

Summary
Mitochondria (MT) participate in most metabolic activities of mammalian cells. A near-unidirectional mitochondrial transfer from T cells to cancer cells was recently observed to “metabolically empower” cancer cells while “depleting immune cells”, providing new insights into tumor-T cell interaction and immune evasion. Here, we leverage single-cell RNA-seq technology and introduce MERCI, a statistical deconvolution method for tracing and quantifying mitochondrial trafficking between cancer and T cells. Through rigorous benchmarking and validation, MERCI accurately predicts the recipient cells and their relative mitochondrial compositions. Application of MERCI to human cancer samples identifies a reproducible MT transfer phenotype, with its signature genes involved in cytoskeleton remodeling, energy production and TNFα signaling pathways. Moreover, MT transfer is associated with increased cell cycle activity and poor clinical outcome across different cancer types. In summary, MERCI enables systematic investigation of an understudied aspect of tumor-T cell interactions that may lead to the development of therapeutic opportunities.
eTOC blurb:
Recent work observes mitochondrial transfer from T cells to cancer cells. Zhang et al. systematically investigate this process using single cell sequencing data from human cancers and identify a distinct phenotype related to mitochondrial transfer. Genes and pathways associated with this phenotype may serve as future therapeutic targets.
Graphical Abstract
Introduction
The mitochondrion (MT) is a subcellular organelle that participates in critical physiological processes, such as energy production, calcium signaling, and programmed cell death1–3. Mitochondria have their own 16.6Kb genome (16.3Kb for mouse) of circular DNA (mtDNA) with high mutation rate4–6. Each somatic cell contains 102–104 copies of MT genomes7, with a subset of mutations reaching high levels of heteroplasmy8 due to cytoplasmic inheritance or random drift9. A few recent studies thus use the single nucleotide variations (SNVs) in the mtDNA as endogenous genetic barcodes to trace lineage relationships and clonal dynamics10,11. For example, new methods have been developed to reconstruct shared lineages with cellular heteroplasmy of MT variants from single cell RNA-seq samples12–14. These studies provide evolutionary insights in the fields of developmental biology15,16, stem cell research17 and oncology18.
In the meanwhile, a growing body of literature reports that mitochondria are not only inherited through lineage, but also laterally shared between different mammalian cells19–21 primarily through tunneling nanotubes (TNT)22–24. This phenomenon was first observed during coculture of mitochondria-deprived A549 lung cancer cells with mesenchymal stem cells, where the latter transfer mitochondria to the former to restore their aerobic respiratory functions23. Follow-up studies further demonstrate that this intercellular transfer occurs among diverse mammalian cell types in vitro and in vivo22,24,25. A recent work examines the nanotube-based communication between cancer and immune cells, and reports unidirectional mitochondrial trafficking from T cells to neighboring cancer cells26. As mitochondria provide both energy and the essential metabolites for T cell activation27,28, this finding may point to a different mechanism of T cell dysfunction.
Many exciting questions follow this discovery, such as how nanotubes initiate in selected cancer cells, what genes or pathways cause the preferential MT transfer from T cells to cancer cells, and what the consequences of this unidirectional MT transfer are. Currently, most related studies have been conducted in vitro20,29, which limited the generalizability of their findings. To better understand the mechanism of tumor mitochondrial interception and to identify precise, druggable targets, it is imperative to investigate this process under physiological conditions. However, in vivo tracking of MT transfer relies on genetic engineered mice with photo-activatable mitochondria30, which cannot be implemented in humans. Consequently, there is no practical approach to investigate this process in the human tumor microenvironment.
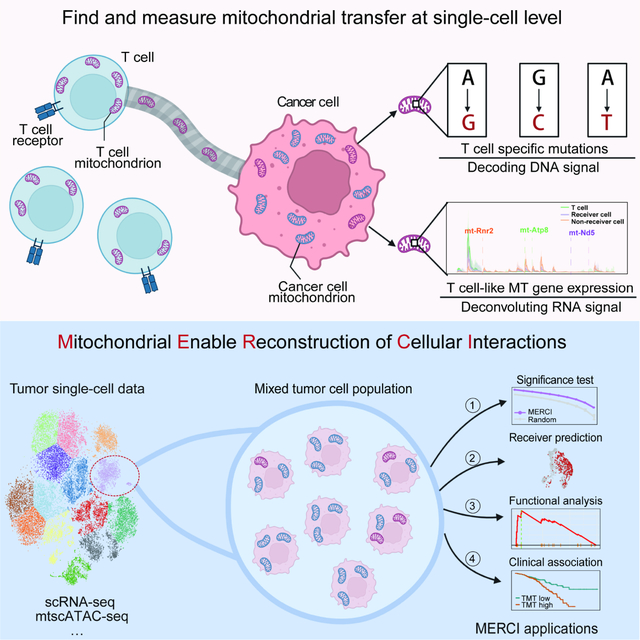
To bridge this gap, we developed a single cell deconvolution method, MERCI (Mitochondrial-Enabled Reconstruction of Cellular Interactions), to computationally infer the mitochondrial receiver cells in the patient tumor samples. MERCI takes the single cell RNA-seq (scRNA-seq) data as input, identifies the lineage-specific MT mutations through a verified mutational calling pipeline, and predicts receiver cells based on mutation and mitochondrial gene expression profiles. Application of MERCI to human tumor samples uncovered a reproducible cancer cell phenotype of MT transfer, which is associated with multiple pathways of nanotube formation and energy metabolism. The signature was also predictive of worse clinical outcome for diverse cancer types.
Results
Cancer mitochondrial hijack recapitulated via scRNA-seq data
To reproduce the observation of mitochondrial transfer, we first conducted the in vitro experiment described in the recent literature but using a different cancer cell line20,26. Specifically, the murine KrasG12D/p53ko (KP) lung cancer cells31 were transfected with pDsRed2-Mito vector to label the mitochondria with red fluorescent signal. Primary autologous CD8+ T cells were isolated from mice spleen with mitochondria labeled with MitoTracker™ green. Coculture experiments were conducted to visualize and quantify the transfer of mitochondria between cancer and T cells (Figure 1A). Fluorescence imaging revealed a dominant translocation of T cell mitochondria into the cytoplasm of KP cells (Figures 1B, S1A and S1B). 24 hours after coculture, over 30% KP cells carried high level of T cell mitochondria, while very few of T cells were weakly positive for the cancer signal (Figures 1B and S1B). We repeated experiments with different time spans and made similar observations (Figure 1C). We also tested a range of cancer to T cell ratios and observed higher level of mitochondrial transfer with more T cells in the coculture (Figures S1C). These results confirmed the near unidirectional T cell to cancer mitochondrial flow reported by Saha et al26. Further, we repeated the coculture experiment using a different cell line, the MC38 colon cancer cells, and observed quantitatively similar MT transfer events (Figures S1D). Our observations suggested that mitochondrial hijacking is a common process initiated by diverse malignant cells.
