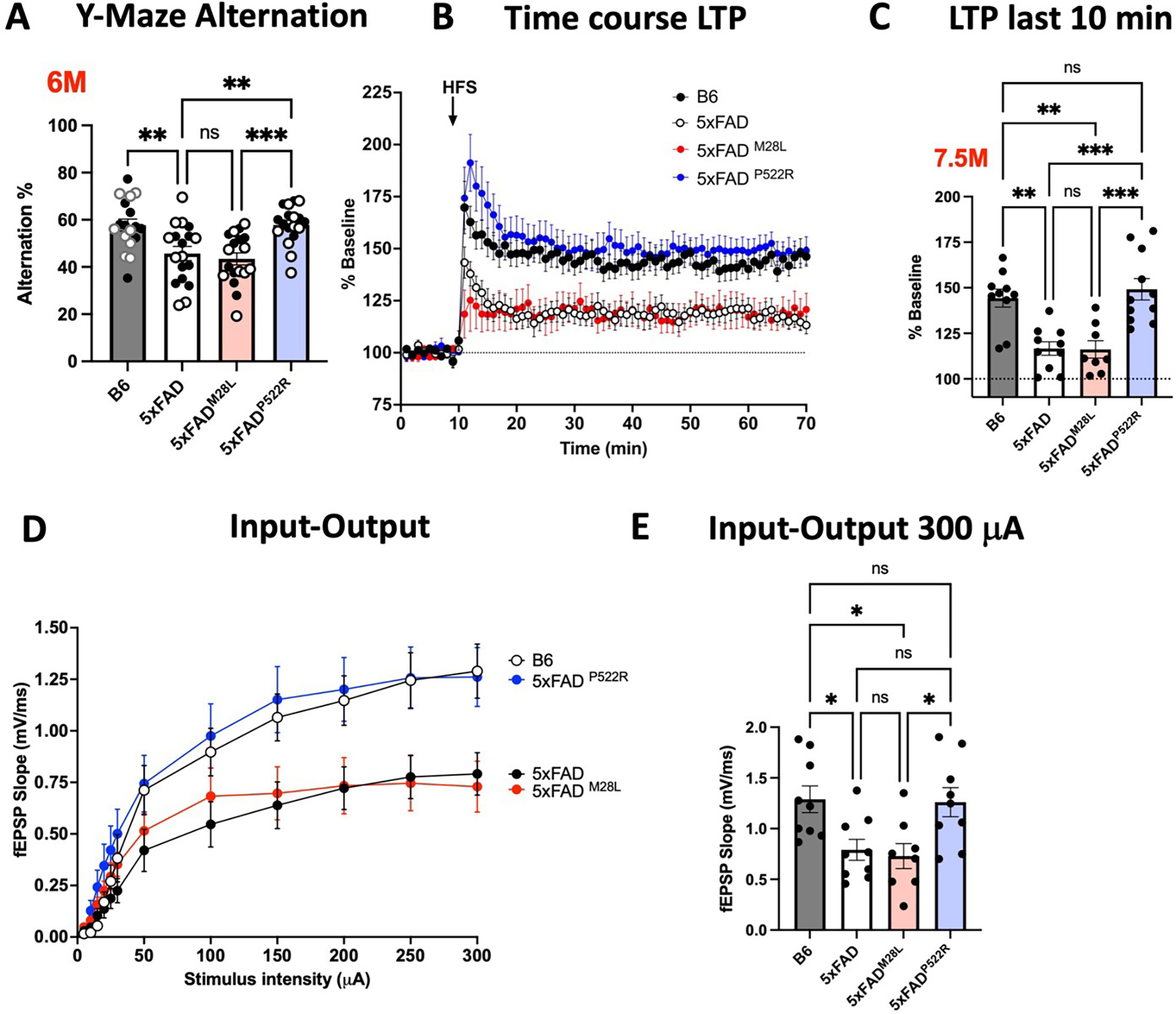
(A) Working memory of 6-month-old B6, 5xFAD, 5xFADM28L, and 5xFADP522R mice assessed by percent spontaneous alternation in the Y-maze task (n=18 mice per group; 9 male and 9 female mice). (B) The PLCG2P522R variant ameliorated impaired LTP in 7.5-month-old male 5xFAD mice. (C) Data show an average of normalized fEPSP slope for the final 10 min of recording (60 to 70 min) relative to 10 min baseline average. (D) Input/output curves were obtained by plotting the slope of fEPSPs in the CA1 area of the hippocampus. (E) Input/output curves showed diminished basal synaptic transmission in 5xFAD and 5xFADM28L mice. Statistical analyses were performed by one-way ANOVA followed by Dunnett’s multiple comparison test. The results of individual values from the slices are shown in the scatter plot. Each genotype data set shows at least 4 male mice. All data are expressed as the mean values ± SEM (*P < 0.05, **P < 0.01, and ***P < 0.001; ns: not significant). Male mice: •; female mice: ∘. See also Figure S3.
LTP Long-term potentiation, fEPSPs Extracellular recordings of field excitatory postsynaptic potential, ANOVA Analysis of variance
PLCG2 variants elicit distinct transcriptional programs in the 5xFAD mouse brain
PLCG2 is a critical signaling intermediate subserving the actions of several immune receptors that selectively alter the microglial transcriptome 32. We performed gene expression analysis using bulk RNA-seq data from cortex of 7.5-month-old AD mice to evaluate the impact of PLCG2 variants on gene expression in the brain. In the comparison between 5xFADM28L and 5xFAD mice, we identified 47 significant differentially expressed genes (DEGs); 20 upregulated (15 genes with fold changes greater than 1.5) and 27 downregulated (25 genes with fold changes greater than 1.5) (Figure 5A). The pathway analysis identified several Gene Ontology (GO) terms, including “antigen processing and presentation, tumor necrosis factor production, interleukin-1 beta production, inflammatory response, endocytosis and phagocytosis” (Figure 5B), suggesting an important role for PLCG2M28L in inflammation-and endocytosis/phagocytosis-related pathways in AD. In the comparison between 5xFADP522R and 5xFAD mice, we identified 763 significant DEGs; 568 upregulated (384 genes with fold changes greater than 1.5) and 195 downregulated (61 genes with fold changes greater than 1.5) (Figure 5C). The pathway analysis identified several GO terms, including “multicellular organism metabolic process, glial cell development, regulation of phagocytosis/endocytosis, inflammatory response, and lipid metabolic process” (Figure 5D), supporting our results indicating that PLCG2 variants play an important role in phagocytosis/endocytosis and inflammatory responses in AD.
Figure 5. PLCG2 variants elicit distinct transcriptional programs in 5xFAD.
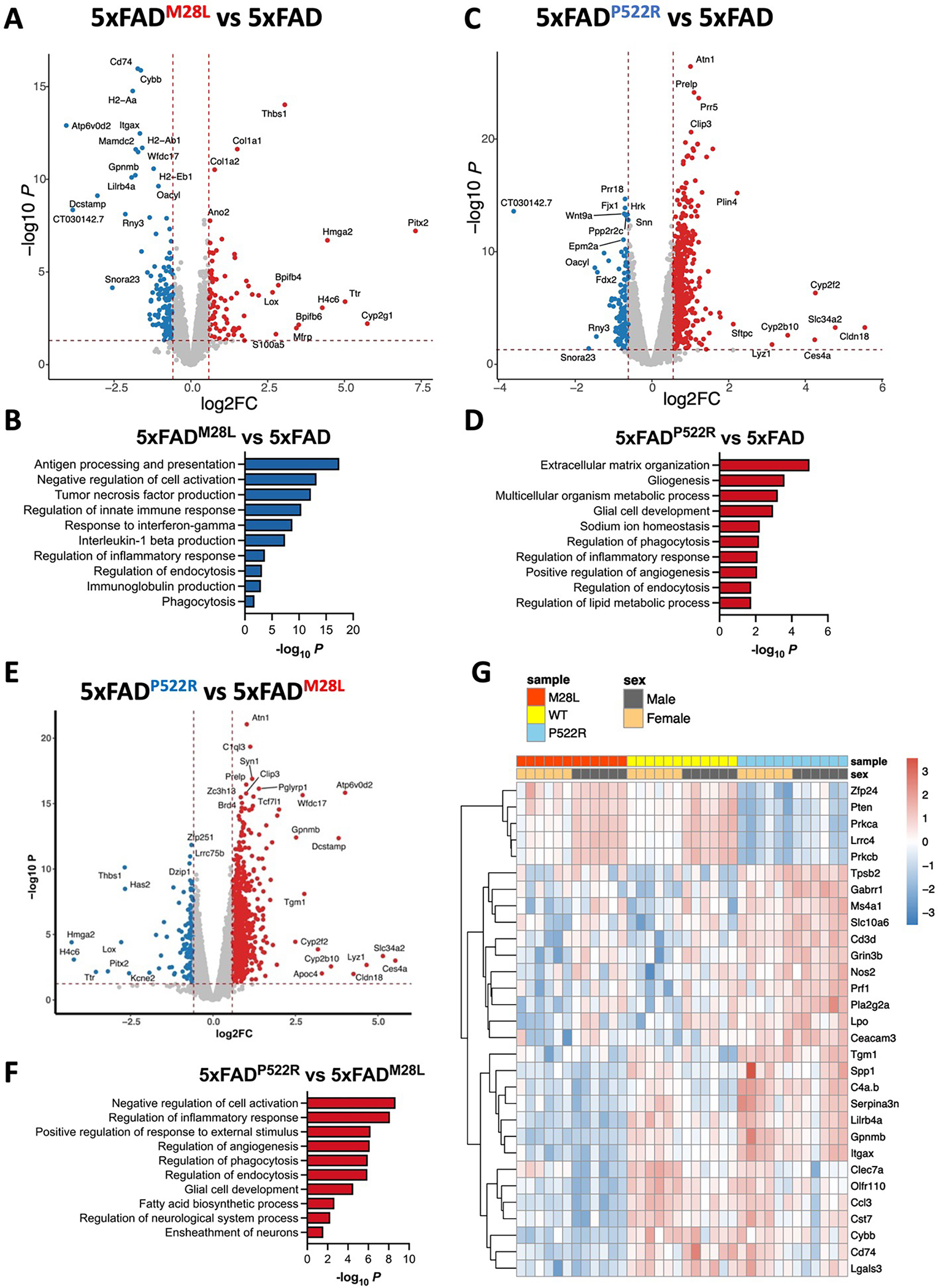
Bulk RNA sequencing was performed on the cortex of 7.5-month-old mice of the indicated genotype. (A) The volcano plot shows significant DEGs (FDR<0.05, FC>1.5) in the cortex from 5xFADM28L mice (n=8, 4 male and 4 female mice) versus 5xFAD mice (n=8, 4 male and 4 female mice). (B) Top 10 Gene Ontology biological processes identified through analysis of the DEGs between 5xFADM28L and 5xFAD mice. (C) The volcano plot shows significant DEGs (FDR<0.05, FC>1.5) in the cortices from 5xFADP522R mice (n=8, 4 male and 4 female mice) versus 5xFAD mice. (D) Top 10 Gene Ontology biological processes identified through analysis of the DEGs between 5xFADP522R and 5xFAD mice. (E) The volcano plot shows significant DEGs in the cortices from 5xFADM28L mice versus 5xFADP522R mice. (F) Top 10 Gene Ontology biological processes identified through analysis of the DEGs between 5xFADM28L and 5xFADP522R mice. (G) Nanostring nCounter Gial Profiling and Neuropathology panels were employed to analyze cortical RNA. The gene expression heatmap shows selected DEGs derived from the NanoString analysis of cortices of 7.5-month-old mice (each genotype n=12, 6 male and 6 female; 1 experiment).
