

Summary
Single-cell clonal selection is a critical procedure for generating a homogeneous population of human pluripotent stem cells. Here, we present a protocol that repurposes the STRIPPER Micropipetter, normally used for in vitro fertilization, to pick single stem cells. We describe steps for tool and reagent preparation, single-cell picking, and colony passaging. We then detail procedures for amplification and analysis. Our protocol does not require cell sorting and produces homogenous clonal cultures with more than 50% survival rate.
For complete details on the use and execution of this protocol, please refer to Deng et al.1
Subject areas: CRISPR, Neuroscience, Stem Cells
Graphical abstract
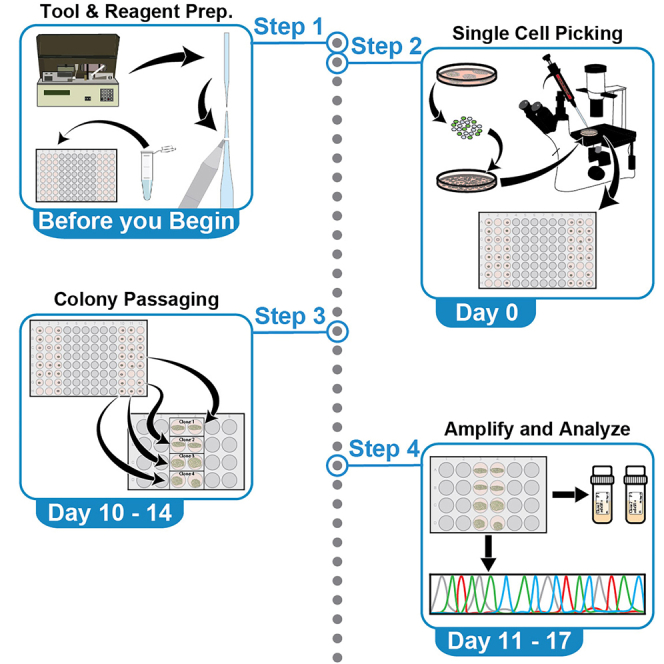
Highlights
-
•
Detailed step-by-step guide for single-cell selection of hPSCs
-
•
Isolation of single hPSC using the STRIPPER Micropipetter
-
•
Cell sorting and serial-dilution-free approach for generating homogeneous hPSC clones
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Single-cell clonal selection is a critical procedure for generating a homogeneous population of human pluripotent stem cells. Here, we present a protocol that repurposes the STRIPPER Micropipetter, normally used for in vitro fertilization, to pick single stem cells. We describe steps for tool and reagent preparation, single-cell picking, and colony passaging. We then detail procedures for amplification and analysis. Our protocol does not require cell sorting and produces homogenous clonal cultures with more than 50% survival rate.
Before you begin
Institutional permissions
All hPSC experiments were conducted following prior approval from the University of Michigan Human Pluripotent Stem Cell Research Oversight (HPSCRO) Committee and Institutional Review Board. Readers should acquire all necessary permissions from the relevant institutions when working with human pluripotent stem cells.
CRISPR/CAS9 editing and fluorescent reporter integration
Timing: 2–3 days
hPSCs are transfected to generate CRIPSR/CAS9 edited or GFP labeled lines.
Since the transfection methods are not the focus of this protocol, they will be not described in the current study. Detailed information about generating and validating the POGZ hPSC CRISPR lines, including genomic integrity analysis using SNP-ChIP analysis can be found in Deng et al. 2022.1 In brief, single guide RNAs (sgRNAs) were either cloned into pSpCas9(BB)-2A-Puro V2.0 vector (Addgene)2 or were generated using the Precision gRNA Synthesis Kit (Thermo Fisher Scientific). The pSpCas9(BB)2A-Puro vector containing the desired sgRNA sequences was transfected into cells using Mirus Bio TransIT-LT1 Transfection Reagent (Mirus Bio LLC). Ribonuclear protein (RNP) complex containing guide RNA and CAS9 protein (Invitrogen) was electroporated into hPSCs using a Neon transfection electroporation system (Thermo Fisher Scientific). Reporter hPSC lines were generated either using a Lenti-EV-GFP-VSVG lentivirus or using transcription activator-like effector nuclease (TALEN) gene editing to knock in a ubiquitous GFP reporter into the AAVS1 (safe harbor) locus.3,4
Prepare glass pipettes
Borosilicate glass capillary tubes are pulled and cut into micropipettes with a bore of 15–30 μm (Methods video S1).
-
1.
Set up micropipette puller using the setting listed in below.
| Heat | 365 |
| Pull | 250 |
| Velocity (Vel) | 150 |
| Time | 150 |
| Pressure | 200 |
-
2.
Insert borosilicate glass into right puller bar and tighten clamp knob.
-
3.
Depress the spring stop on each puller bar and pull each puller bar towards each other so the left end of the glass can be inserted into the left puller bar.
-
4.
Fasten the left clamp knob securely around the left end of the capillary tube.
Note: The borosilicate glass should be evenly positioned between the two clamps for optimal pulling.
-
5.
Close the lid of the micropipette puller and press the start button.
-
6.
Remove the pulled borosilicate glass (i.e., pulled pipette) and store them in a 10 cm petri dish on putty.
Note: Pulling step is optional. Pre-pulled glass pipettes can be purchased from World Precision Instruments.
-
7.Cut pulled pipettes to the desired diameter of 15–30 μm (Figure 1A) using two approaches listed in below.
-
a.Use Microforge: (Manufacturer’s Video; Troubleshooting 1).
-
i.Create a glass bead on the tip of the heating filament by touching an unpulled borosilicate glass capillary to the filament.Note: Heat should be set to high at this step.