Figure 1. MT transfer signal captured by scRNA-seq data.
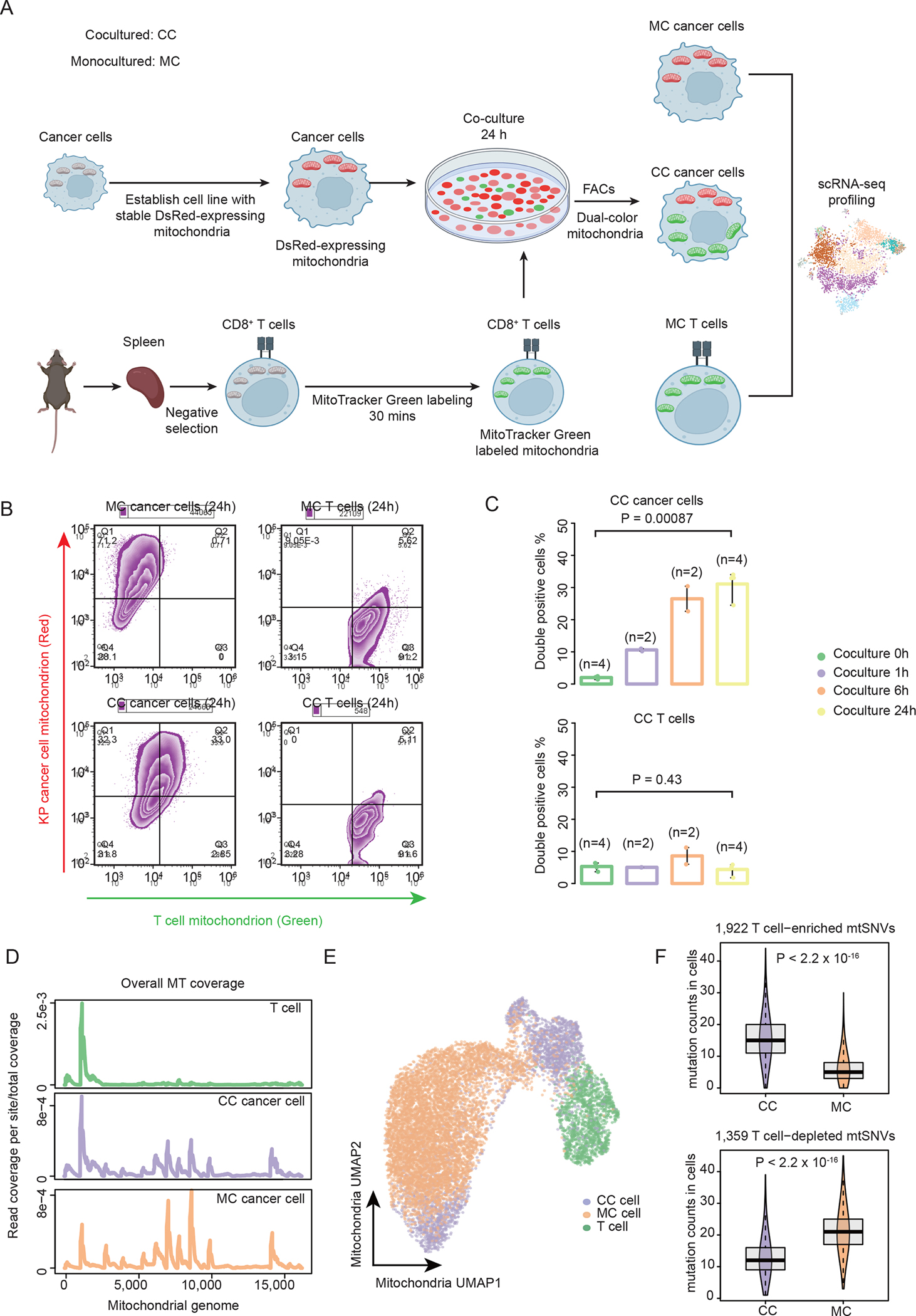
(A) Cartoon illustrating the process of generating the ground-truth data by coculturing KP cancer cells and CD8+ T cells, followed by single cell RNA-seq library construction and next generation sequencing. (B) The contour plots showing the percentage of double positive cells (left: KP cancer cells, right: T cells) after 24 hours of coculturing (lower) and monoculturing (upper). Double positive cells are those carrying both endogenous and donor cell-derived mitochondria. (C) Bar plots showing the percentage of double positive cells for the KP cancer cells (upper) and the T cells (lower) at different coculture time points. Statistical significance was evaluated using two-sided Student t test. Error bars indicate the range (min to max) of the data. (D) The distribution of per site read coverage in mitochondrial genome for CC, MC and T cells. Read depth is normalized by the total reads mapped to the mtDNA region. (E) UMAP plot showing mitochondrial transcriptional profiles of cells. Cells are colored by different experimental groups. (F) Boxplots showing the counts of T cell enriched (upper) and T cell depleted (lower) mtSNVs in CC and MC cancer cells with the MT read-depth range 1000–2000. Lower and upper box hinges represent 25th to 75th percentiles, central line the median and the whisker extend to highest and smallest values no greater than 1.5× interquartile range; the violin component refers to the kernel probability density and encompasses all cells. Two-sided Wilcoxon rank sum test was applied to calculate the P values. See also Figures S1–S2.
We next performed single cell RNA sequencing (scRNA-seq) of three experimental groups: cocultured (CC) cancer cells and T cells as the treatment group and two controls including the monocultured (MC) cancer cells and primary CD8+ T cells (Figure 1A). Culture medium was diluted by a factor of 105 prior to loading to 10x chips to mitigate the potential contamination from cell-free DNA/RNA during cell culture (Figure S1E). Half of the CD8+ T cells were reserved for scRNA-seq data preparation before coculture with cancer cells to ensure the same mitochondrial genetic background. All the sequenced CC cells were positive for the T cell mitochondrial signal. In total, we obtained 12,145 high quality cells with over 1,000 reads mapped to mtDNA, including 2,904 CC cells, 7,572 MC cells and 1,669 T cells (Table S1). Interestingly, the mtDNA read coverage profile of the CC cancer cells already exhibited an ‘intermediate’ pattern between MC cancer and T cells on the overall (Figure 1D) and cellular levels (Figure S2A). For example, CC cells expressed higher protein-coding gene mt-Co3 than the T cells but lower than the MC cells (Figure S2A). Uniform manifold approximation and projection (UMAP)32 analysis using mitochondrial transcriptomic profile also confirmed that most CC cells displayed a mixed phenotype of the two monocultured cell populations (Figure 1E). These results indicated that mitochondria transfer can be manifested on the single cell gene expression level.