DEGs differentially expressed genes, FDR false discovery rate, FC fold change
We performed differential gene expression analyses using bulk RNA-seq data from 5xFADM28L and 5xFADP522R mice to further characterize the differences between the transcriptional programs of the risk and protective variants. We identified 593 DEGs, 439 of which were upregulated (356 genes with fold changes greater than 1.5) and 154 downregulated (61 genes with fold changes greater than 1.5) (Figure 5E). The pathway analysis identified several GO terms, including “regulation of inflammatory response, phagocytosis/endocytosis, glial cell development, fatty acid biosynthesis process and neurological system process” (Figure 5F). These findings indicate that distinct transcriptional programs in 5xFAD mouse brains are elicited by PLCG2 variants, altering amyloid pathogenesis.
Our results demonstrated that PLCG2 variants modulate disease pathology by inducing specific microglial phenotypes and molecular signatures. RNA from the cortices of 7.5-month-old AD mice was analyzed to validate these findings using the nCounter Glial Profiling Panel and Neuropathology Panel from array-based amplification-free NanoString technologies comprising 1,266 genes involved in glial biology, neuro-glial interactions, and neurodegeneration, to increase the overall rigor of the analysis. Thirty-four DEGs (all downregulated, 11 with fold changes greater than 1.5) and several genes associated with disease-associated microglia (DAM), including Itgax, Gpnmb, Ccl3, Cd74, Cybb, Lgals3, Spp1, and Lpl (highlighted in Figure 5G), were found in 5xFADM28L but not 5xFAD mice, supporting our findings that PLCG2M28L impaired the ability of microglia to transition into a disease-associated phenotype. Furthermore, 579 DEGs, 350 upregulated (108 genes with fold changes greater than 1.5) and 229 downregulated (71 genes with fold changes greater than 1.5) were found in 5xFADP522R mice versus 5xFAD mice. Most of these DEGs were related to neuronal connectivity, transmitter response, and structure of axon and dendrite, including Gabrr1, Prl, Grin3b, Prkca, Pten, Lrrc4, and Cacnb4, supporting our findings that PLCG2P522R protects synaptic function in 5xFAD mice.
Single-nuclei RNA-seq distinguishes the cell type–specific effects of PLCG2 variants in the AD brain
We performed snRNA-seq on 7.5-month-old 5xFAD, 5xFADM28L, and 5xFADP522R mice (4 mice per genotype) to investigate the impact of PLCG2 variants and amyloid pathology across cell types. NeuN-negative nuclei were sorted from the mouse cortex to characterize the enrichment of glial cell types and states involved in AD, and 62,588 individual nuclei were obtained. Visualization in uniform manifold approximation and projection (UMAP) space separated nuclei into distinct clusters across all samples, which we mapped to 6 major cell types (Figure 6A). These clusters were manually identified based on the expression of known cell-type-specific markers, including oligodendrocytes (Oligo; Cluster 0), oligodendrocyte progenitor cells (OPCs; Cluster 1), microglia (Cluster 2), astrocytes (Ast; Cluster 3), neurons (Neuron; Cluster 4), and endothelial cells (Endo; Cluster 5) (Figure 6B). The cell type distribution differed across genotypes (Figure 6C), and the PLCG2 variant-associated microglial signatures were analyzed (Figure 7). Only microglial signatures in the brain were differentially altered by the PLCG2 variants, suggesting that PLCG2 variants modulate disease pathologies through microglia-specific transcriptional programs.
Figure 6. Single nuclei RNA-seq distinguishes the cell type–specific effects of PLCG2 variants -in the AD brain.
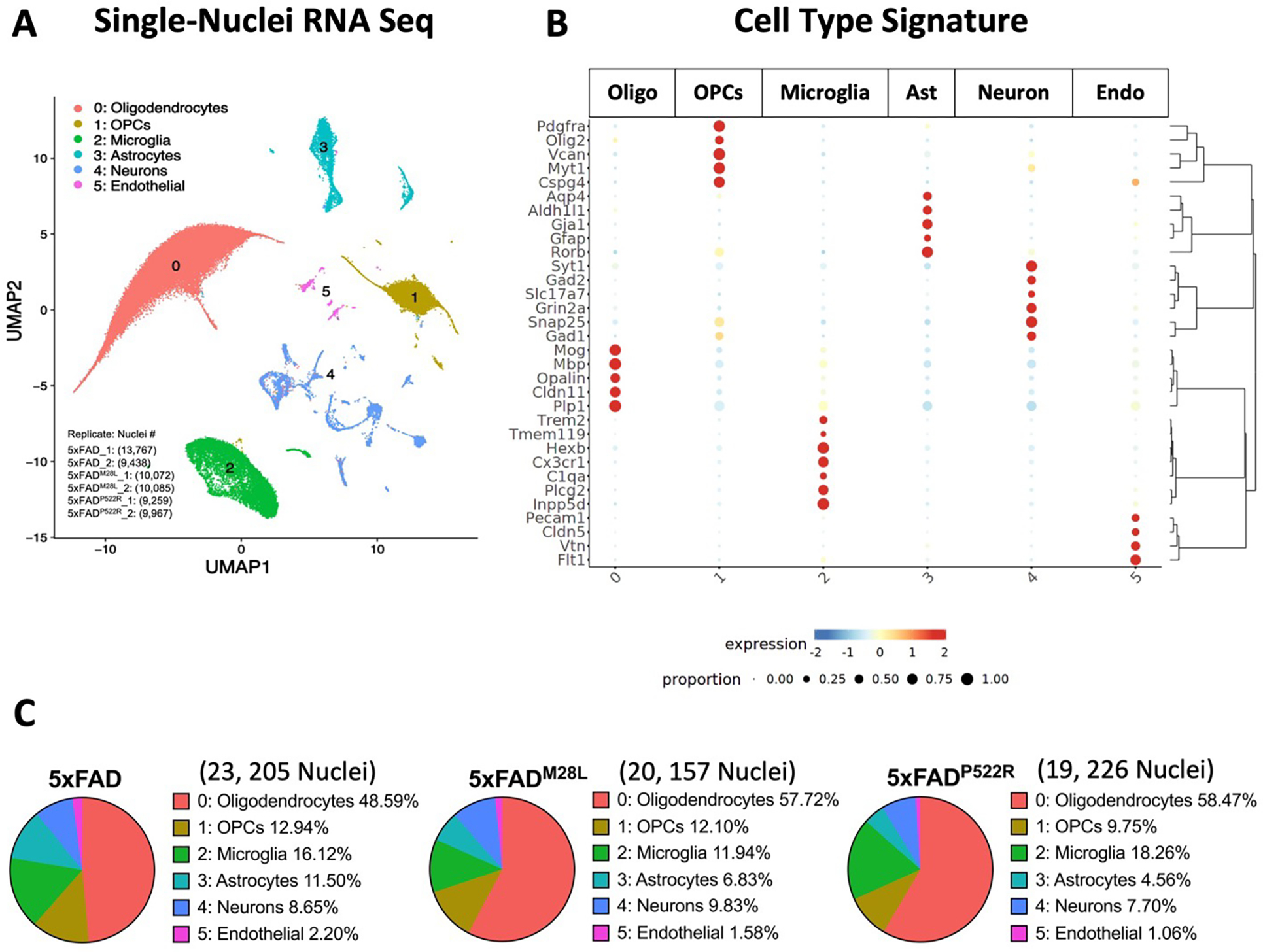
(A) Uniform Manifold Approximation and Projection (UMAP) of 62,588 nuclei captured from 12 cortical samples across three genotypes of AD mice, annotated and colored by cell type (two replicates from each genotype and each replicate includes 1 male and 1 female mouse cortical sample). (B) Heatmap showing the expression of specific markers in each sample, identifying each cluster in A. (C) Pie chart showing the percentage of clusters in each genotype. Oligo Oligodendrocytes, OPCs Oligodendrocyte progenitor cells, Ast Astrocytes, Endo Endothelial cells
Figure 7. Single nuclei RNA-seq identifies PLCG2 variant-specific microglial signatures in AD.