-
ii.Maintain heat until the glass melts over the filament and round bead forms.
-
iii.Insert pulled pipette into clamp and adjust its orientation so that the tip of the pipette and glass bead are in proximity.
-
iv.Adjust the position of the pipette to the desired diameter by aligning tip with the micrometer scale in the eyepiece.
-
v.Set the temperature to low-medium heat.
-
vi.Simultaneously touch pipette to glass bead while activating heat with foot pedal.
-
vii.Quickly release foot pedal to snap the pulled pipette at the desired diameter.Note: If pulled pipette bends before it breaks off, the pipette should be discarded.
-
viii.Store the cut pipette with the desired diameter in a new 10 cm dish for future use.
-
i.
-
b.Use diamond scribes: (Methods video S1; Troubleshooting 2).
-
i.Place the diamond tipped pen on the pulled pipette where the bore of the tip will be approximately 15–30 μm in diameter.
-
ii.Rotate the pipette between fingers while in contact with the diamond tipped pen to allow the pen to score the glass at the desired location.Note: The scoring process should be gentle. Pushing the pen too hard against the pipette may shatter or break the pipette at an undesired location.
-
i.
-
a.
Figure 1.

Single-cell selection using the STRIPPER Micropipetter results in homogenous hPSC clones
(A) Phase and brightfield image panels show a single cell picked by the micropipetter tool. Top Panel: sparse single cell suspension. Scale bar: 200 μm. Bottom panel: a single cell (red arrow) being picked by the micropipetter. White arrows show the cells not being picked. Scale bar: 400 μm.
(B) Phase image panels show the establishment of healthy hPSC clones. hPSC clones with a differentiated cell population (top left of dotted line) were first dissociated and then single-cell selected using micropipetter. A suboptimal colony (red) or a colony with healthy cell morphology (green) forms 10 days after single cell selection. Scale bar: 200 μm.
(C) Fluorescence image panel shows the whole process of a GFP reporter hPSC line formed from a single cell. A ubiquitous GFP reporter was knocked in the AAVS1 (safe harbor) locus in a male control iPSC line (CON2E)5 using transcription activator-like effector nucleases (TALEN) gene editing. Top panel shows a colony containing both GFP positive and negative cells before single cell picking. Bottom panel shows colonies 1-, 2-, 5- and 8-days post picking. Images were taken using an EVOS FL Auto Imaging System (Thermo Fisher Scientific). Scale bar: 200 μm.
(D and E) The percentages of surviving clones of multiple hPSC lines including hESCs (in circles) and hiPSCs (in squares) after single-cell cloning. Female lines: WA-09, and SCN1B 6b/4A (unpublished cell line); Male lines: WA-01, and UM-139-2/4–66). (D) On average, 51.2% of single cells of reporter hPSC clones resulted in viable clones (n = 19 experiments, SEM = 3.5%). (E) On average, 53.9% of single cells resulted in viable clones after CRISPR/CAS9 genome editing. Gene targets included POGZ,1FMR1,6KCNH2,7RAI1 (unpublished cell line), KDM5C (unpublished cell line), ANK3 (unpublished cell line), STXBP1 (unpublished cell line) and SYNGAP1 (unpublished cell line) (n = 18 experiments; SEM = 3.8%).
Reagent preparation
-
8.
Aliquot Laminin-521 and Clone R2 and store the aliquots at −20°C for long-term storage.
Note: Aliquot 72 μL of the Laminin-521 (500 μg) into a 1.5 mL Eppendorf tube (enough for coating 24 wells of a 96-well plate). Aliquot 1 mL of the CloneR2 into a 1.5 mL Eppendorf tube. This will reduce freeze-thaw cycles.
-
9.
Prepare 1:100 Geltrex solution by adding 500 μL of 100% Geltrex to 49.5 mL DMEM/F-12.
Note: Dilution should be done on ice with pre-chilled pipette tips to prevent Geltrex forming aggregates.
-
10.
Prepare 0.8 mM EDTA (PH = 7.2) by adding 80 μL 0.5 M EDTA solution to 49.92 mL 1xDPBS without Ca++/ Mg++.
Thawing reagents – Day -2
-
11.
Thaw frozen aliquots of Laminin-521, CloneR2, and chill 96-well plates at 2°C–8°C overnight.
Cell culture – Day -1
Cells are grown to a desired confluency that allows for a large population of cells to be selected.
-
12.
Culture cells to 50–60% confluent on the day of single-cell dissociation.
-
13.
Remove spontaneously differentiated cells using a p10 pipette tip under an inverted microscope (Methods video S2).
Note: hPSC clones with spontaneously differentiated cells do not have defined borders (Figure 1B).
Coat 96-well plate – Day -1
96-well plates are coated with Laminin-521 to facilitate cell attachment.
-
14.
Prepare 5 μg/mL Laminin solution containing 72 μL Laminin-521 and 1368 μL 1XDPBS with Ca++/Mg++.
Note: This volume is enough to coat 24 wells of a 96-well plate.
-
15.
Add 60 μL of the Laminin-521 solution to each well of the pre-chilled 96-well plates.
Note: The 60 μL volume results in 1 μg/cm2 of Laminin-521 per well. Less than 60 μLof the Laminin-521 solution could lead to poor cell attachment if each coat is not evenly spread.
CRITICAL: All components for this step (96-well plate, pipette tips, DPBS, Laminin-521) should be pre-chilled at 2°C–8°C overnight and kept on ice until the 96-well plate is transferred to the incubator to prevent matrix from premature polymerization.
Optional: Use the first and last three columns (48 wells in total) of a 96-well plate as it is easier to track which wells are used to receive single cells.
-
16.
