

Abstract
Background
The efficacy of anti‐programmed cell death protein 1 (PD‐1) immunotherapy in various cancers, including gastric cancer (GC), needs to be potentiated by more effective targeting to enhance therapeutic efficacy or identifying accurate biomarkers to predict clinical responses. Here, we attempted to identify molecules predicting or/and promoting anti‐PD‐1 therapeutic response in advanced GC (AGC).
Methods
The transcriptome of AGC tissues from patients with different clinical responses to anti‐PD‐1 immunotherapy and GC cells was analyzed by RNA sequencing. The protein and mRNA levels of the major facilitator superfamily domain containing 2A (MFSD2A) in GC cells were assessed via quantitative real‐time polymerase chain reaction, Western blotting, and immunohistochemistry. Additionally, the regulation of anti‐PD‐1 response by MFSD2A was studied in tumor‐bearing mice. Cytometry by Time‐of‐Flight, multiple immunohistochemistry, and flow cytometry assays were used to explore immunological responses. The effects of MFSD2A on lipid metabolism in mice cancer tissue and GC cells was detected by metabolomics.
Results
Higher expression of MFSD2A in tumor tissues of AGC patients was associated with better response to anti‐PD‐1 immunotherapy. Moreover, MFSD2A expression was lower in GC tissues compared to adjacent normal tissues, and its expression was inversely correlated with GC stage. The overexpression of MFSD2A in GC cells enhanced the efficacy of anti‐PD‐1 immunotherapy in vivo by reprogramming the tumor microenvironment (TME), characterized by increased CD8+ T cell activation and reduced its exhaustion. MFSD2A inhibited transforming growth factor β1 (TGFβ1) release from GC cells by suppressing cyclooxygenase 2 (COX2)‐prostaglandin synthesis, which consequently reprogrammed TME to promote anti‐tumor T cell activation.
Conclusions
MFSD2A potentially serves as a predictive biomarker for anti‐PD‐1 immunotherapy response in AGC patients. MFSD2A may be a promising therapeutic target to potentiate the efficacy of anti‐PD‐1 immunotherapy by reprogramming the TME to promote T cells activation.
Keywords: MFSD2A, immunotherapy, anti‐PD‐1, gastric cancer, TME, T cell activation, TGFβ1
List Of Abbreviations
- AGC
advanced gastric cancer
- ATCC
American type culture collection
- CircDLG1
circular RNA discs large MAGUK scaffold protein 1
- COX2
cyclooxygenase 2
- CyTOF
Cytometry by Time‐of‐Flight
- DAB
3,3′‐diaminobenzidine
- DC
dendritic cell
- ECL
enhanced chemiluminescence
- ELISA
enzyme‐linked immunosorbent assay
- EP2
prostaglandin E receptor 2
- EP4
prostaglandin E receptor 4
- FACS
fluorescence‐activated cell sorting
- FBS
fetal bovine serum
- FPKM
fragments per kilobase of transcript per million fragments mapped
- FDR
false discovery rate
- GC
gastric cancer
- ICB
immune checkpoint blockade
- IL10
interleukin 10
- IHC
immunohistochemistry
- IFNγ
interferon gamma
- ITGB2
integrin beta 2
- MMR
mismatch repair
- MDSC
myeloid‐derived suppressor cell
- MFSD2A
major facilitator superfamily domain containing 2A
- mIHC
multiplex immunohistochemistry
- MSI‐H
high microsatellite instability
- MUC4
mucin 4, cell surface associated
- MUC16
mucin 16, cell surface associated
- NK
natural killer cell
- ORR
objective response rate
- PD‐1
programmed cell death protein 1
- PD‐L1
programmed death‐ligand 1
- PBMC
peripheral blood mononuclear cells
- PD
progressive disease
- PR
partial response
- PVR
PVR cell adhesion molecule
- PGE2
prostaglandin E2
- PGB2
prostaglandin B2
- PGJ2
prostaglandin J2
- PTEN
phosphatase and tensin homolog
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- SD
stable disease
- SETDB1
SET domain bifurcated histone lysine methyltransferase 1
- SEM
standard error of the mean
- TMB
tumor mutation burden
- Treg
regulatory T cell
- TAM
tumor‐associated macrophage
- TME
tumor microenvironment
- TGFβ
transforming growth factor β
- TIL
tumor‐infiltrating lymphocyte
- TPM
transcripts per million
- TTN
titin
1. BACKGROUND
Immunotherapy, including the use of immune checkpoint blockade (ICB) [1], adoptive cell transfer therapy [2], and therapeutic cancer vaccines [3] and antibodies [4], has emerged as the forefront of cancer therapy with robust effectiveness in recent decades. Among immune checkpoint inhibitors, programmed cell death protein 1 (PD‐1) antibodies have demonstrated promising efficacies in the treatment of multiple hematological and solid cancers [5, 6]. Despite its clinical success, PD‐1 blockade responses are often observed in only a proportion of patients, making it important to identify accurate biomarkers for predicting treatment response and patient selection. Increasing evidence has demonstrated positive correlations between patients' response to anti‐PD‐1 immunotherapy and high tumor mutation burden (TMB), deficient DNA mismatch repair (MMR), high programmed death‐ligand 1 (PD‐L1) expression and DNA polymerase epsilon and delta 1 mutations in tumor cells [5, 7–10]. However, not all patients whose tumors have those biomarkers respond well to anti‐PD‐1 immunotherapy [11, 12]. For instance, only 40%‐70% of patients with MMR‐deficient tumors respond to the PD‐1 inhibitor pembrolizumab [5]. Besides, only 44.8% of non‐small‐cell lung cancer patients with high PD‐L1 expression in tumors respond to pembrolizumab [13]. Therefore, this urges the need to identify more accurate biomarkers for predicting clinical responsiveness to anti‐PD‐1 immunotherapy.
Gastric cancer (GC) is the fifth most common cancer worldwide [14]. Despite recent advances in this field, therapeutic options for advanced GC (AGC) cases remain limited. The anti‐PD‐1 immunotherapy has been applied to treat AGC patients [15, 16, 17]; however, most AGC patients were found to respond poorly to single‐agent PD‐1 antibodies with an objective response rate (ORR) to pembrolizumab and nivolumab of only 11.6% [18] and 11.2% [19], respectively. Unsatisfactory overall clinical benefit made investigations of the mechanism of anti‐PD‐1 monotherapy resistance a necessity.
Manipulation of effector T cells by cancer cells in the tumor microenvironment (TME) is a major mechanism of anti‐PD‐1 immunotherapy resistance [20]. In TME, immunosuppressive cells, including regulatory T cells (Tregs), tumor‐infiltrating myeloid‐derived suppressor cells (MDSCs), and tumor‐associated macrophages (TAM), as well as certain suppressive cytokines produced by cancer cells, such as transforming growth factor β (TGFβ) and interleukin 10 (IL10), suppress the activation and infiltration of tumor‐infiltrating lymphocytes (TILs) and cannot be abolished by anti‐PD‐1 immunotherapy [21, 22, 23]. Moreover, the dysregulated cancer cell metabolism, known to promote tumor progression and confer suppression on immune cells in TME, is considered a crucial hindrance to anti‐tumor immunotherapies [24, 25]. For instance, tumor oxidative metabolism was reported to hamper PD‐1 antibody treatment in melanoma and was associated with increased T cell exhaustion and decreased immune activity [26]. Although targeting metabolic pathways, such as glycolysis, amino acids, and lipids metabolism, may overcome the limitations of immunotherapy by reprogramming TME [24, 27], it is unclear whether metabolism modulates the efficacy of anti‐PD‐1 immunotherapy in GC. Therefore, a thorough understanding of how cancer metabolism in TME affects anti‐PD‐1 immunotherapy could help improve the therapeutic response of AGC patients.
Major facilitator superfamily domain containing 2A (MFSD2A) is a lipid metabolism‐related protein that plays a key role in maintaining the blood‐brain barrier [28, 29]. MFSD2A deficiency was shown to enhance blood‐brain barrier leakage and developmental brain disorders [28, 30]. MFSD2A is considered a tumor suppressor as it was reported to inhibit extracellular matrix attachment, regulate the cell cycle of lung cancer cells [31], and prevent brain cancer cell metastasis by restoring intracellular docosahexaenoic acid transportation [32]. Besides, MFSD2A was shown to be essential for maintaining memory T cells and reducing secondary response to repeated infections [33]. However, the role of MFSD2A in anti‐tumor immunity remains unknown.
In this study, we screened potential biomarkers for predicting the efficacy of anti‐PD‐1 immunotherapy in AGC patients. Then we explored how the candidate biomarker functions during anti‐PD‐1 immunotherapy, providing the promising predictive biomarker for determining anti‐PD‐1 therapy effect and mechanic insights into how cancer cell metabolic reprogramming of TME reverses the immunosuppression to enhance the therapeutic effects of anti‐PD‐1 immunotherapy in AGC.
2. MATERIALS AND METHODS
2.1. Clinical cohort
Twenty‐seven patients with evaluable response to anti‐PD‐1 immunotherapy and RNA sequencing (RNA‐seq) data in a cohort of 58 refractory AGC patients treated with PD‐1 antibody toripalimab monotherapy (3 mg/kg, d1, Q2W; Junshi Biosciences, Shanghai, China) in a previously reported open‐label multicenter phase Ib/II trial (NCT02915432) [34] were included in this present study. Tumor biopsies were obtained before toripalimab treatment and subjected to RNA‐seq as previously described [35]. Gene expression was represented by transcripts per million (TPM), and differentially expressed genes were identified using Student's or Welch's t‐test as well as FDR corrections.
2.2. Cell lines
The human GC cell line MGC803 was obtained from American type culture collection (ATCC, Manassas, VA, USA), and mouse GC cell line MFC from Kebai Biotechnology Co., Ltd (Nanjing, Jiangsu, China). MGC803 cells were mycoplasma free and cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco, Gaithersburg, MD, USA). MFC cells were also mycoplasma free and cultured in 1640 Medium (Gibco, Gaithersburg, MD, USA) supplemented with 10% heat‐inactivated fetal bovine serum (FBS, Gibco), 100 U/mL penicillin G (Sangon Biotech, Shanghai, China), and 100 μg/mL streptomycin (Sangon Biotech) under a humidified atmosphere of 5% CO2 at 37°C.
