

ABSTRACT
Recently, the remarkable success of chimeric antigen receptor T cell (CAR-T) therapy in treating certain tumors has led to numerous studies exploring its potential application to treat non-oncology diseases. This review discusses the progress and evolution of CAR-T cell therapies for treating non-oncology diseases over the past 5 years. Additionally, we summarize the advantages and disadvantages of CAR-T cell therapy in treating non-oncological diseases and identify any difficulties that should be overcome. After conducting an in-depth analysis of the most recent studies on CAR-T technology, we discuss the key elements of CAR-T therapy, such as developing an effective CAR design for non-oncological diseases, controlling the rate and duration of response, and implementing safety measures to reduce toxicity. These studies provide new insights into different delivery strategies, the discovery of new target molecules, and improvements in the safety of CAR-T therapy for non-oncological diseases.
KEYWORDS: Chimeric antigen receptor, chimeric T cell receptors, CAR-T cell therapy, non-oncology diseases, immunotherapy
Introduction
Traditional therapies yield suboptimal outcomes due to the immune system’s inability to recognize tumor antigens and effectively eliminate cancerous cells. Consequently, cancer immunotherapy has been developed to bolster the immune system’s potency. The advancement of chimeric antigen receptor T cells (CAR-T) and their gene editing techniques has created abundant opportunities for the identification of tumor antigens, bypassing the need for immune checkpoints and the major histocompatibility complex (MHC). Notably, significant progress has been made in the development of diverse CAR-T cell therapies specifically designed to target tumor cells.1 Traditionally,CAR-therapy is a form of gene therapy where T cells, which are types of immune cells, are genetically modified to recognize and attack target cells. This is accomplished by inserting a genetic material that codes for an antigen-recognizing protein known as a single-chain antibody or single-chain fragment variable (scFv) and a T cell activation signal into the T cells. Additionally, T cells can be isolated from the blood of patients or donors to generate CAR-T cells. These cells are genetically modified using a lentiviral or retroviral vector that introduces a gene that provides the cells with certain antigen specificity. Subsequently, the modified cells are multiplied and infused into the patient, allowing them to target and eliminate cells with the target antigen.2 When modified T cells encounter target cells with the targeted antigen on their surface, they bind to the antigen and release perforin and granzyme B, which kill the target cells (Figure 1). scFv is derived from the variable regions of both light and heavy antibodies, whereas the linker between scFv and the T cell activation domain is frequently made of repeated glycine and serine residues.2 CAR-T cells can be further classified into first-generation (without co-stimulatory molecules), second-generation (with one co-stimulatory molecule), and third-generation (with two co-stimulatory molecules) based on the presence of intracellular co-stimulatory molecules.3 Fourth-generation CAR-T cells have been modified to include the interleukin 12 (IL-12) gene in the CAR cassette, which enhances T cell activation and stimulates innate immune cells to remove antigen-negative targeted cells from the target area.4 Although this form of therapy has been used to treat various types of cancers, including leukemia and lymphoma, its use in non-oncological diseases is still in its early stages. Research is currently being conducted on the possibility of using CAR-T cells to treat various non-oncological illnesses, including infectious, immune, and cardiovascular diseases (Table 1). We reviewed the current and potential applications of CAR-T therapy in non-oncological diseases and explored advancements and designs in CAR-T treatment sessions to enhance their suitability for treating non-oncological diseases.
Figure 1.
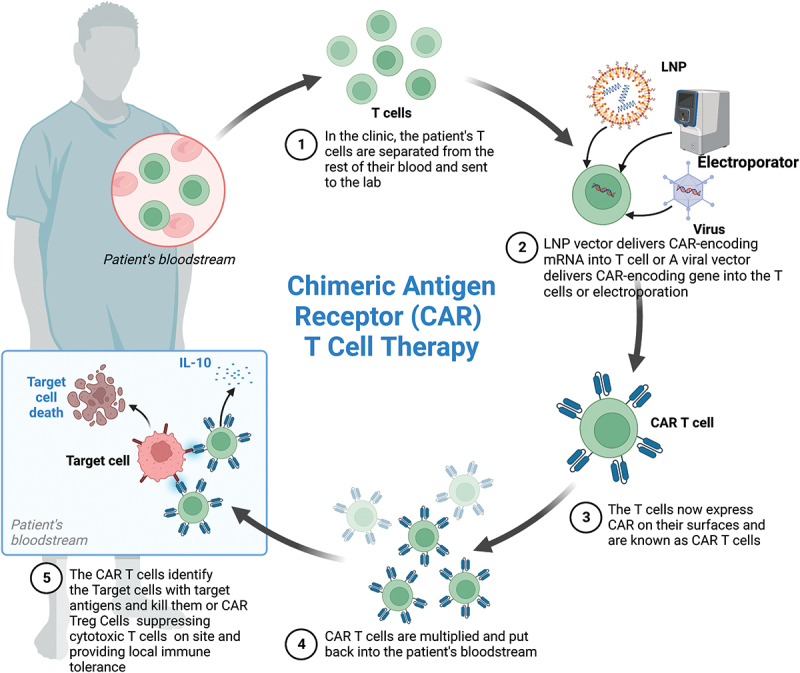
Schematic of CAR-T cell therapy(Created with BioRender.com).
Table 1.
Non-oncology diseases treated with CAR-T cell therapy.
| Disease category | Diseases |
|---|---|
| Infectious diseases | COVID-19 HIV infection*(NCT03617198, NCT03240328,NCT01013415, NCT03980691, NCT04648046) Hepatitis Invasive fungal infections Human cytomegalovirus infections |
| Cardiovascular disease | Myocardial fibrosis Hypercholesterolemia |
| Autoimmune disease | Systemic lupus erythematosus *(NCT03030976, NCT05030779) Type 1 diabetes mellitus Pemphigus Vulgaris Rheumatoid Arthritis Vitiligo Systemic sclerosis*(NCT05085444) Multiple sclerosis |
| others | Senescence Hemophilia A Graft rejection*(NCT04817774) |
*Clinical trials included.
Materials and methods
Based on an initial scoping exercise, qualitative research literature displayed significant heterogeneity, making it unsuitable for conducting a systematic review of qualitative studies following the approach proposed by Dixon-Woods,5 or for conducting a theoretical qualitative meta-synthesis using the methods suggested by Sandelowski.6 Therefore, a narrative review was conducted by searching for relevant articles in the PubMed database between 2017 and 2023. This review focuses on this period because the first CAR-T cell therapy was officially launched in 2017 with United States Food and Drug Administration approval and was subsequently established in practice. This study primarily aimed to assess advances in CAR technology for treating non-oncological diseases. The studies selected for the review were identified through keyword searches, which included ‘‘CAR-T cell,’’ ‘‘engineered T cell,’’ ‘‘chimeric T cell receptor,’’ ‘‘artificial T cell receptors’’ or ‘‘chimeric antigen receptor T cell’’ in the title and abstract. The terms ‘‘tumor,’’ ‘‘neoplasm,’’ ‘‘malignancy,’’ ‘‘cancer,’’ ‘‘lymphoma,’’ and ‘‘leukemia’’ were excluded (Figure 2).
Figure 2.
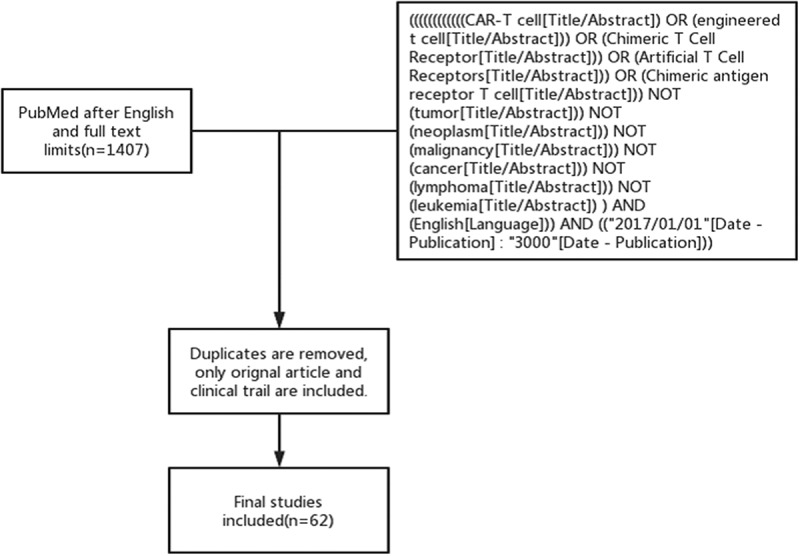
Flowchart of this review.