Incubate the Laminin-521 coated plates at 37°C, 5% CO2 for 2 h or overnight.
Optional: A minimum of 2 h is required for Laminin solution to adhere to the bottom of the plate. However, we observed that incubating the coating for less than 2 h resulted in a significant reduction in clone attachment.
Note: Laminin-521 was chosen as it showed the greatest attachment efficiency. Early attempts using Geltrex, Matrigel or Vitronectin-coated surfaces yielded poor survival (data not shown).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Lenti-EV-GFP-VSVG | University of Michigan Vector Core | Lenti-EV-GFP-VSVG |
| Chemicals, peptides, and recombinant proteins | ||
| CellAdhere Laminin-521 | STEMCELL Technologies | Cat#77003 |
| CloneR2 | STEMCELL Technologies | Cat#100-0691 |
| Geltrex LDEV-Free, hESC-Qualified, Reduced growth factor basement membrane matrix | Thermo Fisher Scientific | Cat#A1413302 |
| AccuGENE 0.5 M EDTA Solution | Lonza | Cat#51201 |
| Dulbecco’s phosphate-buffered saline (DPBS) no calcium, no magnesium | Thermo Fisher Scientific | Cat#14190144 |
| Dulbecco’s phosphate-buffered saline (DPBS) calcium, magnesium | Thermo Fisher Scientific | Cat#14040133 |
| Ambion Nuclease Free Water | Invitrogen | Cat#AM9930 |
| Critical commercial assays | ||
| Q5 High-Fidelity 2× Master Mix | New England Biolabs | Cat#M0492S |
| QIAquick PCR Purification Kit | QIAGEN | Cat#28104 |
| DNeasy Blood & Tissue Kit | QIAGEN | Cat#69504 |
| Infinium CoreExome-24 v1.4 Kit | Illumina Inc | Cat#20039222 |
| TrueCut Cas9 Protein v2 | Invitrogen | Cat#A36498 |
| Mirus Bio TransIT-LT1 Transfection Reagent | Mirus Bio LLC | Cat#MIR2300 |
| Precision gRNA Synthesis Kit | Thermo Fisher Scientific | Cat#A29377 |
| Experimental models: Cell lines | ||
| WA01 (H1) Male Embryonic Stem Cells | WiCell | Cat#WA01 |
| WA09 (H9) Female Embryonic Stem Cells | WiCell | Cat#WA09 |
| UM4-6 | Haenfler et al.6 | N/A |
| UM139-2 | Haenfler et al.6 | N/A |
| CON2E | Tidball et al.5 | N/A |
| SCN1B-4A | This paper | Louis Dang unpublished cell line |
| SCN1B-6B | This paper | Louis Dang unpublished cell line |
| Oligonucleotides | ||
| POGZ sgRNA sequence 1: TCGCTCCTCACTCTACTCTG | Deng et al.1 | N/A |
| POGZ sgRNA sequence 2: CCTCCTCACATTCCATGAAC | Deng et al.1 | N/A |
| Primer to confirm sgRNA1 editing: POGZ Primer Forward 1: GCCTAGTACCTGGTGCCTCA | Deng et al.1 | N/A |
| Primer to confirm sgRNA1 editing: POGZ Primer Reverse1: GGCCTCTCAGTTGTTCACTTC | Deng et al.1 | N/A |
| Primer to confirm sgRNA2 editing: POGZ Primer Forward 2: TCAGAAGAGCTTTTGTTACCTGTG | Deng et al.1 | N/A |
| Primer to confirm sgRNA2 editing: POGZ Primer Reverse 2: CCAAGGAGGTAGGTTACCAATG |
Deng et al.1 | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro V2.0 (PX459) | Ran et al.2 | Addgene plasmid #62988 |
| AAVS1-TALEN-L | Gonzalez et al.3 | Addgene plasmid #59025 |
| AAVS1-TALEN-R | Gonzalez et al.3 | Addgene plasmid #59026 |
| AAVS1-CAG-hrGFP | Qian et al.4 | Addgene plasmid #52344 |
| Software and algorithms | ||
| ICE CRISPR Analysis Tool | Synthego | https://www.synthego.com/products/bioinformatics/crispr-analysis |
| Genome Viewer v2.0 | Illumina | https://support.illumina.com/array/array_software/genomestudio/downloads.html |
| Other | ||
| Accutase Cell Detachment Solution | Innovative Cell Technologies | Cat#AT104 |
| Dulbecco’s Modified Eagle Medium/ Nutrient Mixture F-12 (DMEM/F-12), HEPES | Thermo Fisher Scientific | Cat#11330032 |
| mTeSR Plus | STEMCELL Technologies | Cat#100-0276 |
| Biological Safety Cabinet model: 1300 series A2 | Thermo Fisher Scientific | Cat#1323TS |
| Horizontal Laminar Flow Cabinet model: HeraGuard ECO | Thermo Fisher Scientific | Cat#51029701 |
| CO2 incubator model: HERAcell vios 160i | Thermo Fisher Scientific | Cat#51033557 |
| Inverted fluorescence cell culture microscope model: CKX53F3 | Olympus | Cat#CKX53 |
| EVOS FL Auto Imaging System | Thermo Fisher Scientific | Cat#EVOSFL |
| Countess 3 Automated Cell counter | Invitrogen | Cat# A49865 |
| The STRIPPER Micropipetter with Free Standing Comfort Grip | Cooper surgical | Cat#MXL3-STR-CGR https://fertility.coopersurgical.com/micropipettes/the-stripper/ |
| Fisherbrand Imetera Diamond Scribes | Fisher Scientific | Cat#08-675 https://www.fishersci.com/shop/products/imetra-diamond-scribes/08675#?keyword=08-675 |
| Borosilicate glass capillary 1 mm × 0.58 cm × 10 cm | Sutter Instrument | Cat#B100-58-10 https://www.sutter.com/MICROPIPETTE/glass.html |
| Micropipette puller model: P-87 | Sutter Instrument | Cat#P-87 https://www.sutter.com/MICROPIPETTE/p-97.html |
| Microforge Model: MF2 | Narishige | Cat#MF2 https://products.narishige-group.com/group1/MF2/pipette/english.html |
| 35 mm Petri dish | Fisher Scientific | Cat#08-757-100A |
| Falcon Standard Tissue Culture Dishes | Fisher Scientific | Cat#08-772E |
| 96-well cell culture, flat bottom microplate | Fisher Scientific | Cat#08-772-2C |
| Sterile 15-mL centrifuge tubes | ASI Scientific | Cat#CN50601 |
| 1-Channel mechanical pipette | Fisher Scientific | F123600TG, F123602G |
| Sterile micropipette tips | Fisher Scientific | Cat#02-707-432, 1111-2721 |
Materials and equipment
-
•
Cloning medium: 90% mTeSR Plus + 10% CloneR2.