2.3. Immunohistochemistry (IHC)
The human tissue arrays containing 75 GC and 74 normal adjacent gastric tissues (Xinchao Biotechnology Co., Ltd., Shanghai, China), and paraffin sections of mouse tumor tissues were fixed with formalin, embedded with paraffin and immuno‐stained for MFSD2A or TGFβ1 using standard IHC protocol as previously described [36]. Briefly, the slides were deparaffinized in xylene, rehydrated through graded ethanol, quenched for endogenous peroxidase activity in 3% hydrogen peroxide (Abcam, MA, USA), and processed for antigen retrieval by microwave heating for 7 min in citrate buffer (pH 6.0; Abcam). The primary anti‐MFSD2A (1:500, ab117618, Abcam, MA, USA), anti‐TGFβ1 (1:400, ab215715, Abcam) were diluted in PBS containing 1% bovine serum albumin (BSA) and incubated at 4°C, followed by the incubation with secondary antibody (Dako, Carpinteria, CA, USA) for 60 min at room temperature and stained with (3,3′‐diaminobenzidine, DAB; Dako) solution. The IHC results for MFSD2A or TGFβ1 were determined by adding the scores for staining intensity (no staining, 0; light yellow, 1; yellow, 2; brown, 3) and positive cell percentage (≤ 10%, 1; 11%‐50%, 2; 51%‐75%, 3; > 75%, 4).
2.4. Cell transfection
pcDNA3.1‐puromycin‐flag‐MFSD2A cDNA (mouse or human origin, pMFSD2A) or control pcDNA3.1‐puromycin‐flag vector (mock) was constructed by homologous recombinase (Vazyme, Nanjing, China). MFC or MGC803 cells were transfected with pMFSD2A or mock vectors using JetPEI (Polyplus‐transfection, Illkirch, France) for 48 h and screened under puromycin (Gibco) for 2 weeks. Stable overexpression efficacy was confirmed by Western blotting.
The MFSD2A shRNA (sh‐MFSD2A#1, 5′‐AGCCGGAACGTGTCAAGTTTA‐3′; sh‐MFSD2A#2, 5′‐CACGGCCCATACATCAAACTT‐3′; sh‐MFSD2A#3, 5′‐CACGGCCCATACATCAAACTT‐3′) or control shRNA (sh‐Non, 5′‐CCTAAGGTTAAGTCGCCCTCG‐3′) were cloned into lentivirus vector pSLenti‐U6 ‐CMV‐EGFP‐F2A‐Puro‐WPRE (Obio Technology, Shanghai, China) and packed into lentivirus. MGC803 cells were infected with lentivirus sh‐MFSD2A or sh‐Non for 48 h and selected by puromycin for two weeks. Stable inference efficacy was confirmed by Western blotting.
The MFSD2A siRNAs (si‐MFSD2A#1, 5′‐AAUAGGUCUGGCCGUGUGGTT‐3′; si‐MFSD2A#2, 5′‐UUGAUGUAUGGGCCGUGGCTT‐3′; si‐MFSD2A#3, 5′‐AAGCCCAAGGUGUAGGUGCTT‐3′) and control siRNA (si‐Non, 5′‐AUCUUAGGCAGAUCGUCGCdTdT‐3′) were synthesized by GenePharma Biotechnology (Shanghai, China). siRNA was delivered into MGC803 cells using INTEFERin (Polyplus‐transfection).
2.5. Animal experiments
All animal experiments were performed following the National Institute of Health Guide for the Care and Use of Laboratory Animals, with the approval of Suzhou Institute of Systems Medicine, Animal Science Center, Jiangsu, China. Mice were housed in a virus‐free facility and maintained in a standard temperature‐ and light‐controlled animal facility, mice had free access to food and water during the experimental period.
A total of 66 mice were used for this study. Syngeneic mice model was conducted on 6‐week old female 615 mice obtained from the Blood Institute of the Chinese Academy of Medical Sciences (Tianjin, China) to evaluate the effects of MFSD2A on the efficacy of anti‐PD‐1 immunotherapy in vivo. Briefly, MFSD2A‐overexpressed or control MFC cells (5 × 105 cells in 100 μL PBS) were implanted subcutaneously in the right flanks of the mice. After 9 days, when the tumor grew to 100 mm3, the mice were intraperitoneally injected with PD‐1 antibody (3 mg/kg, BE0273, Bio X Cell, West Lebanon, NH, USA) and control IgG (BE0089, Bio X Cell) every 3 days. Tumor volume was monitored every 3 days using a digital caliper according to the formula: Tumor volume (mm3) = 0.5 × length × width2. After 27 days, the mice were euthanized, and the tumors were isolated and weighed, and the tumor immune microenvironment was analyzed by Cytometry by Time‐of‐Flight (CyTOF). For survival study, after the tumor‐bearing mice models were established and the antibodies were injected according to the above method, the survival of the mice was observed. The evaluation criteria for mouse death are "natural mice death" or "mice tumor volume exceeding 2000mm3". Mice survival status were assessed every day.
In vivo depletion of CD8+ T cells was performed as previously described [37]. Briefly, 615 mice were intraperitoneal administration with 200 μg of CD8α antibody (BE0061, Bio X Cell) or control IgG. After 3 days of primary administration, the mice were subcutaneously bearing MFSD2A‐overexpressed or control MFC cells (as day 0), followed by administration on day 0, 3, 8, 13, 18, 23. Tumor volume was monitored every 3 days. After 30 days, the mice were euthanized, and the tumors were isolated and tumor infiltrating immune cells were selected by CD45 magnetic beads (130‐110‐618, Miltenyi, Bergisch Gladbach, Germany), and then labeled by CD3 antibody (1:50; 553063, BD Biosciences, San Jose, CA, USA) and CD8α antibody (1:50; 553030, BD Biosciences). The proportion of tumor infiltrating CD8+ T cells was analyzed by flow cytometry.
2.6. T cell function assay
Tumors were dissected from tumor‐bearing 615 mice and disassociated into single‐cell suspensions in PBS on ice, as previously described [38]. The cells were washed with PBS, centrifuged at 500 g and 4°C, then sorted by CD8 magnetic beads (130‐116‐478, Miltenyi, Cologne, Germany). CD8+ T cells derived from the mice were co‐cultured with 5 × 104 MFSD2A‐overexpressed or control MFC cells at an E:T ratio of 10:1 in a 24‐well plate for 24 h. T cell function was assayed as previously described [39]. CD8+ T cells were resuspended in 100 μL of PBS containing 2% FBS and stained with CD8 (100750, Biolegend, California, USA), Granzyme B (372208, Biolegend) and interferon γ (IFNγ; 557735, BD Biosciences) flow antibody for 15 min. CD8+ T cell viability was detected by flow cytometry (C6 plus Flow Cytometer, BD Biosciences) analysis.
Human peripheral blood mononuclear cells (PBMCs) were obtained from ORIBIOTECH (Shanghai, China). CD8+ T cells from PBMCs were isolated by magnetic‐activated cell sorting, then co‐incubated with MFSD2A‐silenced or MFSD2A‐overexpressed MGC803 cells and control cells at an E:T ratio of 10:1 in a 24‐well plate with or without 10 μmol/L Disitertide (P144, MedChemExpress, Monmouth Junction, NJ, USA), or 50 μmol/L Celecoxib (S1261, Selleck, Houston, TX, USA) treatment. T cell function was assayed after 24 h, as previously described [40, 41]. CD8+ T cells were resuspended in 100 μL of PBS containing 2% FBS and stained with CD8 (1:50, 557085, BD Biosciences), Granzyme B (1:50, 372204, Biolegend) and IFNγ (1:50, 502509, Biolegend) flow antibody for 15 min. CD8+ T cell viability was detected by flow cytometry (C6 plus Flow Cytometer, BD Biosciences) analysis.
2.7. In vitro cytotoxicity assay
After MFSD2A‐overexpressed or MFSD2A‐silenced MGC803 cells were co‐cultured with CD8+ T cells from human PBMC for 24 h, or MFSD2A‐overexpressed MFC cells were co‐cultured with CD8+ T cells derived from tumor tissues for 24 h, 50 μL of the supernatant was extracted for cytotoxicity assessment. Cytotoxicity was measured using the CytoTox 96 Non‐Radioactive Cytotoxicity Assay (G1781, Promega, Madison, WI, USA) using an Infinite F50 reader (Tecan Group LTD, Männedorf, Switzerland) at 490 nm according to the manufacturer's instructions.
2.8. Multiplex immunohistochemistry (mIHC)
The tissue microarray slide containing human 96 GC tissues (Xinchao Biotechnology Co., Ltd.) was performed with mIHC. A total of 4 antibodies including anti‐MFSD2A (1:500, ab117618, Abcam), anti‐TGFβ1 (1:400, ab215715, Abcam), anti‐CD8 (1:600, 85336S, Cell Signaling Technology, Beverly, MA, USA) and anti‐Ki67 (1:600, 9449S, Cell Signaling Technology) were used. Briefly, the tissue microarray slide was dewaxed and hydrated and endoperoxidase activity was eliminated with 3% H2O2. After the blockade, the slide was incubated for 30 min with the primary antibody and was treated with HRP‐labeled secondary antibody (Perkin Elmer, MA, USA) for 10min. The staining process was repeated sequentially for each primary antibody. After the final staining round, 1 × 4′,6‐diamidino‐2‐phenylindole (DAPI; PerkinElmer) working solution was added dropwise on the glass slide, incubated for 5 min at room temperature, dipped with 1 × TBST (Sangon Biotech), and cover slipped with anti‐fluorescence quenching mounting medium (Sangon Biotech). MFSD2A, TGFβ1, and CD8 positive cell proportion scores were calculated by HALO v3.3.2541.301 (Indica Lab, Albuquerque, USA).
2.9. Transcriptome analysis of MGC803 cells
The RNA of MFSD2A‐silenced MGC803 cells or control cells was extracted. Strand‐specific libraries were constructed using the Stranded mRNA‐seq (NR602; Vazyme), and sequencing was conducted using the Illumina Novaseq 6000 instruments (San Diego, CA, USA) by Neo‐Biotecnology Co., Ltd (Shanghai, China). The number of transcripts in each sample was calculated based on the number of fragments per kilobase of transcript per million fragments mapped (FPKM). The Cuffnorm software was used to calculate the FPKM value of each sample, and the values were log2 transformed. The DESeq software was used to calculate the differential gene transcripts between different samples. For KEGG pathway analysis, the entire set of transcripts was used as the background list, the differential transcripts were used as the candidate list, and P was calculated.
2.10. Western blotting
Western blotting was performed as previously described [42]. Briefly, the extracted proteins were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The membrane was blocked with 5% non‐fat milk and incubated with primary antibodies for anti‐MFSD2A (1:2000, ab117618, Abcam), anti‐cyclooxygenase 2 (COX2; 1:2000, ab179800, Abcam), and anti‐β‐actin (1:3000, 4967S, Cell Signaling Technology), followed by incubation with anti‐rabbit secondary antibodies (1:5000, 7074S, Cell Signaling Technology). The blots were visualized using enhanced chemiluminescence (ECL) detection reagents (Thermo Fisher Scientific, Waltham, MA, USA).
