

Abstract
Background
Immune cell heterogenicity is known to determine the therapeutic response to cancer progression. Neoadjuvant chemoimmunotherapy (NACI) has shown clinical benefits in some patients with advanced head and neck squamous cell carcinoma (HNSCC), but the underlying mechanism behind this clinical response is unknown. The efficacy of NACI needs to be potentiated by identifying accurate biomarkers to predict clinical responses. Here, we attempted to identify molecules predicting NACI response in advanced HNSCC.
Methods
We performed combined single‐cell RNA sequencing (scRNA‐seq) and multiplex immunofluorescence (mIHC) staining with tumor samples derived from NACI‐treated HNSCC patients to identify a new tumor‐infiltrating cell (TIL) subtype, CD103+CD8+ TILs, associated with clinical response, while both in vitro and in vivo assays were carried out to determine its antitumor efficiency. The regulatory mechanism of the CD103+CD8+ TILs population was examined by performing cell‐cell interaction analysis of the scRNA‐seq data and spatial analysis of the mIHC images.
Results
We established intratumoral CD103+CD8+ TILs density as a determinant of NACI efficacy in cancers. Our scRNA‐seq results indicated that the population of CD103+CD8+ TILs was dramatically increased in the responders of NACI‐treated HNSCC patients, while mIHC analysis confirmed the correlation between intratumoral CD103+CD8+ TILs density and NACI efficacy in HNSCC patients. Further receiver operating characteristic curve analysis defined this TIL subset as a potent marker to predict patient response to NACI. Functional assays showed that CD103+CD8+ TILs were tumor‐reactive T cells, while programmed cell death protein‐1 (PD‐1) blockade enhanced CD103+CD8+ TILs cytotoxicity against tumor growth in vivo. Mechanistically, targeting the triggering receptor expressed on myeloid cells 2‐positive (TREM2+) macrophages might enhance the population of CD103+CD8+ TILs and facilitate antitumor immunity during NACI treatment.
Conclusions
Our study highlights the impact of intratumoral CD103+CD8+ TILs density on NACI efficacy in different cancers, while the efforts to elevate its population warrant further clinical investigation.
Keywords: CD103, CD8, head and neck squamous cell carcinoma, neoadjuvant chemoimmunotherapy, predictive marker, tumor‐infiltrating lymphocyte
List of abbreviations
- HNSCC
head and neck squamous cell carcinoma
- scRNA‐seq
single‐cell RNA sequencing
- NACI
neoadjuvant chemoimmunotherapy
- TIL
tumor‐infiltrating cell
- mIHC
multiplex immunofluorescent
- ROC
receiver operating characteristic
- PD‐1
programmed cell death protein‐1
- TREM2
triggering receptor expressed on myeloid cells 2
- RECIST
response evaluation criteria in solid tumors
- CR
complete response
- PR
partial response
- SD
stable disease
- PD
progressive disease
- MPR
major pathologic response
- IPR
incomplete pathological response
- PBMCs
peripheral blood mononuclear cells
- PBS
phosphate‐buffered saline
- FBS
fetal bovine serum
- qRT‐PCR
quantitative real‐time PCR
- PD‐L1
programmed death‐ligand 1
- NK
natural killer
- NSCLC
non‐small cell lung cancer
- TNBC
triple‐negative breast cancer
- GO
gene ontology
- KEGG
Kyoto encyclopedia of genes and genomes
- PID
pathway interaction database
- ICB
immune‐checkpoint blockade
- FFPE
formalin‐fixed, paraffin‐embedded
- TRM
tissue‐resident memory t
- MIF
migration inhibitory factor
- SPP1
secreted phosphoprotein 1
- DCA
decision curve analysis
- CPS
combined positive score
- OS
overall survival
- PFS
progression‐free survival
- RVT
residual viable tumor
- AUC
area under curve
1. BACKGROUND
Head and neck squamous cell carcinoma (HNSCC) is the seventh most common malignancy worldwide, with approximately 64% of patients present with locally advanced disease at diagnosis [1, 2]. Immunotherapy has been widely tested for treating patients with solid tumors, including HNSCC [3, 4]. However, recent clinical trials including neoadjuvant mono‐ or dual‐ immunotherapy in advanced HNSCC only showed modest clinical benefits, which prompted us to explore the use of immunotherapy in combination with chemotherapy [5, 6]. Indeed, Zhang et al. [7] showed that combining paclitaxel plus cisplatin with camrelizumab, an anti‐programmed cell death protein‐1 (PD‐1) inhibitor, can improve the clinical outcome of HNSCC; however, a significant proportion of patients do not respond to it. Although the combined positive score (CPS), a measure of programmed death‐ligand 1 (PD‐L1) expression, has been developed to predict therapeutic response and patient prognosis in solid cancers (including HNSCC) after treatment with PD‐1 inhibitors [8], its performance in predicting immunotherapy response remains unsatisfactory for clinical application [9]. This finding indicated the urgency of identifying proper markers to effectively predict patients’ responses to neoadjuvant chemoimmunotherapy (NACI) in advanced HNSCC.
Accumulating evidence indicates that CD8+ tumor‐infiltrating lymphocytes (TILs) are the key players in recognizing and eliminating cancer cells in antitumor immunity, which shapes the fundamental clinical benefit of immunotherapies in different cancers [10]. However, the prognostic value of CD8+ T cell infiltration is still controversial, primarily due to the functional heterogenicity of CD8+ TILs [11]. Emerging evidence suggests that only a small fraction of tumor‐infiltrating CD8+ T cells play a role in tumor control [12, 13], while the majority of CD8+ T cells are considered “bystanders” T cells that recognize cancer‐unrelated antigens, including common viral antigens, and/or are cancer ignorant [14, 15]. Therefore, it is important to identify the tumor‐specific T cell subset that can predict immunotherapy efficacy. Recent studies have demonstrated CD103 as a marker of T cell activation, which could be used to classify a subset of tumor‐reactive CD8+ T cells, and its expression correlated with better prognosis in various solid tumors [16, 17, 18]. CD103+CD8+ TILs also play a crucial role in HNSCC [19, 20]. Nevertheless, the clinical significance and characteristics of CD103+CD8+T cells remain poorly understood in HNSCC patients treated with NACI.
In this study, we confirmed promising biomarkers for predicting the NACI effect in advanced HNSCC patients. Then we explored the biomarker functions and mechanic insights into how to reverses the immunosuppression microenvironment to enhance the therapeutic effects of NACI treatment in advanced HNSCC patients.
2. MATERIALS AND METHODS
2.1. Patient cohort characteristics
All patients, comprising a discovery cohort of 8 advanced HNSCC patients and a validation cohort of 39 advanced HNSCC patients, were enrolled in this study. Eligible patients were selected based on the following inclusion criteria: 1) confirmed diagnosis of HNSCC by two experienced pathologists; 2) clinical stage III‐IVB according to the eighth edition of the American Joint Committee on Cancer (AJCC) guideline. These patients had no previous history of anticancer therapy and received a treatment regimen of paclitaxel and carboplatin plus pembrolizumab, administered every 3 weeks for up to 6 weeks before surgery. The human papillomavirus (HPV)‐negative HNSCC tissue samples obtained from tumor biopsies before NACI treatment. The efficacy of NACI was evaluated based on the clinical responses and pathologic responses. Clinical responses were evaluated based on radiologic assessments of tumor sizes by magnetic resonance imaging (MRI) before and after neoadjuvant therapy according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) [21]. Patients were classified into responders (complete response [CR] plus partial response [PR]) or non‐responders (stable disease [SD]), and progressive disease [PD]) [16]. Pathologic responses were evaluated based on the percentage of residual viable tumor [22]. We classified each advanced HNSCC patient into major pathologic response (MPR) (defined as ≤10% (residual viable tumor cells, RVT) in the resected tumor specimens) [23] or incomplete pathologic response (IPR; defined as >10% (residual viable tumor cells, RVT) in the resected tumor specimens) [7].
2.2. Cell lines
Cal‐27 cells were purchased from American Type Culture Collection (ATCC). SAS cells were purchased from the Chinese Academy of Sciences. Cal‐27 cells and SAS cells were cultured in DMEM with 10% fetal bovine serum. All cells were cultured at 37 °C with 5% CO2 and grown according to standard protocols.
2.3. Human sample preparation
All 8 tumor biopsy samples were collected with regular clinical practice as a discovery cohort and then were stored in MACS Tissue Storage Solution (Miltenyi, Bergisch Gladbach, Germany). Next, all samples were enzymatically digested with a gentleMACS Tumor Dissociation Kit (Miltenyi) at 37°C for 60 min according to the manufacturer's protocol. Subsequently, the dissociated cells were passed through a 40 μm cell strainer (Biosharp, Hefei, Anhui, China) and centrifuged at 300 × g for 10 min. The pelleted cells were suspended in red blood cell lysis buffer and then resuspended in phosphate‐buffered saline (PBS, Gibco, Carlsbad, CA, USA) supplemented with 2% fetal bovine serum (FBS, ExCell, Shanghai, China) after washing twice with PBS. Peripheral blood mononuclear cells (PBMCs) were isolated according to the manufacturer's protocol [24]. Briefly, 5 mL of fresh peripheral blood was collected before treatment initiation in EDTA anticoagulant tubes and subsequently extracted by Lymphoprep™ (Stem Cell, Vancouver, Canada) solution. After centrifugation, lymphocyte cells were carefully transferred to a new 50 mL tube and washed with PBS. Next, the lymphocyte cells were incubated on ice for 10 min to lyse red blood cells. Finally, the cells were resuspended in a sorting buffer with PBS plus 2% FBS.
2.4. Single‐cell RNA sequencing (scRNA‐seq) and library construction
According to the manufacturer's protocol, scRNA‐seq libraries were constructed [25]. In summary, the density of cells was determined after washing once with PBS containing 0.04% bovine serum albumin (BSA, Invitrogen, Carlsbad, CA, USA). Next, 2 × 105 cells were loaded on a 10x Genomics GemCode Single‐cell instrument (10x Genomics, San Francisco, CA, USA) that generates single‐cell Gel Bead‐In‐EMlusion (GEMs). Libraries were generated and sequenced from the cDNAs with the Chromium Next GEM Automated Single Cell 3’ cDNA Kit v3.1 (10x Genomics, San Francisco, CA, USA). Upon dissolution of the Gel Bead in a GEM, primers containing an Illumina® R1 sequence (read 1 sequencing primer), a 16 nt 10x Barcode, a 10 nt Unique Molecular Identifier (UMI), and a poly‐dT primer sequence were released and mixed with cell lysate and Master Mix. Barcoded, full‐length cDNAs were then reverse‐transcribed from polyadenylated mRNA. Silane magnetic beads were used to remove leftover biochemical reagents and primers from the post‐GEM reaction mixture. Full‐length, barcoded cDNAs were then amplified by quantitative real‐time PCR (qRT‒PCR) to generate sufficient mass for library construction. Finally, single‐cell RNA libraries were sequenced by an Illumina HiSeq X‐Ten sequencer with 150 bp paired‐end reads.