Results
Infectious diseases
Based on the ability of CAR-T cells to recognize specific surface antigens and direct immune cells to eradicate them when injected into patients, this approach has been proposed for treating infectious diseases other than cancer, including coronavirus disease 2019 (COVID-19), human immunodeficiency virus (HIV) infection, hepatitis, invasive fungal infections, human cytomegalovirus (HCMV) infections.
Antiviral drugs, symptomatic drugs, and neutralizing antibody therapies form the basis of the current COVID-19 treatment; however, the prognosis of critically ill patients is poor. CAR-T therapy for individuals with COVID-19 is designed to provide a safe and effective treatment in combination with standard therapy. Al-Utaibial et al. proposed and tested the hypothesis that the N protein binds to severe acute respiratory syndrome 2 (SARS-2) viral RNA as a target of CAR treating COVID-19.7 Furthermore, Zhu et al. constructed a programmed nanovesicle derived from bispecific CAR-T cells expressing two scFvs to targeting the spike protein of SARS coronavirus 2 (SARS-CoV-2). This study indicates that specially created nano vectors can neutralize SARS-CoV-2 spike-pseudo-typed viruses and effectively directs antiviral drugs to areas of viral infection, thereby increasing antiviral efficiency by hindering intracellular viral replication and diminishing unfavorable drug reactions.8 Another recent animal study has demonstrated that CAR-T cell technology is a viable approach for treating COVID-19. In this study, 2 × 106 NIH/3T3-S1 and 4 × 106 CR3014-28Z/CR3022-28Z CAR-T cells were injected into mice, and the study’s results revealed that CAR-T cells could eradicate spike protein-expressing cells from the mice.9 However, CAR-T cell therapy should be combined with other therapies to avoid excessive inflammation while eliminating infected respiratory cells. Therefore, further research is required to determine how to produce potent immune cells and antibodies in a limited time to prevent mild COVID-19 from progressing to more severe cases. As clinical trials continue, this therapy is believed to be an effective treatment option for patients infected with the virus.
HIV has been responsible for the spread of acquired immune deficiency syndrome over the past few decades, resulting in the deaths of millions of individuals worldwide. Although combination antiretroviral therapy has made significant progress in suppressing HIV replication, it has failed to eliminate latently infected cells, and infected individuals remain HIV-positive for life, necessitating lifelong antiretroviral therapy to maintain the control of viral replication. The impaired immune system in individuals with HIV is primarily caused by the ineffectiveness of HIV-1-specific cytotoxic T lymphocytes (CTLs), which play a crucial role in defending against HIV infection by triggering anti-HIV immune responses.10 It is increasingly evident that the virus hampers the ability of CTLs to eradicate HIV reservoirs within the body.11 Thus, the primary objective in managing HIV is to effectively stimulate dormant HIV reservoirs and reinstate HIV-specific CTL responses. However, redirecting immune cells toward HIV antigens using CARs has garnered considerable attention as a strategy for reviving CTL responses against HIV. Since the CAR’s introduction in 1989,12 four generations of CARs have been developed to treat various diseases. This review summarizes the relevant literature regarding the use of CAR technology in HIV treatment recently, where CD8+ T cells are collected from patients with HIV and transduced with the CAR gene, followed by the reinfusion of CAR-T cells with HIV specificity and efficacy validated in vitro to kill HIV-infected cells.13 Therefore, equipping CD8+ T cells with CARs that can recognize multiple HIV antigens is essential to fight the virus because HIV can rapidly mutate to escape detection by CTL.14 CD4 receptors and bNAbs are currently used to construct anti-HIV CARs.11,13 For example, Anthony-Gonda et al. demonstrated that primary T cells transduced with a multispecific CAR (targeting both the gp120 CD4-binding and co-receptor-binding sites) strongly inhibited HIV-infected cells, and this multitargeted CAR is expected to address some of the shortcomings of single-targeted CAR.15 Clinical trials are ongoing to determine the long-term effectiveness of CAR-T cell therapy and its effects on HIV viral reservoirs.16,17 Despite the potential drawbacks of CAR-T cell expansion, persistence, off-target effects, and severe cytokine release syndrome, a recent study has suggested that HIV-1-specific CAR-T cells do not recognize and eliminate follicular dendritic cells in vitro HIV reservoirs, which may be a fundamental limitation of CAR-T cell therapy for HIV eradication.18 However, CAR-T cell therapy can revolutionize how we treat HIV.
Chronic viral hepatitis is a long-term liver inflammation caused by a viral infection. It is usually caused by the hepatitis B or C virus but can also be caused by other viruses, including hepatitis A, E, and G. Symptoms may include fatigue, jaundice, abdominal pain, and loss of appetite. Complications included cirrhosis, liver cancer, and liver failure. Treatment typically involves lifestyle changes, medication, and antiviral therapy. Current research on CAR-T therapy has significantly focused on chronic hepatitis B. The accepted treatment for hepatitis B virus (HBV) currently suppresses viral replication but does not result in complete recovery. The immune system of patients with chronic hepatitis B cannot effectively eliminate viruses from the body because of the presence of multiple tolerance mechanisms. Kah et al. demonstrated that HBV-T-cell receptor (TCR) T cells could destroy HBV-containing hepatoma cells in a culture system and that three injections of HBV-TCR T cells into HBV-infected human liver chimeric mice decreased viral load.19 Kruse et al. showed a mean decrease of 4.7 times in serum hepatitis B surface antigen (HBsAg), a mean reduction of 3.0 times in the viral load, and an average 70% decrease in HBcAg-positive hepatocytes after transplanting HBsAg-CAR-T cells to human liver chimeric mice infected with HBV.20 A recent study showed that murine CD8+ T cells engineered to express the human CAR receptor-targeting protein S (S-CAR), which is composed of a human B-cell-derived single-chain antibody fragment, a human immunoglobulin G spacer, and CD28- and CD3-signaling immunogenic domains, could be transferred into HBV transgenic mice. After this transfer, the S-CAR-T cells exhibited strong antiviral activity and caused a significant decrease in serum HBsAg, HBeAg, and HBV DNA levels and an increase in anti-HBs without any side effects. However, HBsAg-CAR-T cells did not eradicate HBV infection in mice entirely.21 Wisskirchen et al. demonstrated the potential therapeutic value and safety of T cells that stably express high-affinity TCRs, which recognize the Asian and European human leukocyte antigen-A2 (HLA-A2) subtypes of the HBV envelope and core. After grafting with HBV-specific TCRs, both CD4+ and CD8+ T cells from healthy donors and patients with chronic hepatitis B became polyfunctional effector cells that eliminated the virus from infected HepG2-NTCP cell cultures. Additionally, a single transfer of TCR-grafted T cells into HBV-infected humanized mice controlled the infection, with virological markers declining by 4–5 logs or lower than the detection limit. Furthermore, when the situation is similar to a typical clinical setting, where only a minority of hepatocytes are infected, engineered T cells can efficiently clear the infected hepatocytes without damaging non-infected cells.22
Individuals with weakened immune systems are particularly susceptible to chronic liver disease when infected with the hepatitis E virus (HEV). Current hepatitis E treatments are ineffective and can cause numerous adverse effects. Soon et al. screened and designed TCR-engineered T cells targeting HEV-1527 and HEV-1116. These engineered cells were validated in vitro for their ability to recognize virus-specific epitopes and mediate the destruction of target cells in patients with chronic hepatitis E.23
Therefore, several factors should be considered when using CAR-T cells to treat hepatitis. First, excessive T cell-mediated killing can result in severe liver damage and acute liver failure. Second, it should be demonstrated that engineered T cells possess enhanced functionalities and can remain functional despite immune-tolerizing conditions in the liver. Further research is needed to determine whether HBV-specific T cells can effectively suppress HBV infection without directly harming hepatocytes.
The inhalation of dehydrated yeasts or spores of Cryptococcus spp. can result in cryptococcosis, an infection that affects both immunocompromised and immunocompetent hosts. Cryptococcosis affects the lungs and central nervous system (CNS) and presents with symptoms, including chest pain, dyspnea, headache, confusion, and fever.24 Currently, antifungal drugs are the primary treatment for cryptococcosis. The major virulence factor in Cryptococcus spp. capsules is glucuronoxylomannan (GXM);25 therefore, GXM can be targeted by the CAR because of its importance as a virulence factor. Notably, mAb 18B7 was developed to bind to the GXM of Cryptococcus spp. Subsequently, the scFv derived from 18B7 was used to construct a GXM-specific chimeric antigen receptor (GXMR-CAR).26 After incubating with Cryptococcus neoformans yeast, GXMR-CAR-T cells significantly reduced the number of titanium cells in the CAR-T cell-treated group compared to the other groups. This was due to GXMR-CAR mediating recognition of soluble GXM and triggering the secretion of granzyme and interferon-gamma (IFN-γ).26 The ubiquitous mold, Aspergillus fumigatus, can cause grave infections in immunocompromised individuals, most frequently presenting with invasive pulmonary aspergillosis (IPA). Seif et al. generated a CAR-targeting domain, AB90-E8, which recognizes a conserved protein antigen in the cell walls of A. fumigatus hyphae. T cells with A. fumigatus-CAR (Af-CAR) were shown to recognize A. fumigatus strains and clinical isolates and were effective in directly killing hyphae. Furthermore, in an in vivo mouse model of IPA, CD8+ Af-CAR-T cells were localized to the site of infection, interacted with innate immune cells, and significantly reduced the lung fungal burden.27 These findings demonstrate the potential of genetically engineered T cells to treat infectious diseases and support the clinical development of CAR-T cell therapy to treat infectious diseases. However, the design of CARs should be evaluated across various fungal species, and the antigen recognition moiety and its ligands should be thoroughly characterized.