2.11. Quantitative real‐time polymerase chain reaction (qRT‐PCR)
qRT‐PCR was performed as previously described [43]. Briefly, total RNA was extracted from cells by Trizol (Invitrogen), and the cDNA was generated by reverse transcription using the RevertAid TM First Strand cDNA Synthesis Kit (TOYOBO, Kita‐ku, Osaka, Japan). Primer sequences for MFSD2A, TGFβ1, and COX2 are listed in Supplemental Table S1. qRT‐PCR was conducted using a LightCycler480 II Real‐Time PCR system (Roche, Indianapolis, IN, USA) with the SYBR‐Green‐based method according to the manufacturer's instructions. The PCR reaction consisted of an initial denaturation at 94°C for 10 min, followed by denaturation at 94°C for 30 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 30 seconds in each PCR cycle for a total of 45 cycles. Relative expression levels of MFSD2A, TGFβ1, and COX2 mRNA were analyzed by the 2−△△CT method, following normalization to β‐actin.
2.12. CyTOF analysis
Single‐cell suspensions derived from freshly isolated tumors were prepared by mechanical dissociation and enzymatic digestion with collagenase I (V900891, Vetec, St. Louis, MO, USA), collagenase IV (V900893, Vetec) and DNase I (60852700, Roche, Basel, Switzerland). CD45+ immune cells were separated by magnetic beads (130‐110‐618, Miltenyi). The CyTOF staining panels are detailed in Supplemental Table S2. Surface and intracellular staining cocktail master mixes (Fluidigm, San Francisco, CA, USA) were prepared prior to each experiment. Briefly, 3 × 106 cells per sample were incubated with Cell‐ID Cisplatin (Fluidigm, San francisco, CA, USA) at 0.5 μmol/L for 2 min for viability staining, followed by washing, fixation using 1.6% polyformaldehyde and permeabilized, and incubation with Palladium barcodes (Fluidigm) for 30 min. The samples were subsequently washed and pooled together, resuspended with Fc receptor block (BioLegend) for 10 min and stained with a cocktail of surface staining antibodies for 15 min. They were then washed, fixed and permeabilized for 30 min. The cells were stained with a cocktail of intracellular staining antibodies for 1 h, washed twice with MaxPar Cell Staining Buffer (Fluidigm), and fixed with MaxPar Fix and Perm Buffer (Fluidigm) containing 0.125 μmol/L Cell‐ID Intercalator‐Ir (Fluidigm) for 15 min to stain the nuclei. The samples were acquired on CyTOF2 Helios (Fluidigm) equipped with a SuperSampler fluidics system (Victorian Airships) at an event rate of < 500 events/second. Gating analysis was performed using Cytobank (Mountain View, CA, USA). Data analysis was performed using FlowJo (BD Biosciences, La Jolla, CA, USA).
2.13. Multicolor flow cytometry analysis
Single‐cell suspensions derived from freshly isolated tumors were prepared by mechanical dissociation and enzymatic digestion with collagenase I (V900891, Vetec), collagenase IV (V900893, Vetec) and DNase I (60852700, Roche). CD45+ immune cells were separated by magnetic beads (130‐110‐618, Miltenyi). Cell suspensions were stained using fluorochrome‐conjugated antibodies specific for mouse CD4, CD8, CD11b, CD25, and Gr‐1 (eBioscience, San Diego, CA, USA). The samples were analyzed on a FACS Calibur (BD Bioscience), and the data were analyzed using FlowJo.
2.14. Enzyme‐linked immunosorbent assay (ELISA)
MFSD2A‐overexpressed MGC803 cells or MFSD2A‐silenceing MGC803 cells and control cells were treated with a complete medium containing 20 μmol/L prostaglandin E2 (MedChemExpress) or arachidonic acid (ARA; MedChemExpress), COX2 inhibitor (Ibuprofen, MedChemExpress) or 10 μmol/L lipoprotein‐associated phospholipase A2 (Lp‐PLA2) inhibitor (Darapladib, MedChemExpress) for 48 h. The content of TGFβ1 in the cell culture supernatant was detected using ELISA Kits (Dakewe, Shanghai, China) as described previously [44]. In addition, 1 × 105 MFSD2A‐silenced GC cells were treated with a medium containing Disitertide for 48 h. The level of TGFβ1 in the cell culture supernatant was analyzed according to the manufacturer's protocol. All samples were measured in triplicate, and three independent experiments were performed for each platelet concentrate.
2.15. Metabonomic analysis
MFSD2A‐overexpressed or MFSD2A‐silenced MGC803 cells or tumor tissues from tumor‐bearing mice were washed with pre‐cooled PBS. A total of 1 × 107 of tumor cells or 50 mg tumor tissue are rapidly frozen in liquid nitrogen for 10 min. Fatty acid metabolites were detected by MetWare (http://www.metware.cn/) based on the Agilent 8890‐5977B GC‐MS platform.
2.16. Statistical analysis
The data in this study are shown as the mean ± standard error of the mean (SEM) of three independent experiments. Differences between groups were assessed by paired t‐test or one‐way analysis of variance (ANOVA) using the GraphPad Prism v8.0 software (Graphpad Software, La Jolla, CA, USA). The CyTOF data were analyzed by the Wilcoxon rank‐sum test. Statistical significance was indicated as * P < 0.05, ** P < 0.01, and *** P < 0.001. For mouse survival analysis, Kaplan‐Meier survival curves were generated and analyzed using the GraphPad Prism v8.0 software.
3. RESULTS
3.1. Higher MFSD2A expression in GC correlates with better clinical response to anti‐PD‐1 immunotherapy
Our clinical cohort comprising 27 patients was divided into 3 groups according to their best overall response (progressive disease [PD], stable disease [SD], or partial response [PR]) to toripalimab. The baseline patient demographics, clinical characteristics, and evaluable response are presented in Table 1. All of them had advanced‐stage disease, and the majority were heavily pretreated, with 20 out of 27 (74.1%) patients having at least 2 prior lines of systemic treatment. 5 out of 27 (18.5%) patients had positive PD‐L1 expression, and 8 out of 27 (29.6%) patients had high TMB, whereas only 1 (3.7%) patient had high microsatellite instability (MSI‐H) GC. 7 (25.9%) patients obtained PR from anti‐PD‐1 immunotherapy, while 9 (33.3%) were primarily resistant to PD‐1 antibodies as they demonstrated PD at first evaluation. Therefore, it was of great interest to compare the transcriptomic profile of PR and PD tumors to determine the underlying molecular mechanism associated with anti‐PD‐1 immunotherapy sensitivity in GC.
TABLE 1.
Baseline demographics and clinical characteristics of 27 GC patients.
| Characteristic | Case (%) |
|---|---|
| Age | |
| ≥ 65 years | 6 (22.2) |
| < 65 years | 21 (77.8) |
| Sex | |
| Male | 21 (77.8) |
| Female | 6 (22.2) |
| MSI status | |
| MSS/MSI‐L | 26 (96.3) |
| MSI‐H | 1 (3.7) |
| PD‐L1 status | |
| Negative | 21 (77.8) |
| Positive | 5 (18.5) |
| N/A | 1 (3.7) |
| EBV status | |
| Negative | 16 (59.3) |
| Positive | 3 (11.1) |
| N/A | 8 (29.6) |
| Lauren classification | |
| Diffuse | 1 (3.7) |
| Intestinal | 4 (14.8) |
| Mixed | 3 (11.1) |
| N/A | 19 (70.4) |
| Liver metastasis | |
| No | 19 (70.4) |
| Yes | 8 (29.6) |
| ECOG performance status | |
| 0 | 12 (44.4) |
| 1 | 15 (55.6) |
| Baseline LDH | |
| Normal | 19 (70.4) |
| Abnormal | 6 (22.2) |
| N/A | 2 (7.4) |
| Tumor burden a | |
| ≤ 100 mm | 18 (66.7) |
| > 100 mm | 9 (33.3) |
| Prior Lines of treatment | |
| 1 | 7 (25.9) |
| 2 | 8 (29.6) |
| ≥ 3 | 12 (44.4) |
| TMB status | |
| ≥ 12 Muts/Mb | 8 (29.6) |
| < 12 Muts/Mb | 19 (70.4) |
| Best Response | |
| PR | 7 (25.9) |
| SD | 11 (40.7) |
| PD | 9 (33.3) |
Indicated as the sum of the longest diameter of the target lesions at baseline.
Abbreviations: MSI, microsatellite instability; PD‐L1, programmed death‐ligand 1; EBV, Epstein‐Barr virus; ECOG, electrocorticography; LDH, lactate dehydrogenase; TMB, tumor mutation burden; PD, progressive disease; SD, stable disease; PR, partial response; GC, gastric cancer; N/A, not available.
By performing KEGG pathway analysis of the differentially up‐regulated genes of AGC tissues from patients before receiving toripalimab between the PR group and PD group, we found that these genes were significantly enriched in certain metabolic pathways (Supplemental Figure S1A), implying the involvement of metabolic molecules in regulating the efficacy of anti‐PD‐1 immunotherapy. Among those metabolic‐related genes, the expression of MFSD2A was the most significantly increased in AGC patients of the PR group compared to the PD group (Supplemental Figure S1B), suggesting that MFSD2A might be a potential predictor for anti‐PD‐1 immunotherapy response.
To clarify whether MFSD2A expression was associated with GC progression without anti‐PD‐1 immunotherapy, we detected MFSD2A by performing IHC on 149 samples of GC and adjacent normal tissues. The results showed that the expression level of MFSD2A was significantly lower in GC tissues than in adjacent normal tissues (Supplemental Figure S2A‐B). Specifically, 29.3% (22/75) of GC samples and 40.5% (30/74) of adjacent normal tissue samples were positive for MFSD2A staining. Moreover, we found that MFSD2A expression was inversely correlated with GC stage (Supplemental Figure S2C‐D). Altogether, these results suggested that the low MFSD2A expression in GC tissues might be related to the immunosuppressive condition in the TME, which is conducive to tumor progression.
3.2. MFSD2A enhances the anti‐PD‐1 efficacy in GC‐bearing mice
A syngeneic mouse GC model was used to evaluate the effect of MFSD2A on the efficacy of anti‐PD‐1 immunotherapy. After subcutaneously injecting MFSD2A‐overexpressed or control MFC cells (Supplemental Figure S3A) into syngeneic 615 mice for 9 days, the mice were treated with an anti‐PD‐1 antibody or a control IgG. The overexpression of MFSD2A was confirmed by IHC (Figure 1A). We observed that anti‐PD‐1 immunotherapy did not significantly inhibit the growth of control tumors. Comparatively, MFSD2A overexpression significantly inhibited tumor growth, which, importantly, enhanced the efficacy of anti‐PD‐1 immunotherapy (Figure 1B‐C). Moreover, anti‐PD‐1 immunotherapy alone, MFSD2A overexpression alone, and MFSD2A overexpression in combination with anti‐PD‐1 immunotherapy significantly prolonged the survival of mice compared to mice inoculated with control GC cells and treated with control IgG (Figure 1D). Altogether, these findings suggested that MFSD2A could enhance the efficacy of anti‐PD‐1 immunotherapy for GC.