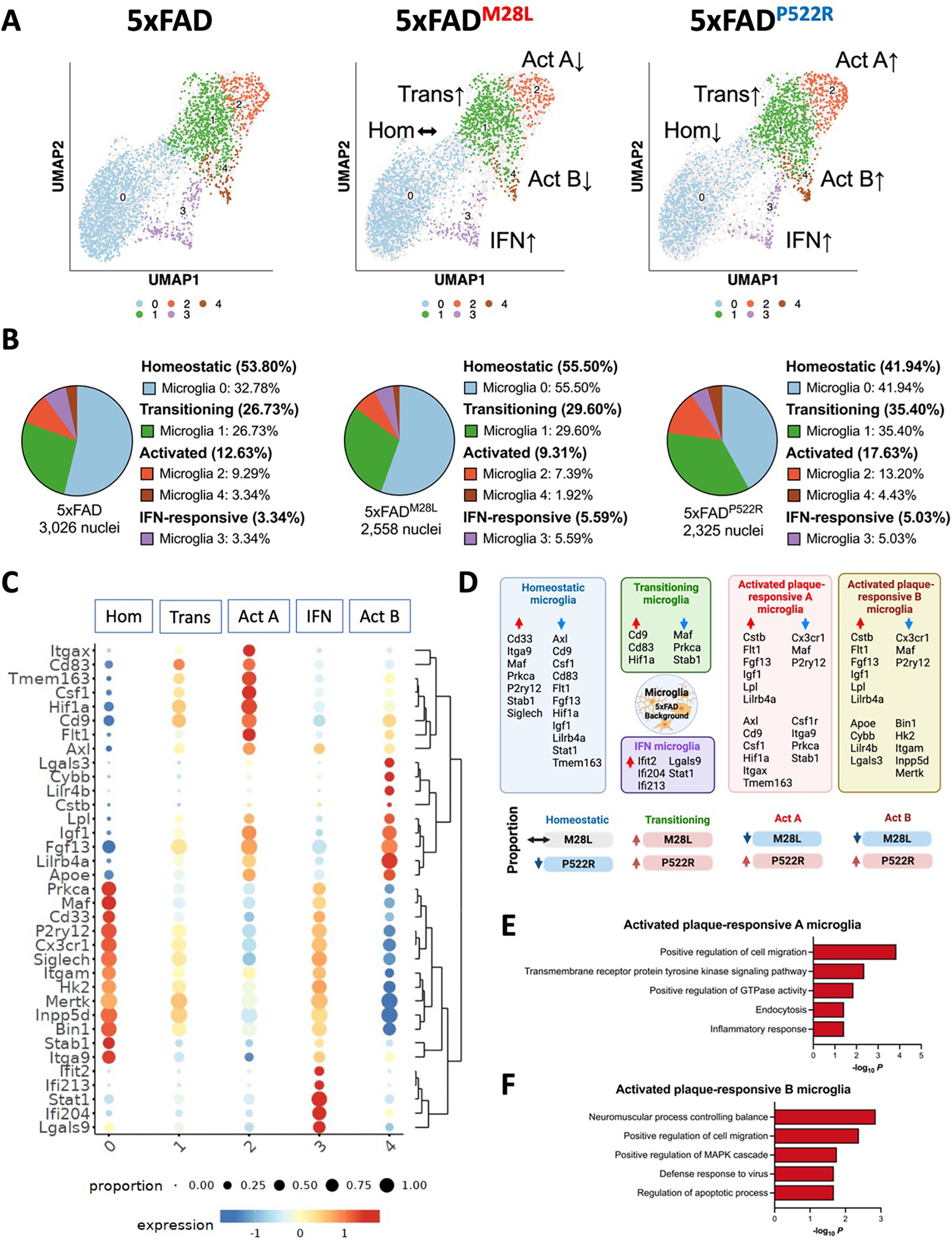
(A) UMAP plot of 7,909 nuclei showing the re-clustered microglia (from cluster 2 in Figure 6A) annotated and colored by microglial subcluster. (B) Pie chart showing the percentage of microglial subclusters. (C) Heatmap showing the expression of canonical microglial genes in each microglial subclusters, including homeostatic (Hom), transitioning (Trans), activated plaque-responsive (Act A and Act B), and IFN-responsive (IFN) for each genotype. (D) Schematic illustration showing microglial signature altered by PLCG2 variants switching between homeostatic, transitioning, activated plaque-responsive, and IFN-responsive microglial subclusters. Key genes involved in each microglial population are shown. The arrows indicate upregulated (red) or downregulated (blue) genes or proportions. (E) Top 5 Gene Ontology biological processes identified through analysis of the upregulated DEGs in activated plaque-responsive microglia A subcluster. (F) Top 5 Gene Ontology biological processes identified through analysis of the upregulated DEGs in activated plaque-responsive microglia B subcluster. See also Figure S4, S5, and S6.
Hom homeostatic, Trans transitioning, Act activated plaque-responsive, IFN interferon-responsive
Single-nuclei RNA-seq identifies PLCG2 variant-associated microglial signatures in AD
Robust induction of microglial PLCG2 expression was observed in the brains of 5xFAD mice and AD patients 12. We next focused on the 7,909 nuclei mapped in the microglial cluster and examined DEGs in distinct microglial subpopulations. The analysis revealed 5 discrete microglial clusters, each representing a different microglial state, including previously characterized homeostatic, transitioning, activated plaque-responsive (Act), and interferon (IFN)-responsive microglial subsets 1,3,33 (Figure 7A and 7B). Our results demonstrated a subpopulation of microglia, cluster 0, expressing homeostatic microglial genes, such as P2ry12 and Cx3cr1 (Figure 7C). In contrast, cluster 2 microglia (termed Act A microglia) expressed more genes upregulated in endocytosis and inflammatory response microglia, including Itgax, Cd9, and Axl (Figure 7C and 7E; Figure S4A and S4B). Cluster 1 microglia were transitioning microglia exhibiting slightly increased expression of genes enriched in cluster 2 and slightly decreased expression of genes enriched in cluster 1. Cluster 4 microglia (termed Act B microglia) were enriched in genes related to apoptotic processes, lipid metabolism and plaque compaction, such as Lpl, Apoe, Lgals3 and Lilr4b (Figure 7C and 7F). The transcriptional differences between Act A and Act B microglia are shown in Figure S5. In addition, IFN-responsive microglia (cluster 3) demonstrated enrichment of genes such as Ifit2, Ifi204, and Ifi213 and were mapped to cluster 3 (Figure 7C). Compared to 5xFAD mice, 5xFADM28L mice show reduced nuclei percentages in clusters 2 and 4; 5xFADP522R mice show increased nuclei percentages in clusters 1, 2, and 4 and reduced nuclei percentages in clusters 0 (Figure 7B). 5xFAD mice with the protective PLCG2P522R variant exhibited an increased number of microglia associated with cell migration, endocytosis, inflammatory responses, and apoptotic process pathways; these microglia were reduced in 5xFADM28L mice. Our single-nuclei analysis revealed that risk and protective PLCG2 variants elicit differential transcriptome programming (Figure 7D and Figure S6) linked to their association with plaques and Aβ clearance leading to opposite effects on disease pathogenesis in 5xFAD mice.
Discussion
Genetic studies have linked the PLCG2P522R variant with reduced AD risk; however, the functionality of PLCG2 in AD pathophysiology remains poorly defined. Notably, we identified the loss-of-function PLCG2M28L variant associated with increased AD risk, allowing us to investigate the mechanisms of PLCG2 variants and identify molecular signatures and pathways that discriminate the divergent effects of genetic variants of PLCG2 in AD mouse models. Characterizing specific microglial phenotypes imparted by the PLCG2 AD risk variants allowed the discrimination of microglial mechanisms subserving altered disease risk. Moreover, the phenotypic effects of the variants validate PLCG2 as a critical hub gene that regulates numerous effectors.
Missense mutations exhibit modest effects on PLCG2 enzymatic activity. PLCG2P522R shows slightly elevated activity 11,16,34, whereas M28L variant and WT enzyme activity demonstrate no differences 23. How the variants affect PLCG2 functionality needs to be revisited in a more rigorous analysis of its interactions with other signaling elements.
We found that the expression of the different PLCG2 variants in the brain differed, likely contributing to the functional differences in the phenotypes. PLCG2P522R mRNA and protein expression in the 5xFAD brain did not differ from that in the WT brain (Figures 1C and 1D). However, 5xFADM28L mice exhibited lower protein expression without a measurable change in mRNA (Figure 1D). We verified the reduced protein expression in the spleen (Figure 1E). Reduced PLCG2M28L protein expression confers a loss-of-function effect, and the observed phenotypes are consistent with those observed in PLCG2-deficient microglia 5,11. The basis of the reduced PLCG2M28L protein expression is unknown.
PLCG2 is a critical participant in the immune response induced by amyloid plaques in AD patients and animal models 35. PLCG2 is robustly induced in a subset of plaque-associated microglia, and PLCG2 variants differ in their response to amyloid plaques (Figure 3) regarding the density of plaque-associated microglia and their ability to remodel plaque size and compaction (Figure 2). The PLCG2M28L variant is associated with an overall greater plaque burden, whereas PLCG2P522R mice have a reduced plaque burden (Figure 2D). PLCG2M28L mice demonstrate a substantial reduction in the number of plaque-associated microglia, suggesting that PLCG2M28L microglia may be unable to shift their phenotypes to a more plaque-responsive microglial state, similar to TREM2 loss-of-function microglia, as they cannot efficiently mobilize a robust response to the deposited amyloid 36.