2.5. scRNA‐seq data analysis
The Cell Ranger toolkit (version 3.1.0) provided by 10x Genomics was applied to aggregate raw data, filter low‐quality reads, align reads to the human reference genome (GRCh38), assign cell barcodes, and generate the UMI matrix. A Python‐based toolkit, Scanpy (version 2.3.4) [25], was used to analyze the scRNA‐seq data. The raw data were first processed to filter out low‐quality reads by the Cell Ranger toolkit (version 3.1.0) from 10x Genomics and then mapped with the human reference genome (GRCh38), cell barcodes were assigned, and the UMI matrix was generated. Next, the data were analyzed by Scanpy (version 2.3.4), and Scanpy was used for analyzing dimension reduction and unsupervised clustering. After clustering results were obtained, to identify differentially expressed genes, ANOVA was performed using the R package limma and the difference between each cluster pair was tested using the P value of Tukey's Honest Significant Difference test < 0.01 implemented in R function. Furthermore, the R package was applied to perform Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Pathway Interaction Database (PID) analyses. Of note, the uniform manifold approximation and projection (UMAP) method was used for the visualization of cell distance in the reduced 2D space. To identify potential cell–cell interactions, the R package Cell Chat (version 1.1.3) was used to evaluate the expression of pairs of ligands and receptors within cell populations. Briefly, we first pooled cells by subtype across all tumor samples. To determine available ligands‐receptors that were enriched in individual subtypes, we performed differential expression analysis of ligand‐receptors genes by a one‐sided Wilcoxon rank‐sum test.
2.6. Multicolor immunohistochemistry (mIHC)
According to the manufacturer's instructions, mIHC staining of formalin‐fixed, paraffin‐embedded (FFPE) tumor samples from 39 advanced HNSCC patients was performed using an Opal 7‐Color Kit (Cat#0004100100, Panovue, Beijing, China). The following antibodies were purchased from Cell Signaling Technology (CST, Danvers, MA, USA) or Abcam (Waltham, MA, USA): CD8 (1:200, #85336, CST), CD103 (1:200, EPR22590‐27, Abcam), cytokeratin (1:250, #67306, CST), CD68 (1:1000, #76437, CST), and TREM2 (1:500, #91068, CST). For each staining, a secondary horseradish peroxidase‐conjugated antibody (Panovue, Beijing, China) was incubated with a tyramide‐coupled fluorophore: Opal 690, Opal 620, Opal 570, Opal 520, and Opal 480. The selected field of mIHC images was captured by using Akoya Vectra Polaris (Akoya Biosciences, Marlborough, MA, USA), and then the InForm software (Akoya Biosciences, Marlborough, MA, USA) was used to analyze the images for spectral unmixing, tissue segmentation (epithelial versus stromal areas), and cell segmentation followed by cell phenotyping for identification of cell populations defined by the combination of individual markers. The density (number of cells per mm2) of CD8+, CD103+CD8+, and CD103−CD8+ cells was determined for each tumor sample in the total tumor area, as well as in the stromal and intraepithelial compartments, based on tissue segmentation. The total CD103+CD8+ cells in the tumor microenvironment were defined as intratumoral CD8+CD103+ TILs, and the CD103+CD8+ cells in epithelial regions were defined as intraepithelial CD8+CD103+ TILs. Images were also analyzed by HALO® platform v3.3 (Indica Labs, Albuquerque, NM, USA), which allows for the identification of marker of interest colocalization and cellular spatial relation analysis. HALO® density heatmaps of CD103+CD8+ TIL compartments were created to visually compare immune infiltrate patterns. To further investigate the infiltration pattern of CD103+CD8+ TILs, we quantified the expression of CD103 and CD8 in both tumor and stromal compartments by using the HALO® (Indica Labs) v3.3 platform, an immune cell spatial analysis software [26]. Spatial relationships between CD103+CD8+ TILs and triggering receptor expressed on myeloid cells 2‐positive (TREM2+) CD68+ macrophage cells were also evaluated.
2.7. T cell activation and cell sorting
Naive CD8 T cell subsets were isolated by CD8 MicroBeads (Miltenyi, Cat#: 130‐045‐201), and then 2 × 106 naive T cells were cultured in complete RPMI‐1640 (Gibco, Carlsbad, CA, USA) containing 10% FBS supplemented with CD3/CD28 MicroBeads (Miltenyi, Cat#: 130‐050‐101 and Cat#: 130‐093‐247) and 2 ng/mL TGFβ‐1 (Peprotech, #100‐21, Cranbury, NJ, USA) according to the manual. After blocking with Fc Receptor Blocking Solution (Cat# 422301, Biolegend, San Diego, CA, USA), the CD8 subsets were sorted as CD103− and CD103+ (#350216, Biolegend) by flow cytometry.
2.8. Flow cytometry (FACS) analysis
After tissue processing and cell counting, approximately 2 × 106 human cell suspensions were stained with a fixable live/dead dye (Biolegend) for 10 min at room temperature and then stained with fluorophore‐conjugated antibodies for 30 min at 4°C after blocking with Fc receptor. The fluorescently labeled antibodies were as follows: FITC anti‐CD8 (1:100; #980908; Biolegend), BV421 anti‐CD103 (1:100; #350214; Biolegend), BV650 anti‐PD‐1 (1:100; #367430; Biolegend), FITC anti‐IFN‐γ (1:50; #552887; BD Biosciences), APC anti‐CXCL13 (eBioscience; 1:100; #DS8CX13, California, USA), APC anti‐TNF‐α (1:200; # 502912; Biolegend), PE anti‐granzyme B (1:100; #561142; BD Biosciences), BV605 anti‐CD69 (1:50; #310937; Biolegend), APC anti‐IL‐2 (1:100; #339008; Biolegend), and PE anti‐Ki‐67 (1:100; #567720; BD Biosciences; Franklin Lakes, NJ, USA).
2.9. Cytotoxic experiments
Cancer cells (2 × 103) were seeded in 96‐well plates, and then T cells were added to the cocultures at different ratios (E:T ratios of 1.563:1, 3.125:1, 6.25:1, 12.5:1, 25:1, or 50:1) without the addition of exogenous cytokines. The cytolytic activity was analyzed by LDH‐Glo™ Cytotoxicity Assay (Promega, Woods Hollow Road, WI, USA) according to the manual. The mean percentage of specific cell lysis in triplicate wells was calculated using the following formula: mean percentage of specific cell lysis = test release‐spontaneous release/maximal release‐spontaneous release × 100.
2.10. PD‐L1 immunohistochemistry (IHC)
The experiments were performed as previously described. All fixed tissue samples were deparaffinized using xylene and graded concentrations of ethanol, subjected to antigen retrieval with EDTA at pH 9.0, and incubated with 30% hydrogen peroxide for 10 min at room temperature. Then, they were blocked with goat serum for 1 hour at 37°C and incubated with the indicated antibody at 4°C overnight. PD‐L1 expression was evaluated using a PD‐L1 22C3 pharmDx assay (Dako North America, Carpinteria, CA, USA). CPS was defined as the total number of PD‐L1‐stained cells (including tumor cells, tumor‐associated lymphocytes, and macrophages) divided by the total number of viable tumor cells multiplied by 100.
2.11. Animal experiments
Female NOD/ShiLtJGpt‐Prkdcem26Cd52Il2rgem26Cd22/Gpt mice (NCG) at 3 to 4 weeks of age were purchased from Gempharmatech (Nanjing, Jiangsu, China). All animal experiments were approved by the Institutional Animal Care and Use Committee of Sun Yat‐sen University. Mice were bred in a specific pathogen‐free (SPF) animal house at 28°C and 50% humidity. For the tumor suppression experiments, the mice were randomly divided into 3 groups (n = 3 per group). SAS cells (2 × 106) were subcutaneously injected into the dorsum of the mice. Tumor growth was measured every 5 days during a 5‐week period with tumor volume (tumor volume = length × width2 × 0.5). When the tumors grew to 50 mm3, the mice were injected with PBS, 2 × 107 CD103+CD8+ T cells, or 2 × 107 CD103+CD8+ T cells combined with a PD‐1 inhibitor (pembrolizumab; Merck & Co., Inc., Rahway, NJ, USA) at a 5 μg/g dose per mouse once every week 4 times via the tail vein. Tumor weight and size of mice were observed at set time points over a 35‐day period, and then animals were euthanized and prepared for FACS.
2.12. Statistical analysis
Comparisons between patient characteristics were performed using the chi‐square or Fisher's exact test for discrete variables and unpaired 2‐tailed Student's t‐test. Receiver operating characteristic (ROC) curves were constructed, and the areas under the curves (AUCs) were calculated to evaluate the predictive abilities of the CPS and CD103+CD8+ TILs. Meanwhile, decision curve analysis (DCA) was used to validate the clinical utility of the model based on the net benefit [27]. Survival analyses were performed using the Kaplan‐Meier method and log‐rank test. Progression‐free survival (PFS) was calculated from the date of first immunotherapy until disease progression or death due to any cause. Overall survival (OS) was calculated from the date of the first immunotherapy administration until death due to any cause. Statistical significance was determined with the paired or unpaired 2‐tailed Student's t‐test or with one‐way analysis of variance (ANOVA) with Bonferroni correction. Pearson correlation was used to estimate correlations between two groups. Statistical analyses were performed with GraphPad Prism software 8 (GraphPad Software Inc., San Diego, CA). Statistical significance was indicated as P < 0.05, P < 0.01, P < 0.001.
3. RESULTS
3.1. Identification of a unique T cell subset, CD8+CD103+ T cells, in tumors derived from NACI‐responsive patients with advanced HNSCC
To exploit the complexity of the tumor microenvironment in determining NACI efficacy, we performed scRNA‐seq on tumor biopsies obtained from 8 patients with advanced HNSCC who were either responsive or non‐responsive (based on RECIST, version 1.1) [25]. The regimen for NACI was 2 courses of 3 weeks, and the response to NACI was evaluated after the end of treatment. In this study, tumor samples were collected from patients before receiving NACI and were then subjected to scRNA‐seq analysis, and the results were validated by subsequent FACS and mIHC analysis as described in the workflow chart (Figure 1A). After quality filtering of our scRNA‐seq data, we obtained 54,955 high‐quality cells, and the single‐cell transcriptome data were then visualized following UMAP dimensionality reduction and clustered using the Seurat Louvain algorithm [25]. A total of 17 distinct major clusters representing epithelial, immune, and stromal populations were identified (Figure 1B). In comparison with the non‐responders, the responders showed a significant increase in the level of T and natural killer (NK) cell clusters, suggesting that these cells could be the key players in modulating NACI efficacy (Figure 1C). Based on our high‐resolution T and NK subset map in HNSCC tumors, we identified 4 CD8+ T cell clusters (CD8‐CD103+, CD8‐GZMK+, CD8‐LEF1+, and CD8‐ISG+), 2 CD4+ T cell clusters (Treg‐TNFRSF4+ and CD4‐CXCL13+), an NK cell cluster (NK‐FCGR3A+), a gamma delta T cell cluster (gd T‐TRDC+), a double‐positive T cell cluster (DP‐MKI67+), and a double‐negative T cell cluster (DN‐TRDV2+) (Figure 1D). Interestingly, our results indicated that the cell density of the CD103+CD8+ T cell subtype was significantly higher in responders than in non‐responders (Figure 1E), which was consistent with the previously observed predictive role of CD103+CD8+ T cells in the immunotherapy response in non‐small cell lung cancer (NSCLC) [16]. Since the application of NACI has been approved for NSCLC [28] and triple‐negative breast cancer (TNBC) patients [29], we further explored the role of CD103+CD8+ T cells in patients with NSCLC and TNBC by reanalyzing the scRNA‐seq data obtained from published papers [25, 30]. As expected, our bioinformatic results revealed that the increased CD103+CD8+ T cell density strongly correlated with a better response to NACI in TNBC patients (Supplementary Figure S1A‐C). In contrast, we did not observe an association between CD103+CD8+ T cells and NACI efficacy in NSCLC patients (Supplementary Figure S1D‐F).