Allogeneic hematopoietic stem cell transplantation carries the risk of CMV infection, which can be a significant complication. With the aid of advanced genetic engineering, it is currently feasible to swiftly generate transgenic virus-specific T cells from either the patient’s cells (autologous) or a seronegative donor. These engineered T cells can be used to redirect a patient’s virus-specific T cell response and enhance adoptive cell transfer therapy. Furthermore, T cells can be genetically modified to recognize antigens independent of the major histocompatibility complex (MHC) and retain their protective functions during simultaneous treatments.28 The use of engineered T cells, specifically CAR-T cells, to treat CMV was first investigated in 2016.29 Recently, Olbrich et al. engineered T cells with a CAR-T that specifically targeted the human CMV glycoprotein B (gB) molecule, which is expressed during viral reactivation. In vitro and in vivo comparisons were conducted to demonstrate the specificity and strength of gB-CAR-T cells. Moreover, CAR-T cells engineered with an scFv design featuring a high-affinity anti-gB antibody linked to 4-1BB have yielded positive results in both in vitro and in vivo studies to control HCMV infection.30 CAR-T cells represent a potential therapeutic avenue for managing HCMV reactivation, particularly in cases where donor memory T cells are unavailable.
Cardiovascular disease
However, the potential use of CAR-T cell therapy in human heart disease remains uncertain. Thus, researchers have conducted trials using CAR-T cells to treat myocardial fibrosis and hypercholesterolemia to explore this possibility.
Fibrosis, a condition that stiffens the myocardium and severely impairs cardiomyocyte health and function, affects millions of people with cardiac disease.31 Epstein et al. developed CAR-T cells that target activated cardiac fibroblasts associated with cardiac fibrosis. They developed a technique that uses lipid nanoparticles containing mRNA encoding the fibroblast activation protein receptor expressed by cardiac fibroblasts to generate CAR-T cells in vivo to make them safer and more controllable. When this technology was applied to mice with cardiac fibrosis, a significant reduction in fibrosis and a marked improvement were observed in cardiac function.32 Further clinical trials are needed to confirm the safety and efficacy of this technology in humans.
Familial hyperlipidemia is a hereditary metabolic disorder of blood lipids characterized by a tendency to cluster within families. In hypercholesterolemia, CAR-T cell therapy is envisioned for the reconstitution of LDL-R in genetically defective conditions, where injected CAR-T cells return to the hematopoietic ecology and locally promote the establishment of new cell lines expressing LDL-R.33 However, the clinical application of this technique is still limited; therefore, further research is needed to better understand the capacity of CAR-T cells to identify their intended targets, the concentration of reinjected cells in circulation, and the proliferation rate of CAR-T cells.
Autoimmune disease
Immunotherapy based on CARs has attracted considerable interest as a potential therapeutic approach for autoimmune disorders. Currently, conventional and common treatments for autoimmune diseases involve using immunosuppressive agents, including steroids, cytostatic drugs, analgesics, non-steroidal anti-inflammatory drugs, and glucocorticoids. These medications are usually effective in controlling the production of autoantibodies but cannot usually eradicate diseases.34 CARs can direct regulatory T cells (Tregs) to the autoimmune environment, enabling them to move, multiply, and exert their regulatory effects.35
Moreover, CAR-modified T cells can target and eliminate aberrant immune cells associated with autoimmune diseases, including B and antibody-producing plasma cells.
Systemic lupus erythematosus (SLE) is a systemic autoimmune disorder characterized by producing numerous autoantibodies in the blood, causing damage to multiple organs and systems. The primary cause of SLE is an autoimmune response due to an imbalance in the immune system, with B cells playing a key role in the dysregulated activation of self-reactive antibodies.36 Self-reactive B cells are the target of SLE therapy, and anti-cd19 CAR-T cell approaches can induce B cell depletion, particularly in tissues where engineered cells are more readily accessible. Kansal et al. explored the preventive effects of CD8+ anti-CD19 CAR-T cells in two mouse models of SLE. This study showed that CD8+ anti-CD19 CAR-T cells effectively reduced B cell numbers over a prolonged period, thereby preventing the onset of SLE in the two models.37 Jin et al. also examined the efficacy of anti-CD19 CAR-T cell therapy for treating active SLE in an MLR-lpr lupus mouse model. The results showed that this therapy successfully achieved a complete remission rate of 60% in preexisting life-threatening cases of SLE.38 The first use of anti-CD19 CAR-T cell therapy in patients with refractory SLE resulted in rapid clinical remission with minimal adverse effects, accompanied by a prolonged reduction in circulating B cells and a rapid decrease in anti-double-stranded DNA antibodies in the serum.39 In a case of SLE complicated by Stage IV diffuse large B-cell lymphoma, Zhang et al. demonstrated successful treatment with BCMA-CD19 dual-target CAR-T cell reinfusion.40 Moreover, Mougiakakos et al.39 and Zhang et al.40 showed that total-body irradiation could be replaced by fludarabine and glucocorticoids as an alternative preconditioning regimen for lymphocyte depletion, enabling CAR-T cell expansion and successful SLE treatment, potentially making it a viable option for clinical use. The results of all trials are encouraging regarding efficacy and safety; however, further research is needed to determine the persistence of the response during B cell regeneration and to identify the best target populations for this therapy. Immunosuppressive mechanisms, particularly those mediated by regulatory lymphocytes, are crucial for tolerance. Notably, an imbalance in the quantity and/or functionality of regulatory cells can lead to the development of various autoimmune diseases, including SLE, rheumatoid arthritis (RA), type 1 diabetes (T1D), and multiple sclerosis (MS).41 Adoptive therapy using Tregs has been implemented in patients diagnosed with SLE. These findings revealed that this treatment approach led to an augmentation of activated Tregs within the inflamed skin and facilitated the transition from Th1 to Th17 immune responses.42 However, treating autoimmune diseases using induced Tregs has a major disadvantage. This occurs because induced regulatory T cells (iTregs) are polyspecific, implying that they can suppress desired immune processes when administered to a patient. The initial step was to generate iTregs that specifically targeted a specific autoantigen. In this context, molecular engineering plays a crucial role in creating Tregs equipped with CAR.41 CARs are a promising solution for redirecting Treg cell specificity. Multiple preclinical studies have provided evidence for the efficacy of CAR-Treg cells in the context of autoimmune diseases.43–45 However, no CAR-Treg cells are specifically engineered for SLE treatment. Considering the intricate pathogenesis of SLE, which involves self-reactive immune responses to various self-antigens, identifying suitable targets for adoptive Treg cell therapy poses a therapeutic challenge. CARs, designed to target molecules expressed by pathogenic cells, including CD19 for B cells39 or B cell maturation antigen46 for plasma cells, offer a potential solution. However, these targets lack specificity for autoreactive cells as their protective counterparts also express them. Type 1 diabetes mellitus (T1DM) is an autoimmune disorder caused by a person’s immune system attacking and destroying the β cells that produce insulin. This leads to a decrease in insulin production, which is the disease’s main symptom. Insulin supplementation and a tailored dietary plan are the current approaches for managing T1DM. The ongoing research on T1DM primarily aims to develop a successful treatment based on targeted antigens. When transferred to a non-obese diabetic mouse model, the five-module CAR-T cells were observed to rapidly localize to the inflamed pancreas and reduce the symptoms of T1DM by targeting autoimmune CD4+ T cells.47 A previous study showed that mAb287 could target IAg7-B:9–23 complexes, thereby inhibiting T1DM progression. The study further revealed that CAR-T cells carrying 287-CAR selectively migrated to the pancreatic lymph nodes after adoptive transfer, resulting in delayed disease.48 However, it is evident that a single dose of the current treatment can delay T1DM progression. Therefore, further research should be conducted to identify strategies to improve the durability of transferred cells. In another study, Radichev et al. engineered CAR to recognize the human pancreatic endocrine marker HPi2. After transducing human T cells with the resultant HPi2-CAR, these cells proliferated and showed increased granzyme B accumulation when co-cultured with human β cells. Additionally, when exposed to the islets, CD8+ lymphocytes demonstrated increased CD107a (LAMP-1) expression, whereas CD4+ cells produced higher levels of IL-2. However, HPi2-CAR-Tregs could not be expanded, as CAR engagement resulted in tonic signaling caused by the unexpected presence of the HPi2 antigen on Tregs.49 This study revealed the ineffectiveness of HPi2-CAR and emphasized the importance of selecting an appropriate CAR recognition driver for the continual functioning and scalability of engineered T cells.