Note: Storage condition: 4°C, maximum storage time: 1 week.
-
•
5 μg/mL Laminin solution: 3 μL CellAdhere Laminin-521 in 57 μL 1XDPBS with Ca++/ Mg++ per well (96-well plate).
Note: Storage condition: use immediately after dilution of CellAdhere Laminin-521.
-
•
1:100 Geltrex matrix solution: add 1 part Geltrex to 100 parts DMEM/F-12.
Note: Storage temperature: 4°C, maximum storage time: 1 month.
-
•
0.8 mM EDTA solution (pH ∼7.2): add 80 μL of AccuGENE 0.5 M EDTA to 49.92 mL of 1XDPBS without Ca++/Mg++
Note: Storage temperature: room temperature; maximum storage time: 1 year.
Step-by-step method details
96-Well plate preparation – Day 0
-
1.
Aspirate the Laminin solution from each well of the 96 well plate.
-
2.
Add 100 μL of the prewarmed cloning medium into each well.
Note: Gently dispense cloning medium to each well to avoid disrupting the Laminin-521 coating.
-
3.
Place in an incubator at 37°C 5% CO2 for 1 h before starting experiment.
Single cell dissociation – Day 0
Cells are gently dissociated into single-cell solution using accutase cell dissociation reagent (Methods video S3).
-
4.
Remove tissue culture vessels from incubator and place them in biosafety cabinet.
-
5.
Aspirate medium from the vessels, and wash cells with 1 mL room temperature (RT) 1X DPBS without Ca++/Mg++.
Note: the volume is for each well of a 6-well plate or a 35 mm tissue culture dish.
-
6.
Aspirate DPBS from previous step and replace with 1 mL of accutase cell dissociation reagent and place the plate back into incubator for 5–7 min.
-
7.
Remove the plate from incubator and place it in biosafety cabinet.
Note: At this point most colonies should be lift in suspension.
-
8.Liberate remaining colonies from the bottom of the plate.
-
a.Draw up cell suspension using a p1000 micropipette and spray remaining adherent colonies off the plate.
-
b.Gently pipette up and down twice to break up cell aggregates into single cells.
-
c.Transfer cell mixture into a 15 mL conical tube, and then immediately add 9 mL DMEM F-12 medium to neutralize the accutase reaction (Methods video S3).
-
a.
-
9.
Centrifuge at 200 g for 5 min at RT, aspirate supernatant, and gently resuspend cell pellet in 1 mL of mTeSR Plus medium.
-
10.
Count viable cells using Trypan blue.
Note: Do not continue if cell viability is below 80%.
-
11.
Dilute single-cell solution to approximately 1000 cells/mL with pre-warmed mTeSR Plus medium in a 35 mm untreated petri dish.
-
12.
Rock the petri dish back and forth to separate single cells.
-
13.
Place single cell suspension in a 37°C incubator for 5 min to allow cells to settle, but not attach to the bottom of the petri dish.
Single cell picking – Day 0
Single-cell selection into 96-well plates using the pulled pipettes (Figure 1A, Methods video S3, Troubleshooting 3).
Note: An experienced cell picker can expect to pick 48 clones (one 96-well plate) in 15–20-min.
-
14.
Attach a pre-cut borosilicate glass micropipette tip to the STRIPPER Micropipetter in the horizontal laminar flow cabinet/clean bench.
-
15.
Retrieve the 35 mm petri dish containing single-cell suspension and the Laminin-521 coated 96-well plate containing cloning medium from the incubator, and place them in the horizontal laminar flow cabinet/clean bench.
-
16.Pick single cells using the STRIPPER Micropipetter under an inverted microscope at 4× or 10× magnification.
-
a.Depress the plunger on STRIPPER micropipette to first stop, carefully place tip in front of desired cell and aspirate (∼1 μL).
-
b.Place the pipette tip into one well of the Laminin-521 precoated 96-well plate containing mTesR Plus with CloneR2.
-
c.Depress the pipette plunger to the second stop to expel the cell into the desired well.
-
a.
Note: These pipette tips are very fragile, so be careful not to scratch the bottom of the plate or wells.
Note: Early attempts of this protocol used Y-27632 Rho Kinase inhibitor, but this yielded poor efficiency. The use of CloneR2 was recommended to us by the manufacturer. However, a recent publication using a cocktail containing chroman 1, emricasan, polyamines, trans-ISRIB (CEPT) was show to have greater efficiency compared to CloneR during fluorescence activated cell sorting (FACS) of hPSCs, and therefore may increase the efficiency of this single cell picking protocol.8
-
17.
