

Abstract
G protein-coupled receptors (GPCRs) represent ~30% of current drug targets. Ligand binding to these receptors activates G proteins and arrestins, which function in different signaling pathways. Because functionally selective or biased ligands preferentially activate one of these two groups of pathways, they may be superior medications for certain diseases states. The identification of such ligands requires robust drug screening assays for both G protein and arrestin activity. This unit describes protocols for assays that monitor reversible arrestin recruitment to GPCRs in living cells using either bioluminescence resonance energy transfer (BRET) or nanoluciferase complementation (NanoLuc). Two types of assays can be used – one configuration directly measures arrestin recruitment to a GPCR fused to a protein tag at its intracellular C-terminus, whereas the other configuration detects arrestin translocation to the plasma membrane in response to the activation of an umodified GPCR. Together, these assays are powerful tools for studying dynamic interactions between GPCRs and arrestins.
Keywords: G protein-coupled receptors (GPCRs), arrestin, bioluminescence resonance energy transfer (BRET)
INTRODUCTION
The G protein-coupled receptors (GPCRs), which are targeted by ~30% of drugs currently on the market, signal through both G proteins and arrestins (Kolb et al., 2022; Wingler & Lefkowitz, 2020). Under certain circumstances, it may be advantageous to selectively activate only one of these branches of signaling (Overington, Al-Lazikani, & Hopkins, 2006; Violin, Crombie, Soergel, & Lark, 2014). Despite certain inherent limitations of this approach (Gurevich & Gurevich, 2020; Seyedabadi, Gharghabi, Gurevich, & Gurevich, 2022), significant efforts are being made to identify compounds that preferentially activate either G proteins or arrestins (i.e., functionally selective or biased ligands). Some studies in cultured cells suggest that arrestin-dependent signaling can only be observed in the presence of functional G proteins (Grundmann et al., 2018), and findings in genetically modified mice suggest that physiological effects previously ascribed to arrestin-mediated signaling are actually mediated by G proteins (Kliewer et al., 2020; Kliewer et al., 2019). Nevertheless, biasing GPCR signaling has substantial potential in research and therapy. Commercial high-throughput assays available for identifying drug-dependent arrestin recruitment to GPCRs (e.g., PathHunter, DiscoveRx, Tango) employ modified receptors and arrestins fused to accessory protein fragments (van der Lee et al., 2009; Zhang & Xie, 2012). The main drawback of these assays is that the signals generated are irreversible, in sharp contrast to the highly dynamic and reversible biologically relevant interactions of arrestins with receptors in cells. These assay approaches are also limited by the fact that necessary modifications may alter normal signaling and that irreversible complementation- and proteolysis-based assays may artificially augment the pharmacological activity of the test agents (e.g., efficacy, kinetic profile).
Bioluminescence resonance energy transfer (BRET) and reversible complementation of nanoluciferase (NanoBiT), two techniques used to dynamically measure protein-protein interactions in living cells (Dixon et al., 2016; Hamdan, Percherancier, Breton, & Bouvier, 2006; Kocan & Pfleger, 2011; Pfleger, Seeber, & Eidne, 2006; Salahpour et al., 2012), have been used to monitor arrestin recruitment to GPCRs in real time. To study protein interactions using BRET, one partner is genetically fused with a donor protein, typically a variant of Renilla luciferase, and the other partner is fused to an acceptor protein, typically a variant of green fluorescent protein (GFP). When the donor and acceptor are within ~10 nm of each other, instead of emiting light at characteristic wavelength, the donor transfers energy to the acceptor, thereby exciting it. This results in emission from the acceptor protein at a longer wavelength. In the NanoBiT assay, one protein is fused to a small fragment of nanoluciferase (small bit, ~1.3 kDa) and the other protein is fused to the rest of nanoluciferase (large bit, ~18 kDa). The interaction between the two protein fragments results in the formation of a functional nanoluciferase, which is detected by its enzymatic activity. Both assays detect reversible arrestin-GPCR interactions in cells, thereby allowing us to monitor interactions in real time.
Described in this unit are two BRET-based assays that are useful for monitoring specific arrestin recruitment to a wide variety of GPCRs. Included are details of the traditional approach in which the receptor is labeled with a donor molecule (referred to here as the receptor-arrestin BRET assay; Basic Protocol 1), and a procedure that does not require receptor labeling (referred to here as the arrestin translocation BRET assay; Alternative Protocol 1). Two NanoBiT complementation-based assays are also described. In one, Small BiT (SmBiT) is fused to arrestin, and Large BiT (LgBiT) to the intracellular C-terminus of a GPCR (Basic Protocol 2). In the other variation, unmodified GPCRs are used, and the large bit is tethered to the plasma membrane (Alternative Protocol 2). In both protocols, HEK293 cells where the two nonvisual arrestin subtypes are knocked out (Alvarez-Curto et al., 2016; Grundmann et al., 2018) can be used, which eliminates the competition of endogenous arrestins with the expressed tagged ones and makes the expressed arrestin the only one present (which is particularly useful for studies of mutant arrestins). A Support Protocol is also provided for optimization of polyethylenimine (PEI) concentration for transfection.
BASIC PROTOCOL 1
RECEPTOR-ARRESTIN BRET ASSAY TO MEASURE LIGAND-INDUCED RECRUITMENT OF ARRESTIN TO RECEPTORS
A widely used cell-based assay for studying ligand-induced recruitment of arrestin to receptors utilizes receptors fused to a BRET donor (typically Renilla luciferase; e.g., Rluc or Rluc8) and arrestins fused to an acceptor (typically variants of GFP; e.g., YFP or Venus) (Hamdan, Audet, Garneau, Pelletier, & Bouvier, 2005) (Fig. 1A). Receptors are most often linked to Rluc8 (Loening, Fenn, Wu, & Gambhir, 2006). The Rluc must be fused to the cytoplasmic C-terminus of receptors, either directly or through linkers (e.g., the amino acids SGGGS) that facilitate proper folding of both proteins and that prevent steric hindrance between the receptor, luciferase, and bound arrestin. Acceptors fused to the N- or C-terminus of arrestins have been described previously (Hamdan et al., 2005; Klewe et al., 2008; Vishnivetskiy et al., 2011). We found that fusing Venus (or other modification of GFP) to the arrestin N-terminus is preferable, as N-terminal modifications do not interfere with the arrestin ability to bind cognate receptors, as evidenced by all solved structures of the arrestin-GPCR complexes, in which the N-terminus of bound arrestin points away from the receptor (Bous et al., 2022; Cao et al., 2022; Huang et al., 2020; Kang et al., 2015; Lee et al., 2020; Staus et al., 2020; Yin et al., 2019; Zhou et al., 2017).
Figure 1. BRET-based arrestin-GPCR interaction assay.
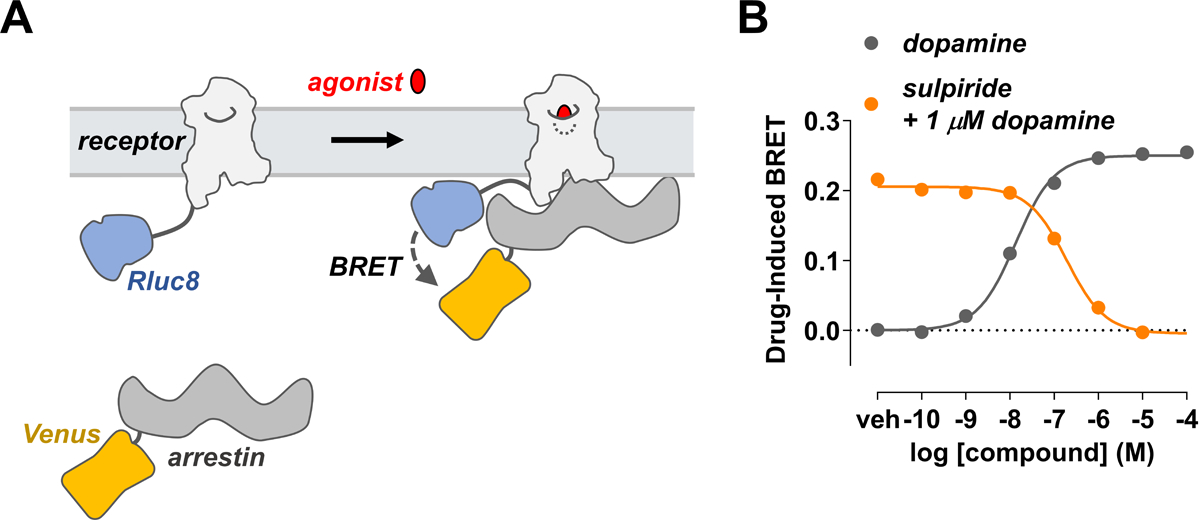
(A) Schematic of the receptor-arrestin BRET assay. A receptor is C-terminally fused to the donor Rluc8 and arrestin is fused to the acceptor Venus. Receptor activation recruits arrestin and results in an increased BRET between the donor and acceptor molecules. (B) Representative dose-response curves of a receptor-arrestin BRET assay. In the agonist mode, the increase in BRET signal with increasing dopamine concentration indicates that arrestin is recruited to the receptor D2R-Rluc8. In antagonist mode, the loss in BRET signal with increasing concentrations of the antagonist sulpiride indicates that antagonist inhibits recruitment of arrestin.
Both the BRET- and NanoBiT-based assays of the receptor-arrestin interactions can be used to screen compounds in a medium- to high-throughput manner in immortalized mammalian cell lines including HEK293 and CHO (Hamdan et al., 2005). Receptor and arrestin biosensors can be transiently or stably transfected, and can be co-expressed with G-protein receptor kinases (GRKs) to enhance agonist-dependent phosphorylation of the receptor and consequent arrestin recruitment (Clayton, Donthamsetti, Lambert, Javitch, & Neve, 2014). These protocols may be employed to study a wide variety of GPCRs. In BRET-based protocols of this unit, we describe the use of dopamine D2 receptor (D2R) fused C-terminally with Rluc8 (D2R-Rluc8) to recruit arrestin-3 with Venus fused to the N-terminus (Venus-arrestin3) in transiently transfected HEK293T cells (Fig. 1B).