Importantly, the PLCG2P522R variant demonstrates robust effects in sustaining synaptic function and working memory, which are impaired in 5xFAD and 5xFADM28L mice. Specifically, LTP and glutamatergic and GABAergic transmission are impaired in 5xFAD and 5xFADM28L mice (Figure 4 and Figure S3). We hypothesized that the loss of LTP is more likely a product of reduced glutamate transmission than enhanced GABA transmission. This hypothesis is consistent with previous studies showing that Aβ deposition contributes to disturbed glutamatergic neurotransmission; however, further studies are required for verification 37,38. Our transcriptional analysis revealed many GO terms associated with synaptic function and structure that support our physiological measures and provide clues regarding the mechanistic underpinnings of the synaptic function associated with the different PLCG2 variants. These findings also require more directed analyses to determine causative mechanisms. Synaptic function could be protected by the stimulation of a protective microglial response in the PLCG2P522R variant or through PLCG2P522R variant-enhanced phagocytic microglial actions associated with plaque remodeling and synapse pruning. Overall, the related role of the PLCG2P522R variant in mediating excitatory and inhibitory synaptic function might be important for determining the mechanisms by which microglia modulate disease progression. However, the molecular signatures of neuronal populations among genotypes in AD mice and the exact mechanism by which PLCG2 variants regulate synaptic function in the absence of amyloid plaques require further assessment.
PLCG2 regulates the expression of genes governing pathways related to inflammation, lipid metabolism, microglial viability and phagocytosis 12,16. However, the effects of its inactivation or genetic variants on transcriptional programming in AD have yet to be extensively explored in AD animal models. It is of particular interest to determine the unique microglial subpopulations differentially affected by PLCG2 genotypes and linked to altered disease risk. Our single-nuclei RNA-seq analysis indicated that PLCG2 variants regulate microglial cluster composition in the AD brain, consistent with previous findings in AD chimeric mice with xenografted human iPSC-derived microglia expressing PLCG2P522R 39, in which Spp1-, Fabp5-, and Cd74-enriched microglia were elevated. Moreover, 5xFADP522R mice exhibit a plaque-responsive microglial subcluster enriched in Hcar2, a microglial receptor we recently identified as required for efficient and neuroprotective microglial responses to amyloid pathology 40.
Here, we identified specific microglial subclusters enriched in phagocytic and plaque-responsive genes conferring both reduced and elevated AD risk, enabling us to identify distinct stages in the plaque-responsive microglial phenotypes associated with the PLCG2 variants. Initially, plaque deposition and microglia recruitment activate a set of genes, including Cd9, Cd83, and Hif1a, within a subset of microglia; concomitantly, a set of homeostatic genes is downregulated in non-plaque-associated microglia, such as Maf, Prkca, and Stab1, shifting the microglial phenotype from cluster 0 (homeostatic) to cluster 1 (transitioning). The second phase of plaque-responsive microglia activation includes the induction of lipid metabolism or phagocytic disease-associated pathways (e.g., Cstb, Flt1, Fgf13, Igf1, Lpl, and Lilrb4a), shifting the microglial phenotype from cluster 1 (transitioning) to cluster 2 (activated plaque-responsive A; Act A) or cluster 4 (activated plaque-responsive B; Act B). Compared to 5xFAD mice, 5xFADM28L mice with increased amyloid burdens have increased percentages of transitioning microglia; however, the phenotypes were not shifted to a more activated stage than that of activated plaque-responsive microglia (Act microglia). In contrast, 5xFADP522R mice exhibit reduced percentages of microglia-enriched homeostatic markers and increased percentages of transitioning and Act microglia. Notably, we identified two types of Act microglia (Act A and Act B; Figures 6C and 6D). Nuclei mapped to Act A microglia were enriched endocytosis and inflammatory response genes and highly expressed Axl, Cd9, Csf1, Hif1a, Itgax, and Tmem163. Moreover, nuclei mapped into Act B microglia were enriched migration and apoptotic process genes and highly expressed Apoe, Cybb, Lilr4b, and Lgals3. Together, our results highlight the role of PLCG2 variants in microglial processes associated with AD risk and amyloid pathologies.
In summary, we validated the human genetic findings that loss-of-function and gain-of-function variants of PLCG2 have opposite effects on transcriptional mechanisms, conferring altered microglial phenotypes and differential pathogenesis in AD mice. The data are consistent with the conclusion that PLCG2M28L is a loss-of-function variant due to reduced expression, impairing the microglial response to plaques, suppressing cytokine release, downregulating plaque-associated and disease-associated microglial genes, and increasing plaque deposition. Conversely, PLCG2P522R appears to be a mild hypermorph causing a broad range of positive effects in 5xFAD mice, including a reduction in plaque burden, amelioration of impaired synaptic function, and rescue of working memory deficits, indicating that promoting a neuroprotective microglial response to amyloid pathology could limit AD progression. Overall, PLCG2-directed therapeutics may provide strategies to induce neuroprotective microglial responses and attenuate AD pathogenesis.
Limitations of the study
It is encouraging that the PLCG2P522R variant ameliorated impaired LTP in 5xFAD mice. However, our models cannot provide insight into whether PLCG2 variants directly affect neuronal function. Human genetic studies have postulated that the PLCG2M28L variant confers an increased risk for AD. Although the analysis of data from the recent AD GWAS studies showed unimpressive p-values, our findings in 5xFAD mice suggest that the association between this putative risk variant of PLCG2 and diagnosis may not be strong in all datasets, but may be robustly associated with amyloid pathology. Unlike the PLCG2P522R variant, this risk variant has not been functionally validated in human samples. PLCG2 protein expression is reduced in 5xFADM28L mice, but comparable data from AD patients carrying the PLCG2M28L variant will be difficult to obtain owing to its low minor allele frequency. Future studies are needed to address these limitations and provide strategies for PLCG2-targeted therapeutics that may attenuate AD pathogenesis.
STAR METHODS
RESOURCE AVALIBILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by lead contact Gary E. Landreth (glandret@iu.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Bulk RNA-Seq, snRNA-Seq and gene expression data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-PLCG2 | Cell Signaling Technology | Cat#3872S; RRID:AB_2299586 |
| Mouse monoclonal anti- β-Actin | Santa Cruz Biotechnology | Cat#sc-47778; RRID:AB_626632 |
| Goat polyclonal anti-AIF1/Iba1 | Novus Biologicals | Cat#NB100-1028; RRID:AB_521594 |
| Mouse monoclonal anti-Aβ1-16 (6E10) | BioLegend | Cat#803001; RRID:AB_2564653 |
| Rabbit polyclonal anti-NeuN | Abcam | Cat#ab104225; RRID:AB_10711153 |
| Rat monoclonal anti-Clec7a/Dectin1 | InvivoGen | Cat#mabg-mdect; RRID:AB_2810285 |
| Rabbit polyclonal anti-P2ry12 | AnaSpec | Cat#AS-55043A; RRID:AB_2298886 |
| Goat polyclonal anti-Axl | R&D Systems | Cat#AF854; RRID:AB_355663 |
| Rat anti-mouse CD16/CD32 (Fc Block) | BD Biosciences | Cat#553142; RRID: AB_394656 |
| Rabbit monoclonal anti-NeuN | Abcam | Cat#ab190565; RRID:AB_2732785 |
| Chemicals, peptides, and recombinant proteins | ||
| Beta-Amyloid (1-42), HiLyte™ Fluor 555-labeled | AnaSpec | Cat#AS-60480-01 |
| X-34 | Milipore Sigma | Cat#SML1954 |
| Critical commercial assays | ||
| Taqman Gene Expression Assay: Plcg2, Mm00549424_ml | Applied Biosystems | Cat#4331182 |
| Taqman Gene Expression Assay: Gapdh, Mm99999915_g1 | Applied Biosystems | Cat#4331182 |
| V-PLEX Proinflammatory Panel 1 Mouse Kit | Meso Scale Discovery | Cat#K15048D |
| Nuclei isolation Kit: Nuclei EZ Prep | Millipore Sigma | Cat#NUC101 |
| Deposited data | ||
| Raw data (NanoString Glial Profiling and Neuropathology Panels) | This paper | GEO: GSE221806 |
| Raw data (Bulk RNA Seq and snRNA Seq | This paper | GEO: GSE237495 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain#000664; RRID:IMSR_JAX:000664 |
| Mouse: 5xFAD | The Jackson Laboratory | Strain#034848-JAX; RRID:MMRRC_034848-JAX |
| Mouse: plcg2R522 | The Jackson Laboratory | Strain#029598; RRID:IMSR_JAX:029598 |
| Mouse: Plcg2*M28L/APOE4/Trem2*R47H | The Jackson Laboratory | Strain#030674; RRID:IMSR_JAX:030674 |
| Mouse: 5×FADM28L | This paper | N/A |
| Mouse: 5×FADP522R | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad Software | Version 9.3.1 |
| PyMOL | The PyMOL Molecular Graphics System | Version 2.0 |
| limma | R Package | Version 4.3 |
| edgeR | R Package | Version 3.6.3 |
| CellRanger | R Package | Version 6.1.3 |
| ImageJ | Schneider et al. | https://imagej.nih.gov/ij/ |
| ANY-maze | Stoelting Co. | Version 7.0 |
| MiniAnalysis | Synaptosoft | Version 6.0 |
| nSolver Analysis Software | NanoString Technologies | Version 4.0 |
| Advanced Analysis Software | NanoString Technologies | Version 2.0.115 |
| Other | ||
| RNA STAT-60 | amsbio | Cat#CS-502 |
| KIMBLE Dounce tissue grinder set | Millipore Sigma | Cat#D8938 |
| Antigen Decloaker, 10x | Biocare Medical | Cat#CB910M |
| DMEM medium | Gibco | Cat#10566016 |
| Advanced DMEM/F12 medium | Gibco | Cat#12634028 |
| Fetal Bovine Serum | Gibco | Cat#16000044 |
| L-Glutamine | Gibco | Cat#25030081 |
| Penicillin/Streptomycin | Gibco | Cat#15140122 |
| EDTA-Trypsin | Gibco | Cat#25-200-072 |
| Nunc Lab-Tek chamber slide system | Thermo Scientific | Cat#154453 |
Experimental model and study participant details
Whole-genome sequencing (WGS)
Pre-processed whole-genome sequencing data were obtained from the Accelerating Medicines Partnership for Alzheimer’s Disease (AMP-AD) Consortium (https://adknowledgeportal.synapse.org/Explore/Programs/DetailsPage?Program=AMP-AD) through the Synapse database (https://www.synapse.org/) 12. Whole-genome sequencing libraries were prepared using the KAPA Hyper Library Preparation Kit per the manufacturer’s instructions. Libraries were sequenced on an Illumina HiSeq X sequencer using paired-end read chemistry and read lengths of 150bp. The paired-end 150bp reads were aligned to the NCBI reference human genome (GRCh37) using the Burrows-Wheeler Aligner (BWA-MEM) 41 and processed using the GATK best practices workflow that marks potential duplicates, locally realigns any suspicious reads, and re-calibrates the base-calling quality scores using Genome Analysis Toolkit (GATK) 42. The resulting BAM files were analyzed to identify variants using the HaplotypeCaller module of GATK for multi-sample variant callings 43.
