

Abstract
Uncontrolled inflammation is linked to poor outcomes in sepsis and wound healing, both of which proceed through distinct inflammatory and resolution phases. Eicosanoids are a class of bioactive lipids that recruit neutrophils and other innate immune cells. The interaction of ceramide 1-phosphate (C1P) with the eicosanoid biosynthetic enzyme cytosolic phospholipase A2 (cPLA2) reduces the production of a subtype of eicosanoids called oxoeicosanoids. We investigated the effect of shifting the balance in eicosanoid biosynthesis on neutrophil polarization and function. Knockin mice expressing a cPLA2 mutant lacking the C1P binding site (cPLA2αKI/KI mice) showed enhanced and sustained neutrophil infiltration into wounds and the peritoneum during the inflammatory phase of wound healing and sepsis, respectively. The mice exhibited improved wound healing and reduced susceptibility to sepsis, which was associated with an increase in anti-inflammatory N2-type neutrophils demonstrating proresolution behaviors and a decrease in proinflammatory N1-type neutrophils. The N2 polarization of cPLA2αKI/KI neutrophils resulted from increased oxoeicosanoid biosynthesis and autocrine signaling through the oxoeicosanoid receptor OXER1 and partially depended on OXER1-dependent inhibition of the pentose phosphate pathway (PPP). Thus, C1P binding to cPLA2α suppresses neutrophil N2 polarization, thereby impairing wound healing and the response to sepsis.
INTRODUCTION
Wound healing is a multi-stage physiological response comprised of four distinct phases: coagulation, inflammation, proliferation, and remodeling1. A hallmark of healthy wound progression is the transition from the inflammatory phase to proliferation and, ultimately, tissue remodeling1. During the inflammatory stage of mammalian wound healing, circulating neutrophils are recruited into damaged tissues to rapidly eliminate pathogens and remove damaged cells and debris from the wound site2. The infiltration of neutrophils initiates inflammation, and their timely clearance by macrophages through efferocytosis is crucial for proper progression into subsequent phases of wound healing2. The physiologic condition of sepsis is also initially characterized by a hyperinflammatory stage, as shown by the presence of fever, tachycardia, tachypnea, and altered leukocyte numbers along with a known site of infection2. As with wound healing, neutrophil infiltration, in this case to the site of infection, is also required to clear pathogens and damaged tissue3. Efferocytosis is also a key step in the septic response for eventual resolution and recovery4,5. However, both wounds and the septic response can become stalled in the inflammatory stage causing delayed healing or resolution as well as sustained hyperinflammation linked to the overproduction of inflammatory mediators such as interleukin 6 (IL-6) and tumor necrosis factor (TNF)6,7. Because neutrophils rely on many proinflammatory mechanisms to combat pathogens, the specific behaviors of neutrophils in a wound site or at a site of infection are of great importance. Indeed, previous research suggests that moderate suppression of neutrophil infiltration is generally a hallmark of better healing outcomes and survival in sepsis models8,9. Furthermore, prolonged and sustained increases in neutrophils, known as neutrophilia, in the wound or infected environment are strongly linked to poor outcomes in both wound healing and sepsis10,11.
Neutrophils are recruited to the site of injury or infection by cytokines, chemokines, and bioactive lipid mediators such as eicosanoids, including the subclasses of leukotriene B4 (LTB4) and other leukotrienes8,12. Once at the site of injury or infection, neutrophils use a combination of phagocytosis and secretion of antimicrobials to kill invading pathogens and remove damaged cells12,13. Prolonged neutrophil activity also induces damage to healthy tissue14,15, and much of this neutrophilia-induced damage results from the activity of reactive oxygen species (ROS) secreted by neutrophils during a persistent and sustained inflammatory response16. In addition to ROS, neutrophils secrete chromatin “traps” termed neutrophil extracellular traps (NETs) comprised of chromosomal DNA, active proteases, and globular proteins to ensnare extracellular invaders and deliver a concentrated dose of antimicrobial components17. However, evidence suggests that NETs can also participate in the pathogenesis of autoimmune and inflammatory disorders18 as well as occlude the vasculature, causing delayed wound healing19,20.
Neutrophilia-induced damage may be related to the subtype of neutrophil because a proinflammatory neutrophil would foster ROS and NET production. Although macrophage polarization is well established, neutrophil polarization is a relatively new scientific field initially characterized by Fridlender et al. in 200921. Indeed, neutrophils were presumed to be metabolically inert due to their transient appearance during infection and relatively short lifespan, but it is now accepted that neutrophil behavior switches between a proinflammatory, cytotoxic phenotype, termed the N1 subtype, and an anti-inflammatory, proresolution phenotype, termed the N2 subtype21. These distinct neutrophil populations are typically determined by their functional phenotype22,23. For example, proinflammatory N1 neutrophils show higher ROS, NET generation, and production of inflammatory cytokines, such as TNF, compared to N2 neutrophils, which present with lower amounts of these inflammatory phenotypes while also demonstrating increased production of the proresolution cytokine vascular endothelial growth factor (VEGF)22,23 Specific cell surface receptors are also reported markers for these two neutrophil subtypes, intercellular adhesion molecule-1 (ICAM-1) and C-X-C motif chemokine ligand 10 (CXCL10, also known as IFN-γ-inducible protein-10) for N1 and interleukin 8 (IL-8) and C-X-C motif chemokine receptor 2 (CXCR2) for N222,23.
Although eicosanoids have not been characterized for specific roles in neutrophil polarization, various eicosanoids have been implicated in pro-inflammatory and pro-resolution phases of sepsis and wound healing, both of which are mediated by neutrophil behavior8,12,16,24–27. Ceramide 1-phosphate (C1P) is a sphingolipid that promotes the production of prostaglandins, a subset of eicosanoids, by binding to the enzyme that mediates the first rate-determining step in eicosanoid biosynthesis, group IVA cytosolic phospholipase A2 (cPLA2α)24,28–37. The association of cPLA2α with C1P enhances its association with cellular membranes, specifically the Golgi apparatus24,28,31–37. Mutations in cPLA2α that block C1P binding (R57A, K58A, R59A) decrease the production of prostaglandins such as PGE2 in vitro and in vivo24,28,31–37. Furthermore, loss of association of cPLA2α with C1P in fibroblasts redirects the cellular localization of the enzyme to the cytoplasm and induces a class switch in eicosanoid biosynthesis leading to enhanced 5-HETE biosynthesis24,31. In this study, we observed a positive outcome for sepsis survival and chronic wound healing in a genetically engineered mouse model24,31 in which the C1P interaction site in cPLA2 was ablated by knockin (KI) of a cPLA2α allele encoding a protein with three amino acid substitutions in the C1P binding site (cPLA2αKI/KI mice). These positive outcomes correlated with neutrophilia in acute wounds and in the peritoneum following sepsis induction despite neutrophilia at these sites being generally associated with poor outcomes10,11. This “neutrophil conundrum” provided the opportunity to answer fundamental questions in the field of wound healing and inflammatory responses. Specifically, why do sustained and prolonged increases in neutrophils occur, and why is neutrophilia unresolved in non-healing wounds? Our study sheds light on these questions because we show that loss of the C1P-cPLA2α interaction increased the biosynthesis of a specific class of eicosanoids, 5-hydroxyeicosatetraenoic acid (5-HETE) and 5-oxo-eicosatetraenoic acid (5-oxo-ETE), in primary neutrophils, which activated oxoeicosanoid receptor 1 (OXER1) to enhance neutrophil infiltration, suppress the pentose phosphate pathway (PPP), and simultaneously polarize neutrophils to an anti-inflammatory, N2 phenotype. Thus, C1P, a bioactive sphingolipid generated by direct phosphorylation of ceramide by ceramide kinase (CERK)28–30, is shown as a negative regulator of neutrophil polarization to the N2 subtype through association with cPLA2α and suppression of 5-HETE biosynthesis. Overall, our study suggests the model that enhancement of 5-HETE and 5-oxo-ETE biosynthesis or CERK inhibition are potential avenues for therapeutic approaches to treat chronic, non-healing wounds and sepsis by reducing the induction of inflammatory neutrophils, thereby allowing for progression of these multi-phasic physiologic responses to resolution. Our study further suggests that the type of polarization of the neutrophils is more important than the neutrophilia itself.
RESULTS
Ablation of the C1P-cPLA2α interaction enhances neutrophil infiltration and promotes wound healing and survival outcome in sepsis
Our laboratory previously demonstrated improved healing of acute wounds by constraining C1P production through genetic ablation or pharmacological inhibition of CERK or ablation of the C1P interaction site in the direct target of this bioactive lipid, cPLA2α24,31. The increased healing rate in these models is suggestive of compression of one or more of the phases of wound healing. The inflammatory phase, in particular, is associated with high amounts of C1P, and this is the phase linked to the stalled healing of chronic wounds due to neutrophilia38. To examine the inflammatory phase of wound healing, we utilized knockin mice expressing enhanced green fluorescent protein (EGFP) from the Lysozyme M (MLys) locus (MLys-EGFP mice), which labels neutrophils as well as other granulocytes and macrophages, in an acute model of wound healing39,40. In vivo fluorescence imaging of EGFP-expressing cells in cPLA2α+/+ and cPLA2αKI/KI mice following acute dermal wounding demonstrated significantly increased immune cell infiltration into cPLA2αKI/KI wounds as early as 4 hours (hrs) post-injury and remained higher for over 24 hrs (Fig. 1A). To determine the ratios of neutrophils to macrophages, we employed immunohistological staining for well-established, cell-specific markers41–44, and by the 6-hr time point, cPLA2αKI/KI mice showed a significantly higher presence of neutrophils in the wound tissues compared to cPLA2α+/+ mice (Fig. 1B). Similarly, macrophage markers CD6845 and F4/8046 were higher in cPLA2α+/+ mice compared to cPLA2αKI/KI mice (Fig. 1B), indicating lower macrophage-mediated inflammatory response in the mutants.
Fig. 1. Loss of the C1P-cPLA2α interaction enhances neutrophil infiltration and accumulation in acute wounds.
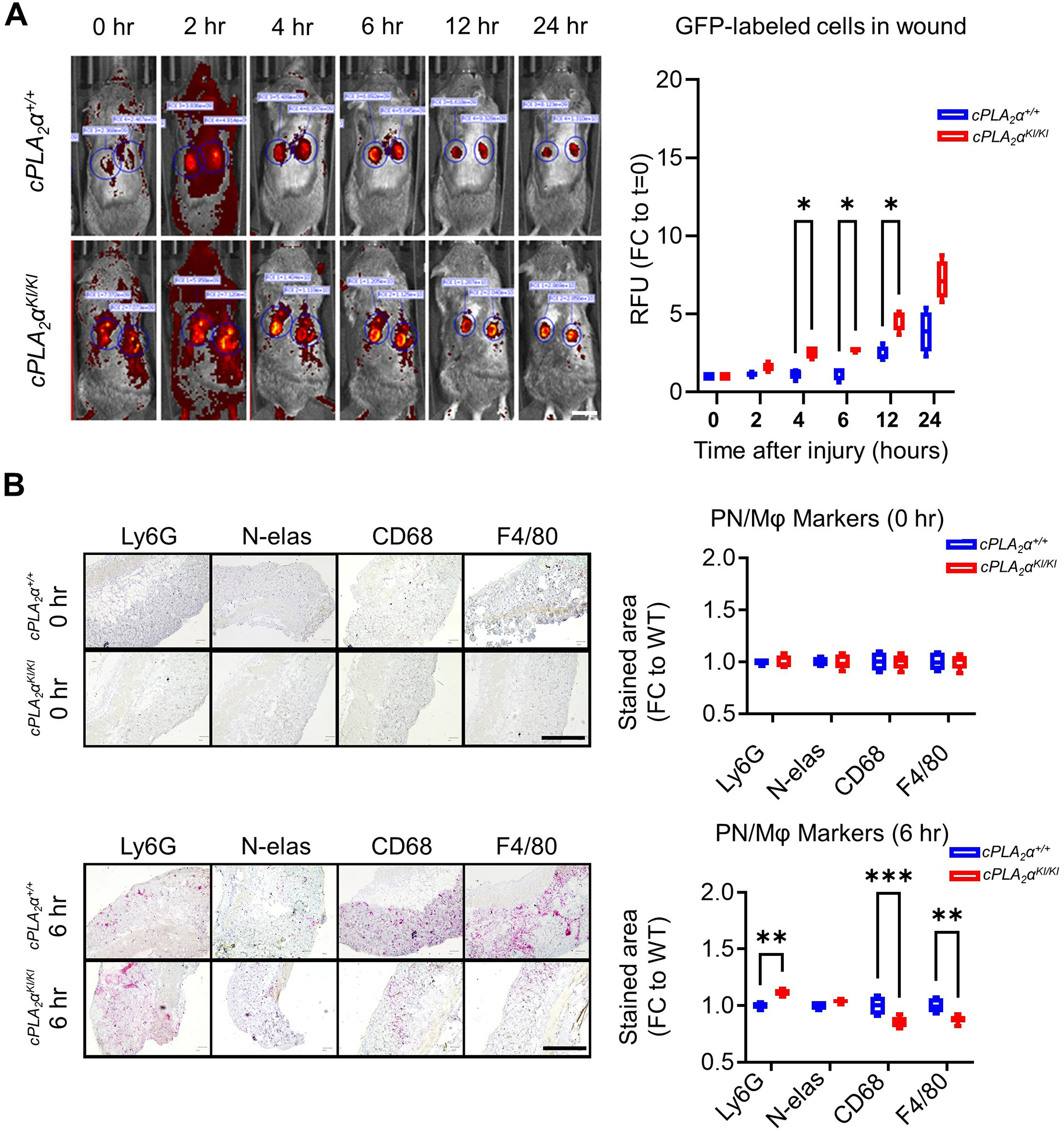
(A) The cPLA2α+/+;MLys-EGFP and cPLA2αKI/KI;MLys-EGFP mice were subjected to acute dorsal wounding and fluorescence imaging at the indicated times after injury. Fluorescence intensity was measured using Living Image Software (IVIS Imaging Systems) for fluorescent MLys-EGFP cells for each time point. The graph shows the quantification of fluorescence as fold-change relative to t=0. n = 5 (5 mice per genotype; 2 wounds/mouse), which was repeated on at least two separate occasions. Boxplots show mean ± SD. Data were analyzed using Repeated Measures ANOVA; *p < 0.05. Scale bar, 1 cm. (B) Tissue sections from acute dorsal wounds in cPLA2α+/+;MLys-EGFP and cPLA2αKI/KI;MLys-EGFP mice at 0 and 6 h post-injury stained with neutrophil markers Ly6G+ and Neutrophil elastase (N-elas) and the macrophage markers CD68 and F4/80. Neutrophils and macrophages were quantified by stained area per tissue field as fold-change relative to wild-type (cPLA2α+/+). Each time point represents n = 10 wounds from 5 separate mice per genotype and was repeated in at least two separate occasions. Data are displayed as boxplots showing mean ± SD. Two-way ANOVA with Šídák’s multiple comparisons test **p<0.01, ***p<0.001). Scale bar, 500 μm.