FIGURE 1.
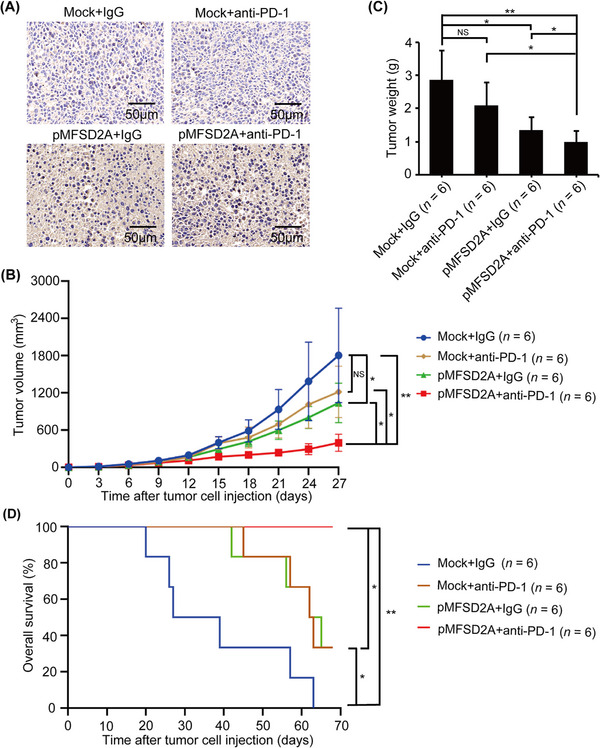
MFSD2A enhances the efficacy of anti‐PD‐1 immunotherapy in GC‐bearing mice. MFSD2A‐overexpressed MFC cells or control cells (5 × 105 cells in 100 μL PBS) were injected subcutaneously into 615 mice. When the tumor grew to 100 mm3, the mice were injected intraperitoneally with PD‐1 antibody or control IgG every 3 days. (A) The expression of MFSD2A in indicated tumor tissues was analyzed by IHC. (B) Growth curves of subcutaneously transplanted tumors in indicated groups of mice. (C) The weight of tumors in indicated groups was calculated and analyzed on the 27th day. (D) The overall survival of tumor‐bearing mice in indicated groups. The mice were considered "dead" when the tumor grew to 2000 mm3 or natural death. All values are presented as the mean ± SEM. * P < 0.05; ** P < 0.01; NS, not significant. Abbreviations: MFSD2A, Major Facilitator Superfamily Domain Containing 2A; PD‐1, programmed cell death protein 1; IHC, immunohistochemistry; GC, gastric cancer; SEM, standard error of the mean.
3.3. MFSD2A overexpression in GC cells reprograms TME to be immunogenic
To understand how MFSD2A enhanced the efficacy of anti‐PD‐1 immunotherapy, we used CyTOF to profile tumor‐infiltrating immune cell populations in the TME of the above tumors using antibodies in the myeloid panel and the lymphoid panel against various lineage and functional markers of immune cells (Supplemental Table S2). Unsupervised clustering analysis of CD45+ immune cells from tumors using the R software identified 25 major immune cell populations, or clusters, using the myeloid panel (Figure 2A, Supplemental Figure S3B). We determined the number of cell subsets in the TME according to the expression of different markers (Figure 2B), including T cells (CD3+ cells), B cells (CD19+ and B220+ cells), natural killer cells (NK; NKp46+ cells), dendritic cells (DC; CD11c+ and MHCII+ cells), macrophage (CD11b+ and F4/80+ cells), MDSC (CD11b+ and Ly6C+ cells), neutrophil cells (CD11b+ and Ly6G+ cells), and others (CD44+ and CD103+ cells). We evaluated the correlation between MFSD2A level and the percentage of tumor‐infiltrating immune cell populations. The results showed the T cell population was increased whereas the TAM population was decreased in MFSD2A overexpression alone or combination with anti‐PD‐1 immunotherapy TME compared with control TME. However, the other cell populations were not significantly affected by MFSD2A overexpression (Figure 2C). We further classified immune cells according to their expression of various markers (Figure 2B, Supplemental Figure S3C). T cells in cluster 22 (CD3+, CD8+, Ly6C+, and CD86+ cells) were significantly enriched whereas T cells in cluster 1 (CD3+, CD8+, and Ly6C‐ cells), M2‐like macrophages (cluster 17) (CD11b+, F4/80+, CD206+, and CD163+ cells), and CD11b+, F4/80+, and PD‐L1+ macrophages (cluster 4) were significantly reduced in MFSD2A‐overexpressed tumor samples, alone or in combination with anti‐PD‐1 immunotherapy (Figure 2D). To further elucidate the reprogramming of T cells, we analyzed CyTOF data from the lymphoid panel, and identified and extracted lymphoid cells (T cells, NK cells, and B cells) for further clustering (Figure 2E). The clusters were annotated using specific markers (Figure 2F). Exhausted CD8+ T cells, including cluster 1 (CD3+, CD8+, PD‐1+, TIM3+, and GZMBlo cells) and cluster 7 (CD3+, CD8+, PD‐1+, and TIM3+ cells), were significantly reduced in MFSD2A‐overexpressed samples, without or with anti‐PD‐1 immunotherapy tumor (Figure 2G). Effector CD8+ T cells (cluster 9) with high Perforin and Granzyme B levels and relatively low PD‐1 and TIM3 levels were increased after MFSD2A overexpression, without or with anti‐PD‐1 immunotherapy (Figure 2G, Supplemental Figure S4). The increase in CD8+ T cells population in MFSD2A‐overexpressed tumor samples was also confirmed by multicolor FACS (Supplemental Figure S5A) and IHC (Supplemental Figure S5B) analyses.
FIGURE 2.
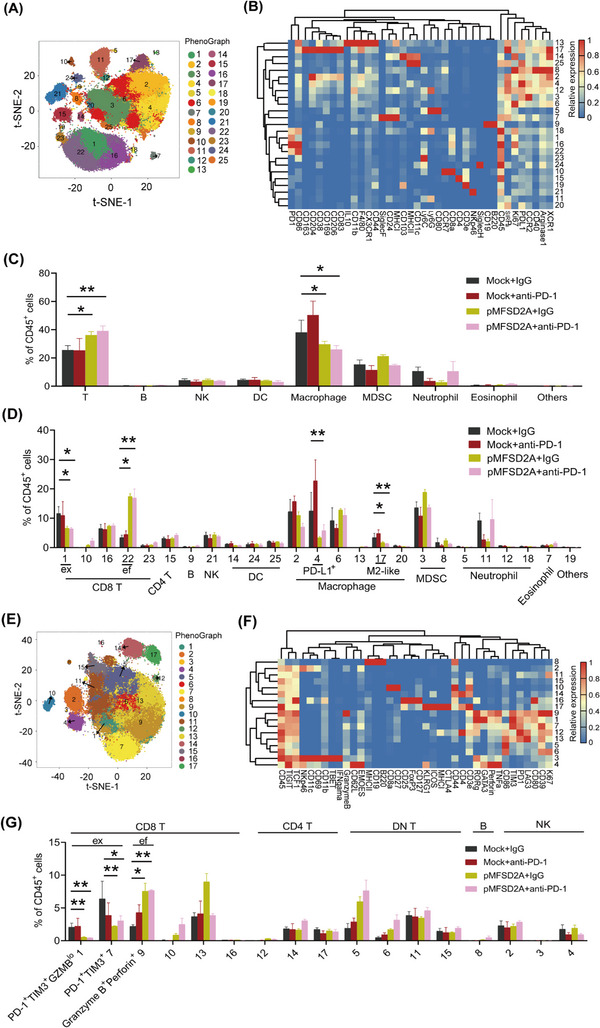
MFSD2A reprograms TME in GC‐bearing mice. (A‐D) Immune single‐cell suspensions of tumor samples from MFSD2A‐overexpressed MFC cells‐bearing mice treated with an anti‐PD‐1 antibody or control IgG were analyzed by CyTOF (n = 3 each study group). Cell populations defined by the manual gating strategy were projected onto t‐SNE space and assigned specific colors (A). Heatmap showing differential marker expression in CD45+ TIL clusters identified by analysis of CyTOF data (B). The frequency of T cells, B cells, NK cells, DC, macrophages, MDSCs, and other cells in total CD45+ TILs (C). Proportion of different cell subsets in total CD45+ cells based on differential marker expression shown in (D). (E‐G) Analysis of T cell, B cell, and NK cell subsets by lymphocyte panel. The lymphocyte population defined by the manual gating strategy is projected into the t‐SNE space and assigned specific colors (E). Expression of markers of different cell subsets (F). Proportion of CD8+ T cells including exhausted T cells cluster 1 (PD‐1+ cells, TIM3+ cells, and GZMBlo cells) and cluster 7 (PD‐1+ cells and TIM3+ cells), and effector T cells (cluster 9; Perforin+ cells and GranzymeB+ cells) were analyzed (G). All values are presented as the mean ± SEM. * P < 0.05 and ** P < 0.01. Abbreviations: tSNE, t‐Distributed Stochastic Neighbor Embedding; MFSD2A, Major Facilitator Superfamily Domain Containing 2A; PD‐1, programmed cell death protein 1; IL10, interleukin 10; C‐X3‐C motif chemokine receptor 1; SiglecF, sialic acid binding Ig‐like lectin F; SiglecH, sialic acid binding Ig‐like lectin H; MHCI, major histocompatibility complex‐1; MHCII, major histocompatibility complex‐1; Ly6C, lymphocyte antigen 6 complex, locus C; Ly6G, lymphocyte antigen 6 complex, locus G; CCR7, C‐C motif chemokine receptor 7; SIRPa, signal regulatory protein alpha; PD‐L1, programmed death‐ligand 1; CCR2, C‐C motif chemokine receptor 2; XCR1, X‐C motif chemokine receptor 1; MDSC, Myeloid‐derived suppressor cells; CyTOF, Cytometry by Time‐of‐Flight; CD8 T, CD8+ T cells; CD4 T, CD4+ T cells; DC, dendritic cells; MDSC, myeloid‐derived suppressor cells; GZMB, granzyme B; TIM3, T cell immunoglobulin and mucin domain 3; TME, tumor microenvironment; GC, gastric cancer.