We then investigated single nucleotide variants (SNVs) in the mtDNA, which have been used as genetic barcodes to track cell lineage11,12. We customized a variant-calling pipeline (MERCI-mtSNP) for fast retrieving mitochondrial genotypes on cellular level from single cell sequencing data (Figure S2B, STAR Methods). We tested MERCI-mtSNP by examining the downloaded benchmark scRNA-seq data33, which is a modified 10x scRNA-seq data with targeted enrichment to increase the coverage of MT genome. There are 17 bona fide mitochondrial variants in this dataset that differentiate two pre-defined cell lineages. MERCI-mtSNP successfully captured all 17 MT mutations in the correct cell populations (Figure S2C).
We then used MERCI-mtSNP to examine scRNA-seq samples derived from KP/T cells and identified 21,341 passed-filtered mitochondrial SNVs (mtSNVs) across all the cells. Next, we sought to trace mitochondrial transfer in the CC cancer cells using T cell enriched mutations, which occurred at significantly higher frequencies in the primary T cells compared to the MC cancer cells (FDR<0.1). To be precise, ‘frequency’ refers to the percentage of cells carrying the mutation in a given population. In this analysis, we used half of the MC cells to define these mtSNVs, holding out the other half for testing to avoid data leakage. A total of 1,922 T cell enriched variants were identified. As expected, the depth-normalized counts of T cell enriched mtSNVs were significantly higher in the CC cells compared to the held-out MC cells (Figures 1F and S2D). This difference was reversed when using the T cell depleted mtSNVs (Figures 1F and S2E). Here, the depth-normalized count refers to the number of mutations observed in a cell within a given MT read count range (Methods), which is applied to avoid the bias caused by read coverage difference (Figure S2F). Consistently, 9 out of the top 10 most enriched mtSNVs were significantly more frequent in the CC population (Figure S2G). We provided a table listing all the T cell enriched variants used in this work (Table S2), which had been selected in the regions with ≥5X coverage in both cell types by MERCI-mtSNP (Figure S2H). Together, our data indicated that the genetic variants of the transferred mitochondrial genome can be captured by the scRNA-seq data as well.
Tracing MT receivers through single cell deconvolution
Following the above results, we developed MERCI to identify the MT receiver cells through quantifying the donor-derived mtSNVs and estimating the relative abundance of exogenous mitochondria (Figure 2A, STAR Methods). For a given cell, MERCI combines the MT gene expression and mtSNV profiles to predict if it is a receiver and infer the relative fraction of donor-derived mitochondria. Specifically, MERCI first defines T cell-enriched mtSNVs based on the reference (cancer and T cell) populations and calculate an ‘effective count statistic’ (Neff) for each cancer cell to assign each cell with a DNA rank score (Figure S3A). It then uses the averaged MT gene expression profiles of cancer and T cell populations as reference and applies support vector regression (SVR)34,35 to estimate the relative abundance of transferred MT in the target cancer cells (Figure S3B). Each cell of interest is assigned with an RNA rank score based on the ordering of the SVR coefficient: the relative proportion of T cell-derived MT. Rank transformation is necessary to make the scores robust to outliers and less sensitive to sequencing depth or cancer type. Cells with scores passing a predefined cutoff for DNA or RNA rank are considered as candidate MT receivers.
Figure 2. Overview of MERCI and application to the ground-truth data.
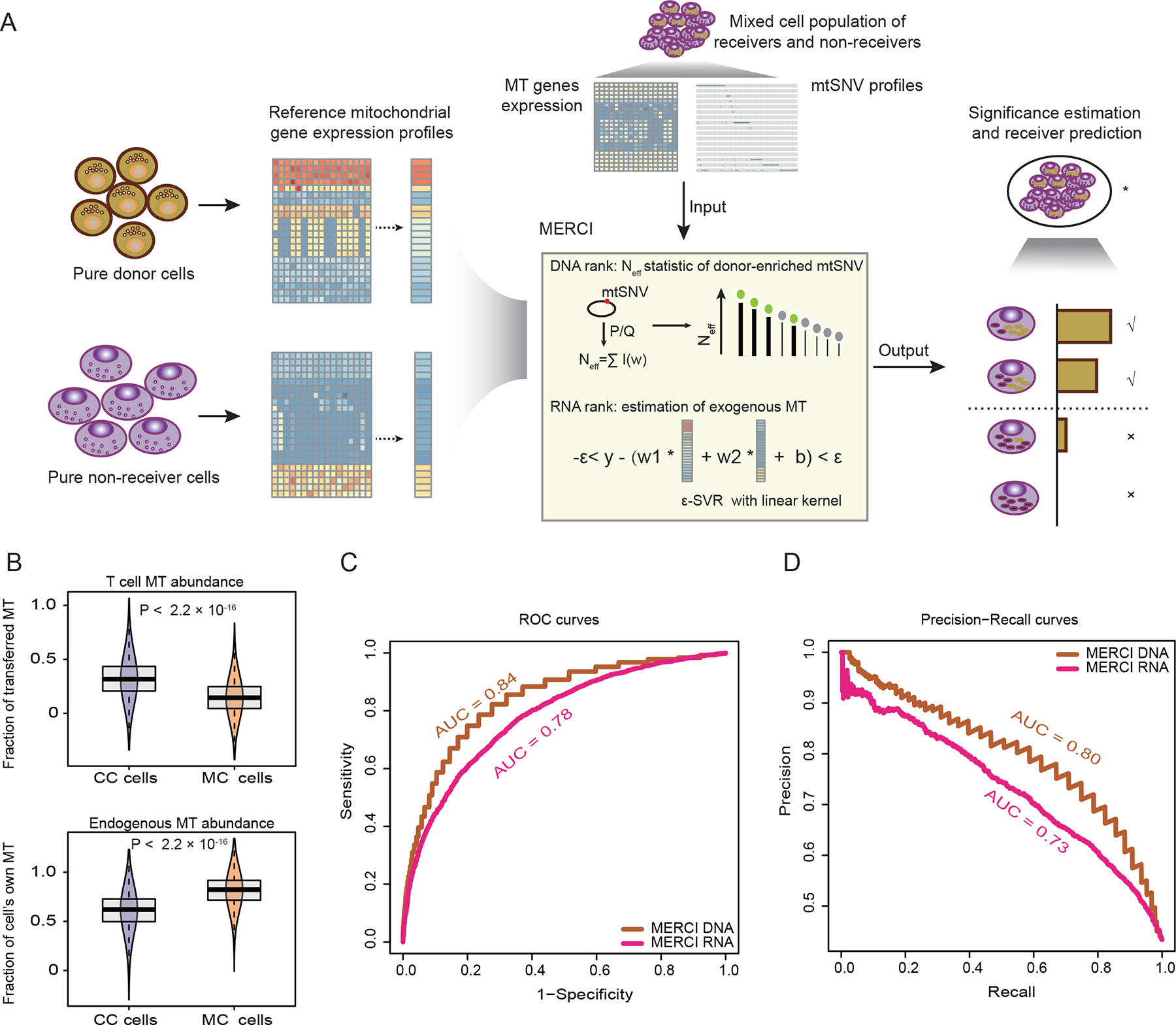
(A) Schematic illustration of MERCI. Single cell RNA-seq data from reference donor and non-receiver cells were used to deconvolute MT fractions in the cancer cell population. By combining ranks from DNA and RNA information, MERCI estimates the statistical significance of the existence of true MT receivers, and evaluates each candidate cancer cell as a receiver or not. (B) Boxplots showing the estimated abundance (SVR coefficients) of T cell transferred (upper) and endogenous mitochondria (lower) in CC and MC cancer cells. Lower and upper box limits represent 25th to 75th percentiles, central line the median and the whiskers extend to highest and lowest values no greater than 1.5× interquartile range; the violin component refers to the kernel probability density and encompasses all cells. Statistical significance was evaluated using two-sided Wilcoxon rank sum test. (C-D) Receiver operating characteristic (ROC) (C) and precision-recall (PR) (D) curves using MERCI-derived rank scores as predictors of mitochondrial receivers, i.e. CC cells. Area under the ROC and PR curves (AUC) were labeled. See also Figure S3.
