

Abstract
Intracellular phosphate is critical for cellular processes such as signaling, nucleic acid synthesis, and membrane function. Extracellular phosphate (Pi) is an important component of the skeleton. Normal levels of serum phosphate are maintained by the coordinated actions of 1,25-dihydroxyvitamin D3, parathyroid hormone and fibroblast growth factor-23, which intersect in the proximal tubule to control the reabsorption of phosphate via the sodium-phosphate cotransporters Npt2a and Npt2c. Furthermore, 1,25-dihydroxyvitamin D3 participates in the regulation of dietary phosphate absorption in the small intestine. Clinical manifestations associated with abnormal serum phosphate levels are common and occur as a result of genetic or acquired conditions affecting phosphate homeostasis. For example, chronic hypophosphatemia leads to osteomalacia in adults and rickets in children. Acute severe hypophosphatemia can affect multiple organs leading to rhabdomyolysis, respiratory dysfunction, and hemolysis. Patients with impaired kidney function, such as those with advanced CKD, have high prevalence of hyperphosphatemia, with approximately two-thirds of patients on chronic hemodialysis in the United States having serum phosphate levels above the recommended goal of 5.5 mg/dL, a cutoff associated with excess risk of cardiovascular complications. Furthermore, patients with advanced kidney disease and hyperphosphatemia (>6.5 mg/dL) have almost one-third excess risk of death than those with phosphate levels between 2.4 and 6.5 mg/dL. Given the complex mechanisms that regulate phosphate levels, the interventions to treat the various diseases associated with hypophosphatemia or hyperphosphatemia rely on the understanding of the underlying pathobiological mechanisms governing each patient condition.
Phosphorus is a fundamental component of many of the normal cellular processes, as well as an integral part of the skeleton. Although often used interchangeably, the terms phosphorus and phosphate are not the same. Phosphorus is a basic chemical element with a molar mass of 30.97 g/mol, which in biologic systems is present as phosphate ions (PO43−), mass of 94.97 g/mol. In the body, phosphate (Pi) exists as inorganic phosphate, such as HPO42− and H2PO4−, in the organic form as part of nucleotides, phospholipids, adenosine 5ʹ-triphosphate or forming hydroxyapatite crystals with calcium in the skeleton, where approximately 90% of the Pi in the body resides.1,2
Intracellular Pi is required for cellular signaling, nucleic acid synthesis, and membrane function among others. Inorganic, extracellular Pi is measured clinically as an estimation of total body phosphate balance, and used to define hyperphosphatemia and hypophosphatemia.3 Chronic Pi deficiency leads to rickets in children or osteomalacia in adults, while acute hypophosphatemia might affect different organs with clinical manifestations such as rhabdomyolysis, respiratory dysfunction, or hemolysis.4 Not only low Pi levels are pathologic but also elevated serum Pi is associated with adverse clinical outcomes. For example, failure to adequately regulate Pi levels can lead to kidney stones and hyperphosphatemia, the latter associated with increased mortality in patients with advanced kidney failure.5 Individuals with serum Pi > 6.5 mg/dL have a 27% higher mortality risk than those with Pi levels between 2.4 and 6.5 mg/dL.5 One should note that the association of hyperphosphatemia in patients with CKD and mortality does not imply causality, as additional underlying factors in such patients can affect both Pi levels and mortality. When kidney function significantly decreases in advanced CKD, urinary excretion of Pi is no longer effective to maintain Pi balance and, therefore, prevention and mitigation of hyperphosphatemia are important therapeutic targets in CKD.6,7
Thus, maintaining normal serum Pi levels is important and involves the coordinated actions of several hormones including parathyroid hormone (PTH), fibroblastic growth factor 23 (FGF-23), and 1,25-dihydroxyvitamin vitamin D (1,25-dihydroxyvitamin D3). In addition, it depends on the normal functioning of several organs such as the parathyroid gland, small intestine, kidneys, and bones8 (Fig 1). In this review, we provide a summary of principles of Pi homeostasis under normal physiology and key aspects of the pathophysiology of hypophosphatemia and hyperphosphatemia, as well as a commentary on phosphate-driven interventions with emphasis in patients with advanced CKD.
Figure 1.
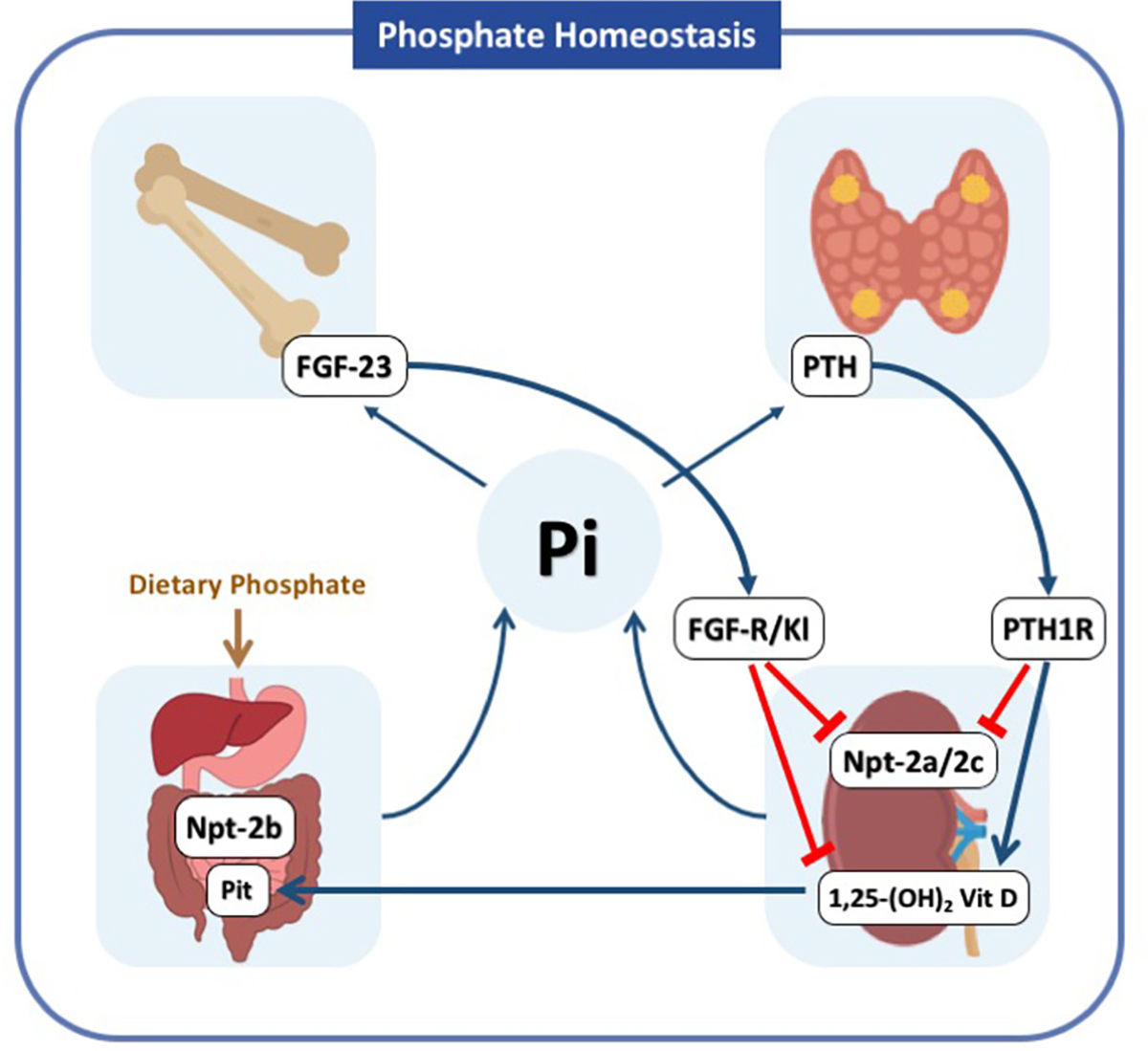
Phosphate Homeostasis, Maintaining normal serum Pi levels involves the coordinated actions of the parathyroid hormone (PTH), fibroblastic growth factor-23 (FGF-23), 1,25-dihydroxyvitamin D3 on target organs such as the parathyroid gland, small intestine, kidneys and bones. When Pi levels increase FGF-23 is secreted from the bone and PTH is secreted from the parathyroid gland. FGF-23 binds to the FGF receptor and its coreceptor Klotho in the proximal renal tubule, while PTH binds to the PTH receptor (PTH1R). The actions of FGF-23 include inhibition of 1α-hydroxylase and the Pi transporters Npt2a and Npt2c with the net effect of decreasing Pi reabsorption in the kidneys and indirectly reducing Pi absorption in the intestine due to reduced levels of 1,25-dihydroxyvitamin D3. PTH also reduces the abundance of Npt2a and Npt2c; however, it has a stimulatory effect on 1α-hydroxylase, which is critical to maintain normal levels of calcium. Abbreviations: 1,25 (OH)2 vit D, 1,25-dihydroxyvitamin D3; PTH, parathyroid hormone; FGF-23, fibroblast growth factor-23; PTH1R, parathyroid hormone–related peptide receptor type 1; Npt2a, type II sodium phosphate cotransporter a; Npt2b, type II sodium phosphate cotransporter b; Npt2c, type II sodium phosphate cotransporter c; FGF-R, fibroblast growth factor receptor; Kl, klotho.