Neutrophil infiltration was also examined using an Escherichia coli intraperitoneal (IP) injection model of sepsis. As with acute wound healing, neutrophil infiltration, in this case in the peritoneum, was increased significantly at both early (4 hrs) and late (12 hrs) time points after IP injection in the cPLA2αKI/KI mice (Fig. 2A). The cPLA2αKI/KI mice also demonstrated a shift toward neutrophil-mediated inflammatory response at time points T=4 hours and T=12 hours post-IP injection as opposed to the macrophage-mediated inflammation observed in cPLA2α+/+ mice (Fig. 2A). Thus, the t½ for the neutrophil resolution profile for the cPLA2αKI/KI mice was significantly increased to ~16 hrs versus ~12 hrs for the cPLA2α+/+ mice although the numbers of neutrophils were trending lower in cPLA2αKI/KI mice versus cPLA2α+/+ mice at 24 hours (Fig. 2A). Because increased neutrophil infiltration is linked to reduced survival in the cecal ligation and puncture (CLP) model of sepsis, we examined whether the cPLA2αKI/KI mice showed a worse survival outcome in this model. Survival was significantly increased in cPLA2αKI/KI mice versus cPLA2α+/+ and cPLA2α−/− mice (Fig. 2B). Furthermore, cPLA2αKI/KI mice also demonstrated significantly increased closure rates in regard to the healing of pressure ulcers using the Stadler model47,48(Fig. 2C–D). These data demonstrated that C1P is a negative regulator of neutrophil infiltration, acute and chronic wound healing, and sepsis outcome through direct association with cPLA2α. Furthermore, our data demonstrate that neutrophilia induced by the loss of the C1P-cPLA2α interaction positively correlates with better healing and survival outcomes, which is in stark contrast to the expected and reported clinical and pre-clinical manifestations for these disease states10,11.
Fig. 2. Loss of the C1P-cPLA2α interaction induces peritoneal neutrophilia in E. coli infection, improves survival outcomes in sepsis, and enhances the closure rate of pressure ulcers.
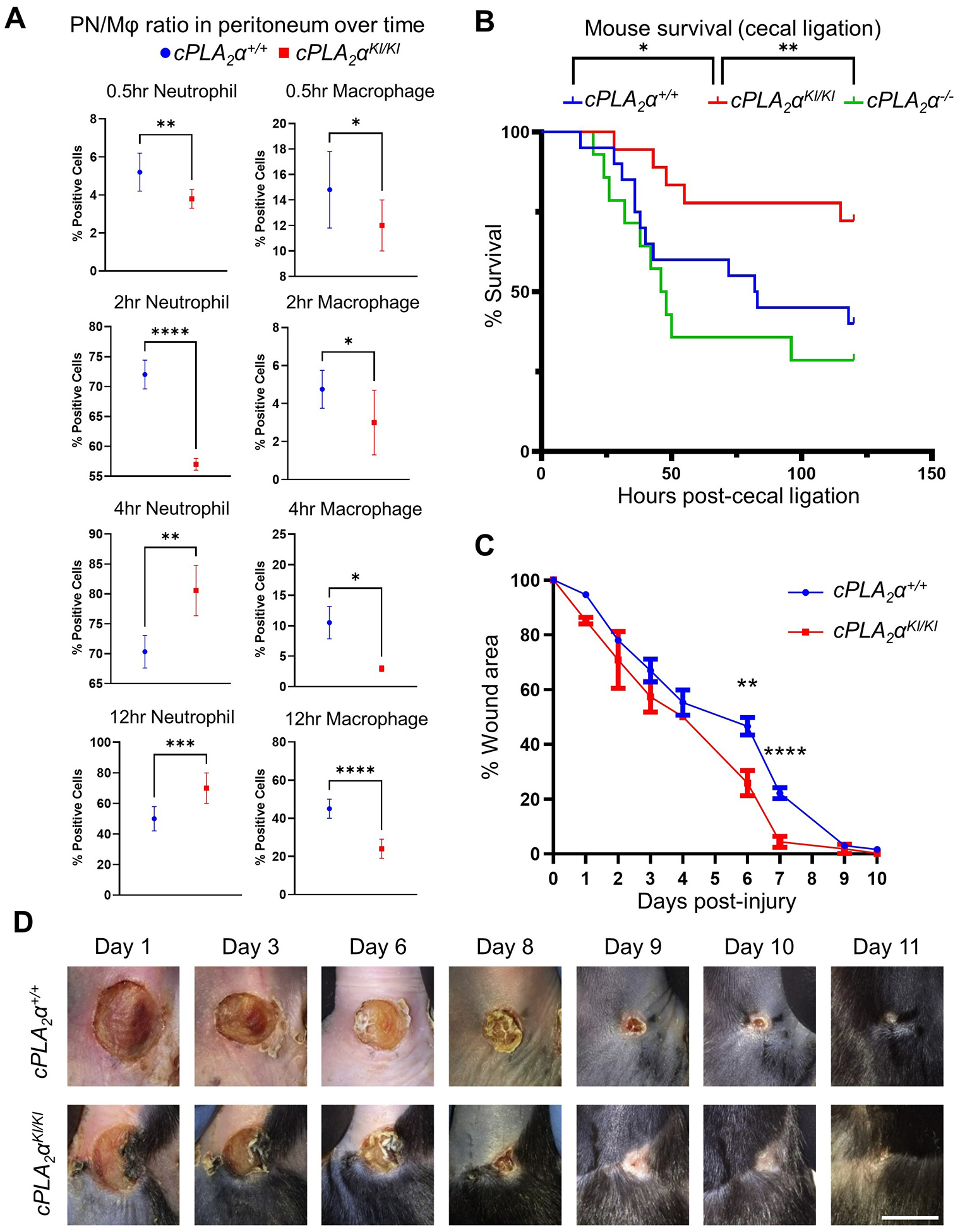
(A) Quantification of neutrophils and macrophages in peritoneal cells isolated from cPLA2α+/+, cPLA2αKI/KI, and cPLA2α−/− mice after intraperitoneal (IP) injection of E. coli. At the indicated times, peritoneal cells were extracted, sorted for neutrophil and macrophage markers (LY6G++ high and F4/80++ high, respectively), and quantified by FACS analysis. Data are displayed as mean ± SD and were statistically analyzed by unpaired t-test with Welch’s correction; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n=4–8 mice per genotype for each noted time point and repeated on at least two separate occasions. (B) Survival of cPLA2α+/+, cPLA2αKI/KI, and cPLA2α−/− mice subjected to cecal ligation and puncture to induce sepsis. The Kaplan-Meier method was used to estimate survival and statistically analyzed with respect to survival using the Log-rank Mantel-Cox test (cPLA2αKI/KI vs cPLA2α−/− **p = 0.0034, cPLA2αKI/KI vs cPLA2α+/+ *p = 0.0430, n=15–20 mice per genotype repeated on more than two occasions). (C and D) Quantification (C) and representative images (D) of wound closure in cPLA2α+/+ and cPLA2αKI/KI mice subjected to the Stadler model of pressure ulcers by ischemia and reperfusion. At the indicated times, wound area was assessed by change in area versus day 0. Boxplots are displayed. Data analyzed using Repeated Measures ANOVA; **p < 0.01, ****p<0.0001); n= 5–8 mice per genotype repeated on more than two occasions). Scale bar, 5 mm.
C1P attenuates neutrophil infiltration by suppressing 5-oxo-ETE biosynthesis and OXER1 activation
Because ablation of the C1P-cPLA2α interaction enhanced neutrophil infiltration in vivo, we examined the migration of neutrophils across an inflamed human endothelial monolayer in response to the chemoattractant lipolysaccharide (LPS) in trans-endothelial migration (NTEM) assays. In this regard, cPLA2αKI/KI mouse primary neutrophils (PNs) displayed a nearly 4-fold increase in NTEM over cPLA2α+/+ and cPLA2α−/− neutrophils after only 4 hours (Fig. 3A). Neutrophil chemotaxis is strongly controlled by eicosanoids, specifically lipoxygenase-derived eicosanoids such as leukotrienes, the biosynthesis of which depends on the activation of a phospholipase A2 such as cPLA2α39. Whereas leukotrienes were not detected in our NTEM system, cPLA2αKI/KI neutrophils possessed a lipidomic profile of increased 5-HETE and 5-oxo-ETE compared to cPLA2α+/+ and cPLA2α−/− cells, whereas cPLA2αKI/KI and cPLA2α−/− had decreased prostaglandin D2 (PGD2) and prostaglandin E2 (PGE2) compared to cPLA2α+/+ cells (Fig. 3B,C). These modulations in eicosanoid biosynthesis were biologically relevant, because inhibiting 5-HETE and 5-oxo-ETE production with a 5-lipoxygenase–activating protein (FLAP) inhibitor, MK886, or blocking OXER1, the target receptor of 5-HETE and 5-oxo-ETE, with Gue1654 reduced the NTEM of cPLA2αKI/KI PNs to that of cPLA2α+/+ PNs (Fig. 3D). The addition of biologically relevant amounts of 5-HETE rescued the effect of MK886, but not that of Gue1654, demonstrating the specificity of the inhibitors for 5-HETE biosynthesis or OXER1 (Fig. 3D). The lipid profile of cPLA2α+/+ or cPLA2αKI/KI PNs treated with MK886 showed reduced 5-HETE and 5-oxo-ETE, but PNs treated with Gue1654 maintained HETE and PGE2 abundances similar to their DMSO-treated controls (Fig. 3E) despite having decreased migration equal to cPLA2α+/+ cells. These data further demonstrate the specificity of these compounds for particular aspects of either 5-HETE biosynthesis or 5-HETE and 5-oxo-ETE receptor (OXER1) activation. These data also show that loss of the C1P-cPLA2α interaction in primary neutrophils enhances NTEM through the induction of 5-HETE and 5-oxo-ETE biosynthesis and subsequent activation of OXER1.
Fig. 3. Loss of the C1P-cPLA2α interaction enhance trans-endothelial migration of primary neutrophils through the induction of 5-HETE and 5-oxo-ETE biosynthesis and OXER1 activation.
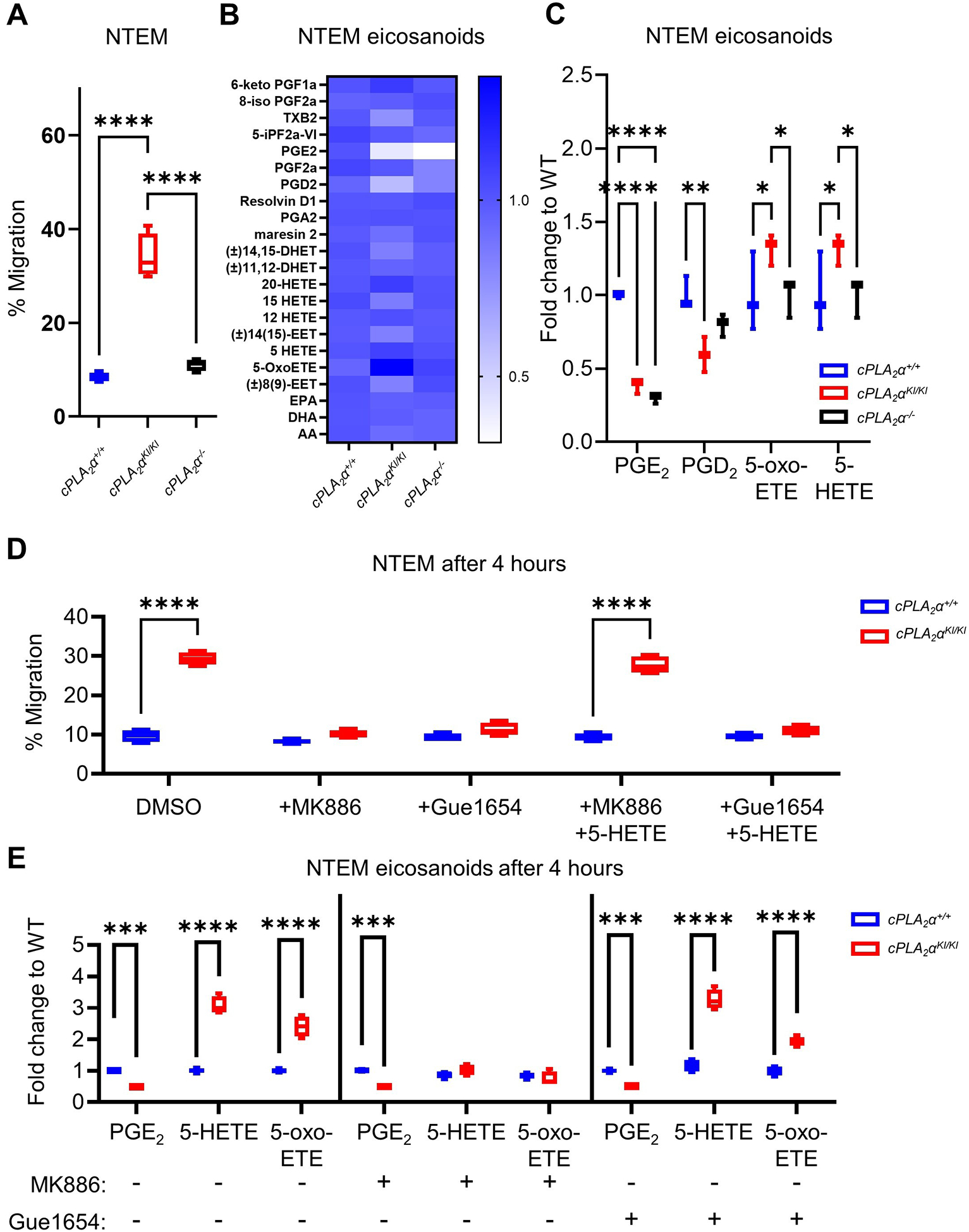
(A) Quantification of neutrophil trans-endothelial migration (NTEM) of primary neutrophils (PNs) from cPLA2α+/+, cPLA2αKI/KI, and cPLA2α−/− mice. (B) Eicosanoid profile of cytokine- and chemoattractant-activated neutrophils after 4 hours of NTEM. (C) Quantification of the eicosanoid, PGD2, PGE2, 5-HETE, and 5-oxo-ETE after 4 hours NTEM. (D) NTEM after 4 hours in the presence of vehicle (DMSO), the FLAP inhibitor MK886, the OXER1 antagonist Gue1654, and 5-HETE as indicated. (E) Quantification of eicosanoids in cPLA2α+/+ and cPLA2αKI/KI PNs treated with DMSO vehicle (−), MK886, and Gue1654 as indicated. Data for all panels are displayed as boxplots (one-way ANOVA with Tukey’s multiple comparisons test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). NTEM assays were n=6 biological replicates/group, and eicosanoid analyses were n=4 biological replicates/group; all experiments were repeated on at least two separate occasions.