FIGURE 1.
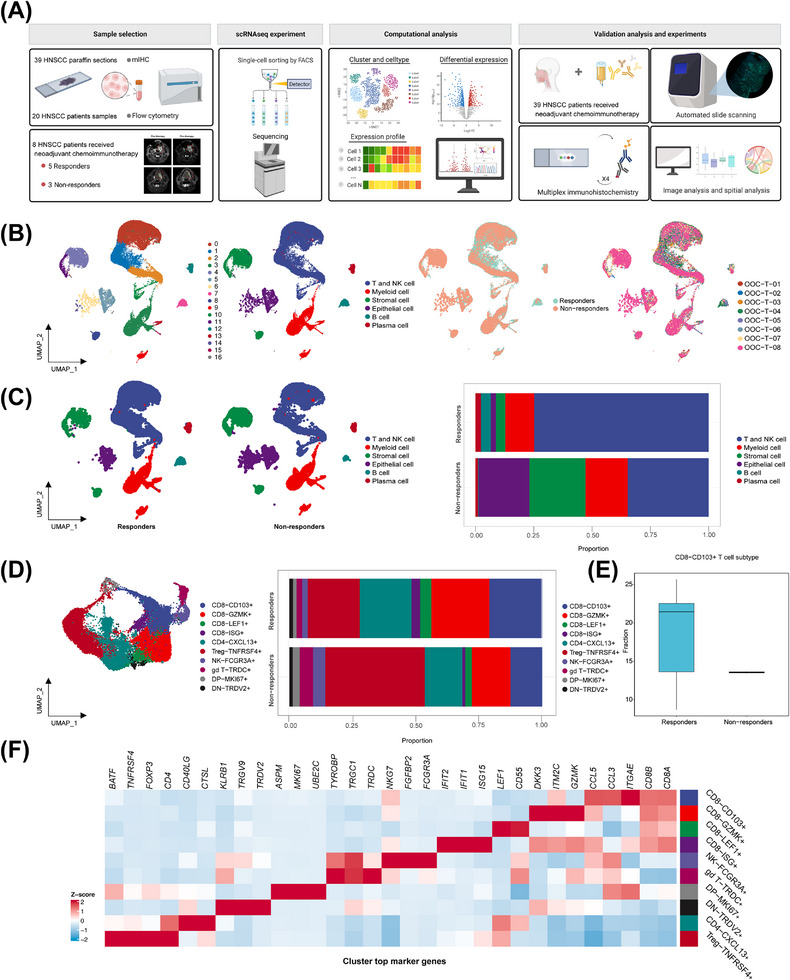
Immune cell profile of advanced HNSCC at single‐cell atlas. (A) Schematic overview of the experimental design and analytical workflow. We performed combined scRNA‐seq and mIHC staining with tumor samples derived from NACI‐treated HNSCC patients to identify CD103+CD8+ TILs, associated with clinical response. (B) A total of 17 mega‐clusters in advanced HNSCC. The clusters are visualized and labeled by cell type via UMAP visualization. The distributions of cell clusters between the responders and non‐responders are visualized by UMAP visualization. The distributions of cell clusters from 8 tumor samples are visualized respectively by UMAP visualization. (C) All cell type clusters are visualized via UMAP visualization in the responders versus non‐responders. Boxplots showing the alterations of cell clusters between the responders and non‐responders. (D) UMAP visualization of T and NK cell subclusters. The clusters are visualized and labeled by cell type. The bar graph represents the proportion of these subclusters in responders and non‐responders. (E) The bar graph represents the proportion of CD103+CD8+ T cells in the T cell expression between the responders and non‐responders. (F) Heatmap depicting the cell‐type‐specific markers in the T and NK cell subclusters of scRNA‐seq analysis. Abbreviations: HNSCC, head and neck squamous cell carcinoma; scRNA‐seq, single‐cell RNA sequencing; NK, natural killer; GZMK, Granzyme K; LEF1, lymphoid enhancer binding factor 1; ISG, interferon stimulated exonuclease gene; TNFRSF4, TNF receptor superfamily member 4; CXCL13, C‐X‐C motif chemokine ligand 13; FCGR3A, Fc gamma receptor IIIa; TRDC, T cell receptor delta constant; MKI67, Marker of proliferation Ki‐67; TRDV2, T cell receptor delta variable 2.
To generate the comprehensive T cell atlas of our scRNA‐seq data, we presented a heatmap to visualize the heterogeneous T cell subtypes that exhibited distinct molecular signatures, indicative of their unique cellular identities (Figure 1F). Overall, our work identified a unique CD8+ T cell subset, known as CD8+CD103+ T cells, in which its density may correlate with NACI efficacy in advanced HNSCC patients.
3.2. Characterization of CD103+CD8+ T cells across molecular features
To explore the biological states of CD103+CD8+ T cells in the tumor microenvironment, we performed GO, KEGG, and PID analyses to investigate the functional characteristics of this T cell subset (Figure 2A‐C), indicating that the CD103+CD8+ T cell subset was more enriched in biological processes associated with immunomodulation, such as “antigen processing and presentation” and “NK cell‐mediated cytotoxicity”, compared to the other T cell subsets. Consistent with these findings, CD103+CD8+ T cells had relatively high expression of genes involved in T cell activation and tumor‐reactive cytotoxic markers (Figure 2D).
FIGURE 2.
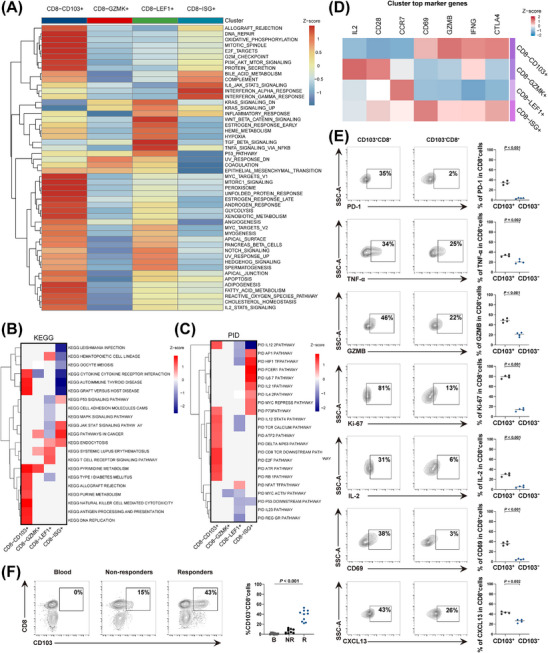
The molecular characterization of CD103+CD8+ TILs. (A) GO analysis of gene set from the CD8+ TILs subtypes. (B) KEGG analysis of gene set from CD8+ TILs subtypes. (C) PID analysis of gene set from the CD8+ TILs subtypes. (D) Heatmap depicting the cell‐type‐specific markers in the CD8+ TILs subtypes. (E) FACS analysis of gene expression in CD103+CD8+ T cells and CD103−CD8+ T cells from advanced HNSCC tissues (n = 4). (F) FACS analysis of CD103+CD8+ T cell population in PBMC (B), responders (R) and non‐responders (NR) tumor tissues, respectively (n = 10). Horizontal bar indicates the median. Significance was determined with an unpaired 2‐tailed student's t‐test or one‐way ANOVA with Bonferroni post hoc test. Abbreviations: TIL, tumor‐infiltrating cell; HNSCC, head and neck squamous cell carcinoma; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PID, Pathway Interaction Database; IL‐2, Interleukin 2; TNF‐α, tumor necrosis factor; GZMB, Granzyme B; CXCL13, C‐X‐C motif chemokine ligand 13; PD‐1, programmed cell death protein‐1; CCR7, C‐C Motif Chemokine Receptor 7; PBMC, peripheral blood mononuclear cell; FACS, flow cytometry.
To gain further insight into the molecular characteristics of CD103+CD8+ T cells, we conducted FACS analysis with tumor samples obtained from advanced HNSCC, and the characteristics of CD103+CD8+ T cells were further screened and examined (Supplementary Figure S2A). As expected, our results showed that the expression of proliferation (Ki‐67 and IL‐2), activation (CD69), effector (TNF‐α, GZMB, and CXCL13) and immune checkpoint (PD‐1) markers were elevated in CD103+CD8+ T cells compared to CD103−CD8+ T cells (Figure 2E), indicating that this subset could be capable of killing cancer cells more efficiently in conjunction with immune checkpoint blockade (ICB) responses. Interestingly, our FACS results also found that CD103+CD8+ T cells were more enriched in the responders, which was consistent with the distribution profile displayed in the scRNA‐seq data (Figure 2F). In contrast, our FACS analysis indicated that CD103+CD8+ T cells were hardly detected in the peripheral blood, suggesting they could be tumor‐infiltrating lymphocytes (TILs) with a tissue‐resident memory phenotype. Next, we performed in vitro cytotoxicity assays with CD103− CD8+ or CD103+CD8+ T cells, in which they were first stimulated by TGF‐β and then cocultured with tumor cells at different effector:target (E:T) ratios. The results indicated that CD103+CD8+ T cells displayed a stronger cytotoxic capacity against cancer cells than the CD103−CD8+ T cells, while their cytotoxicity was also enhanced with an increasing E:T ratio (Supplementary Figure S2B). To further explore the function of human CD103+CD8+ T cells in antitumor immunity in vivo, we subcutaneously injected human HNSCC cells (SAS) into NCG mice (a mouse model with triple immunodeficiency and a lack of functional/mature T, B, and NK cells), which were then treated with either PBS, solely CD103+CD8+ T cells, or a combination of CD103+CD8+ T cells and the anti‐PD‐1 (Supplementary Figure S3A). Our data revealed that CD103+CD8+T cell treatment reduced tumor growth in SAS tumor‐bearing mice, while coadministration of anti‐PD‐1 and CD103+CD8+ T cells suppressed tumor growth even further compared to the tumor growth suppression in mice treated with CD103+CD8+ T cells alone or PBS (Supplementary Figure S3B‐D). Further FACS analysis of the above tumor samples revealed a significant increase in the release of the cytokines IFN‐γ, GZMB and TNF‐α as well as the CD103+CD8+ T cell population in the combinatorial treatment group compared to the PBS or solely CD103+CD8+ T cell groups (Supplementary Figure S3E). Collectively, these results showed that the infiltrated CD103+CD8+ T cells, known as CD103+CD8+ TILs, in HNSCC tumors were tumor‐reactive T cells, which might provide an effective antitumor function for immunotherapy.
