

Abstract
High‐risk endometrial cancer has poor prognosis and is increasing in incidence. However, understanding of the molecular mechanisms which drive this disease is limited. We used genetically engineered mouse models (GEMM) to determine the functional consequences of missense and loss of function mutations in Fbxw7, Pten and Tp53, which collectively occur in nearly 90% of high‐risk endometrial cancers. We show that Trp53 deletion and missense mutation cause different phenotypes, with the latter a substantially stronger driver of endometrial carcinogenesis. We also show that Fbxw7 missense mutation does not cause endometrial neoplasia on its own, but potently accelerates carcinogenesis caused by Pten loss or Trp53 missense mutation. By transcriptomic analysis, we identify LEF1 signalling as upregulated in Fbxw7/FBXW7‐mutant mouse and human endometrial cancers, and in human isogenic cell lines carrying FBXW7 mutation, and validate LEF1 and the additional Wnt pathway effector TCF7L2 as novel FBXW7 substrates. Our study provides new insights into the biology of high‐risk endometrial cancer and suggests that targeting LEF1 may be worthy of investigation in this treatment‐resistant cancer subgroup.
Keywords: Driver genes, Endometrial cancer, Fbxw7, Functional models, GEMM
Subject Categories: Cancer, Urogenital System
The consequences of common endometrial driver mutations were studied in mice and found to exert different functional effects alone and in combination.
The paper explained.
Problem
Endometrial cancer (EC) is the most common gynaecological malignancy in the developed world. Mechanistic insights into high‐risk EC may help identify novel therapeutic targets against this poor prognosis subgroup.
Results
Using genetically modified mouse models (GEMM) we show that driver mutations common in high‐risk EC exert different effects alone and in combination. Trp53 missense mutation reliably induces EC, while Trp53 loss does not. Cancer‐associated missense mutation of the ubiquitin ligase Fbxw7 does not cause EC in isolation, but potently accelerates EC caused by Pten loss or Trp53 missense mutation. Fbxw7‐mutant mouse EC display enrichment of a gene signature of the Wnt pathway effector Lef1, and the same signature is evident in human EC and human isogenic cell lines with FBXW7 mutation. Using immunoprecipitation, we confirm LEF1 and the related Wnt effector TCF7L2 as novel FBXW7 substrates, thus identifying a novel mechanism of FBXW7 mediated tumour suppression.
Impact
This study sheds new lights on the mechanistic basis of EC and suggests that targeting of the Wnt pathway may be worthy of investigation in EC carrying FBXW7 mutations.
Introduction
Endometrial cancer (EC) is the most common gynaecological malignancy in the developed world, with more than 150,000 cases each year in Europe and the United States (Sung et al, 2021). While most are low‐grade endometrioid tumours of early stage with favourable prognosis, the 10–20% of ECs classified as high‐risk have considerably worse outcomes (Creasman et al, 2006; León‐Castillo et al, 2020; Crosbie et al, 2022). This group includes high‐grade (grade 3) endometrioid tumours, cases of non‐endometrioid histologies including serous, clear‐cell and undifferentiated histology, and de‐differentiated carcinosarcomas; metaplastic tumours derived from epithelial progenitors (Toboni et al, 2021).
Tumour sequencing studies, including those performed by The Cancer Genome Atlas (TCGA) have characterised the somatic mutational landscape of EC and its variation between subgroups (Gallo et al, 2012; Cancer Genome Atlas Research Network et al, 2013; Zhao et al, 2013). These have confirmed the high mutation frequency of known drivers such as PTEN and TP53 in endometrioid and non‐endometrioid tumours respectively, and identified over 30 other significantly recurrently mutated genes (Gallo et al, 2012; Cancer Genome Atlas Research Network et al, 2013; Zhao et al, 2013; Martincorena et al, 2017; Bailey et al, 2018; Martínez‐Jiménez et al, 2020). Among the most commonly mutated of these, particularly in non‐endometrioid tumours, is FBXW7—the substrate recognition component of an SCF complex responsible for the ubiquitylation and degradation of multiple oncogenic substrates, including cyclin E, mTOR and Jun (Welcker & Clurman, 2008). Mechanistically, FBXW7 recognises substrates through interaction of arginine residues in its WD40 domain beta propellors with a conserved Cdc4 phosphodegron (CPD) sequence (Orlicky et al, 2003; Hao et al, 2007). Cancer‐associated FBXW7 mutations are often heterozygous substitutions of these residues, rather than the truncating mutations predominant among other tumour suppressors (Davis & Tomlinson, 2012). This finding has been attributed to the fact that FBXW7 exists as a homodimer, meaning that these missense mutations exert dominant negative effects (Davis et al, 2014a). However, whether this is the case in EC is unknown.
The functional consequences of manipulation of several genes identified as EC drivers has been investigated using genetically engineered mouse models (GEMM). Conditional uterine deletion of Pten reliably caused endometrial cancer in mice, and this phenotype was accelerated by concomitant Trp53 deletion (Daikoku et al, 2008). While this study did not report the effects of Trp53 deletion alone, another study which used an alternative Cre recombinase showed this caused EC with long latency (typically > 65 weeks; Wild et al, 2012). The effects of deletion of Fbxw7 in the mouse endometrium were recently reported (Cuevas et al, 2019). While deletion of Fbxw7 alone did not induce neoplasia by 52 weeks age, co‐deletion with Pten caused aggressive tumours resembling carcinosarcomas in all cases by 40–67 weeks age (Cuevas et al, 2019). These tumours showed frequent Trp53 mutation or p53‐mutant immunostaining, suggesting functional interplay between the Pten‐PI3K, Trp53 and Fbxw7 pathways (Cuevas et al, 2019).
While these preclinical studies have substantially advanced our understanding of EC biology, several gaps remain. The functional consequences of Fbxw7 and Trp53 missense mutations typical in human cancer are currently uncharacterised. The effects of combined defects in Fbxw7 and Trp53 common in high‐risk human EC await definition. And the substrates dysregulated as a result of endometrial Fbxw7 mutation in vivo are unknown. Given that FBXW7 missense (but not truncating) mutations are common in normal endometrial glands (Moore et al, 2020) and that its substrates appear to vary by tissue (Davis et al, 2014a), addressing these is critical if FBXW7 is to be a target for endometrial cancer prevention and therapy.
In this study, we sought to do this by the generation and phenotyping of GEMM with endometrial expression of a cancer hotspot Fbxw7 mutation alone, in combination with Pten deletion, and in combination with either deletion or mutation of Trp53.
Results
FBXW7 hotspot mutations, PTEN and TP53 mutation are common in high‐risk endometrial cancer
We first examined FBXW7, TP53 and PTEN mutation frequency and type relative to other IntOgen EC drivers (Martínez‐Jiménez et al, 2020) in the TCGA uterine corpus endometrial cancer (UCEC; Cancer Genome Atlas Research Network, 2013) and carcinosarcoma (UCS; Cherniack et al, 2017) cohorts, focusing on the copy number high UCEC subgroup and carcinosarcomas—hereafter referred to as high‐risk endometrial cancer—in view of their exaggerated morbidity and mortality (Crosbie et al, 2022) (note that this definition excludes clear cell and undifferentiated tumours, which have poor prognosis but were excluded from the TCGA analyses). As expected, TP53 was the most frequently mutated driver (84.0% cases), with PIK3CA (32.0%), PPP2R1A (24.2%) and FBXW7 (21.5%), the next most commonly mutated genes. Interestingly, despite its association with low‐grade disease, PTEN was the 5th most commonly mutated driver (15.1% cases) in this high‐risk subset (Fig 1A). Known and predicted oncogenic mutations in TP53 and PTEN included recurrent hotspot missense mutations and protein‐truncating variants (PTVs), with preponderance of hotspots in TP53 (146 of 184; 79.3%) and PTVs in PTEN (20 of 33; 60.6%). In contrast, nearly 90% (42 of 47) of oncogenic FBXW7 mutations were hotspot missense alterations – typically arginine residues in the WD40 domain—suggesting strong selection for these over PTVs in high‐risk EC (Fig 1A). This was supported by cross‐cancer comparison, which revealed that the proportion of FBXW7 missense driver mutations to PTVs in high‐risk EC was significantly greater than all other cancer types in which FBXW7 is mutated in more than 5% cases (P < 1.0e−04 to 7.2e−03, Fisher exact test) (Appendix Fig S1). Oncogenic mutations in FBXW7 in this cohort of high‐risk endometrial cancers tended to mutual exclusivity with those in TP53 (OR = 0.53, unadjusted P = 0.12) and particularly PTEN (OR = 0.32, unadjusted P = 0.067).
Figure 1. Functional analysis of high‐risk endometrial cancer drivers in combination reveals interactions between Fbxw7, Pten and p53.
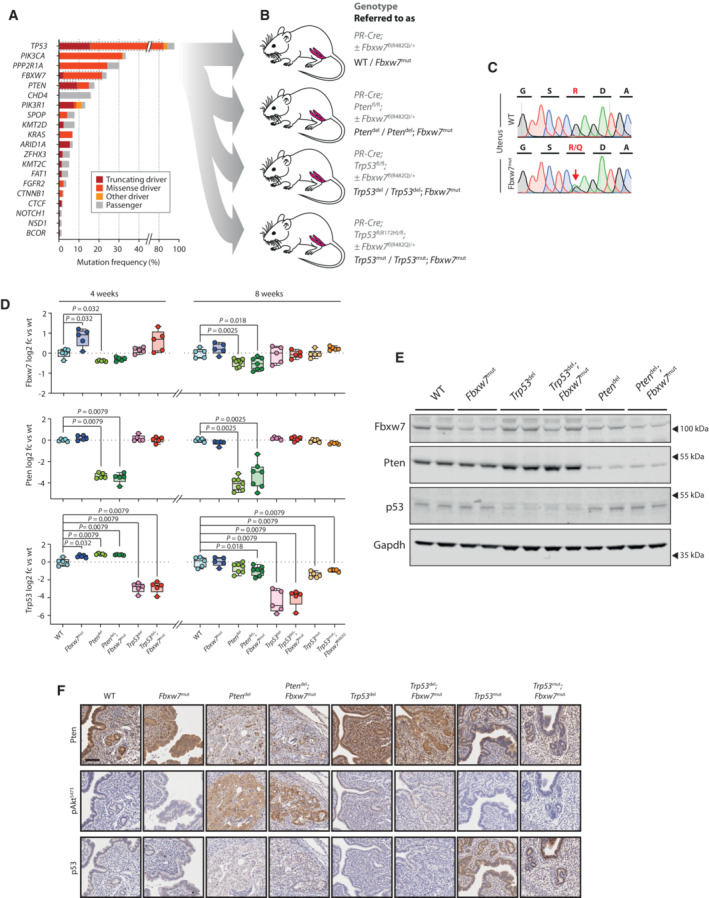
- Frequency and type of somatic mutations in top 20 driver genes in high‐risk endometrial cancer (EC) from TCGA UCEC and UCS cohorts. Details of mutation classification are provided in Materials and Methods.
- Genetically engineered mouse models (GEMM) used in this study comprising conditional knock‐in of heterozygous Fbxw7 R482Q missense mutation (corresponding to human hotspot FBXW7 R479Q), homozygous Pten loss, homozygous Trp53 loss and heterozygous Trp53 R172H missense mutation (corresponding to human hotspot TP53 R175H) alone and in combination. Full genotypes are provided in grey text, with abbreviations used in this study in black text. Further details are provided in Table 1.