C1P associates with cPLA2α to polarize neutrophils to an N1 subtype and suppress polarization to the N2 subtype
Ablation of the C1P-cPLA2α interaction enhanced neutrophil migration in vitro and in vivo, inducing neutrophilia in the inflammatory phase of wound healing and sepsis that was unexpectedly linked to improved healing and survival, outcomes that appear to contradict the reported effects of neutrophilia8–11,38. To investigate this apparent conundrum, our laboratory explored the hypothesis that the neutrophils from the cPLA2αKI/KI mice were less inflammatory, which might explain why the neutrophilia was more prohealing and proresolution than proinflammatory. To explore this concept of neutrophil polarity, we employed cellular assays for key characteristics of N1- vs N2-subtype neutrophils and demonstrated that activated cPLA2αKI/KI neutrophils exhibited the proresolution N2 phenotypes of reduced NETs (Fig. 4A), lower ROS (Fig. 4B), higher VEGF (Fig. 4C), and decreased TNF (Fig. 4D) compared to activated cPLA2α+/+ or cPLA2α−/− neutrophils. cPLA2αKI/KI neutrophils also uniquely showed enhanced phagocytosis for bacterial removal (Fig. 4E). Lastly, cPLA2αKI/KI neutrophils produced less epithelial damage across all time points evaluated (Fig. 4F). These N2-associated behaviors could be recapitulated in cPLA2α+/+ PNs and reversed in cPLA2αKI/KI PNs by inhibition of 5-HETE biosynthesis and OXER1 signaling or addition of exogenous 5-HETE or 5-oxo-ETE (Fig. S1A–F). Generation of NETs (NETosis) and ROS production were both increased in cPLA2αKI/KI neutrophils (to amounts similar to cPLA2α+/+ neutrophils) by inhibiting 5-HETE production using MK886 or OXER1 signaling using Gue1654 and could be suppressed in cPLA2α+/+ neutrophils by adding 5-HETE or 5-oxo-ETE (Fig. S1A,B). Similarly, proresolution VEGF production was increased in cPLA2αKI/KI PNs as well as enhanced in cPLA2α+/+ PNs by the presence of 5-HETE, whereas the opposite was observed for the inflammatory cytokine TNF, which was reduced by 5-HETE and 5-oxo-ETE in neutrophils from both genotypes (Fig. S1C,D). Phagocytosis was increased in cPLA2αKI/KI PNs compared to cPLA2α+/+ PNs. Inhibition of OXER1 signaling using Gue1654 also induced a large reduction in phagocytosis in both the cPLA2αKI/KI PNs and the cPLA2α+/+ PNs, whereas 5-HETE and 5-oxo-ETE treatment induced significant increases in cPLA2α+/+ PNs (Fig. S1E). Lastly, decreased epithelial damage caused by cPLA2αKI/KI PNs in comparison to cPLA2α+/+ PNs was also linked to 5-HETE and 5-oxo-ETE production and OXER1 signaling (Fig. S1F). These data demonstrate that the increases in 5-HETE and 5-oxo-ETE biosynthesis observed in cPLA2αKI/KI PNs enhanced PN functions associated with a less inflammatory N2 phenotype while suppressing proinflammatory phenotypes linked to the N1 phenotype.
Fig. 4. cPLA2αKI/KI primary neutrophils exhibit proresolution and anti-inflammatory phenotypes and polarization to the N2 subtype.
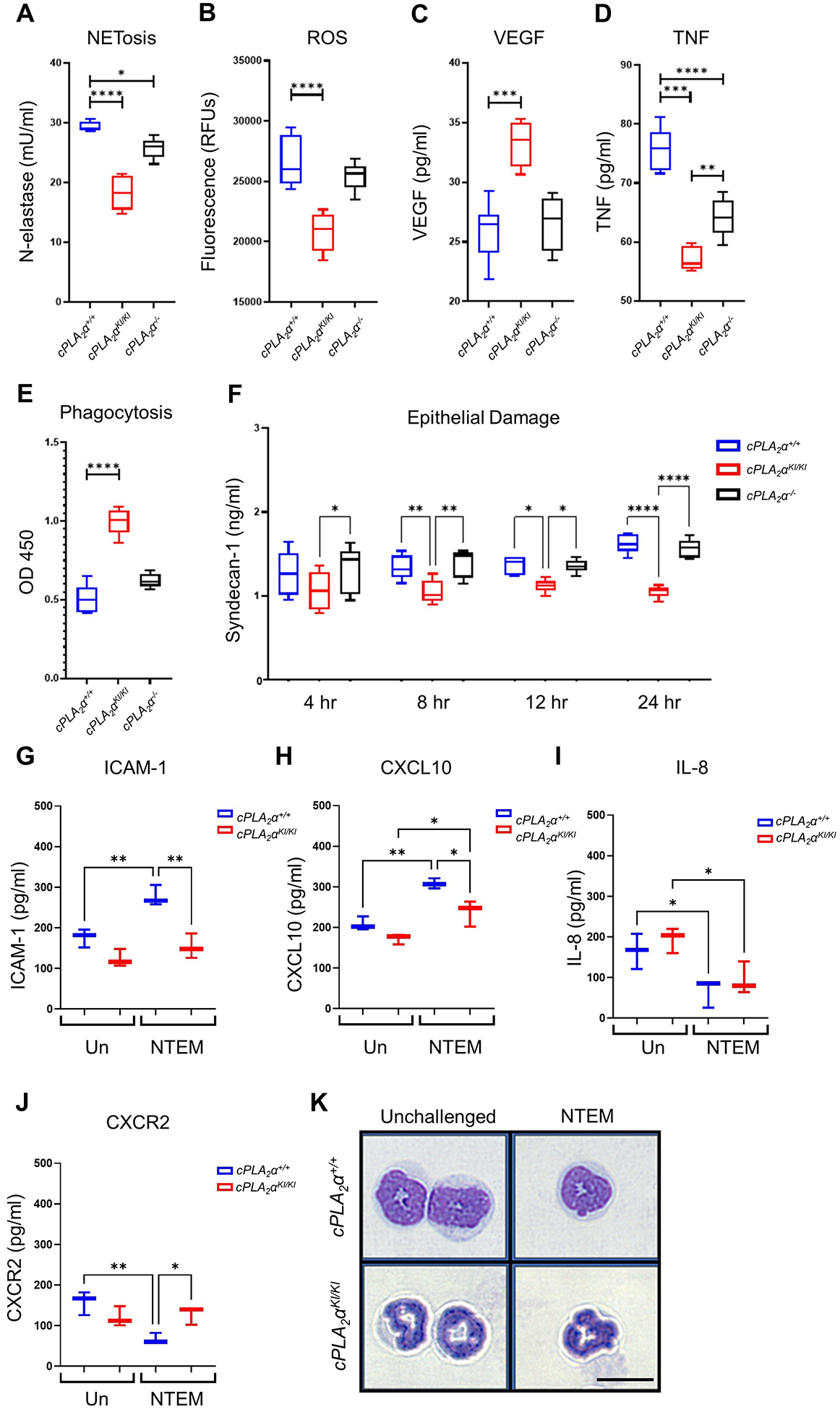
(A) NETosis assays measuring neutrophil elastase activity, (B) fluorescence-based ROS assays, (C) VEGF secretion, (D) TNF secretion, and (E) phagocytosis of E. coli by cPLA2α+/+, cPLA2αKI/KI, and cPLA2α−/− primary neutrophils after 4 hours of NTEM. (F) Quantification of Syndecan-1, a marker of endothelial damage, in culture media from cPLA2α+/+, cPLA2αKI/KI, and cPLA2α−/− primary neutrophils after NTEM for the indicated durations. Data in (A) to (F) were analyzed by one-way ANOVA with Tukey post-hoc; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n=6 biological replicates/group/time point, repeated on at least two separate occasions. (G to J) Quantification of neutrophil polarization markers in primary neutrophils from cPLA2α+/+ and cPLA2αKI/KI mice that were unchallenged (Un) or after 6 hours NTEM. N1 markers ICAM-1 (G) and CXCL10 (H) and N2 markers IL-8 (I) and CXCR2 (J) were assayed. Data in (G) to (J) were analyzed by one-way ANOVA with Tukey’s multiple comparisons test; *p < 0.05, **p < 0.01; n=4 biological replicates/group, repeated on two separate occasions. (K) Hematoxylin and eosin staining showing morphology of primary neutrophils from cPLA2α+/+ and cPLA2αKI/KI mice before challenge and after 6 hours of NTEM. Images are representative of n = 3 independent experiments. Scale bar, 10 μm.
To confirm that cPLA2αKI/KI PNs were more polarized toward the proresolution N2 subtype, we examined reported markers for the N1 and N2 subtypes23 in NTEM assays. Compared to cPLA2αKI/KI PNs, the cytotoxic N1-subtype neutrophil marker ICAM-123 was increased in cPLA2α+/+ PNs upon activation with LPS (Fig. 4G). Similarly, the perfusion recovery marker, CXCL10, another N1-subtype marker23, was also increased in activated cPLA2α+/+ PNs significantly more than in cPLA2αKI/KI PNs (Fig. 4H). In contrast, the neutrophil attractant IL-8, a reported N2-subtype marker23, was decreased in both cPLA2α+/+ and cPLA2αKI/KI PNs (Fig. 4I). On the other hand, the chemokine receptor, CXCR2, another reported N2-subtype marker23, was decreased significantly in cPLA2α+/+ PNs upon activation, but in contrast to IL-8, the amounts were unaffected and sustained in cPLA2αKI/KI PNs (Fig. 4J). Thus, cPLA2αKI/KI PNs demonstrated lower abundance of N1-subtype markers with concomitant maintenance of the N2-subtype marker, CXCR2, following activation. Lastly, cPLA2αKI/KI PNs had a thinner, less granulated nuclei compared to cPLA2α+/+ cells (Fig. 4K), a morphology that favors diapedesis and cell mobility during migration through tight tissue spaces50,51. Unlike cPLA2αKI/KI PNs, the nuclei of cPLA2α+/+ PNs presented with a reduced tubular shape, which is indicative of chromatin decondensation and granular protein disintegration commonly associated with increased NET production and the N1 phenotype52,53. Collectively, these data demonstrate that cPLA2αKI/KI PNs are polarized to a less inflammatory, N2-subtype.
The N2 phenotype induced by the ablation of the C1P-cPLA2α interaction is associated with suppression of the pentose phosphate pathway
To investigate the mechanism by which OXER1 signaling contributed to the N2 phenotype, we subjected cPLA2α+/+ and cPLA2αKI/KI PNs to unbiased proteomics because neutrophils tend to respond to stimuli in a rapid and transient fashion by modulating their proteome. Proteomic analysis of these PNs revealed five factors that were significantly differentially abundant in cPLA2α+/+ and cPLA2αKI/KI neutrophils (Fig. 5A). One of them, 6-phosphogluconolactolase (PGLS), which was decreased in cPLA2αKI/KI PNs, is directly linked to the oxidative branch of the pentose phosphate pathway (PPP), which generates NADPH and ribose 5-phosphate from glucose-6-phosphate (Fig. 5B). An additional key PPP enzyme, 6-phosphogluconate dehydrogenase (PGD), was also significantly decreased in cPLA2αKI/KI PNs, which could be recapitulated in cPLA2α+/+ PNs by inhibiting 5-HETE and 5-oxo-ETE biosynthesis or signaling (Fig. 5C).
Fig. 5. Loss of the C1P-cPLA2α interaction in primary neutrophils suppresses the pentose phosphate pathway.

(A) Quantification of pentose phosphate pathway (PPP) components Sucla2, PGLS, Cope, Tmx1, and Ebp in unchallenged cPLA2α+/+ and cPLA2αKI/KI primary neutrophils from unbiased proteomics analysis. LFQ, label-free quantification. Unpaired t-test with Welch’s correction; *p<0.05, **p<0.01, n=5 samples from 5 different mice for each genotype obtained on two separate occasions. (B) In the oxidative PPP, G6PD utilizes glucose-6-phosphate to generate NADPH and 6-phosphoglucolactone, which gluconolactolase (PGLS) converts to 6-phosphogluconate. PGD utilizes 6-phosphogluconate to generate ribulose-5-phosphate and NADPH. (C) Quantification of and immunoblotting for PGD and G6PD in protein extracts from cPLA2α+/+ and cPLA2αKI/KI primary neutrophils treated with DMSO vehicle, 5-HETE, or MK886 for 2 hours. FC to WT POI = fold change relative to wild-type protein of interest. POI was further quantified relative to the loading control (HSP90). (D) Enzyme activity assays for PGD and G6PD using protein extracts from cPLA2α+/+ and cPLA2αKI/KI primary neutrophils treated with DMSO vehicle or physcion. (E) NTEM of cPLA2α+/+ and cPLA2αKI/KI primary neutrophils treated with DMSO vehicle, physcion, or a combination of physcion plus 5-HETE. All data are displayed as boxplots (C-E analyzed by Two-way ANOVA with Šídák’s multiple comparisons; *p<0.05, **p<0.01, ****p<0.0001; n=3 to 4 biological replicates/group repeated on two separate occasions for each genotype.
We next examined whether modulation of PGD affected the polarization of neutrophils using physcion, an inhibitor of PGD. Physcion reduced PGD activity in cPLA2α+/+ PNs, effectively reproducing the cPLA2αKI/KI PPP phenotype of reduced PGD abundance, without further reducing PGD activity in cPLA2αKI/KI PNs or influencing the activity of glucose 6-phosphate dehydrogenase (G6PD), an enzyme that functions upstream of PGLS and PGD in the PPP (Fig. 5D). Inhibition of PGD did not significantly affect the migration of cPLA2α+/+ neutrophils in NTEM assays (Fig. 5E). Although the enhanced NTEM phenotype of cPLA2αKI/KI PNs was not recapitulated in cPLA2α+/+ PNs by physcion, inhibition of PGD with this compound decreased ROS and TNF production (Fig. 6A,B) and increased VEGF production in cPLA2α+/+ PNs (Fig. 6C), mimicking the abundance of these markers in cPLA2αKI/KI PNs. NETosis and epithelial damage were unaffected by blocking the PPP through PGD inhibition (Fig. 6D,E), and phagocytotic activity was diminished in cPLA2αKI/KI PNs when PGD was inhibited, whereas exogenous 5-HETE could overcome this effect and enhance phagocytosis (Fig. 6F).
Fig. 6. The PPP is linked to specific neutrophil behaviors.