As a major population of anti‐tumor immunity, T cells in the TME play a crucial role in host responsiveness to anti‐PD‐1 immunotherapy [45, 46]. To further clarify whether T cells was involved in MFSD2A‐mediated anti‐tumor immunity, we treated MFSD2A‐overexpressed MFC cells‐bearing mice with CD8α antibody and found that depletion of CD8+ T cells almost abolished MFSD2A‐mediated anti‐tumor effects (Figure 3A‐B), suggesting that the anti‐tumor effect of MFSD2A was dependent on CD8+ T cells. To further investigate the possible function of MFSD2A in T cell activation, MFC cells were co‐cultured with CD8+ T cells from MFSD2A‐overexpressed or control MFC tumor samples. The results showed that the levels of granzyme B and IFNγ were increased in CD8+ T cells when MFSD2A was overexpressed (Figure 3C‐D). Similar result was also observed when CD8+ T cells from human PBMCs were incubated with MFSD2A‐overexpressed MGC803 cells (Figure 3E‐F, Supplemental Figure S6A). In contrast, the levels of granzyme B and IFNγ of CD8+ T cells from human PBMCs were decreased when incubated with MFSD2A‐silenced MGC803 cells (Figure 3G‐H, Supplemental Figure S6B). Moreover, the cytotoxicity of CD8+ T cells from MFSD2A‐overexpressed tumors or human PBMCs was enhanced when incubated with MFSD2A‐overexpressed GC cells (Figure 3I‐J) and was reduced when incubated with MFSD2A‐silenced MGC803 cells (Figure 3K). These results suggested that MFSD2A could reprogram the TME of GC and promote T cell activation to enhance the therapeutic effect of anti‐PD‐1 immunotherapy.
FIGURE 3.
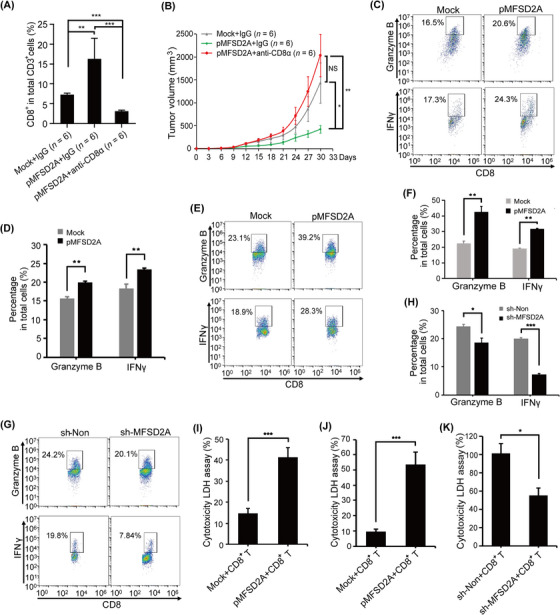
MFSD2A expression in GC cells enhances CD8+ T cell activation. (A) Flow cytometry analysis of the proportion of tumor infiltrating CD8+ T cells in tumor tissue from MFSD2A‐overexpressed MFC cells bearing 615 mice or control mice administrated with CD8α antibody or control IgG. (B) Depletion of CD8+ T cells by CD8α antibody rescued the inhibitory effect of MFSD2A overexpression on GC growth. (C‐D) Tumor‐infiltrating CD8+ T cells were co‐cultured with MFSD2A‐overexpressed or control MFC cells for 24 h, and the expression of Granzyme B and IFNγ produced by T cells was analyzed by flow cytometry. (E‐H) CD8+ T cells from human PBMCs were co‐cultured with MFSD2A‐overexpressed or control MGC803 cells (E and F), MFSD2A‐silenced MGC803 cells or control cells (G and H) for 24 h. The expression of granzyme B and IFNγ produced by T cells was analyzed by flow cytometry. (I‐K) The cytotoxicity of CD8+ T cells from MFSD2A‐overexpressed MFC tumor (I) and of PBMCs (J and K) after co‐cultured with MFSD2A‐overexpressed MFC or MGC803 cells or control cells (I and J) or with MFSD2A‐silenced MGC803 cells or control MGC803 cells (K) for 24 h was assayed by LDH analysis. All values are presented as the mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, NS, not significant. Abbreviations: MFSD2A, Major Facilitator Superfamily Domain Containing 2A; IFNγ, Interferon‐gamma; LDH, lactate dehydrogenase; PBMCs, peripheral blood mononuclear cells; GC, gastric cancer; SEM, standard error of the mean.
3.4. MFSD2A suppresses GC cell production of TGFβ1 to enhance CD8+ T cell activation
Transcriptome analysis showed that the expression of TGFβ1 was significantly increased while the expressions of other immunosuppressive factors, such as PD‐L1, PVR cell adhesion molecule (PVR), and IL10, were not significantly changed in MFSD2A‐silenced MGC803 cells (Figure 4A, Supplemental Figure S6C‐D). These results were confirmed by qRT‐PCR in MFSD2A‐silenced MGC803 cells (Figure 4B). Consistently, the secretion of TGFβ1 was increased in MFSD2A‐silenced MGC803 cells (Figure 4C) but decreased in MFSD2A‐overexpressed MGC803 cells (Figure 4D) and MFC cells (Figure 4E), and tumor tissue from MFSD2A‐overexpressed MFC cells bearing mice (Figure 4F‐G).
FIGURE 4.
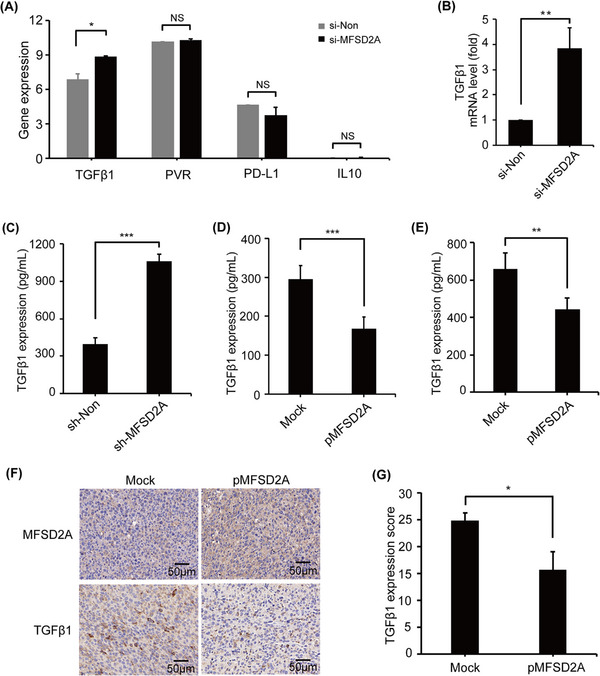
MFSD2A inhibits TGFβ1 release from GC cells. (A) Expressions of TGFβ1, PVR, PD‐L1, and IL10 genes were analyzed in MFSD2A‐silenced or control MGC803 cells by transcriptome sequencing. (B) TGFβ1 mRNA expression in MFSD2A‐silenced or control MGC803 cells was examined by qRT‐PCR. (C‐E) TGFβ1 protein levels in the supernatant of MFSD2A‐silenced or control MGC803 cells (C), MFSD2A‐overexpressed MGC803 cells (D), or MFSD2A‐overexpressed MFC cells (E) was examined by ELISA. (F‐G) The expressions of MFSD2A and TGFβ1 in tumor tissues derived from MFSD2A‐overexpressed MFC cells‐bearing mice or mock control mice were analyzed by IHC. All values are presented as the mean ± SEM. * P < 0.05, ** P < 0.01, and *** P < 0.001, NS, not significant. Abbreviations: GC, gastric cancer; MFSD2A, Major Facilitator Superfamily Domain Containing 2A; TGFβ1, transforming growth factor‐beta 1; qRT‐PCR, quantitative real‐time polymerase chain reaction; ELISA, enzyme‐linked immunosorbent assay; PVR, PVR cell adhesion molecule; PCD‐L1, Programmed cell death ligand 1; IL10, interleukin 10; IHC, immunohistochemistry; SEM, standard error of the mean.
Further experiments indicated that the blockade of TGFβ1 signaling with Disitertide, a TGFβ1‐specific blocker, effectively rescued MFSD2A deficiency‐induced inhibitory effect on CD8+ T cells (Figure 5A‐B). In addition, IHC analysis showed that TGFβ1 expression was inversely correlated with MFSD2A expression and CD8+ T cell infiltration in GC tissues (Figure 5C‐D). Together, our findings suggested that MFSD2A in GC cells enhanced CD8+ T cell activation by suppressing the production of TGFβ1.
FIGURE 5.
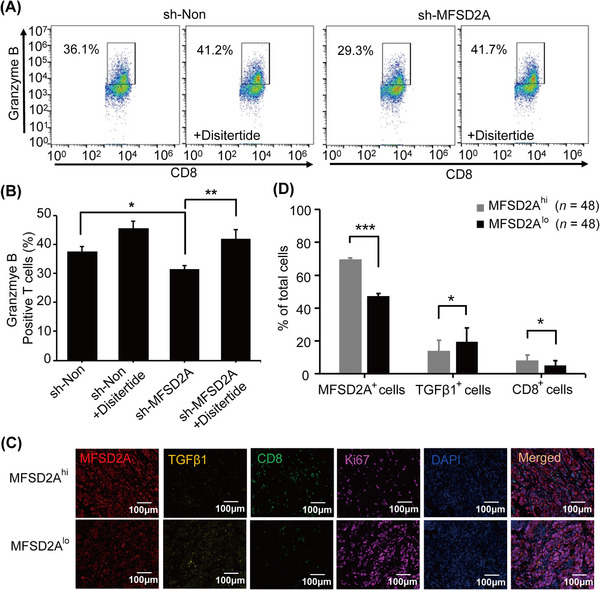
MFSD2A promotes CD8+ T cell activation by inhibiting GC cell expression of TGFβ1. (A‐B) The expression of granzyme B in CD8+ T cells co‐cultured with MFSD2A‐silenced or control MGC803 cells pretreated with Disitertide (20 μmol/L) analyzed by flow cytometry. (C‐D) mIHC analysis of the distribution (C) and proportion (D) of MFSD2A (red), TGFβ1 (yellow), and CD8 (green) in MFSD2Ahi and MFSD2Alo GC tissues (samples with MFSD2A expression above the median are considered high expression groups, while samples with MFSD2A expression below the median are considered low expression groups). Ki67 (purple) was used to label tumor cells, and DAPI (blue) was used to locate cells. All values are presented as mean ± SEM. * P < 0.05, ** P < 0.01, and *** P < 0.001. Abbreviations: GC, gastric cancer; MFSD2A, Major Facilitator Superfamily Domain Containing 2A; TGFβ1, transforming growth factor‐beta 1; DAPI, 4',6‐diamidino‐2‐phenylindole; mIHC, multiplex immunohistochemistry; SEM, standard error of the mean.