We evaluated the performance of MERCI with the ground-truth scRNA-seq data. T cells and half of the MC populations were used to generate the reference MT gene expression and T cell enriched mtSNVs. CC and the held-out MC cells were mixed to produce an in silico sample to test MERCI. After deconvolution, we observed significantly higher proportion of T cell-derived mitochondria in the cocultured cancer cells (Figure 2B), consistent with our previous observation (Figure 1B). We next used the receiver operating characteristic (ROC) and precision-recall curves (PRC) to measure the prediction accuracy of MERCI. Both DNA and RNA rank scores reached a moderate level of area under the curve (AUC) ranging between 0.7 to 0.85 (Figures 2C and 2D), yet neither is accurate enough as a single predictor. As DNA and RNA ranks were estimated from independent sources, it is intuitive to combine them to achieve better performance. One simple approach is to take the averaged rank score as a predictor, which indeed reached an improved AUC of 0.88 for the ROC curve, and 0.85 for the PRC (Figures S3C and S3D). However, this strategy can become problematic when applied to real-world data, because a rank predictor will always make positive calls under a given cutoff even without any true positives in the data. Therefore, in the next section, we assessed a different approach to: 1) test if a sample contains any true MT receivers and 2) isolate the receiver cells based on MERCI rank scores.
Benchmark MERCI rank scores for real-world application
We implemented a simple strategy to call a ‘positive’ only if both ranks are higher than the given cutoff, thus putting an ‘AND’ gate after the DNA and RNA scores. Assume the cutoff is top rank 10%: in a null sample with no true positives, DNA and RNA ranks will be independent, following the same Uniform distribution. AND gate selection is expected to make 1% of positive calls. Alternatively, if the sample contains true positives, the two ranks will be correlated by MT receivers, and AND gate will predict higher percentage of positive calls. This rationale can be used to avoid making calls from disqualified samples. We first tested this strategy using the in-silico mixture sample with ~40% true positive cocultured cancer cells, and confirmed that DNA and RNA ranks are indeed correlated, leading to higher-than-expected positive calls across a range of cutoffs (Figure 3A). In contrast, when using the null sample (only monocultured cells) as the input, the reported positives were within the range established by 10,000 randomly permutated ranks (Figure 3B). These results confirmed that the existence of MT receivers in the sample generates statistical dependence between the two MERCI ranks. To quantify this signal, we defined the ‘ratio of cell number’ () as MERCI reported number of positive calls over the maximum number from 10,000 random permutations. In MERCI, we used (empirical p value <0.0001) as the criterion to determine if the input sample contained any MT receivers. This criterion performed well as we applied it to the mixture and null samples: at all non-trivial cutoffs was greater than 1 with FDR<0.05 for the mixture sample, yet insignificant and smaller than 1 for the null sample (Figure 3C).
Figure 3. Benchmarking of MERCI for real-world application.
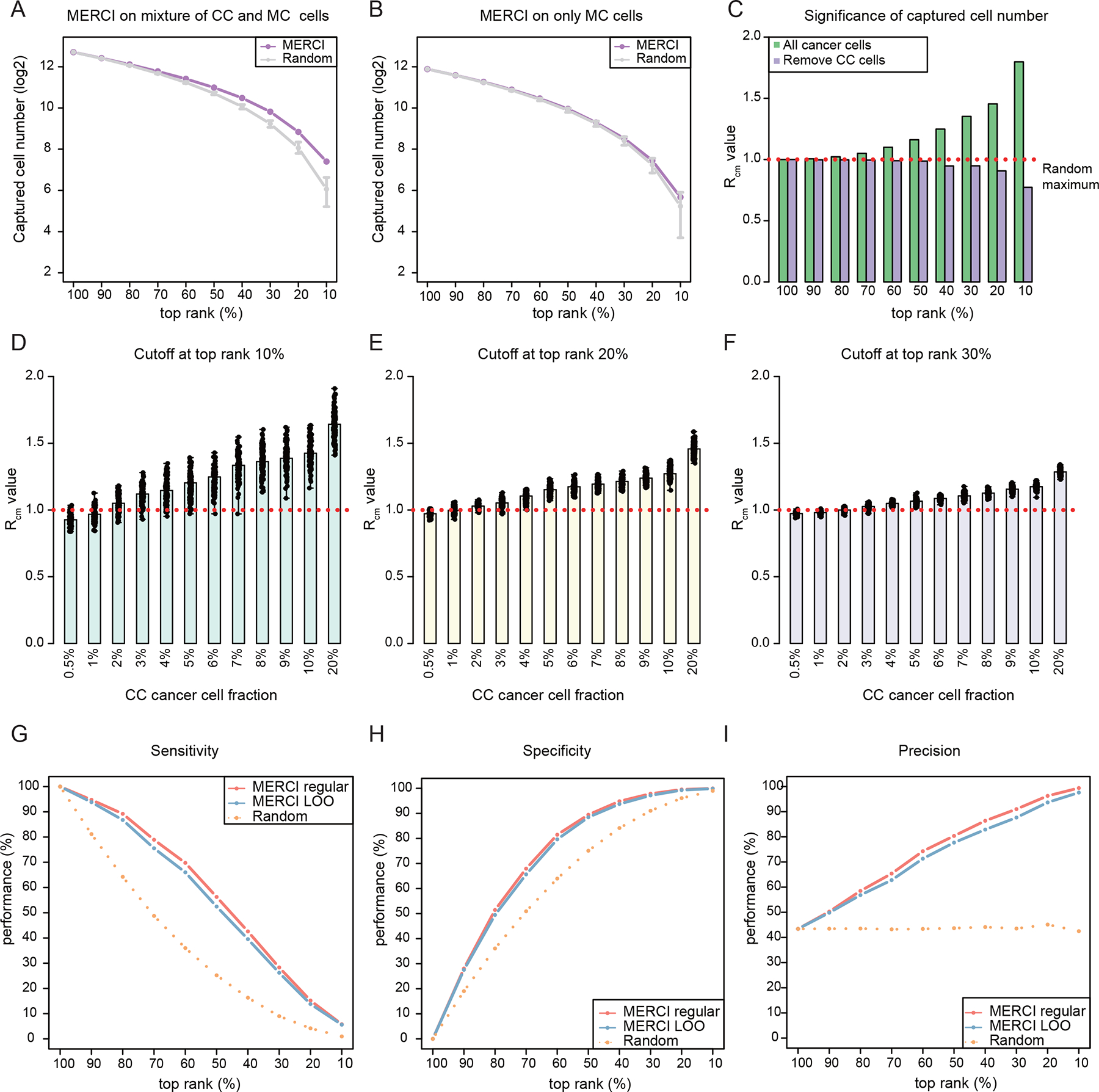
(A-B) Dot-line plots showing the number of positive calls captured by using AND gate to MERCI DNA and RNA rank scores across a range of cutoffs. Purple dots represent the results of in silico mixture sample and gray intervals indicate the ranges established by 10,000 randomly permutated ranks. Error bars indicate the range (min to max) of the data. (C) Barplots showing the values at different rank cutoffs. Red dotted line indicates . (D-F) Significance estimation of positive calls when different fractions of true receivers are included. Barplots showing the averaged values reported by MERCI for down-sampled datasets at rank cutoffs at top 10% (D), 20% (E) and 30% (F) respectively. Black dots indicate the values of down-sampled datasets. (G-I) The sensitivity, specificity and precision of MERCI when using different rank cutoffs to predict the MT receivers. See also Figure S3.