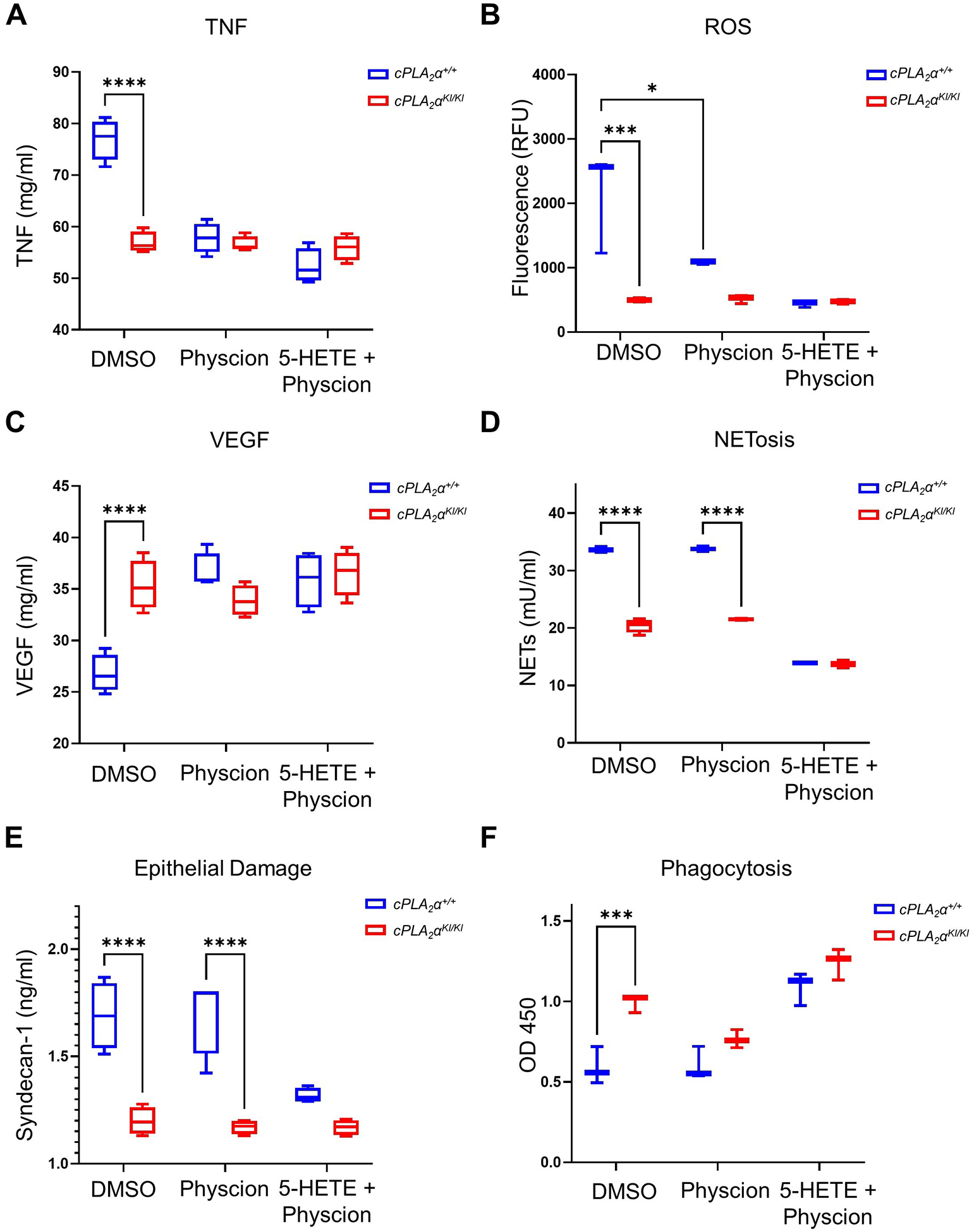
(A) TNF production, (B) ROS production, (C) VEGF production, and (D) NET production was quantified in cPLA2α+/+ and cPLA2αKI/KI primary neutrophils pre-treated with DMSO vehicle, physcion, or a combination of physcion plus 5-HETE for 30 minutes and collected after 6 hours NTEM. (E) Quantification of the damage marker Syndecan-1 in cPLA2α+/+ and cPLA2αKI/KI primary neutrophils after 30 minute pre-treatment with DMSO vehicle, physcion, or a combination of physcion plus 5-HETE followed by 12 hours NTEM. (F) Phagocytosis of bacteria was measured in cPLA2α+/+ and cPLA2αKI/KI primary neutrophils pre-treated for 30 minutes with DMSO vehicle, physcion, or a combination of physcion plus 5-HETE and collected after 6 hours NTEM. Data displayed are mean ± SD. Two-way ANOVA with Šídák’s multiple comparisons; *p<0.05, ***p<0.001, ****p<0.0001; n=4 biological replicates/group for each genotype repeated on two separate occasions.
The ability of the 5-HETE and 5-oxo-ETE biosynthetic pathway to suppress PGD production and activity translated to the HL-60 cell model of neutrophil polarization. Specifically, treatment of HL-60 cells, a human granulocyte precursor cell line, with 5-oxo-ETE mimicked the effect of the reported N2 polarization factor transforming growth factor–β (TGF-β)23,54 on significantly reducing PGD abundance (Fig.7A) but did not affect the response to the neutrophil polarization factor, all-trans retinoic acid (ATRA) (Fig. 7A), which is reported to induce an N1 phenotype, as shown by enhancement of ICAM-1 abundance23, 55–59. The effect of 5-oxo-ETE on the suppression of PGD required the OXER1 receptor, because Gue1654 blocked this effect (Fig. 7B). Additionally, 5-oxo-ETE induced CXCR2 production while suppressing ICAM-1 production (Fig. 7A). In congruence with our findings with physcion in the PNs, modulation of PGD expression using genetic approaches showed that PGD is a major upstream regulator of neutrophil polarization. PGD overexpression induced an N1 phenotype, as shown by significantly enhanced production of ICAM-1, ROS, and TNF with concomitant decreases in CXCR2 and VEGF (Fig. 7C and S2A). In contrast, PGD knockdown induced the opposite effect, provoking an N2 phenotype (Fig. 7D and S2B). These data demonstrated that suppression of the PPP is downstream of OXER1 signaling reducing specific neutrophil functions such as ROS generation and TNF production while stimulating VEGF production, all of which are key elements of neutrophil polarization (Fig. 7, C and D). Furthermore, enhanced NTEM, suppressed NETosis, and a reduction in neutrophil-mediated epithelial damage linked to the N2 phenotype of cPLA2αKI/KI PNs were PPP-independent but downstream of OXER1 signaling.
Fig. 7. 5-oxo-ETE–dependent activation of OXER1 polarizes HL-60 cells to an N2 phenotype, in part by suppressing the PPP.
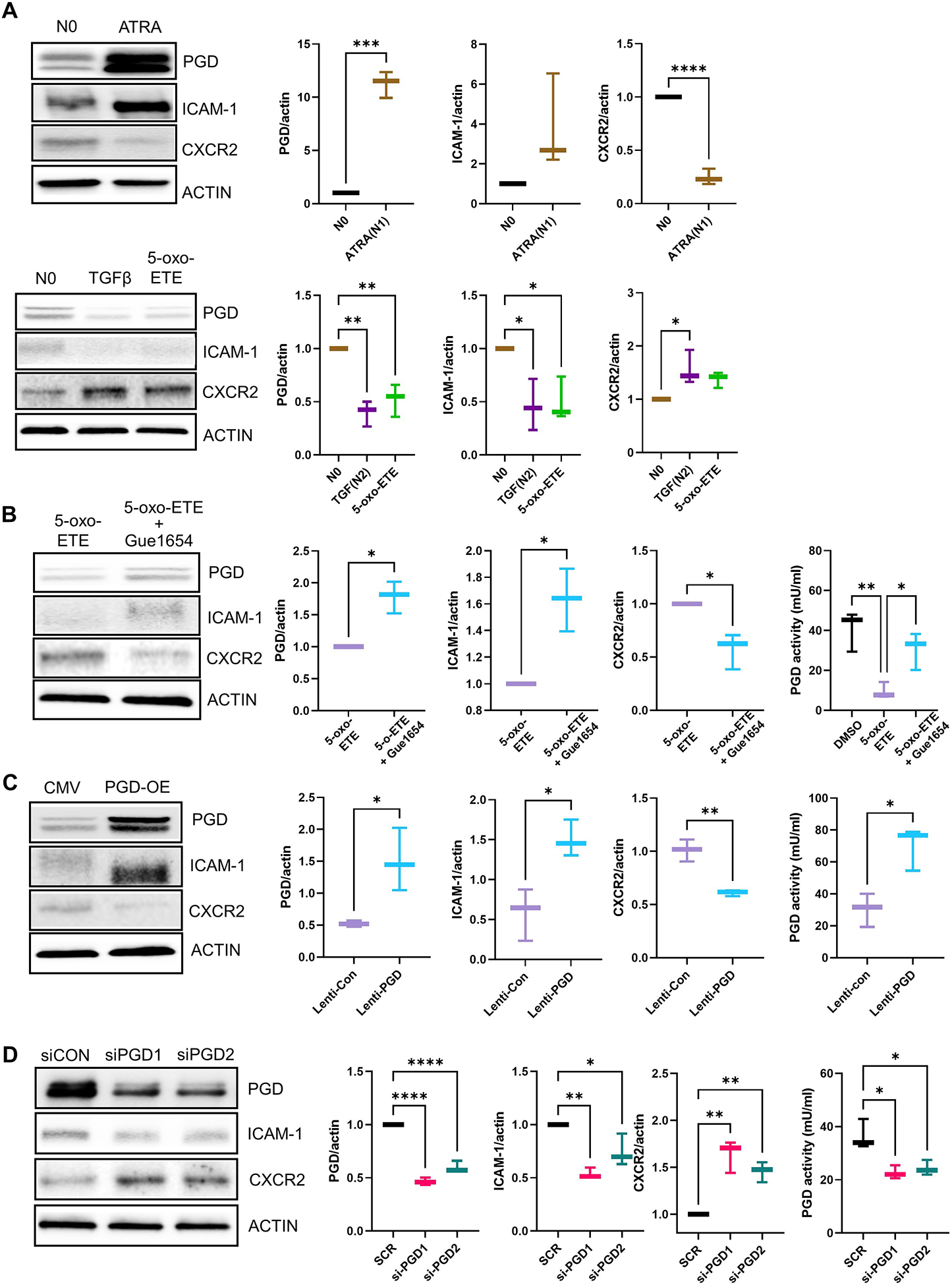
(A and B) HL-60 cells were treated with DMSO (no polarization, N0), the N1 polarization inducer all-trans retinoic acid (ATRA) for 3 days, the N2 polarization inducer TGFβ for 3 days, or with 5-oxo-ETE or 5-oxo-ETE plus Gue1654 for 12 hrs. Gue1654 was pre-treated for 30 min pior to the addition of 5-oxo-ETE for 12 hrs. Protein extracts were used for immunoblotting and quantification of PGD, the N1 marker ICAM-1, the N2 marker CXCR2, and β-actin (ACTIN). In (B), protein extracts were also used to measure PGD enzymatic activity. Data are displayed as boxplots (mean ± SD) analyzed by either unpaired t-test with Welch’s correction (graphs with two groups) or one-way ANOVA with Tukey’s multiple comparisons test (graph with three groups); *p<0.05, **p<0.01, n=3 to 4 biological replicates/group repeated on at least two separate occasions. (C and D) Lentiviral-mediated PGD overexpression (A) and siRNA-mediated PGD knockdown (B) in HL-60 cells. Cells were treated with high-titer control lentivirus (CMV), PGD lentivirus (PGD-OE), scrambled control siRNA (siCON), or PGD-targeting siRNAs (siPGD1 and siPGD2) for 48 hrs. Protein extracts were used for immunoblotting and quantification of PGD, ICAM-1, CXCR2, and β-actin and for PGD activity assays. Data for PGD enzymatic activity are displayed as boxplots (mean ± SD) and were analyzed by one-way ANOVA with Tukey’s multiple comparisons test; *p < 0.05, , n=4 (biological replicates/group) repeated on two separate occasions. Data for protein quantification are displayed as boxplots (mean ± SD) and were analyzed by one-way ANOVA with Tukey’s multiple comparisons test; *p<0.05, **p<0.01, ****p<0.0001, (p > 0.05), n=3 to 4 (biological replicates/group) repeated on two separate occasions.
Suppression of the PPP by 5-oxo-ETE signaling contributes to neutrophil N2 polarization
There are no reports specifically linking OXER1 signaling to regulation of the PPP pathway, but the OXER1 pathway has been reported to induce the mitogen-activated protein kinase (MAPK) pathway60, which has been reported as an upstream activation pathway for the PPP61–64. Treatment of HL-60 cells with 5-oxo-ETE induced the activation of the MAPK pathway, as shown by enhanced phosphorylation of MAPK kinase MEK (Fig. S3). Pre-treatment of HL-60 cells with the MEK inhibitor PD-325901 prior to 5-oxo-HETE treatment reduced the phosphorylation of MEK, blocked the induction of CXCR2, and partially alleviated the suppression of PGD and ICAM-1 (Fig. S3). The MEK inhibitor also reduced the ability of 5-oxo-ETE to reduce ROS and TNF and increase VEGF (Fig. S3). These data show that 5-oxo-ETE, through OXER1, promotes activation of the MAPK pathway, which contributes to the N2 polarization of neutrophils by inhibiting the PPP (Fig. 8).
Fig. 8. Schematic representation of the proposed model of the C1P-cPLA2α interaction modulating neutrophil polarization.
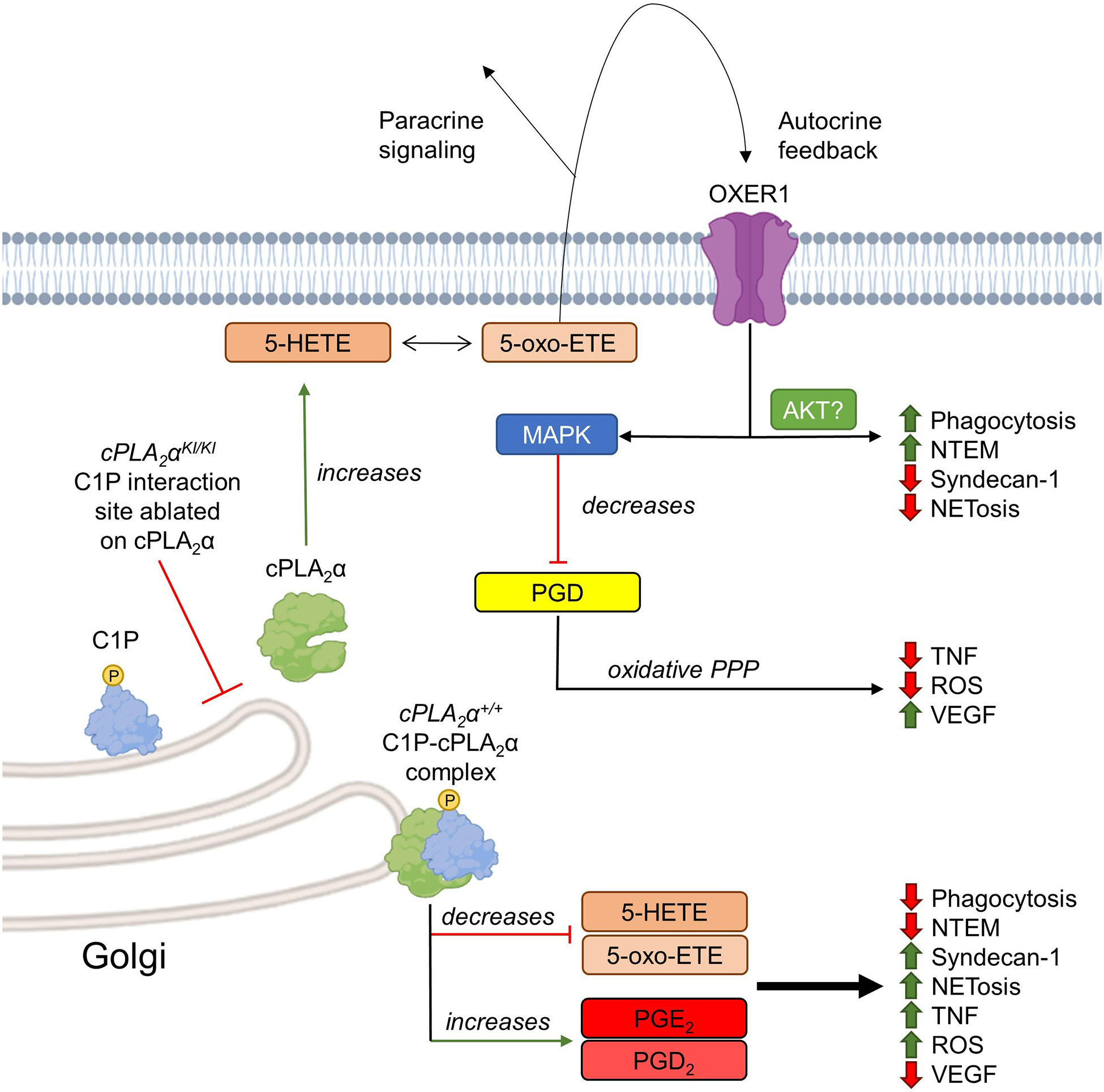
cPLA2α not bound to C1P increases 5-HETE and 5-oxo-ETE production and paracrine and autocrine signaling through OXER1, resulting in activation of the MAPK pathway and downstream suppression of the PPP. This lipid profile correlates with decreased endothelial damage (Syndecan-1), NETosis, ROS production, and TNF production and increased neutrophil phagocytosis, VEGF production, and NTEM. Alternatively, C1P recruits cPLA2α to the Golgi apparatus and increases COX-2–derived eicosanoids (PGE2 and PDE2), decreases 5-HETE and 5-oxo-ETE biosynthesis, and results in pro-inflammatory neutrophil behaviors.