- Sanger sequencing of cDNA from indicated tissues showing expression of Fbxw7 R482Q knock‐in allele in mouse uterus.
- Real‐time reverse transcription quantitative PCR (RT–qPCR) showing Fbxw7, Pten and Trp53 expression in mouse uteri at 4 and 8 weeks postnatal age (NB Trp53 R172H uteri were not analysed at the 4 week timepoint). Box extends from 25th to 75th percentiles, with line at median, whiskers indicate minimum and maximum values, and dots individual values (n = 5 samples per group with exception of Pten del and Pten del, Fbxw7 mut genotypes at 8 weeks which contain n = 7 samples each). Statistical comparison between WT controls and other genotypes was performed by unadjusted, unpaired Mann–Whitney test; *P < 0.05, **P < 0.01.
- Western blot of mouse uteri at 8 weeks postnatal age (NB Trp53 R172H uteri were not analysed by Western blotting). Values indicate densitometric units (mean ± SD) normalised to control group of WT samples in case of Fbxw7 protein and both WT and Fbxw7 mut samples in case of Pten and p53 proteins. Image is representative of duplicate technical replicates. Statistical comparison between controls and other groups was performed by unadjusted, unpaired Mann–Whitney test; and are shown next to densitometry results where statistically significant.
- Immunohistochemical (IHC) analysis of mouse uteri at 8 weeks postnatal age. Images are representative of minimum of 3 biological replicates for each genotype and protein. Scale bar indicates 100 μm.
Source data are available online for this figure.
Variable effects of high‐risk endometrial cancer drivers alone and in combination in GEMM
We sought to understand the mechanisms of tumorigenesis caused by EC‐associated FBXW7, PTEN, and TP53 mutations through functional analysis of GEMM. We bred mice expressing Cre recombinase from the progesterone receptor locus (PR‐Cre; Soyal et al, 2005; which drives recombination in the endometrium and female reproductive tract from 2 weeks post‐natal age) with mice carrying Pten fl or Trp53 fl conditional knockout alleles and/or Trp53 fl(R172H) (corresponding to human TP53 R175H) and Fbxw7 fl(R482Q) (corresponding to human FBXW7 R479Q) conditional knock‐in alleles (Marino et al, 2000; Suzuki et al, 2001; Olive et al, 2004; Davis et al, 2011 ) carrying EC hotspot missense mutations to generate control and experimental mice (Table 1 and Fig 1B) with the following alterations in the uterus:
Cre expression only (WT)
Knock‐in of Fbxw7 hotspot missense mutation (Fbxw7 mut)
Pten deletion (Pten del)
Pten deletion plus Fbxw7 hotspot missense mutation (Pten del; Fbxw7 mut)
p53 deletion (Trp53 del)
p53 deletion plus Fbxw7 hotspot missense mutation (Trp53 del; Fbxw7 mut)
p53 hotspot missense mutation (Trp53 mut)
p53 hotspot missense and Fbxw7 hotspot missense mutations (Trp53 mut; Fbxw7 mut)
Table 1.
Genetically engineered mouse models (GEMM) used in study.
| Allelic composition a | Genetic modification post‐recombination | Predicted alteration in uterus | Referred to as |
|---|---|---|---|
| PRcre/+ | Unaltered | Nil | WT |
| PRcre/+; Fbxw7 fl(R482Q)/+ | Fbxw7 R482Q/+ | Knock‐in of heterozygous Fbxw7 hotspot mutation | Fbxw7mut |
| PRCre/+; Pten fl/fl | Pten del/del | Pten deletion | Ptendel |
| PRCre/+; Pten fl/fl; Fbxw7 fl(R482Q)/+ | Pten del/del; Fbxw7 R482Q/+ | Pten deletion plus heterozygous Fbxw7 hotspot mutation | Ptendel; Fbxw7mut |
| PRCre/+; Trp53 fl/fl | Trp53 del/del | p53 deletion | p53del |
| PRCre/+; Trp53 fl/fl; Fbxw7 fl(R482Q)/+ | Trp53 del/del; Fbxw7 R482Q/+ | p53 deletion plus heterozygous Fbxw7 hotspot mutation | p53del; Fbxw7mut |
| PRCre/+; Trp53 fl(R172H)/fl | Trp53 R172H/del | Knock‐in of heterozygous Trp53 hotspot mutation | p53mut |
| PRCre/+; Trp53 fl(R172H)/fl; Fbxw7 fl(R482Q)/+ | Trp53 R172H/del; Fbxw7 R482Q/+ | Knock‐in of heterozygous Trp53 and Fbxw7 hotspot mutations | p53mut; Fbxw7mut |
All alleles are wild‐type unless explicitly stated otherwise.
Females were culled at four and eight weeks (4w, 8w) for phenotyping and also aged for survival analysis. Analysis of uteri at 4w and 8w confirmed allelic recombination, expression of Fbxw7 R482Q and Trp53 R172H mutations and reduction in Pten and Trp53 mRNA and protein in Pten del and Trp53 del mice respectively (Fig 1B–F; Appendix Fig S2A and B). This also revealed apparent interactions between Fbxw7, Pten and Trp53 at the level of transcription. Fbxw7 mut caused an increase in global Fbxw7 expression (P < 0.05, Mann–Whitney test) which was lost with concomitant Pten del but not Trp53 del. Fbxw7 mut and Pten del uteri showed increased Trp53 expression at 4w, although by 8w this effect was lost or reversed (Fig 1D). We also noted possible interactions at the post‐transcriptional level. Trp53 del uteri had increased Pten protein on immunoblotting (Fig 1E), despite similar Pten expression, while Tp53 mut uteri showed substantially increased p53 immunostaining to controls despite similar Trp53 expression (Fig 1F).
Consistent with previous work (Daikoku et al, 2008), Pten deletion caused endometrial cancer as early as 4 weeks age (Appendix Fig S3), with mice requiring sacrifice for uterine tumours from 11 weeks age (Fig 2A–C). A single Pten del mouse developed an extrauterine tumour in the form of an inclusion cyst containing a squamous cell carcinoma (Fig 2D). In contrast, Trp53 del mice showed no evidence of endometrial neoplasia at early timepoints, but instead developed external tumours requiring sacrifice from 24 weeks age, most of which were found to be soft tissue sarcomas on pathologist review (Fig 2C–E). One Trp53 del female culled for an external tumour at 57 weeks age was found to also have an endometrial carcinoma on pathological review, and another two Trp53 del mice culled for external tumours at 51 and 68 weeks age had uterine sarcomas (Fig 2A). While some Trp53 mut females also required sacrifice for external tumours, an appreciable fraction were culled for abdominal distension or genital bleeding, found to be secondary to uterine tumours on necropsy. Although survival of Trp53 mut mice was not significantly different to that of Trp53 del animals, and pathology review revealed similar frequency of extrauterine tumours (predominantly sarcomas) to Trp53 del mice (Fig 2A, D, and E), the frequency of endometrial carcinoma in Trp53 mut mice was far greater, with 8 of 11 (72.7%) developing epithelial malignancies during the study (P = 3e−04 vs. Trp53 del, Fisher exact test) (Fig 2A–C). Interestingly, Fbxw7 mut mice showed no evidence of endometrial neoplasia up to the study endpoint, although a single animal developed an external angiosarcoma at 72 weeks age (Fig 2A–C).
Figure 2. Fbxw7 hotspot mutation accelerates Pten and Trp53‐driven endometrial cancer but does not accelerate extrauterine tumour development.
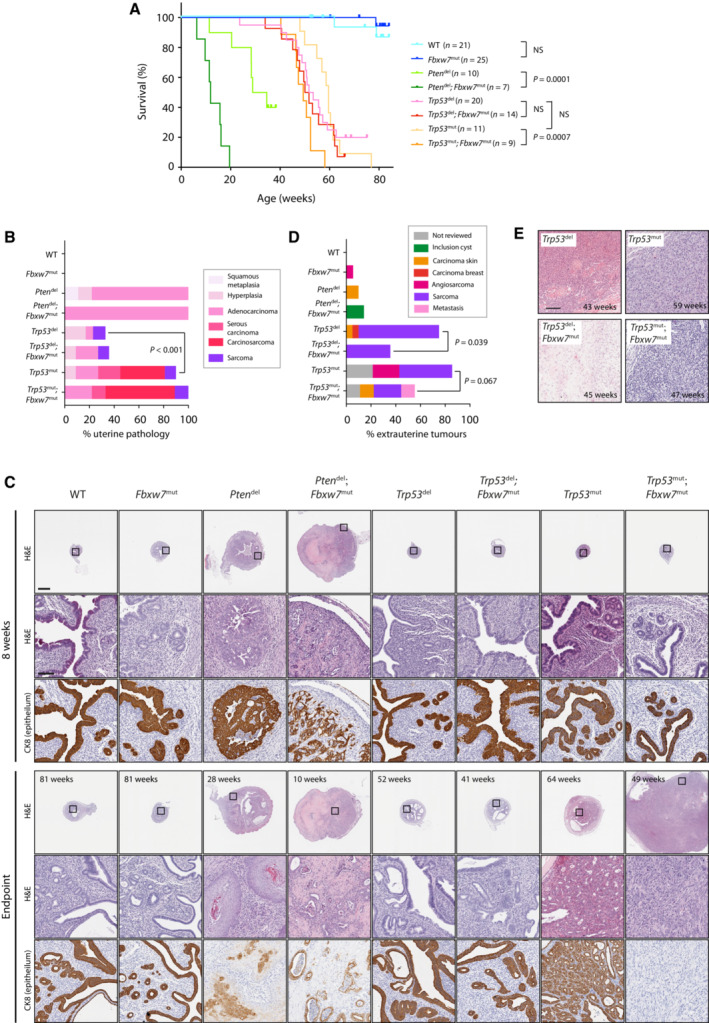
- Kaplan–Meier curves showing survival of genetically engineered mouse models (GEMM) of indicated genotypes and group sizes. Statistical comparison between groups was performed by unadjusted log‐rank test.
- Prevalence of uterine pathology in GEMM shown in (A) at study endpoint as graded by expert pathologist. Statistical comparison between the proportion of mice with epithelial endometrial malignancy (ie carcinoma or carcinosarcoma) in groups was made by unadjusted Fisher exact test.
- Low and high power images of GEMM uteri at 8 weeks age and at study endpoint after H&E staining or IHC for cytokeratin 8 (CK8) (epithelia). Scale bars in low and high magnification panels indicate 1,000 and 100 μm respectively. Images are representative of a minimum of 5 mice per genotype per timepoint, with exception of wild‐type and Fbxw7 mut mice at 8 weeks age where 3 mice were analysed.
- Prevalence and type of external tumours in GEMM shown in (A) at study endpoint as graded by expert pathologist. Statistical comparison of the proportion of mice with external tumours of any histotype between groups was made by Fisher exact test.
- H&E‐stained sections of high‐grade external sarcomas in Trp53del, Trp53 del; Fbxw7mut, Trp53 mut and Trp53 mut; Fbxw7 mut mice. Images are representative of minimum of 3 mice examined for each genotype. Scale bar indicates 100 μm.
Source data are available online for this figure.