The power to detect true positives by MERCI is intuitively affected by the fraction of MT receivers in the sample. To assess the limit of MERCI when this fraction is small, we performed an in silico titration analysis by gradually lowering the percentage of CC cells from 20% to 0.5% in the mixture sample. We found that with over 7% CC cells, values remained robustly above 1 at different rank cutoffs (10~50%) (Figures 3D–3F and S3E–S3F). Further, positive calls made by MERCI were significantly enriched for the true CC cells even with lower fraction (5~15%) of positive cells (Figure S3G). As the fraction of CC cells dropped below 5%, MERCI did not always detect the true signal, yielding smaller and insignificant p values. We concluded that MERCI is sensitive to determine the existence of MT receivers for samples with over 7% of true positive cells. Depending on the cancer type, the fraction of receivers in the tumor could range between 13% to 45%26, which is sufficient for MERCI implementation.
Next, we assessed how rank cutoffs influence the prediction of MERCI using the criteria of sensitivity, specificity and precision. The benchmark in silico sample contained all the CC cells (n=2,904) as true positives and held-out MC cells (n=3,786) as true negatives, which was used to evaluate the performance of MERCI under different cutoffs (Figures 3G–3I). MERCI achieved high specificity and precision with more rigorous cutoffs, at the cost of a reduced sensitivity. When using a less strict cutoff, for example, at the threshold of top rank 50%, MERCI reached approximately 90% specificity and 80% precision, with a moderate sensitivity of 56.3%. Therefore, in our downstream analysis, we consistently used a “lenient” cutoff up to 50% given to balance the sensitivity and precision.
Finally, in a real-world scenario, cancer samples are comprised of both “receiver” and “non-receivers” cells without known class labels. Thus, it is challenging to find the ‘pure’ non-receiver cancer cells to infer the reference MT gene expression and the T cell enriched mtSNVs. To solve this problem, we implemented a ‘Leave One Out’ (LOO) strategy: for a given cancer cell of interest, we considered all the cancer cells except the given one as the non-receiver reference to estimate the rank scores. This approach is reasonable because majority of tumor cells in the cancer population do not receive mitochondria from the T cells26. We assessed the performance of MERCI LOO using the same in silico sample described above (Figures 3G–3I). The slightly lower sensitivity (Figure 3G) is expected as the presence of the true receivers in the reference may diminish the cancer-T cell difference, which does not influence the specificity (Figure 3H). Together, our analysis provided a practical way to apply MERCI to real-world samples.
Evaluation of MT transfer and MERCI performance with mtscATAC-seq
Detection of mitochondrial transfer in our analysis so far is primarily based on 10x Genomics scRNA-seq platform, which, although heavily used in recent clinical studies, is not optimized for mtDNA profiling due to its limited and uneven coverage in this region11,36. Therefore, we repeated the coculture experiments (Figure 1A) and implemented a recently developed, cutting-edge technology for mtDNA single cell profiling (mtscATAC-seq) (Table S1, Figure 4A)10 to profile the MT regions for CC, MC and primary T cells. In brief, mtscATAC-seq repurposed scATAC-seq to deeply sequence mtDNA (Figure S3H), offering significantly improved coverage (per-site, per-cell average 48X) within the mitochondrial region in our analysis (Figure 4B). In the same batch, we also generated matched scRNA-seq data (15X coverage) for each sample to evaluate MERCI.
Figure 4. Independent validation using mtscATAC-seq technique.