Materials
HEK293T cells (ATCC, cat. no. CRL-3216), or HEK293 arrestin-2/3 knockout cells, fully confluent 10-cm plate
HEK293T culture medium (see recipe)
Dulbecco’s phosphate-buffered saline (DPBS; Cellgro, cat. no. 21–031-CV)
Trypsin (Cellgro, cat. no. 25–052-Cl)
- Mammalian expression plasmids (plasmids that are not commercially available can be obtained from the authors for non-profit use):
- Plasmid encoding donor-fused GPCR, e.g., pcDNA3.1-D2R-linker-Rluc8
- Plasmid encoding acceptor-fused arrestin, e.g., pIRES-puro-Venus-linker-arrestin-3
- Plasmid encoding GRK, e.g., pcDNA3.1-GRK2
- Empty vector plasmid, e.g., pcDNA3.1 (Life Technologies)
DMEM (Gibco, cat. no. 11965–092)
1 μg/μl polyethyleimine (PEI; see recipe)
Agonist stock: e.g., 22 mM dopamine in dH2O (dopamine hydrochloride available from Sigma-Aldrich, cat. no. H8502)
Antagonist stock: e.g., 44 mM sulpiride in DMSO (S-(–)-sulpiride available from Sigma-Aldrich, cat. no. S7771)
DPBS with 40 mg/L sodium bisulfite, pH 7.4, used to reduce dopamine oxidation (required only when using dopamine)
5 mM glucose in DPBS, pH 7.4
5 mM coelenterazine H in absolute ethanol (see recipe)
10-cm tissue-culture plates (BD Falcon, cat. no. 353003)
Compound plate, 96-well V-bottom plates (Greiner Bio-One, cat. no. 651101)
BRET assay plate, white 96-well flat bottom plates (Greiner Bio-One, cat. no. 655075) or Black/White 96-well Isoplate (PerkinElmer, cat. no. 6005030)
12-Channel multichannel pipettor, 50–300 μl (Labsystems Finnpipette, cat. no. Z368989) or 5–50 μl (Labsystems Finnpipette, cat. no. Z678031)
Pipette basin (USA Scientific, cat. no. 2320–2620)
Repeater Plus Pipettor (Eppendorf, cat. no. 022260201)
Plate reader for luminescence, fluorescence, and BRET detection (e.g., BMG Labtech, Pherastar FS, or Tecan, Infinite F500)
Software for data analysis (e.g., Microsoft Excel and GraphPad Prism)
-
NOTE: This protocol can be employed for either a single dopamine agonist curve or a sulpiride antagonist curve at a single dose of dopamine. The procedure can be scaled up, depending on the number of compounds for screening.
NOTE: All mammalian tissue culture must be conducted using aseptic techniques in a laminar flow hood. Cells should be maintained in an incubator at 37°C at 5% CO2.
Day 1. Seed cells
-
1
Aspirate media from a fully confluent 10-cm plate of HEK293T cells. Wash cells with 3 ml sterile DPBS and aspirate.
-
2
Add 1 ml trypsin to the plate and incubate at room temperature for 30 sec to 1 min to detach cells.
-
3
Add 5 ml HEK293T culture medium and transfer cells to a 15-ml conical tube.
-
4
Spin for 3 min at 600 × g, room temperature.
-
5
Aspirate medium, then suspend cells in 15 ml HEK293T culture medium.
-
6
Count the cells using a hemocytometer or cell counter.
-
7
Seed 3–4 × 106 HEK293T cells into a 10-cm tissue culture plate in a total volume of 10 ml.
Day 2. Transfect cells
-
8
After 24 hr of incubation, prepare plasmids for transient transfection. Combine the appropriate amounts of each plasmid, including donor-fused receptor, acceptor-fused arrestin, and, if desired, GRK plasmids, in a 1.5-ml microcentrifuge tube. Adjust the total amount of plasmid DNA to 20 μg using the empty vector plasmid.
For example, for the D2R-arrestin-3 BRET, use 0.2 μg D2R-linker-Rluc8; 8 μg Venus-linker-arrestin-3; 5 μg GRK2, if desired; and 6.8 μg pCDNA3.1 (or 11.8 μg pCDNA3.1 without GRK2).
The amount of each plasmid must be optimized to obtain the desired expression level as evidenced by luminescence and fluorescence on the day of the assay. The amounts of DNA will vary depending on the identity of the receptor-donor construct and the efficiency of the transient transfection. In general, the highest signal-to-noise ratio is obtained when the acceptor is in stoichiometric excess.
-
9
In a tissue culture hood, add non-supplemented DMEM to the tube containing plasmid DNA to a final volume of 500 μl.
-
10
Vortex the 1 μg/μl PEI stock solution.
-
11
Into a separate 1.5-ml microcentrifuge tube, add non-supplemented DMEM followed by PEI pipetted directly into the DMEM. The final volume of the DMEM/PEI solution should be 500 μl.
Use optimized PEI ratio as determined by PEI optimization (see Support Protocol).
-
12
Vortex the DMEM/PEI solution well.
-
13
Add 500 μl of DMEM/PEI solution to 500 μl of DMEM/DNA solution.
-
14
Vortex, then incubate at room temperature for 15 min.
-
15
Using a pipet, drip the 1 ml mixture into the medium in the 10-cm plate containing HEK293T cells. Gently rock the 10-cm plate back and forth to mix, and return the plate to the incubator.
Day 3
-
16
After ~20 hr, aspirate the media and replace with 10 ml fresh HEK293T cell culture media.
Day 4. Perform BRET assay to screen agonist activity in a concentration-dependent manner
NOTE: As none of the following steps require aseptic technique, they can all be performed outside of the tissue culture hood.
Prepare agonist dilution series
-
17
Dilute 3 μl of freshly prepared 22 mM dopamine (agonist) stock 100-fold to a final volume of 300 μl in DPBS with sodium bisulfite, to yield a maximum concentration of 220 μM.
-
18
Using this high-concentration solution, prepare a 10-fold dilution series to a minimum concentration of 22 pM (a total of seven dilutions). That is, serially transfer 30 μl to 270 μl DPBS with sodium bisulfite, mixing each dilution well.
45 μl of compound will be added to a final assay volume of 100 μl. Therefore, each dilution must be 2.22-fold higher than the final assay concentration.
-
19
Tranfer 60 μl of each agonist dilution to each of three wells of a V-bottom 96-well assay plate, and transfer 45 μl DPBS with sodium bisulfite to each of three wells to serve as the vehicle control.
This dilution series (24 wells in all) can either be apportioned into three columns or two rows of the assay plate.
Prepare antagonist dilution series
-
20
Dilute 1.5 μl of the 44 mM sulpiride stock 100-fold to a final volume of 150 μl in DPBS with sodium bisulfite, yielding a maximum concentration of 440 μM sulpiride.
22.5 μl of sulpiride will be added to a final assay volume of 100 μl. Thus, each dilution must be 4.44-fold higher than the final assay concentration.
The antagonist dilution series should yield final concentrations that range from ineffective to those that completely suppress receptor activity in the presence of agonist at its EC80 (i.e., the concentration that elicits 80% of the maximum response). If the inhibitory constant (Ki) of a particular compound at the receptor of interest is known, the dilution series should be adjusted based on this concentration. If the antagonist Ki is not known, concentrations ranging from 10 pM to 100 μM should be tested to ensure that antagonist activity is detected.
-
21
Using this concentrated solution prepare a 10-fold dilution series to a minimum concentration of 44 pM (a total of seven dilutions). That is, serially transfer 15 μl to 135 μl DPBS with sodium bisulfite, mixing each dilution well.
-
22
Aliquot 30 μl of each dilution into each of three wells of a V-bottom assay plate, and transfer 30 μl DPBS with sodium bisulfite to each of three wells to serve as the vehicle control.
This dilution series (24 wells in all) can either be apportioned into three columns or two rows of the assay plate. In all dilutions, the concentration of DMSO should be adjusted to 1%.
-
23
Prepare 1 ml of 4.44 μM dopamine in a 1.5-ml microcentrifuge tube. Aliquot 30 μl into each of 24 wells of a 96-well plate using the same pattern as that used for the sulpiride antagonist dilution series (step 20).
Dopamine prepared in step 23 will be used in step 35. Because some agonists or antagonists are dissolved in DMSO or other solvents that may affect the BRET assay, it is critical to adjust each dilution to ensure the concentration of solvent is the same throughout the entire concentration range examined.
Prepare cells
-
24
Aspirate the media from the transfected cells in the 10-cm plate.
-
25
Wash the plate with 3 ml DPBS, being careful not to detach the cells.
-
26
Aspirate the DPBS.
-
27
Add 2.5 ml DPBS containing 5 mM glucose to the cells, scrape the cells from the plate using the pipettor, and transfer the cells to a 15-ml conical tube.
-
28
Using a multichannel pipette, aliquot 45 μl of cells into each well of the BRET assay plate for each experiment (agonist and antagonist mode), either 3 columns or 2 rows.
Perform BRET experiment
-
29
Dilute 10 μl of 5 mM coelenterazine H into 990 μl DPBS, to a final concentration of 50 μM.
Coelenterazine H is light sensitive and must be stored in a dark tube.
Agonist mode
-
30
Use a repeating pipettor to inject 10 μl of 50 μM coelenterazine H into each well of the 96-well BRET assay plate containing cells.
Stagger the injection time for each well to allow for the amount of time it will take the plate reader to read one well (~1 to 2 sec).
Inject in the pattern that the plate reader will read across the plate (e.g., in a serpentine fashion across columns or rows).
-
31
Eight minutes after injecting the coelenterazine H, begin to inject 45 μl of dopamine (agonist) one row or column at a time using a multipipettor.
Stagger the injection into each row by the length of time it will take the plate reader to read an entire row or column (~10 to 20 sec).
-
32
Read the plate 2, 10, and 20 min after dopamine (agonist) injection using a BRET plate reader with detection filters or monochrometers set for Rluc8 (~485 nm) and Venus (~525 nm).
Antagonist mode
-
33
Inject 22.5 μl of sulpiride (antagonist) one row or column at a time using a multichannel pipettor and incubate for a desired amount of time (e.g., 10 min for sulpiride).
Stagger the injection into each row or column ~10 to 20 sec to allow for the amount of time it will take the plate reader to read each well. This pre-incubation allows the antagonist to bind and occupy the receptor. The desired incubation time is defined by equilibrium. In most cases the assay will be at pseudo-equilibrium, consistent with all functional assays.
Sulpiride pre-incubation ends at the time of addition of agonist (dopamine, in this case) in step 35. Thus, the antagonist incubation time overlaps with the 8-min incubation of coelenterazine H.
-
34
Using a repeater pipettor inject 10 μl of 50 μM coelenterazine H into each well of the 96-well BRET assay plate containing cells.
Stagger the injection time for each well to account for the amount of time it will take the plate reader to read one well (~1 to 2 sec).
Inject in the pattern that the plate reader will read across the plate (e.g., in a serpentine fashion across columns or rows).