Pemphigus vulgaris (PV) is a severe antibody-driven autoimmune disorder characterized by the breakdown of cell-to-cell adhesion in the skin and mucous membranes, commonly known as acantholysis. Antibodies against the desmosomal cadherin desmoglein 3 (DSG3) are believed to be the primary cause of this disease. Advancements in CAR technology have enabled the development of a potential new therapy for autoimmune conditions: CAR-T cells targeting antibody-producing B cells.50 Ellebrecht et al. engineered CAR-T cells composed of Dsg-3 fused to CD137-CD3ζ signaling domains and demonstrated that these cells possess specific cytotoxicity in vitro against PV autoantigen-expressing cells, and they successfully eliminated Dsg3-specific B cells in vivo with minimal toxicity in animal models.51 This technology could be a potential model for treating autoimmune diseases by removing harmful cells while avoiding the dangers associated with total immunosuppression. A potential disadvantage of CAR-T cell therapy for patients with PV is the presence of multiple pathogenic autoantibodies that are not specific to Dsg3. These non-Dsg antibodies may act in tandem with one another to exacerbate disease effects of the disease.52
RA is a widespread autoimmune disorder characterized by persistent inflammation of the synovial tissue that typically affects smaller joints.53 Typical approaches to managing RA include administering non-steroidal anti-inflammatory drugs, glucocorticoids, conventional disease-modifying antirheumatic drugs, and biological agents. In a previous study, citrullinated peptide epitopes (citrullinated vimentin, citrullinated type II collagen, citrullinated fibrinogen, and tenascin C) along with cyclic citrulline peptide-1 were selected as target antigens for autoreactive B cells. Subsequently, T cells were genetically modified to express a fixed anti-fluorescein isothiocyanate (FITC) CAR and then evaluated to determine their ability to eliminate specific autoreactive B cells by recognizing FITC-labeled auto-antigenic peptide epitopes. These specially targeted anti-FITC CAR-T cells exhibited the potential to recognize the corresponding FITC-labeled citrullinated peptide epitope and effectively destroyed autoreactive B cell subsets derived from patients with RA under laboratory conditions.54 Furthermore, Whittington et al. engineered HLA-DR1 CAR to create CD8+ CAR-T cells that targeted CD4+ T cells in an antigen-specific manner. These CAR-T cells contain CD3f activation and CD28 signaling domains and a covalently linked autoantigenic peptide from type II collagen (CII;DR1-CII). Through in vitro CTL assays with cloned CD4+ T cells as targets, DR1-CII CAR-T cells effectively recognized and killed CD4+ T cells specific for the CII autoantigen. These results were highly specific because no killing of DR1-restricted CD4+ T cells that recognized other antigens was observed. When tested in a mouse model of autoimmune arthritis, B6.DR1 mice treated with DR1-CII CAR-T cells showed a decrease in the CII-specific autoimmune CD4+ T cell response, a reduction in autoantibody production, and a decrease in the incidence and severity of autoimmune arthritis. This study demonstrates that HLA-DR CAR-T cells provide a targeted approach for treating autoimmune diseases.55
The worldwide prevalence of vitiligo, a common skin disorder that causes depigmentation, is estimated to range from 0.5% to 2%. This disorder is characterized by the loss of melanocytes, leading to the formation of non-scaly, chalk-white patches. Over the years, our understanding of the causes of vitiligo has improved, and vitiligo is currently classified as an autoimmune disease. In a previous study, Tregs were modified to express the GD3-responsive CAR in a mouse model of progressive vitiligo. This modification caused a significant delay in the depigmentation process without causing any major side effects and was effective in protecting melanocytes from destruction by T cells.56
Systemic sclerosis (SSc) is an uncommon disorder that causes an autoimmune response, vasculopathy, and tissue fibrosis. These dysfunctions lead to the progressive deterioration of the microvascular bed and fibrosis of the skin, lungs, and gastrointestinal tract.57 Biological DMARDs and anti-fibrotic therapies have enabled a more precise and efficient treatment of SSc. Nevertheless, these medications usually slow the development of the disease and rarely lead to the reversal of their effects. Clinical trials are underway at clinicaltrials.gov (NCT05085444) to assess the safety and efficacy of CAR-T cell therapies targeting CD19 and B cell maturation antigens in patients with SSc.
The use of genetically engineered Tregs that express CARs or CAR-Tregs may be a possible treatment for autoimmune conditions and demyelinating diseases, such as MS. MS is an autoimmune disease that affects the CNS and causes inflammation, demyelination, and neuronal degeneration. The underlying mechanism is straightforward: CAR-Tregs can identify autoantigens in an MHC-independent manner, thereby suppressing inflammatory responses directed against healthy tissue.58 Major concerns exist regarding CAR-T cells that cause prolonged depletion of B cells and increased infection rates. To avoid this, alternative approaches, including Tregs, could be implemented, as they have less capacity to deplete B cells but can still provide immune-modulating properties. CAR-Tregs may be safer than other CAR-T products regarding potential off-target toxicity and impact on the immune system.59
Myelin-specific CAR-Treg therapy has demonstrated promising outcomes in treating experimental autoimmune encephalomyelitis (EAE), a mouse model that mimics MS. The use of this therapy has shown encouraging results. In a previous study, Tregs were genetically modified to express a TCR specific for myelin basic protein (MBP) peptide. Additionally, human Tregs have been engineered to express functional single-chain chimeric antigen receptors (scFv CAR) that target either MBP or myelin oligodendrocyte glycoprotein. Following long-term expansion under laboratory conditions, these scFv CAR-transduced Tregs expressed the characteristic markers, FoxP3 and Helios, which are associated with Tregs. Moreover, these modified Tregs, with CNS-targeting CARs, effectively suppressed autoimmune damage in EAE, indicating the potential application of these Tregs as a new therapy for patients with MS.60 Mitsdörffer et al. examined the efficacy of an anti-CD19 CAR-T cell approach in a mouse model of MS. A preclinical model of acute lymphoblastic leukemia was used to assess the results. The findings showed that anti-CD19 CAR-T cell-mediated elimination of B cells successfully decreased meningeal B cell aggregates; however, it also had the unintended consequence of worsening disease progression of the disease.57
For autoimmune diseases, eliminating autoreactive cells through CAR-T cells could be more effective and longer-lasting and might reach inflamed tissues that are not accessible to traditional drug-based treatments, whereas CAR-Treg cells can suppress CTL on site and provide local immune tolerance, indicating that CAR-T cell therapy may elicit a rapid response to autoimmune diseases; however, further research is essential to determine its lasting effectiveness. It is crucial to carefully design a tailored epitope to identify corresponding autoantibodies before constructing chimeric autoantibody receptor T (CAAR-T) cells. This step is vital to ensure that the CAAR-T cells recognize the specific antigen of CAAR.61 The success of CAR-T cells against autoimmune conditions may be hindered by several factors, including the absence of a unique antigen in most autoimmune diseases, inefficient CAR-T cell trafficking to the inflamed area, inadequate ex vivo expansion of CAR-T cells for proliferation and maintenance, off-target effects due to heterogeneous antigen expression, immunosuppressive cells’ action against CAR-T cells, lack of growth and supplementary factors, and toxicity from cytokine release syndrome (e.g., tumor necrosis factor-alpha (TNF-α), IL-6, IFN-γ, and IL-2) and neurotoxicity.62
Senescence
Cellular senescence is a crucial mechanism for suppressing tumors and contributes to tissue damage under physiological circumstances.63 As they enter permanent cell cycle arrest, senescent cells (SCs) can significantly impact their local environment and, as a result, affect the fate and development of cells in adult tissue.64 Under pathological conditions, SCs may contribute to the development and progression of tumors by producing various soluble substances, including growth factors, proteases, cytokines, chemokines, and extracellular matrix (ECM) components. This phenomenon is referred to as senescence-associated secretory phenotype (SASP).65 The SASP can have diverse impacts on the microenvironment, encompassing both positive and negative consequences. On the positive side, SASP facilitates the recruitment and activation of immune cells by releasing chemokines (CCL2, CXCL2, and CXCL3) and cytokines (IL-1b, IL-2, IL-6, and IL-8). Conversely, it suppresses the immune system through the secretion of transforming growth factor-beta (TGF-b), triggers fibroblast activation and collagen deposition via profibrotic factors (TGF-b, IL-11, and PAI1), remodels the ECM by secreting matrix metalloproteases, activates and proliferates progenitor cells through growth factors (epidermal and platelet-derived growth factors), and induces paracrine senescence in neighboring cells (TGF-b, TNF-b, and IL-8). In many instances, the overall consequences of the SASP include chronic inflammation and progressive fibrosis.66 SCs induce persistent oxidative stress and inflammation by generating reactive oxygen species and releasing SASP; the pathological buildup of SCs produces an inflammatory environment that can cause ongoing tissue damage, which may lead to several illnesses, such as liver and lung fibrosis, atherosclerosis, diabetes, and osteoarthritis.67,68 Amor et al. demonstrated that urokinase-type plasminogen activator receptor (uPAR) is a cell-surface protein that is highly expressed during senescence. Furthermore, uPAR-specific CAR-T cells effectively eliminated SCs both in vitro and in vivo.69 Moreover, CAR-T cells targeting uPAR can prolong the survival of mice with lung adenocarcinoma treated with senescence-inducing drugs and restore tissue equilibrium in mice with chemically- or diet-induced liver fibrosis. The findings of this study demonstrate that senolytic CAR-T cells may be an effective treatment option for senescence-related diseases. Senolytic CARs may not be subject to the same limitations as other CARs in terms of providing long-term immunological memory, persisting in vivo, and avoiding T-cell exhaustion, because the target cells are non-proliferating. Additionally, transient CAR expression via messenger RNA-based strategies may be a viable option for reducing the cost and complexity of production. Cellular senescence has also been linked to obesity-related inflammation and metabolic disturbances, and the use of emerging senolytic agents may be a potential avenue for treating these metabolic dysfunctions and their associated complications.70 Senolytic treatment, which involves the removal of SCs, may be beneficial for reducing metabolic issues among individuals with severe obesity.71 Other studies have demonstrated that inhibiting chronic inflammation in aging cells can improve metabolism.72,73 Senolytic CAR-T cells can be used to manage metabolic illnesses. The efficacy of targeting uPAR in mice has been encouraging; however, the safety of this approach in humans is yet to be determined. Further research is essential to determine the extent to which this immense potential can be harnessed to develop effective therapies for various age-related diseases.