We speculated that the tumorigenic effect of a cancer‐associated Fbxw7 missense mutation may be unmasked when combined with another driver. Consistent with this prediction, Fbxw7 mut dramatically accelerated endometrial tumorigenesis and reduced survival in Pten del mice, with Pten del; Fbxw7 mut compound mutants requiring sacrifice as early as 6 weeks for large, high‐grade endometrial tumours (Fig 2A–C; Appendix Fig S3). Fbxw7 mut also significantly accelerated endometrial neoplasia and reduced survival of Trp53 mut mice, but interestingly not Trp53 del mice (P INTERACTION = 0.13) (Fig 2A–C). Intriguingly, in contrast to its tumour promotion in the uterus in Trp53 mut females, the addition of Fbxw7 mut resulted in significantly lower frequency of extrauterine tumours in Trp53 del mice (P = 0.039, Fisher exact test), and to a lesser extent in Trp53 mut mice, though the latter was not statistically significant (Fig 2D).
We analysed endometrial tumours from the GEMM in detail. Expert gynaecological pathology review of endometrial cancers from Pten del and Pten del; Fbxw7 mut mice revealed most were adenocarcinomas of endometrioid histotype; with none displaying serous or carcinoscarcomatous morphology, in contrast to the recent study of Cuevas et al (2019) (Fig 2B). Pten del and particularly Pten del; Fbxw7 mut endometrial cancers displayed downregulation of the endometrial tumour suppressor Foxa2 (Appendix Fig S4), and frequent abnormal p53 immunostaining (Appendix Fig S5). The few endometrial carcinomas which occurred in Trp53 del, and Trp53 del; Fbxw7 mut animals were of endometrioid histotype, although one tumour displayed sarcomatoid appearances insufficient for formal diagnosis of carcinosarcoma. In contrast, Trp53 mut (3 of 11) and particularly Trp53 mut; Fbxw7 mut females (5 of 9) developed carcinosarcoma, characterised by typical morphology and loss of the epithelial marker cytokeratin 8 (Krt8; Fig 2B and C). The difference in carcinosarcoma prevalence between Pten del ± Fbxw7 mut and Trp53 mut ± Fbxw7 mut mice was statistically significant (P = 0.0043, Fisher exact test). Trp53 mut mice also developed serous endometrial cancers in several cases, though the difference in prevalence with Pten del mice was not statistically significant. Trp53 mut and Trp53 mut; Fbxw7 mut endometrial tumours also often displayed abnormal p53 immmunostaining, with variable patterns observed (Appendix Fig S5).
Transcriptomic profiling of murine and human tumours, human isogenic cell lines and in silico analysis identifies Wnt pathway effectors LEF1 and TCF7L2 as candidate FBXW7 substrates
We investigated the mechanisms of Fbxw7 mut ‐driven tumorigenesis in the GEMM uteri. Gene expression profiling and gene set enrichment analysis (GSEA) revealed no differentially‐expressed genes (DEGs), and a small number of significantly enriched gene sets between Fbxw7 mut and wild‐type uteri at 8 weeks age, paralleling the minimal perturbation of uterine Fbxw7 and Trp53 expression at this timepoint (Datasets [Link], [Link]; Appendix Fig S6). In contrast, analysis of Pten del and Pten del; Fbxw7 mut compound mutant uteri revealed 788 DEGs (FDR < 0.05) and multiple enriched gene sets (Fig 3A; Datasets [Link], [Link]; Appendix Fig S7). These included those corresponding to epithelial‐mesenchymal transition (EMT) (the most enriched hallmark gene set), dysregulation of p53 signalling (consistent with the increased p53 protein on immunostaining, although we note that enrichment of p53 gene sets is not specific for alterations in p53 itself), signalling by known FBXW7 substrate Myc, and multiple other oncogenic cellular signalling pathways (Fig 3A). Among these, LEF1 signalling caught our attention, owing to its strong upregulation, its critical role in endometrial gland development (adenogenesis; Mericskay et al, 2004; Shelton et al, 2012) and its role as an effector in the Wnt pathway, which is recurrently dysregulated in cancer. We confirmed upregulation of Lef1 targets and significant DEGs Mmp13 and Wisp1 (Ccn4) in Pten del; Fbxw7 mut compound mutant uteri by qRT–PCR, (Appendix Fig S8A and B), and excluded differential infiltration of neoplastic uteri by Lef1‐expressing lymphocytes as a confounder (Appendix Fig S9A and B).
Figure 3. Fbxw7 hotspot mutation drives enrichment of LEF1 signalling in GEMM uteri and human cancer cells.
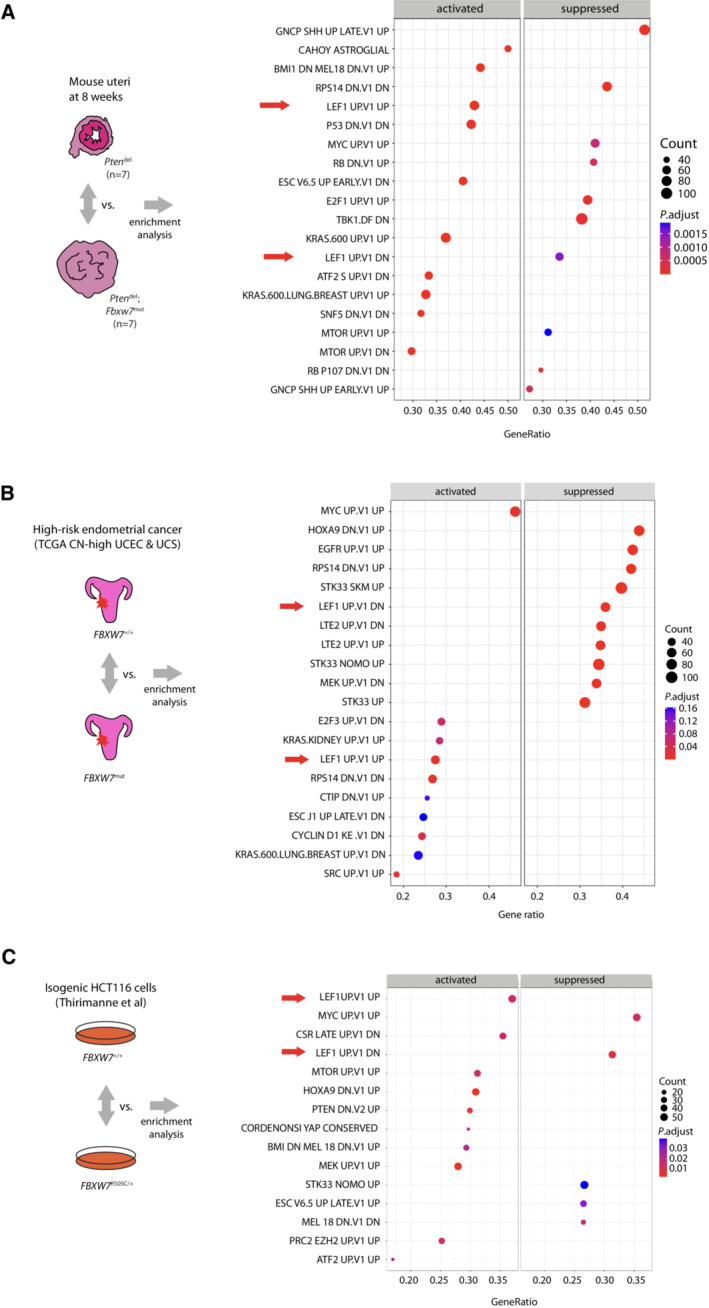
- (Left panel) Schematic illustrating gene expression profiling of mouse uterine samples of indicated genotypes and group sizes at 8 weeks age, with groups compared by enrichment analysis. (Right panel) Dot plot indicating enrichment of MSigDB C6 oncogenic gene sets in Pten del; Fbxw7 mut uteri vs. Pten del uteri. Arrows indicate enrichment of LEF1 signatures.
- (Left panel) Schematic illustrating comparison of high‐risk endometrial cancers from UCEC and UCS cohorts (see Results for definition) by enrichment analysis according to pathogenic FBXW7 missense mutation status. (Right panel) Dot plot indicating enrichment of MSigDB C6 signatures in FBXW7 mutant tumours. Arrows indicate enrichment of LEF1 signatures.
- (Left panel) Schematic illustrating comparison of isogenic parental FBXW7 wild‐type (FBXW7 +/+) and heterozygous WD40 hotspot missense mutant (FBXW7 R505C/+) isogenic HCT116 cells reported by Thirimanne et al (2022) by enrichment analysis following pre‐ranking. (Right panel) Dot plot indicating enrichment of MSigDB C6 oncogenic sets in FBXW7 R505C/+ cells. Arrows indicate enrichment of LEF1 signatures.
Source data are available online for this figure.
We sought to confirm the association of FBXW7 mutation with LEF1 signalling in human samples. GSEA of high‐risk endometrial cancers from TCGA (CN high UCEC and UCS) revealed enrichment of LEF1 signalling in cases with FBXW7 missense driver mutation (Fig 3B; Dataset EV7). Similarly, GSEA of RNAseq data from a recent study (Thirimanne et al, 2022) of isogenic human colon cancer cells with wild‐type or oncogenic missense mutant FBXW7 (FBXW7 R505C) identified LEF1 signalling as the most enriched gene set in FBXW7 R505C cells, supporting a causal relationship. LEF1 signalling was also significantly enriched in isogenic cells with FBXW7 deletion, though this effect was weaker (Fig 3C; Appendix Fig S10). Interestingly, while as in the GEMM tumours FBXW7 mutation was associated with enrichment of MYC signalling in the human endometrial cancers, this was not the case in the colon cancer cells, and dysregulation of p53 gene sets was less evident in both, possibly owing to different p53 targets in mouse and human, or mutations in TP53 and other genes in its pathway in the human samples (Fig 3A–C). To explore the mechanistic basis of the LEF1 pathway enrichment with FBXW7 mutation, we performed an in silico search for novel FBXW7 targets using a CPD sequence derived from experimentally validated substrates (Materials and Methods; Fig 4A; Dataset EV8). The 1,064 candidates (Dataset EV9) contained significant overrepresentation of Wnt pathway components, including LEF1 and other transcriptional effectors TCF7L1 and TCF7L2 (Fig 4B–D), but not β‐catenin – noteworthy given previous data suggesting β‐catenin is an FBXW7 substrate (Jiang et al, 2016). Importantly, the putative CPD in LEF1 and TCF7L2 was highly conserved, consistent with a functional role (Fig 4E and F).
Figure 4. In silico identification of candidate FBXW7 targets reveals enrichment of Wnt pathway components including LEF1 and TCF7L2.