Repeat step 16 until the desired number of cells are selected.
-
18.
Place 96-well plates containing single cells in the back of tissue culture incubator at 37°C 5% CO2 for 2 days.
Colony maintenance – Day 1 - 10/14
Regular medium changes are performed to allow a single cell to attach and form a homogenous colony (Troubleshooting 4).
Note: It is difficult to visualize cell growth between day 1 and day 5. Therefore, it is important to perform medium changes carefully without touching the bottom of each well to avoid disrupting cell attachment.
If single-cell cloning is used for selecting iPSC reporter lines, you may check the fluorescence signal on day 1.
-
19.
On day 2, perform full medium change with prewarmed 100 μL cloning medium.
Note: It is safest to aspirtate the medium out of the well using a 200 μL pipette. But once comfortable with the depth of the well, a vacuum aspirator may be used with the suction set to lowest setting.
-
20.
On day 3, add an additional 25 μL of prewarmed fresh cloning medium (25% of initial volume) to each well.
-
21.
From day 4-10/14, perform full medium changes daily with prewarmed mTeSR Plus.
Note: The colonies may be passaged on day 10 or day 14.
Fluorescent reporter confirmation – Day 1-10/14
Visual inspection under a fluorescent microscope to confirm fluorescence reporter expression.
-
22.
On day 1, check fluorescent signal using an Evos imaging system or other desired system (Figure 1C).
Note: Check each well periodically to ensure that the wells of interest maintain reporter expression. During the recovery and expansion period, it is possible that reporter expression is silenced in some cells.
-
23.
Passage multiple single-cell clones with various reporter expression levels.
Note: Since viral integration occurs randomly, it can cause toxicity if more than one integration event occurs in a cell.
Expansion of single-cell colonies – Day 10–14
Single-cell colonies are expanded for future experiments (Troubleshooting 5).
-
24.
Coat the surfaces of a 24-well plate with 500 μL of Geltrex solution.
Note: Laminin-521 is only needed to enhance the attachment of single cells after picking. Subsequent passaging of single-cell derived colonies can be done with conventional matrices such as Geltrex and Matrigel.
-
25.
Incubate 24-well plates in 37°C incubator for a minimum of 30 min or overnight.
-
26.
Retrieve 96-well plates containing sing-cell colonies from incubator, and place them in a biosafety cabinet about 5 min before the Geltrex incubation time is complete.
-
27.
Aspirate the medium from 96-well plates and wash each well with 100 μL 1XDPBS, without Ca++/Mg++.
-
28.
Add 100 μL of 0.8 mM EDTA solution per well (pH 7.2).
-
29.
Place 96-well plates in 37°C incubator for 5–7 min.
-
30.
During the EDTA incubation, remove the Geltrex solution from 24-well plates, then add 480 μL of mTeSR Plus medium to one well and 420 μL of medium to a second well.
Note: The different volumes reflect the need for screening clones after CRIPSR gene editing. One sparsely populated (480 μL) well is for maintenance and a more densely populated well (420 μL) is for genomic DNA (gDNA) isolation. If single-cell cloning is for isolating iPSC reporter lines or population selections, use the same volume and plate an equal number of cells in each well.
-
31.
Place 96-well plates back into the biosafety cabinet and carefully aspirate EDTA solution.
-
32.
Add 100 μL of mTeSR Plus medium to 96-well plates, and pipette up and down to gently dissociate large cell clumps into smaller pieces.
-
33.
Add 20 μL of cell suspension to one well of 24-well plates containing 480 μL of mTeSR Plus medium to bring the total volume to 500 μL.
-
34.
Add the remaining 80 μL of cell suspension to the second well containing 420 μL of mTeSR Plus medium.
-
35.
Place plates back into the cell culture incubator at 37°C 5% CO2.
Note: Researchers may use their established protocols in their laboratories for routine maintenance of hPSCs such as medium change, passaging, and cryopreservation. If your lab does not use mTeSR Plus medium routinely, hPSCs can be gradually transitioned from mTeSR Plus medium to another desired medium.
Note: Sudden switch between hPSC maintenance medium could reduce cell viability.
Genome editing confirmation – Day 11–17
Genomic DNA (gDNA) is extracted from the densely populated well to assess gene editing efficiency.
-
36.
Liberate cells from plates as outlined in the “Passaging Colonies” section of the protocol (Step 27–32).
Pause point: Cell pellet can be flash frozen and gDNA isolation can be done later.
-
37.
Isolate gDNA using the DNeasy Blood and Tissue Kit (Qiagen) using manufacturer’s protocol.
-
38.
Amplify the region of interest and send out purified PCR products for Sanger sequencing (Figure 2B).
PCR reaction master mix
| Reagent | Amount |
|---|---|
| DNA template | 2 μL |
| Q5 High-Fidelity 2× Master Mix | 25 μL |
| 10 μM Forward Primer (500 mM stock) | 2.5 μL |
| 10 μM Reverse Primer (500 mM stock) | 2.5 μL |
| Ambion Nuclease Free Water | 18 μL |
PCR cycling condition
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 98°C | 30 s | 1 |
| Denaturation | 98°C | 5–10 s | 35 cycles |
| Annealing | 55°C–72°C | 10–30 s | |
| Extension | 72°C | 20–30 s/kb | |
| FinalExtension | 72°C | 2 min | 1 |
| Hold | 12°C | forever | |
Note: We use Eurofins Genomics for Sanger sequencing.
-
39.
Estimate the gene editing efficiency and indel patterns using the online software ICE from Synthego Inc.
Note: To use this software edited lines must be compared to an unedited control (this should be the original line without going through the gene editing process) (Figure 2B).