Mice
The 5xFAD amyloid AD mouse model was used for immunofluorescence, cytokine production, qPCR and differential expression analyses. Mice were maintained on the C57BL/6J (B6) background and purchased from the Jackson Laboratory (JAX MMRRC Stock# 034848). The 5xFAD transgenic mice overexpress the following five FAD mutations under control of the Thy1 promoter: the APP (695) transgene containing the Swedish (K670N, M671L), Florida (I716V), and London (V7171) mutations, and the PSEN1 transgene containing the M146L and L286V FAD mutations 27.
We also used two PLCG2P522R and PLCG2M28L mouse models recently generated by the IU/JAX/UCI MODEL-AD consortium (JAX MMRRC Stock# 029598 and #030674; https://www.model-ad.org/strain-table/). PLCG2P522R and PLCG2M28L mice were generated using CRISPR/cas9 endonuclease-mediated genome editing to introduce the mutations. The APOE4 gene sequence and TREM2R47H mutation from the PLCG2M28L mouse model 44 were moved by crossing with B6 mice. These mice were maintained on the B6 background and crossed with 5xFAD mice to yield the 5xFAD; PLCG2M28L and 5xFAD; PLCG2P522R genotypes (5xFADM28L and 5xFADP522R). The same numbers of male and female mice (7.5-month-old; 223–227 days) were used in the current study. Up to five mice were housed per cage with SaniChip bedding and LabDiet® 5K52/5K67 (6% fat) feed. The colony room was kept on a 12:12 h light/dark schedule with the lights on from 7:00 am to 7:00 pm daily. The mice were bred and housed in specific-pathogen-free conditions. Both male and female mice were used, and the numbers of male and female mice were equally distributed. The number of mice used for each experiment is stated in the corresponding figure legends, and the results of individual values are shown in the scatter plot.
Mice were euthanized by perfusion with ice-cold phosphate-buffered saline (PBS) following full anesthetization with Avertin® (125–250 mg/kg intraperitoneal injection). Animals used in the study were housed in the Stark Neurosciences Research Institute Laboratory Animal Resource Center at Indiana University School of Medicine. All animals were maintained, and experiments were performed, in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at Indiana University School of Medicine.
METHOD DETAILS
Homology modeling
The model of the PLCG2 structure containing all residues from amino acid 14–1190 was built using the template PLCG1 model 45. The cartoons with substitutions of M28L and P522R were generated with PyMol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
Differential gene expression and pathway enrichment analysis
The limma package in R software 46 was used to identify differentially expressed genes and to perform differential expression analyses of bulk RNA-Seq data from different samples. The ClusterProfiler package was used to automate biological-term classification and enrichment analysis for the differentially expressed genes 47.
Immunoblotting
Tissue was extracted and processed as described above, then centrifuged. Protein concentrations were measured with a BCA kit (Thermo Scientific). One hundred micrograms of protein per sample was denatured by heating the samples for 10 min at 95°C. The samples were then loaded into 4–12% Bis-Tris gels (Life Technologies) and run at 100 V for 90 min. The following primary antibodies were used: PLCG2 (CST #3872 1:500, Rabbit mAb) and β-Actin (Santa Cruz #sc-47778). Each sample was normalized to β-Actin, and the graphs represent the values normalized to the mean of the WT mouse group at each time point.
Immunofluorescence and image analysis
Perfused brains from mice at 7.5 months of age were fixed in 4% paraformaldehyde for 24 h at 4°C. Following 24 h fixation, brains were cryoprotected in 30% sucrose at 4°C and embedded. Brains were cut on a microtome into 30-μm free-floating sections. For immunostaining, at least three matched brain sections were used. Free-floating sections were washed and permeabilized in 0.1% TritonX in PBS (PBST), and antigen retrieval was subsequently performed using 1x Reveal Decloaker (Biocare Medical) at 85°C for 10 min. Sections were blocked in 5% normal donkey serum in PBST for 1 h at room temperature (RT). The sections were then incubated with the following primary antibodies in 5% normal donkey serum in PBST overnight at 4°C: Iba1 (Novus Biologicals #NB100–1028, goat, 1:1000); 6E10 (BioLegend #803001, mouse, 1:1000; AB_2564653); NeuN (Abcam, ab104225, rabbit, 1:1000); CLEC7A (InvivoGen, mabg-mdect, rat, 1:500 of 1 mg/ml); P2RY12 (AnaSpec, AS-55043A, rabbit, 1:1000); Axl (R&D Systems. Sections were washed and visualized using the respective species-specific AlexaFluor fluorescent antibodies (diluted 1:1000 in 5% normal donkey serum in PBST for 1 h at RT). Sections were counterstained with antibodies and mounted onto slides. For X34 staining (Sigma, #SML1954, 100 uM), sections were dried at RT, rehydrated in PBST, and stained for ten mins at RT. Sections were then washed five times in double-distilled water and washed again in PBST for five mins 48. Images were acquired on a fluorescence microscope with similar exposure and gains across stains and animals.
Images were taken on a Leica DM6 microscope or a Nikon A1R confocal microscope (Nikon Instruments, Melville, NY) for higher magnification and analyzed using ImageJ software (NIH) 49. The results were obtained from an average of at least three sections per mouse, with a threshold applied across all images. The threshold function in ImageJ was used to determine the percentage of the immunoreactive area (X34, 6E10, IBA1, PR2Y12 and CLEC7A) within the region of interest (ROI). Plaque categorization was performed by the quantification of the 6E10, X34 and 6E10+/X34+ costained area. For quantification of the staining percentage area of X34 plaques, the surface of X34 plaques was created by the create selection function, and the percentage of the immunoreactive area was analyzed within the X34 positive extended surface. Plots were generated using GraphPad Prism (Version 9.3.1).
Magnetic resonance imaging
High-resolution T2*-weighted magnetic resonance (MR) images were acquired using a Bruker BioSpec 9.4T/30 MRI scanner outfitted with a high-sensitivity cryogenic RF surface receive-only coil (Bruker CyroProbe) as previously described 50. Images were acquired using a 3D gradient echo sequence with the following acquisition parameters: TR: 200 ms; TE: 10 ms; Ave: 2; Flip Angle: 45; Spatial resolution 25 μm isotropic. To assess amyloid deposition in the brain of AD mice, ITKSNAP 51 was used to determine the hypo-intense area in MR images.
Primary microglia culture
Microglia were isolated as previously described 52. Briefly, brain tissue from C57BL/6 neonatal mice aged P2–P4 was homogenized in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco™ 10566016), filtered through 250 and 100 μM meshes sequentially, and cultured in Advanced DMEM/F12 (Gibco™ 12634028) supplemented with 10% FBS (Gibco™ 16000–044), 2 mM L-glutamine (Gibco™ 25030081) and Penicillin/Streptomycin (Gibco™ 15140122). After 21 days in culture, the cells were subjected to mild trypsinization using a 1:3 dilution of EDTA-Trypsin (Gibco™ 25–200-072) in DMEM for 20 min. Trypsinization resulted in the detachment of an intact layer of astrocytes, leaving microglia attached to the bottom of the plate, which were used for experiments within 48 h. For the RNA analysis, microglia were cultured in polystyrene 6-well plates (Falcon™ 08–772-1B); for immunofluorescence, microglia were cultured in a chamber slide system (Nunc™ Lab-Tek™ II Chamber Slide™, 154453).
Aβ1–42 aggregation and uptake assay
The HiLyte™ Fluor 555-labeled Aβ1–42 peptide was obtained from AnaSpec (AS-60480–01), reconstituted as suggested by the manufacturer with 1.0% ammonium hydroxide and phosphate-buffered saline (PBS) pH=7.4 up to 1 mg/ml. The peptides were diluted to 0.5 mg/ml in PBS pH=7.4 and aggregated at 37°C for 5 days, similar to a previously described method 40. Incubation of microglia with HiLyte™ Fluor 488-labeled Aβ1–42 aggregates was followed by the immunofluorescence protocol.
Y-maze task
Working memory in mice was assessed by spontaneous alternation in the Y-maze task as previously described 40. Animals were placed in the Y-maze with free access to all three arms and tracked and recorded for 8 min using ANY-maze software (Stoelting Co.). The number of alternations was determined by counting the sequential entries into the three different arms of the maze (same-arm re-entries were possible). The percentage of spontaneous alternation was calculated using the following formula: (number of alternations performed)/(number of possible alternations [total arm entries−2])×100. An entry counted when all four limbs of a mouse entered the arm.