-
35
Eight minutes after injecting coelenterazine H, begin to inject 22.5 μl of dopamine (agonist) prepared in step 23 one row or column at a time using a multichannel pipettor.
Stagger the injection to account for the amount of time it will take the plate reader to read each row or column (~10 to 20 sec).
-
36
Read the plate 2, 10, and 20 min after dopamine (agonist) injection using a BRET plate reader with detection filters set for Rluc8 (~485 nm) and Venus (~525 nm).
Data analysis
-
33
Export the raw data for each filter set, import into a Microsoft Excel spreadsheet, and ensure that the luminescence counts for the 485-nm filter (Rluc8 emission) are not saturated.
Generally, the 485-nm signal should range from ~105 to 106 light units. As this depends on the instrument and exact filter sets employed, the range must be determined empirically. Titrate the donor DNA and calculate the level that achieves an optimal agonist-induced change in BRET.
If signal is saturated, reduce the amount of plasmid DNA encoding for the donor-fused protein.
-
34
Calculate the BRET ratio for each well by dividing the 525-nm signal by that of the 485-nm signal.
-
35
Organize the data according to compound concentration.
-
36
Import the data into a points-only, XY-plot in GraphPad Prism, with three replicate values.
-
37
Fit the data to a non-linear regression curve. For fitting agonist or antagonist curves, use the response vs. log[agonist] or log[antagonist] fits, respectively.
-
38
To calculate the compound-induced effect, transform the Y-values using the Y = Y-K function, with the K-value being the bottom fit of the non-linear regression.
Examples of dopamine agonist and sulpiride antagonist curves are provided on Figure 1B.
BASIC PROTOCOL 2
RECEPTOR-ARRESTIN NANOBIT ASSAY TO MEASURE LIGAND-INDUCED RECRUITMENT OF ARRESTIN TO RECEPTORS
The study of GPCR interactions with signal transducers has been of great interest in the field of drug discovery due to the role of GPCRs in mediating a wide range of physiological processes. However, many methods for studying GPCR interactions with other proteins have been limited by the lack of sensitivity and the level of background signal. Recently, a novel assay technology called NanoLuc Binary Technology (NanoBiT) has emerged as a promising tool for studying arresitn-GPCR interaction in living cells with high sensitivity.
NanoBiT is a bioluminescence-based assay technology that uses NanoLuc luciferase (England, Ehlerding, & Cai, 2016), a small and bright luciferase derived from the deep-sea shrimp Oplophorus gracilirostris (Hall et al., 2012). NanoBiT is a complementation-based split luciferase system, where the small 11-amino acid subunit (Small BiT, SmBiT) and the larger 17.6 kDa subunit (Large BiT, LgBiT) are fused to the proteins of interest. Upon interaction between the target proteins, the SmBiT and LgBiT fragments are brought into close proximity, allowing for efficient complementation and reconstitution of the functional luciferase enzyme (Fig. 2).
Figure 2. Overview of the NanoBiT assay for arrestin-GPCR recruitment.
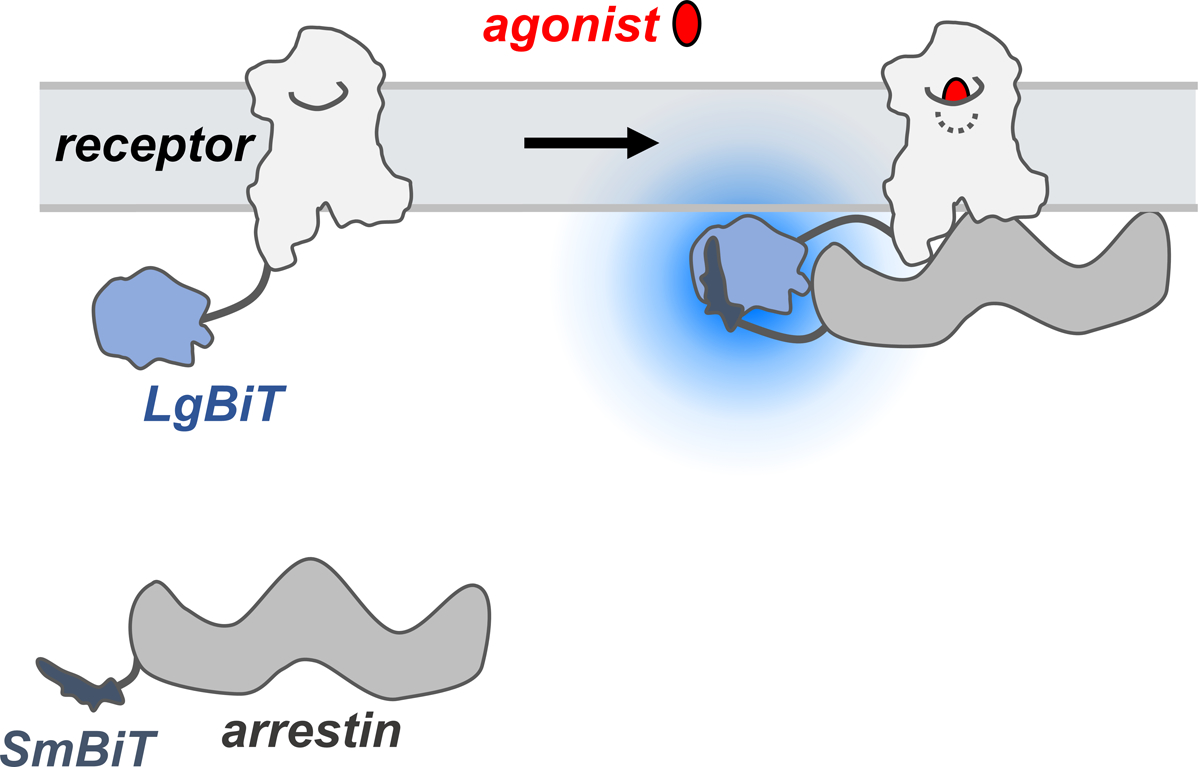
The SmBiT and LgBiT fragments are fused to arrestin and GPCR, respectively, and fusion proteins are expressed in cells. Upon interaction between the fusion partners, the LgBiT and SmBiT fragments complement each other, forming a functional enzyme that emits a luminescent signal.
The NanoBiT assay technology has been successfully applied to the study of protein-protein interactions (Reyes-Alcaraz et al., 2022). NanoBiT is very sensitive due to the high activity of NanoLuc. This is particularly important in the study of GPCR interactions with signal transducers. The recombination of NanoBiT is reversible, allowing for the real-time monitoring of reversible protein-protein interactions in live cells. Moreover, the ease of use and scalability of NanoBiT make it an attractive tool for high-throughput screening of GPCR ligands. In this NanoBiT part of this unit, we describe the use of β2-adrenergic receptor (β2AR) fused C-terminally with LgBiT (β2AR-LgBiT) to recruit SmBiT fused to the N-terminus of arrestin-3 (SmBiT-arrestin3) in transiently transfected HEK293 arrestin-2/3 knockout cells (Fig. 3).
Figure 3. Representative data for arrestin-3 recruitment to β2AR in the NanoBiT assay.
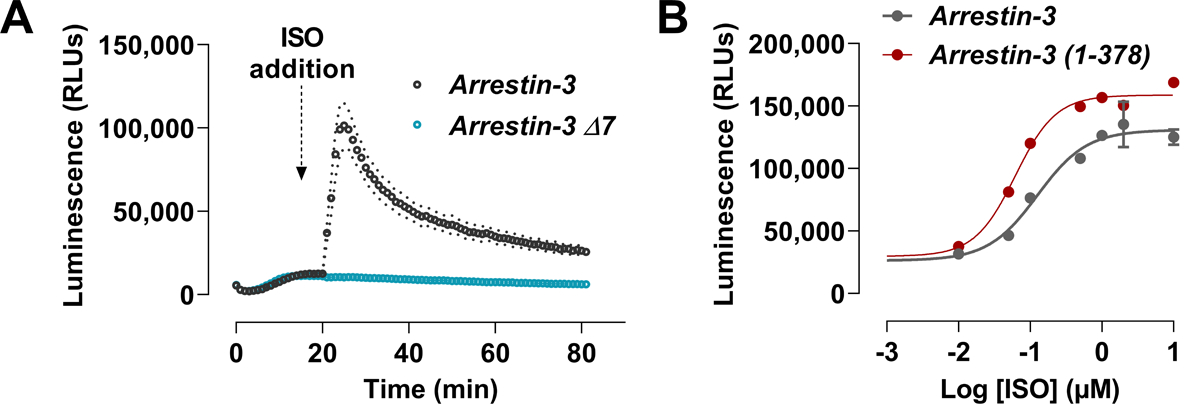
HEK arrestin-2/3 KO cells were transfected with SmBiT-arrestin-3 and β2AR-LgBiT. 48 h post-transfection, Nano-Glo reagent was added, and the luminescence was measured for 20 minutes. Isoproterenol was added after the 20-minute read of the background signal and the luminescence was measured for 60 minutes. (A) Real-time recruitment of arrestin-3 to β2AR. Arrestin-3 Δ7 was used as the negative control, as it does not bind to GPCR. (B) Dose-response curve that compares the recruitment of wildtype arrestin and the “enhanced” arrestin-3 mutant with C-terminus deleted to β2AR upon isoproterenol activation.
Materials:
HEK293 arrestin-2/3 knockout cells, fully confluent 10-cm plate
HEK293 complete culture medium (see recipe)
Dulbecco’s phosphate-buffered saline (DPBS; GIBCO, cat. no. 14190144)
Trypsin-EDTA solution (0.05%, GIBCO, cat. no. 25300062)
Mammalian expression plasmids (plasmids are commercially available from Promega or obtained from the authors for non-profit use):
Plasmid encoding fused GPCR, e.g., pcDNA3-b2AR-linker-LgBiT
Plasmid encoding fused arrestin, e.g., pcDNA3-SmBiT-linker-arrestin-3
Empty vector plasmid, e.g., pcDNA3 (Life Technologies)
Agonist stock: e.g., 10 mM isoproterenol (agonist of β2AR) in DPBS (isoproterenol hydrochloride available from Sigma-Aldrich, cat. no. I6504)
Nano-Glo live cell substrate system (Promega, cat. no N2011) or 5 mM coelenterazine H in DMSO (NanoLight technology, cat. no. 301) (see recipe)
10-cm tissue-culture plates (BD Falcon, cat. no. 353003)
NanoBiT assay measuring plate, white 96-well flat bottom plates (Thermo, cat. no. 136101)
Trans-Hi™ transfection reagent (FormuMax Scientific, cat. no. F90101TH10) or another transfection reagent
8-Channel multichannel pipettor, 50–300 μl (Labsystems Finnpipette, cat. no. Z368989) or 5–50 μl (Labsystems Finnpipette, cat. no. Z678031)
Reagent Reservoirs (Polystyrene, Thermo, cat. no. 8093–11)
Plate reader for luminescence detection (e.g., BioTek Neo plate reader)
Software for data analysis (e.g., Microsoft Excel and GraphPad Prism)
The following protocol is given for measuring arrestin-3 recruitment to β2AR. The cell seeding and transfection was performed in 6-well plate. Adjust cell numbers proportionately for different size plates.