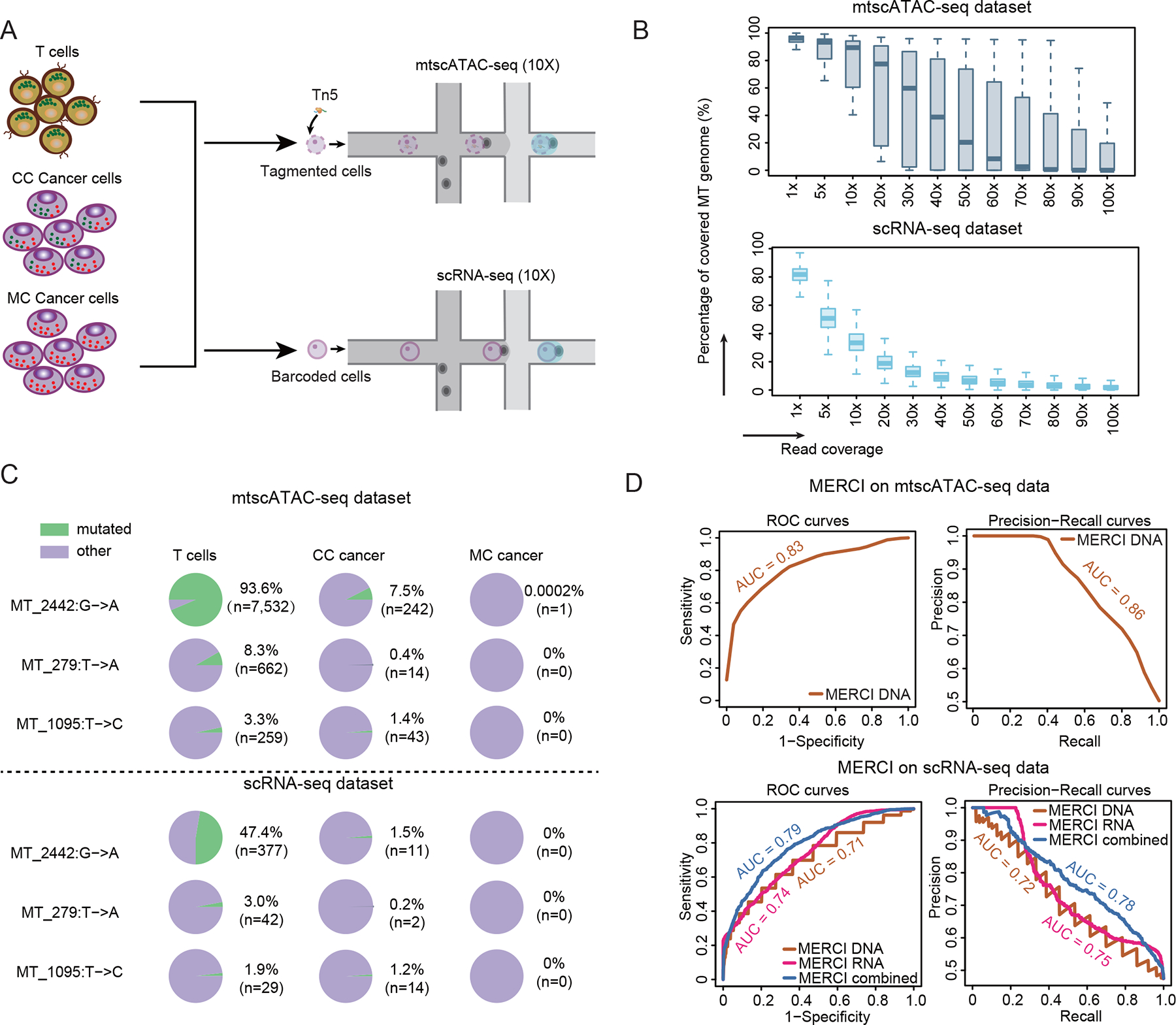
(A) Diagram showing the process of generating mtscATAC-seq and matched scRNA-seq datasets. (B) Boxplots showing the distribution of read coverage per cell in the mtscATAC-seq dataset (upper) and the matched scRNA-seq dataset (lower). Box center line: median; box limits: upper and lower quantiles; box whiskers: 1.5×interquartile range (IQR). (C) The cell frequencies of three T cell-specific variants in T cells, CC and MC cancer cell populations. Label of the variant marks the 0-index coordinate, followed by the nucleotide change. (D) Receiver operating characteristic (ROC) and precision-recall (PR) curves using MERCI-derived rank scores as predictors of mitochondrial receivers. As it is not feasible to estimate the RNA rank scores from mtscATAC-seq dataset, the upper panel only shows the performance DNA rank scores. See also Figure S3 and Tables S1–S2.
We next called mtSNVs for each sample through the MERCI-mtSNP pipeline and identified 1,989 T cell-enriched variants from mtscATAC-seq data (Table S2). As expected, CC cancer cells, the bona fide receivers, harbored significantly more T cell-enriched mtSNVs compared to the negative control (MC cancer cells), a finding also confirmed using matched scRNA-seq data (Figures S3I and S3J). A scrutiny over the variants called from mtscATAC-seq revealed several interesting mtSNVs. For example, MT_2442: G→A, among others, occurred in 94% of the T cells, while almost completely absent in the MC population. We observed that a sizeable fraction (7.5%) of cancer cells cocultured with T cells picked up this mutation (Figure 4C). These variants provided compelling genetic evidence of mitochondrial transfer from T cell to the cancer cell population. In contrast, cocultured T cells (Figure S3K) did not show higher amount of cancer-enriched mtSNVs (Figures S3L and S3M), supporting the nearly unidirectional mitochondrial transfer from T cells to cancer cells.
To further evaluate MERCI’s performance in identifying receiver cells, we applied MERCI to the mtscATAC-seq data from the mixed population of CC and MC cancer cells. The outcomes revealed that MERCI is capable of differentiating CC cancer cells from a blend of cancer cells with both ROC and PRC AUC values exceeding 0.8 (Figure 4D). We repeated this analysis using the scRNA-seq data matched to the mtscATAC-seq samples and confirmed that MERCI reached similar prediction accuracy as in the discovery cohort. These confirmed the ability of MERCI to uncover true MT receivers from single cell RNA-seq samples.
Distinct MT receiver phenotype in human solid tumor uncovered by MERCI
We next implemented MERCI to scRNA-seq samples in the public domain, which were derived from patients with basal cell carcinoma (BCC)37 or esophageal squamous-cell carcinoma (ESCC) 38. Both cohorts provided sufficient cancer and T cells that were ideal for discovery of gene markers related to MT transfer. We first analyzed the samples from three BCC patients and tested the existence of receivers in each sample using the criterion. These samples were selected as they contained sufficient numbers (n>500) of both cancer and T cells. While two samples showed no evidence of MT receivers (, Figures S4A and S4B), positive signals were detected in one sample, ‘su006’ (Figures 5A and 5B). Specifically, its values were significantly greater than 1 at the rank cutoffs of 20%–60% (FDR<0.05, Figure 5B), thus qualified the next step implementation of MERCI. We then used the LOO approach to predict MT receivers using the recommended parameters (rank cutoff=50%), and called a total of 309 tumor cells (~30% of all tumor cells). Interestingly, the predicted receiver cells formed a tight subcluster within the original cancer cell cluster, which was not observed for the predicted non-receivers (Figures 5C and S4C), indicating a converged cancer cell phenotype associated with MT transfer.
Figure 5. Distinct MT receiver phenotype predicted by MERCI in human tumor samples.