3.3. Intratumoral CD103+CD8+ TILs predict favorable responses to NACI in cancer patients
Since our analysis above revealed that CD103+CD8+ TILs might play a central role in antitumor immunity, we established a retrospective tumor cohort (validation cohort) of 39 patients with advanced HNSCC treated with NACI to further examine our findings (Supplementary Table S1). Tumor samples were collected from the patients before receiving NACI treatment, and the density of CD103+CD8+ TILs was evaluated by performing mIHC. We found that CD103+CD8+ TILs were significantly enriched in responders (Figure 3A). To better confirm the contribution of CD103+CD8+ TILs to NACI efficacy, the pathological responses were evaluated based on the percentage of residual viable tumor (RVT) cells. Therefore, we classified each of the 39 patients into the group with either MPR (defined as ≤10% RVT cells) or IPR (defined as >10% RVT cells). Since the MPR met the criteria for a surrogate endpoint after neoadjuvant therapy in a variety of cancers [1, 7, 23], it was strongly associated with improved survival, which is also reflective of the treatment impact and captures the magnitude of the treatment benefit on patient survival [23]. As expected, the density of CD103+CD8+ TILs had a higher fraction in the patients with MPR than in those with IPR. However, CD103−CD8+ TILs showed a weak association with the response to NACI (Figure 3B). In addition, no strong correlation was observed between the density of CD103+CD8+ TILs and the radiologic response or pathologic response of the patients (Supplementary Figure S4A). To decipher the immune cell dynamics during NACI treatment, we performed mIHC staining experiments to examine the modulation of CD103+CD8+ TILs subset in tumor samples obtained from 39 advanced HNSCC patients pre‐ and post‐NACI treatment (Figure 3C). Our results indicated that the dynamic alteration of CD103+CD8+ TILs density was expanded in the responsive patients after treatment with NACI, whereas the population of CD103+CD8+ TILs was not different in the non‐responsive cohort before and after NACI treatment (Figure 3D‐E). Similarly, the density of CD103+CD8+ TILs was inclined to increase in the patients with MPR after receiving NACI treatment (Supplementary Figure S4B). In contrast, no expansion of this TIL subset was observed in the non‐responders and patients with IPR after receiving NACI treatment (Supplementary Figure S4B). Unlike CD103+CD8+ TILs, our results showed that CD103−CD8+ TILs did not respond to NACI treatment in either responders or non‐responders (Figure 3E, Supplementary Figure S4B).
FIGURE 3.
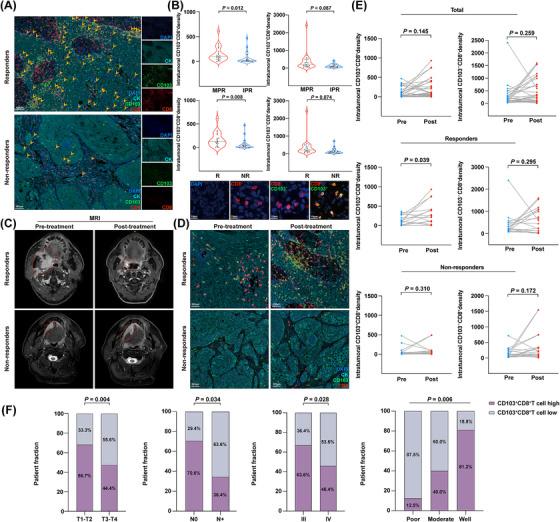
The increased CD103+CD8+ TIL density is associated with better therapeutic response in NACI‐treated HNSCC patients. (A) mIHC image of DAPI, Cytokeratin (CK), CD103, and CD8 staining of advanced HNSCC tissues in the responders and non‐responders. Scale bar, 100 μm. (B) Density of intratumoral CD103+CD8+ TILs and CD103−CD8+ TILs in tumors was assessed in the MPR group (n = 22) and IPR group (n = 17) patients to NACI via mIHC assay. Density of intratumoral CD103+CD8+ TILs and CD103−CD8+ TILs in tumors was assessed in the responders (R; n = 19) and non‐responders (NR; n = 20) patients to NACI. Image shows CD103+CD8+ (yellow), CD103−CD8+ (red), and CD8+ (red) cells in advance HNSCC (lower panel). Scale bar, 10 μm. (C) MRI showing the tumor size in responders and non‐responders before and after NACI, respectively. (D) mIHC image shows the density of CD103+CD8+ TILs in tumors from responders and non‐responders before and after NACI. Scale bar, 200 μm. (E) The temporal alterations of the density of CD103+CD8+ TILs and CD103−CD8+ TILs from pre‐ and post‐NACI in the responders (n = 19) and non‐responders (n = 20) via mIHC assay. (F) Distribution of CD103+CD8+ TILs between different clinical characteristics in patients with advanced HNSCC. Horizontal lines indicate the mean ± SEM. Significance was determined with a 2‐tailed Mann‐Whitney U test. Pearson's chi‐square test was used for studying the distribution of intraepithelial CD103+CD8+ TILs across different subgroups. Abbreviations: TIL, tumor‐infiltrating cell; HNSCC, head and neck squamous cell carcinoma; mIHC, multiplex immunofluorescent; MRI, Magnetic resonance imaging; MPR, major pathologic response; IPR, incomplete pathologic response; R, responders; NR, non‐responders; SEM, standard error of the mean; NACI, neoadjuvant chemoimmunotherapy; MRI, magnetic resonance imaging.
To better explore the potential diagnostic value of CD103+CD8+ TILs for clinical study, we sought to determine the relationship between the CD103+CD8+ TILs density and the clinical characteristics of advanced HNSCC patients in our validation cohort. These results demonstrated that 40.0% (8/20) of the patients with a high density of CD103+CD8+ TILs were at the T1‐T2 stage, but only 21.1% (4/19) of the patients with a low density of CD103+CD8+ TILs were within the T1‐T2 stage, indicating that the T1‐T2 primary tumors had more CD103+CD8+T cell infiltration. Similarly, we found that 21.1% (4/19) of the patients with a low density of CD103+CD8+ TILs and 35.0% (7/20) of the patients with a high density of CD103+CD8+ TILs belonged to clinical stage III. Intriguingly, 12 (70.6%) patients with no lymph node metastasis were defined as having a high density of CD103+CD8+ TILs. However, only 26.3% (5/19) of the patients with a low density of CD103+CD8+ TILs had no lymph node metastasis. Among the poorly differentiated stage, only 12.5% (1/8) of the patients had a high density of CD103+CD8+ TILs. Conversely, 81.3% (13/16) of the patients with good differentiation contained a high density of CD103+CD8+ TILs (Figure 3F, Supplementary Table S2). We then correlated the survival data of these patients with CD103/CD8 scores (the expression of CD103 was divided by CD8 expression). As shown in Supplementary Figure S4C, patients with a higher frequency of CD103+ cells among CD8+ TILs tended to have prolonged PFS or OS compared to patients with a low frequency of the CD103+CD8+ TIL subtype. Collectively, our results indicated that the dynamic alteration of CD103+CD8+ TILs density may dictate the NACI response in advanced HNSCC patients, while the density of CD103+CD8+ TILs also correlated with the clinical characteristics of advanced HNSCC patients.
3.4. The intraepithelial CD103+CD8+ TILs subset functions as a strong indicator of NACI efficacy in HNSCC
Most solid tumors are divided into 3 major immunological phenotypes, termed immune inflamed, immune excluded, and immune desert [31]. Accumulating evidence supports the idea that a higher level of T cell infiltration in tumors is positively correlated with response rates to immunotherapies [32]. Thus, examining the infiltration pattern of CD103+CD8+ T cells could be even more valuable in predicting the patient's response to NACI. To do so, we classified each mIHC‐stained image into tumor nests and stromal areas using InForm software, a supervised image analysis system based on machine learning [33]. Next, we identified the nuclear, cytoplasmic, and membranous outlines using the cell segmentation technique. Cell phenotyping data were obtained based on the positivity and relative intensity of all markers in one panel. Finally, the cell density of CD103+CD8+ TILs was measured separately in the tumor and stromal compartments of the tissue samples obtained from the responders and non‐responders (Figure 4A). Our data showed that the density of intraepithelial CD103+CD8+ TILs was positively correlated with the efficiency of NACI, whether it was based on radiographical or pathological assessment, whereas the increased stromal CD103+CD8+ TILs level did not respond efficiently to NACI treatment (Figure 4B).
FIGURE 4.
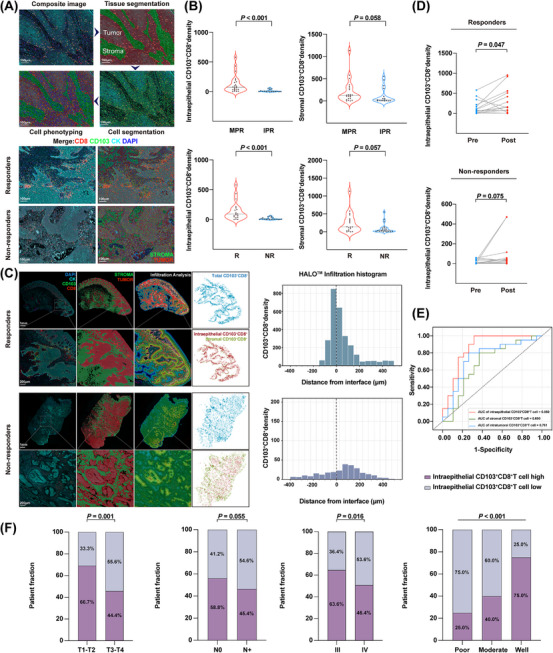
Intraepithelial CD103+CD8+ TIL density has a strong predictive value for therapeutic response in HNSCC patients treated with NACI. (A) Overview of the automated image analysis pipeline via inform analysis (upper panel). The spatial distribution of CD103+CD8+ TILs in the responders and non‐responders (lower panel). Digital markup image shows tumor (red) and stromal (green) zones of the tumor section defined by cytokeratin staining. Digital markup image shows CD103+CD8+ TILs (red). Scale bar, 100 μm. (B) Density of intraepithelial and stromal CD103+CD8+ TILs in tumors were assessed in the MPR group (n = 22) and IPR group (n = 17) patients to NACI via inform analysis. Density of intraepithelial and stromal CD103+CD8+ TILs in tumors were assessed in the responders (R; n = 19) and non‐responders (NR; n = 20) with NACI. (C) HALO® spatial analyses demonstrate the spatial distribution of CD103+CD8+ TILs in the responders and non‐responders. Digital markup image shows tumor (red) and stromal (green) zones of the tumor section defined by cytokeratin staining. Digital markup image shows the density of intratumoral CD103+CD8+ TILs (blue), intraepithelial CD103+CD8+ TILs (red), and stromal CD103+CD8+ TILs (green). Scale bar, 1 mm, 200 μm. Proximity analysis histograms of CD103+CD8+ TILs within an 800 μm radius by progressive segments of 200 μm bands in advanced HNSCC tissues. (D) Temporal alterations in the density of intraepithelial CD103+CD8+ TILs from pre‐ and post‐NACI in the responders (n = 19) and non‐responders via inform analysis (n = 20). (E) Predictive value of intratumoral CD103+CD8+ TILs, intraepithelial CD103+CD8+ TILs, and stromal CD103+CD8+ TILs via ROC analysis. (F) Distribution of intraepithelial CD103+CD8+ TILs between different clinical characteristics in patients with advanced HNSCC. Horizontal lines indicate the mean ± SEM. Significance was determined with a 2‐tailed Mann‐Whitney U test. Pearson's chi‐square test was used for studying the distribution of intraepithelial CD103+CD8+ TILs across different subgroups.