Day 1. Plating cells
Seed appropriate numbers of cells to reach > ~80–90% confluency on the next day.
Aspirate media from a fully confluent 10-cm plate of HEK293 cells. Wash cells with 5 ml sterile DPBS (Ca2+ and Mg2+ free) and aspirate.
Add 1 ml trypsin to the plate and incubate at room temperature for 30 sec to 1 min to detach cells.
Add 5 ml complete culture medium to inactivate the trypsin and gently pipette cells to obtain cell suspension.
Centrifuge cells for 5 minutes at 300 × g. Carefully resuspend cell pellets in 5 ml of complete culture medium.
Count the cells using a hemocytometer or cell counter.
-
Dilute cells at a density of 0.1–0.2 × 106 cells/ml in completed culture medium. Transfer 3 ml cell suspension to each well of 6-well tissue culture plate.
If use 96-well plate, only use inner 60 wells for cell seeding. Add the DPBS/culture medium in outside wells to avoid the evaporation across the plate during overnight incubation.
Day 2. Transfect cells
Use the Table 1 for scaling up/down with different culture format. All amounts and volumes are on a per well basis. For direct binding assay, LgBiT and SmBiT vectors comprise ~5–20% of the DNA and the rest is carrier DNA (empty vector).
Table 1.
Scaling Up or Down for transfection.
| Plate Size | Day 2 | Day 3 | |||||
|---|---|---|---|---|---|---|---|
| SmBiT-Arr3 (ng) | GPCR-LgBiT (ng) | Carrier DNA (ng) | Total DNA (μg) | Trypsin-EDTA (ml) | Resuspending medium (ml) | Incubation medium (ml) | |
| 6-well | 100 | 20 | 880 | 1 | 0.5 | 2 | 2.5 |
| 24-well | 50 | 10 | 440 | 0.5 | 0.2 | 0.8 | 1 |
| 96-well | 10 | 5 | 85 | 0.1 | - | - | - |
30–60 minutes prior to transfection, replace the medium in the culture dish with 1 ml fresh complete culture medium.
Dilute 0.5 μg DNAs containing NanoBiT fusion plasmids in 50 μl of serum-free DMEM with high glucose, vortex the solution for 5 sec and spin down.
Dilute 1.5 μl Trans-Hi™ transfection reagent (at a 3:1 lipid-to-DNA ratio) in 50 μl serum-free DMEM. Pipette the solution to mix well.
Add the diluted Trans-Hi™ immediately to the diluted DNA solution. Pipette the solution to mix well.
Incubate the mixture for 10 minutes at room temperature to allow the lipid-DNA complexes to form.
Dropwise add the mixture onto medium and gently swirl the plate to ensure an even distribution.
12–16 h post-transfection, replace the transfection medium with fresh complete culture medium.
Day 3. Transfer cells into 96-well plate
If cells were seeded in 96-well plate in Day 1, replace the culture medium with 100 μl incubation medium per well. Return the plate to the CO2 incubator overnight. If cells were seeded in other than 96-well plate, aspirate the culture medium from the cell culture dishes. Wash cells with 1ml DPBS.
Add 0.5 ml of trypsin and incubate in a CO2 incubator at 37 °C for 1–2 min, ensuring cells are detached.
-
Add 2 ml of resuspending medium to neutralize trypsin, gently pipetting a few times to break up any cell clumps. Then centrifuge at 400 × g for 5 min and discard the supernatant.
The medium containing up to 10% FBS is helpful to minimize the cell damage when breaking up the cell clumps.
-
Resuspend cell pellets in 2.5 ml of incubation medium. Transfer 100 μl cell suspension into each well of the 96-well plate. Incubate cells overnight (> 12 h) before measurement.
The incubation medium is used for serum-starvation. If serum-starvation is not required, complete culture medium can be used.
As the trypsin may cleave the cell surface proteins (including LgBiT-fused GPCR), it is recommended to incubate cells overnight to allow for receptor recovery before the measurement.
(Optional) Collect the remaining cells by 400 × g for 5 min and lyse cells in NP-40 cell lysis buffer. The samples can be used to evaluate the expression of arrestin and GPCR using western blot.
Day 4. Perform the NanoBiT assay to measure the agonist-induced arrestin-GPCR interactions
NOTE: As none of the following steps require aseptic technique, they can all be performed outside of the tissue culture hood.
Preparation of the substrate dilution and agonist dilution
Either Nano-Glo or coelenterazine H can be used as substrates in NanoBiT assay. Nano-Glo reagent contains furimazine, which was found to be more stable in cell culture medium and to produce less autoluminescence compared to coelenterazine H (Hall et al., 2012). The substrate and agonist dilution need to be prepared fresh on the day of the measurement. Use Table 2 to calculate the proper dilution for substrate and agonist. An example for preparing stock solution for 30 wells assay is given below:
Table 2.
Preparation of substrate and agonist dilution for NanoBiT assay
| Volume / well (μl) | Final Concentration | Stock Concentration | ||
|---|---|---|---|---|
| Medium | 100 | - | - | |
| Substrate dilution | 20 | 25 μM | 5 mM | Coelenterazine H |
| 1/120 dilution | Original stock | nano-Glo | ||
| Agonist dilution | 5 | 10 μM or as desired | 10 mM | |
| Total Volume | 125 | - | - | |
If using Nano-Glo reagent, equilibrate the LCS dilution buffer (provided in the kit) at ambient temperature, then add 30 μl stock solution to 570 μl LCS dilution (20-fold dilution).
If using coelenterazine H, dilute 18 μl stock substrate in 582 μl DPBS.
-
To prepare the agonist dilution for 30-well assay (10 μM final after adding to the well), dilute 3.75 μl (0.125 μl/well × 30 wells) stock agonist in 146.25 μl DPBS.
Adding the agonist solution will result in a dilution of the substrate concentration, leading to a slight reduction in the luminescence signal. Therefore, it is advisable to add small volumes of the agonist solution to prevent any significant fluctuations in the luminescence signal. In case the fluctuations become a problem, it is recommended to add substrate to the agonist solution to maintain constant substrate concentration in the well.
Measurement
If possible, pre-warm the plate reader to 37 °C to ensure optimal temperature conditions for the assay.
Transfer the substrate dilution and agonist solution into separate reagent reservoirs and wrap them to prevent exposure to light and evaporation.
-
Add 20 μl diluted substrate in each well using multi-channel pipettor. Gently shake or swirl the plate to ensure even distribution. Read the total luminescence every minute for 20 min until the signal reaches equilibrium across the entire plate.
The last 2–3 reads (at 18–20 min) reflect the baseline signal (including basal level interaction and background signal) and will be used for normalization described in data analysis.
After the 20 reads, add 5 μl diluted agonist into each well by multichannel pipettor or the plate reader injector, then immediately monitor the luminescence every 30 sec for 30–60 min.
Data analysis
Using the baseline signal to normalize the interaction response of each well can help reduce variability resulting from differences in cell numbers or transfection efficiency between wells, among other factors.
Calculate the average of the last three measurements from the well prior to agonist addition.
Use the measurements of agonist-induced arrestin recruitment (reads after 20 mins) divided by the baseline signal.
ALTERNATIVE PROTOCOL 1
BRET ASSAY TO MEASURE LIGAND-INDUCED RECRUITMENT OF ARRESTIN TO PLASMA MEMBRANE
Fusion of a relatively large protein directly to the C-terminus of the receptor may affect receptor function in the receptor-arrestin BRET assay, as in the commercially available PathHunter and Tango assays. For this reason, we developed a BRET-based assay that does not require receptor modifications (Clayton et al., 2014). Rather than being directly fused to the receptor, the BRET sensor is fused to an unrelated plasma membrane marker. Translocation of arrestin to the plasma membrane due to its interactions with activated receptor increases the proximity of arrestin to this marker, resulting in increased resonance energy transfer. Arrestin translocation to the plasma membrane is dynamically regulated, as receptor antagonists can reverse agonist-induced arrestin translocation. Moreover, this assay is capable of characterizing GPCR ligands with varying efficacy, potency, and kinetic profiles, potentially in a high-throughput manner.
Specifically, in immortalized mammalian cell lines (e.g., HEK293), unlabeled GPCRs are co-expressed with: Rluc8-arrestin-3 fused at its C-terminus with the weak helper peptide Sp1 (Rluc8-arrestin3-Sp1); and a membrane marker composed of a doubly palmitoylated fragment of GAP43 linked to citrine and the weak helper peptide SH3 through a serine- and glycine-rich linker (mem-linker-citrine-SH3). Though the Sp1 and SH3 helper peptides, adapted from the helper-interaction FRET (hiFRET) system (Grunberg et al., 2013), are not essential for the detection of arrestin translocation to the plasma membrane, they enhance the interaction between arrestin recruited to the plasma membrane by the receptor and the membrane marker. This modification only modestly increases the basal signal between the plasma membrane marker and arrestin, while significantly increasing the dynamic range of the assay. As in the receptor-arrestin BRET assay (see Basic Protocol), GRKs can be co-expressed to enhance activation-induced receptor phosphorylation and consequent arrestin recruitment.
Unlike the commercially available assays and the receptor-arrestin BRET assay described in Basic Protocol 1, this procedure can be employed to screen any GPCR by simply co-expressing the unmodified receptor with the arrestin and plasma membrane biosensors, making it unnecessary to generate and optimize a fusion receptor. The translocation of arrestin-3 to the plasma membrane by D2R is used as an example in the protocol described below (Fig. 4).
Figure 4. BRET-based arrestin translocation assay.