-
40.
Confirm indel patterns using Next Generation Sequencing (NGS) on selected clones (Figure 2C).
Note: We use the CRIPSR NGS sequencing service at the Massachusetts General Hospital DNA Core Facility.
Note: NGS generates quantitative readouts of sequence variants in each edited stem cell clone, which can be used to confirm the homogeneity of single-cell selection. For example: If it is a wildtype clone, we expect a nearly 100% (above 97%) variant that matches to the NCBI reference sequence.9 If it is a heterozygous knockout (KO) clone, we expect a ∼50:50 ratio between WT sequence, and a modified sequence containing insertions/deletions (Figure 2C). If it is homozygous KO, we expect a nearly 100% (above 97%) variant that is different from the WT sequence (Figure 2C). If it is a compound heterozygous clone, we expect a ∼50:50 ratio between two different variants, both of which contain insertions/deletions. Large indels can slightly skew this ratio in heterozygous lines to more than 70% WT.
-
41.
Perform genetic integrity analysis to ensure there are no confounding chromosome abnormalities before starting downstream analyses.
Note: We use the Illumina Infinium CoreExome-24 v1.4 kit and the genotyping service at University of Michigan Advanced Genomics Core Facility and analyze the data using Genome Studio v2.0.
Alternatives: We have also used hPSC Genetic Analysis Kit from Stem Cell Technologies (Catalog # 07550). This is an alternative approach to check chromosome abnormalities, which is cost-effective but lower throughput.
-
42.
Perform other downstream analyses such as RT-qPCR, immunoblotting, and immunostaining.
Figure 2.

STRIPPER single cell selection can be used to isolate CRISPR/CAS9 genome edited colonies
(A) Diagram depicting the POGZ gene. Stars on the diagram indicate the locations of mutations for heterozygous POGZ (POGZ+/−; red) and homozygous mutations (POGZ−/−; blue) in the POGZ gene.1
(B) Sanger sequencing confirmation of indels in POGZ+/− and POGZ−/− lines (analysis were done using the ICE software from Synthego Inc).
(C) Next generation sequencing confirmation of indel distribution in POGZ+/− and POGZ−/− lines. Top panel: We observed a ∼50:50 ratio between WT sequence (48.7%) and a modified sequence containing a single nucleotide insertion (51%) in POGZ+/− line. Bottom panel: We observed a nearly 100% (99.81%) variant that is different from the reference sequence in POGZ−/− lines. This figure was modified from Deng et al. 2022.1
Expected outcomes
Frequently used methods for single cell selection include performing serial dilutions or cell sorting, but both methods have shortcomings.10,11 The serial dilution method offers an inexpensive solution, but there is a higher risk of developing heterogenous colonies.12 The use of cell sorting instruments is fast and efficient at selecting single cells, but is expensive and may be a financial barrier to many labs.13,14,15 We have developed a single cell picking assay based on a common in vitro fertilization procedure,16 which is effective in terms of cost and colony formation.1 The precision of single cell selection comes from the STRIPPER Micropipetter’s ability to withdraw only 1–3 μL, making it difficult to carry along unwanted cells. Individually picked cells adhere to the plate/dish surface 2 h after being selected. However, they are sensitive to environmental changes (e.g., temperature, pH, and humidity) at this stage. The best time to view selected cells is five days after selection as the cells have had time to divide and to form colonies. At this stage cells are less sensitive to environmental fluctuations. Once a researcher is comfortable with this protocol, one can expect on average 50% survival rate, with the possibility of achieving over 80% viability (Figures 1D and 1E). This rate is equivalent to and, in some cases, exceeds the efficiency of single cell sorting protocols which utilize similar reagents.8,14
We have demonstrated three applications for which this protocol is optimal. The first application is to remove differentiated cells in human PSC cultures (Figure 1B). Over time spontaneous differentiation occurs in stem cell culture, which reduces the differentiation potential of hPSCs.17 Using this method, we were able to isolate healthy colonies based on the morphology of stem cell colonies (Figure 1B). The second application is to select single cells after performing CRISPR/CAS9 genome editing (Figure 1E and 2). As analyzed by sanger sequencing and NGS, we successfully generated both POGZ heterozygous and homozygous KO H9 human embryonic stem cell lines (Figure 2).1 In addition, we also applied this technique 18 times in CRISPR/CAS9 genome editing projects (Figure 1E). We generated knockout lines for KCNH2,7 FMR1,6 RAI1 (unpublished), KDM5C (unpublished), ANK3 (unpublished), STXBP1(unpublished), and SYNGAP1(unpublished), introduced a KCNH2 point mutation in the H9 ESC line (unpublished), and corrected an ANK3 patient mutation in a patient iPSC line (unpublished). Finally, our protocol can be used to establish stable fluorescent reporter hPSC lines. Using our single cell picking protocol, we have carried out 19 different singe cell picking experiments to isolate hPSC reporter lines, with each line expressing the fluorescence reporter uniformly (Figure 1C). This is particularly helpful for researchers to select series of iPSC reporter lines that display various levels of reporter intensity as high expression levels of some fluorescent reporters have been shown to be toxic to cells.18
Limitations
Our single cell picking method was effective to isolate pure and healthy homogeneous populations of hPSCs. Although single cell picking is more precise than serial dilutions and more accessible than using a cell sorter, using the STRIPPER Micropipetter and pulling glass pipettes could require special training. However, based on our experience, it often takes a couple of hours to learn how to use the STRIPPER tool and pulled glass pipettes may be readily available in any animal transgenic cores at many research institutions. In our hands, it takes about 15 min to pick 48 single cells up to 83% of which are viable (Figures 1D and 1E).