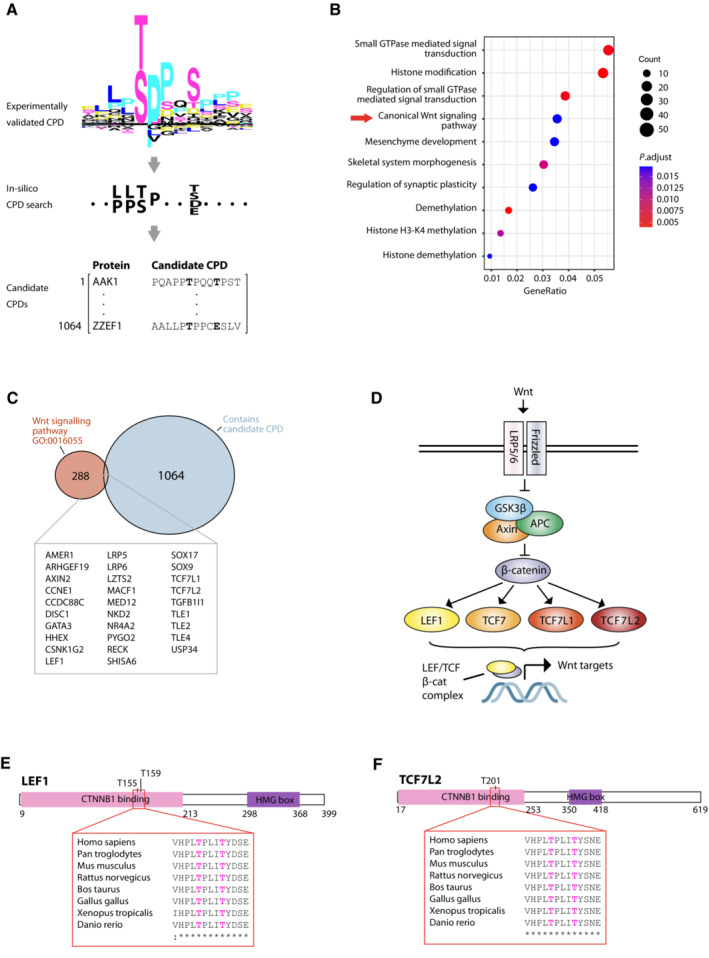
- Schematic indicating workflow for in silico identification of candidate FBXW7 targets carrying putative conserved phosphodegrons (CPDs). Experimentally validated FBXW7 CPDs (top, provided as Dataset EV8) were used to define a consensus motif (middle) for search against the human proteome to identify candidate FBXW7 targets (bottom).
- Dot plot showing enrichment analysis of 1,064 putative FBXW7 substrates against GO biological processes.
- Venn diagram showing overlap between putative FBXW7 substrates identified by in silico analysis and Wnt pathway components curated by the Gene Ontology Consortium (GO:0016055).
- Simplified representation of Wnt signalling pathway. Following stimulation by Wnt ligand, the GSK3β/Axin/APC destruction complex is inhibited and β‐catenin can translocate to the nucleus where it complexes with TCF/LEF family transcription factors to regulate Wnt target gene expression.
- Schematic of putative FBXW7 substrate LEF1 indicating position and conservation of candidate CPD, and phosphorylation sites corresponding to the ‘0’ and ‘+4’ positions curated in PhosphoSitePlus database.
- Corresponding schematic for putative FBXW7 substrate TCF7L2.
Source data are available online for this figure.
LEF1 and TCF7L2 are novel FBXW7 substrates expressed in proliferative endometrial stem cells and dysregulated by Fbxw7/FBXW7 hotspot mutation
We investigated interactions between FBXW7 and these putative novel targets. LEF1 and TCF7L2 were detected in immunoprecipitates following FBXW7 pulldown in HEK293T cells (Fig 5A and B), and a reciprocal experiment confirmed presence of FBXW7 in LEF1 and TCF7L2 immunoprecipitates (Fig 5C and D). Analysis of the interaction of LEF1 with WD40 domain mutant FBXW7, defective in substrate binding, was complicated by robust and reproducibly greater levels of WD40 mutant than wild‐type FBXW7 protein following transfection (Fig 5E). However, densitometric quantification confirmed the amount of LEF1 relative to immunoprecipitated FBXW7 was reproducibly lower with WD40 mutant than wild type FBXW7 (see values under band) (Fig 5E). Interestingly, wild type and WD40 domain mutant FBXW7 protein levels were similar when co‐transfected with TCF7L2, and the reduction in the interaction of TCF7L2 with the WD40 domain‐mutant FBXW7 was more obvious (Fig 5F). The proven FBXW7‐LEF1 interaction, and the absence of a candidate FBXW7 CPD in β‐catenin in the in silico analysis caused us to speculate that the previously reported FBXW7–β‐catenin interaction could in fact be mediated indirectly via LEF1 rather than through direct interaction. In keeping with this hypothesis, immunoprecipitates of full length LEF1 contained both FBXW7 and β‐catenin, while those from a truncated LEF1 lacking the β‐catenin‐interacting N terminal domain contained FBXW7 but not β‐ catenin (Fig 5G).
Figure 5. LEF1 and TCF7L2 are novel FBXW7 substrates whose interaction is disrupted by FBXW7 WD40 hotspot mutation.
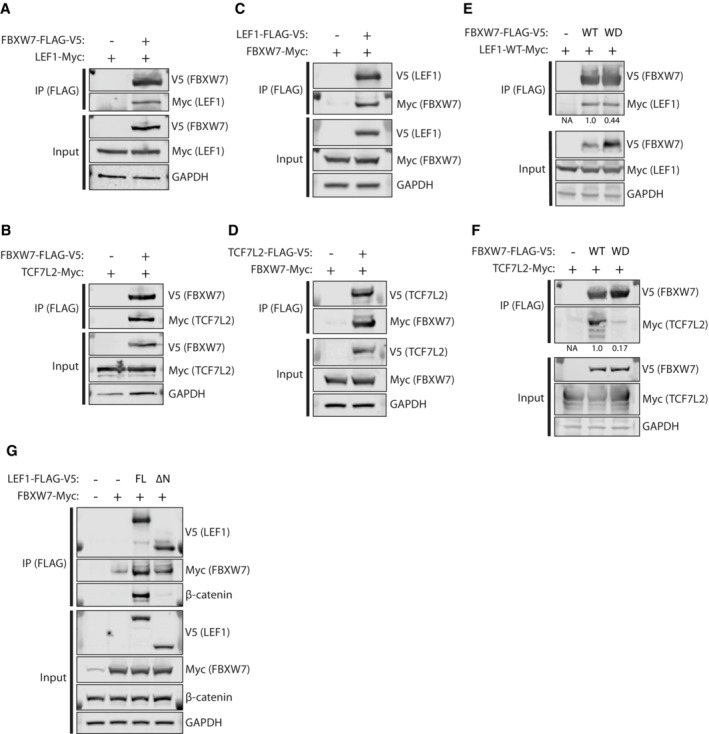
- HEK293T cells were transfected with constructs encoding FLAG‐V5‐tagged FBXW7α and Myc‐tagged LEF1. FLAG‐V5‐tagged FBXW7 α was immunoprecipitated (IP) from cell extracts with anti‐FLAG antibody before immunoblotting for tagged proteins as indicated (upper panels). Lower panels show inputs.
- HEK293T cells were transfected with constructs encoding FLAG‐V5‐tagged FBXW7α and Myc‐tagged TCF7L2. IP was performed as in (A) before immunoblotting for tagged proteins as indicated.
- Reciprocal experiment to (A) in which FLAG‐V5‐tagged LEF1 was pulled down by IP.
- Reciprocal experiment to (B) in which FLAG‐V5‐tagged TCF7L2 was pulled down by IP.
- Complementary experiment to (A) in which HEK293T cells were transfected with either FLAG‐V5‐tagged wild‐type FBXW7α (WT) or a substrate‐binding mutant (WD40), in which three arginine residues within one of the FBXW7 WD40 repeats have been mutated. Numbers below upper bands indicate densitometric quantification of the ratio of LEF1‐Myc to FBXW7 α‐V5 following IP by anti‐FLAG antibody, with WT normalised to 1.
- Complementary experiment to (B), of similar design to (E) but using Myc‐tagged TCF7L2 rather than LEF1.
- HEK293T cells were transfected with Myc‐tagged FBXW7α and either FLAG‐V5‐tagged full length LEF1 (FL) or truncated LEF1 in which the N terminal domain required for β‐catenin binding has been deleted. Immunoblotting for indicated proteins was performed following IP by anti‐FLAG antibody.
Data information: All images shown are representative of a minimum of two independent experiments; densitometry results indicate mean of three independent experiments.
Source data are available online for this figure.
As noted above, LEF1/Lef1 is essential for adenogenesis in several tissues, including the endometrium (van Genderen et al, 1994; Shelton et al, 2012). We investigated its role in endometrial physiology using human scRNAseq data from a recent study (Garcia‐Alonso et al, 2021). LEF1 expression was greatest in proliferating cells defined by the stem cell marker SOX9 (Fig 6A), signatures of which are enriched in human endometrial cancers (Garcia‐Alonso et al, 2021). Epithelial Lef1 expression in GEMM endometria at 8 weeks age was more common in Fbxw7 mut mice than controls (P = 0.02, logistic regression) (Fig 6B and C), with ectopic expression outside the gland base stem/progenitor cell niche (Syed et al, 2020) (e.g. luminal positivity in bottom right image in Fig 6B), and associated nuclear β‐catenin localisation (Morgan et al, 2019) (Fig 6D). Interestingly, analysis of high‐grade human endometrial cancers of no specific molecular profile (NSMP) and p53 mutant subtypes and murine endometrial cancers from Pten del and Trp53 mut GEMM at experimental endpoint revealed near‐universal epithelial LEF1/Lef1 expression, irrespective of FBXW7/Fbxw7 mutation (Appendix Figs S11A and B, and S12A and B), suggesting that FBXW7/Fbxw7 wild‐type tumours may employ alternative mechanisms of Lef1 upregulation, as has been suggested by earlier studies of mouse and human tumours (Shelton et al, 2012; Ruz‐Caracuel et al, 2021).
Figure 6. LEF1 is highly expressed in proliferative endometrial stem cells and dysregulated by Fbxw7 hotspot mutation.
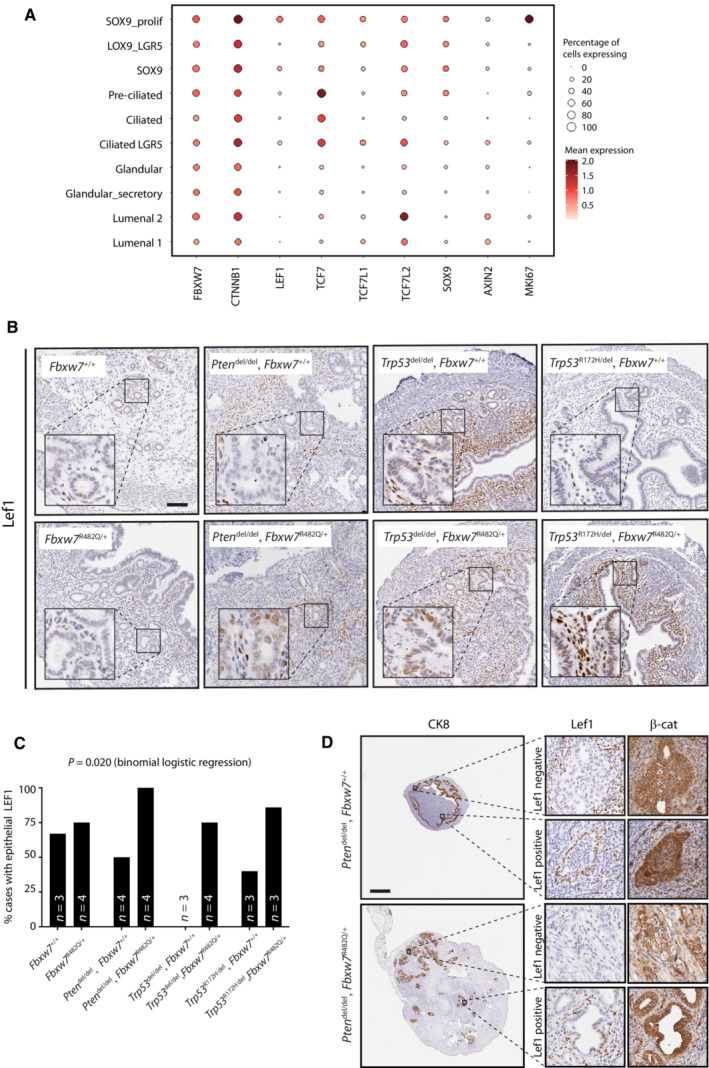
- Dot plot showing log2‐transformed expression of FBXW7, LEF1 and other wnt pathway genes in endometrial epithelial cell subsets defined by single cell RNAseq (scRNAseq) in Garcia‐Alonso et al (2021).