PHOSPHATE HOMEOSTASIS AND FACTORS REGULATING RENAL PHOSPHATE EXCRETION
Pi levels in the body are determined by dietary uptake in the small intestine and urinary excretion by the kidneys (Fig 1). Daily dietary requirements are ~1.2 g in adults, yet the total phosphate pool in the body is ~800 g in a 70-kg person, indicating a small daily turnover that must be precisely regulated to maintain functionality of physiological processes.3,8
Uptake of Pi from the diet occurs in the small intestine. A Westernized diet results in almost complete Pi absorption from the food.6 The majority (>90%) of intestinal Pi transport is estimated to be active, transepithelial, and Na+-dependent,9,10 with the remainder being absorbed passively through paracellular transport (Fig 1).11 Pi transport in the mouse is mostly confined to the ileum where the sodium-phosphate cotransporter Npt2b (SLC34A2) is expressed12; however, it is still unclear how dietary Pi and 1,25-dihydroxyvitamin D3 are able to regulate Npt2b expression.13 Although chronic changes in Pi intake regulate the levels of 1,25-dihydroxyvitamin D3, the dietary adaptation of Npt2b seems to be independent from 1,25-dihydroxyvitamin D3 levels, because it is preserved in animal models in which either the signaling or the expression of 1,25-dihydroxyvitamin D3 is abolished.14,15 Transgenic mice that lack Npt2b exhibit embryonic lethality.16 However, studies using whole body conditional and inducible Npt2b9 and noninducible intestinal mucosaspecific Npt2b knockout mice identified that >90% of Na+-dependent active Pi transport is mediated by Npt2b, indicating that transporters from the phosphate inorganic transport (Pit) family are of limited significance in the intestine. Of note, more recent studies suggest that the paracellular pathway can be of significant importance.17 In contrast to the regulation of Npt2a and Npt2c by PTH in the kidney, Npt2b in the intestine has so far not been described to be regulated by PTH,18 even though PTH receptors have been identified in intestinal epithelia.19
In the proximal tubule, Npt2a (SLC34A1) and Npt2c (SLC34A3) mediate Pi reabsorption.20 The primary hormones that regulate renal Pi transport are PTH and FGF-23, which are produced by the parathyroid gland and osteocytes and osteoblasts in bone, respectively. Normally, an increase in serum Pi concentration induces secretion of PTH and FGF-23. These 2 phosphaturic hormones reduce luminal membrane abundance and the expression of Npt2a and Npt2c, in the proximal tubules, thereby reducing Pi reabsorption and increasing urinary Pi excretion.8,21 Fractional Pi excretion is between 5%–20% dependent on dietary phosphate content. The contribution of Npt2a and Npt2c for renal Pi transport has been delineated in gene targeted mice and mutations of phosphate transporters in humans (Fig 2). Studies in Npt2a knockout mice (Npt2a−/−) identified that Npt2a contributes to 70% of renal phosphate reabsorption.22 Npt2a−/− mice are born with~20% lower body weight and have skeletal abnormalities; however, after 13 weeks the differences in body weight between Npt2a−/− and WT mice disappear.23 Npt2c−/− mice do not have a phosphate phenotype but exhibited hypercalcemia, hypercalciuria, and increased 1,25-dihydroxyvitamin D3 levels. Npt2a/c double knockout mice display a blend of both phenotypes: hypophosphatemia, more severe phosphaturia, increased 1,25-dihydroxyvitamin D3 levels, and hypercalciuria23 (Fig 2). Humans with heterozygous mutations in the Npt2a gene presenting with urolithiasis or bone demineralization have hypophosphatemia and urinary phosphate loss24; however, a clear connection of these mutations to the phenotype has never been clearly established.25,26 Single nucleotide deletions, compound heterozygous deletions and missense mutations in the Npt2c gene are the cause of hereditary hypophosphatemic rickets with hypercalciuria.27 Clinically more common are abnormalities in phosphate homeostasis observed in CKD, which include secondary hyperparathyroidism and elevated FGF-23 levels.28 Circulating concentrations of FGF-23 increase progressively as kidney function declines.28 The latest research on FGF-23 points to a key role of this hormone in the pathogenesis of left ventricular hypertrophy, although studies in humans demonstrating a causal role are still needed.29
Figure 2.
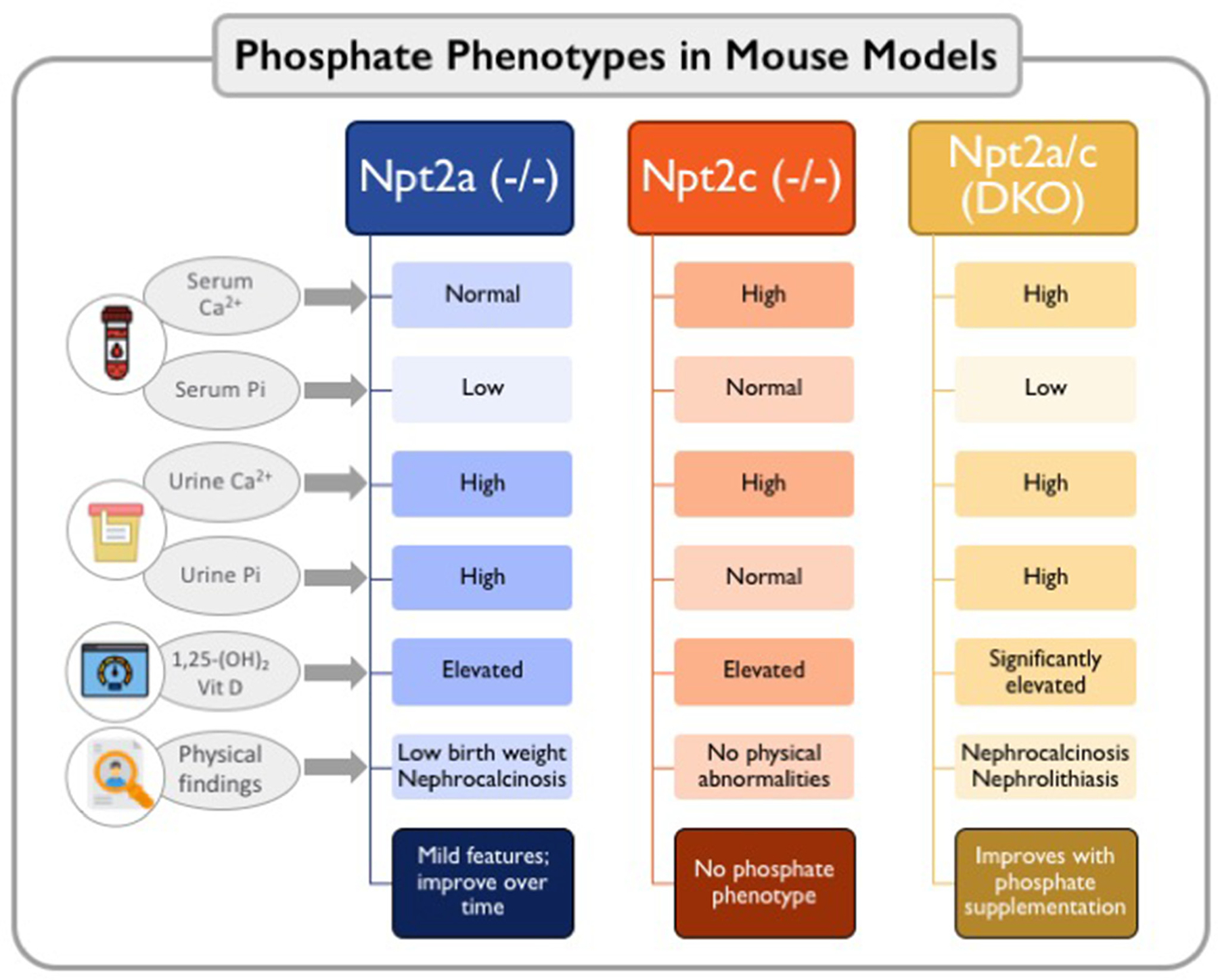
Phosphate phenotypes in genetically modified mouse models, Knockout of Npt2a and Npt2c demonstrate the contribution of each of these transporters for renal Pi transport in mice. Npt2a contributes to 70% of renal Pi reabsorption. Mice Npt2a−/− have hyperphosphaturia and lower levels of serum Pi. Incontrast, Npt2c−/− mice do not have a phosphate phenotype, but exhibit hypercalcemia, hypercalciuria, and increased 1,25-dihydroxyvitamin D3 levels. One should notice that the findings of the relative contributions of Npt2a and Npt2c to Pi homeostasis might differ in humans. Npt2c mutations have been found in patients with hereditary hypophosphatemic rickets with hypercalciuria (HHRH), while Npt2a mutations have not been clearly associated with abnormal Pi handling in humans. Npt2a/c double knockout mice show more severe hypophosphatemia, phosphaturia, increased 1,25-dihydroxyvitamin D3 levels, and hypercalciuria, similar to patients with HHRH. Abbreviations: DKO, double knockout; Npt2a, type II sodium phosphate cotransporter a; Npt2c, type II sodium phosphate cotransporter c.