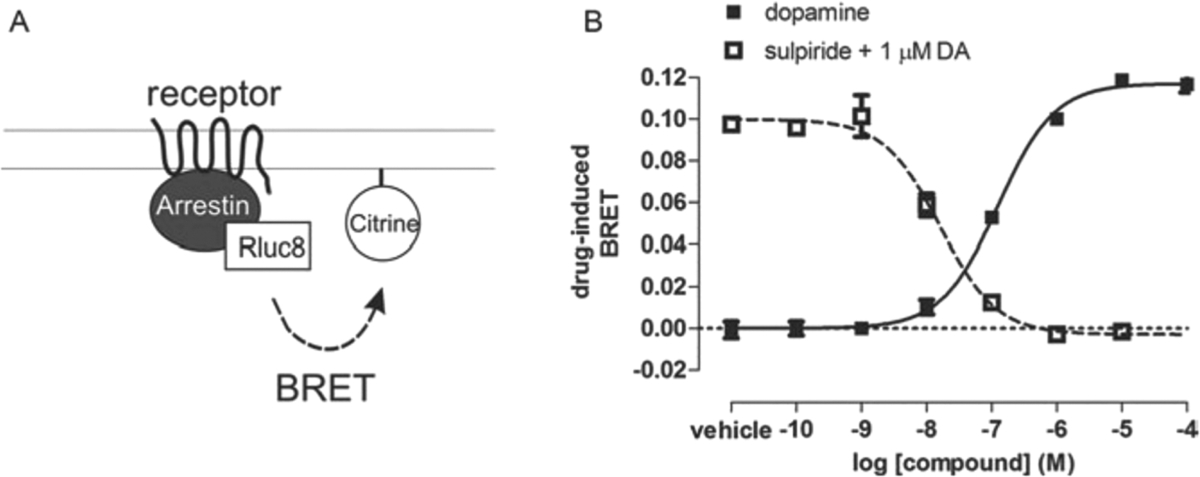
(A) Schematic of arrestin translocation BRET assay. Arrestin is fused to Rluc8 and an unrelated plasma membrane marker is fused to the acceptor citrine. Activation of the unmodified receptor recruits arrestin to the plasma membrane, which increases BRET between the donor and acceptor molecules. (B) Representative concentration-response curves of an arrestin translocation BRET assay.
Additional Materials (also see Basic Protocol)
Mammalian expression plasmids:
Plasmid encoding for GPCR (e.g., pcDNA3.1-D2R)
Plasmid encoding for Rluc8-Arrestin-3-Sp1
- Plasmid encoding for mem-linker-citrine-SH3
- Complete steps 1 to 7 of the Basic Protocol.
-
Combine the appropriate amounts of each plasmid, including donor Rluc8-Arrestin-3-Sp1 fusion, acceptor mem-linker-citrine-SH3, D2R, and, if desired, GRK plasmids, in a 1.5-ml microcentrifuge tube. Adjust the total amount of plasmid DNA to 20 μg using the empty vector plasmid.For example, use 0.25 μg Rluc8-Arrestin-3-Sp1; 5 μg mem-linker-citrine-SH3; 2 μg D2R; 5 μg GRK2, if desired; and 4.8 μg pCDNA3.1 (or 9.8 μg pCDNA3.1 empty vector if GRK2 is excluded).The amount of each plasmid must be optimized to obtain the appropriate expression level as evidenced by luminescence and fluorescence on the day of the assay. The amounts of DNA will vary depending on the identity of the receptor-donor construct and the efficiency of the transient transfection. In general, the highest signal-to-noise ratio is obtained when the acceptor is in stoichiometric excess.
- Complete steps 9 to 42 of the Basic Protocol. Examples of dopamine agonist and sulpiride antagonist curves are provided in Figure 4.
ALTERNATIVE PROTOCOL 2
NANOBIT ASSAY TO MEASURE LIGAND-INDUCED RECRUITMENT OF ARRESTIN TO PLASMA MEMBRANE
One limitation of the NanoBiT assay is that the C-terminus of GPCR must be fused to a NanoBiT fragment, which could interfere with receptor interactions with downstream partners, such as G protein-coupled receptor kinases (GRKs) and arrestins. In addition, modifications of a GPCR may alter its expression, binding selectivity, and other functional parameters.
To overcome these limitations, an alternative approach, called indirect NanoBiT or membrane-tethered NanoBiT, has been developed (Hauge Pedersen et al., 2021; Spillmann et al., 2020). In the indirect NanoBiT assay, one of the NanoBiT fragments is expressed separately anchored to the plasma membrane instead of being attached to a GPCR. When arrestin that is fused to the complementary NanoBiT (SmBiT) translocates from the cytoplasm to the membrane to bind active GPCR, the two nanoBiT fragments recombine into a functional NanoLuc that generates the signal. This approach allows for the use of unmodified GPCRs, thus avoiding any potential disruption of their function or localization associated with fusion of the NanoBiT fragments. This approach also enables the study of GPCRs that cannot be easily fused to the NanoBiT fragments. Both direct and indirect assays are performed using the same protocol, except that instead of using a GPCR that is fused to the large bit at the C-terminus, two plasmids are used — one encoding unmodified GPCR of interest and the other encoding membrane-tethered LgBiT. The translocation of arrestin-3 to the plasma membrane by D2R is used as an example in the protocol detailed below (Fig. 5).
Figure 5. Comparison of direct and indirect NanoBiT assays in measurement of arrestin-3 recruitment to D2R.
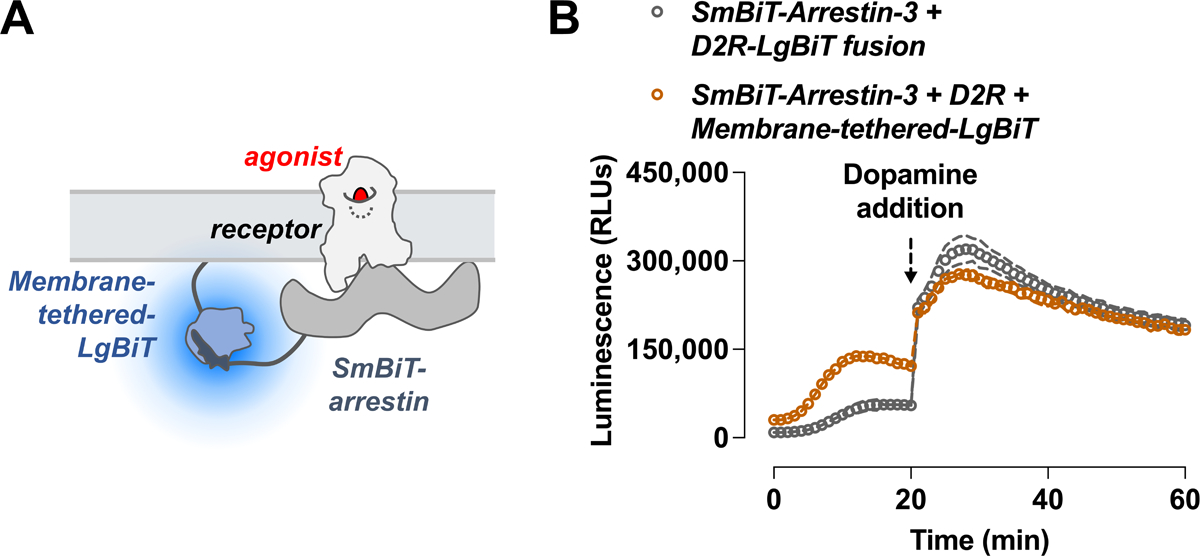
(A) Schematic of arrestin translocation NanoBiT assay. Arrestin is fused to SmBiT and an unrelated plasma membrane marker is fused to the LgBiT. Activation of the unmodified receptor recruits arrestin to the plasma membrane, which increases complementation. (B) HEK293 arrestin-2/3 KO cells were transfected with SmBiT-arrestin-3 and either a D2R fused with LgBiT (blue, direct assay) or a wildtype D2R with LgBiT anchored to the plasma membrane (brown, indirect assay).
The indirect NanoBiT assay has certain disadvantages. First, the indirect assay does not detect the actual binding arrestin to receptor but the translocation of arrestins to the plasma membrane. Second, the expression level of the membrane-tethered large bit may affect the assay sensitivity, and therefore has to be carefully adjusted for maximum signal-to-noise ratio. Additionally, the orientation of the arrestin with respect to the plasma membrane may affect the assay readout. All of these factors introduce higher background signal (Fig. 5B).
In conclusion, both direct and indirect NanoBiT assays have their own advantages and limitations, and the choice of assay depends on the research objectives and the properties of the GPCR of interest. The indirect NanoBiT assay provides a valuable alternative approach, especially in cases where the direct fusion of the NanoBiT fragments is not desirable or feasible.
SUPPORT PROTOCOL
OPTIMIZATION OF POLYETHYLENIMINE (PEI) CONCENTRATION FOR TRANSFECTION
Different lots of PEI, even from the same manufacturer, vary in their efficiency of transfection. Because PEI is cytotoxic to cells, the DNA:PEI ratio must be optimized for each lot to maximize transfection efficiency and minimize cytotoxicity.
Materials
See Basic Protocol
Day 1. Seed cells
-
1
Seed each of six 10-cm plates with 3–4 × 106 HEK293T cells.
Day 2. Transfect cells
-
2
Prepare six 1.5-ml microcentrifuge tubes, each containing 20 μg of plasmid DNA, as described in steps 8 and 9 of the Basic Protocol.
-
3
Prepare six PEI:DNA mixtures by adding 20, 40, 60, 80, 100, or 120 μl PEI to each tube containing 20 μg of plasmid DNA, and use each mixture to transfect a 10-cm plate of cells as described in steps 10 to 15 of the Basic Protocol.
Day 3. Change medium
-
4
After ~20 hr, aspirate the media and replace with 10 ml fresh HEK293T cell culture media.
Day 4. Assess transfection efficiency
-
5
Before preparation of cells, take note of the cell health for each condition. At higher PEI levels cells may become unhealthy (indicated by round shape and small size) or detach from the 10-cm plate.
-
6
Prepare cells as described in steps 24 to 27 of the Basic Protocol, and aliquot 45 μl of cells from each transfection into each of three wells of a 96-well BRET assay plate (18 wells total).
-
7
Adjust each well to a final volume of 90 μl by adding 45 μl DPBS with sodium bisulfite.
-
8
Inject 10 μl 50 μM coelenterazine H in DPBS with sodium bisulfite.
-
9
Eight minutes after coelenterazine H injection, read the plate using a BRET plate reader with detection filters set for Rluc8 (~485 nm) and Venus (~525 nm).
-
10
Compare the luminescence counts from the 485-nm filter, using these data as a measure of transfection efficiency across the PEI titration range. Select the PEI:DNA ratio that yields the highest luminescence with the least cell death for all experiments using this lot of PEI.
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps unless otherwise specified. For common stock solutions, see appendix 2a.