Abbreviations: TIL, tumor‐infiltrating cell; HNSCC, head and neck squamous cell carcinoma; MPR, major pathologic response; IPR, incomplete pathologic response; ROC, Receiver Operating Characteristic; R, responders; NR, non‐responders; SEM, standard error of the mean; NACI, neoadjuvant chemoimmunotherapy.
As shown in Figure 4C, the interface between tumor and stromal cells was first identified, while the density of CD103+CD8+ TILs was then separately quantified within the tumor and stromal compartments using the proximity analysis histogram method. Further data analyses indicated that CD103+CD8+ TILs were enriched among the responders, with a median of 408.1 cells per mm2. More importantly, the density of CD103+CD8+ TILs, ranging from 58.2 cells per mm2 to 849.3 cells per mm2, was higher in the intraepithelial region than in the stroma. However, there were fewer CD103+CD8+ TILs found in the non‐responders, with a median of 22.2 cells per mm2, and their CD103+CD8+ TILs density was only between 6.4 cells per mm2 and 18.8 cells per mm2 in the intraepithelial region, indicating that the density of intraepithelial CD103+CD8+ TILs was more strongly associated with antitumor immunity in cancer patients. Next, we analyzed the clinical value of intraepithelial CD103+CD8+ TILs by evaluating their correlation with radiologic response and pathologic response in HNSCC patients. As shown in Supplementary Figure S5A‐B, the intraepithelial CD103+CD8+ TILs density displayed a positive correlation with radiologic response (Pearson's R2 = 0.211, P = 0.003) and pathologic response (Pearson's R2 = 0.170, P = 0.009) in HNSCC patients. Interestingly, our data showed that most responders possessed a higher density of intraepithelial CD103+CD8+ TILs after receiving NACI treatment. In contrast, no expansion of this subset was observed in the non‐responders (Figure 4D). Similar results were also observed when analyzing the correlation between the dynamic level of intraepithelial CD103+CD8+ TILs and patients’ pathologic responses (Supplementary Figure S5C). Next, we investigated the predictive value of intraepithelial CD103+CD8+ TILs in discriminating responders and non‐responders. Notably, to some extent, intratumoral (including both tumor and stromal compartments) and stromal CD103+CD8+ TILs could be used to discriminate potential NACI responders and non‐responders (area under curve AUC = 0.761 for intratumor and AUC = 0.690 for stroma), whereas the use of intraepithelial CD103+CD8+ TILs improved the probability of correctly separating responders from non‐responders with more than 80% accuracy (AUC = 0.850; Figure 4E). These results demonstrated that the intraepithelial CD103+CD8+ TILs density could be used to predict the response to NACI in HNSCC patients more accurately. Further, we evaluated the correlation between intraepithelial CD103+CD8+ TILs and the clinical characteristics of HNSCC patients. Consistent with the features of intratumoral CD103+CD8+ TILs, our data showed that a high density of intraepithelial CD103+CD8+ TILs was more correlated with primary tumors with T1‐T2, stage IIIand well‐differentiated stages (Figure 4F). In addition, our clinical study indicated that the intraepithelial CD103/CD8 score tended to be positively associated with PFS and OS in advanced HNSCC patients (Supplementary Figure S5D). Collectively, these data suggested that the intraepithelial CD103+CD8+ TILs density had the strongest predictive value for NACI response in HNSCC patients compared to the intratumor or stromal CD103+CD8+ TILs density.
3.5. Incorporating the CPS with intraepithelial CD103+CD8+ TILs level enhances its predictive accuracy of neoadjuvant chemoimmunotherapeutic response in HNSCC
To evaluate the correlation between the density of CD103+CD8+ TILs and the context of distinct clinicopathological factors in our cohort, the waterfall plot of pathological response for individual patients with infiltration profiles is shown in Figure 5A. To date, the CPS has been developed to predict the anti‐PD‐1 (pembrolizumab) therapy response in cancer patients [3]. Although CPS is an evaluating method with a high predictive value of improved survival across solid tumors [3, 34], its performance in predicting the responses of NACI treatments remains unclear especially in the case of advanced HNSCC. Within the cohort in this study, our data showed that responders tended to have high CPSs, whereas patients with low CPS were more likely to be non‐responders (Figure 5B). Similarly, the patients achieving MPR showed higher CPS than those with IPR (Supplementary Figure S6A). However, there was no correlation between the CPS and the radiographic or pathologic response in HNSCC patients (Figure 5C, Supplementary Figure S6B). Moreover, in a paired analysis of pre‐ and post‐NACI‐treated tumor samples, a dynamic change in CPS was not observed in the non‐responders. Interestingly, among responders and the MPR group, the CPS was inclined to decrease after NACI treatment, while this tendency was observed as inversed in the IPR group (Supplementary Figure S6C‐D), suggesting that CPS might be a weak predictor for NACI response in advanced HNSCC patients.
FIGURE 5.
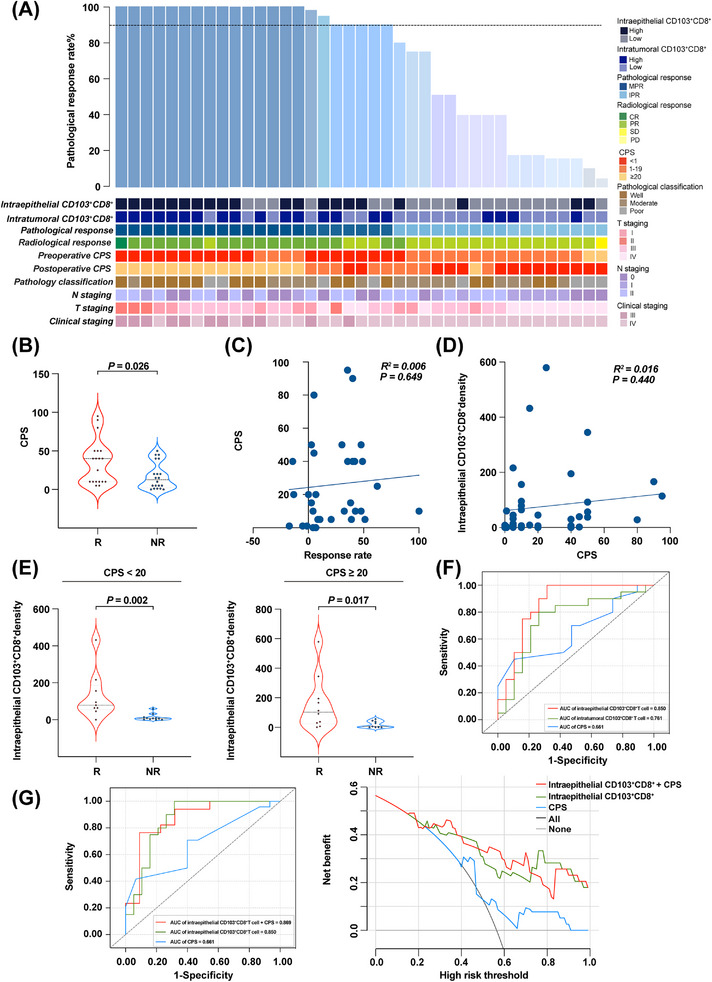
Comparison and association of CPS with CD103+CD8+ TIL level. (A) The waterfall plot depicting the correlation between the density of CD103+CD8+ TILs and clinicopathological features of individual patients who NACI (n = 39). The black horizontal line represents 90% pathological response. (B) CPS was assessed in the responders (R; n = 19) and non‐responders (NR; n = 20) with NACI. (C) Correlation between CPS and radiographic response (n = 39). (D) Correlation between intraepithelial CD103+CD8+ TILs and CPS (n = 39). (E) Intraepithelial CD103+CD8+ TILs were assessed in the responders (R; n = 9) and non‐responders (NR; n = 11) with NACI from CPS < 20 groups (left panel). Intraepithelial CD103+CD8+ TILs were assessed in the responders (R; n = 10) and non‐responders (NR; n = 9) with NACI from CPS ≥ 20 groups (right panel). (F) Predictive value of intratumoral CD103+CD8+ TIL level, intraepithelial CD103+CD8+ TIL level, and CPS via ROC analysis. (G) Predictive value of CPS was compared with intraepithelial CD103+CD8+ TIL level and elevated when combined with intraepithelial CD103+CD8+ TIL level (left panel). Decision curve analysis comparison of CPS, intraepithelial CD103+CD8+ TIL level, and CPS combined with intraepithelial CD103+CD8+ TIL level. Horizontal lines indicate the mean ± SEM. Significance was determined with a 2‐tailed Mann‐Whitney U test. Spearman correlation analyses were used to study the linear association. Abbreviations: TIL, tumor‐infiltrating cell; HNSCC, head and neck squamous cell carcinoma; MPR, major pathologic response; IPR, incomplete pathologic response; ROC, receiver operating characteristic; R, responders; NR, non‐responders; CPS, combined positive score; SEM, standard error of the mean; NACI, neoadjuvant chemoimmunotherapy.