- Representative LEF1 immunohistochemistry in GEMM uteri from mice of indicated genotypes at 8 weeks age. For each of the four columns (i.e genotypes), upper and lower images are taken from littermate females housed in the same cage. Images are representative of 3–4 mice as shown in (C). Scale bar indicates 100 μm.
- Percentage of mice with epithelial LEF1 expression in endometria at 8 weeks age according to genotype. P value was derived from binomial logistic regression with epithelial LEF1 expression as the dependent variable.
- Immunohistochemistry for LEF1 and β‐catenin from endometrial carcinomas in Pten del ± Fbxw7 mut mice. High power images show representative staining in LEF1 negative and positive glands. Scale bar indicates 1,000 μm.
Source data are available online for this figure.
Unfortunately, infertility of Pten del; Fbxw7 mut females (owing to the rapid onset of endometrial neoplasia) precluded breeding to generate GEMM with concomitant Lef1 deletion, meaning that we were unable to examine whether the tumour phenotype was rescued by Lef1 loss. Similarly, our attempts to define the effects of FBXW7 mutation and PTEN loss in normal human endometrial cells proved unsuccessful, as hEM3 cells targeted with FBXW7 mutation failed to expand from single cell colonies.
Discussion
In this study, we used GEMM to define the functional consequences of high‐risk endometrial cancer driver mutations, and human tumours and isogenic cell lines to confirm our key results. We found that while Fbxw7 hotspot missense mutation fails to induce endometrial neoplasia on its own, it significantly accelerates endometrial tumorigenesis caused by Pten deletion or Trp53 hotspot mutation, with many tumours showing characteristic features of human carcinosarcomas in the latter case. Interestingly, Fbxw7 mutation did not potentiate extrauterine tumorigenesis caused by Trp53 deletion, consistent with tissue‐specific variation in the selective advantage it confers in malignancy. Taking an agnostic approach, we identified enrichment of a gene signature corresponding to the Wnt pathway effector LEF1 in Fbxw7 mutant mouse endometrial cancers. We confirmed this association in human endometrial cancers and used isogenic human colorectal cancer cells to provide strong evidence that this relationship is causal. Finally, we confirmed LEF1 and its LEF/TCF family member TCF7L2 as novel FBXW7 substrates by immunoprecipitation. Our results provide new insights into the biology of high‐risk endometrial cancer and suggest that strategies targeting the Wnt pathway may be worthy of investigation in endometrial tumours with FBXW7 mutations.
Cuevas et al (2019) recently reported that co‐deletion of Pten and Fbxw7 in the mouse endometrium using an alternative BAC‐Sprr2f‐Cre recombinase caused EMT and carcinosarcomas with complete penetrance following somatic acquisition of Trp53 mutations (Cuevas et al, 2019). We found similar enrichment of EMT signatures in neoplastic uteri in our Pten del; Fbxw7 mut mice; however none of these animals developed carcinosarcoma. This may relate to the reduced latency of tumorigenesis in our model, or differing effects of Fbxw7 loss and heterozygous missense mutation, as has been demonstrated in other cancer types (King et al, 2013). While formal comparison of Fbxw7 missense mutation and loss was beyond the scope of this study, it is interesting to note that Trp53 missense mutation was a far stronger driver of endometrial tumorigenesis than Trp53 loss in our GEMM. The development of high‐grade endometrial tumours with features of carcinosarcomas in Trp53 mut mice, and their potentiation by Fbxw7 mutation is consistent with a central role of TP53 and FBXW7 in human carcinosarcoma. Furthermore, the failure of Fbxw7 mut to accelerate extrauterine tumours caused by Trp53 loss or mutation supports the hypothesis that Fbxw7 mutation confers greater selective advantage in endometrial malignancy, though this is currently unproven.
The recent discovery that cancer driver mutations are common in normal tissue (Moore et al, 2020) poses important questions about the steps required for malignant transformation. Our initial surprise at the failure of heterozygous Fbxw7 mutation to induce endometrial neoplasia was lessened by the detection of potentially compensatory alterations in gene expression. Upregulation of Fbxw7 expression would be expected to increase the amount of functional Fbxw7 homodimers, thus maintaining physiological levels of substrates, while induction of a p53 response is a well‐characterised block on tumorigenesis (Chen et al, 2005). Whether this is the case in normal human endometrial glands with FBXW7 mutation is unknown. Interestingly, Fbxw7 mut ‐ induced Fbxw7 upregulation was not detected in the background of Pten loss, raising the possibility that Pten may function in a feedback loop, the loss of which contributes to tumorigenesis. Further examination of this, and the mechanisms by which the p53 response is bypassed in mouse and human tumours appears merited.
Since its early characterisation as a ubiquitin ligase for cyclin E (Koepp, 2001; Moberg et al, 2001; Strohmaier et al, 2001), FBXW7 has been shown to regulate a large and increasing number of substrates (Welcker & Clurman, 2008; Davis et al, 2014b). While many have proven oncogenic function, it is currently unclear which drive tumorigenesis in the various cancer types in which FBXW7 is recurrently mutated, as these may vary by tissue (Davis et al, 2014a). LEF1 is a Wnt pathway effector and transcriptional activator essential for endometrial adenogenesis (Shelton et al, 2012). LEF1 is frequently overexpressed in murine and human endometrial cancers and has also been shown to be a key mediator of EMT (Medici et al, 2006; Freihen et al, 2020; Ruz‐Caracuel et al, 2021). Taking these observations together with our data, it appears plausible that failure of Fbxw7 missense‐mutant endometrial cancers to degrade Lef1 may contribute to tumorigenesis and EMT in our GEMM. As we were unable to test whether Lef1 deletion attenuated tumorigenesis in our mice, defining this will be a priority, particularly given that Lef1 ablation in a murine intestinal tumour model unexpectedly increased tumour development (Heino et al, 2021). Whether TCF7L2 dysregulation contributes to FBXW7‐mutant endometrial cancer is less clear. TCF7L2 can act as an activator or repressor of Wnt targets depending on cellular context (Struewing et al, 2010), and has been characterised as a tumour suppressor in colorectal cancer (Wenzel et al, 2020). Understanding the tissue‐specific variation in LEF1 and TCF7L2, along with other FBXW7 substrates will be important topics for future investigation.
Our study has several strengths. These include the large number of alleles examined, our use of clinically relevant hotspot missense Fbxw7 and Trp53 mutations, detailed tumour phenotyping including expert pathological review, agnostic approach to identify novel candidate FBXW7 substrates which were formally confirmed by co‐immunoprecipitation, and confirmation of LEF1 signature in human endometrial cancers with FBXW7 mutation. It also has limitations. Our focus on endometrial tumours meant we did not undertake detailed characterisation of extrauterine lesions beyond basic histopathological analysis. Transcriptomic analyses were of bulk tissue, and it will be important to define the effects of genetic manipulation within the epithelial compartment using single‐cell sequencing and spatial transcriptomic approaches. Logistical considerations caused us to employ a single Cre recombinase and to focus on a single Fbxw7 hotspot missense mutation. It will be important to define whether recombination in non‐epithelial cells with PR‐Cre influenced our results and to examine the impact of substitution of other WD40 propellor‐tip residues (eg R465, R505) in endometria. Similarly, the Trp53 R172H allele used in this study was chosen for pragmatic reasons rather than its prevalence in high‐risk endometrial cancer (6% of oncogenic TP53 point mutations in high‐risk EC in the TCGA studies) and it will be of interest to examine the effects of orthologues of more common mutations such as TP53 R273H (14% of oncogenic TP53 point mutations in TCGA). As noted above, we were unable to test whether LEF1 loss ameliorated tumour development in Pten del; Fbxw7 mut mice. Development of inducible endometrial Cre recombinases would permit this, and the analysis of other genetic alterations in combination, and should be a priority for the field. Finally, our attempts to introduce FBXW7 missense mutation into immortalised human endometrial cells were unsuccessful, and it will be of interest to develop methods to permit this in cells or organoids in the future.
In summary, we show that while Fbxw7 hotspot missense mutation is insufficient to cause endometrial neoplasia in isolation, it potently accelerates tumorigenesis caused by Pten deletion or Trp53 mutation, potentially by failure to degrade the Wnt pathway effector Lef1. Given the morbidity and mortality of high‐risk endometrial cancer (León‐Castillo et al, 2020; Crosbie et al, 2022), further investigation of this pathway as a therapeutic target appears worthwhile.
Materials and Methods
The study confirms to all relevant ethical regulations of the University of Oxford.
Human cancers
Publicly available TCGA data for uterine corpus endometrial cancer (UCEC) and uterine carcinosarcoma (UCS) were downloaded from CBioportal (https://www.cbioportal.org). UCEC data were filtered to include only high‐risk copy number high (CN‐high) cases. Mutations were classified as oncogenic based on the criteria used by TCGA which incorporates data from OncoKB (Chakravarty et al, 2017), CIViC (Griffith et al, 2017), MyCancerGenome (https://www.mycancergenome.org), Cancer Hotspots (Chang et al, 2018) and 3D Cancer Hotspots (Gao et al, 2017). TCGA RNAseq data were downloaded from the Genomic Data Commons Data portal (https://portal.gdc.cancer.gov), details of processing and analysis are provided below. High‐grade (grade 3) endometrial cancers were identified from the databases of the PORTEC2 trial and a prospective cohort of endometrial cancers from the Medisch Spectrum Twente (MST), Enschede (NL). Molecular subtyping of tumours was done as previously reported (Vermij et al, 2023). Ethical approval for analysis of samples was provided by the Leiden‐Den Haag‐Delft medical ethics committee. Participants in the PORTEC 2 trial provided informed consent for sample analysis, for the MST cohort a waiver for informed consent for anonymised analysis of samples was given by the ethics committee. All experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report. Inclusion and exclusion criteria for the PORTEC2 trial have been reported previously. The MST real‐world cohort did not apply specific inclusion or exclusion criteria. Cases were selected for analysis based on known FBXW7 mutation status and availability of FFPE slides for immunohistochemistry. No randomisation was performed.
Mice
Mouse breeding and experiments were performed at the Functional Genomics Facility, Wellcome Centre for Human Genetics, University of Oxford, under UK Home Office Project Licence PPL PDF0B94C3, by appropriately trained, PIL licenced researchers (PILs I8E73D7F0 and ID4B695EE).