BRET assays
Coelenterazine H, 5 mM
-
Prepare 5 mM coelenterzine H (Dalton, cat. no. DC001437; or NanoLight, cat. no. 301) in absolute ethanol under low light conditions, and aliquot into 1.5 ml microcentrifuge tubes. Fill each tube with sufficient argon or nitrogen gas (10 to 15 sec per tube) to cover the solution and displace the atmospheric air. Seal each tube with Parafilm and store for up to 3 years at −20°C in the dark.
Coelenterazine H is sensitive to temperature, light and oxygen and will lose activity over time. Because the amount of coelenterzine H purchased is usually much greater than that required for a single experiment, it must be stored properly to avoid decomposition.
HEK293T culture medium
DMEM (Gibco, cat. no. 11965–092) supplemented with:
10% fetal bovine serum (FBS; Atlanta Biologics, cat. no. D13056)
100 U/ml penicillin-streptomycin (Cellgro, cat. no. 30–002-Cl)
Store for up to 2 months at 4°C
Polyethylenimine (PEI), 1 μg/μl
Suspend 25 mg of PEI (linear, MW 25,000; Polysciences, cat. no. 23966–2); in 25 ml dH2O, and stir continuously with a stir bar. Add concentrated HCl dropwise to the suspension to adjust the pH to less than 2.0. Stir until the PEI is dissolved (30 min to 1 hr), maintain a pH of less than 2.0. Add 10 N NaOH dropwise to the solution to adjust the pH to 7.2. Filter sterilize the solution through a 0.2 micron filter in a tissue culture hood. Dispense 1 ml aliquots into 1.5-ml microcentrifuge tubes. Store for up to 2 years at −20°C.
NanoBit assays
Complete culture medium
DMEM (high glucose, GlutaMAX supplement, cat. no. GIBCO 10566–016) supplemented with:
10% fetal bovine serum
-
1% Penicillin-Streptomycin
Optional: Sodium pyruvate can be added as an additional energy source.
Resuspending medium
DMEM with high glucose (4.5g/L) and without phenol red (GIBCO, cat. no. 31053–028 or 21063–029) supplemented with:
10% fetal bovine serum
Incubation (cell starvation) medium
DMEM with high glucose and without phenol red (GIBCO, cat. no. 21063–029) supplemented with:
-
1% Penicillin-Streptomycin.
A buffered medium (e.g., containing HEPES) is recommended because the NanoBiT assay is performed outside of CO2 incubator.
5 mM coelenterazine H
Dissolve the powder (NanoLight, cat. no. 399; high purity compound from any source is fine) in DMSO or ethanol to make 5 mM stock. Store at −80°C in dark brown tube to protect from light.
NP-40 cell lysis buffer
50 mM Tris-HCl (pH 7.4)
150 mM NaCl
1% NP-40 (RPI, cat. no. N32000)
5 mM EDTA
COMMENTARY
Background Information
Arrestin and drug development
Coupling of GPCRs to heterotrimeric G proteins initiates a number of downstream signaling processes. These receptors also recruit arrestins, which terminate G protein signaling and facilitate receptor internalization (Lefkowitz, 2013). Once recruited to GPCRs, arrestins can act independently of G proteins as scaffolds for a variety of signaling molecules (Lefkowitz, 2013). So-called functionally selective or biased ligands make GPCRs preferentially activate either G proteins or arrestins, and may be superior medications for treatment of conditions in which one pathway downstream of a target receptor is associated with efficacy and the other with side effects (Violin et al., 2014). Significant efforts are being made to develop such selective ligands, some of which have already entered clinical trials (TRV027, TRV130; cf. Trevena; clinicaltrials.gov).
There are a number of commercially available assays for screening G protein activity, both at and downstream of the activated receptor-G protein (Zhang & Xie, 2012). However, screening for arrestin activity is more difficult because of the paucity of reliable readouts specifically downstream of arrestin. Most available arrestin assays (BRET-based assays; PathHunter, DiscoveRx; Tango, Life Technologies) rely on monitoring arrestin recruitment to receptor instead (Zhang & Xie, 2012). Commercially available systems, including PathHunter and Tango, require the use of modified receptors that are fused to accessory proteins that generate a pseudo-irreversible signal upon recruitment. For example, in the PathHunter assay two inactive fragments of β-galactosidase interact to form an active enzyme upon arrestin recruitment to the receptor (van der Lee et al., 2009). The affinity between the accessory proteins may artificially enhance the stability of the receptor-arrestin complex. Additionally, protein complementation is in most cases irreversible. This makes it difficult to accurately characterize pharmacological properties such as efficacy, potency, and kinetics relative to reference agonists, because these assays are performed under non-equilibrium conditions.
BRET as a tool to study GPCR pharmacology
Bioluminescence resonance energy transfer (BRET) is a photophysical phenomenon that involves resonance transfer of energy from a donor (usually the product of a luciferase-catalyzed oxidation reaction) to a nearby acceptor (most often a fluorescent protein). In nature, BRET allows marine organisms to emit bright green rather than blue light (Morin & Hastings, 1971; Morise, Shimomura, Johnson, & Winant, 1974). For pharmacological research BRET has been employed to detect dynamic protein-protein interactions, to monitor conformational changes within individual proteins and multi-protein complexes, and to monitor changes in cellular activity and metabolism (Marullo & Bouvier, 2007). It has been especially useful for studying the structure and function of G protein-coupled receptors (GPCRs), as well as their interactions with G proteins and arrestins. The BRET technique is suitable for medium- to high-throughput drug screening.
Generally, implementation of BRET assays is straightforward, as both donor and acceptor can be genetically encoded as fusions with interacting proteins. BRET assays are also relatively easy to perform, and results can be quantified without specialized equipment. Its extreme sensitivity is another advantage of the BRET assay. Because BRET is based on luminescence, there is no background signal from cellular autofluorescence, so that photons can be collected for long periods of time without a significant accumulation of noise. Another advantage is that unlike Förster (or fluorescence) resonance energy transfer (FRET), there is no possibility of unintended direct excitation of the acceptor, thereby making correction procedures unnecessary. Additionally, because BRET is ratiometric, it is insensitive to variables such as cell number and transfection efficiency. As a consequence, photon emission ratios are measured with high precision and reproducibility, allowing detection even with very weak BRET efficiency.
Critical Parameters and Troubleshooting
Generating fusion proteins
The most widely used BRET-based assay, described above (Basic Protocol), requires the fusion of a BRET donor at the C-terminus of receptors, which, as in constructs used for the commercially available PathHunter and Tango assays, can affect receptor function. BRET requires the fusion of relatively large probes (luciferases or fluorescent proteins) to proteins of interest, and the presence of these probes can change the behavior of the proteins being studied in an unpredictable manner. This makes it necessary to ascertain that luciferase-fused receptors are still active, i.e., can activate G proteins comparably to unfused wildtype receptors (Gimenez, Babilon, Wanka, Beck-Sickinger, & Gurevich, 2014). Full receptor functionality is more likely when receptors are connected to luciferase via a flexible linker (Hamdan et al., 2006).
Poor expression or disruption of the activity of receptor-Rluc fusions requires further optimization, by extending the linker length, introducing a linker with different physical properties (e.g., a rigid linker composed of one or more EAAAK repeats), or by exchanging the donor and acceptor on the receptor and arrestin. These modifications may facilitate the proper folding of the receptor and BRET sensor domains of the fusion protein. However, they can also affect the proximity and orientation of donor and acceptor moieties relative to one another, which determines the efficiency of energy transfer between the receptor and arrestin fusion proteins and, thus, the dynamic range of the assay.
N- or C-terminal acceptor-fused arrestin constructs have been developed that allow for characterization of arrestin recruitment in a subtype-specific manner (Hamdan et al., 2005; Klewe et al., 2008; Vishnivetskiy et al., 2011). Although arrestins with C-terminal GFP or Venus fusions are recruited to receptors, caution must be exercised since the release of the C-terminus of arrestin plays an important role in arrestin transition into high-affinity receptor-binding conformation (Asher et al., 2022; Celver, Vishnivetskiy, Chavkin, & Gurevich, 2002; Gurevich, 1998; Hirsch, Schubert, Gurevich, & Sigler, 1999; Kovoor, Celver, Abdryashitov, Chavkin, & Gurevich, 1999; Shukla et al., 2013; Vishnivetskiy et al., 1999). Perturbation of the regulation of the arrestin C-terminus release may increase basal interactions between arrestin and clathrin or AP-2 (Kim & Benovic, 2002; Nobles, Guan, Xiao, Oas, & Lefkowitz, 2007).
Specific and nonspecific BRET
The energy donor and acceptor must be in close proximity (~10 nm) for BRET to occur. Therefore, as a general rule BRET signals are generated in cells when either: 1) donor- and acceptor-fused proteins are specifically associated, either directly or as part of a macromolecular complex, or 2) when both donor and acceptor are associated with the same membrane, even without a stable direct or indirect interaction between the two proteins. The BRET signals produced by these two mechanisms are referred to as “specific” and “nonspecific” BRET, respectively, with the latter also sometimes referred to as “bystander” BRET. Specific and nonspecific signals are not mutually exclusive, and both are useful for pharmacological assays.
In the case of the receptor-arrestin BRET assay (Basic Protocol 1), the increase in BRET upon activation of the receptor is believed to be due to an increase in the direct interaction between donor-fused receptor and acceptor-fused arrestin. However, it is possible that the increase in BRET is nonspecific because of stimulation of endogenous receptors that are activated by the compound of interest. For example, HEK cells express high levels of chemokine receptor CXCR4 (Atwood, Lopez, Wager-Miller, Mackie, & Straiker, 2011), and off-target activation of this receptor increases the nonspecific bystander interaction between arrestin recruited to the plasma membrane by CXCR4 and the fused receptor of interest. Thus, compounds identified as hits in drug screens must be validated by control experiments with specific antagonists of the receptor of interest that fully inhibit agonist signaling via this particular receptor, and therefore should fully suppress specific BRET.
The arrestin translocation assay (Alternate Protocol 1) takes advantage of nonspecific BRET to obviate the use of fused receptors. Donor-fused arrestin is recruited to the membrane from the cytosol by an unmodified receptor of interest. Due to random proximity, the increased localization of arrestin in this compartment can be sufficient to increase the nonspecific BRET between this sensor and many proteins localized in the plasma membrane (in this instance a fragment of GAP43 that is doubly palmitoylated). This is not the case if the two proteins are localized in different micro-compartments of the plasma membrane, such as clathrin-coated pits.