PATHOPHYSIOLOGY OF HYPOPHOSPHATEMIA
Hypophosphatemia is rare in the general population (<1%),30 but is present in about 3% of hospitalized patients, and one third of patients admitted to the intensive care unit,31 in which it increases morbidity significantly.32
Sustained hypophosphatemia occurs when the renal excretion of Pi is increased relative to the amount of absorbed Pi in the intestine or less commonly when extracellular Pi is shifted into the cells or skeletal tissue, such as during refeeding or hungry bone syndromes.
Decreased Phosphate Intake or Gastrointestinal Losses
Dietary Pi depletion alone rarely causes hypophosphatemia unless this is sustained, accompanied by medication, alcohol use or with concomitant renal phosphate wasting8 (Table 1). Low Pi intake increases Npt2b in epithelial cells of the small intestine14,33 by vitamin D-dependent or independent pathways 14,34 and also increases the abundance of Pi transporters in the brush border membrane of the proximal tubule, namely Npt2a and Npt2c.14 This will efficiently prevent Pi losses during acute Pi depletion.35 The efficiency of renal Pi reabsorption under conditions of chronic low Pi intake or increased gastrointestinal losses is illustrated in mice with inducible knockout of Npt2b.9 Despite a 2-fold increase in fecal Pi excretion, Npt2b−/−mice have normal serum Pi levels of that correlate with a ~2-fold increase in renal Npt2a.9
Table 1.
Causes of Hypophosphatemia
| Decreased Intestinal Absorption | Examples | Pathogenesis | Targeted Interventions |
|---|---|---|---|
|
| |||
| Genetic | |||
| 1–25(OH)2 Vit D deficiency | CYP27B1 and CYP2R1 mutations in infantile forms of vitamin D-dependent rickets | Deficiency of 1-α hydroxylase or 25-α hydroxylase causing lower 1–25(OH)2 resulting in less Pi absorption | Active vitamin D supplementation |
| Acquired | |||
| Low phosphate intake | Chronic low intake (<100 mg/day) | Rarely, chronic low Pi intake may exceed renal capacity for reabsorption | |
| Medication use | Magnesium- and aluminum-based antiacids, niacin | Binding phosphorus by antiacids or inhibition of Npt2b by niacin | |
| Chronic diarrhea | Inflammatory bowel disease, pancreatic insufficiency | Concomitant impairment of vitamin D and Pi intestinal absorption | |
| Increased urinary excretion | |||
| Genetic Npt2c mutations | HHRH | Pi wasting in the proximal tubule, low levels of FGF-23, high vitamin D with increase in calcium-phosphate in renal tubule | Pi supplementation. Avoid vitamin D |
| Elevated iFGF-23 | XLH, ADHR, ARHR | Missense mutations involving the cleavage site at RXXR leading to high levels of intact FGF-23 (ADHR), involving PHEX (XLH) or DMP1, ENPP1, FAM20 C (ARHR). | Vitamin D supplementation. Monoclonal antibody anti-FGF-23 in XLH |
| Acquired | |||
| Elevated iFGF-23 | Tumor-induced osteomalacia, iron infusions | Mesenchymal tumors with unregulated secretion of FGF-23 or certain IV iron preparations | Tumor removal in tumor-induced osteomalacia |
| Proximal tubule dysfunction | Multiple myeloma, tenofovir, severe hypokalemia | Pi wasting in the proximal tubule by direct toxicity with or without Fanconi syndrome | Pi supplementation |
| Extracellular clearance | |||
| Acquired Intracellular redistribution | Refeeding syndrome, respiratory alkalosis | Insulin mediated intracellular redistribution (refeeding). Increased cellular pH and glycolysis (respiratory alkalosis) | |
| Increased skeletal uptake | Hungry bone syndrome | Postparathyroidectomy in patients with tertiary hyperparathyroidism | Control of PTH during CKD |
| Dialysis | Hemodialysis, CRRT (both diffusion and convective clearance modalities) | Nonselective clearance delivered with low Pi dialysate/replacement solution lowers serum Pi | Use of Pi containing replacement fluids, adjusting dialysis clearance, pre-emptive Pi replacement protocols |
| Critical illness | Hospitalized patients, especially in the ICU | Redistribution by glycolysis activation, hormonal triggers. Decreased intestinal absorption of Pi and use of CRRT without pre-emptive Pi replacement | Dynamic monitoring and pre-emptive Pi replacement |
Abbreviations: ADHR, autosomal dominant hypophosphatemic rickets; ARHR, autosomal recessive hypophosphatemic rickets; CRRT, continuous renal replacement therapy, DMP1, dentin matrix acidic phosphoprotein 1; ENPP1, ectonucleotide pyrophosphatase/phosphodiesterase 1; FAM20C, family with sequence similarity 20 member C; FGF-23, fibroblast growth factor 23; Gsa, alpha subunit of stimulatory G protein; HHRH, hereditary hypophosphatemia rickets with hypercalciuria; Npt-2a, type II sodium phosphate cotransporter a; Npt-2b, type II sodium phosphate cotransporter b; Npt-2c, type II sodium phosphate cotransporter c; PHEX, phosphate regulating endopeptidase homolog X-linked; PTH, parathyroid hormone; TIO, tumor-induced osteomalacia; XLH, X-linked hypophosphatemia.