Hippocampal Slice Electrophysiology:
Mice were deeply anesthetized and intracardially perfused with ice-cold artificial cerebrospinal fluid (aCSF) containing (in mM) 124 NaCl, 4.5 KCl, 1.2 NaH2PO4, 26 NaHCO3, 10 glucose, 1 MgCl2, and 2 CaCl2, continuously bubbled with 95% O2/5% CO2; pH 7.4, 310 mOsm. The brains were quickly removed, and hippocampal slices (280 μm) were cut at 0.1 mm/s with a Leica VT1200 vibratome in an ice-cold oxygenated sucrose-based solution containing (in mM) 194 sucrose, 10 glucose, 30 NaCl, 26 NaHCO3, 0.5 NaH2PO4,4.5 KCl, and 1 MgCl2, pH 7.4. After 60 min recovery in an incubation chamber containing oxygenated aCSF solution at 33°C, the slices were then kept at room temperature until they were transferred to a recording chamber, perfused continuously (~2 ml/min) with oxygenated aCSF at ~32°C
Field recordings:
Field excitatory postsynaptic potentials (fEPSP) were recorded with micropipettes filled with 1 M NaCl placed in the stratum radiatum of the CA1 region of the hippocampus. A stimulating stainless steel stereotrode (1 MΩ) was placed in the Schaffer collateral pathway. The intensity of the stimulator was increased stepwise until a maximal response was obtained using a constant current isolated stimulator (Digitimer). Signals were acquired using a Multiclamp 700B amplifier and Clampex software (Molecular Devices). Signals were low pass filtered and digitized at 50 kHz, and the slope of the fEPSP (mV/ms) was measured. Paired-pulse ratios (PPR) were obtained every 20 s at 40 ms increasing inter-stimuli interval (ISI). For LTP recordings, the following protocol was used: after adjusting the stimulation strength to produce 50% of the maximum intensity, a stable 10 min of baseline (30 pulses every 20 s) was recorded, followed by 1 min conditioning trains (10 pulses at 100Hz) repeated 4 times every 20 s; a 60 min post-conditioning was performed at the same baseline stimulation frequency. The synaptic strength change was expressed as the percentage of change with respect to the average baseline.
Whole-cell recordings:
Patch pipettes (2.5–3.5 MΩ) were pulled (Sutter Instruments) from borosilicate glass (World Precision Instruments) and filled with an internal solution containing (in mM) 120 CsMeSO3; 5 NaCl, 10 TEA, 10 HEPES, 5 lidocaine bromide, 1.1 EGTA, 4 Mg-ATP, and 0.3 Na-GTP (pH adjusted to 7.2 and 290 mOsm). Neurons in the CA1 region were visualized with a 40X water-immersion objective with infrared-differential interference contrast video microscopy (BX51WI; Olympus). Spontaneous excitatory postsynaptic currents (sEPSC) were gap-free recorded five mins after breaking for 3 min in voltage-clamp mode with the membrane potential (Vh) held at −70 mV. sIPSCs were recorded with the membrane potential held at +10 mV. Recordings were conducted using a Bessel filter set at 4 kHz and digitized at 50 kHz with a Digidata 1440A A/D interface (Molecular Devices). A negative 5 mV pulse was delivered regularly to monitor access and input resistance. Recordings with access resistance greater than 25 MΩ or with changes in access or greater than 20% were discarded. Recordings of AMPA/NMDA ratios were performed after adding picrotoxin (50 μM) to the aCSF to block inhibitory currents. Evoked synaptic responses were evoked at Vh=+40 mV before and after the addition of d-APV (50 μM) to block NMDA receptors and isolate AMPA currents. pClamp11 (Molecular Devices) and MiniAnalysis (Synaptosoft) were used for quantification. Statistical analyses were conducted using Prism (GraphPad Software). All data are presented as the mean ± SEM. The significance level was set at p<0.05
Mouse RNA isolation for qPCR, NanoString nCounter analysis, and bulk RNA-Seq
Mice were anesthetized with Avertin and perfused with ice-cold PBS. The cortical and hippocampal regions were microdissected and stored at −80°C. Frozen brain tissues were homogenized in T-PER tissue protein extraction reagent (Catalog #78510, Thermo Fisher) and stored in an equal volume of RNA STAT-60 (Amsbio) at −80°C until RNA extraction was performed. RNA was isolated by chloroform extraction and purified using the Purelink RNA Mini Kit (Life Technologies). cDNA was prepared from 750 ng of RNA using the High-Capacity of RNA-to-cDNA kit (Applied Biosystems), and qPCR was performed on the StepOne Plus Real-Time PCR system (Life Technologies) with the Taqman Gene Expression Assay (Plcg2, Mm00549424_m1, Applied Biosystems). Relative gene expression was determined with the ΔΔCT method and was assessed relative to Gapdh (Mm99999915_g1). Statistical analyses of the qPCR results were performed using a one-way analysis of variance (ANOVA) test followed by Tukey’s post hoc to compare genotypes (GraphPad Prism, version 9.3.1).
Glial Profiling and Neuropathology Panels were used for the NanoString nCounter analysis (NanoString Technologies, Seattle, WA, USA). Two hundred nanograms of RNA was loaded per 7.5-month-old male and female mouse from the 5xFAD, 5xFADM28L and 5xFADP522R mouse samples (N=12 per genotype, 6 male and 6 female mice) and hybridized with probes for 16 h at 65°C. The results obtained from the nCounter MAX Analysis System (NanoString Technologies, catalog #NCT-SYST-LS, Seattle WA) were imported into the nSolver Analysis Software (v4.0; NanoString Technologies) for QC verification, normalization, and data statistical analysis using Advanced Analysis software (v2.0.115; NanoString Technologies). All assays were performed according to the manufacturer’s protocols 53,54.
Paired-end RNA sequencing (101bp X2) with ~100 million reads coverage was performed on an Illumina NextSeq 500 instrument. Fastq was aligned to the reference mouse genome with GENCODE annotation (gencode.vM26) using STAR (v.2.7.2b)55, and reads mapped to each gene were counted using featureCounts. Differentially expressed genes were analyzed using the edgeR package in R (version 3.6.3).
Quantification of proinflammatory cytokine concentrations in mouse brains
Protein concentrations of inflammatory cytokines in the mouse brains were quantified using Meso Scale Discovery (MSD) 96-well multispot V-PLEX Proinflammatory Panel I (K15048D, MSD, Gaithersburg, MD, USA), following the manufacturer’s guidelines 56. Frozen brain cortex samples were homogenized in T-PER tissue protein extraction reagent supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich). Total protein concentration was measured using the Pierce BCA Protein Assay Kit (Thermo Scientific). Fifty microliters of protein lysate (500 μg) was used to analyze the proinflammatory cytokine content (N=10 per genotype, 7.5-month-old, 5 male and 5 female mice).
Nuclei isolation and fluorescence-activated cell sorting (FACS)
Nuclei were isolated as previously described 57,58. Nuclei were isolated from the fresh-frozen cortical brain region of the left hemisphere. All reagents were placed on ice. Frozen tissue was minced with a chilled razor blade and then Dounce homogenized 25 times with a loose pastle A followed by 15 times with a tight pastle B (Sigma-Aldrich, St. Louis, USA #D8938), all while simultaneously twisting up and down with lysis buffer from the Nuclei EZ Prep Kit (Sigma-Aldrich, St. Louis, USA, #Nuc101–1KT). The tube was incubated on ice for 5 min. Nuclei were pelleted with 5 min centrifugation at 500 g (4°C). The pellet was then resuspended with 4 ml fresh lysis buffer. Following a subsequent 5 min centrifugation step at 500 g (4°C), the lysis buffer was removed. The pellet was washed with 4 ml PBS and then transferred into a FACS tube.
Nuclei were centrifuged for 10 min at 300 g (4°C) and resuspended in 50 μl FACS buffer (1% BSA, 1× PBS; sterile filtered) containing 2 U/ml Protector RNase Inhibitor (Sigma-Aldrich, St. Louis, USA, #3335402001) with anti-CD16/CD32 Fc Block (BD Biosciences #553142). After 5 min incubation, an additional 50 μl of antibody cocktail with Protector RNase Inhibitor was added: 1 μl NeuN (Abcam #ab190565) in 50 μl FACS buffer. Following Fc blocking, 50 μl of antibody master mix was added to each sample to achieve 1× antibody concentration. Samples were incubated with the staining antibodies on ice with gentle shaking for 30 min and then centrifuged for 10 min at 300 g (4°C) before being resuspended in 700 μl of FACS buffer with Protector RNase Inhibitor. Subsequently, 1 μl Hoechst 33342 was added.
NeuN-positive and NeuN-negative nuclei were separately sorted into 1.5 ml DNA LoBind tubes with 1 ml Dijon buffer (200 μl UltraPure BSA, Thermo, #AM2618, 800 μl PBS, and 5 μl Protector RNase Inhibitor. A total of 50,000 single nuclei were sorted from each sample. Nuclei were counted, and concentrations were adjusted to ~1 mio nuclei/ml.
Chromium 10X library generation and Illumina sequencing
Reagents for the Chromium Single Cell 3′ Library & Gel Bead Kit v3 (10X Genomics, Pleasanton, USA) were thawed and prepared according to the manufacturer’s protocol. The nuclei/master mix solution was adjusted to target 10,000 nuclei per sample (5,000 nuclei from male mice and 5,000 nuclei from female mice) and loaded on a standard Chromium Controller (10X Genomics, Pleasanton, USA), according to the manufacturer’s protocol. All reaction and quality control steps, including library construction (using Chromium Single Cell 3′ Library Construction Kit v3), were conducted according to the manufacturer’s protocol and with the recommended reagents, consumables and instruments. Quality control of cDNA and libraries was conducted using a Bioanalyzer (Agilent, Santa Clara, USA) at the Stanford Protein and Nucleic Acid Facility.