Other diseases
Patients with hemophilia A (HA) have an alteration in the factor VIII (FVIII) gene (F8) located on the X chromosome. This mutation decreases the amount of functional FVIII, which is an essential component of the blood clotting process.74 The standard treatment for HA is a recombinant or plasma-derived FVIII replacement therapy. Unfortunately, some patients have anti-FVIII antibodies that neutralize the therapeutic FVIII, indicating a lack of immunological tolerance for human proteins. Parvathaneni and Scott developed a CAR expressed by both human and murine CTL that specifically targeted FVIII-specific B cells. This T cell construct, known as the FVIII domain engineered B-cell antibody receptor (BAR), could kill FVIII-reactive B-cell hybridomas both in vitro and in vivo. Furthermore, FVIII BAR CD8 + T cells inhibited the formation of specific antibodies in unimmunized spleen cells stimulated with lipopolysaccharide in vitro. Adoptive transfer of FVIII A2- and C2-BAR CD8 T cells also significantly decreased anti-FVIII antibody formation in hemophilic mice, suggesting that BAR-engineered T cells could be a promising prophylactic treatment for patients with severe HA who are at a high risk of having inhibitors.75 A BAR T cell strategy was developed to specifically target B cells containing a B cell receptor (BCR). Thus, this approach is ineffective in targeting plasma cells that have lost their surface BCR expression as they undergo terminal differentiation. Therefore, complementary treatments should be used to target long-lived plasma cells.
Graft rejection is an undesired process caused by alloreactive T cells that identify donor MHC antigens through the direct, indirect, and semi-direct pathways of allorecognition, which can impede organ transplantation’s long-term advantages.76 Although current immunosuppressive regimens effectively reduce acute allograft rejection, they are associated with severe side effects and have not been successful in preventing chronic rejection.77 Boardman et al. employed the CAR-T technology to program human polyclonal Tregs to target donor MHC class I molecules, which are ubiquitous. When exposed to HLA-A2, CAR-Tregs were activated while avoiding cytotoxicity. Additionally, they migrated more efficiently across HLA-A2-expressing endothelial cell monolayers. In a human skin xenograft transplant model, the adoptive transfer of CAR-Tregs was more effective than that of polyclonal Tregs in mitigating alloimmune-mediated skin injury caused by the transfer of allogeneic peripheral blood mononuclear cells. Noyan et al. developed an HLA-specific chimeric antigen receptor (A2-CAR) for transduction into natural Tregs. This modification did not alter their regulatory phenotype or epigenetic stability but stimulated their proliferation and improved their suppressor function. A2-CAR-modified Tregs more effectively controlled strong allospecific immune responses in vitro and in humanized mice than nTregs. Furthermore, A2-CAR-Tregs completely prevented the rejection of allogeneic target cells and tissues in immune-reconstituted humanized mice without the requirement for immunosuppression. Wagner et al. assessed the effectiveness of CAR Tregs targeting HLA-A2 (A2-CAR) in extending the lifespan of heterotopic heart transplants in mice. The findings indicate that CAR Tregs, either used alone or in conjunction with immunosuppressive agents, can safeguard vascularized grafts in fully immunocompetent recipients.78 The study by Muller et al. reveals that A2-CAR+TCR deficient Tregs effectively delayed the onset of graft-versus-host disease, but only when HLA-A2 was present. This can be achieved either through the co-transfer of peripheral blood mononuclear cells or in recipient mice expressing HLA-A2.79 The results of these studies showed that CAR-T technology is a safe and effective approach to improve Treg therapy for organ transplantation. However, more preclinical studies are needed to confirm these findings before they can be applied clinically.
Discussion
CAR technology is an increasingly popular form of immune cell therapy that has witnessed tremendous advances in oncological treatment because of its ability to target T cells or Treg cells expressing chimeric antigens. Moreover, this therapy is being explored as a potential treatment for other medical conditions, including infectious, cardiovascular, autoimmune, and metabolic diseases. Therefore, numerous elements should be considered to effectively use CAR-based therapies for non-oncological diseases, such as stability, durability, trafficking, safety, effectiveness, manufacturing, and persistence. Fortunately, researchers have modified CAR-T cells using various strategies, including lipid nanoparticles to generate transient CAR-T cells in vivo and mRNA electroporation80 to limit the functional activity time and inflammatory capacity of CAR-T cells. In the future, if an antigen is found to be an imperfect target for CAR therapy, there is the potential for exploring strategies to enhance CAR specificity by targeting two or more disease-associated antigens simultaneously. For example, a bivalent synthetic receptor (also known as “tandem CAR”)81 or gated CARs provide a promising approach in the field of immunotherapy.82 And according to a study, the molecular ON-switch has proven effective in regulating the behavior of primary human CAR T cells in laboratory experiments. This implies that lipocalin-based ON-switches hold considerable promise for widespread utilization in the safe pharmacological management of cellular therapeutics.83 Additionally, the modified receptor can be incorporated into a cell-fate control system. This can be achieved by including an epitope that can be targeted by a clinically approved antibody, enabling the acute clearance of cells when necessary. Alternatively, regulatable receptors can be designed to become active only in the presence of a specific drug, providing a means for controlled modulation of cell behavior.84 Therefore, to develop effective and safe strategies, it is crucial to have a precise understanding of their effects on the allorecognition pathways and effector mechanisms that contribute to alloimmune injuries. Furthermore, overcoming the difficulties of maintaining the effectiveness of transplanted T cells, determining the most effective approach to deliver T cells, the ideal number of cells to be used, and the optimum frequency of infusions are all critical issues that should be addressed. We are also concerned about the great promise of CAR-Treg cells in treating autoimmune diseases and preventing allograft rejection. Genetically modified mono antigen-specific A2-CAR Tregs specifically target and accumulate in HLA-A2-expressing grafts and demonstrate antigen-dependent suppression in vivo, regardless of TCR expression. These findings suggest that these strategies have potential applications in developing precise Treg cell therapies for transplant tolerance.79 Expanding upon HLA-A2 CAR-Tregs, researchers have increased the number of cells by introducing the constitutive expression of IL-10 and an imaging reporter as supplementary elements. Tregs expressing the complete construct retained a consistent phenotype following transduction, exhibited specific activation in response to HLA-A2, and potently suppressed alloresponses. IL-10 inclusion further augmented the suppressive capabilities, thereby providing an additional advantage.85 However, numerous questions remain that require further investigation. These include determining the optimal dosing regimen and frequency of CAR Treg infusion and identifying the most effective immunosuppressive protocol to be used alongside CAR Treg therapy. Therefore, identifying assays that can monitor the persistence, longevity, and suppressive capacity of CAR-Tregs is crucial. Additionally, ensuring the stability of the Treg phenotype is vital for successfully translating CAR Treg therapy into clinical practice.