Details of generation of Fbxw7fl(R482Q) conditional knock‐in mice have been reported previously (Davis et al, 2011, 2014a). Ptentm2Mak (Pten fl) mice (Suzuki et al, 2001; MGI: 2182005) were imported from the laboratory of Bass Hassan (University of Oxford). Trp53 tm1Brn (Trp53fl) mice (Marino et al, 2000; MGI:1931011) and B6.129S4(Cg)‐Trp53tm2.1Tyj/J (Trp53 R172H) mice (Olive et al, 2004; MGI:3039264) were imported from the laboratory of Xin Lu (University of Oxford). Pgr tm2(cre)Lyd (PR‐Cre) mice (Soyal et al, 2005) were imported from laboratory of Franco de Mayo (Baylor University). All mice were backcrossed onto a C57BL/6J background for at least 6 generations before use in experimental breeding. Details of primers used for genotyping are provided in Dataset EV10. Mice were housed at 18–24°C, relative humidity 30–70% under a 12 h light/dark cycle (07:00 to 19:00 light, 19:00 to 07:00 dark), with drinking water and chow provided ad libitum. Experimental females were checked daily for general health and tumour development, and sacrificed by Schedule 1 approved method at humane endpoint (hunching, bleeding, weight loss ≥ 15% from baseline, external tumour ≥ 10 mm). Experimental uteri and external tumours were harvested, before snap freezing in liquid nitrogen, embedding in Optimal Cutting Temperature Compound (OCT) (Agar Scientific) and/or fixation in 10% neutral buffered formalin (NBF) depending on sample.
Tissue preparation and molecular/pathological analysis of mouse and human samples
DNA for routine genotyping was extracted from ear snips using the Hot Sodium Hydroxide and Tris (HotSHOT) method. Extraction of DNA and RNA from snap‐frozen tissues was performed using the Qiagen DNeasy and RNAeasy Plus kit respectively (Qiagen, Hilden, Germany) according to manufacturer's protocol and quantified by NanoDrop spectrophotometer (ThermoFisher, Waltham, MA, USA) or by Qubit (ThermoFisher). Confirmation of Cre‐mediated recombination of conditional alleles was done by PCR using primers detailed in Dataset EV10 (reaction conditions available on request). First strand synthesis of RNA was performed by High Capacity cDNA kit (Applied Biosystems, Waltham, MA, USA) using random primers as per manufacturer's instructions. cDNA was quantified by real time, reverse transcription PCR (RT–PCR) using TaqMan primer‐probes (ThermoFisher) on the QuantStudio 6 Flex system (Applied Biosystems). Tissue lysates were made by disruption of frozen mouse samples in RIPA buffer (ThermoFisher Scientific) containing protease (Roche, Basel, Switzerland) and phosphatase inhibitors (Sigma‐Aldrich, St Louis, MO, USA) at manufacturer's recommended concentrations, using a rotor‐stator homogeniser. Lysates were cleared by centrifugation at 16,000 g for 20 min at 4°C, with supernatant quantified using the CBX assay (G Biosciences, St Louis, MO, USA) according to manufacturer's instructions. Mouse samples for immunoblotting were heated to 70°C with NuPage LDS Sample Buffer (Invitrogen), loaded on NuPage Bis‐Tris gels (Invitrogen) and separated by electrophoresis before transfer to PVDF membrane. Membranes were then blocked for 1 h at room temperature (RT) in 5% milk diluted in tris‐buffered saline containing 0.1% Tween (TBST) and washed in TBST before incubation with primary antibody in 5% milk or bovine serum albumin (BSA) (Sigma Aldrich) overnight at 4°C. Membranes were then washed, and incubated with IRDye secondary antibody before imaging using a LI‐COR Odyssey CLx system (LI‐COR, Lincoln, NE, USA). Antibodies and concentrations used for immunoblotting are provided in Dataset EV11. Mouse samples for histological analysis were fixed in NBF at room temperature (RT) for < 24 h before processing and paraffin embedding. 4–5 μm sections were cut by microtome, and H&E staining performed by standard techniques. Pathological grading of H&E‐stained uteri and tumours was performed by two expert gynaecological pathologists (AL, TB) blinded to experimental genotype. Slides for immunohistochemistry were deparaffinised in xylene and rehydrated through graded alcohol series. Antigen retrieval was performed by boiling slides in 10 mM citrate buffer (pH 6.0) for 5 min at 120°C in a pressure cooker. Slides were allowed to cool and washed before peroxidase quench in 3% hydrogen peroxide (Sigma Aldrich) for 10 min, followed by further washes (2× in water and 1× in TBS) and blocking by incubation at RT for 1 h with 5% normal goat serum (Vector Laboratories, Newark, CA, USA). Slides were then incubated with primary antibody for 1 h at RT or overnight at 4°C and washed prior to addition of SignalStain Boost IHC Detection Reagent (Rabbit or Mouse; Cell Signalling Technology, Danvers, MA, USA) for 45 min at RT (details in Dataset EV10). Development of the slides was performed using 3–3′ diaminobenzidine (DAB) (Vector Laboratories) according to manufacturer's instructions. Slides were counterstained with haematoxylin, dehydrated and mounted with coverslips. Stained slides were scanned using Aperio CS2 Slide Scanner and viewed by Aperio ImageScope (version 12.4.3.5008; Leica Biosystems, Wetzlar, Germany).
LEF1 IHC on human endometrial cancers was performed on 4 μm FFPE slides in the Histopathology department of Leiden University Medical Centre (LUMC). Unstained slides were rehydrated via graded ethanol series, followed by endogenic peroxidase activity blocking (0.3% Methanol/H2O2) and antigen retrieval using a microwave oven procedure in 10 mmol/L Tris‐EDTA buffer, pH9.0 for 10 min. Tissue sections were incubated overnight with primary antibodies against LEF1 at 4°. A 30 min incubation with a secondary antibody (Poly‐HRP‐GAM/R/R; DPV0110HRP; ImmunoLogic) was then performed before visualisation with DAB (K3468, DAKO) haematoxylin counterstaining. Staining was reviewed and scored by an expert gyn pathologist (TB).
Transcriptomic analysis
Total RNA samples from mouse uteri at 8 weeks age were analysed by TapeStation 4200 to determine sample RNA Integrity Number (RIN). Samples with a RIN ≥ 7 were analysed by Clariom™ S Expression Assays (Affymetrix, Santa Clara, CA, USA) at the Oxford Genomics Centre, Wellcome Centre for Human Genetics, University of Oxford. Data was obtained in the form of sample level, probe set intensities values, which were clustered by principal component analysis to confirm expected grouping. Data were then RMA normalised using the oligo package for R, and probe sets were annotated to their representative gene using the annotation file provided by the manufacturer. Differentially expressed gene (DEG) analysis was performed using the limma package for R. Gene set enrichment analysis (GSEA; Subramanian et al, 2005) was performed and results visualised using the ClusterProfiler (Yu et al, 2012) package in R.
TCGA RNAseq data were downloaded from the Genomic Data Commons (GDC: https://portal.gdc.cancer.gov), normalised and log2 transformed prior to GSEA using ClusterProfiler (Yu et al, 2012).
Differential gene expression from comparison of isogenic HCT116 wild‐type and FBXW7 hotspot mutant cells was downloaded from the supplementary data from Thirimanne et al (2022). DEGs were used as input for GSEA after pre‐ranking using ClusterProfiler.
In silico CPD search
Experimentally validated FBXW7 phosphodegron sequences were curated from search of published literature using PubMed (Koepp, 2001; Welcker et al, 2004; Sundqvist et al, 2005; Mao et al, 2008; Galli et al, 2010; Liu et al, 2010; Inuzuka et al, 2011; Fukushima et al, 2012; Giráldez et al, 2014; Koo et al, 2015; Kourtis et al, 2015; Lv et al, 2015; Maskey et al, 2015; Chen et al, 2016; Kharat et al, 2016; Zhang et al, 2016; Zhao et al, 2016; Song et al, 2017; Wang et al, 2017; Dataset EV7). Phosphodegrons were tabulated, plotted using Seq2Logo web interface https://services.healthtech.dtu.dk/service.php?Seq2Logo‐2.0 (Thomsen & Nielsen, 2012), and used to derive a ‘validated’ CPD [LP][LP][TS]P..[TSDE] for searching against canonical human sequences listed in SwissProt (Bairoch & Apweiler, 1996) and Ensembl (Cunningham et al, 2022) using Bioconductor (Huber et al, 2015). Details of gene names, number of CPD matches, CPD match position, and match sequence were extracted, and compiled to generate a list of candidate substrates. Enrichment analysis of results against curated gene sets corresponding to biological processes was performed using ClusterProfiler (Yu et al, 2012).
Co‐immunoprecipitation of tagged plasmids
Of the plasmids used in this work: pDONR223_FBXW7_WT was a gift from Jesse Boehm, William Hahn and David Root (Addgene plasmid #81795; http://n2t.net/addgene:81795; RRID: Addgene 81795; Kim et al, 2016), TCF7L2_pLX307 was a gift from William Hahn and Sefi Rosenbluh (Addgene plasmid #98373; http://n2t.net/addgene:98373; RRID: Addgene 98373; Rosenbluh et al, 2016), gateway entry vector containing the LEF1 CDS generated by the Human ORFeome Collection (Accession #EU446873) was obtained from the Cellular High Throughput Screening Group, Target Discovery Institute, University of Oxford, pMH‐MYC was a gift from Michael Huen (Addgene plasmid #101765; http://n2t.net/addgene:101765; RRID: Addgene 101765; An et al, 2017), pcDNA3.1‐3xFLAG‐V5‐ccdB was a gift from Susan Lindquist and Mikko Taipale (Addgene plasmid #87064; http://n2t.net/addgene:87064; RRID: Addgene 87064; Taipale et al, 2012).
A Gateway entry vector, pDONR221‐TCF7L2, was generated by BP recombination cloning of the TCF7L2 CDS from TCF7L2 pLX307 in to the pDONR221 vector (Invitrogen). Entry vectors were recombined into destination vectors pMH‐Myc and pcDNA3.1‐3xFLAG‐V5‐ccdB, to generate plasmids for transfection, using the LR Clonase II enzyme mix (Invitrogen, Waltham, MA, USA), according to the manufacturer's instructions. Where required, site directed mutagenesis was performed using the QuickChange Lightning Kit (Stratagene, La Jolla, CA, USA) on the entry vectors prior to LR recombination, according to manufacturer's instructions. HEK293T immortalised embryonic kidney cells (RRID:CVCL_0063) were confirmed mycoplasma‐free by regular commercial testing and maintained in DMEM with 10% FBS and 1% penicillin and streptomycin (Sigma Aldrich). Cells for transfection were plated at 5 × 106 cells per 10 cm dish in antibiotic‐free medium. After 24 h, cultures were transfected with 10ug total plasmid using 30 μl FuGENE HD transfection reagent (Promega, Madison, WI, US) according to manufacturer's protocol. Transfection efficiency was confirmed the following day by visualisation of GFP by microscopy, and cells split into 2 × 10 cm plates and allowed to grow for a further 24 h before lysis in 500 μl benzonase lysis buffer (20 mM Tris pH 7.5; 40 mM KCl; 2 mM MgCl2; 10% (v/v) glycerol, 0.5% (v/v) IGEPAL‐CA‐630; 50 U/ml Benzonase (25 U/μl); 1× cOmplete EDTA‐free protease inhibitor (Roche); 0.5× phosphatase inhibitor cocktail 2 (Sigma‐Aldrich); 0.5× phosphatase inhibitor cocktail 3 (Sigma‐Aldrich)) on ice for 10 min. KCl was then added to 450 mM final concentration and samples rotated for 30 min at 4°C before clarification and protein quantification by CB‐X assay (G‐Biosciences St Louis, MO, USA). Clarified lysates were diluted to KCl concentration of 150 mM by addition of No‐salt equilibration buffer” (20 mM Tris–HCl pH 7.5; 10% (v/v) glycerol; 0.5 mM dithiothreitol (DTT); 0.5 mM ethylenediaminetetraacetic acid (EDTA); 1× cOmplete EDTA‐free protease inhibitor; 0.5× phosphatase inhibitor cocktail 2; 0.5× phosphatase inhibitor cocktail 3). 2 mg of protein was pre‐cleared by incubation with mouse IgG1 isotype control magnetic beads followed by anti‐FLAG M2 magnetic beads for 2 h. Bead‐substrate complexes were washed five times in wash buffer (20 mM Tris–HCl (pH 7.5); 100 mM KCl; 10% (v/v) glycerol; 0.5 mM DTT; 1X cOmplete EDTA‐free protease inhibitor; 0.5x phosphatase inhibitor cocktail 2; 0.5x phosphatase inhibitor cocktail 3) at 4°C and resuspended in 30 μl LDS elution buffer (50 mM glycine pH 2.8, 1× NuPAGE LDS Sample Buffer, 1× NuPAGE Sample Reducing Agent) before heating at 70°C for 10 min. The supernatant was retained and loaded onto 4–12% Bolt Bis‐Tris mini‐protein gels (Invitrogen) for electrophoretic separation and transfer to PVDF membrane before immunoblotting as described above. Primary and secondary antibodies used are listed in Dataset EV10.