3.5. MFSD2A restricts COX2‐prostaglandin‐mediated TGFβ1 production in GC cells
Considering that MFSD2A plays an important role in intracellular lipid metabolism as a membrane protein that transports long‐chain fatty acids [28], we analyzed the changes in lipid metabolites in MFSD2A‐overexpressed and MFSD2A‐silenced MGC803 cells. The results showed that the synthesis of prostaglandins in MGC803 cells, including prostaglandin E2 (PGE2), prostaglandin B2 (PGB2), and prostaglandin J2 (PGJ2), was significantly inhibited when MFSD2A was overexpressed (Figure 6A‐B) and was enhanced when MFSD2A was silenced (Figure 6C‐D). Similarly, prostaglandins level was significantly reduced in MFSD2A‐overexpressed tumor tissues (Figure 6E‐F), indicating that MFSD2A inhibited prostaglandins synthesis. By analyzing transcriptional data, we further found that COX2, a prostaglandin synthesis rate‐limiting enzyme, was increased in MFSD2A‐silenced MGC803 cells (Supplemental Figure S7A). MFSD2A silencing up‐regulated the expression of COX2 in MGC803 cells, while MFSD2A overexpression down‐regulated the expression of COX2 in MGC803 cells and MFC cells (Figure 6G‐H, Supplemental Figure S7B‐D), suggesting that MFSD2A might function as the suppressor of COX2‐prostaglandin expression. Moreover, we found the inhibitory effect on CD8+ T cell activation when co‐culturing with MFSD2A‐silenced MGC803 cells was largely rescued by pretreatment with celecoxib, an inhibitor of COX2 (Figure 6I‐J). Inhibition of COX2 with another COX2 inhibitor, Ibuprofen, largely abolished MFSD2A silencing‐induced TGFβ1 production. Similar results were also observed when MFSD2A‐silenced MGC803 cells were treated with Darapladib, an inhibitor of Lp‐PLA2, which could inhibit the synthesis of ARA and its metabolite prostaglandins (Figure 6K). In addition, when MFSD2A‐overexpressed MGC803 cells were pretreated with PGE2 or its upstream metabolite ARA, the reduction of TGFβ1 release by MFSD2A overexpression was largely eliminated (Figure 6L). Therefore, these findings demonstrated that MFSD2A restricts COX2‐prostaglandin‐mediated TGFβ1 production and promotes T cell activation (Figure 7).
FIGURE 6.
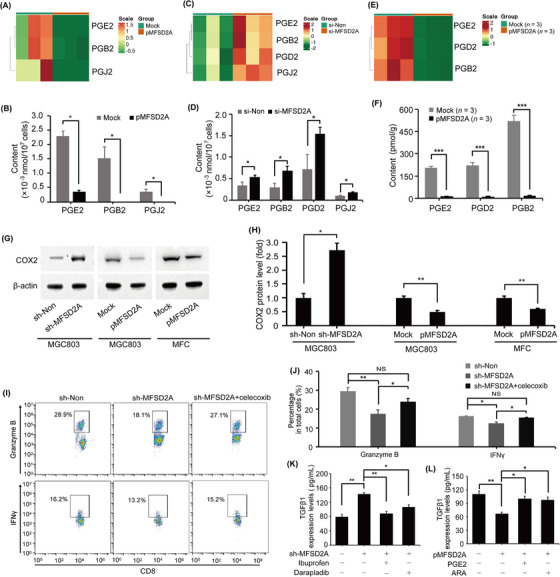
MFSD2A promotes T cell activation by restricting COX2‐prostaglandin‐TGFβ1 axis. (A‐D) Heatmaps and quantification of indicated prostaglandins in MFSD2A‐overexpressed (A and B) and MFSD2A‐silenced (C and D) MGC803 cells compared with control cells analyzed by lipid metabolomics. (E‐F) Heatmaps (E) and quantification of indicated prostaglandins (F) from MFSD2A‐overexpressed or control MFC tumor tissues analyzed by lipid metabolomics. (G‐H) COX2 expression in MFSD2A‐silenced MGC803 cells, MFSD2A‐overexpressed MGC803 cells, MFSD2A‐overexpressed MFC cells and control cells was detected by Western blotting. (I‐J) Flow cytometry analysis of the expression of granzyme B and IFNγ in CD8+ T cells after co‐cultured with MFSD2A‐silenced or control MGC803 cells pretreated with celecoxib (50 μmol/L). (K) TGFβ1 production was examined by ELISA in MFSD2A‐silenced MGC803 cells pretreated with COX2 inhibitor ibuprofen or Lp‐PLA2 inhibitor darapladib. (L) TGFβ1 production was examined by ELISA in MFSD2A‐overexpressed MGC803 cells pretreated with prostaglandin E2 or arachidonic acid. All values are presented as the mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviations: MFSD2A, Major Facilitator Superfamily Domain Containing 2A; TGFβ1, transforming growth factor‐beta 1; COX2, cyclooxygenase 2; Lp‐PLA2, lipoprotein‐associated phospholipase A2; ELISA, enzyme‐linked immunosorbent assay; PGE2, prostaglandin E2; ARA, arachidonic acid; SEM, standard error of the mean.
FIGURE 7.
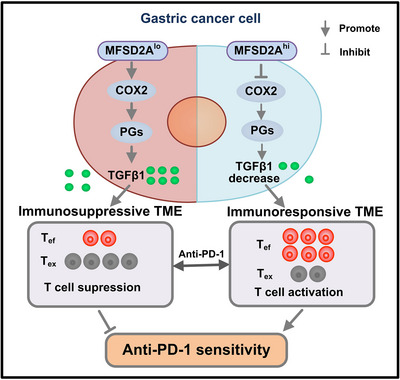
Working model depicting the mechanism of MFSD2A for enhancing anti‐PD‐1 therapeutic efficacy in GC. Abbreviations: MFSD2A, Major Facilitator Superfamily Domain Containing 2A low expression; COX2, cyclooxygenase 2; PGs, prostaglandin; TGFβ1, transforming growth factor‐beta 1; TME, tumor microenvironment; PD‐1, programmed cell death protein 1; GC, gastric cancer; Tef, effector T cell; Tex, exhausted T cell.
4. DISCUSSION
ICB targeting PD‐1 or PD‐L1 has achieved remarkable clinical benefits against various cancers [47, 48]. However, due to tumor heterogeneity, the low response rate of PD‐1 blockade remains a major challenge [49]. Therefore, it is important to predict a patient's response to PD‐1 inhibition to optimize treatment outcomes. In addition to the PD‐L1 expression level [50], DNA MMR capacity [51], TMB [52], immune cell infiltration [53] and an increasing number of molecules have been reported to correlate with the efficacy of anti‐PD‐1 immunotherapy. For instance, metastatic uterine leiomyosarcoma with phosphatase and tensin homolog (PTEN) loss responds poorly to anti‐PD‐1 immunotherapy compared to those with normal PTEN expression, suggesting a correlation between PTEN and tumor responsiveness to PD‐1 blockade [54]. Additionally, integrin beta 2 (ITGB2) was reported as a prognostic indicator for patients with glioma and a predictive biomarker for those treated with a PD‐1 inhibitor because patients with elevated ITGB2 expression in gliomas barely responded to ICB immunotherapy [55]. Moreover, a recent study showed that small‐cell lung cancer patients whose tumors inactivated the Notch signaling pathway had reduced clinical response to PD‐1 inhibitors [56]. Similarly, our previous study showed that mutations in the mucin 4, cell surface associated (MUC4), mucin 16, cell surface associated (MUC16), and titin (TTN) genes in tumors could predict TMB and serve as a more economical and convenient immunotherapy biomarker [57]. Here, we reported that MFSD2A could be a potential predictor of anti‐PD‐1 treatment response in AGC patients as it was related to enhancing the efficacy of anti‐PD‐1 immunotherapy. However, the benefit of MFSD2A alone or in combination with other predictors should be further analyzed. Besides, whether MFSD2A maintains similar functions in other cancers must be clarified in future studies.
The importance of the TME in regulating cancer progression and immunotherapeutic outcomes has been widely recognized [58, 59, 60]. Reprogramming the immunosuppressive TME state to activate anti‐tumor immune responses was shown to potentiate anti‐PD‐1 immunotherapy efficacy [61]. For instance, knocking down the circular RNA discs large MAGUK scaffold protein 1 (CircDLG1) was reported to increase the number of CD8+ T cells and IFNγ+ cells and decrease the number of MDSCs in mouse TME, which promoted anti‐PD‐1 efficacy [62]. DNA‐methyltransferase inhibitor decitabine enhances the effect of PD‐1 blockade by modulating TME in colorectal cancer [63]. Repression of SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) promotes anti‐PD‐1 immunotherapy by activating CD8+ T cells [64, 65]. In this present study, we found that MFSD2A enhanced anti‐PD‐1 immunotherapy efficacy by inhibiting TGFβ1 secretion from GC cells, resulting in the activation of CD8+ T and the reduction of immunosuppressive cells in the TME. TGFβ1 is a well‐accepted major immunoinhibitory cytokine in the TME and can impair the viability of multiple cytotoxic immune cells, including CD4+ T cells, CD8+ T cells, and NK cells [66, 67]. An excessive TGFβ1 production in the TME can suppress anti‐tumor immune responses, leading to the resistance of tumor cells to immune checkpoint inhibitors. Blockade of TGFβ1 production can significantly promote the efficacy of anti‐PD‐1 immunotherapy in colon cancer [68]. TGFβ1 in the TME is mainly derived from tumor cells and inhibitory immune cells, such as MDSC and CD4+ Tregs [69]. The production of TGFβ1 is controlled by multiple modulators, such as epigenetic modification, LncRNA and high glucose intake [70, 71, 72]. Here, we found that MFSD2A inhibited the COX2‐prostaglandin‐mediated TGFβ1 secretion by GC cells into the TME to enhance anti‐tumor immune responses to GC cells, suggesting a new regulatory mechanism of TGFβ1 signaling.
A previous study showed that MFSD2A suppresses tumor progression by promoting the transportation of docosahexaenoic acid and lipid metabolism [32]. Here, we found that prostaglandin anabolism was highly correlated with MFSD2A expression level in GC cells. Additionally, it has been reported that PGE2 inhibited the survival and function of TIL to attenuate the response to anti‐PD‐1 immunotherapy [73] and that inhibition of COX2‐mediated PGE2 release enhanced the effect of PD‐1 blockade [74]. Our study showed that the MFSD2A‐enhanced efficacy of the anti‐PD‐1 immunotherapy was associated with limiting COX2‐prostaglandin synthesis. Previous studies showed that PGE2 suppressed TGFβ1 signaling by acting on prostaglandin E receptor 2 (EP2) and prostaglandin E receptor 4 (EP4), induced tolerance characteristics in DCs [75] and mediated the effects of TGFβ on the migration and invasion of prostate cancer cells by activating the PI3K/AKT/mTOR pathway [76]. Our findings uncovered that MFSD2A‐induced TGFβ1 release inhibition was due to the reduced COX2‐prostaglandin synthesis in the TME. Supplementing PGE2 or its upstream metabolite ARA reversed MFSD2A‐mediated inhibition of TGFβ1 whereas COX2 and Lp‐PLA2 inhibitors abolished MFSD2A deficiency‐induced TGFβ1 production, implying the causal relationship between prostaglandin metabolism and TGFβ1 regulation. However, further investigations are required to fully understand the underlying mechanism.