Decreased Phosphate Reabsorption in the Proximal Tubule
Homozygous or compound heterozygous (2 distinct mutations inherited from each parent in the same gene) loss-of-function mutations in the human gene SLC34A3 are present in patients with hereditary hypophosphatemic rickets with hypercalciuria, where the renal capacity of Pi reabsorption is significantly reduced.36,37 Patients with hypophosphatemic rickets with hypercalciuria develop hypophosphatemia in the first decade of life despite appropriately suppressed levels of FGF-23. These patients have also a compensatory increased activity of 1-alpha hydroxylase, leading to high levels of 1,25-dihydroxyvitamin D3. In addition to enhanced intestinal Pi absorption, 1,25-dihydroxyvitamin D3 downregulates PTH synthesis and secretion. These vitamin D–mediated physiologic changes result in a partial compensatory effect related to circulating Pi levels, but also an increase in calcium absorption in the intestine, which leads to an increase in urinary calcium excretion. The corollary is that patients with genetic defects that affect the proximal tubule Pi transporters have an elevated risk of nephrolithiasis, in addition to their bone abnormalities secondary to hypophosphatemia. In such patients, phosphate supplementation without additional vitamin D can mitigate the risk of osteomalacia.36,38
Under conditions of high Pi intake, PTH and FGF-23 are physiologic regulators of Npt2a and Npt2c.39 However, pathologic elevations of FGF-23 downregulate Npt2a and Npt2c despite normal or even low serum Pi levels.40 Indeed, FGF-23 was first identified when 2 independent groups studied patients with the genetic condition of autosomal dominant hypophosphatemic rickets (ADHR) or the acquired condition of tumor-induced osteomalacia.41,42 ADHR is caused by mutations in FGF-23 that render it cleavage resistant, thereforeFGF-23in patients with ADHR show predominantly intact FGF-23 (iFGF-23), which is biologically active and leads to hypophosphatemia. Tumor-induced osteomalacia is caused by elevated production of FGF-23 by tumor cells.42 Shortly after these discoveries, elevated levels of iFGF-23 were also found to be causative of hypophosphatemia in patients with X-linked hypophosphatemia,43 carrying mutations in the phosphate regulating endopeptidase homolog X-linked (PHEX) protein.44 PHEX is a cell surface-bound protein-cleaving enzyme found in bone and teeth, but its role in phosphate metabolism is related to modulating levels of FGF-23. Thus, loss-of-function mutations in PHEX result in increased levels of FGF-23 by unclear mechanisms.45 Inhibition of FGF-23 by using the monoclonal anti-FGF-23 antibody (Burosumab), improved Pi handling and bone abnormalities in patients with X-linked hypophosphatemia,46 which is in line with studies in mice where double deletion of PHEX and FGF-23 prevented hypophosphatemia.47 In ADHR and X-linked hypophosphatemia, the elevated iFGF-23 suppresses the synthesis of 1,25-dihydroxyvitamin D3. Therefore, the combination of hypophosphatemia and low vitamin D results in severe osteomalacia, but the presence of nephrolithiasis is less characteristic than in hypophosphatemic rickets with hypercalciuria and mostly related to Pi supplementation.48
Elevated iFGF-23 can also result from acquired conditions other than tumor-induced osteomalacia, such as with the use of certain intravenous formulations for treatment of iron deficiency anemia, particularly ferric carboxymaltose.49 While iron deficiency elevates FGF-23 via HIFα, this is typically only C-terminal and not active iFGF-23.50 In contrast, via inhibition of FGF-23 cleavage,51 some iron preparations have been commonly associated with hypophosphatemia.49 In a pooled analysis of clinical trials with a total of 6879 treated with ferric carboxymaltose, 41% of patients developed hypophosphatemia (<2.5 mg/dL), predominantly within 2 weeks of treatment.52 Of note, ferric derisomaltose shows a significantly lower rate (~8%) than other iron preparations.53 In addition to hormonal-mediated inhibition of Pi reabsorption, direct tubular injury in the proximal tubule as seen inFanconisyndrome54 is often accompanied by increased Pi excretion. Severe hypokalemia can also decrease the activity of Npt2 cotransporters in the kidney,55 which could be misinterpreted as being associated with renal tubular acidosis.
Hypophosphatemia in Critically Ill Patients and Those Treated with Continuous Renal Replacement Therapy
Scenarios such as critical illness with limited exposure to dietary Pi and organ dysfunction (e.g., gastrointestinal dysfunction, acute kidney injury, and so on) affect the compensatory mechanisms of Pi handling, predisposing to hypophosphatemia.56,57 Patients with a history of alcoholism are particularly prone to develop hypophosphatemia during hospitalizations due to a combination of decreased intake, vitamin D efficiency and intracellular shifts promoted by glucose containing solutions or respiratory alkalosis.58
In critically ill patients with kidney dysfunction, hypophosphatemia is especially common when continuous extracorporeal clearance of Pi is provided through continuous renal replacement therapy (CRRT).32,57 Indeed, hypophosphatemia is a frequent complication of CRRT use, especially when using dialysate/replacement solutions without Pi. A recent retrospective review of hospitalized patients revealed that among 343 patients undergoing CRRT with Pi-free solutions, 58% had at least 1 episode of hypophosphatemia.32 Catabolism during acute illnesses and malnutrition in the context of increased Pi clearance 32 are proposed possible mechanisms. The use of commercially available phosphate-containing solutions with 1 to 1.2 mmol/L of phosphate might help to maintain normal levels of phosphate and mitigate complications related to hypophosphatemia during CRRT.32
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
In contrast to hypophosphatemia, there is a relatively high prevalence (~15%) of high serum Pi levels (≥4.4 mg/dL) in the general population.30 In patients with CKD, the mean levels of Pi increase significantly only in advanced stages of CKD (estimated glomerular filtration rate <20 mL/min) as compared to earlier CKD stages.59 Both, in the general population and in patients with CKD, hyperphosphatemia is associated with adverse cardiovascular outcomes.60–62
Increased Extracellular Load of Phosphate as a Cause of Hyperphosphatemia
Conditions that acutely increase the extracellular amount of Pi may exceed the kidney capacity for excretion, such as the now rare cases of Pi load by ingestion of Pi containing laxatives.63 Given that Pi is predominantly in the bones bound to calcium (85%) or intracellularly (14%),64 a shift from these sites to the extracellular compartment can rapidly increase serum Pi levels, especially with a concomitant decrease in kidney function, as is the case during tumor lysis syndrome or rhabdomyolysis (Table 2).65
Table 2.
Causes of Hyperphosphatemia
| Increased Extracellular Phosphate Load | Examples | Pathogenesis | Targeted Interventions |
|---|---|---|---|
|
| |||
| Acquired | |||
| High phosphate intake | Acute Pi intake, phosphate-containing laxatives | Decreased GFR or large acute intakes of phosphate can overwhelm renal Pi excretion capacity | |
| Tissue or cell injury | Tumor lysis syndrome, rhabdomyolysis, ketoacidosis, lactic acidosis | Pi is the major intracellular anion. Acute cellular release of Pi due to cellular injury or acid-base disturbances can result in extracellular Pi increase | Intravenous fluids to increase GFR |
| Increased intestinal absorption of phosphate | |||
| Acquired 1–25(OH)2 Vit D excess | High intake of vitamin D supplements | Vitamin D increases Pi absorption and decreases PTH secretion, thereby also limiting Pi excretion | |
| Decreased urinary excretion | |||
| Genetic Familial tumoral calcinosis | Mutations in FGF-23, GALNT3 or Klotho genes | Mutations in GALNT3 result in lower levels of biologically active FGF-23 by increased cleavage. Mutations in FGF-23 or its co-receptor klotho, directly impair FGF-23 mediated Pi excretion | |
| PTH insufficiency | PTH mutations, GNAS mutations, PTH1R mutations | PTH mutations result in hypoparathyroidism, while downstream mutations such as the PTH1R or Gsα result in PTH resistance (PHP) | |
| Acquired | |||
| PTH insufficiency | Surgical or autoimmune hypoparathyroidism | Increased reabsorption of Pi due to impaired PTH mediated Pi excretion. Associated with hypocalcemia | PTH analogs in hypoparathyroidism |
| Lower GFR | AKI, CKD | Occurs mainly at GFR falls below 20 to 25 mL/min | Phosphate binders, dialysis |
| Medications | FGFR inhibitors, etidronate | Increased reabsorption of Pi due to pharmacological blockage of FGF-23 actions | |
Abbreviations: FGF-23, fibroblast growth factor-23; GALNT3, uridine diphosphate-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylga-lactosaminyltransferase 3; Gsα, alpha subunit of stimulatory G protein; PHP, pseudohypoparathyroidism; PTH, parathyroid hormone; PTH1R, parathyroid hormone/parathyroid hormone-related peptide receptor type 1.