Recent studies have reported that TILs can regulate PD‐L1 expression through various signaling pathways [9, 35, 36]. Therefore, we performed a thorough correlation analysis of PD‐L1 and intraepithelial CD103+CD8+ TILs. Notably, the CPS showed a negligible correlation with intraepithelial CD103+CD8+ TILs density in our cohort when analyzed as a continuous variable (Pearson's R2 = 0.016, P = 0.440) (Figure 5D). Meanwhile, we found that the infiltrated intraepithelial CD103+CD8+ TILs level was significantly associated with the response to NACI in both the CPS < 20 and CPS ≥ 20 groups (Figure 5E, Supplementary Figure S6E). These results demonstrated that the intraepithelial CD103+CD8+ TILs density could function as an independent predictor of NACI, which was not affected by PD‐L1 expression. More importantly, our data showed that 70.0% (14/20) and 75.0% (15/20) of the patients with a high density of CD103+CD8+ TILs could be identified as responders and patients with MPR, respectively (Supplementary Table S3). However, only 52.7% (10/19) of the patients within the responders had CPS ≥ 20 and 54.4% (12/22) of the patients with CPS ≥ 20 reached MPR (Supplementary Table S4). In addition, further statistical analyses showed that the density of intraepithelial CD103+CD8+ TILs displayed an enhanced predictive value for NACI in comparison with intratumoral CD103+CD8+ TILs (including both tumor and stromal compartments) and the CPS method alone (AUC = 0.850 for intraepithelial CD103+CD8+ TILs; AUC = 0.761 for intratumoral CD103+CD8+ TILs; AUC = 0.661 for CPS; Figure 5F). Intriguingly, ROC curve analyses showed that combining CPS with intraepithelial CD103+CD8+ TILs level elevated the predictive accuracy of CPS even more than either of them individually. We also revealed that the area under the decision curve of “intraepithelial CD103+CD8+ TILs plus CPS” was larger than that of any single variable (Figure 5G). This finding indicated that incorporating CPS with intraepithelial CD103+CD8+ TILs level significantly improved the predictive value of CPS, suggesting that this combined predictive model could be a promising diagnostic tool to identify advanced HSNCC patients who might be responsive to NACI treatment.
3.6. The TREM2+ macrophage subset may negatively regulate the density of CD103+CD8+ TILs in advanced HNSCC patients
To further dissect the functional characterization of CD103+CD8+ TILs in the tumor microenvironment, we utilized a cell‐cell interaction analysis to gain insights into the regulatory mechanism of CD103+CD8+ TILs (Figure 6A). We observed an extensive interaction of CD103+CD8+ TILs with various antitumor immunity cell subsets. In particular, the TREM2+ macrophage subset showed the strongest potential correlation with CD103+CD8+ TILs. Consistent with this finding, recent studies have shown that TREM2+ macrophages possess a potentially immunosuppressive role in multiple cancer types [37, 38], suggesting that the interaction between CD103+CD8+ TILs and TREM2+ macrophages could be negatively correlated in the tumor microenvironment. In Figure 6B, the bar plot shows the relative proportion of interaction strength for each signaling pathway between the NACI responders and non‐responders. The top signaling pathways colored red were enriched in the non‐responders (NR), and signaling pathways colored blue were enriched in the responders (R). Intriguingly, the pathways involved in the regulation of macrophage activity, such as secreted phosphoprotein 1 (SPP1) and macrophage migration inhibitory factor (MIF) signaling [39, 40], were significantly active in the non‐responders. Consistently, our results found that the ligand SPP1 with the multisubunit receptor ITGAV and the ligand MIF with the multisubunit receptor CD74 were active from TREM2+ macrophages to CD103+CD8+ TILs in the increased signaling of the non‐responders (Supplementary Figure S7). Our results showed that the interactions between TREM2+ macrophages and CD103+CD8+ TILs were associated with these specific pathways, suggesting that TREM2+ macrophages might employ the SPP1‐ITGAV or MIF‐CD74 immunosuppressive paracrine signaling axis to regulate CD103+CD8+ TILs. More importantly, our mIHC staining results revealed that the number of CD103+CD8+ TILs was significantly reduced in the tumor areas proximal to TREM2+ macrophages, whereas an increase in CD103+CD8+ TILs was observed in the tumor areas distal to TREM2+ macrophages (Figure 6C). To better investigate the spatial relationships between CD103+CD8+ TILs and TREM2+ macrophages, the image was analyzed through the HALO® platform (Figure 6D). Notably, proximity analysis histograms further illustrated the infiltration patterns among CD103+CD8+ TILs and TREM2+ macrophages, indicating that TREM2+ macrophages might abrogate the proliferative capacity of CD103+CD8+ TILs (Figure 6E). Consistent with this finding, we also found that a negative correlation between the CD103+CD8+TILs and TREM2+macrophages in both TNBC (Supplementary Figure S8) and NSCLC (Supplementary Figure S9). These results further supported the conclusion that CD103+CD8+ TILs subset might be negatively regulated by TREM2+ macrophages within the tumor microenvironment.
FIGURE 6.
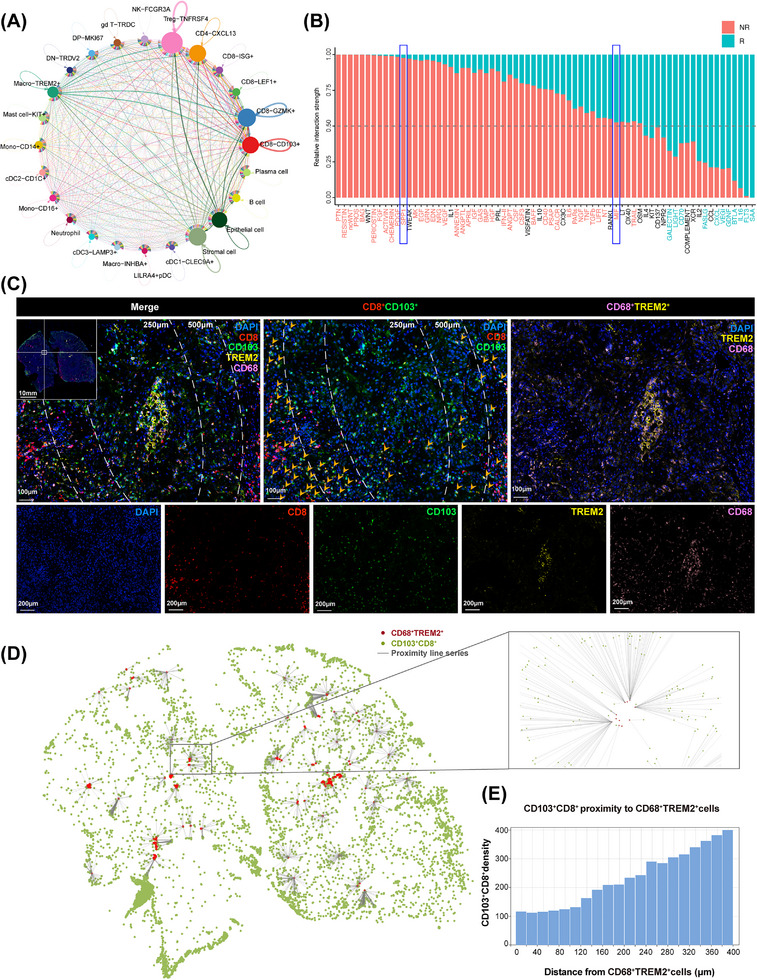
Correlation between CD103+CD8+ TILs and TREM2+ macrophages. (A) Correlative network showing interactions of cell subsets identified in advanced HNSCC tissues based on scRNA‐seq. The arrow width indicates the degree of correlation between two clusters. (B) Bar plot shows the relative proportion of interaction strength for each signaling pathway between responders and non‐responders. The pathways labeled blue are enriched in responders, and labeled red are enriched in non‐responders. R: responders; NR: non‐responders. (C) mIHC staining of TREM2+ macrophages marker CD68 (pink) and TREM2 (yellow), and CD103+CD8+ TILs marker CD103 (green) and CD8 (red), and DAPI staining (blue) in the advanced HNSCC with NACI. Scale bars, 100 μm. Quantification of CD103+CD8+ TILs at a distance of 250 μm or 500 μm from TREM2+ macrophages. (D) Proximity line series between CD103+CD8+ TILs and TREM2+ macrophages. (E) Proximity analysis histograms of CD103+CD8+ TILs to TREM2+ macrophages within a 400 μm radius by progressive segments of 20 μm bands. Abbreviations: TIL, tumor‐infiltrating cell; HNSCC, head and neck squamous cell carcinoma; mIHC, multiplex immunofluorescent; TREM2, triggering receptor expressed on myeloid cells 2; R, responders; NR, non‐responders; NACI, neoadjuvant chemoimmunotherapy; scRNA‐seq, single‐cell RNA sequencing.
4. DISCUSSION
We showed that advanced HNSCC patients with high levels of intratumoral CD103+CD8+ T cell infiltration possessed an increased response rate to NACI (Figure 7). The role of CD103+CD8+ T cells were further analyzed in patients with NSCLC and TNBC. Interestingly, the expression level of CD103+CD8+ T cell also increased in the responders of TNBC patients. These results indicated that CD103+CD8+ T cells might be important for predicting the NACI response. We successfully defined CD103+CD8+ TILs as tumor‐specific T cells that could work synergistically with anti‐PD‐1 therapy to repress tumor growth. We showed that incorporating CPS with intraepithelial CD103+CD8+ TILs level in a diagnostic model could improve its prediction accuracy of the NACI response in HNSCC patients. Overall, these findings offer a new guideline for the application of NACI treatment in advanced HNSCC patients.
FIGURE 7.
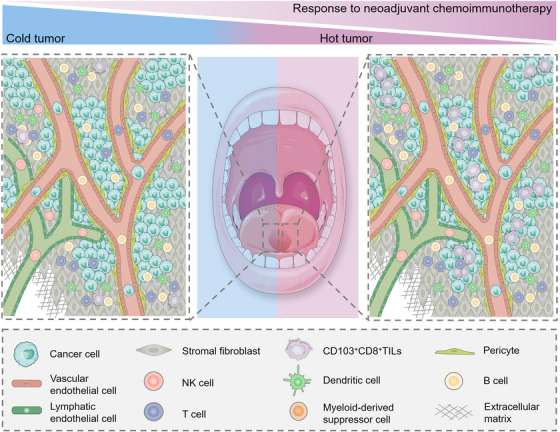
Schematic diagram represents the role of CD8+CD103+ TILs in regulating NACI efficacy. For advanced HNSCC patients treated with NACI, the responders exhibit a “hot tumor” (immune‐inflamed) phenotype, in which the density of intratumoral CD103+CD8+ TILs were increased, and these cells were mostly found in the cancer nest. In contrast, the non‐responders may have a “cold tumor” (immune‐desert or immune‐excluded) phenotype, in which the cell number of intratumoral CD103+CD8+ TILs were lower, and these cells were frequently located in the stroma. Abbreviations: TIL, tumor‐infiltrating cell; HNSCC, head and neck squamous cell carcinoma; NACI, neoadjuvant chemoimmunotherapy; NACI, neoadjuvant chemoimmunotherapy.