Despite CAR technology’s remarkable response, various possible drawbacks should be considered and resolved, such as enduring cytopenia, cytokine storms, neurotoxicity, off-target effects, and destruction of all contaminated cells that may harm vital organs.86–89 As this technology continues to evolve, further efforts to reduce the side effects of CAR technology will make it more viable for various applications for different diseases. Nowadays CAR T-cell immunotherapy is transitioning from being a subject of scientific and clinical interest to gaining technological and commercial significance.90 The clinical development of this therapy over the next few years will help determine the true extent of its potential benefits. We also concern about the emerging role of natural killer cells (NK cells) in the field of immunotherapy. CAR-NK cells offer a valuable advantage with their ability to engage in dual killing mechanisms, both dependent on CAR signaling and independent of it, all while manifesting minimal side effects.91 The rapid advancements in technology, such as the augmentation of NK cell-specific signaling, the utilization of split or adapter CAR systems, and the incorporation of CRISPR/Cas9-based genome editing, will enable the augmentation of NK cell persistence and/or the suppression of immune inhibitory mechanisms.92
Conclusion
In conclusion, as CAR-T cell therapy expands, CAR-T cells’ design and manufacturing processes will become increasingly complex. Therefore, future research should address these underlying challenges to ensure that the benefits of this effective therapy for treating various human diseases can reach a larger population. Simultaneously, the growing understanding of Treg subset specificity and functionality, combined with notable achievements in CAR-T cell manufacturing and the nascent stage of CAR-Treg development, presents a distinctive opportunity to advance the research field of CAR-Tregs. Continued enhancements will involve refining CAR constructs and delivery techniques (utilizing viral or non-viral systems), assessing potency, and implementing manufacturing automation platforms. Once the identified challenges and unresolved issues are properly addressed, the therapeutic potential of using CAR technology to alleviate or cure various debilitating diseases will become limitless. Given that this manuscript is a narrative review, and the scope of our literature search was limited to the period between 2017 and 2023, non-oncological diseases reported in the literature prior of this period are not listed in the manuscript. To achieve a more thorough understanding of CAR-T therapy for a specific disease, it may be essential to expand the search timeframe to access a broader spectrum of information.
Funding Statement
This work was funded by the National High Level Hospital Clinical Research Funding [Grant. No: 2022-PUMCH-B-054] and Whole People Nutrition Research Fund [CNS-NNSRG2022-227].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions
MYS contributed literature retrieval and paper writing. WC contributed project oversight and paper revisiting. WYL contributed a paper revisiting. All authors read and approved the final manuscript.
References
- 1.Hosseinkhani N, Derakhshani A, Kooshkaki O, Abdoli Shadbad M, Hajiasgharzadeh K, Baghbanzadeh A, Safarpour H, Mokhtarzadeh A, Brunetti O, Yue S, et al. Immune checkpoints and CAR-T cells: the pioneers in future cancer therapies? Int J Mol Sci. 2020;21(21):8305. doi: 10.3390/ijms21218305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larson RC, Maus MV.. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21(3):145–12. doi: 10.1038/s41568-020-00323-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kenderian SS, Porter DL, Gill S. Chimeric antigen receptor T cells and hematopoietic cell transplantation: how not to put the CART before the horse. Biol Blood Marrow Transplant. 2017;23(2):235–46. doi: 10.1016/j.bbmt.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chmielewski M, Abken H. Trucks: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–54. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 5.Dixon-Woods M, Agarwal S, Jones D, Young B, Sutton A. Synthesising qualitative and quantitative evidence: a review of possible methods. J Health Serv Res Policy. 2005;10(1):45–53. doi: 10.1177/135581960501000110. [DOI] [PubMed] [Google Scholar]
- 6.Sandelowski M, Barroso J, Voils CI. Using qualitative metasummary to synthesize qualitative and quantitative descriptive findings. Res Nurs Health. 2007;30(1):99–111. doi: 10.1002/nur.20176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Utaibi KA, Nutini A, Sohail A, Arif R, Tunc S, Sait SM. Forecasting the action of CAR-T cells against SARS-corona virus-II infection with branching process. Model Earth Syst Environ. 2022;8(3):3413–21. doi: 10.1007/s40808-021-01312-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu T, Xiao Y, Meng X, Tang L, Li B, Zhao Z, Tan Q, Shan H, Liu L, Huang X, et al. Nanovesicles derived from bispecific CAR-T cells targeting the spike protein of SARS-CoV-2 for treating COVID-19. J Nanobiotechnol. 2021;19(1):391. doi: 10.1186/s12951-021-01148-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo X, Kazanova A, Thurmond S, Saragovi HU, Rudd CE. Effective chimeric antigen receptor T cells against SARS-CoV-2. iScience. 2021;24:103295. doi: 10.1016/j.isci.2021.103295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, Winckelmann A, Palmer S, Dinarello C, Buzon M, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. 2014;1(1):e13–e21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 11.Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, Scharenberg AM, Rawlings DJ, Wagner TA. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol Ther. 2017;25(3):570–9. doi: 10.1016/j.ymthe.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–8. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qi J, Ding C, Jiang X, Gao Y. Advances in developing CAR T-cell therapy for HIV cure. Front Immunol. 2020;11:361. doi: 10.3389/fimmu.2020.00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, Lai J, McHugh HL, Hao H, Zhang H, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. 2015;517(7534):381–5. doi: 10.1038/nature14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anthony-Gonda K, Bardhi A, Ray A, Flerin N, Li M, Chen W, Ochsenbauer C, Kappes JC, Krueger W, Worden A, et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci Transl Med. 2019;11(504):eaav5685. doi: 10.1126/scitranslmed.aav5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, Macken C, Richman DD, Christopherson C, June CH, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther. 2002;5(6):788–97. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- 17.Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, Bakker A, Roberts MR, June CH, Jalali S, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4ζ gene-modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus–infected subjects. Blood. 2000;96(3):785–93. doi: 10.1182/blood.V96.3.785.015k10_785_793. [DOI] [PubMed] [Google Scholar]
- 18.Ollerton MT, Berger EA, Connick E, Burton GF, Silvestri G. HIV-1-specific chimeric antigen receptor T cells fail to recognize and eliminate the follicular dendritic cell HIV reservoir in vitro. J Virol. 2020;94(10). doi: 10.1128/JVI.00190-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kah J, Koh S, Volz T, Ceccarello E, Allweiss L, Lutgehetmann M, Bertoletti A, Dandri M. Lymphocytes transiently expressing virus-specific T cell receptors reduce hepatitis B virus infection. J Clin Invest. 2017;127(8):3177–88. doi: 10.1172/JCI93024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kruse RL, Shum T, Tashiro H, Barzi M, Yi Z, Whitten-Bauer C, Legras X, Bissig-Choisat B, Garaigorta U, Gottschalk S, et al. HBsAg-redirected T cells exhibit antiviral activity in HBV-infected human liver chimeric mice. Cytotherapy. 2018;20(5):697–705. doi: 10.1016/j.jcyt.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Festag MM, Festag J, Frassle SP, Asen T, Sacherl J, Schreiber S, Mück-Häusl MA, Busch DH, Wisskirchen K, Protzer U, et al. Evaluation of a fully human, hepatitis B virus-specific chimeric antigen receptor in an immunocompetent mouse model. Mol Ther. 2019;27(5):947–59. doi: 10.1016/j.ymthe.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wisskirchen K, Kah J, Malo A, Asen T, Volz T, Allweiss L, Wettengel JM, Lütgehetmann M, Urban S, Bauer T, et al. T cell receptor grafting allows virological control of hepatitis B virus infection. J Clin Invest. 