Hyperphosphatemia because of Abnormal PTH or FGF-23 Signaling
PTH acts in the proximal tubule by binding the parathyroid hormone receptor (PTH1R), a class B G protein-coupled receptor. Upon ligand binding, PTH1R acts as a guanine factor exchange which dissociates the alpha stimulatory portion of the G-protein (Gsα) and Gq/11 from their heterotrimeric complex, a required step to generate second messengers such as cyclic adenosine monophosphate, generated by adenylyl cyclase 6,66 and calcium.67 Patients with surgical or congenital hypoparathyroidism commonly have hyperphosphatemia and hypocalcemia which respond favorably to treatment with PTH analogs.68 On the other hand, patients with pseudohypoparathyroidism (PHP) have similar mineral disorders as patients with hypoparathyroidism, but do not respond to PTH supplementation. PTH resistance in PHP is most commonly caused by maternally inherited mutations in the GNAS locus encoding Gsα,69 and mutations in the PTH1R have only recently been identified as possible cause of PTH resistance.70,71 In patients with PHP, active vitamin D supplementation restored the normal levels of serum calcium and might mitigate the bone abnormalities without a substantial increase in the risk of nephrolithiasis.72
Loss of function mutations affecting FGF-23 signaling pathway occur due to mutations in FGF-23 itself,73 increased cleavage of FGF-23 due to GALNT3 mutations74 or genetic absence of the FGF-23 co-receptor αklotho.75 Collectively, these mutations are associated with hyperphosphatemia in the clinical syndrome of familial tumoral calcinosis. Biologically active iFGF-23 is therefore of significant importance for hyperphosphatemia prevention in patients with normal kidney function, but it is perhaps more critical as a compensatory mechanism when GFR declines. For example, in mice with adenine-induced CKD, conditional deletion of FGF-23 results in more profound aortic calcification and cardiac hypertrophy than in CKD mice with elevated intact FGF-23.76
Hyperphosphatemia in Patients with Reduced Kidney Function
Of the 4–8 g of Pi that are daily filtered by the kidneys, less than 20% are normally excreted in the urine. As glomerular filtration decreases during CKD, the reabsorption of Pi also is suppressed by concomitant increase of FGF-23 and PTH, such that the levels of serum Pi typically remain normal until GFR falls below 20 mL/min.59
Patients with CKD have also lower levels of αklotho. Klotho is the co-receptor of FGF-23 and is also encountered in a soluble form. Klotho deficiency in CKD, as demonstrated by low serum and urine levels, might further limit the actions of FGF-23 in the renal tubule as kidney function de-creases.77
Once patients with CKD become severely oliguric or anuric, they depend on dialysis for efficient Pi removal from the extracellular pool. Patients on a low phosphate diet would have a daily intake of ~12.6 mg of phosphate per Kg of body weight, which corresponds to ~880 mg per day or ~6.2 g per week in a 70 kg person.78 A single 4-hour hemodialysis session removes only ~1 g of Pi (~3g removal with 3/weekly schedule) available in the blood compartment during hemodialysis, which is comparable to the weekly removal of Pi by different peritoneal dialysis modalities.79 Thus, hyperphosphatemia is a common problem in patients with end-stage kidney disease, 60% of patients on hemodialysis in the United States have serum Pi levels above the recommended goal of 5.5 mg/dL.6,7
CLINICAL DATA OF PHOSPHATE-DRIVEN INTERVENTIONS IN PATIENTS WITH CKD
Hyperphosphatemia has many deleterious clinical consequences. Although most attention is focused on the associations between hyperphosphatemia and cardiovascular complications among patients with advanced CKD,62 elevated serum Pi levels have been shown to increase the risk of clinical complications even among individuals without advanced CKD. Moreover, hyperphosphatemia also increases the risk for hyperparathyroidism, skeletal fractures, CKD progression, skeletal muscle atrophy, and immune dysfunction.80
One large community-based cohort study evaluated serum Pi as a risk factor for cardiovascular events in the general population.81 The cohort comprised of 24,184 individuals with normal kidney function, 20,356 with CKD stages 1–2, and 13,292 with CKD stages 3–5. In models adjusted for cardiovascular risk factors, serum Pi levels were associated with a graded increase in the risk of a composite outcome of all-cause mortality, cardiovascular events, and advanced coronary artery disease. In the sub-cohort of individuals with normal kidney function, even high normal Pi levels ranging between 3.9 mg/dL and 4.6 mg/dL were observed to have ~35% increased risk for the composite outcome when compared to levels ranging between 2.3 mg/dL and 3.1 mg/dL. Target Pi levels and the efficacy and safety of Pi-based interventions; however, remain unconfirmed among individuals with normal kidney function.
Among patients with CKD stages 3–5 including those treated with dialysis, the Kidney Disease: Improving Global Outcomes 2017 guideline and Kidney Disease Outcomes Quality Initiative commentary recommend lowering elevated Pi levels toward the normal range.82,83 Dietary Pi restriction to 0.9 g/day and Pi binder therapies are mainstays of interventions for hyperphosphatemia among patients with CKD (Fig 3). More stringent dietary restrictions may invite the risk of protein-energy malnutrition and are also frequently impractical. Phosphate binder therapies can be effective for lowering serum Pi levels but are frequently associated with gastrointestinal adverse events and contribute significantly toward the pill burden (Table 3).84 A novel treatment option includes an intestinal-specific NHE3 inhibitor. Tenapanor was shown to reduce serum Pi levels with a mean decrease of ~1.4 mg/dL in patients on maintenance dialysis after 26 weeks of treatment.85 However, studies in intestinal-specific NHE3 knockout mice do not support this Pi lowering effect.86
Figure 3.
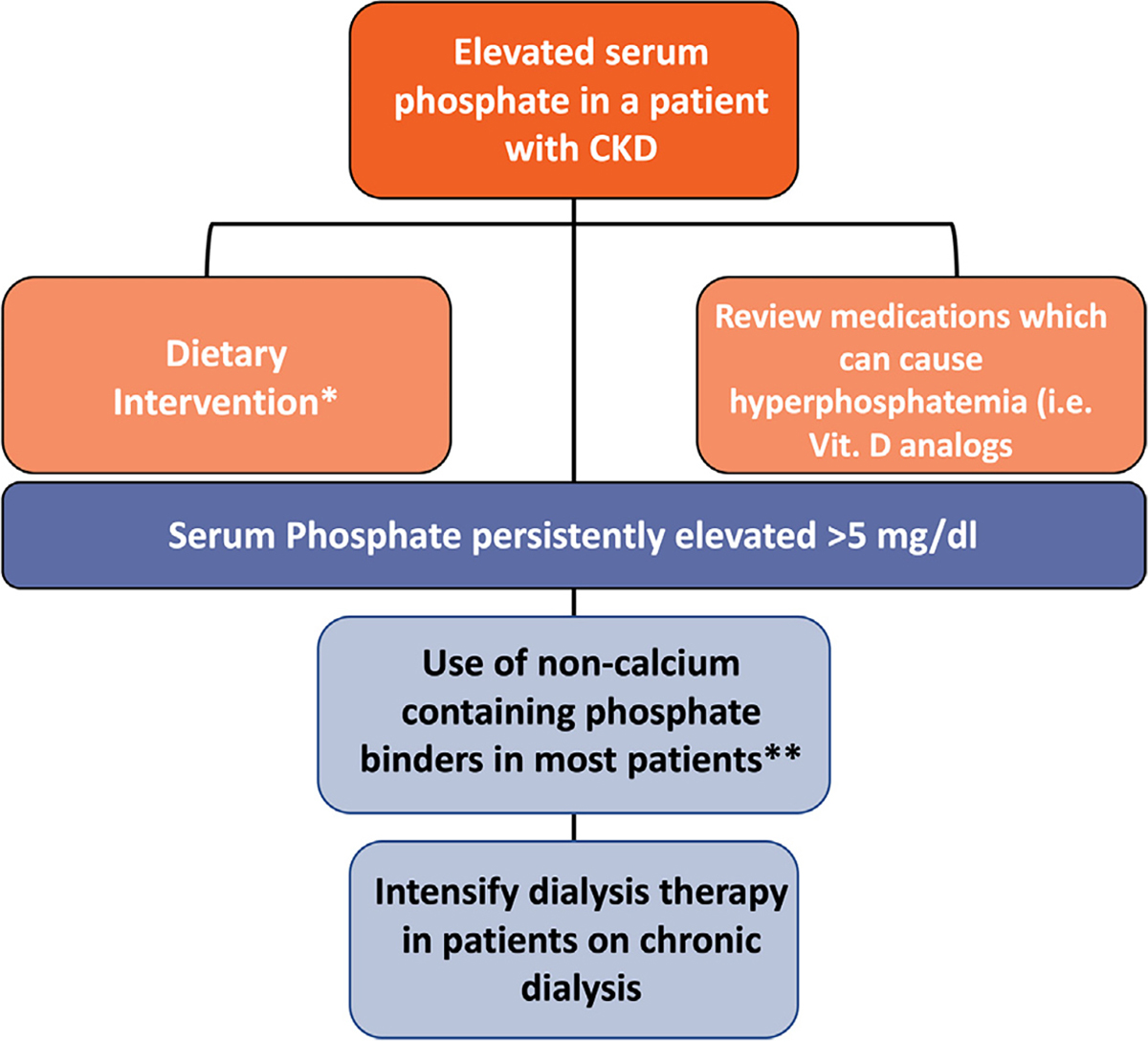
Approach to hyperphosphatemia management in patients with CKD. KDIGO 2017 guidelines suggest lowering elevated phosphate levels toward the normal range in patients with CKD G3a–G5D rather than preventive phosphate lowering. Initial intervention includes dietary modification and medication management. *Reasonable to consider phosphate source in making dietary recommendation as phosphate derived from plants is less absorbed than that from animals or food additives. **The use of calcium-containing phosphate binders is generally restricted to a selected group of patients with CKD without hypercalcemia, vascular calcification or adynamic bone disease.
Table 3.