The magnitude of the nonspecific interaction can be increased by: 1) increasing the expression level of the unmodified receptor of interest, thereby increasing the total amount of cytosolic arrestin that can be recruited to the membrane upon receptor activation; 2) overexpression of GRKs that phosphorylate the receptor, increasing its affinity for arrestin; or 3) increasing the expression level of the acceptor-fused plasma membrane marker, thereby increasing the probability that arrestins recruited to the plasma membrane will be in close proximity to, or randomly collide with, the membrane marker. Because this assay is also subject to nonspecific BRET associated with off-target activation of endogenous receptors it is critical to use an appropriate antagonist control to ensure agonist effect on BRET is mediated solely by the receptor of interest.
Negative controls
Regardless of the assay selected, it is helpful to include negative controls such as arrestin mutants that are incapable of binding GPCRs. These include arrestin-KNC mutants, which contain substitutions of 12 key receptor-binding residues, as well as mutants that have a seven-residue deletion in the inter-domain hinge of arrestin, which prevents domain twisting (Gimenez et al., 2014; Gimenez et al., 2012; Hanson et al., 2007; O’Hayre et al., 2017; Vishnivetskiy et al., 2011). These non-receptor-binding mutants have the same size as wildtype arrestins, and therefore are expected to diffuse in the cytoplasm similarly. Thus, they yield the same level of background signal that results from random encounters while being insensitive to receptor activation. Ideally, a full set of controls would also include receptor mutants that do not bind G proteins or arrestins, some of which are now available (Donthamsetti et al., 2018; Wess, Nakajima, & Jain, 2013).
Interpretation of arrestin recruitment
The commercially available PathHunter and Tango assays, as well as BRET- and nanoluc-based arrestin assays, measure the degree of recruitment of arrestin from the cytosol to the plasma membrane. However once recruited to the receptor, arrestin can adopt multiple conformations that may be involved differentially in various arrestin fuctions, i.e., inhibition of G protein-mediated signaling, internalization, and activation of individual downstream signaling pathways (Gurevich & Gurevich, 2020; Lefkowitz, 2013). As shown using an alternative BRET assay, in which arrestin is doubly fused with both a donor and acceptor sensor, these confirmations can be differentially stabilized in a ligand-specific manner (Charest, Terrillon, & Bouvier, 2005). Upon activation of the receptor, arrestin is recruited to it and undergoes a conformational change, which causes a change in BRET. Unfortunately, this assay has a poor dynamic range relative to both BRET-based arrestin-receptor interaction assays, making it unsuitable for robust drug screening. Additionally, there is a paucity of available screening assays that detect the signaling that is unambiguously downstream of arrestin. For example, while phospho-ERK (pERK) assays are often used to study arrestin activity, the interpretation of results is complicated by the contribution of G protein-mediated and other signaling pathways to the measured endpoint (Coffa, Breitman, Spiller, & Gurevich, 2011; Eishingdrelo & Kongsamut, 2013; O’Hayre et al., 2017). Thus, caution must be exercised in interpreting arrestin recruitment as a readout of arrestin activity, because arrestin-dependent inhibition of G protein signaling, internalization of the receptor, and activation of downstream signaling proteins are not measured.
Anticipated Results
The background BRET ratio observed in the assay using membrane-targeted, rather than receptor-fused, component is generally higher than that in the receptor-arrestin BRET assay. This is indicative of a higher basal interaction between arrestin and the plasma membrane (~0.5 to 0.7 and ~0.4 to 0.5, respectively, depending on the expression level of the biosensors and BRET filter sets specific to each plate reader). This is due to the hiBRET peptides (Sp1 and SH3) used in this assay. The maximal drug-induced BRET values (ranging from 0 to ~0.3) depend on the ability of the receptor to recruit arrestin, the expression level of receptor and the other components of the assay, as well as the presence or absence of overexpressed GRKs.
Time Considerations
For transient transfections, both assays are generally performed over a period of four days and include cell seeding, transfection and expression over a period of 24 to 48 hr, and processing and assaying. When stable cell lines are available, only cell seeding, processing, and assay are needed, reducing the overall time by 24 to 48 hr. Assaying can require anywhere from 10 min to 2 hr, depending on the number of samples and plates being run. The time it takes to read a full 96-well plate can be anywhere from 1 to 5 min, depending on the plate reader.
Designing the NanoBiT constructs
The strategic positioning of the NanoBiT fragments within the fusion proteins is crucial for optimizing the sensitivity and accuracy of NanoBiT assays. As both SmBiT and LgBiT can be fused to the protein of interest at either the N- or C-terminus, theoretically there are four combinations that can be used to study the interactions of two proteins. However, when measuring the recruitment of arrestins to GPCRs, only the C-terminal fusion is feasible, as the GPCR N-terminus is extracellular. Additionally, it is advantageous to fuse to the N-terminus of arrestins for two reasons: (a) the arrestin C-terminus is released upon GPCR binding (Gurevich & Gurevich, 2006); (b) several N-terminal residues in arrestins appear to be highly flexible, as they are never resolved in crystal structures (Granzin et al., 1998; Han, Gurevich, Vishnivetskiy, Sigler, & Schubert, 2001; Hirsch et al., 1999; Milano, Kim, Stefano, Benovic, & Brenner, 2006; Sutton et al., 2005; Zhan, Gimenez, Gurevich, & Spiller, 2011); (c) the N-terminus does not participate in GPCR binding, as the structures of arrestin-1 and −2 with cognate receptors demonstrate (Bous et al., 2022; Cao et al., 2022; Huang et al., 2020; Kang et al., 2015; Lee et al., 2020; Staus et al., 2020; Yin et al., 2019; Zhou et al., 2017). Consequently, only two fusion combinations need to be considered: SmBiT-arrestin with GPCR-LgBiT or LgBiT-arrestin with GPCR-SmBiT. In general, as the SmBiT is much smaller than LgBiT and therefore less likely to affect the fusion protein’s diffusion in the cytoplasm, the choice of fusion combination is dependent on the structural features of the receptor and the objectives of the study.
The design of the linker sequence between the NanoBiT fragments and the protein of interest is also important for a successful assay. The length and composition of the linker sequence can affect the flexibility of the fusion protein. Flexible amino acid sequences, containing (Gly-Ser), (Gly-Ala), and (Gly-Pro), are commonly employed as linkers. The optimal linker sequence for a given fusion protein depends on the specific protein domains or subunits being fused and needs to be validated in the NanoBiT assay.
Acknowledgements
Supported by NIH grants GM122491, EY011500 (VVG), MH54137 (JAJ), , GM145284 (NAL) and by the Hope for Depression Research Foundation (JAJ).
Literature Cited
- Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, & Milligan G (2016). Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling. J Biol Chem, 291(53), 27147–27159. doi: 10.1074/jbc.M116.754887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher WB, Terry DS, Gregorio GGA, Kahsai AW, Borgia A, Xie B, … Javitch JA (2022). GPCR-mediated β-arrestin activation deconvoluted with single-molecule precision. Cell, 185(10), 1661–1675.e1616. doi: 10.1016/j.cell.2022.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Lopez J, Wager-Miller J, Mackie K, & Straiker A (2011). Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics, 12, 14. doi: 10.1186/1471-2164-12-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bous J, Fouillen A, Orcel H, Trapani S, Cong X, Fontanel S, … Bron P (2022). Structure of the vasopressin hormone-V2 receptor-beta-arrestin1 ternary complex. Sci Adv, 8(35), eabo7761. doi: 10.1126/sciadv.abo7761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Barros-Alvarez X, Zhang S, Kim K, Damgen MA, Panova O, … Roth BL (2022). Signaling snapshots of a serotonin receptor activated by the prototypical psychedelic LSD. Neuron, 110(19), 3154–3167 e3157. doi: 10.1016/j.neuron.2022.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celver J, Vishnivetskiy SA, Chavkin C, & Gurevich VV (2002). Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem, 277(11), 9043–9048. doi: 10.1074/jbc.M107400200 [DOI] [PubMed] [Google Scholar]
- Charest PG, Terrillon S, & Bouvier M (2005). Monitoring agonist-promoted conformational changes of beta-arrestin in living cells by intramolecular BRET. EMBO Rep, 6(4), 334–340. doi: 10.1038/sj.embor.7400373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton CC, Donthamsetti P, Lambert NA, Javitch JA, & Neve KA (2014). Mutation of Three Residues in the Third Intracellular Loop of the Dopamine D2 Receptor Creates an Internalization-Defective Receptor. J Biol Chem. doi: 10.1074/jbc.M114.605378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffa S, Breitman M, Spiller BW, & Gurevich VV (2011). A single mutation in arrestin-2 prevents ERK1/2 activation by reducing c-Raf1 binding. Biochemistry, 50(32), 6951–6958. doi: 10.1021/bi200745k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, … Wood KV (2016). NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem Biol, 11(2), 400–408. doi: 10.1021/acschembio.5b00753 [DOI] [PubMed] [Google Scholar]
- Donthamsetti P, Gallo EF, Buck DC, Stahl EL, Zhu Y, Lane JR, … Javitch JA (2018). Arrestin recruitment to dopamine D2 receptor mediates locomotion but not incentive motivation. Mol Psychiatry. doi: 10.1038/s41380-018-0212-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eishingdrelo H, & Kongsamut S (2013). Minireview: Targeting GPCR Activated ERK Pathways for Drug Discovery. Curr Chem Genomics Transl Med, 7, 9–15. doi: 10.2174/2213988501307010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- England CG, Ehlerding EB, & Cai W (2016). NanoLuc: A Small Luciferase Is Brightening Up the Field of Bioluminescence. Bioconjug Chem, 27(5), 1175–1187. doi: 10.1021/acs.bioconjchem.6b00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Babilon S, Wanka L, Beck-Sickinger AG, & Gurevich VV (2014). Mutations in arrestin-3 differentially affect binding to neuropeptide Y receptor subtypes. Cell Signal, 26(7), 1523–1531. doi: 10.1016/j.cellsig.2014.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Kook S, Vishnivetskiy SA, Ahmed MR, Gurevich EV, & Gurevich VV (2012). Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G protein-coupled receptors. J Biol Chem, 287(12), 9028–9040. doi: 10.1074/jbc.M111.311803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzin J, Wilden U, Choe HW, Labahn J, Krafft B, & Büldt G (1998). X-ray crystal structure of arrestin from bovine rod outer segments. Nature, 391(6670), 918–921. doi: 10.1038/36147 [DOI] [PubMed] [Google Scholar]