Previous studies have suggested that CD103+CD8+ T cells retain tissue‐resident properties and play an indispensable role in immune defense [41, 42, 43]. Interestingly, tissue‐resident memory T cells (TRM) have been shown to represent a lineage of T cells that specialize in function within tissues and play an important role in the control of solid tumors [44]. Emerging insights also showed that a high frequency of TRM cells correlates with favorable disease progression in cancer patients [44], while TRM cells are known to express high levels of immune checkpoint proteins, thereby exhibiting an exhausted and tumor‐reactive characterization in multiple solid tumors [45]. Indeed, we showed that CD103+CD8+ T cells had stronger cytotoxic activity and higher PD‐1 coinhibitory receptor expression than CD103−CD8+ T cells, while they were predominantly found in tumor tissues but not in PMBCs, confirming their tumor‐infiltrating nature. Further functional assays also confirmed CD103+CD8+T cells’ tumor reactive nature, as they could more effectively eliminate cancer cells compared to the CD103−CD8+ T cells. We also showed that administration of CD103+CD8+ T cells could enhance anti‐PD‐1 therapy efficacy against tumor growth in vivo, while FACS analyses further demonstrated that the strategy of anti‐PD‐1 treatment combined with CD103+CD8+ T cells effectively increased the release of antitumor immune response‐related cytokines. Based on these observations, we speculated that the increased CD103+CD8+ TILs density could help to restore antitumor T cell responses, thereby becoming an optimal immunological control of solid tumors in advanced HNSCC. However, recent studies have shown that CD103+CD39+CD8+ TILs are enriched for tumor‐reactive cells in HNSCC [46, 47]. This subset had a distinct T cell receptor (TCR) repertoire, with T cell clones expanded in the tumor. CD103+CD39+CD8+ TILs also efficiently kill autologous tumor cells in an MHC‐I‐dependent manner. Meanwhile, higher frequencies of CD103+CD39+CD8+ TILs in HNSCC patients are associated with better OS and response to immunotherapy [19]. Therefore, we will further analyze the proportion of CD39 coexpression on the CD103+CD8+ TILs populations in our future studies in an effort to explore the role of CD103+CD39+CD8+ TILs in HNSCC patients with NACI treatment.
By performing a spatial image analysis of CD103‐ and CD8‐immunostained tissue sections, we showed that the density of CD103+CD8+ TILs within cancer islands was more significantly associated with the efficacy of NACI in advanced HNSCC patients. The discovery of the intratumoral CD103+CD8+ T cell infiltration pattern highlighted the importance of lymphocyte infiltration in advanced HNSCC. Recent studies have defined 3 basic immune profiles that correlate with the response to immunotherapy [48]. The first profile was the immune‐inflamed phenotype, where immune cells infiltrated the nests of malignant cells as well as the surrounding and intervening stroma. The second profile was the immune‐excluded phenotype, in which most immune cells were retained in the stroma. The third profile, the immune‐desert phenotype, was characterized by a paucity of T cells in either the parenchyma or the tumor stroma [31]. In particular, the immune‐inflamed phenotype generally correlated with higher response rates to immunotherapy in cancer patients [32]. Therefore, the spatial feature of CD103+CD8+ TILs tended to be the immune‐inflamed phenotype, indicating a favorable response to NACI treatment. To further confirm the superior predictive power of CD103+CD8+ TILs for NACI, we evaluated the predictive value of CD103+CD8+ TILs level and CPS. To date, CPS has been used as a method to predict the clinical response of immunotherapy in HNSCC [8]. Interestingly, our data established intraepithelial CD103+CD8+ TILs as an independent predictor, which was superior to the CPS method. Notably, combining the CPS value with the intraepithelial CD103+CD8+ TILs level statistically elevated its predictive value and accuracy of NACI efficacy when compared with either CPS or CD103+CD8+ TILs alone. Collectively, our results suggested that the construction of a comprehensive, precise, and multivariable model using both CPS and CD103+CD8+ TILs evaluation data to predict NACI response in HNSCC is needed in clinical treatment if we want to improve patient outcome.
Considering the importance of CD103+CD8+ TILs, an in‐depth understanding of the regulatory mechanisms for CD103+CD8+ TILs could provide the possibility of converting non‐responsive cold tumors into responsive hot tumors, subsequently allowing non‐responders to benefit from NACI. We found that the density of CD103+CD8+ TILs and TREM2+ macrophages might be negatively correlated in the tumor microenvironment of HNSCC. Interestingly, consistent with this finding, we also demonstrated that CD103+CD8+ TILs showed a negative correlation with TREM2+ macrophages in both TNBC and NSCLC. Recent studies have demonstrated that TREM2+ macrophages exhibit an immunosuppressive phenotype in multiple solid tumors. Additionally, coculturing CD8+ T cells with TREM2+ macrophages has been shown to impede T cell proliferation, while knockdown of TREM2 in these macrophages led to favorable preclinical outcomes [49]. Therefore, we speculated that TREM2+ macrophages could suppress the expansion of CD103+CD8+ TILs in the tumor microenvironment. Furthermore, our ligand‐receptor network analyses revealed the cellular crosstalk between TREM2+ macrophages and CD103+CD8+ TILs, indicating that the SPP1 and MIF signaling pathways were specifically active from TREM2+ macrophages to CD103+CD8+ TILs. Previous work showed that SPP1 is an integrin‐binding glycophosphoprotein secreted by a variety of cell types, including macrophages, and mediates several signaling networks that participate in cancer progression through interactions with the integrin family. Integrins are a family of heterodimeric transmembrane glycoproteins mediating cell‐cell adhesion, migration, proliferation, and survival. The expression of SPP1 by macrophages has recently attracted considerable attention. Liu et al. [50] showed that blocking SPP1 or macrophage‐specific deletion of Spp1 in mice could increase cytotoxic T cell infiltration and sensitize hepatocellular carcinoma cells to immunotherapy. In addition, the knockdown of SPP1 in macrophages mitigated lung cancer progression and activated T cells, while SPP1 was shown to be highly expressed in glioma‐associated macrophages and was associated with poor prognosis in human glioblastoma [50]. Meanwhile, MIF is a ligand for membrane receptors, namely, the cluster of differentiation 74 (CD74) and chemokine (CXC motif) receptor (CXCR) 4 [51]. Based on the inflammatory context and cell types, MIF binds to individual receptors or receptor complexes that determine the functional activity of MIF. The established signaling receptor complexes include CD74/CXCR4 [52]. Macrophages are the predominant source of MIF. A recent study indicated that MIF activates myeloid‐derived suppressor cells (MDSCs) and tumor‐associated macrophages (TAMs), which act together to inhibit cytotoxic T cell and NK cell activity and thus allow tumor cells to escape antitumor immunity. Activation of MIF signaling has also been demonstrated to induce the expression of inflammatory cytokines and tumor progression in melanoma [40]. Thus, these SPP1‐ITGAV or MIF‐CD74 immunosuppressive signaling pathways may play a key role in regulating CD103+CD8+ TILs density by TREM2+ macrophages. Overall, these results demonstrated the potential of targeting TREM2+ macrophages as an effective strategy for enhancing the population of CD103+CD8+ TILs, which could ultimately reactivate the antitumor immune response in conjunction with chemoimmunotherapy.
5. CONCLUSIONS
In summary, our study demonstrated that patients with a high density of intratumoral CD103+CD8+ TILs had a superior response to NACI in advanced HNSCC, while these cells exhibited a strong cytotoxic phenotype against tumor cells in vitro and in vivo. Moreover, incorporating CPS with the CD103+CD8+ TILs level in a combined predictive model enhanced diagnosis with potential implications for personalized patient counseling and chemoimmunotherapy treatment selection. Thus, with an in‐depth understanding of the molecular mechanisms of intratumoral CD103+CD8+ TILs formation, function, and therapeutic benefit, we anticipated strategies that might help guide personalized precision medicine in NACI.
DECLARATIONS
AUTHORS’ CONTRIBUTIONS
Bowen Li designed and supervised the study, conducted the experiments, analyzed the data, and wrote the manuscript. Jinsong Li and Zhaoyu Lin designed the study, analyzed the data, and revised the manuscript. Siqi Ren, Tianjun Lan, Fan Wu, Suling Chen, and Xue Jiang conducted the experiments and analyzed the data. Chuying Huo, Zitian Li, Shule Xie, Donghui Wu, Ruixin Wang, Yanyan Li, Lin Qiu, Guoxin Huang, Shurui Li, Xiaojuan Wang, Meifeng Cen, Tingting Cai conducted experiments and provided reagents. All authors read and approved the final manuscript.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING INFORMATION
This work was supported by the National Natural Science Foundation of China (82272788, 82072990, 81903045, and 82072988), China Postdoctoral Science Foundation (2021M703692), Department of Health of Guangdong Province Science Foundation (A2022165 and A2021142), Guangzhou Municipal Science and Technology Project (202201011479), and Guangdong Science and Technology Development Fund (2019A1515011867 and 2020A1515010405).
AVAILABILITY OF DATA AND MATERIALS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All surgical tumor and blood samples used in this study were obtained from patients treated at Sun Yat‐sen Memorial Hospital. This study was approved by the Ethical Committee of Sun Yat‐sen Memorial Hospital (SYSKY‐2022‐503‐01), and the informed written consent of all participants was obtained for research with the collection of tissue and blood samples. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Sun Yat‐sen University (SYSU‐IACUC‐2020‐B0483).
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors have nothing to report.
Ren S, Lan T, Wu F, Chen S, Jiang X, Huo C, et al. Intratumoral CD103+CD8+ T cells predict response to neoadjuvant chemoimmunotherapy in advanced head and neck squamous cell carcinoma. Cancer Commun. 2023;43:1143–1163. 10.1002/cac2.12480
Siqi Ren, Tianjun Lan, Fan Wu, Suling Chen and Xue Jiang contributed equally to this work.
Contributor Information
Zhaoyu Lin, Email: linzhy35@mail.sysu.edu.cn.
Jinsong Li, Email: lijins@mail.sysu.edu.cn.
Bowen Li, Email: libw9@mail2.sysu.edu.cn.