5. CONCLUSIONS
This study reported that MFSD2A inhibited TGFβ1 production in GC cells by suppressing COX2‐prostaglandin synthesis, promoted CD8+ T cell activation in the TME, and enhanced the efficacy of anti‐PD‐1 immunotherapy in GC patients. Our data unveiled a new predictive marker for anti‐PD‐1 immunotherapy and described a novel mechanism for sensitization to anti‐PD‐1 immunotherapy. However, the underlying mechanism of MFSD2A in regulating the prostaglandin metabolic pathway requires further investigation.
DECLARATIONS
AUTHOR CONTRIBUTIONS
Chun‐Mei Wang, Rui‐Hua Xu, and Xue‐Tao Cao designed and supervised the study. Bin Zhang, Chun‐Mei Wang, Hao‐Xiang Wu, Feng Wang, Bing‐Jing Wang, Zhou Yu, Ye Hu, and Yang‐Yang Chai performed and completed the experiments. Hao‐Xiang Wu and Feng Wang collected and analyzed clinical tissues. Rong‐Hua Xia, Ye Hu, and Yang‐Yang Chai analyzed the data. Bin Zhang, Chun‐Mei Wang, Rui‐Hua Xu, and Xue‐Tao Cao analyzed the data and wrote the manuscript. All authors have approved this manuscript.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
This study was supported by grants from the NCTIB Fund for R&D Platform for Cell and Gene Therapy, the CAMS Innovation Fund for Medical Sciences (No. 2022‐I2M‐2‐004 and No. 2021‐I2M‐1‐074), National Natural Science Foundation of China (No. 82001677, No. 82102921, and No. 82388201), Fundamental Research Funds for the Central Universities (No. 3332021075), and Jiangsu Innovative and Entrepreneurial Talent Programme (No. 2020‐30084).
AVAILABILITY OF DATA AND MATERIALS
The omics data of this study are available from the corresponding author (Xue‐Tao Cao, caoxt@immunol.org) upon reasonable request.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All animal experiments were approved by the Animal Science Center of Suzhou Institute of Systems Medicine (SYXK [Su] 2019‐0029, ISM‐IACUC‐AUP‐202303001). All procedures involving human participants in this study were approved by the Institutional Ethical Boards of Sun Yat‐sen University Cancer Center (A2016‐046‐01). All patients provided written informed consent before enrollment.
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
We thank Qi‐Jiang Cheng ((National Key Laboratory of Immunity and Inflammation, Suzhou Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, Suzhou, China), for technical assistance and Dr. Qian Zhang, Dr. Yan Gu, and Dr. Yan‐Fang Liu(Institute of Immunology, Second Military Medical University, Shanghai, China), for technical assistance and helpful discussions.
Zhang B, Wang C‐M, Wu H‐X, Wang F, Chai Y‐Y, Hu Y, et al. MFSD2A potentiates gastric cancer response to anti‐PD‐1 immunotherapy by reprogramming the tumor microenvironment to activate T cell response. Cancer Commun. 2023;43:1097–1116. 10.1002/cac2.12476
Contributor Information
Chun‐Mei Wang, Email: chunmeiwang@cams.cn.
Rui‐Hua Xu, Email: xurh@sysucc.org.cn.
Xue‐Tao Cao, Email: caoxt@immunol.org.
REFERENCES
- 1. Chowell D, Yoo SK, Valero C, Pastore A, Krishna C, Lee M, et al. Improved prediction of immune checkpoint blockade efficacy across multiple cancer types. Nat Biotechnol. 2022;40(4):499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kirtane K, Elmariah H, Chung CH, Abate‐Daga D. Adoptive cellular therapy in solid tumor malignancies: review of the literature and challenges ahead. J Immunother Cancer. 2021;9(7):e002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu J, Fu M, Wang M, Wan D, Wei Y, Wei X. Cancer vaccines as promising immuno‐therapeutics: platforms and current progress. J Hematol Oncol. 2022;15(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lou H, Cao X. Antibody variable region engineering for improving cancer immunotherapy. Cancer Commun (Lond). 2022;42(9):804–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD‐1 Blockade in Tumors with Mismatch‐Repair Deficiency. N Engl J Med. 2015;372(26):2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang L, Mai W, Jiang W, Geng Q. Sintilimab: A Promising Anti‐Tumor PD‐1 Antibody. Front Oncol. 2020;10:594558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cho J, Kang SY, Kim KM. MMR protein immunohistochemistry and microsatellite instability in gastric cancers. Pathology. 2019;51(1):110–3. [DOI] [PubMed] [Google Scholar]
- 8. Li W, Zhou K, Li M, Hu Q, Wei W, Liu L, et al. Identification of SCN7A as the key gene associated with tumor mutation burden in gastric cancer. BMC Gastroenterol. 2022;22(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patel SP, Kurzrock R. PD‐L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther. 2015;14(4):847–56. [DOI] [PubMed] [Google Scholar]
- 10. Valero C, Lee M, Hoen D, Zehir A, Berger MF, Seshan VE, et al. Response Rates to Anti‐PD‐1 Immunotherapy in Microsatellite‐Stable Solid Tumors With 10 or More Mutations per Megabase. JAMA Oncol. 2021;7(5):739–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sahin IH, Akce M, Alese O, Shaib W, Lesinski GB, El‐Rayes B, et al. Immune checkpoint inhibitors for the treatment of MSI‐H/MMR‐D colorectal cancer and a perspective on resistance mechanisms. Br J Cancer. 2019;121(10):809–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou X, Yao Z, Bai H, Duan J, Wang Z, Wang X, et al. Treatment‐related adverse events of PD‐1 and PD‐L1 inhibitor‐based combination therapies in clinical trials: a systematic review and meta‐analysis. Lancet Oncol. 2021;22(9):1265–74. [DOI] [PubMed] [Google Scholar]
- 13. Reck M, Rodriguez‐Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD‐L1‐Positive Non‐Small‐Cell Lung Cancer. N Engl J Med. 2016;375(19):1823–33. [DOI] [PubMed] [Google Scholar]
- 14. Seidlitz T, Merker SR, Rothe A, Zakrzewski F, von Neubeck C, Grutzmann K, et al. Human gastric cancer modelling using organoids. Gut. 2019;68(2):207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim J, Kim B, Kang SY, Heo YJ, Park SH, Kim ST, et al. Tumor Mutational Burden Determined by Panel Sequencing Predicts Survival After Immunotherapy in Patients With Advanced Gastric Cancer. Front Oncol. 2020;10:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zeng D, Wu J, Luo H, Li Y, Xiao J, Peng J, et al. Tumor microenvironment evaluation promotes precise checkpoint immunotherapy of advanced gastric cancer. J Immunother Cancer. 2021;9(8):e002467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun YT, Guan WL, Zhao Q, Wang DS, Lu SX, He CY, et al. PD‐1 antibody camrelizumab for Epstein‐Barr virus‐positive metastatic gastric cancer: a single‐arm, open‐label, phase 2 trial. Am J Cancer Res. 2021;11(10):5006–15. [PMC free article] [PubMed] [Google Scholar]
- 18. Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE‐059 Trial. JAMA Oncol. 2018;4(5):e180013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, et al. Nivolumab in patients with advanced gastric or gastro‐oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO‐4538‐12, ATTRACTION‐2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;390(10111):2461–71. [DOI] [PubMed] [Google Scholar]
- 20. Granier C, De Guillebon E, Blanc C, Roussel H, Badoual C, Colin E, et al. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open. 2017;2(2):e000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meyer C, Cagnon L, Costa‐Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63(3):247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019;79(18):4557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xue G, Wang Z, Zheng N, Fang J, Mao C, Li X, et al. Elimination of acquired resistance to PD‐1 blockade via the concurrent depletion of tumour cells and immunosuppressive cells. Nat Biomed Eng. 2021;5(11):1306–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang Z, Hsu JL, Li Y, Hortobagyi GN, Hung MC. Cancer Cell Metabolism Bolsters Immunotherapy Resistance by Promoting an Immunosuppressive Tumor Microenvironment. Front Oncol. 2020;10:1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shi Q, Shen Q, Liu Y, Shi Y, Huang W, Wang X, et al. Increased glucose metabolism in TAMs fuels O‐GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer Cell. 2022;40(10):1207–22 e10. [DOI] [PubMed] [Google Scholar]
- 26. Najjar YG, Menk AV, Sander C, Rao U, Karunamurthy A, Bhatia R, et al. Tumor cell oxidative metabolism as a barrier to PD‐1 blockade immunotherapy in melanoma. JCI Insight. 2019;4(5):e124989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Augustin RC, Delgoffe GM, Najjar YG. Characteristics of the Tumor Microenvironment That Influence Immune Cell Functions: Hypoxia, Oxidative Stress, Metabolic Alterations. Cancers (Basel). 2020;12(12):3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cater RJ, Chua GL, Erramilli SK, Keener JE, Choy BC, Tokarz P, et al. Structural basis of omega‐3 fatty acid transport across the blood‐brain barrier. Nature. 2021;595(7866):315–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berger JH, Charron MJ, Silver DL. Major facilitator superfamily domain‐containing protein 2a (MFSD2A) has roles in body growth, motor function, and lipid metabolism. PLoS One. 2012;7(11):e50629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ben‐Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, et al. Mfsd2a is critical for the formation and function of the blood‐brain barrier. Nature. 2014;509(7501):507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spinola M, Falvella FS, Colombo F, Sullivan JP, Shames DS, Girard L, et al. MFSD2A is a novel lung tumor suppressor gene modulating cell cycle and matrix attachment. Mol Cancer. 2010;9:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tiwary S, Morales JE, Kwiatkowski SC, Lang FF, Rao G, McCarty JH. Metastatic Brain Tumors Disrupt the Blood‐Brain Barrier and Alter Lipid Metabolism by Inhibiting Expression of the Endothelial Cell Fatty Acid Transporter Mfsd2a. Sci Rep. 2018;8(1):8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Piccirillo AR, Hyzny EJ, Beppu LY, Menk AV, Wallace CT, Hawse WF, et al. The Lysophosphatidylcholine Transporter MFSD2A Is Essential for CD8(+) Memory T Cell Maintenance and Secondary Response to Infection. J Immunol. 2019;203(1):117–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang F, Wei XL, Wang FH, Xu N, Shen L, Dai GH, et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo‐refractory gastric cancer treated with toripalimab, a PD‐1 antibody in phase Ib/II clinical trial NCT02915432. Ann Oncol. 2019;30(9):1479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sheng X, Yan X, Chi Z, Si L, Cui C, Tang B, et al. Axitinib in Combination With Toripalimab, a Humanized Immunoglobulin G(4) Monoclonal Antibody Against Programmed Cell Death‐1, in Patients With Metastatic Mucosal Melanoma: An Open‐Label Phase IB Trial. J Clin Oncol. 2019;37(32):2987–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hou J, Zhou Y, Zheng Y, Fan J, Zhou W, Ng IO, et al. Hepatic RIG‐I predicts survival and interferon‐alpha therapeutic response in hepatocellular carcinoma. Cancer Cell. 2014;25(1):49–63. [DOI] [PubMed] [Google Scholar]