DISCUSSION
Previously our laboratory showed that C1P increased during the inflammatory phase of human wound healing and then decreased during the initiation of the proliferation phase65. Further, loss of the ability of cPLA2α to bind C1P or inhibition of C1P biosynthesis enhanced wound maturation showing C1P to be a negative regulator of the later stages of acute wound healing24,31,65. Herein, we show that C1P, through association with cPLA2α, also plays a key role in the inflammatory phases of multiphasic physiologies such as the immune response to septic challenge as well as the wound healing process, specifically the enhancement of inflammatory phenotypes. This was shown by experiments with knockin mice expressing a form cPLA2α that is unable to bind C1P, which provided beneficial survival for mice subjected to the cecal ligation and puncture model of sepsis as well as to the healing of pressure ulcers. Poor outcomes in these disease states are correlated with neutrophilia in the inflammatory phase leading to tissue destruction10,11,38. Unexpectedly, we observed increased and sustained neutrophil numbers in the wound site and the peritoneum in these mice despite the common association of neutrophilia with poor outcomes in patients10,11,38. This unexpected positive outcome caused what we term the “neutrophil conundrum” and raised the key mechanistic question as to how neutrophilia was correlated with better healing and survival outcomes. Our model suggests that C1P is an important mediator of polarizing neutrophils to an N1 subtype through association with its direct target, cPLA2α. Indeed, blockage of this interaction polarized neutrophils to an N2 subtype that was less inflammatory and more proresolution regardless of the number of neutrophils attracted to the wound site, and thus, neutrophilia, in this context, was beneficial.
Our findings also suggest the plausible hypothesis that the stalling of wounds in the healing process is caused by sustained increases in C1P in the wound, which would drive the production of inflammatory, cyclooxygenase (COX)-derived prostaglandins such as PGE224,28,31–37,65–69. Indeed, inhibition of CERK or ablation of the cPLA2α-C1P interaction led to the loss of these inflammatory mediators and induction of 5-oxo-ETE biosynthesis, which is a plausible key mechanism regulating the transition from the inflammatory stage to proresolution and induction of the proliferation and remodeling stages. These findings may also explain reported observations, such as the link between amounts of PGE2 in non-healing pressure ulcers70, as well as the link between aging and poor healing outcomes, because C1P amounts increase with aging71. Our laboratory previously demonstrated a critical role for C1P in recruiting cPLA2α to the Golgi apparatus in response to inflammatory agonists to induce the formation of prostaglandins by providing arachidonic acid to COX24,27,31–37,65–69. Indeed, C1P drives PGE2 biosynthesis at the expense of 5-oxo-ETE biosynthesis through differential cellular localization24,27,31–37,65–69,72, but our findings show that suppression of C1P biosynthesis could reverse the process and suppress inflammation, fostering the transition to resolution and healing31. Thus, the clinical implications of this finding are high because our data imply that either inhibition of CERK or induction of 5-oxo-ETE biosynthesis will suppress the N1 subtype of neutrophils and induce a prohealing and proresolution phenotype leading to the healing of stalled pressure ulcers. In regard to sepsis, this same therapeutic manipulation may reduce the hyperinflammatory stage, allow for resolution and less tissue and immune system damage, and limit the negative transition to the immune suppressive stage, which is linked to increased mortality73,74. CERK inhibitors are still in the stage of 1st generation molecules such as the nanomolar CERK inhibitors, NVP-231 (Novartis) and SYR382141 (Takeda), but treatment of mice post-wounding with SYR382141 enhances acute wound healing and is well-tolerated by the mice with no noticeable side effects31. Therefore, the inhibition of CERK may be a plausible avenue to treat disease states linked to hyperinflammatory stalling, and evolution of the current CERK inhibitors could make this a future therapeutic reality.
As noted previously, the observed phenotypic differences in neutrophils were linked to neutrophil polarization (N1 vs N2) through specific phenotypic presentations and cell surface markers. As to the latter, the N1-subtype marker ICAM-1 increased nearly 50% in cPLA2α+/+ neutrophils after 6 hours of NTEM but remained relatively unchanged in cPLA2αKI/KI cells. ICAM-1 is cleaved in a dose-dependent manner by neutrophil elastase in humans75, which correlates to the slight increase in neutrophil elastase present in wound tissue of cPLA2αKI/KI mice. Similarly, both cPLA2α+/+ and cPLA2αKI/KI neutrophils showed increases in CXCL10 over 6 hours, but cPLA2αKI/KI cells produced significantly less CXCL10 despite having more active neutrophil recruitment. This is in contrast to reports that antibodies recognizing CXCL10 reduce neutrophil recruitment to oxidative stress–induced neutrophilic airway inflammation by up to 72%76 and suggest context-driven recruitment of N1 vs N2 neutrophils. For markers of anti-inflammatory N2 neutrophils, we first examined IL-8. Unlike ICAM-1, which is induced by proinflammatory cytokines, IL-8 is a neutrophil attractant produced by mononuclear phagocytic cells and other noninflammatory cells77. Although some research suggests that IL-8 can itself cause neutrophil-dependent tissue damage78, work in tumor identification has established IL-8 as a standard proresolution N2 marker that enhances the immunosuppressive microenvironment23,79. In this study, cPLA2α+/+ neutrophils decreased IL-8 production after 6 hours of NTEM, which was also observed for cPLA2αKI/KI neutrophils. In contrast, CXCR2 decreased significantly over time in cPLA2α+/+ neutrophils, which was sustained in cPLA2αKI/KI cells. CXCR2 ligands, CXCL1 and CXCL2/3, are chemotactic for neutrophils, thus CXCR2 is thought to recruit neutrophils independently of the presence of cytokines with proinflammatory properties80. This fortifies the hypothesis that cPLA2αKI/KI neutrophils can recruit more neutrophils to a wound without incurring a hyperinflammatory cytokine storm resulting in tissue damage. Indeed, previous work in bovine models demonstrate that increased CXCR2 corresponds to enhanced neutrophil phagocytosis of bacterial pathogens81, which our data showed is reliant upon OXER1 signaling. However, the existing research on N1 and N2 polarization is not unanimous on phenotypic observations. For instance, murine tumor models suggest that neutrophils high in CXCR2 and IL-8 have a decreased ability to kill parasitic protozoa23. Thus, the complex microenvironment appears to influence neutrophil behavior in a manner that requires further understanding. Additionally, our data showed that the N2-subtype marker IL-8 did not differ between the genotypes, which suggests that either IL-8 is not a marker for N2-subtype neutrophils in all contexts or the cPLA2αKI/KI PNs are a mixed N1/N2 subtype analogous to an M1/M2 macrophage82,83. Because the biological phenotypes match the N2-subtype, the former possibility is more likely in this context, but future research is needed within the neutrophil polarization field in this regard. Furthermore, the question remains as to what is the normal physiological response where N2 neutrophils are recruited and needed. Our study suggests that for wound healing, a logical mechanism would be that at the end of the inflammatory stage of wound healing as C1P amounts decline, 5-HETE biosynthesis is activated in the neutrophils present in the wound site. 5-oxo-ETE would strongly recruit N2 neutrophils just prior to or during efferocytosis to complete the clearance of pathogens, apoptotic bodies, and damaged tissue while simultaneously producing VEGF to initiate the revascularization that is linked to the proliferation and remodeling stages.
Mechanistically, proteomic analysis indicated cPLA2αKI/KI neutrophils had decreased PGLS, which is part of the oxidative branch of the PPP. This enzyme is responsible for the production of 6-phosphogluconate, which the second of two NADPH-producing enzymes in the oxidative PPP, PGD, utilizes as a substrate. Accordingly, PGD amounts were also significantly decreased in cPLA2αKI/KI neutrophils before and after chemokine challenge. The PPP has been linked to some neutrophil behaviors such as NETosis57, ROS production84, and phagocytosis85. For example, Amar et al. showed that activated human neutrophils shunted metabolic activity away from pyruvate-generating glycolysis in favor of the PPP, thereby enhancing the antimicrobial function of neutrophils85. Additionally, the PPP enzymes G6PD and PGD produce NADPH to fuel superoxide production through NADPH oxidase during NET formation86. Various aspects of the cPLA2αKI/KI N2 phenotype could be recapitulated in cPLA2α+/+ PNs by inhibiting PGD with the compound physcion, and our findings are in line with physcion reported as having anti-inflammatory87 and antimicrobial activities88. Whereas physcion alone was unable to decrease NETosis or epithelial damage in cPLA2α+/+ PNs, inhibition of the PPP reduced ROS production in line with other studies. Suppression of PGD with physcion also decreased TNF production and increased VEGF production in cPLA2α+/+ PNs analogous to cPLA2αKI/KI PNs and an N2 phenotype. Physcion reduced cPLA2αKI/KI phagocytosis to that of cPLA2α+/+ neutrophils, but this phenotype was only recovered by treatment with a combination of physcion and 5-HETE. Whereas PGD knockdown stimulated the same phenotypes as physcion in the HL-60 model, increased expression of PGD induced the opposite phenotype. These data confirm the major role that this enzyme plays in regulating neutrophil polarization. Thus, neutrophil behaviors in regard to ROS, TNF, and VEGF production as well as ICAM-1 and CXCR2 production can be tilted in favor of an anti-inflammatory phenotype by decreasing PGD using physcion, and these PN behaviors show complete dependence on the PPP downstream of OXER1 signaling. On the other hand, NETosis, endothelial damage assayed by increases in Syndecan-1, and phagocytosis were PPP-independent in this context although rigidly dependent upon OXER1 signaling (Fig. 8).
This study also explored the cell signaling pathway mediating OXER1-induced suppression of PGD. Our laboratory found that inhibition of MEK, a key enzyme in the MAPK pathway, blocked the ability of 5-oxo-ETE to reduce PGD and drive polarization of the HL-60 cells to an N2 phenotype. Our results support the previous report by Konya and co-workers showing that that the activation of OXER1 induced activation of the MAPK pathway60. Our findings suggest that FDA-approved therapeutics that target the MAPK pathway may be effective in enhancing both wound healing and the recovery of patients from sepsis89. Aiding these types of therapeutics would be the knowledge of mechanisms downstream of OXER1 signaling linked to the PPP-independent phenotypes, such as those damaging the endothelial layer and driving NTEM. As to the latter, OXER1 activation has been reported to induce migration and chemotaxis of breast cancer cells and Chinese Hamster Ovary cells through the AKT pathway90,91, which makes this signaling pathway a major candidate for future studies92 on 5-oxo-ETE–induced NTEM.
The data presented in this study strongly suggest that 5-HETE and 5-oxo-ETE metabolism and OXER1 signaling are potent regulators of neutrophil behavior that tips the scale toward proresolution mechanisms allowing wounds to progress to proliferation and remodeling stages at a much faster rate as well as promoting the survival of mice to sepsis. Suppression of the PPP downstream of OXER1 signaling was required for key aspects of the polarization of neutrophils to the N2 subtype. Lastly, this study strongly supports a proinflammatory role for C1P through direct binding of cPLA2α to divert arachidonic acid generation to the production of proinflammatory prostaglandins at the expense of 5-HETE and 5-oxo-ETE biosynthesis, which may provide a serendipitous opportunity to induce this eicosanoid “class-switch” for the development of new therapies, thereby providing better clinical outcomes for both sepsis and chronic wound patients.
MATERIALS AND METHODS
Reagents
Materials and sources are provided in the Supplementary Materials (table S1).
Genotyping of cPLA2α+/+, cPLA2αKI/KI, and cPLA2α−/− mice
In this study, both cPLA2α knockout (KO) mice that globally lacked cPLA2α (cPLA2α−/−) and cPLA2a knock-in (KI) mice that globally lacked the C1P interaction site in cPLA2α (cPLA2αKI/KI) were utilized as previously described by us24,31,93,94. These mouse models were designed using a construct with an inserted cassette containing puromycin resistance, a premature stop codon flanked by loxP sites, and three mutated amino acids (R57A, K58A, R59A) in the endogenous cPLA2α locus24. This design enabled the generation of cPLA2α KO mice producing the truncated protein in all tissues, and cPLA2α KI mice by the addition of cytomegalovirus (CMV)-driven expression of CRE recombinase24,31. Genotyping of cPLA2α+/+, cPLA2αKI/KI, and cPLA2α−/− mice was performed by first collecting genomic DNA using an Accustart II genotyping kit followed by polymerase chain reaction (PCR) as previously described by us24,31,93,94 using the following primers: KO: PLA2 58534-58556 (P1), PLA2 58780-58757 (P2), and KI: PLA2 58534-58556 (P1), PLA2 58780-58757 (P2), and SV40 2590-2567 (P3). 50ng of genomic DNA was used for each reaction at the following reaction cycles repeated 33 times, 94°C for 1 min, 59°C for 1 min, and 72°C for 2 min. Reaction products expected are as follows cPLA2α- cPLA2α+/+: 237bp, cPLA2αKI/KI: 412bp, and cPLA2α−/−: 361bp and were examined using a 2% agarose gel.
PGD gene gain of or loss of function
HL-60 cells (2×106) in 5ml 5% FBS medium were plated in a 10-cm cell culture dish and transfected with Silencer Select Negative Control No.1 siRNA (Thermo Fisher Scientific, 4390844) or two different, Silencer select siRNA-PGDs (Thermo Fisher Scientific, 4390824, ID: s10394 (siPGD#1) and s10395 (siPGD#2)) using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, 13778150) following manufacturer instructions. For overexpression of PGD, cells were transduced with Control Lentiviral Activation Particles (Santa Cruz, sc-437282) or PGD synthase Lentiviral Activation Particles (Santa Cruz, sc-405437-LAC and sc-405437-LAC-2) with 5ug/ml polybrene (Millipore Sigma, TR-1003-G) in a 5% FBS medium. 18 hours of post-transfection or post-transduction, the medium was replaced with a full medium for additional 30 hours.