Pharmacologic Interventions for the Management of Hyperphosphatemia
| Class | Name | Mechanism | Advantages | Disadvantages |
|---|---|---|---|---|
|
| ||||
| Calcium containing binders | Calcium carbonate, calcium phosphate | Calcium combines with phosphate in the bowel and excreted as insoluble complex in the feces | Low cost | Potential risk of hypercalcemia, adynamic bone disease, and vascular calcification |
| Non-calcium-containing binders | Sevelamer carbonate or hydrochloride | Cationic polymers that bind phosphate in the intestine through ion exchange | Avoids positive calcium balance | Cost, pill burden. Sevelamer hydrochloride might induce metabolic acidosis |
| Lanthanum carbonate | Phosphate binding in the gastrointestinal tract | Avoids positive calcium balance. Chewable, less pill burden | Cost | |
| Other mechanism | Sucroferric oxyhydroxide, ferric citrate | Phosphate binding in the gastrointestinal tract | Avoids calcium, relatively less pill burden, small iron absorption | Cost |
| Nicotinamide | Decreases sodium-dependent phosphate transporter in the intestine | Intolerance (flushing) | ||
| Tenapanor | Inhibitor of intestinal sodium/hydrogen exchanger 3 (NHE3), reduce paracellular phosphate transport in the intestine | Inhibits principal route of phosphate absorption, potentially less pill burden | Diarrhea, cost | |
| Aluminum hydroxide | Phosphate binding in the gastrointestinal tract | Low cost | Rarely used chronically dueto riskof aluminum exposure in patients with CKD | |
A large meta-analysis that included 104 randomized controlled trials (1467 participants with nondialysis-dependent CKD and 10,364 participants with ESRD) concluded that among adults with dialysis-dependent CKD, sevelamer may lower all-cause mortality compared to calcium-based binders. No clinical benefit from any Pi binder on cardiovascular death, myocardial infarction, stroke, fracture, or coronary artery calcification was noted among dialysis-dependent and nondialysis-dependent patients with CKD in this meta-analysis.87 Phosphate-binder use; however, remains highly prevalent with over 80% of dialysis-dependent patients in the United States being prescribed this treatment.88
Hypophosphatemia has also been linked with increased mortality among patients with advanced CKD. It is however, challenging to discern whether the deleterious effects are driven specifically by low serum Pi versus nutritional impairments or underlying comorbidities that most of these patients with chronic persistent hypophosphatemia suffer from. Similarly, acute hypophosphatemia in critical care settings has been recognized as a potential therapeutic target, although evidence to support best practices – particularly during CRRT – is still needed.
ONGOING TRIALS AND FUTURE DIRECTIONS
Several fundamental questions related to how treatment of hyperphosphatemia may impact clinical outcomes in patients with CKD remain unanswered.
One such question is what the optimal Pi level is and whether treatment of hyperphosphatemia in patients with ESRD leads to clinical benefit or harm. An ongoing pragmatic randomized controlled trial in patients with ESRD aims to enroll 4400 patients undergoing 3-times-weekly, in-center hemodialysis and compare the effect of phosphate binder prescriptions and dietary recommendations to achieve serum Pi target of ≥6.5 mg/dL versus <5.5 mg/dL on all-cause mortality and all-cause hospitalization.89 This important trial that follows many innovative design features including eConsent and remote study monitoring will not, however, account for the use of calcium-based versus non–calcium-based binders, and will not provide adjudicated causes of hospitalizations. Furthermore, clinical trials of phosphate-driven interventions could potentially address the heterogeneity of treatment effect based on subphenotyping according to circulating levels of FGF-23, PTH, αklotho – among other proteins or strategies – to get 1 step closer to precision medicine phosphate-driven interventions.
A novel Npt2a inhibitor could also be used to lower serum Pi levels. This drug has been shown to be effective in lowering serum Pi in rodents with and without CKD.90–92 Another major area to address in future research inquiries is the effect of Pi lowering interventions on patient-oriented outcomes and quality of life. Furthermore, the impact of social determinants of health on phosphate-driven interventions should be also evaluated.
CONCLUSIONS
In this review, we highlighted the importance of both intracellular and extracellular Pi for normal human physiology, while describing the orchestrated actions of 1,25-dihydroxyvitamin D3, PTH, and FGF-23 for Pi homeostasis at the level of the kidney and small intestine. We summarized genetic and acquired clinical conditions predisposing to hypophosphatemia and hyperphosphatemia. We included clinical scenarios associated with hypophosphatemia that are not commonly described in literature such as critical illness and the effect of extracorporeal removal of Pi with continuous renal replacement therapy. Finally, we commented on the rationale of phosphate-driven interventions in patients with advanced CKD and potential areas of future research that include novel therapies but also a multifocal selection of risk factors, outcomes and the recognition of heterogeneity of treatment effect. One should note that even when our understanding of Pi physiology has significantly improved over the past decades, testing and implementation of novel therapies to prevent and/or mitigate Pi imbalances/dysregulation in humans under different clinical scenarios are still scarce.
CLINICAL SUMMARY.
Phosphate levels in the body are regulated by FGF-23, PTH, vitamin D and klotho.
Hypophosphatemia is common in critically ill patients with acute kidney injury undergoing continuous renal replacement therapies in the ICU.
Hyperphosphatemia is more prevalent with chronic kidney dysfunction and is associated with increased mortality in patients with advanced chronic kidney disease.
Pharmacologic strategies to treat hyperphosphatemia include calcium-containing and non–calcium-containing phosphate binders, as well as novel therapies targeting paracellular phosphate transport in the intestine.
ACKNOWLEDGMENTS
JAN is currently supported by NIDDK grants R01DK128208, U01DK12998, and P30 DK079337.
TR was supported by NIDDK grant R01DK110621, VA Merit Review Award IBX004968 A, American Heart Association Transformational Project Award 19TPA34850116, and by the NIDDK Diabetic Complications Consortium (RRID:SCR_001415, www.diacomp.org), grants DK076169 and DK115255.
SN is supported by the National Institute of Biomedical Imaging and Bioengineering (1R01EB031813-01A1) and by the National Institute of Diabetes and Digestive and Kidney Diseases (1U01DK123818-01).
Footnotes
Financial Disclosure: IPC and TR have no relevant disclosures. SN has received grant support from Alnylam Pharmaceuticals, Hope Pharmaceuticals, Inozyme Pharma, Laboratoris Sanifit, and and has served as a consultant to Ardelyx, Inc, Epizon Pharma, Inozyme Pharma, Laboratoris Sanifit, and Fresenius Medical Therapies Group. JAN has served as a consultant for Baxter Healthcare, Outset Medical, and Vifor Pharma.
Contributor Information
Ignacio Portales-Castillo, Division of Nephrology, Department of Medicine, Massachusetts General Hospital, and Harvard Medical School, Boston, MA; Endocrine Unit, Massachusetts General Hospital, and Harvard Medical School, Boston, MA.
Timo Rieg, Department of Molecular Pharmacology and Physiology, University of South Florida, Tampa, FL; James A. Haley Veterans’ Hospital, Tampa, FL; Center for Hypertension and Kidney Research, University of South Florida, Tampa, FL.