Statistical analysis
Sample sizes were not predetermined and animals were not randomised. With the exception of pathological grading, researchers were not blinded to experimental genotype. Data were analysed using GraphPad Prism (GraphPad, San Diego, CA, USA) or R (R Core Team, 2022). Statistical tests used for comparison of groups are indicated in the text or figure legends in all cases. Survival curves were generated according to the Kaplan–Meier method and groups compared by the log‐rank test. Testing for Fbxw7*Trp53 interaction in survival analysis was performed by analysis of the cross‐product term in a Cox proportional hazards model including mutation status of both genes (Fbxw7 +/+ or Fbxw7 R482Q/+ and Trp53 del/del or Trp53 R172H/del). All statistical tests were two‐sided. Statistical significance was accepted at P < 0.05 or FDR < 0.05 depending on the analysis.
Author contributions
Matthew Brown: Conceptualization; resources; data curation; formal analysis; validation; investigation; visualization; methodology; project administration; writing – review and editing. Alicia Leon: Formal analysis; investigation; writing – review and editing. Katarzyna Kedzierska: Formal analysis; investigation; writing – review and editing. Charlotte Moore: Data curation; investigation; writing – review and editing. Hayley L Belnoue‐Davis: Resources; writing – review and editing. Susanne Flach: Investigation; writing – review and editing. John P Lydon: Resources; writing – review and editing. Francesco J DeMayo: Resources; writing – review and editing. Annabelle Lewis: Resources; writing – review and editing. Tjalling Bosse: Formal analysis; supervision; validation; writing – review and editing. Ian Tomlinson: Resources; supervision; funding acquisition; writing – review and editing. David N Church: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
For more information
Author website: https://www.well.ox.ac.uk/research/research‐groups/church‐group.
Endometrial cancer driver mutations: https://www.intogen.org/search?cancer=UCEC.
Endometrial cancer information and patient support: https://peachestrust.org.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Dataset EV11
Source Data for Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was funded by a Cancer Research UK Advanced Clinician Scientist Fellowship (C26642/A27963) to DNC and supported by the Oxford NIHR Comprehensive Biomedical Research Centre (BRC). KK is supported by a Genomic Medicine and Statistics Studentship from the Wellcome Trust. SF is supported by an award from the Munich Clinician Scientist Programme (Verein zur Förderung von Wissenschaft und Forschung an der Medizinischen Fakultät der LMU München e.V.). JPL is supported by the NIH extramural research program of the National Institute of Child Health and Human Development grant no. RO1 HD042311 (JPL). FJD is supported by NIH project nos. Z1AES103311. AL was supported by a Medical Research Council New Investigator Research Grant (MR/P000738/1). TB is supported by a KWF Kankerbestrijding Young Investigator Award. IT is supported by a Cancer Research UK Programme Award. The cost of open access publication was provided by core funding to the Wellcome Centre for Human Genetics from the Wellcome Trust (203141/Z/16/Z). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, the Department of Health or the Wellcome Trust. We are grateful to Phalguni Rath, Transenic Core Facility, Wellcome Centre for Human Genetics, University of Oxford for attempted gene editing of immortalised human endometrial epithelial cell line hEM3. We also thank Matthieu Miossec, Bioinformatics Core Facility, Wellcome Centre for Human genetics, University of Oxford for assistance with GEO upload of microarray data. We also thank Noelia Che, Wellcome Centre for Human Genetics, University of Oxford, for technical assistance with mouse immunohistochemistry and Natalja Ter Haar, Department of Pathology, Leiden University Medical Centre, Netherlands for technical assistance with LEF1 immunohistochemistry on human samples.
EMBO Mol Med (2023) 15: e17094
See also: LD Mayo (October 2023)
Data availability
Microarray data from this study have been deposited in GEO under accession number GSE232356: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE232356.
References
- An L, Jiang Y, Ng HH, Man EP, Chen J, Khoo US, Gong Q, Huen MS (2017) Dual‐utility NLS drives RNF169‐dependent DNA damage responses. Proc Natl Acad Sci USA 114: E2872–E2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta‐Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B et al (2018) Comprehensive characterization of cancer driver genes and mutations. Cell 173: 371–385.e318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R (1996) The SWISS‐PROT protein sequence data bank and its new supplement TREMBL. Nucleic Acids Res 24: 21–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network , Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R et al (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497: 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH et al (2017) OncoKB: a precision oncology knowledge base. JCO Precis Oncol 2017: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, Chakravarty D, Phillips S, Kandoth C, Penson A et al (2018) Accelerating discovery of functional mutant alleles in cancer. Cancer Discov 8: 174–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin H‐K, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W et al (2005) Crucial role of p53‐dependent cellular senescence in suppression of Pten‐deficient tumorigenesis. Nat Cell Biol 436: 725–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Li Y, Xue J, Gong A, Yu G, Zhou A, Lin K, Zhang S, Zhang N, Gottardi CJ et al (2016) Wnt‐induced deubiquitination FoxM1 ensures nucleus β‐catenin transactivation. EMBO J 35: 668–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherniack AD, Shen H, Walter V, Stewart C, Murray BA, Bowlby R, Hu X, Ling S, Soslow RA, Broaddus RR et al (2017) Integrated molecular characterization of uterine carcinosarcoma. Cancer Cell 31: 411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creasman WT, Odicino F, Maisonneuve P, Quinn MA, Beller U, Benedet JL, Heintz AP, Ngan HY, Pecorelli S (2006) Carcinoma of the corpus uteri. FIGO 26th annual report on the results of treatment in gynecological cancer. Int J Gynaecol Obstet 95: S105–S143 [DOI] [PubMed] [Google Scholar]
- Crosbie EJ, Kitson SJ, McAlpine JN, Mukhopadhyay A, Powell ME, Singh N (2022) Endometrial cancer. Lancet 399: 1412–1428 [DOI] [PubMed] [Google Scholar]
- Cuevas IC, Sahoo SS, Kumar A, Zhang H, Westcott J, Aguilar M, Cortez JD, Sullivan SA, Xing C, Hayes DN et al (2019) Fbxw7 is a driver of uterine carcinosarcoma by promoting epithelial‐mesenchymal transition. Proc Natl Acad Sci USA 116: 25880–25890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, Allen JE, Allen J, Alvarez‐Jarreta J, Amode MR, Armean IM, Austine‐Orimoloye O, Azov AG, Barnes I, Bennett R et al (2022) Ensembl 2022. Nucleic Acids Res 50: D988–D995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikoku T, Hirota Y, Tranguch S, Joshi AR, Demayo FJ, Lydon JP, Ellenson LH, Dey SK (2008) Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res 68: 5619–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis H, Tomlinson I (2012) CDC4/FBXW7 and the ‘just enough’ model of tumourigenesis. J Pathol 227: 131–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis H, Lewis A, Spencer‐Dene B, Tateossian H, Stamp G, Behrens A, Tomlinson I (2011) FBXW7 mutations typically found in human cancers are distinct from null alleles and disrupt lung development. J Pathol 224: 180–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis H, Lewis A, Behrens A, Tomlinson I (2014a) Investigation of the atypical FBXW7 mutation spectrum in human tumours by conditional expression of a heterozygous propellor tip missense allele in the mouse intestines. Gut 63: 792–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ, Welcker M, Clurman BE (2014b) Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26: 455–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freihen V, Rönsch K, Mastroianni J, Frey P, Rose K, Boerries M, Zeiser R, Busch H, Hecht A (2020) SNAIL1 employs β‐Catenin‐LEF1 complexes to control colorectal cancer cell invasion and proliferation. Int J Cancer 146: 2229–2242 [DOI] [PubMed] [Google Scholar]
- Fukushima H, Matsumoto A, Inuzuka H, Zhai B, Lau AW, Wan L, Gao D, Shaik S, Yuan M, Gygi SP et al (2012) SCF(Fbw7) modulates the NFkB signaling pathway by targeting NFkB2 for ubiquitination and destruction. Cell Rep 1: 434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli F, Rossi M, D'Alessandra Y, De Simone M, Lopardo T, Haupt Y, Alsheich‐Bartok O, Anzi S, Shaulian E, Calabrò V et al (2010) MDM2 and Fbw7 cooperate to induce p63 protein degradation following DNA damage and cell differentiation. J Cell Sci 123: 2423–2433 [DOI] [PubMed] [Google Scholar]
- Gallo ML, O'Hara AJ, Rudd ML, Urick ME, Hansen NF, O'Neil NJ, Price JC, Zhang S, England BM, Godwin AK et al (2012) Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin‐remodeling and ubiquitin ligase complex genes. Nat Genet 44: 1310–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Chang MT, Johnsen HC, Gao SP, Sylvester BE, Sumer SO, Zhang H, Solit DB, Taylor BS, Schultz N et al (2017) 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Med 9: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Alonso L, Handfield LF, Roberts K, Nikolakopoulou K, Fernando RC, Gardner L, Woodhams B, Arutyunyan A, Polanski K, Hoo R et al (2021) Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro . Nat Genet 53: 1698–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Genderen C, Okamura RM, Fariñas I, Quo RG, Parslow TG, Bruhn L, Grosschedl R (1994) Development of several organs that require inductive epithelial‐mesenchymal interactions is impaired in LEF‐1‐deficient mice. Genes Dev 8: 2691–2703 [DOI] [PubMed] [Google Scholar]
- Giráldez S, Herrero‐Ruiz J, Mora‐Santos M, Japón M, Tortolero M, Romero F (2014) SCF(FBXW7α) modulates the intra‐S‐phase DNA‐damage checkpoint by regulating Polo like kinase‐1 stability. Oncotarget 5: 4370–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M, Spies NC, Krysiak K, McMichael JF, Coffman AC, Danos AM, Ainscough BJ, Ramirez CA, Rieke DT, Kujan L et al (2017) CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet 49: 170–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP (2007) Structure of a Fbw7‐Skp1‐Cyclin E complex: multisite‐phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 26: 131–143 [DOI] [PubMed] [Google Scholar]