- Grunberg R, Burnier JV, Ferrar T, Beltran-Sastre V, Stricher F, van der Sloot AM, … Serrano L (2013). Engineering of weak helper interactions for high-efficiency FRET probes. Nat Methods, 10(10), 1021–1027. doi: 10.1038/nmeth.2625 [DOI] [PubMed] [Google Scholar]
- Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, … Kostenis E (2018). Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun, 9(1), 341. doi: 10.1038/s41467-017-02661-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV (1998). The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem, 273(25), 15501–15506. doi: 10.1074/jbc.273.25.15501 [DOI] [PubMed] [Google Scholar]
- Gurevich VV, & Gurevich EV (2006). The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol Ther, 110(3), 465–502. doi: 10.1016/j.pharmthera.2005.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, & Gurevich EV (2020). Biased GPCR signaling: Possible mechanisms and inherent limitations. Pharmacol Ther, 211, 107540. doi: 10.1016/j.pharmthera.2020.107540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, … Wood KV (2012). Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol, 7(11), 1848–1857. doi: 10.1021/cb3002478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Audet M, Garneau P, Pelletier J, & Bouvier M (2005). High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J Biomol Screen, 10(5), 463–475. doi: 10.1177/1087057105275344 [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Percherancier Y, Breton B, & Bouvier M (2006). Monitoring protein-protein interactions in living cells by bioluminescence resonance energy transfer (BRET). Curr Protoc Neurosci, Chapter 5, Unit 5 23. doi: 10.1002/0471142301.ns0523s34 [DOI] [PubMed] [Google Scholar]
- Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, & Schubert C (2001). Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane Translocation. Structure, 9(9), 869–880. doi: 10.1016/s0969-2126(01)00644-x [DOI] [PubMed] [Google Scholar]
- Hanson SM, Cleghorn WM, Francis DJ, Vishnivetskiy SA, Raman D, Song X, … Gurevich VV (2007). Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J Mol Biol, 368(2), 375–387. doi: 10.1016/j.jmb.2007.02.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauge Pedersen M, Pham J, Mancebo H, Inoue A, Asher WB, & Javitch JA (2021). A novel luminescence-based β-arrestin recruitment assay for unmodified receptors. J Biol Chem, 296, 100503. doi: 10.1016/j.jbc.2021.100503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, & Sigler PB (1999). The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell, 97(2), 257–269. doi: 10.1016/s0092-8674(00)80735-7 [DOI] [PubMed] [Google Scholar]
- Huang W, Masureel M, Qu Q, Janetzko J, Inoue A, Kato HE, … Kobilka BK (2020). Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature, 579(7798), 303–308. doi: 10.1038/s41586-020-1953-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, … Xu HE (2015). Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature, 523(7562), 561–567. doi: 10.1038/nature14656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, & Benovic JL (2002). Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. J Biol Chem, 277(34), 30760–30768. doi: 10.1074/jbc.M204528200 [DOI] [PubMed] [Google Scholar]
- Klewe IV, Nielsen SM, Tarpo L, Urizar E, Dipace C, Javitch JA, … Christensen KV (2008). Recruitment of beta-arrestin2 to the dopamine D2 receptor: insights into anti-psychotic and anti-parkinsonian drug receptor signaling. Neuropharmacology, 54(8), 1215–1222. doi: 10.1016/j.neuropharm.2008.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer A, Gillis A, Hill R, Schmiedel F, Bailey C, Kelly E, … Schulz S (2020). Morphine-induced respiratory depression is independent of β-arrestin2 signalling. Br J Pharmacol, 177(13), 2923–2931. doi: 10.1111/bph.15004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer A, Schmiedel F, Sianati S, Bailey A, Bateman JT, Levitt ES, … Schulz S (2019). Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat Commun, 10(1), 367. doi: 10.1038/s41467-018-08162-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocan M, & Pfleger KD (2011). Study of GPCR-protein interactions by BRET. Methods Mol Biol, 746, 357–371. doi: 10.1007/978-1-61779-126-0_20 [DOI] [PubMed] [Google Scholar]
- Kolb P, Kenakin T, Alexander SPH, Bermudez M, Bohn LM, Breinholt CS, … Gloriam DE (2022). Community guidelines for GPCR ligand bias: IUPHAR review 32. Br J Pharmacol, 179(14), 3651–3674. doi: 10.1111/bph.15811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovoor A, Celver J, Abdryashitov RI, Chavkin C, & Gurevich VV (1999). Targeted construction of phosphorylation-independent beta-arrestin mutants with constitutive activity in cells. J Biol Chem, 274(11), 6831–6834. doi: 10.1074/jbc.274.11.6831 [DOI] [PubMed] [Google Scholar]
- Lee Y, Warne T, Nehme R, Pandey S, Dwivedi-Agnihotri H, Chaturvedi M, … Tate CG (2020). Molecular basis of beta-arrestin coupling to formoterol-bound beta(1)-adrenoceptor. Nature, 583(7818), 862–866. doi: 10.1038/s41586-020-2419-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ (2013). Arrestins come of age: a personal historical perspective. Prog Mol Biol Transl Sci, 118, 3–18. doi: 10.1016/B978-0-12-394440-5.00001-2 [DOI] [PubMed] [Google Scholar]
- Loening AM, Fenn TD, Wu AM, & Gambhir SS (2006). Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng Des Sel, 19(9), 391–400. doi: 10.1093/protein/gzl023 [DOI] [PubMed] [Google Scholar]
- Marullo S, & Bouvier M (2007). Resonance energy transfer approaches in molecular pharmacology and beyond. Trends Pharmacol Sci, 28(8), 362–365. doi: 10.1016/j.tips.2007.06.007 [DOI] [PubMed] [Google Scholar]
- Milano SK, Kim YM, Stefano FP, Benovic JL, & Brenner C (2006). Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding. J Biol Chem, 281(14), 9812–9823. doi: 10.1074/jbc.M512703200 [DOI] [PubMed] [Google Scholar]
- Morin JG, & Hastings JW (1971). Energy transfer in a bioluminescent system. J Cell Physiol, 77(3), 313–318. doi: 10.1002/jcp.1040770305 [DOI] [PubMed] [Google Scholar]
- Morise H, Shimomura O, Johnson FH, & Winant J (1974). Intermolecular energy transfer in the bioluminescent system of Aequorea. Biochemistry, 13(12), 2656–2662. [DOI] [PubMed] [Google Scholar]
- Nobles KN, Guan Z, Xiao K, Oas TG, & Lefkowitz RJ (2007). The active conformation of beta-arrestin1: direct evidence for the phosphate sensor in the N-domain and conformational differences in the active states of beta-arrestins1 and −2. J Biol Chem, 282(29), 21370–21381. doi: 10.1074/jbc.M611483200 [DOI] [PubMed] [Google Scholar]
- O’Hayre M, Eichel K, Avino S, Zhao X, Steffen DJ, Feng X, … Gutkind JS (2017). Genetic evidence that β-arrestins are dispensable for the initiation of β(2)-adrenergic receptor signaling to ERK. Sci Signal, 10(484). doi: 10.1126/scisignal.aal3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, & Hopkins AL (2006). How many drug targets are there? Nat Rev Drug Discov, 5(12), 993–996. doi: 10.1038/nrd2199 [DOI] [PubMed] [Google Scholar]
- Pfleger KD, Seeber RM, & Eidne KA (2006). Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat Protoc, 1(1), 337–345. doi: 10.1038/nprot.2006.52 [DOI] [PubMed] [Google Scholar]
- Reyes-Alcaraz A, Lucero Garcia-Rojas EY, Merlinsky EA, Seong JY, Bond RA, & McConnell BK (2022). A NanoBiT assay to monitor membrane proteins trafficking for drug discovery and drug development. Commun Biol, 5(1), 212. doi: 10.1038/s42003-022-03163-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahpour A, Espinoza S, Masri B, Lam V, Barak LS, & Gainetdinov RR (2012). BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Front Endocrinol (Lausanne), 3, 105. doi: 10.3389/fendo.2012.00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedabadi M, Gharghabi M, Gurevich EV, & Gurevich VV (2022). Structural basis of GPCR coupling to distinct signal transducers: implications for biased signaling. Trends Biochem Sci, 47(7), 570–581. doi: 10.1016/j.tibs.2022.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, … Lefkowitz RJ (2013). Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature, 497(7447), 137–141. doi: 10.1038/nature12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillmann M, Thurner L, Romantini N, Zimmermann M, Meger B, Behe M, … Berger P (2020). New Insights into Arrestin Recruitment to GPCRs. Int J Mol Sci, 21(14). doi: 10.3390/ijms21144949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Hu H, Robertson MJ, Kleinhenz ALW, Wingler LM, Capel WD, … Skiniotis G (2020). Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature, 579(7798), 297–302. doi: 10.1038/s41586-020-1954-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, … Gurevich VV (2005). Crystal structure of cone arrestin at 2.3A: evolution of receptor specificity. J Mol Biol, 354(5), 1069–1080. doi: 10.1016/j.jmb.2005.10.023 [DOI] [PubMed] [Google Scholar]
- van der Lee MM, Blomenrohr M, van der Doelen AA, Wat JW, Smits N, Hanson BJ, … Zaman GJ (2009). Pharmacological characterization of receptor redistribution and beta-arrestin recruitment assays for the cannabinoid receptor 1. J Biomol Screen, 14(7), 811–823. doi: 10.1177/1087057109337937 [DOI] [PubMed] [Google Scholar]
- Violin JD, Crombie AL, Soergel DG, & Lark MW (2014). Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci, 35(7), 308–316. doi: 10.1016/j.tips.2014.04.007 [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS, & Gurevich VV (2011). Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J Biol Chem, 286(27), 24288–24299. doi: 10.1074/jbc.M110.213835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, & Gurevich VV (1999). How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem, 274(17), 11451–11454. doi: 10.1074/jbc.274.17.11451 [DOI] [PubMed] [Google Scholar]
- Wess J, Nakajima K, & Jain S (2013). Novel designer receptors to probe GPCR signaling and physiology. Trends Pharmacol Sci, 34(7), 385–392. doi: 10.1016/j.tips.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler LM, & Lefkowitz RJ (2020). Conformational Basis of G Protein-Coupled Receptor Signaling Versatility. Trends Cell Biol, 30(9), 736–747. doi: 10.1016/j.tcb.2020.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Li Z, Jin M, Yin YL, de Waal PW, Pal K, … Eric Xu H (2019). A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res, 29(12), 971–983. doi: 10.1038/s41422-019-0256-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Gimenez LE, Gurevich VV, & Spiller BW (2011). Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J Mol Biol, 406(3), 467–478. doi: 10.1016/j.jmb.2010.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, & Xie X (2012). Tools for GPCR drug discovery. Acta Pharmacol Sin, 33(3), 372–384. doi: 10.1038/aps.2011.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XE, He Y, de Waal PW, Gao X, Kang Y, Van Eps N, … Xu HE (2017). Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell, 170(3), 457–469 e413. doi: 10.1016/j.cell.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]