Sheikh B. Khalid, Department of Internal Medicine, The Indus Hospital, Lahore. Pakistan
Sagar U. Nigwekar, Division of Nephrology, Department of Medicine, Massachusetts General Hospital, and Harvard Medical School, Boston, MA
Javier A. Neyra, Department of Internal Medicine, Division of Nephrology, University of Alabama at Birmingham, Birmingham, AL
REFERENCES
- 1.Michigami T Advances in understanding of phosphate homeostasis and related disorders. Endocr J. 2022;69(8):881–896. [DOI] [PubMed] [Google Scholar]
- 2.Iheagwara OS, Ing TS, Kjellstrand CM, Lew SQ. Phosphorus, phosphorous, and phosphate. Hemodial Int. 2013;17(4):479–482. [DOI] [PubMed] [Google Scholar]
- 3.Bansal VK. Serum inorganic phosphorus. Clinical methods: The History, Physical, and Laboratory Examinations. 3rd ed. Boston, MA: Butterworth Publishers; 1990. Chapter: 198. [PubMed] [Google Scholar]
- 4.Christov M, Jüppner H. Phosphate homeostasis disorders. Best Pract Res Clin Endocrinol Metab. 2018;32(5):685–706. [DOI] [PubMed] [Google Scholar]
- 5.Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis. 1998;31(4):607–617. [DOI] [PubMed] [Google Scholar]
- 6.Lee GJ, Marks J. Intestinal phosphate transport: a therapeutic target in chronic kidney disease and beyond? Pediatr Nephrol. 2015;30(3):363–371. [DOI] [PubMed] [Google Scholar]
- 7.Pohlmeier R, Vienken J. Phosphate removal and hemodialysis conditions. Kidney Int. 2001;59(suppl 78):S190–S194. [DOI] [PubMed] [Google Scholar]
- 8.Bergwitz C, Jüppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabbagh Y, O’Brien SP, Song W, et al. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009;20(11):2348–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernando N, Myakala K, Simona F, et al. Intestinal depletion of NaPi-IIb/Slc34a2 in mice: renal and hormonal adaptation. J Bone Miner Res. 2015;30(10):1925–1937. [DOI] [PubMed] [Google Scholar]
- 11.Miyamoto K-I, Haito-Sugino S, Kuwahara S, et al. Sodium-dependent phosphate cotransporters: lessons from gene knockout and mutation studies. J Pharm Sci. 2011;100(9):3719–3730. [DOI] [PubMed] [Google Scholar]
- 12.Radanovic T, Wagner CA, Murer H, Biber Jr. Regulation of Intestinal Phosphate Transport I. Segmental expression and adaptation to low-Pi diet of the type IIb Na+-Pi cotransporter in mouse small intestine. Am J Physiol Gastrointest Liver Physiol. 2005;288(3):G496–G500. [DOI] [PubMed] [Google Scholar]
- 13.Sabbagh Y, Giral H, Caldas Y, Levi M, Schiavi SC. Intestinal phosphate transport. Adv Chron Kidney Dis. 2011;18(2):85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Capuano P, Radanovic T, Wagner CA, et al. Intestinal and renal adaptation to a low-Pi diet of type II NaPi cotransporters in vitamin D receptor-and 1αOHase-deficient mice. Am J Physiol Cell Physiol. 2005;288(2):C429–C434. [DOI] [PubMed] [Google Scholar]
- 15.Segawa H, Kaneko I, Yamanaka S, et al. Intestinal Na-Pi cotransporter adaptation to dietary Pi content in vitamin D receptor null mice. Am J Physiol Renal Physiol. 2004;287(1):F39–F47. [DOI] [PubMed] [Google Scholar]
- 16.Shibasaki Y, Etoh N, Hayasaka M, et al. Targeted deletion of the tybe IIb Na+-dependent Pi-co-transporter, NaPi-IIb, results in early embryonic lethality. Biochem Biophys Res Commun. 2009;381(4):482–486. [DOI] [PubMed] [Google Scholar]
- 17.Knöpfel T, Himmerkus N, Günzel, D, Bleich M, Hernando N, Wagner CA. Paracellular transport of phosphate along the intestine. Am J Physiol Gastrointest Liver Physiol. 2019;317(2):G233–G241. [DOI] [PubMed] [Google Scholar]
- 18.Karim-Jimenez Z, Hernando N, Biber J, Murer H. A dibasic motif involved in parathyroid hormone-induced down-regulation of the type IIa NaPi cotransporter. Proc Natl Acad Sci U S A, 97. 2000;(23),:12896–12901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watson P, Fraher L, Hendy G, et al. Nuclear localization of the type 1 PTH/PTHrP receptor in rat tissues. J Bone Miner Res. 2000;15(6):1033–1044. [DOI] [PubMed] [Google Scholar]
- 20.Virkki LV, Biber J, Murer H, Forster IC. Phosphate transporters: a tale of two solute carrier families. Am J Physiol Ren Physiol. 2007;293(3):F643–F654. [DOI] [PubMed] [Google Scholar]
- 21.Thomas L, Dominguez Rieg JA, Rieg T. Npt2a as a target for treating hyperphosphatemia. Biochem Soc Trans. 2022;50(1):439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U S A. 1998;95(9):5372–5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Segawa H, Onitsuka A, Furutani J, et al. Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am J Physiol Ren Physiol. 2009;297(3):F671–F678. [DOI] [PubMed] [Google Scholar]
- 24.Prié D, Huart V, Bakouh N, et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium–phosphate cotransporter. N Engl J Med. 2002;347(13):983–991. [DOI] [PubMed] [Google Scholar]
- 25.Virkki LV, Forster IC, Hernando N, Biber J, Murer H. Functional characterization of two naturally occurring mutations in the human sodium-phosphate cotransporter type IIa. J Bone Miner Res. 2003;18(12):2135–2141. [DOI] [PubMed] [Google Scholar]
- 26.Lapointe J-Y, Tessier J, Paquette Y, et al. NPT2a gene variation in calcium nephrolithiasis with renal phosphate leak. Kidney Int. 2006;69(12):2261–2267. [DOI] [PubMed] [Google Scholar]
- 27.Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, Econs MJ. Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab. 2006;91(10):4022–4027. [DOI] [PubMed] [Google Scholar]
- 28.Kuro-o M Klotho and calciprotein particles as therapeutic targets against accelerated ageing. Clin Sci. 2021;135(15):1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wojcicki JM. Hyperphosphatemia is associated with anemia in adults without chronic kidney disease: results from the National Health and Nutrition Examination Survey (NHANES): 2005–2010. BMC Nephrol. 2013;14(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaasbeek A, Meinders AE. Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118(10):1094–1101. [DOI] [PubMed] [Google Scholar]
- 32.Bastin MLT, Stromberg AJ, Nerusu SN, et al. Association of phosphate-containing versus phosphate-free solutions on ventilator days in patients requiring continuous kidney replacement therapy. Clin J Am Soc Nephrol. 2022;17(5):634–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pastor-Arroyo EM, Knöpfel T, Imenez Silva PH. Intestinal epithelial ablation of Pit-2/Slc20a2 in mice leads to sustained elevation of vitamin D3 upon dietary restriction of phosphate. Acta Physiol. 2020;230(2):e13526. [DOI] [PubMed] [Google Scholar]
- 34.Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1, 25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146(12):5358–5364. [DOI] [PubMed] [Google Scholar]
- 35.Lötscher M, Kaissling B, Biber J, Murer H, Levi M. Role of microtubules in the rapid regulation of renal phosphate transport in response to acute alterations in dietary phosphate content. J Clin Invest. 1997;99(6):1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergwitz C, Miyamoto K-I. Hereditary hypophosphatemic rickets with hypercalciuria: pathophysiology, clinical presentation, diagnosis and therapy. Pflügers Arch. 2019;471(1):149–163. [DOI] [PubMed] [Google Scholar]
- 37.Chi Y, Zhao Z, He X, et al. A compound heterozygous mutation in SLC34A3 causes hereditary hypophosphatemic rickets with hypercalciuria in a Chinese patient. Bone. 2014;59:114–121. [DOI] [PubMed] [Google Scholar]
- 38.Dhir G, Li D, Hakonarson H, Levine MA. Late-onset hereditary hypophosphatemic rickets with hypercalciuria (HHRH) due to mutation of SLC34A3/NPT2c. Bone. 2017;97:15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volk C, Schmidt B, Brandsch C, et al. Acute effects of an inorganic phosphorus additive on mineral metabolism and cardiometabolic risk factors in healthy subjects. J Clin Endocrinol Metab. 2022;107(2):e852–e864. [DOI] [PubMed] [Google Scholar]
- 40.Gattineni J, Bates C, Twombley K, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Ren Physiol. 2009;297(2):F282–F291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.White KE, Evans WE, O’Riordan JL, et al. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26(3):345–348. [DOI] [PubMed] [Google Scholar]
- 42.Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98(11):6500–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonsson KB, Zahradnik R, Larsson T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348(17):1656–1663. [DOI] [PubMed] [Google Scholar]
- 44.Francis F, Hennig S, Korn B, et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X–linked hypophosphatemic rickets. Nat Genet. 1995;11(2):130–136. [DOI] [PubMed] [Google Scholar]
- 45.Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone. 2004;35(2):455–462. [DOI] [PubMed] [Google Scholar]
- 46.Linglart A, Imel EA, Whyte MP, et al. Sustained efficacy and safety of burosumab, a monoclonal antibody to FGF23, in children with X-linked hypophosphatemia. J Clin Endocrinol Metab. 2022;107(3):813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291(1):E38–E49. [DOI] [PubMed] [Google Scholar]