Cecal Ligation and Puncture (CLP) Surgical Procedure
Male mice (6–12 weeks of age) were anesthetized with isoflurane at 5% for induction and at 3% for surgical procedures. A longitudinal midline incision was made with a scalpel, being careful not to penetrate into the peritoneal cavity. After the initial incision, entry into the peritoneal cavity (mice, 1.5–2 cm was completed to gain access to the cecum. The cecum was ligated with 2.0 silk ties at the designated position for the desired severity grade (High-grade/Severe ~75% ligation). The cecum was perforated with a 26-gauge needle by single through-and-through puncture midway between the ligation and the tip of the cecum in a mesenteric-to-antimesenteric direction. After removing the needle, extrude a small amount (droplet) of feces from both the mesenteric and antimesenteric penetration holes. Peritoneum and abdominal musculature were closed by applying simple running sutures. Mice were injected with prewarmed normal saline (37 °C; 5 ml per 100 g body weight) subcutaneously and monitored every 30 minutes for 3 hours. Post- surgical recovery monitoring occurred every 6–12 hours for 5 days.
IP Injection and FACS Neutrophil and Macrophage Sorting
cPLA2α+/+, cPLA2αKI/KI, and cPLA2α−/− mice (male, 6–12 weeks of age) were subjected to a 1×104 E. coli injection IP (Thermo Fisher Scientific #E13231). At the indicated times, peritoneal cells were extracted, centrifuged, and re-suspended in FACS buffer with 0.5 μL Ly6G+ (BD biosciences #551461) and F4/80 (Invitrogen/Thermo Fisher Scientific #MF48000/BD biosciences#553142) antibodies for flow cytometry.
Chronic Wound Healing in Mice
Chronic would closure rate was examined in male mice (8–12 weeks of age) using a previously described pressure ulcer model of ischemia-reperfusion injury47. Specifically, dorsal skin was shaved, gently pulled and placed between two round ceramic magnetic plates (1000 G magnetic force). The magnets were cycled (on for 12 hours, then removed for 12 hours) 3 times, leaving a 5-mm pressure ulcer. Wound images were analyzed using the Fiji image J bundle. Wounds were tracked as percent of initial wound size over 10 days.
Acute Wound Healing in Mice
The acute wound closure rate was examined in male mice (10–14 weeks of age) as recently reported by us24,31. Specifically, a 5 mm biopsy punch was performed on the dorsum of each mouse. Wounds were dressed using Tegaderm (3M Medical) and imaged over the course of 72 hours using an in vivo imaging system (IVIS), in particular, the Xenogen IVIS Spectrum system. Wound images were analyzed using the Living Image software (Perkin Elmer) and GFP fluorescence (RFUs) of regions of interest (ROI) were measured with excitation wavelength of 480 nm and emission wavelength of 550 nm. Wounds were tracked as percent of RFUs at time 0 over 72 hours for cPLA2α+/+ or cPLA2αKI/KI mice.
Western Immunoblotting
Total cellular protein (5–10 μg) was electrophoretically separated on 10% SDS-polyacrylamide gels. Samples were electrophoretically transferred to PVDF membranes, blocked with non-fat milk, and probed with the indicated antibodies for proteins of interest as previously done95–99. Antibodies were purchased from Thermo Fisher Scientific (G6PD polyclonal, #PA5-27359; RRID_AB_2544835, 1:1000; PGD polyclonal, #PA5-83188; RRID_AB_2790344, 1:1000; TMX polyclonal, #PA5-116605, 1:1000; heat shock protein 90 (HSP90) monoclonal, #MA1-10372, 1:1000, and β-actin monoclonal antibody, #MA1-140; RRID_AB_2536844, 1:1000); from Cell signaling Technology (Total MEK antibody, #9126s and phospho-MEK antibody, #3958s); and from Proteintech (CXCR2 antibody, #20634-1-AP, 1:1000; ICAM-1 antibody, #10831-1-AP).
Cell Culture and Reagents
Human umbilical vein endothelial cells (HUVECs) (Lonza C2519A) were cultured in EBM-2 endothelial cell growth basal media (Lonza CC-3156) and SingleQuots™ supplement kit (Lonza CC-4176). Cells were maintained in a 95% air / 5% CO2 incubator at 37 °C. Cells were passaged every 3–5 days at > 90% confluency, and all experiments were performed within 8 passages. HL-60 cells (ATCC, CC240, Human) were cultured in ATCC-formulated Iscove’s Modified Dulbecco’ Medium (ATCC, 30–2005) with 20% fetal bovine serum and maintained in a 95% air/5% CO2 incubator at 37 °C. Cells were passaged and maintained at ~5×10^5 cells per ml density. The medium was renewed every 3 days. All experiments were performed under 15 passages. Cell lines were tested every two months for mycoplasma (universal mycoplasma detection kit, ATCC, #30-1012K) throughout the study starting two months after thawing cells received from ATCC. Parental cell lines were authenticated by STR (short tandem repeat) profiling.
HL-60 cells underwent N1 differentiation with ATRA (Cayman Chemical, #11017, 50 μM) for 3 days or N2 differentiation with TGFβ (Abcam, ab50036, 100 ng/ml) for 3 days. N0 (untreated, naïve cells) control cells were with 0.1% DMSO which is with respect to the solvent of inhibitors (0.1% DMSO). Neutrophil-liked HL-60 cells used in the assays were from 1.25% DMSO treatment for 3 days. For combination treatment, Gue1654 (25 μM) with 5-oxo-ETE or MEK inhibitor, PD0325901 (25 μM) (TOCRIS, #4192) with 5-oxo-ETE, cells were pre-treated with inhibitor for 30min before adding 5-oxo-ETE(1 μM).
Exogenous Addition of Small Molecule Compounds to Neutrophils
Eicosanoids and/or inhibitors (from Cayman Chemical) were applied at the following concentrations: 1.0 nM 5-HETE (#34210), 1.0 nM 5-oxo-ETE (#34250), 7.5 nM MK886 (#21753), 25 μM Gue1654 (#29686), 100 nM physcion (Santa Cruz Biotech SC-205805). Cells were allowed to equilibrate with inhibitors for 30 minutes before experimentation.
Assays for PGD, G6PD, Cytokines, Phagocytosis, ROS Production, Cell Surface Markers, and NETosis
The following were utilized in this study: G6PD activity assay kit (Abcam #ab176722), PGD activity assay kit (Abcam #ab273328), NETosis assay kit (Cayman #601010), ROS detection assay kit (Cayman #601290), mouse VEGF kit (Thermo Fisher Scientific, #KHG0111 & #MMV00), mouse TNFα kit (Thermo Fisher Scientific, #BMS223-4 & #MTA00B), Phagocytosis assay kit (Cell Biolabs #CBA-222), Mouse Syndecan-1 ELISA kit (Abcam #ab273165), ICAM-1 ELISA kit (R&D Systems #MIC100), CXCL10 ELISA kit (R&D Systems #DY466-05), IL-8 ELISA kit (MyBioSource #MBS7606860), and CXCR2 ELISA kit (MyBioSource #MBS726530). All experiment assays followed the manufacturers’ instructions as done by us previously95. Treated cells were put in a serum-free medium and stimulated with LPS (1 μg/ml) for 3 hrs before assay ROS, VEGF, and TNF.
Neutrophil trans-endothelial Migration Assay (NTEM)
HUVECs were seeded on the upper wells Corning® Transwell® polycarbonate membrane inserts with 3.0 μm pores (Sigma-Aldrich CLS3415) inserted into a 24-well tissue culture plate at a density of 1.6 × 106 cells per ml in 250 μl volume. Inflammatory response was induced by adding TNFα (Cayman #32020, 20 ng/ml) 16–18 hours before addition of PNs. PNs were then isolated from mouse bone marrow and added to the upper well while HUVEC media containing lipopolysaccharides (LPS) from E. coli (Millipore Sigma #L3023, 1 μg/ml) was added to the bottom well as a chemoattractant. After time indicated (6 hours), cells from the lower well were counted using a hemocytometer to determine the number of neutrophils successfully migrated through the endothelial layer toward the chemoattractant.
Analysis of Eicosanoids by UPLC ESI-MS/MS
Eicosanoids were separated using a Shimadzu Nexara X2 LC-30AD coupled to a SIL-30AC auto injector, coupled to a DGU-20A5R degassing unit in the following way as previously described100–111. A 14-minute reversed phase LC method utilizing an Acentis Express C18 column (150mm × 2.1mm, 2.7μm) was used to separate the eicosanoids at a 0.5 ml/min flow rate at 40°C as previously described by us100–111. The column was equilibrated with 100% Solvent A [acetonitrile:water:formic acid (20:80:0.02, v/v/v)] for 5 min and then 10 μl of sample was injected. 100% Solvent A was used for the first two minutes of elution. Solvent B [acetonitrile:isopropanol:formic acid (20:80:0.02, v/v/v)] was increased in a linear gradient to 25% Solvent B at 3 min, to 30% at 6 min., to 55% at 6.1 min, to 70% at 10 min, and to 100% at 10.10 min. 100% Solvent B was held constant until 13.0 min, where it was decreased to 0% Solvent B and 100% Solvent A from 13.0 min to 13.1 min. From 13.1 min to 14.0 min. Solvent A was held constant at 100%. Eicosanoids were analyzed by mass spectrometry using an AB SCIEX Triple Quad 5500 Mass Spectrometer as previously described by us100–117. Q1 and Q3 were set to detect distinctive precursor and product ion pairs. Ions were fragmented in Q2 using N2 gas for collisionally induced dissociation. Analysis used multiple-reaction monitoring in negative-ion mode. Eicosanoids were monitored using precursor → product MRM pairs. The mass spectrometer parameters as previously described by us100–117 were: Curtain Gas: 20; CAD: Medium; Ion Spray Voltage: −4500V; Temperature: 300°C; Gas 1: 40; Gas 2: 60; Declustering Potential, Collision Energy, and Cell Exit Potential vary per transition.
Isolation of Murine Bone Marrow-Derived Neutrophils
Mouse neutrophils were harvested from bone marrow from both male and female mice (10–14 weeks of age) as previously described118. In brief, bone marrow was flushed using 1x Hank’s Balanced Salt Solution (HBSS) (Thermo Fisher Scientific #14185052), washed, and centrifuged at 1545 x g for 30 minutes over a Percoll™ gradient (Fisher #45001748) consisting of 72%, 64%, and 52% solutions. After centrifugation, a pure, mature neutrophil population will be retained at the 72%−64% interface. Non-neutrophil layers were decanted and the neutrophil population is washed in 1x HBSS and counted using a hemocytometer. Neutrophils were used for experimentation immediately after isolation and were not maintained or passaged in media.
Unbiased Proteomics Analyses
Proteins were solubilized with 5% SDS, 50mM TEAB (pH 7.6), incubated at 95°C for 5 minutes, and sonicated at 20% amplitude. Protein concentrations will be determined using the Pierce 660 Assay (Thermo Fisher Scientific, # 23227), and equal amounts of protein will be digested using S-traps (Protifi). Briefly, proteins are reduced with dithiothreitol (DTT), alkylated with iodoacetamide (IAA), acidified using phosphoric acid, and combined with s-trap loading buffer (90% MeOH, 100mM TEAB). Proteins are loaded onto s-traps, washed, and finally digested with Trypsin/Lys-C overnight at 37°C. Peptides are eluted and dried with a vacuum concentrator. Peptides are resuspended in H2O/0.1% formic acid for global proteome LC-MS/MS analysis. Peptides are separated using a 75 μm × 50 cm C18 reversed-phase-HPLC column (Thermo Fisher Scientific) on an Ultimate 3000 UHPLC (Thermo Fisher Scientific) with a 120-minute gradient (2–32% ACN with 0.1% formic acid) and analyzed on a hybrid quadrupole-Orbitrap instrument (Q Exactive Plus, Thermo Fisher Scientific). Full MS survey scans were acquired at 70,000 resolution. The top 10 most abundant ions were selected for MS/MS analysis. Raw data files are processed in MaxQuant (www.maxquant.org) and searched against the current Uniprot human protein sequences database. Search parameters include constant modification of cysteine by carbamidomethylation and the variable modification, methionine oxidation. Proteins are identified using the filtering criteria of 1% protein and peptide false discovery rate. Label free quantitation analysis was performed using Perseus, software developed for the analysis of omics data119,120. Briefly, intensities were Log2-transformed, and then filtered to include proteins containing at least 60% valid values (reported LFQ intensities) in at least one experimental group. Finally, the missing values in the filtered dataset were replaced using the imputation function in Perseus with default parameters120. Statistical analyses were carried out using the filtered and imputed protein groups file. Statistically significant changes in protein abundance were determined using Welch’s t-test p-values and z-scores.
Histology and Immunohistochemistry
6 mm samples of wound tissue were excised at indicated time points (0- and 6-hours post-injury) from male mice (10–14 weeks of age); wounds were prepared for histological evaluation using the following procedure, as previously described by us121,122. Excised wounds were fixed by placing them in 4% paraformaldehyde for 24 hours, following fixation the wound was placed in a cassette that allowed for the dehydration of the tissue, followed by clearing of the tissue using xylene (Fisher brand), and finally imbedding the tissue in paraffin wax. Sections (5 μm) of the paraffin block were placed on clear glass slides for further treatment and staining. Staining with Masson’s trichrome and hematoxylin & eosin was performed. Rabbit monoclonal anti-Ly6g antibody (Abcam ab238132; RRID AB_2923218, 1:1000), rabbit polyclonal anti-neutrophil elastase (Abcam ab68672; RRID AB_1658868, 1:200), rabbit monoclonal anti-F4/80 (Abcam ab111101; RRID AB_10859466, 1:100), and rabbit polyclonal anti-CD86 (Abcam ab125212; RRID AB_10975465, 1 μg/ml) were used in immunohistochemical staining followed by anti-Rabbit from Vectastain Elite kit (VectorLabs PK-6100) secondary and avidin-biotin complex enhancement. All sections were visualized with Vector NovaRED Chromogen kit (VectorLabs SK-4800) and counterstained with hematoxylin. Slides were viewed on Keyence BZ-X710 microscope and analyzed using the Fiji image J bundle for watershed cell counting or high contrast stained area calculation, where appropriate.
Statistical analysis
Graphing and statistics were performed using Prism GraphPad (Prism Software, San Diego, CA). The following models were used for statistical analysis as described, unpaired t-test with Welch’s correction were used when only two experimental groups were being compared. Repeated measures ANOVA were used when analyzing comparison groups that contained a dependent variable that was measured several times over the course of the experiment. One-way ANOVA with Tukey’s multiple comparisons test were used when comparing three or more independent groups. Two-way ANOVA with Šídák’s multiple comparisons were used when an examination of how the combination of two independent variables affected a dependent variable. Kaplan—Meier and Log-rank Mantel-Cox test were performed in order to determine if a statistical difference in the mortality of two groups was present, All data reported as boxplot or mean ± standard deviation (SD) where applicable; p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgements:
The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the United States Government. We would like to thank Dr. Alpha A. Fowler III for critical reading of the manuscript and suggestion of examining endothelial cell damage. We would also like to thank Dr. Kellie Archer for consultation and critical analysis of our statistical approaches.
Funding:
This work was supported by the Veteran’s Administration (VA Merit Review, I BX001792 (CEC); and a Research Career Scientist Award, IK6BX004603 (CEC); the National Institutes of Health by way of R01s HL125353 (CEC), AI139072 (CEC), and GM137394 (CEC). The cPLA2α knock-in and knockout mice were created using funds from the Paul M. Corman, M.D. Endowed Chair in Cancer Research held by CEC when located at Virginia Commonwealth University, Richmond, VA. This work was also supported by funds from the University of Virginia Cancer Center and from the Department of Medicine in the University of Virginia-School of Medicine.