One of the most common methods for hPSC cloning is cell sorting. It is a quick and efficient strategy for isolating single cells but the machines are costly to purchase and maintain.13 This protocol offers a much economic solution for single cell cloning compared to the use of a higher priced cell sorting machine. Nevertheless, we recognize that the machines used to pull the pipette tips may not be accessible at all institutions and may provide a significant financial restriction to some labs. As an alternative approach, pre-pulled glass pipettes can be purchased from World Precision Instruments and then be cut to an approximate diameter of 15–30 μm using the Diamond Scribe tool.
One major benefit of cell sorter is the ability to quickly select hundreds of cells, which makes it useful when performing genetic or pharmacological screens. Such experiments may not be feasible using our manual method. However, if selecting more than 48 cells is required for a specific experiment, the researcher should avoid keeping the single cell suspension out of the incubator for extended periods of time, and we recommend performing the single cell picking in batches. Finally, with the recent development of chemical cocktails to enhance hPSC viability,8 our current protocol may be further optimized to enhance cell viability during the single-cell selection process.
Troubleshooting
Problem 1
Difficulty with cutting pulled glass tips to the desired diameter using Microforge (related to before beginning Step 7a).
Potential solution
-
•
Check the glass bead. The size and shape of the glass bead changes after repeated usage. In the case of the bead becoming too large, or too small, it will not cut the pulled glass pipette correctly. Use manufacturer’s protocols for adjusting the size and shape.
-
•
Timing and temperature of heating application. If the heat is too low, too high or is left on for too long before the bead touches the pulled glass pipette, the pipette will melt instead of snapping. Timing operation can be learned via a video found on the manufacturer’s website.
Problem 2
Difficulty with cutting pulled glass tips to the desired diameter using Diamond Scribe tool (related to before beginning Step 7b).
Potential solution
-
•
It can be tedious to cut the tip with the Diamond Scribe tool. Make sure the end of the diamond tip is perpendicular to the surface of the pulled pipette when scoring the glass. It can be helpful to view the scoring more closely under a stereoscope.
-
•
Use even pressure at the stem of the pipette to prevent the tip from breaking. Also be gentle when applying pressure once the tip that has been scored to prevent cracking or an uneven break.
Problem 3
At single cell picking stage, it is difficult to pick only one cell at a time (related to Step 14–18).
Potential solution
-
•
If cells are too dense, perform an additional dilution in a 2nd petri dish.
-
•
If the bore of the needle is too big (diamond scribe cutting), try cutting a new pipette closer to the tip.
-
•
Cells could float out of the focus plane, which results in multiple cells drawn into the pipette. Be sure to wait for the cells to settle before starting the selection process. In addition, handle the petri dish gently, as disruption to the petri dish can cause the cells to drift in and out of the field of focused view.
Problem 4
Low cell attachment (related to Step 19–21).
Potential solution
-
•
The 5 μg/mL Laminin-521 solution is not reconstituted properly, 96-well plate is not coated with the correct volume of Laminin, or coating time is too short. Make sure the laminin aliquots are fresh and are diluted to a minimum of 5 μg/mL. Coating the wells with less than 60 μL of Laminin solution and/or for less than 2 h could result in a significant reduction in attachment.
-
•
Cells after picking are not healthy. This can be caused by multiple factors: (1) cells are not healthy preceding the use of the protocol, (2) excessive single cell dissociation with Accutase, (3) cells are kept at room temperature for an extended period of time. To increase cell viability, be sure that the starting cell population is healthy (i.e., no differentiated cells and not overgrown). Be sure to optimize the accutase dissociation procedure before performing this protocol. Finally, picking single cells should be done at an approximate interval of 30 min out of the incubator and 30 min in the incubator.
-
•
Cloning solutions are not freshly made. Repeated freeze-thaw cycles should be avoided.
Problem 5
Difficulty passaging single-cell derived colonies (related to Step 27–37).
Potential solution
-
•
Cell colonies were not dissociated after 0.8 mM EDTA treatment. First, make sure that the solution is made at the proper concentration. Second, wash cells with 1xDPBS without Ca++/Mg++ before EDTA dissociation. The salts found in the culture medium can prevent the EDTA lifting the cells off the bottom of the plate.
-
•
Cell colonies were left too long in 0.8 mM EDTA. Passaging solutions such as EDTA can be harsh on cells and the length of incubation and concentration can vary between cell lines. The incubation time in this solution should be tested before starting this protocol.
-
•
24-well plate was poorly coated with the Geltrex solution. Since Geltrex polymerizes at temperatures above 10°C, it is critical that this solution is prepared on ice with pre-chilled pipettes. Before each usage, make sure that there are no clumps in the solution. If there are large clumps, the solution will not coat the plate evenly and will lead to poor attachment of stem cells. If the Geltrex solution is left on the plate for less than 30 min or more than 48 h in incubator, the matrix will either not be fully polymerized or will be desiccated, eventually leading to poor cell attachment.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wei Niu (wei.niu@utoledo.edu).
Materials availability
POGZ-hESC cell lines generated in the study are available upon request from the lead contact via email.
Acknowledgments
This work was supported by the Taubman Institute Scholars Fund at Michigan Medicine, Pandemic Research Recovery (PRR) Program at Michigan Medicine, and U54NS117170 from the National Institute of Neurological Disorders and Stroke (NINDS). Parts of this study were conducted through the Michigan Medicine Human Stem Cell and Gene Editing Core (HSCGE). The design of POGZ sgRNA and the generation of POGZ KO H9 ESC lines were described in Deng et al., 2022. We thank Drs. Peter Todd, Shigeki Iwase, David Jones, Paul Jenkins, and Louis Dang for allowing us to use the viability data of single-cell cloning of their unpublished cell lines. We thank the Advanced Genomics Core at the University of Michigan (Ann Arbor, MI) and the Center for Computational and Integrative Biology DNA Core Facility at Massachusetts General Hospital (Cambridge, MA) for providing sequencing services. We would like to thank the Transgenic Animal Models Core, specifically Galina Gavrilina, for her assistance and allowing us to use their micropipette manufacturing equipment. We also thank Dr. Zhonggang Hou for his insightful comments on the protocol.