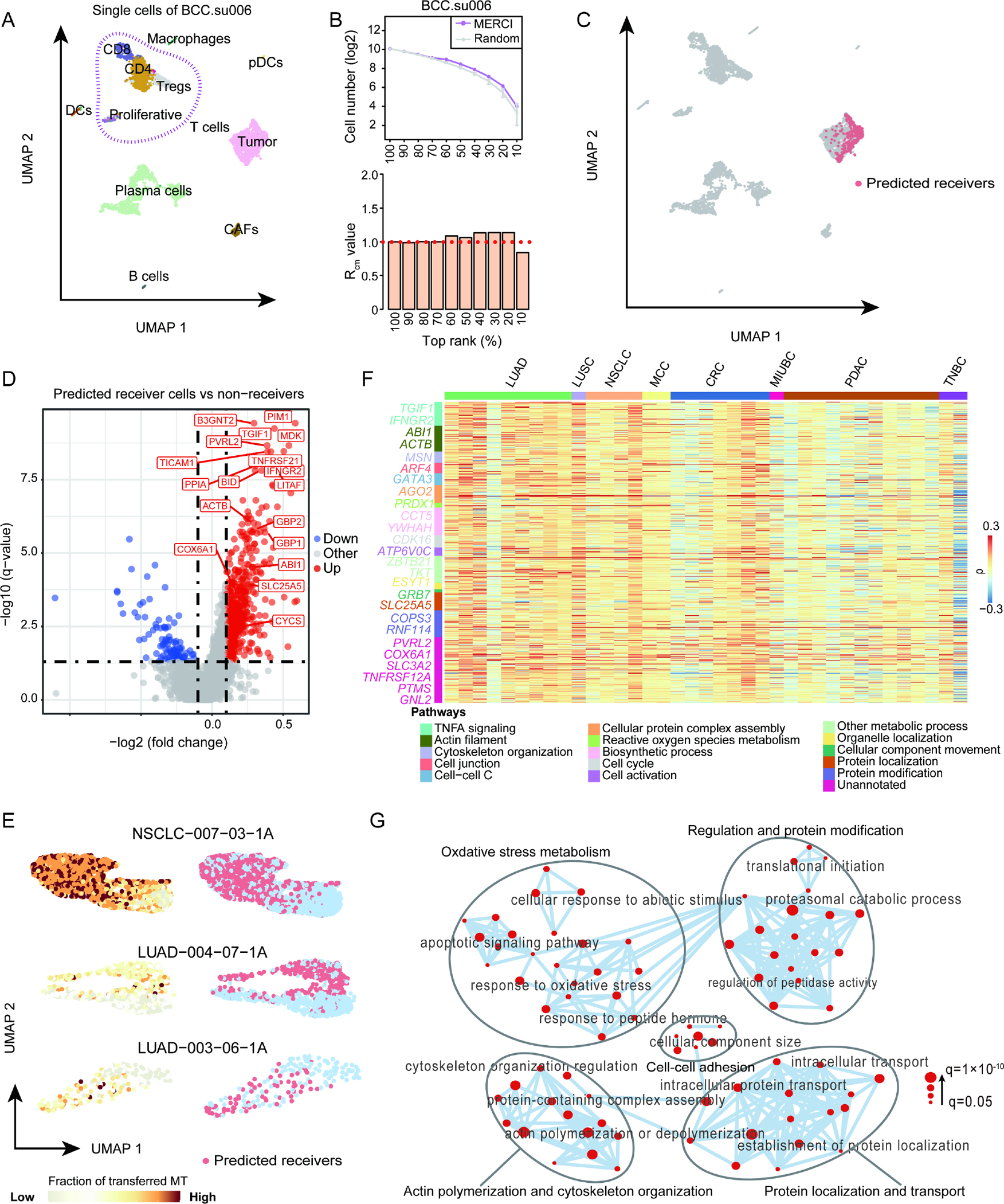
(A) UMAP plot showing the cell clusters and distribution of single cells from BCC patient ‘su006’. (B) Significance estimation of the number of positive calls reported by MERCI for scRNA-seq data of BCC patient ‘su006’. (C) UMAP plots showing the projection of MERCI predicted receiver cells. (D) Volcano plot showing DEGs of the predicted receiver cells versus non-receivers, with fold change calculated using the mean values of the two groups. Statistical significance was evaluated using two-sided Wilcoxon rank sum test, with FDR corrected using Benjamini-Hochberg procedure. (E) UMAP plots illustrating the estimated fraction of transferred MT in cancer cells (left) and the distribution of predicted receivers (right) for three selected cancer patients. These patients were chosen based on the highest Pearson correlations between the fraction of transferred mitochondria (Fr.T-Mito) and the gene expression phenotype measured with UMAP (either umap1 or umap2). (F) Heatmap showing the Spearman correlation coefficients between the expression of 608 DEGs identified in BCC dataset and the Fr.T-Mito in cancer cells across 37 samples of different cancer types. LUAD: lung adenocarcinoma, LUSC: lung squamous cell carcinoma, NSCLC: non-small cell lung cancer, MCC: merkel cell carcinoma, CRC: colorectal cancer, MIUBC: muscle-invasive urothelial bladder cancer, PDAC: pancreatic ductal adenocarcinoma, TNBC: triple negative breast cancer. The selected representative genes of different pathways were marked with different colors. (G) Gene ontology network based on the commonly enriched GO terms of the 95 MT transfer-related genes. Each node represents a gene ontology. Node size corresponds to gene ratio of each GO term vs total analyzed genes in human BCC sample. See also Figures S4–S5 and Tables S3–S5.
We proceeded to investigate this phenotype through differentially expressed genes (DEGs) of the MT receivers against the other cancer cells. The genes associated with ATP synthesis and oxidative phosphorylation, such as BID, COX6A1, CYCS and SLC25A5, were significantly upregulated in the predicted receivers (Table S3, Figure 5D), supporting that these cells carried more mitochondrial-mediated energy production. As expected, genes involved in cytoskeleton regulation and actin polymerization, such as PIM1, ACTB, PFN1, PVRL2 and GBP1/2 etc., were also expressed at higher levels, consistent with the mechanism of nanotube formation. Interestingly, essential components of the tumor necrosis factor alpha (TNFα) pathway, including TGIF1, IFNGR2 and LITAF, were among the top upregulated genes. Functional enrichment analysis of the 608 DEGs also revealed ‘TNFα signaling via NFκB’ was the most enriched pathway (Table S4, Figure S4D). Previous studies have reported that TNFα pathway is closely involved in nanotube initiation and intercellular nanotube connectivity39,40, which is in line with our observations. This phenotype was further reproduced when we analyzed the scRNA-seq data of another three patients with ESCC, where MERCI-predicted receivers also showed distinct gene expression pattern from non-receivers (Figure S4E). The signature genes of the MT receivers from the ESCC patients significantly overlapped with those from the BCC sample (Figure S4F). Specifically, genes involved in cytoskeleton remodeling, cell junction and TNFα pathways, such as ACTB, PFN1, PVRL2 and TGIF1, were also upregulated (Figures S4G–S4J).
To verify the above findings, we extended MERCI analysis to incorporate more cancer samples from the public domain by releasing the requirements for sample inclusion: ≥100 counts for both T cells and cancer cells. We assessed 156 samples with both cancer and T cells from recent clinical studies, as cataloged in CancerSCEM database41, and subsequently filtered in 61 samples from lung, gastric, skin, pancreatic, bladder, colorectal, and breast cancers (Table S1). We applied MERCI on each sample and found that the fraction of T cell-derived mitochondria in cancer cells was significantly associated the gene expression variations on UMAP plot in 61% (37/61) of these samples (|ρ| > 0.2, P < 0.001, Table S5, Figures 5E and S5A). Further, we observed that the 608 DEGs identified in BCC were also positively correlated with the estimated fraction of T cell-derived mitochondria in most of these 37 samples. Specifically, genes in pathways related to biological processes such as biosynthesis, cell activation, TNF-α signaling, and cell cytoskeleton remodeling were also significantly correlated with this fraction (Figure 5F). These findings suggested that the gene markers identified by MERCI from human samples are reproducible across multiple cancer types.