Footnotes
Ethical considerations
All mouse studies were undertaken under the supervision and approval of the University of South Florida (USF) Institutional Animal Care and Use Committee (Protocol# IS00008261 and IS00008191) and the University of Virginia (UVA) Institutional Animal Care and Use Committee (Protocol# 4373) following standards set by the Federal and State government. USF and UVA are fully accredited by AAALAC International as program #000434 (USF) and #000468 (UVA) and are registered with OLAW with PHS assurance, A4100-01 (USF) and A3245-01 (UVA).
Conflict of Interest Statement: The authors declare that they have no conflicts of interest.
Data and Materials Availability:
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://www.proteomexchange.org/) through the PRIDE partner repository with the dataset identifier PXD039415. All mass spectrometry lipidomics data have been deposited to NIH Common Fund’s National Metabolomics Data Repository (NMDR) website, the Metabolomics Workbench (https://www.metabolomicsworkbench.org), where it has been assigned Study ID ST002445. The data can be accessed directly through the Project DOI: http://dx.doi.org/10.21228/M80709, and this NMDR repository is supported by NIH grant U2C-DK119886. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The mouse models are available from the Chalfant Laboratory at the University of Virginia through a material transfer agreement Virginia Commonwealth University. All other reagents are commercially available.
REFERENCES AND NOTES
- 1.Krizanova O, Penesova A, Sokol J, Hokynkova A, Samadian A, Babula P. Signaling pathways in cutaneous wound healing. Front Physiol. 25, 13:1030851. (2022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nathan C, Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol 6, 173–182 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Dal-Pizzol F, de Medeiros GF, Michels M, Mazeraud A, Bozza FA, Ritter C, Sharshar T, What Animal Models Can Tell Us About Long-Term Psychiatric Symptoms in Sepsis Survivors: a Systematic Review. Neurotherapeutics 18, 1393–1413 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Körner A, Schlegel M, Theurer J, Frohnmeyer H, Adolph M, Heijink M, Giera M, Rosenberger P, Mirakaj V, Resolution of inflammation and sepsis survival are improved by dietary Ω−3 fatty acids. Cell Death & Differentiation 25, 421–431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jun J-I, Kim K-H, Lau LF, The matricellular protein CCN1 mediates neutrophil efferocytosis in cutaneous wound healing. Nature Communications 6, 7386 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel S, Maheshwari A, Chandra A, Biomarkers for wound healing and their evaluation. Journal of Wound Care 25(1), 46–55 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Zhang F, Liu A-L, Gao S, Ma S, Guo S-B, Neutrophil dysfunction in sepsis. Chin Med J 129(22), 2741–2744 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolaczkowska E, Kubes P, Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol 13, 159–175 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Qin W, Song M, Zhang Y, Sun B, Exogenous carbon monoxide inhibits neutrophil infiltration in LPS-induced sepsis by interfering with FPR1 via p38 MAPK but not GRK2. Oncotarget 7(23), 34250–34265 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dovi JV, Szpaderska AM, DiPietro LA, Neutrophil function in the healing wound: adding insult to injury? Thromb Haemost. 92(20), 275–280 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Hoesel LM, Neff TA, Neff SB, Younger JG, Olle EW, Gao H, Pianko M, Bernacki KD, Sarma JV, Ward PA, Harmful and protective roles of neutrophils in sepsis. Shock 24(1), 40–47 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A, The Neutrophil. Immunity 54(7), 1377–1391 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Guimaraes-Costa AB, Nascimento MTC, Wardini AB, Pinto-da-Silva LH, Saraiva EM, NETosis: A microbial mechanism beyond cell death. Immunity to Protozoan Parasites 2012, Article 929743 (2012). [Google Scholar]
- 14.Kruger P, Saffarzadeh M, Weber ANR, Rieber N, Radsak M, von Bernuth H, Benarafa C, Roos D, Skokowa J, Hartl D, Neutrophils: Between Host Defence, Immune Modulation, and Tissue Injury. PLOS Pathogens 11(3), e1004651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilgus TA, Roy S, McDaniel JC, Neutrophils and wound repair: Positive actions and negative reactions. Advances in Wound Care 2(7), 379–388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soehnlein O, Steffens S, Hidalgo A, Weber C, Neutrophils as protagonists and targets in chronic inflammation. Nature Reviews Immunology 17, 248–261 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria Science 303, 1532–1535 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Cooper PR, Palmer LJ, Chapple ILC, Neutrophil extracellular traps as a new paradigm in innate immunity: friend or foe? Periodontology 63(1), 165–197 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD, Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nature Medicine 21, 815–819 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meegan JE, Yang X, Coleman DC, Jannaway M, Yuan SY, Neutrophil-mediated vascular barrier injury: Role of neutrophil extracellular traps. Microcirculation 24(3), e12352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM, Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2”, TAN. Cancer Cell. 16,183–194. (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coffelt SB, Wellenstein MD, de Visser KE, Neutrophils in cancer: neutral no more. Nat Rev Cancer. 16, 431–446 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Ohms M, Möller S, Laskay T, An Attempt to Polarize Human Neutrophils Toward N1 and N2 Phenotypes in vitro. Front. Immunol 11, 532 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacKnight HP, Stephenson DJ, Hoeferlin LA, Benusa SD, DeLigio JT, Maus KD, Ali AN, Wayne JS, Park MA, Hinchcliffe EH, Brown RE, Ryan JJ, Diegelmann RF, Chalfant CE, The interaction of ceramide 1-phosphate with group IVA cytosolic phospholipase A(2) coordinates acute wound healing and repair. Sci Signal. 12(610), eaav5918 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serhan CN Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serhan CN, and Levy BD Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J. Clin. Invest 128, 2657–2669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serhan CN, Chiang N, Dalli J, and Levy BD Lipid mediators in the resolution of inflammation. Cold Spring Harb. Perspect. Biol 7, a016311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcu AC, Chalfant CE, Ceramide-1-phosphate, a new bioactive sphingolipid in regulating cell signaling. Future Lipidology 2(1), 75–84 (2017). [Google Scholar]
- 29.Sugiura M, Kono K, Liu H, Shimizugawa T, Minekura H, Spiegel S, Kohama T. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J Biol Chem. 277(26), 23294–23300 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Bajjalieh SM, Martin TF, Floor E, Synaptic vesicle ceramide kinase: A calcium-stimulated lipid kinase that co-purifies with brain synaptic vesicles. J Biol Chem. 264(24), 14354–14360 (1989). [PubMed] [Google Scholar]
- 31.Maus KD, Stephenson DJ, Ali AN, MacKnight HP, Huang H-J, Serrats J, Kim M, Diegelmann RF, Chalfant CE, Ceramide kinase regulates acute wound healing by suppressing 5-oxo-ETE biosynthesis and signaling via its receptor OXER1. J Lipid Res. 63(4) 100187 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamour NF, Subramanian P, Wijesinghe DS, Stahelin RV, Bonventre JV, Chalfant CE. Ceramide 1-phosphate is required for the translocation of group IVA cytosolic phospholipase A2 and prostaglandin synthesis. J Biol Chem. 284(39), 26897–907 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamour NF, Stahelin RV, Wijesinghe DS, Maceyka M, Wang E, Allegood JC, Merrill AH Jr, Cho W, Chalfant CE. Ceramide kinase uses ceramide provided by ceramide transport protein: localization to organelles of eicosanoid synthesis. J Lipid Res. 48(6), 1293–304 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Stahelin RV, Subramanian P, Vora M, Cho W, Chalfant CE. Ceramide-1-phosphate binds group IVA cytosolic phospholipase α2 via a novel site in the C2 domain. J Biol Chem. 282(28), 20467–74 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Subramanian P, Stahelin RV, Szulc Z, Bielawska A, Cho W, Chalfant CE. Ceramide 1-phosphate acts as a positive allosteric activator of group IVA cytosolic phospholipase A2 alpha and enhances the interaction of the enzyme with phosphatidylcholine. J Biol Chem. 280(18), 17601–7 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Pettus BJ, Bielawska A, Subramanian P, Wijesinghe DS, Maceyka M, Leslie CC, Evans JH, Freiberg J, Roddy P, Hannun YA, Chalfant CE. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J Biol Chem. 279(12), 11320–6 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Pettus BJ, Bielawska A, Spiegel S, Roddy P, Hannun YA, Chalfant CE. Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J Biol Chem. 278(40), 38206–13 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Diegelmann RF, Excessive neutrophils characterize chronic pressure ulcers. Wound Repair Regen. 11(6), 490–495 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Faust N, Varas F, Kelly LM, Heck S, Graf T, Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood 96(2), 719–726 (2000). [PubMed] [Google Scholar]
- 40.Wang X, Ge J, Tredget EE, Wu Y, The mouse excisional wound splinting model, including applications for stem cell transplantation. Nature Protocols 8, 302–309 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Kunisch E, Fuhrmann R, Roth A, Winter R, Lungershausen W, Kinne RW, Macrophage specificity of three anti-CD68 monoclonal antibodies (KP1, EBM11, and PGM1) widely used for immunohistochemistry and flow cytometry. Ann Rheum Dis 63, 774–784 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amanzada A, Malik IA, Nischwitz M, Sultan S, Naz N, Ramadori G, Myeloperoxidase and elastase are only expressed by neutrophils in normal and in inflammed liver. Histochemistry and Cell Biology 135, 305–315 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu T, Wang W, Nassiri S, Kwan T, Dang C, Liu W, Spiller KL, Temporal and spatial distribution of macrophage phenotype markers in the foreign body response to glutaraldehyde-crosslinked gelatin hydrogels. Journal of Biomaterials Science, Polymer Edition 27(8), 721–742 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou X, Yang L, Fan X, Zhao X, Chang N, Yang L, Li L, Neutrophil Chemotaxis and NETosis in Murine Chronic Liver Injury via Cannabinoid Receptor 1/Gαi/o/ROS/p38 MAPK Signaling Pathway. Cells 9(2), 373 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kunz-Schughart LA, Weber A, Rehli M, Gottfried E, Brockhoff G, Krause SW, Andreesen R, Kreutz M, [The “classical” macrophage marker CD68 is strongly expressed in primary human fibroblasts]. Verhandlungen der Deutschen Gesellschaft fur Pathologie. 87, 215–223 (2003). [PubMed] [Google Scholar]
- 46.dos Anjos Cassado A, F4/80 as a Major Macrophage Marker: The Case of the Peritoneum and Spleen. In: Kloc M (eds) Macrophages. Results and Problems in Cell Differentiation, vol 62. Springer, Cham. (2017). [DOI] [PubMed] [Google Scholar]
- 47.Stadler I, Zhang RY, Oskoui P, Whittaker MS, Lanzafame RJ, Development of a simple, noninvasive, clinically relevant model of pressure ulcers in the mouse. J Invest Surg. 17(4), 221–227 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Stadler I, Lanzafame RJ, Evans R, Narayan V, Dailey B, Buehner N, Naim JO, 830-nm irradiation increases the wound tensile strength in a diabetic murine model. Lasers Surg Med. 28(3), 220–226 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Pettus B,J, Kitatani K, Chalfant CE, Taha TA, Kawamori T, Bielawski J, Obeid LM, Hannun YA, The coordination of prostaglandin E2 production by sphingosine-1-phosphate and ceramide-1-phosphate. Mol Pharmacol. 68(2), 330–335 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Hoffmann K, Sperling K, Olins AL, Olins DE, The granulocyte nucleus and lamin B receptor: Avoiding the ovoid. Chromosoma 116(3), 227–235 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Gaines P, Tien CW, Olins AL, Olins DE, Shultz LD, Carney L, Berliner N, Mouse neutrophils lacking lamin B-receptor expression exhibit aberrant development and lack critical functional responses. Exp Hematol 36, 965–976 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brinkmann V, Zychlinsky A, Beneficial suicide: Why neutrophils die to make NETs. Nat Rev Microbiol 5(8), 577–582 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Carvalho LO, Aquino EN, Neves ACD, Fontes W, The neutrophil nucleus and its role in neutrophilic function. J Cell Biochem 116, 1831–1836 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P, The polarization of immune cells in the tumour environment by TGFβ. Nature Reviews Immunology. 10, 554–567 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waxman S, Huang Y, Scher BM, Scher M, Enhancement of differentiation and cytotoxicity of leukemia cells by combinations of fluorinated pyrimidines and differentiation inducers: development of DNA double-strand breaks. Biomed Pharmacother. 46(5–7), 183–92 (1992). [DOI] [PubMed] [Google Scholar]
- 56.Zou H, Li L, Han Y, Ma R, Liao Q, Tian J, Zhang X, Ren X, Song G, Guo Q, Li X, Ding H, Jiang G, Upregulation of CD54 and downregulation of HLA ABC contribute to the novel enhancement of the susceptibility of HL-60 cells to NK cell-mediated cytolysis induced by ATRA plus VPA. Oncol Rep. 37(1), 105–114 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Barber N, Belov L, Christopherson RI, All-trans retinoic acid induces different immunophenotypic changes on human HL60 and NB4 myeloid leukaemias. Leuk Res. 32(2), 315–22 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Congleton J, Jiang H, Malavasi F, Lin H, Yen A, ATRA-induced HL-60 myeloid leukemia cell differentiation depends on the CD38 cytosolic tail needed for membrane localization, but CD38 enzymatic activity is unnecessary. Experimental Cell Research. 317(7), 910–919 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tasseff R, Jensen HA, Congleton J, Dai D, Rogers KV, Sagar A, Bunaciu RP, Yen A, Varner JD, An Effective Model of the Retinoic Acid Induced HL-60 Differentiation Program. Sci Rep. 7, 14327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Konya V, Blättermann S, Jandl K, Platzer W, Ottersbach PA, Marsche G, Gütschow M, Kostenis E, Heinemann A, A biased non-Gαi OXE-R antagonist demonstrates that Gαi protein subunit is not directly involved in neutrophil, eosinophil, and monocyte activation by 5-oxo-ETE. J Immunol. 192(10), 4774–82 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Santana-Codina N, Roeth AA, Zhang Y, Yang A, Mashadova O, Asara JM, Wang X, Bronson RT, Lyssiotis CA, Ying H, Kimmelman AC, Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun. 9(1), 4945 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Balayssac S, Malet-Martino M, Dekiouk S, Joncquel Chevalier Curt M, Maboudou P, Garçon G, Ravasi L, Guerreschi P, Mortier L, Quesnel B, Marchetti P, Kluza J, Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 9(3), 325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Su Z, Gao A, Li X, Zou S, He C, Wu J, Ding WQ, Zhou J, DNA Polymerase Iota Promotes Esophageal Squamous Cell Carcinoma Proliferation Through Erk-OGT-Induced G6PD Overactivation. Front Oncol. 11, 706337 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu J, Maki D, Stapleton SR, Mediation of cadmium-induced oxidative damage and glucose-6-phosphate dehydrogenase expression through glutathione depletion. J Biochem Mol Toxicol. 17(2), 67–75 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Wijesinghe DS, Brentnall M, Mietla JA, Hoeferlin LA, Diegelmann RF, Boise LH, Chalfant CE. Ceramide kinase is required for a normal eicosanoid response and the subsequent orderly migration of fibroblasts. J Lipid Res. 55(7), 1298–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ward KE, Bhardwaj N, Vora M, Chalfant CE, Lu H, Stahelin RV, The molecular basis of ceramide-1-phosphate recognition by C2 domains. J Lipid Res. 54(3), 636–648 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lamour NF, Chalfant CE, Ceramide kinase and the ceramide-1-phosphate/cPLA2alpha interaction as a therapeutic target. Curr Drug Targets. 9(8), 674–82 (2008). [DOI] [PubMed] [Google Scholar]