Illumina sequencing of 10X snRNA-seq libraries was performed by Novogene Co. Inc. (Sacramento, USA; https://en.novogene.com/). Multiplexed libraries were sequenced with 2 × 150-bp paired-end (PE) reads in a single S4 lane on an Illumina Novaseq S4 (Illumina, San Diego, USA) targeting 100 million reads per library. Novogene conducted base-calling, demultiplexing, and the generation of FastQ files. Raw snRNA-seq data were processed using CellRanger (v.6.1.3) with –include-introns commands, and downstream analysis was performed using the Seurat package in R. Low quality cells with <200 features, >5% mitochondrial UMI counts were discarded. Dimension reduction and clustering were performed following the Seurat integration pipeline. UMAP-projected clusters were manually annotated based on their expression of canonical marker genes. Differentially expressed genes between cell types and conditions were identified using the FindMarker function in Seurat.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests were performed using GraphPad Prism 9 (GraphPad Software, La Jolla, CA). All data analyses were performed blindly. All data are presented as the mean ± SEM, analyzed by an ordinary one-way ANOVA followed by Tukey’s multiple comparison test. * P <0.05; ** P< 0.01; *** P< 0.001; ns: not significant. Male mice are marked with a solid circle (•), and the female mice are marked with a hollow circle (∘). Detailed information on the statistical method for each quantitative data was described in each figure legend.
Supplementary Material
Highlights.
M28L variant of PLCG2 is associated with an increased risk for Alzheimer’s disease (AD)
In an amyloidogenic AD mouse model, PLCG2M28L exacerbates disease pathogenesis
Conversely, PLCG2P522R, a protective PLCG2 variant, attenuates AD pathogenesis
The PLCG2 variants uniquely alter the microglial transcriptome and phenotypes
Acknowledgments:
The authors thank the members of the Landreth and Lamb laboratory for feedback and support throughout the study. We thank Louise Pay for her critical comments on the manuscript and Teaya N. Thomas for the help with taking care of the mice. This work was supported by NIA grant K01 AG054753 (A.L.O), NINDS grant R01 NS125020 (N.W), NIA grant R03 AG063250 (K.N), NIH grant NLM R01 LM012535 (K.N), NIA grant U54 AG054345 (B.T.L et al.), and NIA grant RF1 AG074566 (B.T.L, S.J.B, and G.E.L).
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a gender minority in their field of research. We worked to ensure sex balance in the selection of non-human subjects.
Footnotes
Declaration of interests:
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, et al. (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun 11, 6129. 10.1038/s41467-020-19737-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, Scheiwe C, Nessler S, Kunz P, van Loo G, et al. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392. 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- 3.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e1217. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 4.Lewcock JW, Schlepckow K, Di Paolo G, Tahirovic S, Monroe KM, and Haass C (2020). Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer’s Disease. Neuron 108, 801–821. 10.1016/j.neuron.2020.09.029. [DOI] [PubMed] [Google Scholar]
- 5.Andreone BJ, Przybyla L, Llapashtica C, Rana A, Davis SS, van Lengerich B, Lin K, Shi J, Mei Y, Astarita G, et al. (2020). Alzheimer’s-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nature Neuroscience 23, 927–938. 10.1038/s41593-020-0650-6. [DOI] [PubMed] [Google Scholar]
- 6.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, et al. (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49, 1373–1384. 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bunney TD, Opaleye O, Roe SM, Vatter P, Baxendale RW, Walliser C, Everett KL, Josephs MB, Christow C, Rodrigues-Lima F, et al. (2009). Structural insights into formation of an active signaling complex between Rac and phospholipase C gamma 2. Mol Cell 34, 223–233. 10.1016/j.molcel.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 8.Walliser C, Tron K, Clauss K, Gutman O, Kobitski AY, Retlich M, Schade A, Röcker C, Henis YI, Nienhaus GU, and Gierschik P (2015). Rac-mediated Stimulation of Phospholipase Cγ2 Amplifies B Cell Receptor-induced Calcium Signaling. J Biol Chem 290, 17056–17072. 10.1074/jbc.M115.645739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falasca M, Logan SK, Lehto VP, Baccante G, Lemmon MA, and Schlessinger J (1998). Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J 17, 414–422. 10.1093/emboj/17.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulze-Luehrmann J, and Ghosh S (2006). Antigen-receptor signaling to nuclear factor kappa B. Immunity 25, 701–715. 10.1016/j.immuni.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Jing H, Reed A, Ulanovskaya OA, Grigoleit JS, Herbst DM, Henry CL, Li H, Barbas S, Germain J, Masuda K, and Cravatt BF (2021). Phospholipase Cγ2 regulates endocannabinoid and eicosanoid networks in innate immune cells. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2112971118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsai AP, Dong C, Lin PB, Messenger EJ, Casali BT, Moutinho M, Liu Y, Oblak AL, Lamb BT, Landreth GE, et al. (2022). PLCG2 is associated with the inflammatory response and is induced by amyloid plaques in Alzheimer’s disease. Genome Med 14, 17. 10.1186/s13073-022-01022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romero-Molina C, Garretti F, Andrews SJ, Marcora E, and Goate AM (2022). Microglial efferocytosis: Diving into the Alzheimer’s disease gene pool. Neuron 110, 3513–3533. 10.1016/j.neuron.2022.10.015. [DOI] [PubMed] [Google Scholar]
- 14.van der Lee SJ, Conway OJ, Jansen I, Carrasquillo MM, Kleineidam L, van den Akker E, Hernández I, van Eijk KR, Stringa N, Chen JA, et al. (2019). A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol 138, 237–250. 10.1007/s00401-019-02026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleineidam L, Chouraki V, Próchnicki T, van der Lee SJ, Madrid-Márquez L, Wagner-Thelen H, Karaca I, Weinhold L, Wolfsgruber S, Boland A, et al. (2020). PLCG2 protective variant p.P522R modulates tau pathology and disease progression in patients with mild cognitive impairment. Acta Neuropathol 139, 1025–1044. 10.1007/s00401-020-02138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takalo M, Wittrahm R, Wefers B, Parhizkar S, Jokivarsi K, Kuulasmaa T, Makinen P, Martiskainen H, Wurst W, Xiang X, et al. (2020). The Alzheimer’s disease-associated protective Plcgamma2-P522R variant promotes immune functions. Mol Neurodegener 15, 52. 10.1186/s13024-020-00402-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maguire E, Menzies GE, Phillips T, Sasner M, Williams HM, Czubala MA, Evans N, Cope EL, Sims R, Howell GR, et al. (2021). PIP2 depletion and altered endocytosis caused by expression of Alzheimer’s disease-protective variant PLCgamma2 R522. EMBO J 40, e105603. 10.15252/embj.2020105603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu S, Huo J, Lee KG, Kurosaki T, and Lam KP (2009). Phospholipase Cgamma2 is critical for Dectin-1-mediated Ca2+ flux and cytokine production in dendritic cells. J Biol Chem 284, 7038–7046. 10.1074/jbc.M806650200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Obba S, Hizir Z, Boyer L, Selimoglu-Buet D, Pfeifer A, Michel G, Hamouda MA, Gonçalvès D, Cerezo M, Marchetti S, et al. (2015). The PRKAA1/AMPKα1 pathway triggers autophagy during CSF1-induced human monocyte differentiation and is a potential target in CMML. Autophagy 11, 1114–1129. 10.1080/15548627.2015.1034406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, Subramanian N, Bunney TD, Baxendale RW, Martins MS, et al. (2012). Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med 366, 330–338. 10.1056/NEJMoa1102140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, Xue L, Li DH, Steggerda SM, Versele M, et al. (2014). Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med 370, 2286–2294. 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nature Genetics 51, 414–430. 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walliser C, Hermkes E, Schade A, Wiese S, Deinzer J, Zapatka M, Desire L, Mertens D, Stilgenbauer S, and Gierschik P (2016). The Phospholipase Cgamma2 Mutants R665W and L845F Identified in Ibrutinib-resistant Chronic Lymphocytic Leukemia Patients Are Hypersensitive to the Rho GTPase Rac2 Protein. J Biol Chem 291, 22136–22148. 10.1074/jbc.M116.746842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keaney J, Gasser J, Gillet G, Scholz D, and Kadiu I (2019). Inhibition of Bruton’s Tyrosine Kinase Modulates Microglial Phagocytosis: Therapeutic Implications for Alzheimer’s Disease. J Neuroimmune Pharmacol 14, 448–461. 10.1007/s11481-019-09839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olive C, Ibanez L, Farias FHG, Wang F, Budde JP, Norton JB, Gentsch J, Morris JC, Li Z, Dube U, et al. (2020). Examination of the Effect of Rare Variants in TREM2, ABI3, and PLCG2 in LOAD Through Multiple Phenotypes. J Alzheimers Dis 77, 1469–1482. 10.3233/jad-200019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magno L, Bunney TD, Mead E, Svensson F, and Bictash MN (2021). TREM2/PLCγ2 signalling in immune cells: function, structural insight, and potential therapeutic modulation. Mol Neurodegener 16, 22. 10.1186/s13024-021-00436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140. 