REFERENCES
- 1. Vos JL, Elbers JBW, Krijgsman O, Traets JJH, Qiao X, van der Leun AM, et al. Neoadjuvant immunotherapy with nivolumab and ipilimumab induces major pathological responses in patients with head and neck squamous cell carcinoma. Nat Commun. 2021;12(1):7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: a time‐trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond). 2021;41(10):1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang J, Huang D, Saw PE, Song E. Turning cold tumors hot: from molecular mechanisms to clinical applications. Trends Immunol. 2022;43(7):523–45. [DOI] [PubMed] [Google Scholar]
- 4. Wang Y, Wang M, Wu HX, Xu RH. Advancing to the era of cancer immunotherapy. Cancer Commun (Lond). 2021;41(9):803–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Uppaluri R, Campbell KM, Egloff AM, Zolkind P, Skidmore ZL, Nussenbaum B, et al. Neoadjuvant and Adjuvant Pembrolizumab in Resectable Locally Advanced, Human Papillomavirus–Unrelated Head and Neck Cancer: A Multicenter, Phase II Trial. Clin Cancer Res. 2020;26(19):5140–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schoenfeld JD, Hanna GJ, Jo VY, Rawal B, Chen YH, Catalano PS, et al. Neoadjuvant Nivolumab or Nivolumab Plus Ipilimumab in Untreated Oral Cavity Squamous Cell Carcinoma: A Phase 2 Open‐Label Randomized Clinical Trial. JAMA Oncol. 2020;6(10):1563–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Z, Wu B, Peng G, Xiao G, Huang J, Ding Q, et al. Neoadjuvant Chemoimmunotherapy for the Treatment of Locally Advanced Head and Neck Squamous Cell Carcinoma: A Single‐Arm Phase 2 Clinical Trial. Clin Cancer Res. 2022;28(15):3268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burtness B, Harrington KJ, Greil R, Soulieres D, Tahara M, de jr Castro G, , et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE‐048): a randomised, open‐label, phase 3 study. Lancet. 2019;394(10212):1915–28. [DOI] [PubMed] [Google Scholar]
- 9. Smith JD, Bellile EL, Ellsperman SE, Heft‐Neal ME, Mann JE, Birkeland AC, et al. Prognostic value of CD103(+) tumor‐infiltrating lymphocytes and programmed death ligand‐1 (PD‐L1) combined positive score in recurrent laryngeal squamous cell carcinoma. Oral Oncol. 2022;135:106226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu B, Zhang Y, Wang D, Hu X, Zhang Z. Single‐cell meta‐analyses reveal responses of tumor‐reactive CXCL13(+) T cells to immune‐checkpoint blockade. Nat Cancer. 2022;3(9):1123–36. [DOI] [PubMed] [Google Scholar]
- 11. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706):575–9. [DOI] [PubMed] [Google Scholar]
- 12. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. [DOI] [PubMed] [Google Scholar]
- 13. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour‐specific mutant antigens. Nature. 2014;515(7528):577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, et al. Virus‐specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun. 2019;10(1):567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reuben A, Zhang J, Chiou SH, Gittelman RM, Li J, Lee WC, et al. Comprehensive T cell repertoire characterization of non‐small cell lung cancer. Nat Commun. 2020;11(1):603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Corgnac S, Malenica I, Mezquita L, Auclin E, Voilin E, Kacher J, et al. CD103(+)CD8(+) T(RM) Cells Accumulate in Tumors of Anti‐PD‐1‐Responder Lung Cancer Patients and Are Tumor‐Reactive Lymphocytes Enriched with Tc17. Cell Rep Med. 2020;1(7):100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anadon CM, Yu X, Hänggi K, Biswas S, Chaurio RA, Martin A, et al. Ovarian cancer immunogenicity is governed by a narrow subset of progenitor tissue‐resident memory T cells. Cancer Cell. 2022;40(5):545–57.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Banchereau R, Chitre AS, Scherl A, Wu TD, Patil NS, de Almeida P, et al. Intratumoral CD103+ CD8+ T cells predict response to PD‐L1 blockade. J Immunother Cancer. 2021;9(4):e002231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duhen R, Ballesteros‐Merino C, Frye AK, Tran E, Rajamanickam V, Chang SC, et al. Neoadjuvant anti‐OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen‐specific tumor‐infiltrating T cells. Nat Commun. 2021;12(1):1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Redman JM, Friedman J, Robbins Y, Sievers C, Yang X, Lassoued W, et al. Enhanced neoepitope‐specific immunity following neoadjuvant PD‐L1 and TGF‐β blockade in HPV‐unrelated head and neck cancer. J Clin Invest. 2022;132(18):e161400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47. [DOI] [PubMed] [Google Scholar]
- 22. Cottrell TR, Thompson ED, Forde PM, Stein JE, Duffield AS, Anagnostou V, et al. Pathologic features of response to neoadjuvant anti‐PD‐1 in resected non‐small‐cell lung carcinoma: a proposal for quantitative immune‐related pathologic response criteria (irPRC). Ann Oncol. 2018;29(8):1853–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hellmann MD, Chaft JE, jr William WN, , Rusch V, Pisters KM, Kalhor N, et al. Pathological response after neoadjuvant chemotherapy in resectable non‐small‐cell lung cancers: proposal for the use of major pathological response as a surrogate endpoint. Lancet Oncol. 2014;15(1):e42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lei X, Lin H, Wang J, Ou Z, Ruan Y, Sadagopan A, et al. Mitochondrial fission induces immunoescape in solid tumors through decreasing MHC‐I surface expression. Nat Commun. 2022;13(1):3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Chen H, Mo H, Hu X, Gao R, Zhao Y, et al. Single‐cell analyses reveal key immune cell subsets associated with response to PD‐L1 blockade in triple‐negative breast cancer. Cancer Cell. 2021;39(12):1578–93.e8. [DOI] [PubMed] [Google Scholar]
- 26. Lopez DC, Robbins YL, Kowalczyk JT, Lassoued W, Gulley JL, Miettinen MM, et al. Multi‐spectral immunofluorescence evaluation of the myeloid, T cell, and natural killer cell tumor immune microenvironment in chordoma may guide immunotherapeutic strategies. Front Oncol. 2022;12:1012058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rong H, Cai T, Peng Y, Wang X, Lan T, Ou Z, et al. Correlation Between TCF7(+) T Cells and Prognosis of Patients With Oral Squamous Cell Carcinoma. Front Oncol. 2022;12:782058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Forde PM, Spicer J, Lu S, Provencio M, Mitsudomi T, Awad MM, et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N Engl J Med. 2022;386(21):1973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schmid P, Dent R, O'Shaughnessy J. Pembrolizumab for Early Triple‐Negative Breast Cancer. Reply. N Engl J Med. 2020;382(26):e108. [DOI] [PubMed] [Google Scholar]
- 30. Liu B, Hu X, Feng K, Gao R, Xue Z, Zhang S, et al. Temporal single‐cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti‐PD‐1 therapy in lung cancer. Nat Cancer. 2022;3(1):108–21. [DOI] [PubMed] [Google Scholar]
- 31. Chen DS, Mellman I. Elements of cancer immunity and the cancer‐immune set point. Nature. 2017;541(7637):321–30. [DOI] [PubMed] [Google Scholar]
- 32. Salmon H, Remark R, Gnjatic S, Merad M. Host tissue determinants of tumour immunity. Nat Rev Cancer. 2019;19(4):215–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Y, Jia K, Sun Y, Zhang C, Li Y, Zhang L, et al. Predicting response to immunotherapy in gastric cancer via multi‐dimensional analyses of the tumour immune microenvironment. Nat Commun. 2022;13(1):4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de jr Castro G, , et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE‐048): a randomised, open‐label, phase 3 study. Lancet. 2019;394(10212):1915–28. [DOI] [PubMed] [Google Scholar]
- 35. Saliba M, Shaheen M, Hajj RE, Abbas F, Bashir S, Sheikh UN, et al. PD‐L1 expression in sebaceous carcinomas. Cancer Immunol Immunother. 2021;70(7):1907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu SM, Shi CJ, Xia RH, Wang LZ, Tian Z, Ye WM, et al. Analysis of Immunological Characteristics and Genomic Alterations in HPV‐Positive Oropharyngeal Squamous Cell Carcinoma Based on PD‐L1 Expression. Front Immunol. 2021;12:798424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, et al. Coupled scRNA‐Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell. 2020;182(4):872–85 e19. [DOI] [PubMed] [Google Scholar]
- 38. Nalio Ramos R, Missolo‐Koussou Y, Gerber‐Ferder Y, Bromley CP, Bugatti M, Nunez NG, et al. Tissue‐resident FOLR2(+) macrophages associate with CD8(+) T cell infiltration in human breast cancer. Cell. 2022;185(7):1189–207 e25. [DOI] [PubMed] [Google Scholar]
- 39. Qi J, Sun H, Zhang Y, Wang Z, Xun Z, Li Z, et al. Single‐cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat Commun. 2022;13(1):1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fukuda Y, Bustos MA, Cho SN, Roszik J, Ryu S, Lopez VM, et al. Interplay between soluble CD74 and macrophage‐migration inhibitory factor drives tumor growth and influences patient survival in melanoma. Cell Death Dis. 2022;13(2):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, et al. Fatty Acid Oxidation Controls CD8(+) Tissue‐Resident Memory T‐cell Survival in Gastric Adenocarcinoma. Cancer Immunol Res. 2020;8(4):479–92. [DOI] [PubMed] [Google Scholar]
- 42. Wang T, Shen Y, Luyten S, Yang Y, Jiang X. Tissue‐resident memory CD8(+) T cells in cancer immunology and immunotherapy. Pharmacol Res. 2020;159:104876. [DOI] [PubMed] [Google Scholar]
- 43. Corgnac S, Boutet M, Kfoury M, Naltet C, Mami‐Chouaib F. The Emerging Role of CD8(+) Tissue Resident Memory T (T(RM)) Cells in Antitumor Immunity: A Unique Functional Contribution of the CD103 Integrin. Front Immunol. 2018;9:1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mueller SN, Mackay LK. Tissue‐resident memory T cells: local specialists in immune defence. Nat Rev Immunol. 2016;16(2):79–89. [DOI] [PubMed] [Google Scholar]
- 45. Amsen D, van Gisbergen K, Hombrink P, van Lier RAW. Tissue‐resident memory T cells at the center of immunity to solid tumors. Nat Immunol. 2018;19(6):538–46. [DOI] [PubMed] [Google Scholar]
- 46. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co‐expression of CD39 and CD103 identifies tumor‐reactive CD8 T cells in human solid tumors. Nat Commun. 2018;9(1):2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Darragh LB, Knitz MM, Hu J, Clambey ET, Backus J, Dumit A, et al. A phase I/Ib trial and biological correlate analysis of neoadjuvant SBRT with single‐dose durvalumab in HPV‐unrelated locally advanced HNSCC. Nat Cancer. 2022;3(11):1300–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kandalaft LE, Dangaj Laniti D, Coukos G. Immunobiology of high‐grade serous ovarian cancer: lessons for clinical translation. Nat Rev Cancer. 2022;22(11):640–56. [DOI] [PubMed] [Google Scholar]
- 49. Obradovic A, Chowdhury N, Haake SM, Ager C, Wang V, Vlahos L, et al. Single‐cell protein activity analysis identifies recurrence‐associated renal tumor macrophages. Cell. 2021;184(11):2988–3005e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu L, Zhang R, Deng J, Dai X, Zhu X, Fu Q, et al. Construction of TME and Identification of crosstalk between malignant cells and macrophages by SPP1 in hepatocellular carcinoma. Cancer Immunol Immunother. 2022;71(1):121–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schwartz V, Lue H, Kraemer S, Korbiel J, Krohn R, Ohl K, et al. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009;583(17):2749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Alampour‐Rajabi S, El Bounkari O, Rot A, Müller‐Newen G, Bachelerie F, Gawaz M, et al. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP‐70 signaling, and lymphocyte chemotaxis. Faseb j. 2015;29(11):4497–511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.