- 68.Wijesinghe DS, Subramanian P, Lamour NF, Gentile LB, Granado MH, Bielawska A, Szulc Z, Gomez-Munoz A, Chalfant CE, Chain length specificity for activation of cPLA2alpha by C1P: use of the dodecane delivery system to determine lipid-specific effects. J Lipid Res. 50(10), 1986–95 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mietla JA, Wijesinghe DS, Hoeferlin LA, Shultz MD, Natarajan R, Fowler AA 3rd, Chalfant CE, Characterization of eicosanoid synthesis in a genetic ablation model of ceramide kinase. J Lipid Res. 54(7), 1834–47 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Romana-Souza B, Santos JS, Bandeira LG, Monte-Alto-Costa A, Selective inhibition of COX-2 improves cutaneous wound healing of pressure ulcers in mice through reduction of iNOS expression. Life Sci 153, 82–92 (2016). [DOI] [PubMed] [Google Scholar]
- 71.Trayssac M, Hannun YA, Obeid LM, Role of sphingolipids in senescence: implication in aging and age-related diseases. J Clin Invest. 128(7), 2702–2712 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Powell WS, Rokach J, Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, 1851(4), 340–355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A, The new normal: immunomodulatory agents against sepsis immune suppression. Trends in Molecular Medicine. 20(4), 224–233 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brady J, Horie S, Laffey JG, Role of adaptive immune response in sepsis. Intensive Care Medicine Experimental. 8, 20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Champagne B, Tremblay P, Cantin A, St. Pierre Y, Proteolytic cleavage of ICAM-1 by human neutrophil elastase. J Immunol 161(11), 6398–6405 (1998). [PubMed] [Google Scholar]
- 76.Michalec L, Choudhury BK, Postlethwait E, Wild JS, Alam R, Lett-Brown M, Sur S, CCL7 and CXCL10 orchestrate oxidative stress-induced neutrophilic lung inflammation. J Immunol 168, 846–852 (2002). [DOI] [PubMed] [Google Scholar]
- 77.Kunkel SL, Standiford T, Kasahara K, Strieter RM, Interleukin-8 (IL-8): The Major Neutrophil Chemotactic Factor in the Lung. Experimental Lung Research 17, 17–23 (1991). [DOI] [PubMed] [Google Scholar]
- 78.Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K, Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol 56, 559–564 (1994). [PubMed] [Google Scholar]
- 79.David JM, Dominguez C, Hamilton DH, Palena C, The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 4(3), 22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS, Ley K, Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. Pulmonology 116(3), 695–702 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rambeaud M, Pighetti GM, Impaired neutrophil migration associated with specific bovine CXCR2 genotypes. Infection and Immunity 73(8), 4955–4959 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bonecchi R, Facchetti F, Dusi S, Luini W, Lissandrini D, Simmelink M, Locati M, Bernasconi S, Allavena P, Brandt E, Rossi F, Mantovani A, Sozzani S, Induction of Functional IL-8 Receptors by IL-4 and IL-13 in Human Monocytes. J Immunol. 164(7), 3862–3869 (2000). [DOI] [PubMed] [Google Scholar]
- 83.Giese MA, Hind LE, Huttenlocher A, Neutrophil plasticity in the tumor microenvironment. Blood 133(20), 2159–2167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Britt EC, Fan J, Reversed upper glycolysis and rapid activation of oxidative pentose phosphate pathway supports the oxidative burst in neutrophils. bioRxiv 26 November 2020. ( 10.1101/2020.11.25.396838). [DOI] [Google Scholar]
- 85.Amara N, Cooper MP, Voronkova MA, Webb BA, Lynch EM, Kollman JM, Ma T, Yu K, Lai Z, Sangaraju D, Kayagaki N, Newton K, Bogyo M, Staben ST, Dixit VM, Selective activation of PFKL suppresses the phagocytic oxidative burst. Cell 184(17), 4480–4494.e15 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Azevedo EP, Rochael NC, Guimaraes-Costa AB, de Souza-Vieira TS, Ganilho J, Saraiva EM, Palhano FL, Foguel D, A Metabolic Shift toward Pentose Phosphate Pathway Is Necessary for Amyloid Fibril- and Phorbol 12-Myristate 13-Acetate-induced Neutrophil Extracellular Trap (NET) Formation. J Biol Chem 290(36), 22174–22183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ghosh S, Sarma MD, Patra A, Hazra B, Anti-inflammatory and anticancer compounds isolated from Ventilago madraspatana Gaertn., Rubia cordifolia Linn. and Lantana camara Linn. J Pharm Pharmacol 62, 1158–1166 (2010). [DOI] [PubMed] [Google Scholar]
- 88.Tamokou JDD, Tala MF, Wabo HK, Kuiate JR, Tane P, P, Antimicrobial activities of methanol extract and compounds from stem bark of Vismia rubescens. J Ethnopharmacol, 124, 571–575 (2009). [DOI] [PubMed] [Google Scholar]
- 89.Khojasteh Poor F, Keivan M, Ramazii M, Ghaedrahmati F, Anbiyaiee A, Panahandeh S, Khoshnam SE, Farzaneh M, Mini review: The FDA-approved prescription drugs that target the MAPK signaling pathway in women with breast cancer. Breast Dis. 40(2), 51–62 (2021). [DOI] [PubMed] [Google Scholar]
- 90.Hosoi T, Sugikawa E, Chikada A, Koguchi Y, Ohnuki T, TG1019/OXE, a G-alpha(i/o)-protein-coupled receptor, mediates 5-oxo-eicosatetraenoic acid-induced chemotaxis. Biochem Biophys Res Commun. 334(4), 987–95 (2005). [DOI] [PubMed] [Google Scholar]
- 91.Masi M, Garattini E, Bolis M, Di Marino D, Maraccani L, Morelli E, Grolla AA, Fagiani F, Corsini E, Travelli C, Govoni S, Racchi M, Buoso E, OXER1 and RACK1-associated pathway: a promising drug target for breast cancer progression. Oncogenesis. 9(12), 105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grant GE, Rokach J, Powell WS, 5-Oxo-ETE and the OXE receptor. Prostaglandins & Other Lipid Mediators. 89(3–4), 89–104 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C, The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol 176(7), 929–939 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gowda SGB, Gowda D, Kain V, Chiba H, Hui S-P, Chalfant CE, Parcha V, Arora P, Halade GV, Sphingosine-1-phosphate interactions in the spleen and heart reflect extent of cardiac repair in mice and failing human hearts. American Journal of Physiology-Heart and Circulatory Physiology 321(3), H599–H611 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lamour NF, Wijesinghe DS, Mietla JA, Ward KE, Stahelin RV, Chalfant CE, Ceramide kinase regulates the production of tumor necrosis factor α (TNFα) via inhibition of TNFα-converting enzyme. J Biol Chem. 286(50), 42808–42817 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Patel NA, Apostolatos HS, Mebert K, Chalfant CE, Watson JE, Pillay TS, Sparks J, Cooper DR, Insulin regulates protein kinase CbetaII alternative splicing in multiple target tissues: development of a hormonally responsive heterologous minigene. Mol Endocrinol. 18(4), 899–911 (2004). [DOI] [PubMed] [Google Scholar]
- 97.Patel NA, Chalfant CE, Yamamoto M, Watson JE, Eichler DC, Cooper DR, Acute hyperglycemia regulates transcription and posttranscriptional stability of PKCbetaII mRNA in vascular smooth muscle cells. FASEB J. 13(1), 103–13 (1999). [DOI] [PubMed] [Google Scholar]
- 98.Acevedo-Duncan M, Collins J, Zhang R, Haller E, Chalfant CE, Cooper DR, In situ effects of interferon on human glioma protein kinase C-alpha and -beta ultrastructural localization. Cell Growth Differ. 6(11), 1353–1365 (1995). [PubMed] [Google Scholar]
- 99.Johnson RM, Vu NT, Griffin BP, Gentry AE, Archer KJ, Chalfant CE, Park MA, The Alternative Splicing of Cytoplasmic Polyadenylation Element Binding Protein 2 Drives Anoikis Resistance and the Metastasis of Triple Negative Breast Cancer. J Biol Chem. 290(42), 25717–25727 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Simanshu DK, Kamlekar RK, Wijesinghe DS, Zou X, Zhai X, Mishra SK, Molotkovsky JG, Malinina L, Hinchcliffe EH, Chalfant CE, Brown RE, Patel DJ, Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature 500, 463–467 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dhall S, Do D, Garcia M, Wijesinghe DS, Brandon A, Kim J, Sanchez A, Lyubovitsky J, Gallagher S, Nothnagel EA, Chalfant CE, Patel RP, Schiller N, Martins-Green M, A novel model of chronic wounds: importance of redox imbalance and biofilm-forming bacteria for establishment of chronicity. PLoS One. 9(10), e109848 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stephenson DJ, MacKnight HP, Hoeferlin LA, Park M, Allegood J, Cardona CL, Chalfant CE, A rapid and adaptable lipidomics method for quantitative UPLC-mass spectrometric analysis of phosphatidylethanolamine and phosphatidylcholine in vitro, and in cells. Anal Methods. 11(13), 1765–1776 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dhall S, Wijesinghe DS, Karim ZA, Castro A, Vemana HP, Khasawneh FT, Chalfant CE, Martins-Green M, Arachidonic acid-derived signaling lipids and functions in impaired healing. Wound Repair Regen. 23(5), 644–56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wijesinghe DS, Mayton EK, Mietla JA, Mukherjee A, Wu J, Fang X, Chalfant CE, Characterization of lysophosphatidic acid subspecies produced by autotaxin using a modified HPLC ESI-MS/MS method. Anal Methods. 3(12), 2822–2828 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wijesinghe DS, Allegood JC, Gentile LB, Fox TE, Kester M, Chalfant CE, Use of high performance liquid chromatography-electrospray ionization-tandem mass spectrometry for the analysis of ceramide-1-phosphate levels. J Lipid Res. 51(3), 641–51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vu NT, Kim M, Stephenson DJ, MacKnight HP, Chalfant CE, Ceramide Kinase Inhibition Drives Ferroptosis and Sensitivity to Cisplatin in Mutant KRAS Lung Cancer by Dysregulating VDAC-Mediated Mitochondria Function. Mol Cancer Res. 20(9), 1429–1442 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Priyadarsini S, McKay TB, Sarker-Nag A, Allegood J, Chalfant C, Ma JX, Karamichos D, Complete metabolome and lipidome analysis reveals novel biomarkers in the human diabetic corneal stroma. Exp Eye Res. 153, 90–100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hirano Y, Gao YG, Stephenson DJ, Vu NT, Malinina L, Simanshu DK, Chalfant CE, Patel DJ, Brown RE, Structural basis of phosphatidylcholine recognition by the c2–domain of cytosolic phospholipase a2α. eLife 8, e44760 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mishra SK, Stephenson DJ, Chalfant CE, Brown RE, Upregulation of human glycolipid transfer protein (GLTP) induces necroptosis in colon carcinoma cells. Biochim Biophys Acta Mol Cell Biol Lipids 1864(2), 158–167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nelson AJ, Stephenson DJ, Bone RN, Cardona CL, Park MA, Tusing YG, Lei X, Kokotos G, Graves CL, Mathews CE, Kramer J, Hessner MJ, Chalfant CE, Ramanadham S, Lipid mediators and biomarkers associated with type 1 diabetes development. JCI Insight 5(16), e138034 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stephenson DJ, MacKnight HP, Hoeferlin LA, Washington SL, Sawyers C, Archer KJ, Strauss JF 3rd, Walsh SW, Chalfant CE. Bioactive lipid mediators in plasma are predictors of preeclampsia irrespective of aspirin therapy. J Lipid Res. 64(6), 100377 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Caslin HL, Abebayehu D, Abdul Qayum A, Haque TT, Taruselli MT, Paez PA, Pondicherry N, Barnstein BO, Hoeferlin LA, Chalfant CE, Ryan JJ, Lactic Acid Inhibits Lipopolysaccharide-Induced Mast Cell Function by Limiting Glycolysis and ATP Availability. J Immunol. 203(2), 453–464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Spijkers LJ, Janssen BJ, Nelissen J, Meens MJ, Wijesinghe D, Chalfant CE, De Mey JG, Alewijnse AE, Peters SL, Antihypertensive treatment differentially affects vascular sphingolipid biology in spontaneously hypertensive rats. PLoS One. 6(12), e29222 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Spijkers LJ, van den Akker RF, Janssen BJ, Debets JJ, De Mey JG, Stroes ES, van den Born BJ, Wijesinghe DS, Chalfant CE, MacAleese L, Eijkel GB, Heeren RM, Alewijnse AE, Peters SL, Hypertension is associated with marked alterations in sphingolipid biology: a potential role for ceramide. PLoS One. 6(7), e21817 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mitra P, Maceyka M, Payne SG, Lamour N, Milsten S, Chalfant CE, Spiegel S, Ceramide kinase regulates growth and survival of A549 human lung adenocarcinoma cells. FEBS Lett 581, 581–793 (2007). [DOI] [PubMed] [Google Scholar]
- 116.Chalfant CE, Ogretmen B, Galadari S, Kroesen B-J, Pettus BJ, Hannun YA, FAS Activation Induces Dephosphorylation of SR Proteins. J Biol Chem 276(48), 44848 – 44855 (2001). [DOI] [PubMed] [Google Scholar]
- 117.Wijesinghe DS, Chalfant CE, Systems-Level Lipid Analysis Methodologies for Qualitative and Quantitative Investigation of Lipid Signaling Events During Wound Healing. Advances in Wound Care 2(9), 538–548 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ubags NDJ, Suratt BT, Isolation and Characterization of Mouse Neutrophils. Methods Mol Biol. 1809, 45–57 (2018). [DOI] [PubMed] [Google Scholar]
- 119.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M, Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ*. Molecular & Cellular Proteomics. 13(9), 2513–2526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J, The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 121.Hill K, Messina J, Wu J, Chalfant CE, Kim M, PTPN11 plays oncogenic roles and is a therapeutic target for BRAF wild-type melanomas. Cancer Res. 79(13_Supplement), LB-085 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim M, Vu NT, Wang X, Bulut GB, Wang MH, Uram-Tuculescu C, Pillappa R, Kim S, Chalfant CE. Caspase 9b Drives Cellular Transformation, Lung Inflammation, and Lung Tumorigenesis. Mol Cancer Res. 20(8), 1284–1294. (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://www.proteomexchange.org/) through the PRIDE partner repository with the dataset identifier PXD039415. All mass spectrometry lipidomics data have been deposited to NIH Common Fund’s National Metabolomics Data Repository (NMDR) website, the Metabolomics Workbench (https://www.metabolomicsworkbench.org), where it has been assigned Study ID ST002445. The data can be accessed directly through the Project DOI: http://dx.doi.org/10.21228/M80709, and this NMDR repository is supported by NIH grant U2C-DK119886. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The mouse models are available from the Chalfant Laboratory at the University of Virginia through a material transfer agreement Virginia Commonwealth University. All other reagents are commercially available.