- 48.Seton M, Jüppner H. Autosomal dominant hypophosphatemic rickets in an 85 year old woman: characterization of her disease from infancy through adulthood. Bone. 2013;52(2):640–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schaefer B, Tobiasch M, Wagner S, et al. Hypophosphatemia after intravenous iron therapy: Comprehensive review of clinical findings and recommendations for management. Bone. 2022;154:116202. [DOI] [PubMed] [Google Scholar]
- 50.David V, Martin A, Isakova T, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016;89(1):135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res. 2013;28(8):1793–1803. [DOI] [PubMed] [Google Scholar]
- 52.Rosano G, Schiefke I, Göhring U-M, Fabien V, Bonassi S, Stein J. A pooled analysis of serum phosphate measurements and potential hypophosphataemia events in 45 interventional trials with ferric carboxymaltose. J Clin Med. 2020;9(11):3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf M, Rubin J, Achebe M, et al. Effects of iron isomaltoside vs ferric carboxymaltose on hypophosphatemia in iron-deficiency anemia: two randomized clinical trials. JAMA. 2020;323(5):432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Portales-Castillo I, Mount DB, Nigwekar S, Elaine WY, Rennke H, Gupta S. Zoledronic acid–associated Fanconi syndrome in patients with cancer. Am J Kidney Dis. 2022;80(4):555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zajicek HK, Wang H, Puttaparthi K, et al. Glycosphingolipids modulate renal phosphate transport in potassium deficiency. Kidney Int. 2001;60(2):694–704. [DOI] [PubMed] [Google Scholar]
- 56.Wadsworth R, Siddiqui S. Phosphate homeostasis in critical care. BJA Educ. 2016;16(9):305–309. [Google Scholar]
- 57.Berger M, Appelberg O, Reintam-Blaser A, et al. Prevalence of Hypophosphatemia in the ICU–results of an international one-day point prevalence survey. Clin Nutr. 2021;40(5):3615–3621. [DOI] [PubMed] [Google Scholar]
- 58.Knochel JP. Hypophosphatemia in the alcoholic. Arch Intern Med.1980;140(5):613–615. [PubMed] [Google Scholar]
- 59.Levin A, Bakris G, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007;71(1):31–38. [DOI] [PubMed] [Google Scholar]
- 60.Foley RN. Phosphate levels and cardiovascular disease in the general population. Clin J Am Soc Nephrol. 2009;4(6):1136–1139. [DOI] [PubMed] [Google Scholar]
- 61.Foley RN, Collins AJ, Herzog CA, Ishani A, Kalra PA. Serum phosphorus levels associate with coronary atherosclerosis in young adults. J Am Soc Nephrol. 2009;20(2):397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011;305(11):1119–1127. [DOI] [PubMed] [Google Scholar]
- 63.Fass R, Do S, Hixson LJ. Fatal hyperphosphatemia following Fleet Phospo-Soda in a patient with colonic ileus. Am J Gastroenterol. 1993;88(6):929–932. [PubMed] [Google Scholar]
- 64.Prié D, Friedlander G. Genetic disorders of renal phosphate transport. N Engl J Med. 2010;362(25):2399–2409. [DOI] [PubMed] [Google Scholar]
- 65.Llach F, Felsenfeld AJ, Haussler MR. The pathophysiology of altered calcium metabolism in rhabdomyolysis-induced acute renal failure: interactions of parathyroid hormone, 25-hydroxycholecalciferol, and 1, 25-dihydroxycholecalciferol. N Engl J Med. 1981;305(3):117–123. [DOI] [PubMed] [Google Scholar]
- 66.Fenton RA, Murray F, Rieg JAD, Tang T, Levi M, Rieg T. Renal phosphate wasting in the absence of adenylyl cyclase 6. J Am Soc Nephrol. 2014;25(12):2822–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu Y, He Q, Aydin C, et al. Ablation of the stimulatory G protein α-subunit in renal proximal tubules leads to parathyroid hormone-resistance with increased renal Cyp24a1 mRNA abundance and reduced serum 1, 25-dihydroxyvitamin D. Endocrinology. 2016;157(2):497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mannstadt M, Bilezikian JP, Thakker RV, et al. Hypoparathyroidism. Nat Rev Dis Primers. 2017;3(1):1–21. [DOI] [PubMed] [Google Scholar]
- 69.Jüppner H Molecular definition of pseudohypoparathyroidism variants. J Clin Endocrinol Metab. 2021;106(6):1541–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Demaret T, Wintjens R, Sana G, et al. Case Report: Inactivating PTH/PTHrP signaling disorder type 1 presenting with PTH resistance. Front Endocrinol. 2022;13:928284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guerreiro R, Bras J, Batista S, et al. Pseudohypoparathyroidism type I-b with neurological involvement is associated with a homozygous PTH1R mutation. Gene Brain Behav. 2016;15(7):669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mantovani G, Bastepe M, Monk D, et al. Recommendations for diagnosis and treatment of pseudohypoparathyroidism and related disorders: an updated practical tool for physicians and patients. Horm Res Paediatr. 2020;93(3):182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Benet-Pages A, Orlik AP, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14(3):385–390. [DOI] [PubMed] [Google Scholar]
- 74.Ichikawa S, Sorenson AH, Austin AM, et al. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology. 2009;150(6):2543–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ichikawa S, Imel EA, Kreiter ML, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117(9):2684–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clinkenbeard EL, Noonan ML, Thomas JC, et al. Increased FGF23 protects against detrimental cardio-renal consequences during elevated blood phosphate in CKD. JCI Insight. 2019;4(4):e123817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barker SL, Pastor J, Carranza D, et al. The demonstration of αKlotho deficiency in human chronic kidney disease with a novel synthetic antibody. Nephrol Dial Transplant. 2015;30(2):223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsai W-C, Wu H-Y, Peng Y-S, et al. Short-term effects of very-low-phosphate and low-phosphate diets on fibroblast growth factor 23 in hemodialysis patients: a randomized crossover trial. Clin J Am Soc Nephrol. 2019;14(10):1475–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kuhlmann MK. Phosphate elimination in modalities of hemodialysis and peritoneal dialysis. Blood Purif. 2010;29(2):137–144. [DOI] [PubMed] [Google Scholar]
- 80.Komaba H, Fukagawa M. Phosphate—a poison for humans? Kidney Int. 2016;90(4):753–763. [DOI] [PubMed] [Google Scholar]
- 81.McGovern AP, de Lusignan S, van Vlymen J, et al. Serum phosphate as a risk factor for cardiovascular events in people with and without chronic kidney disease: a large community based cohort study. PLoS One. 2013;8(9):e74996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Isakova T, Nickolas TL, Denburg M, et al. KDOQI US commentary on the 2017 KDIGO clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease–mineral and bone disorder (CKD-MBD). Am J Kidney Dis. 2017;70(6):737–751. [DOI] [PubMed] [Google Scholar]
- 83.Wheeler DC, Winkelmayer WC. KDIGO 2017 clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD) foreword. Kidney Int Suppl. 2017;7(1):1–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chiu Y-W, Teitelbaum I, Misra M, De Leon EM, Adzize T, Mehrotra R. Pill burden, adherence, hyperphosphatemia, and quality of life in maintenance dialysis patients. Clin J Am Soc Nephrol. 2009;4(6):1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Block GA, Bleyer AJ, Silva AL, et al. Safety and efficacy of Tenapanor for Long-term serum phosphate control in maintenance dialysis: a 52-week randomized Phase 3 trial (PHREEDOM). Kidney360 2021;2(10):1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xue J, Thomas L, Murali SK, et al. Enhanced phosphate absorption in intestinal epithelial cell-specific NHE3 knockout mice. Acta Physiol. 2022;234(2):e13756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruospo M, Palmer SC, Natale P, et al. Phosphate binders for preventing and treating chronic kidney disease-mineral and bone disorder (CKD-MBD). Cochrane Database Syst Rev. 2018;(8):1465–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fishbane SN, Nigwekar S. Phosphate absorption and hyperphosphatemia management in kidney disease: a physiology-based review. Kidney Med. 2021;3(6):1057–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Edmonston DL, Isakova T, Dember LM, et al. Design and rationale of HiLo: a pragmatic, randomized trial of phosphate management for patients receiving maintenance hemodialysis. Am J Kidney Dis. 2021;77(6):920–930. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thomas L, Xue J, Tomilin VN, Pochynyuk OM, Dominguez Rieg JA, Rieg T. PF-06869206 is a selective inhibitor of renal Pi transport: evidence from in vitro and in vivo studies. Am J Physiol Ren Physiol. 2020;319(3):F541–F551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thomas L, Xue J, Murali SK, Fenton RA, Rieg JAD, Rieg T. Pharmacological Npt2a inhibition causes phosphaturia and reduces plasma phosphate in mice with normal and reduced kidney function. J Am Soc Nephrol. 2019;30(11):2128–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Clerin V, Saito H, Filipski KJ, et al. Selective pharmacological inhibition of the sodium-dependent phosphate cotransporter NPT2a promotes phosphate excretion. J Clin Invest. 2020;130(12):6510–6522. [DOI] [PMC free article] [PubMed] [Google Scholar]