- 37. Zheng X, Fang Z, Liu X, Deng S, Zhou P, Wang X, et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J Clin Invest. 2018;128(5):2104‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, et al. Tumor Exosomal RNAs Promote Lung Pre‐metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell. 2016;30(2):243–56. [DOI] [PubMed] [Google Scholar]
- 39. Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, et al. EZH2 inhibition activates a dsRNA‐STING‐interferon stress axis that potentiates response to PD‐1 checkpoint blockade in prostate cancer. Nat Cancer. 2021;2(4):444–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. He W, Zhang H, Han F, Chen X, Lin R, Wang W, et al. CD155T/TIGIT Signaling Regulates CD8(+) T‐cell Metabolism and Promotes Tumor Progression in Human Gastric Cancer. Cancer Res. 2017;77(22):6375–88. [DOI] [PubMed] [Google Scholar]
- 41. Kong LY, Wei J, Sharma AK, Barr J, Abou‐Ghazal MK, Fokt I, et al. A novel phosphorylated STAT3 inhibitor enhances T cell cytotoxicity against melanoma through inhibition of regulatory T cells. Cancer Immunol Immunother. 2009;58(7):1023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yu Z, Li X, Yang M, Huang J, Fang Q, Jia J, et al. TRIM41 is required to innate antiviral response by polyubiquitinating BCL10 and recruiting NEMO. Signal Transduct Target Ther. 2021;6(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Y, Wang P, Zhang Y, Xu J, Li Z, Li Z, et al. Decreased Expression of the Host Long‐Noncoding RNA‐GM Facilitates Viral Escape by Inhibiting the Kinase activity TBK1 via S‐glutathionylation. Immunity. 2020;53(6):1168–81 e7. [DOI] [PubMed] [Google Scholar]
- 44. Xu J, Wang P, Li Z, Li Z, Han D, Wen M, et al. IRF3‐binding lncRNA‐ISIR strengthens interferon production in viral infection and autoinflammation. Cell Rep. 2021;37(5):109926. [DOI] [PubMed] [Google Scholar]
- 45. Kim KH, Kim HK, Kim HD, Kim CG, Lee H, Han JW, et al. PD‐1 blockade‐unresponsive human tumor‐infiltrating CD8(+) T cells are marked by loss of CD28 expression and rescued by IL‐15. Cell Mol Immunol. 2021;18(2):385–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang X, Wang C, Wang J, Hu Q, Langworthy B, Ye Y, et al. PD‐1 Blockade Cellular Vesicles for Cancer Immunotherapy. Adv Mater. 2018;30(22):e1707112. [DOI] [PubMed] [Google Scholar]
- 47. de Streel G, Bertrand C, Chalon N, Lienart S, Bricard O, Lecomte S, et al. Selective inhibition of TGF‐beta1 produced by GARP‐expressing Tregs overcomes resistance to PD‐1/PD‐L1 blockade in cancer. Nat Commun. 2020;11(1):4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shen X, Zhao B. Efficacy of PD‐1 or PD‐L1 inhibitors and PD‐L1 expression status in cancer: meta‐analysis. BMJ. 2018;362:k3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cheng B, Ding K, Chen P, Ji J, Luo T, Guo X, et al. Anti‐PD‐L1/TGF‐betaR fusion protein (SHR‐1701) overcomes disrupted lymphocyte recovery‐induced resistance to PD‐1/PD‐L1 inhibitors in lung cancer. Cancer Commun (Lond). 2022;42(1):17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Valentinuzzi D, Simoncic U, Ursic K, Vrankar M, Turk M, Jeraj R. Predicting tumour response to anti‐PD‐1 immunotherapy with computational modelling. Phys Med Biol. 2019;64(2):025017. [DOI] [PubMed] [Google Scholar]
- 51. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair‐deficient or microsatellite instability‐high colorectal cancer (CheckMate 142): an open‐label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tu C, Zeng Z, Qi P, Li X, Guo C, Xiong F, et al. Identification of genomic alterations in nasopharyngeal carcinoma and nasopharyngeal carcinoma‐derived Epstein‐Barr virus by whole‐genome sequencing. Carcinogenesis. 2018;39(12):1517–28. [DOI] [PubMed] [Google Scholar]
- 53. Jin K, Wang S, Zhang Y, Xia M, Mo Y, Li X, et al. Long non‐coding RNA PVT1 interacts with MYC and its downstream molecules to synergistically promote tumorigenesis. Cell Mol Life Sci. 2019;76(21):4275–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN Is Associated with Resistance to Anti‐PD‐1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity. 2017;46(2):197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu H, Zhang A, Han X, Li Y, Zhang Z, Song L, et al. ITGB2 as a prognostic indicator and a predictive marker for immunotherapy in gliomas. Cancer Immunol Immunother. 2022;71(3):645–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Roper N, Velez MJ, Chiappori A, Kim YS, Wei JS, Sindiri S, et al. Notch signaling and efficacy of PD‐1/PD‐L1 blockade in relapsed small cell lung cancer. Nat Commun. 2021;12(1):3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang Y, Zhang J, Chen Y, Xu R, Zhao Q, Guo W. MUC4, MUC16, and TTN genes mutation correlated with prognosis, and predicted tumor mutation burden and immunotherapy efficacy in gastric cancer and pan‐cancer. Clin Transl Med. 2020;10(4):e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gu Y, Liu Y, Fu L, Zhai L, Zhu J, Han Y, et al. Tumor‐educated B cells selectively promote breast cancer lymph node metastasis by HSPA4‐targeting IgG. Nat Med. 2019;25(2):312–22. [DOI] [PubMed] [Google Scholar]
- 59. Liu Y, Cao X. Characteristics and Significance of the Pre‐metastatic Niche. Cancer Cell. 2016;30(5):668–81. [DOI] [PubMed] [Google Scholar]
- 60. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med (Berl). 2016;94(5):509–22. [DOI] [PubMed] [Google Scholar]
- 61. Shi Y, Shi Q, Shen Q, Zhang Q, Cao X. Dicer‐independent snRNA/snoRNA‐derived nuclear RNA 3 regulates tumor‐associated macrophage function by epigenetically repressing inducible nitric oxide synthase transcription. Cancer Commun (Lond). 2021;41(2):140–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen DL, Sheng H, Zhang DS, Jin Y, Zhao BT, Chen N, et al. The circular RNA circDLG1 promotes gastric cancer progression and anti‐PD‐1 resistance through the regulation of CXCL12 by sponging miR‐141‐3p. Mol Cancer. 2021;20(1):166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu G, Wu Y, Wang W, Xu J, Lv X, Cao X, et al. Low‐dose decitabine enhances the effect of PD‐1 blockade in colorectal cancer with microsatellite stability by re‐modulating the tumor microenvironment. Cell Mol Immunol. 2019;16(4):401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Griffin GK, Wu J, Iracheta‐Vellve A, Patti JC, Hsu J, Davis T, et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature. 2021;595(7866):309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang Z, Wang B, Cao X. Epigenetic checkpoint blockade: new booster for immunotherapy. Signal Transduct Target Ther. 2021;6(1):281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang J, Wang J, Gu Q, Yang Y, Ma Y, Zhang Q. TGFbeta1: An Indicator for Tumor Immune Microenvironment of Colon Cancer From a Comprehensive Analysis of TCGA. Front Genet. 2021;12:612011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Knudson KM, Hicks KC, Luo X, Chen JQ, Schlom J, Gameiro SR. M7824, a novel bifunctional anti‐PD‐L1/TGFbeta Trap fusion protein, promotes anti‐tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology. 2018;7(5):e1426519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Larson C, Oronsky B, Carter CA, Oronsky A, Knox SJ, Sher D, et al. TGF‐beta: a master immune regulator. Expert Opin Ther Targets. 2020;24(5):427–38. [DOI] [PubMed] [Google Scholar]
- 69. Hargadon KM. Dysregulation of TGFbeta1 Activity in Cancer and Its Influence on the Quality of Anti‐Tumor Immunity. J Clin Med. 2016;5(9):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sheng W, Liu Y, Chakraborty D, Debo B, Shi Y. Simultaneous Inhibition of LSD1 and TGFbeta Enables Eradication of Poorly Immunogenic Tumors with Anti‐PD‐1 Treatment. Cancer Discov. 2021;11(8):1970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang D, Jin W, Wu R, Li J, Park SA, Tu E, et al. High Glucose Intake Exacerbates Autoimmunity through Reactive‐Oxygen‐Species‐Mediated TGF‐beta Cytokine Activation. Immunity. 2019;51(4):671–81 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dong X, Wang J, Li T, Xu YP, Li SY. Down regulation of lncRNA MEG3 promotes colorectal adenocarcinoma cell proliferation and inhibits the apoptosis by up‐regulating TGF‐beta1 and its downstream sphingosine kinase 1. Eur Rev Med Pharmacol Sci. 2018;22(23):8265–72. [DOI] [PubMed] [Google Scholar]
- 73. Chen JH, Perry CJ, Tsui YC, Staron MM, Parish IA, Dominguez CX, et al. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med. 2015;21(4):327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liu W, Fan T, Li M, Zhang G, Guo W, Yang X, et al. Andrographolide potentiates PD‐1 blockade immunotherapy by inhibiting COX2‐mediated PGE2 release. Int Immunopharmacol. 2020;81:106206. [DOI] [PubMed] [Google Scholar]
- 75. Remes Lenicov F, Paletta AL, Gonzalez Prinz M, Varese A, Pavillet CE, Lopez Malizia A, et al. Prostaglandin E2 Antagonizes TGF‐beta Actions During the Differentiation of Monocytes Into Dendritic Cells. Front Immunol. 2018;9:1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vo BT, jr Morton D, , Komaragiri S, Millena AC, Leath C, Khan SA. TGF‐beta effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology. 2013;154(5):1768–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The omics data of this study are available from the corresponding author (Xue‐Tao Cao, caoxt@immunol.org) upon reasonable request.