10.1523/jneurosci.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, and Grutzendler J (2016). TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 90, 724–739. 10.1016/j.neuron.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masuda T, Sankowski R, Staszewski O, and Prinz M (2020). Microglia Heterogeneity in the Single-Cell Era. Cell Rep 30, 1271–1281. 10.1016/j.celrep.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 30.Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, and Lamb BT (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 4, 575–590. 10.1016/j.trci.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, and Rowan MJ (2005). Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med 11, 556–561. 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- 32.Jackson JT, Mulazzani E, Nutt SL, and Masters SL (2021). The role of PLCγ2 in immunological disorders, cancer, and neurodegeneration. J Biol Chem 297, 100905. 10.1016/j.jbc.2021.100905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e569. 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Magno L, Lessard CB, Martins M, Lang V, Cruz P, Asi Y, Katan M, Bilsland J, Lashley T, Chakrabarty P, et al. (2019). Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther 11, 16. 10.1186/s13195-019-0469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai AP, Dong C, Lin PB-C, Messenger EJ, Casali BT, Moutinho M, Liu Y, Oblak AL, Lamb BT, Landreth GE, et al. (2022). PLCG2 is associated with the inflammatory response and is induced by amyloid plaques in Alzheimer’s disease. Genome Medicine 14, 17. 10.1186/s13073-022-01022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng-Hathaway PJ, Reed-Geaghan EG, Jay TR, Casali BT, Bemiller SM, Puntambekar SS, von Saucken VE, Williams RY, Karlo JC, Moutinho M, et al. (2018). The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Molecular Neurodegeneration 13, 29. 10.1186/s13024-018-0262-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, and Huerta PT (2006). AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci U S A 103, 3410–3415. 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Danysz W, and Parsons CG (2012). Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine--searching for the connections. Br J Pharmacol 167, 324–352. 10.1111/j.1476-5381.2012.02057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Claes C, England WE, Danhash EP, Kiani Shabestari S, Jairaman A, Chadarevian JP, Hasselmann J, Tsai AP, Coburn MA, Sanchez J, et al. (2022). The P522R protective variant of PLCG2 promotes the expression of antigen presentation genes by human microglia in an Alzheimer’s disease mouse model. Alzheimers Dement. 10.1002/alz.12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moutinho M, Puntambekar SS, Tsai AP, Coronel I, Lin PB, Casali BT, Martinez P, Oblak AL, Lasagna-Reeves CA, Lamb BT, and Landreth GE (2022). The niacin receptor HCAR2 modulates microglial response and limits disease progression in a mouse model of Alzheimer’s disease. Sci Transl Med 14, eabl7634. 10.1126/scitranslmed.abl7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498. 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nho K, West JD, Li H, Henschel R, Bharthur A, Tavares MC, and Saykin AJ (2014). Comparison of Multi-Sample Variant Calling Methods for Whole Genome Sequencing. IEEE Int Conf Systems Biol 2014, 59–62. 10.1109/ISB.2014.6990432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oblak AL, Kotredes KP, Pandey RS, Reagan AM, Ingraham C, Perkins B, Lloyd C, Baker D, Lin PB, Soni DM, et al. (2022). Plcg2(M28L) Interacts With High Fat/High Sugar Diet to Accelerate Alzheimer’s Disease-Relevant Phenotypes in Mice. Front Aging Neurosci 14, 886575. 10.3389/fnagi.2022.886575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hajicek N, Keith NC, Siraliev-Perez E, Temple BR, Huang W, Zhang Q, Harden TK, and Sondek J (2019). Structural basis for the activation of PLC-gamma isozymes by phosphorylation and cancer-associated mutations. Elife 8. 10.7554/eLife.51700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Styren SD, Hamilton RL, Styren GC, and Klunk WE (2000). X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer’s disease pathology. J Histochem Cytochem 48, 1223–1232. 10.1177/002215540004800906. [DOI] [PubMed] [Google Scholar]
- 49.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maharjan S, Tsai AP, Lin PB, Ingraham C, Jewett MR, Landreth GE, Oblak AL, and Wang N (2022). Age-dependent microstructure alterations in 5xFAD mice by high-resolution diffusion tensor imaging. Front Neurosci 16, 964654. 10.3389/fnins.2022.964654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, and Gerig G (2006). User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage 31, 1116–1128. 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 52.Saura J, Tusell JM, and Serratosa J (2003). High-yield isolation of murine microglia by mild trypsinization. Glia 44, 183–189. 10.1002/glia.10274. [DOI] [PubMed] [Google Scholar]
- 53.Reilly AM, Tsai AP, Lin PB, Ericsson AC, Oblak AL, and Ren H (2020). Metabolic Defects Caused by High-Fat Diet Modify Disease Risk through Inflammatory and Amyloidogenic Pathways in a Mouse Model of Alzheimer’s Disease. Nutrients 12. 10.3390/nu12102977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Preuss C, Pandey R, Piazza E, Fine A, Uyar A, Perumal T, Garceau D, Kotredes KP, Williams H, Mangravite LM, et al. (2020). A novel systems biology approach to evaluate mouse models of late-onset Alzheimer’s disease. Mol Neurodegener 15, 67. 10.1186/s13024-020-00412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oblak AL, Lin PB, Kotredes KP, Pandey RS, Garceau D, Williams HM, Uyar A, O’Rourke R, O’Rourke S, Ingraham C, et al. (2021). Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study. Front Aging Neurosci 13, 713726. 10.3389/fnagi.2021.713726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hahn O, Fehlmann T, Zhang H, Munson CN, Vest RT, Borcherding A, Liu S, Villarosa C, Drmanac S, Drmanac R, et al. (2021). CoolMPS for robust sequencing of single-nuclear RNAs captured by droplet-based method. Nucleic Acids Res 49, e11. 10.1093/nar/gkaa1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods 14, 955–958. 10.1038/nmeth.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA-Seq, snRNA-Seq and gene expression data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-PLCG2 | Cell Signaling Technology | Cat#3872S; RRID:AB_2299586 |
| Mouse monoclonal anti- β-Actin | Santa Cruz Biotechnology | Cat#sc-47778; RRID:AB_626632 |
| Goat polyclonal anti-AIF1/Iba1 | Novus Biologicals | Cat#NB100-1028; RRID:AB_521594 |
| Mouse monoclonal anti-Aβ1-16 (6E10) | BioLegend | Cat#803001; RRID:AB_2564653 |
| Rabbit polyclonal anti-NeuN | Abcam | Cat#ab104225; RRID:AB_10711153 |
| Rat monoclonal anti-Clec7a/Dectin1 | InvivoGen | Cat#mabg-mdect; RRID:AB_2810285 |
| Rabbit polyclonal anti-P2ry12 | AnaSpec | Cat#AS-55043A; RRID:AB_2298886 |
| Goat polyclonal anti-Axl | R&D Systems | Cat#AF854; RRID:AB_355663 |
| Rat anti-mouse CD16/CD32 (Fc Block) | BD Biosciences | Cat#553142; RRID: AB_394656 |
| Rabbit monoclonal anti-NeuN | Abcam | Cat#ab190565; RRID:AB_2732785 |
| Chemicals, peptides, and recombinant proteins | ||
| Beta-Amyloid (1-42), HiLyte™ Fluor 555-labeled | AnaSpec | Cat#AS-60480-01 |
| X-34 | Milipore Sigma | Cat#SML1954 |
| Critical commercial assays | ||
| Taqman Gene Expression Assay: Plcg2, Mm00549424_ml | Applied Biosystems | Cat#4331182 |
| Taqman Gene Expression Assay: Gapdh, Mm99999915_g1 | Applied Biosystems | Cat#4331182 |
| V-PLEX Proinflammatory Panel 1 Mouse Kit | Meso Scale Discovery | Cat#K15048D |
| Nuclei isolation Kit: Nuclei EZ Prep | Millipore Sigma | Cat#NUC101 |
| Deposited data | ||
| Raw data (NanoString Glial Profiling and Neuropathology Panels) | This paper | GEO: GSE221806 |
| Raw data (Bulk RNA Seq and snRNA Seq | This paper | GEO: GSE237495 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain#000664; RRID:IMSR_JAX:000664 |
| Mouse: 5xFAD | The Jackson Laboratory | Strain#034848-JAX; RRID:MMRRC_034848-JAX |
| Mouse: plcg2R522 | The Jackson Laboratory | Strain#029598; RRID:IMSR_JAX:029598 |
| Mouse: Plcg2*M28L/APOE4/Trem2*R47H | The Jackson Laboratory | Strain#030674; RRID:IMSR_JAX:030674 |
| Mouse: 5×FADM28L | This paper | N/A |
| Mouse: 5×FADP522R | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad Software | Version 9.3.1 |
| PyMOL | The PyMOL Molecular Graphics System | Version 2.0 |
| limma | R Package | Version 4.3 |
| edgeR | R Package | Version 3.6.3 |
| CellRanger | R Package | Version 6.1.3 |
| ImageJ | Schneider et al. | https://imagej.nih.gov/ij/ |
| ANY-maze | Stoelting Co. | Version 7.0 |
| MiniAnalysis | Synaptosoft | Version 6.0 |
| nSolver Analysis Software | NanoString Technologies | Version 4.0 |
| Advanced Analysis Software | NanoString Technologies | Version 2.0.115 |
| Other | ||
| RNA STAT-60 | amsbio | Cat#CS-502 |
| KIMBLE Dounce tissue grinder set | Millipore Sigma | Cat#D8938 |
| Antigen Decloaker, 10x | Biocare Medical | Cat#CB910M |
| DMEM medium | Gibco | Cat#10566016 |
| Advanced DMEM/F12 medium | Gibco | Cat#12634028 |
| Fetal Bovine Serum | Gibco | Cat#16000044 |
| L-Glutamine | Gibco | Cat#25030081 |
| Penicillin/Streptomycin | Gibco | Cat#15140122 |
| EDTA-Trypsin | Gibco | Cat#25-200-072 |
| Nunc Lab-Tek chamber slide system | Thermo Scientific | Cat#154453 |