- Heino S, Fang S, Lähde M, Högström J, Nassiri S, Campbell A, Flanagan D, Raven A, Hodder M, Nasreddin N et al (2021) Lef1 restricts ectopic crypt formation and tumor cell growth in intestinal adenomas. Sci Adv 7: eabj0512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T et al (2015) Orchestrating high‐throughput genomic analysis with Bioconductor. Nat Methods 12: 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW et al (2011) SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471: 104–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JX, Sun CY, Tian S, Yu C, Chen MY, Zhang H (2016) Tumor suppressor Fbxw7 antagonizes WNT signaling by targeting β‐catenin for degradation in pancreatic cancer. Tumour Biol 37: 13893–13902 [DOI] [PubMed] [Google Scholar]
- Kharat SS, Tripathi V, Damodaran AP, Priyadarshini R, Chandra S, Tikoo S, Nandhakumar R, Srivastava V, Priya S, Hussain M et al (2016) Mitotic phosphorylation of Bloom helicase at Thr182 is required for its proteasomal degradation and maintenance of chromosomal stability. Oncogene 35: 1025–1038 [DOI] [PubMed] [Google Scholar]
- Kim E, Ilic N, Shrestha Y, Zou L, Kamburov A, Zhu C, Yang X, Lubonja R, Tran N, Nguyen C et al (2016) Systematic functional interrogation of rare cancer variants identifies oncogenic alleles. Cancer Discov 6: 714–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, Aranda‐Orgilles B, Perez‐Garcia A, Shi J, Vakoc C et al (2013) The ubiquitin ligase FBXW7 modulates leukemia‐initiating cell activity by regulating MYC stability. Cell 153: 1552–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepp DM (2001) Phosphorylation‐dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294: 173–177 [DOI] [PubMed] [Google Scholar]
- Koo J, Wu X, Mao Z, Khuri FR, Sun SY (2015) Rictor undergoes glycogen synthase kinase 3 (GSK3)‐dependent, FBXW7‐mediated ubiquitination and proteasomal degradation. J Biol Chem 290: 14120–14129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtis N, Moubarak RS, Aranda‐Orgilles B, Lui K, Aydin IT, Trimarchi T, Darvishian F, Salvaggio C, Zhong J, Bhatt K et al (2015) FBXW7 modulates cellular stress response and metastatic potential through HSF1 post‐translational modification. Nat Cell Biol 17: 322–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- León‐Castillo A, de Boer SM, Powell ME, Mileshkin LR, Mackay HJ, Leary A, Nijman HW, Singh N, Pollock PM, Bessette P et al (2020) Molecular classification of the PORTEC‐3 trial for high‐risk endometrial cancer: impact on prognosis and benefit from adjuvant therapy. J Clin Oncol 38: 3388–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Li H, Li S, Shen M, Xiao N, Chen Y, Wang Y, Wang W, Wang R, Wang Q et al (2010) The Fbw7/human CDC4 tumor suppressor targets proproliferative factor KLF5 for ubiquitination and degradation through multiple phosphodegron motifs. J Biol Chem 285: 18858–18867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv XB, Wu W, Tang X, Wu Y, Zhu Y, Liu Y, Cui X, Chu J, Hu P, Li J et al (2015) Regulation of SOX10 stability via ubiquitination‐mediated degradation by Fbxw7α modulates melanoma cell migration. Oncotarget 6: 36370–36382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J‐H, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A (2008) FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321: 1499–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A (2000) Induction of medulloblastomas in p53‐null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14: 994–1004 [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, Davies H, Stratton MR, Campbell PJ (2017) Universal patterns of selection in cancer and somatic tissues. Cell 171: 1029–1041.e1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐Jiménez F, Muiños F, Sentís I, Deu‐Pons J, Reyes‐Salazar I, Arnedo‐Pac C, Mularoni L, Pich O, Bonet J, Kranas H et al (2020) A compendium of mutational cancer driver genes. Nat Rev Cancer 20: 555–572 [DOI] [PubMed] [Google Scholar]
- Maskey D, Marlin MC, Kim S, Kim S, Ong EC, Li G, Tsiokas L (2015) Cell cycle‐dependent ubiquitylation and destruction of NDE1 by CDK5‐FBW7 regulates ciliary length. EMBO J 34: 2424–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici D, Hay ED, Goodenough DA (2006) Cooperation between snail and LEF‐1 transcription factors is essential for TGF‐beta1‐induced epithelial‐mesenchymal transition. Mol Biol Cell 17: 1871–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mericskay M, Kitajewski J, Sassoon D (2004) Wnt5a is required for proper epithelial‐mesenchymal interactions in the uterus. Development 131: 2061–2072 [DOI] [PubMed] [Google Scholar]
- Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK (2001) Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature 413: 311–316 [DOI] [PubMed] [Google Scholar]
- Moore L, Leongamornlert D, Coorens THH, Sanders MA, Ellis P, Dentro SC, Dawson KJ, Butler T, Rahbari R, Mitchell TJ et al (2020) The mutational landscape of normal human endometrial epithelium. Nature 580: 640–646 [DOI] [PubMed] [Google Scholar]
- Morgan RG, Ridsdale J, Payne M, Heesom KJ, Wilson MC, Davidson A, Greenhough A, Davies S, Williams AC, Blair A et al (2019) LEF‐1 drives aberrant β‐catenin nuclear localization in myeloid leukemia cells. Haematologica 104: 1365–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T (2004) Mutant p53 gain of function in two mouse models of Li‐Fraumeni syndrome. Cell 119: 847–860 [DOI] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Willems A, Tyers M, Sicheri F (2003) Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell 112: 243–256 [DOI] [PubMed] [Google Scholar]
- R Core Team (2022) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; [Google Scholar]
- Rosenbluh J, Mercer J, Shrestha Y, Oliver R, Tamayo P, Doench JG, Tirosh I, Piccioni F, Hartenian E, Horn H et al (2016) Genetic and proteomic interrogation of lower confidence candidate genes reveals signaling networks in β‐catenin‐active cancers. Cell Syst 3: 302–316.e304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruz‐Caracuel I, López‐Janeiro Á, Heredia‐Soto V, Ramón‐Patino JL, Yébenes L, Berjón A, Hernández A, Gallego A, Ruiz P, Redondo A et al (2021) Clinicopathological features and prognostic significance of CTNNB1 mutation in low‐grade, early‐stage endometrial endometrioid carcinoma. Virchows Arch 479: 1167–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton DN, Fornalik H, Neff T, Park SY, Bender D, DeGeest K, Liu X, Xie W, Meyerholz DK, Engelhardt JF et al (2012) The role of LEF1 in endometrial gland formation and carcinogenesis. PloS One 7: e40312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Lai L, Chong Z, He J, Zhang Y, Xue Y, Xie Y, Chen S, Dong P, Chen L et al (2017) E3 ligase FBXW7 is critical for RIG‐I stabilization during antiviral responses. Nat Commun 8: 14654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyal SM, Mukherjee A, Lee KY, Li J, Li H, DeMayo FJ, Lydon JP (2005) Cre‐mediated recombination in cell lineages that express the progesterone receptor. Genesis 41: 58–66 [DOI] [PubMed] [Google Scholar]
- Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI (2001) Human F‐box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 413: 316–322 [DOI] [PubMed] [Google Scholar]
- Struewing I, Boyechko T, Barnett C, Beildeck M, Byers SW, Mao CD (2010) The balance of TCF7L2 variants with differential activities in Wnt‐signaling is regulated by lithium in a GSK3beta‐independent manner. Biochem Biophys Res Commun 399: 245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundqvist A, Bengoechea‐Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J (2005) Control of lipid metabolism by phosphorylation‐dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab 1: 379–391 [DOI] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71: 209–249 [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, Yoshida R, Wakeham A, Higuchi T, Fukumoto M et al (2001) T cell‐specific loss of Pten leads to defects in central and peripheral tolerance. Immunity 14: 523–534 [DOI] [PubMed] [Google Scholar]
- Syed SM, Kumar M, Ghosh A, Tomasetig F, Ali A, Whan RM, Alterman D, Tanwar PS (2020) Endometrial Axin2(+) cells drive epithelial homeostasis, regeneration, and cancer following oncogenic transformation. Cell Stem Cell 26: 64–80.e13 [DOI] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S (2012) Quantitative analysis of HSP90‐client interactions reveals principles of substrate recognition. Cell 150: 987–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirimanne HN, Wu F, Janssens DH, Swanger J, Diab A, Feldman HM, Amezquita RA, Gottardo R, Paddison PJ, Henikoff S et al (2022) Global and context‐specific transcriptional consequences of oncogenic Fbw7 mutations. Elife 11: e74338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MC, Nielsen M (2012) Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two‐sided representation of amino acid enrichment and depletion. Nucleic Acids Res 40: W281–W287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toboni MD, Crane EK, Brown J, Shushkevich A, Chiang S, Slomovitz BM, Levine DA, Dowdy SC, Klopp A, Powell MA et al (2021) Uterine carcinosarcomas: From pathology to practice. Gynecol Oncol 162: 235–241 [DOI] [PubMed] [Google Scholar]
- Vermij L, Jobsen JJ, León‐Castillo A, Brinkhuis M, Roothaan S, Powell ME, de Boer SM, Khaw P, Mileshkin LR, Fyles A et al (2023) Prognostic refinement of NSMP high‐risk endometrial cancers using oestrogen receptor immunohistochemistry. Br J Cancer 128: 1360–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang P, Wang Y, Zhan P, Liu C, Mao JH, Wei G (2017) Distinct interactions of EBP1 isoforms with FBXW7 elicits different functions in cancer. Cancer Res 77: 1983–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Clurman BE (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8: 83–93 [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE (2004) The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation‐dependent c‐Myc protein degradation. Proc Natl Acad Sci USA 101: 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel J, Rose K, Haghighi EB, Lamprecht C, Rauen G, Freihen V, Kesselring R, Boerries M, Hecht A (2020) Loss of the nuclear Wnt pathway effector TCF7L2 promotes migration and invasion of human colorectal cancer cells. Oncogene 39: 3893–3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild PJ, Ikenberg K, Fuchs TJ, Rechsteiner M, Georgiev S, Fankhauser N, Noske A, Roessle M, Caduff R, Dellas A et al (2012) p53 suppresses type II endometrial carcinomas in mice and governs endometrial tumour aggressiveness in humans. EMBO Mol Med 4: 808–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16: 284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Karnak D, Tan M, Lawrence TS, Morgan MA, Sun Y (2016) FBXW7 facilitates nonhomologous end‐joining via K63‐linked polyubiquitylation of XRCC4. Mol Cell 61: 419–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S et al (2013) Landscape of somatic single‐nucleotide and copy‐number mutations in uterine serous carcinoma. Proc Natl Acad Sci USA 110: 2916–2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Hirota T, Han X, Cho H, Chong LW, Lamia K, Liu S, Atkins AR, Banayo E, Liddle C et al (2016) Circadian amplitude regulation via FBXW7‐targeted REV‐ERBα degradation. Cell 165: 1644–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]