2019;129(7):2932–45. doi: 10.1172/JCI120228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soon CF, Behrendt P, Todt D, Manns MP, Wedemeyer H, Sallberg Chen M, Cornberg M. Defining virus-specific CD8+ TCR repertoires for therapeutic regeneration of T cells against chronic hepatitis E. J Hepatol. 2019;71(4):673–84. doi: 10.1016/j.jhep.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 24.May RC, Stone NR, Wiesner DL, Bicanic T, Nielsen K. Cryptococcus: from environmental saprophyte to global pathogen. Nat Rev Microbiol. 2016;14(2):106–17. doi: 10.1038/nrmicro.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dyląg M, Colon-Reyes RJ, Kozubowski L. Titan cell formation is unique to Cryptococcus species complex. Virulence. 2020;11(1):719–29. doi: 10.1080/21505594.2020.1772657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.da Silva TA, Hauser PJ, Bandey I, Laskowski T, Wang Q, Najjar AM, da Silva TA, Kumaresan PR. Glucuronoxylomannan in the Cryptococcus species capsule as a target for chimeric antigen receptor T-cell therapy. Cytotherapy. 2021;23(2):119–30. doi: 10.1016/j.jcyt.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seif M, Kakoschke TK, Ebel F, Bellet MM, Trinks N, Renga G, Pariano M, Romani L, Tappe B, Espie D, et al. CAR T cells targeting aspergillus fumigatus are effective at treating invasive pulmonary aspergillosis in preclinical models. Sci Transl Med. 2022;14(664):eabh1209. doi: 10.1126/scitranslmed.abh1209. [DOI] [PubMed] [Google Scholar]
- 28.Mehdizadeh M, Karami S, Ghaffari Nazari H, Sankanian G, Hamidpour M, Hajifathali A. Immunotherapy with adoptive cytomegalovirus-specific T cells transfer: summarizing latest gene engineering techniques. Health Sci Rep. 2021;4(3):e322. doi: 10.1002/hsr2.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stripecke R, Gerasch L, Theobald S, Sundarasetty BS, Mamonkin M, Shum T, Rooney CM. CAR T cells targeted with a high affinity Scfv against the HCMV glycoprotein Gb as adoptive T cell therapy after hematopoietic stem cell transplantation. Blood. 2016;128(22):5721. doi: 10.1182/blood.V128.22.5721.5721. [DOI] [Google Scholar]
- 30.Olbrich H, Theobald SJ, Slabik C, Gerasch L, Schneider A, Mach M, Shum T, Mamonkin M, Stripecke R. Adult and cord blood-derived high-affinity gB-CAR-T cells effectively react against human cytomegalovirus infections. Hum Gene Ther. 2020;31(7–8):423–39. doi: 10.1089/hum.2019.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.González A, Schelbert EB, Díez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol. 2018;71(15):1696–706. doi: 10.1016/j.jacc.2018.02.021. [DOI] [PubMed] [Google Scholar]
- 32.Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L, Kimura T, Soliman OY, Papp TE, Tam YK, et al. CAR T cells produced in vivo to treat cardiac injury. Sci. 2022;375(6576):91–6. doi: 10.1126/science.abm0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bellosta S, Rossi C, Alieva AS, Catapano AL, Corsini A, Baragetti A. Cholesterol lowering biotechnological strategies: from monoclonal antibodies to antisense therapies. A pre-clinical perspective review. Cardiovasc Drugs Ther. 2022;37(3):585–98. doi: 10.1007/s10557-021-07293-w. [DOI] [PubMed] [Google Scholar]
- 34.Abdo AIK, Tye GJ. Interleukin 23 and autoimmune diseases: current and possible future therapies. Inflamm Res. 2020;69(5):463–80. doi: 10.1007/s00011-020-01339-9. [DOI] [PubMed] [Google Scholar]
- 35.Maldini CR, Ellis GI, Riley JL. CAR T cells for infection, autoimmunity and allotransplantation. Nat Rev Immunol. 2018;18(10):605–16. doi: 10.1038/s41577-018-0042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin X, Han Y, Wang JQ, Lu L. CAR-T cell therapy: new hope for systemic lupus erythematosus patients. Cell Mol Immunol. 2021;18(12):2581–2. doi: 10.1038/s41423-021-00787-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kansal R, Richardson N, Neeli I, Khawaja S, Chamberlain D, Ghani M, Ghani QUA, Balazs L, Beranova-Giorgianni S, Giorgianni F, et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Transl Med. 2019;11(482):eaav1648. doi: 10.1126/scitranslmed.aav1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin X, Xu Q, Pu C, Zhu K, Lu C, Jiang Y, Xiao L, Han Y, Lu L. Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell Mol Immunol. 2021;18(8):1896–903. doi: 10.1038/s41423-020-0472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mougiakakos D, Kronke G, Volkl S, Kretschmann S, Aigner M, Kharboutli S, Böltz S, Manger B, Mackensen A, Schett G, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med. 2021;385(6):567–9. doi: 10.1056/NEJMc2107725. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W, Feng J, Cinquina A, Wang Q, Xu H, Zhang Q, Sun L, Chen Q, Xu L, Pinz K, et al. Treatment of systemic lupus erythematosus using BCMA-CD19 compound CAR. Stem Cell Rev Rep. 2021;17(6):2120–3. doi: 10.1007/s12015-021-10251-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buc M. Mechanisms of action of regulatory lymphocytes and a possibility to use them in the treatment of autoimmune diseases and GvH reactions. Bratisl Lek Listy. 2020;121(1):3–7. doi: 10.4149/BLL_2020_001. [DOI] [PubMed] [Google Scholar]
- 42.Dall’era M, Pauli ML, Remedios K, Taravati K, Sandova PM, Putnam AL, Lares A, Haemel A, Tang Q, Hellerstein M, et al. Adoptive treg cell therapy in a patient with systemic lupus erythematosus. Arthritis Rheumatol. 2019;71(3):431–40. doi: 10.1002/art.40737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, Marelli-Berg FM, Watt FM, Lechler RI, Maher J, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. 2017;17(4):931–43. doi: 10.1111/ajt.14185. [DOI] [PubMed] [Google Scholar]
- 44.Yoon J, Schmidt A, Zhang AH, Königs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood. 2017;129(2):238–45. doi: 10.1182/blood-2016-07-727834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang AH, Yoon J, Kim YC, Scott DW. Targeting antigen-specific B cells using antigen-expressing transduced regulatory T cells. J Immunol. 2018;201(5):1434–41. doi: 10.4049/jimmunol.1701800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng D, Sun J. Overview of anti-BCMA CAR-T immunotherapy for multiple myeloma and relapsed/refractory multiple myeloma. Scand J Immunol. 2020;92(2):e12910. doi: 10.1111/sji.12910. [DOI] [PubMed] [Google Scholar]
- 47.Kobayashi S, Thelin MA, Parrish HL, Deshpande NR, Lee MS, Karimzadeh A, Niewczas MA, Serwold T, Kuhns MS. A biomimetic five-module chimeric antigen receptor (5M CAR) designed to target and eliminate antigen-specific T cells. Proc Natl Acad Sci U S A. 2020;117(46):28950–9. doi: 10.1073/pnas.2012495117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Sosinowski T, Cox AR, Cepeda JR, Sekhar NS, Hartig SM, Miao D, Yu L, Pietropaolo M, Davidson HW, et al. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II: peptide complex modulate the progression of autoimmune diabetes. J Autoimmun. 2019;96:50–8. doi: 10.1016/j.jaut.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Radichev IA, Yoon J, Scott DW, Griffin K, Savinov AY. Towards antigen-specific tregs for type 1 diabetes: construction and functional assessment of pancreatic endocrine marker, HPi2-based chimeric antigen receptor. Cell Immunol. 2020;358:104224. doi: 10.1016/j.cellimm.2020.104224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellebrecht CT, Payne AS. Setting the target for pemphigus vulgaris therapy. JCI Insight. 2017;2(5):e92021. doi: 10.1172/jci.insight.92021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Sci. 2016;353(6295):179–84. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yanovsky RL, McLeod M, Ahmed AR. Treatment of pemphigus vulgaris: part 2 – emerging therapies. Expert Rev Clin Immunol. 2019;15(10):1061–71. doi: 10.1080/1744666X.2020.1672539. [DOI] [PubMed] [Google Scholar]
- 53.Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, Sawa S, Nitta T, Takayanagi H. Osteoimmunology: the conceptual framework unifying the immune and skeletal systems. Physiol Rev. 2017;97(4):1295–349. doi: 10.1152/physrev.00036.2016. [DOI] [PubMed] [Google Scholar]
- 54.Zhang B, Wang Y, Yuan Y, Sun J, Liu L, Huang D, Hu J, Wang M, Li S, Song W, et al. In vitro elimination of autoreactive B cells from rheumatoid arthritis patients by universal chimeric antigen receptor T cells. Ann Rheum Dis. 2021;80(2):176–84. doi: 10.1136/annrheumdis-2020-217844. [DOI] [PubMed] [Google Scholar]