Author contributions
S.P.M.P. conceptualized the methodology. S.P.M.P. and K.S. developed the protocol and wrote the original draft. S.P.M.P., K.S., D.C.J., S.J., and W.N. conducted the experiments and analyzed the data. All authors reviewed and revised the manuscript. W.N., J.M.P., and M.U. acquired the funding support and supervised the study.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2023.102629.
Contributor Information
Jack M. Parent, Email: parent@umich.edu.
Wei Niu, Email: wei.niu@utoledo.edu.
Data and code availability
This study did not generate or analyze any dataset or code.
References
- 1.Deng L., Mojica-Perez S.P., Azaria R.D., Schultz M., Parent J.M., Niu W. Loss of POGZ alters neural differentiation of human embryonic stem cells. Mol. Cell. Neurosci. 2022;120 doi: 10.1016/j.mcn.2022.103727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.González F., Zhu Z., Shi Z.D., Lelli K., Verma N., Li Q.V., Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qian K., Huang C.T.L., Chen H., Blackbourn L.W., 4th, Chen Y., Cao J., Yao L., Sauvey C., Du Z., Zhang S.C. A simple and efficient system for regulating gene expression in human pluripotent stem cells and derivatives. Stem Cell. 2014;32:1230–1238. doi: 10.1002/stem.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tidball A.M., Dang L.T., Glenn T.W., Kilbane E.G., Klarr D.J., Margolis J.L., Uhler M.D., Parent J.M. Rapid Generation of Human Genetic Loss-of-Function iPSC Lines by Simultaneous Reprogramming and Gene Editing. Stem Cell Rep. 2017;9:725–731. doi: 10.1016/j.stemcr.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haenfler J.M., Skariah G., Rodriguez C.M., Monteiro da Rocha A., Parent J.M., Smith G.D., Todd P.K. Targeted Reactivation of FMR1 Transcription in Fragile X Syndrome Embryonic Stem Cells. Front. Mol. Neurosci. 2018;11:282. doi: 10.3389/fnmol.2018.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain A., Stack O., Ghodrati S., Sanchez-Conde F.G., Ukachukwu C.U., Salwi S., Jimenez-Vazquez E.N., Jones D.K. KCNH2 encodes a nuclear-targeted polypeptide that mediates hERG1 channel gating and expression. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2214700120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tristan C.A., Hong H., Jethmalani Y., Chen Y., Weber C., Chu P.H., Ryu S., Jovanovic V.M., Hur I., Voss T.C., et al. Efficient and safe single-cell cloning of human pluripotent stem cells using the CEPT cocktail. Nat. Protoc. 2023;18:58–80. doi: 10.1038/s41596-022-00753-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armstrong L.C., Westlake G., Snow J.P., Cawthon B., Armour E., Bowman A.B., Ess K.C. Heterozygous loss of TSC2 alters p53 signaling and human stem cell reprogramming. Hum. Mol. Genet. 2017;26:4629–4641. doi: 10.1093/hmg/ddx345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y.H., Pruett-Miller S.M. Improving single-cell cloning workflow for gene editing in human pluripotent stem cells. Stem Cell Res. 2018;31:186–192. doi: 10.1016/j.scr.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Vallone V.F., Telugu N.S., Fischer I., Miller D., Schommer S., Diecke S., Stachelscheid H. Methods for Automated Single Cell Isolation and Sub-Cloning of Human Pluripotent Stem Cells. Curr. Protoc. Stem Cell Biol. 2020;55:e123. doi: 10.1002/cpsc.123. [DOI] [PubMed] [Google Scholar]
- 12.Gross A., Schoendube J., Zimmermann S., Steeb M., Zengerle R., Koltay P. Technologies for Single-Cell Isolation. Int. J. Mol. Sci. 2015;16:16897–16919. doi: 10.3390/ijms160816897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beers J., Gulbranson D.R., George N., Siniscalchi L.I., Jones J., Thomson J.A., Chen G. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 2012;7:2029–2040. doi: 10.1038/nprot.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh A.M. An Efficient Protocol for Single-Cell Cloning Human Pluripotent Stem Cells. Front. Cell Dev. Biol. 2019;7:11. doi: 10.3389/fcell.2019.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicholas C.R., Chen J., Tang Y., Southwell D.G., Chalmers N., Vogt D., Arnold C.M., Chen Y.J.J., Stanley E.G., Elefanty A.G., et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell. 2013;12:573–586. doi: 10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reichman D.E., Politch J., Ginsburg E.S., Racowsky C. Extended in vitro maturation of immature oocytes from stimulated cycles: an analysis of fertilization potential, embryo development, and reproductive outcomes. J. Assist. Reprod. Genet. 2010;27:347–356. doi: 10.1007/s10815-010-9416-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romito A., Cobellis G. Pluripotent Stem Cells: Current Understanding and Future Directions. Stem Cells Int. 2016;2016 doi: 10.1155/2016/9451492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu H.S., Jan M.S., Chou C.K., Chen P.H., Ke N.J. Is green fluorescent protein toxic to the living cells? Biochem. Biophys. Res. Commun. 1999;260:712–717. doi: 10.1006/bbrc.1999.0954. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze any dataset or code.