- 55.Whittington KB, Prislovsky A, Beaty J, Albritton L, Radic M, Rosloniec EF. CD8(+) T cells expressing an HLA-DR1 chimeric antigen receptor target autoimmune CD4(+) T cells in an antigen-specific manner and inhibit the development of autoimmune arthritis. J Immunol. 2022;208(1):16–26. doi: 10.4049/jimmunol.2100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mukhatayev Z, Dellacecca ER, Cosgrove C, Shivde R, Jaishankar D, Pontarolo-Maag K, Eby JM, Henning SW, Ostapchuk YO, Cedercreutz K, et al. Antigen specificity enhances disease control by tregs in vitiligo. Front Immunol. 2020;11:581433. doi: 10.3389/fimmu.2020.581433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360(19):1989–2003. doi: 10.1056/NEJMra0806188. [DOI] [PubMed] [Google Scholar]
- 58.Sedaghat N, Etemadifar M. Inducing chimeric antigen receptor (CAR) regulatory T cells in-vivo: a novel concept for a potential feasible cure of demyelinating diseases. Mult Scler Relat Disord. 2022;57:103341. doi: 10.1016/j.msard.2021.103341. [DOI] [PubMed] [Google Scholar]
- 59.Sadeqi Nezhad M, Seifalian A, Bagheri N, Yaghoubi S, Karimi MH, Adbollahpour-Alitappeh M. Chimeric antigen receptor based therapy as a potential approach in autoimmune diseases: how close are we to the treatment? Front Immunol. 2020;11:603237. doi: 10.3389/fimmu.2020.603237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Paula Pohl A, Schmidt A, Zhang AH, Maldonado T, Königs C, Scott DW. Engineered regulatory T cells expressing myelin-specific chimeric antigen receptors suppress EAE progression. Cell Immunol. 2020;358:104222. doi: 10.1016/j.cellimm.2020.104222. [DOI] [PubMed] [Google Scholar]
- 61.Chen Y, Sun J, Liu H, Yin G, Xie Q. Immunotherapy deriving from CAR-T cell treatment in autoimmune diseases. J Immunol Res. 2019;2019:5727516. doi: 10.1155/2019/5727516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Srivastava S, Riddell SR. Chimeric antigen receptor T cell therapy: challenges to bench-to-bedside efficacy. J Immunol. 2018;200(2):459–68. doi: 10.4049/jimmunol.1701155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burton DG, Krizhanovsky V. Physiological and pathological consequences of cellular senescence. Cell Mol Life Sci. 2014;71(22):4373–86. doi: 10.1007/s00018-014-1691-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chin AF, Elisseeff JH. Senescent cells in tissue engineering. Curr Opin Biotechnol. 2022;76:102737. doi: 10.1016/j.copbio.2022.102737. [DOI] [PubMed] [Google Scholar]
- 65.Alimirah F, Pulido T, Valdovinos A, Alptekin S, Chang E, Jones E, Diaz DA, Flores J, Velarde MC, Demaria M, et al. Cellular senescence promotes skin carcinogenesis through p38MAPK and p44/42MAPK signaling. Cancer Res. 2020;80(17):3606–19. doi: 10.1158/0008-5472.CAN-20-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186(2):243–78. doi: 10.1016/j.cell.2022.11.001. [DOI] [PubMed] [Google Scholar]
- 67.He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169(6):1000–11. doi: 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Childs BG, Gluscevic M, Baker DJ, Laberge R-M, Marquess D, Dananberg J, van Deursen JM. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017;16(10):718–35. doi: 10.1038/nrd.2017.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, Mansilla-Soto J, Boyer JA, Li X, Giavridis T, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583(7814):127–32. doi: 10.1038/s41586-020-2403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, Prata LG, Dijk TH, Verkade E, Casaclang‐Verzosa G, et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell. 2019;18(3):e12950. doi: 10.1111/acel.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rouault C, Marcelin G, Adriouch S, Rose C, Genser L, Ambrosini M, Bichet J-C, Zhang Y, Marquet F, Aron-Wisnewsky J, et al. Senescence-associated β-galactosidase in subcutaneous adipose tissue associates with altered glycaemic status and truncal fat in severe obesity. Diabetologia. 2021;64(1):240–54. doi: 10.1007/s00125-020-05307-0. [DOI] [PubMed] [Google Scholar]
- 72.Marín-Aguilar F, Lechuga-Vieco AV, Alcocer-Gómez E, Castejón-Vega B, Lucas J, Garrido C, Peralta‐Garcia A, Pérez‐Pulido AJ, Varela‐López A, Quiles JL, et al. NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell. 2020;19(1):e13050. doi: 10.1111/acel.13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, Ridker P, Lorenzatti A, Krum H, Varigos J. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10105):1833–42. doi: 10.1016/S0140-6736(17)32247-X. [DOI] [PubMed] [Google Scholar]
- 74.Scott DW, Pratt KP, Miao CH. Progress toward inducing immunologic tolerance to factor VIII. Blood. 2013;121(22):4449–56. doi: 10.1182/blood-2013-01-478669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parvathaneni K, Scott DW. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv. 2018;2(18):2332–40. doi: 10.1182/bloodadvances.2018018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sagoo P, Lombardi G, Lechler RI. Relevance of regulatory T cell promotion of donor-specific tolerance in solid organ transplantation. Front Immunol. 2012;3:184. doi: 10.3389/fimmu.2012.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pasquet L, Douet JY, Sparwasser T, Romagnoli P, van Meerwijk JP. Long-term prevention of chronic allograft rejection by regulatory T-cell immunotherapy involves host Foxp3-expressing T cells. Blood. 2013;121(21):4303–10. doi: 10.1182/blood-2012-08-452037. [DOI] [PubMed] [Google Scholar]
- 78.Wagner JC, Ronin E, Ho P, Peng Y, Tang Q. Anti-HLA-A2-CAR Tregs prolong vascularized mouse heterotopic heart allograft survival. Am J Transplant. 2022;22(9):2237–45. doi: 10.1111/ajt.17063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Muller YD, Ferreira LMR, Ronin E, Ho P, Nguyen V, Faleo G, Zhou Y, Lee K, Leung KK, Skartsis N, et al. Precision engineering of an anti-HLA-A2 chimeric antigen receptor in regulatory T cells for transplant immune tolerance. Front Immunol. 2021;12:686439. doi: 10.3389/fimmu.2021.686439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oh HL, Chia A, Chang CX, Leong HN, Ling KL, Grotenbreg GM, Gehring AJ, Tan YJ, Bertoletti A. Engineering T cells specific for a dominant severe acute respiratory syndrome coronavirus CD8 T cell epitope. J Virol. 2011;85(20):10464–71. doi: 10.1128/JVI.05039-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schönfeld K, Koch J, Dotti G, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ebert LM, Yu W, Gargett T, Brown MP. Logic-gated approaches to extend the utility of chimeric antigen receptor T-cell technology. Biochem Soc Trans. 2018;46(2):391–401. doi: 10.1042/BST20170178. [DOI] [PubMed] [Google Scholar]
- 83.Zajc CU, Dobersberger M, Schaffner I, Mlynek G, Pühringer D, Salzer B, Djinović-Carugo K, Steinberger P, De Sousa Linhares A, Yang NJ, et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc Natl Acad Sci U S A. 2020;117(26):14926–35. doi: 10.1073/pnas.1911154117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, Harris RA, Magnusson PU, Brittebo E, Loskog AS, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. 2012;9(1):112. doi: 10.1186/1742-2094-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mohseni YR, Saleem A, Tung SL, Dudreuilh C, Lang C, Peng Q, Volpe A, Adigbli G, Cross A, Hester J, et al. Chimeric antigen receptor-modified human regulatory T cells that constitutively express IL-10 maintain their phenotype and are potently suppressive. Eur J Immunol. 2021;51(10):2522–30. doi: 10.1002/eji.202048934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321–30. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rubin DB, Danish HH, Ali AB, Li K, LaRose S, Monk AD, Cote DJ, Spendley L, Kim AH, Robertson MS, et al. Neurological toxicities associated with chimeric antigen receptor T-cell therapy. Brain. 2019;142(5):1334–48. doi: 10.1093/brain/awz053. [DOI] [PubMed] [Google Scholar]
- 88.Bachanova V, Perales MA, Abramson JS. Modern management of relapsed and refractory aggressive B-cell lymphoma: a perspective on the current treatment landscape and patient selection for CAR T-cell therapy. Blood Rev. 2020;40:100640. doi: 10.1016/j.blre.2019.100640. [DOI] [PubMed] [Google Scholar]
- 89.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–54. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jürgens B, Clarke NS. Evolution of CAR T-cell immunotherapy in terms of patenting activity. Nat Biotechnol. 2019;37(4):370–5. doi: 10.1038/s41587-019-0083-5. [DOI] [PubMed] [Google Scholar]
- 91.Karadimitris A. Cord blood CAR-NK cells: favorable initial efficacy and toxicity but durability of clinical responses not yet clear. Cancer Cell. 2020;37(4):426–7. doi: 10.1016/j.ccell.2020.03.018. [DOI] [PubMed] [Google Scholar]
- 92.Reindl LM, Albinger N, Bexte T, Müller S, Hartmann J, Ullrich E. Immunotherapy with NK cells: recent developments in gene modification open up new avenues. Oncoimmunology. 2020;9(1):1777651. doi: 10.1080/2162402X.2020.1777651. [DOI] [PMC free article] [PubMed] [Google Scholar]