To further investigate cross-species conservation of MT transfer13, 26, we searched for all the genes positively correlated with the fraction of T cell-transferred mitochondria in cancer cells (Spearman ρ>0 and FDR<0.05) in the human BCC sample and the murine benchmark sample. We observed 95 overlapped genes as MT transfer related. The number of overlapped genes between human and mouse data is significantly higher than expected (p=1.28×10−8, Figure S5B), suggesting that these overlapped genes might be evolutionary conserved. Gene set enrichment analysis confirmed that MT transfer-related genes are mainly enriched in the pathways related to nanotube formation, such as cell-cell adhesion, actin polymerization and cytoskeleton organization (Figures 5G, S5C and S5D). These results indicated that nanotube-related genes could potentially serve as a marker of MT transfer.
Pan-cancer analysis of MT transfer in large human patient cohorts
To quantify the MT transfer activity in cancer cells or bulk tumors, we generated a 17-gene signature from the 95 evolutionary conserved MT transfer-related genes (Table S6). Selection of the 17 signature genes was guided by prior knowledge of gene function, as these genes are directly involved in the pathways associated with nanotube formation or mitochondrial activity (Figure S6A). We excluded genes involved in various metabolic pathways, such as “Oxidative phosphorylation” and “Regulation of ATP synthesis”, as they are more likely the consequences, not the cause of mitochondrial transfer42. As our goal is to find markers related to the initiation of this process, these genes were not suitable to serve as signatures. We defined the gene set enrichment score43 based on the mRNA expression of these 17 genes as tumor MT transfer (TMT) score, which served as a measure of MT transfer in tumor. To access its performance, we first calculated TMT score for each cancer cell in the murine benchmark and human BCC sample, and confirmed that the MT receivers showed significantly higher TMT scores than the non-receivers (Figures 6A and 6B). Next, the TMT score was highly correlated with the exogenous MT abundance in the cancer cells with a Spearman correlation coefficient higher than any of the 17 genes alone (ρ=0.34 for benchmark data and ρ=0.40 for BCC data, Figures S6B and S6C).
Figure 6. Functional and clinical impact of TMT score across different cancer types.
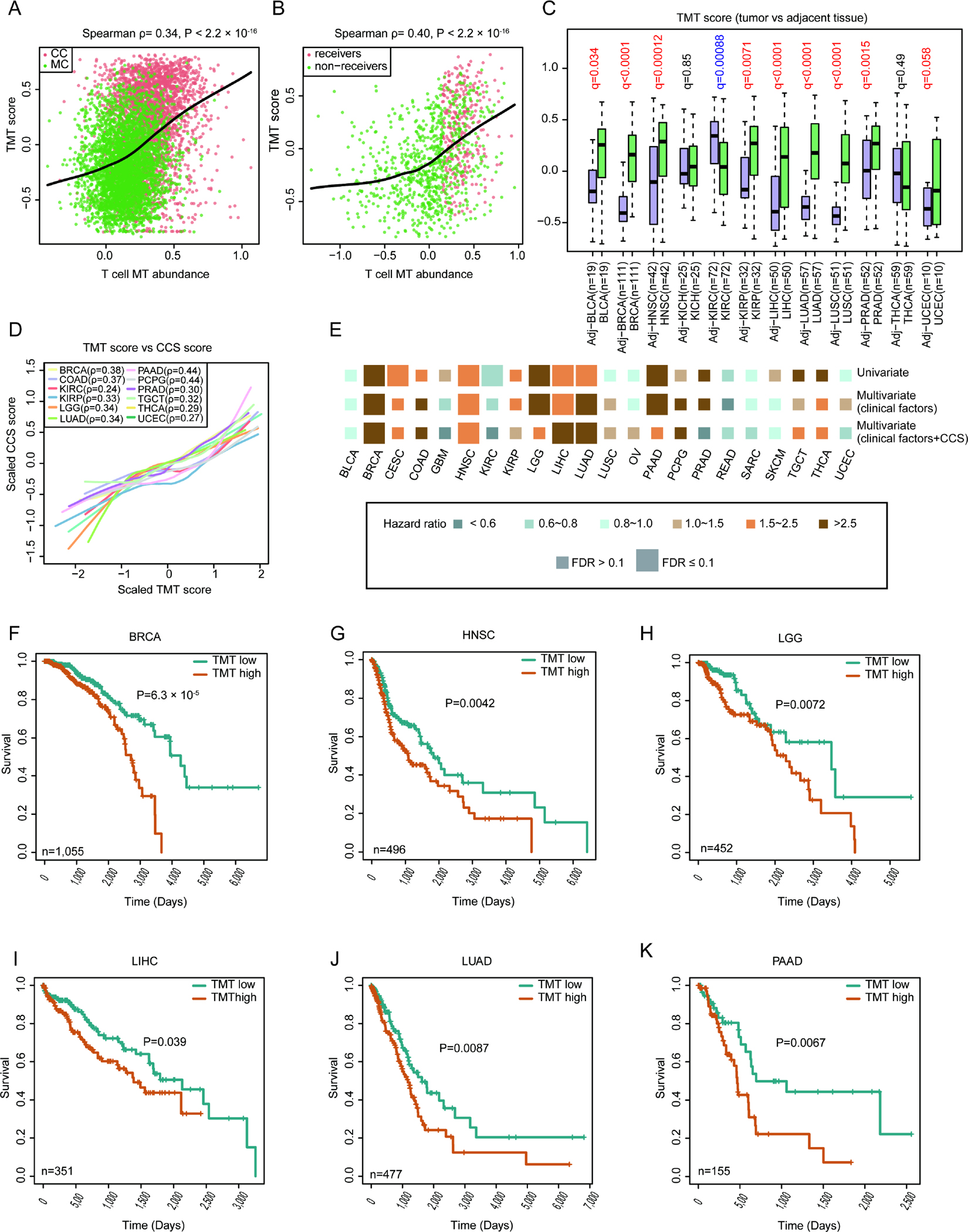
(A-B) TMT scores of cancer cells showing high correlation with the estimated foreign mitochondrial abundance for both murine training (A) and human BCC (B) data. The Spearman correlation test was used to calculate the P values. (C) The TMT scores of primary tumor and adjacent (Adj) samples in 12 cancer types. BLCA: bladder urothelial carcinoma, BRCA: breast invasive carcinoma, HNSC: head and neck cancer, KICH: kidney chromophobe, KIRC: kidney renal clear cell carcinoma, KIRP: kidney renal papillary cell carcinoma, LIHC: liver hepatocellular carcinoma, PRAD: prostate adenocarcinoma, THCA: thyroid carcinoma, UCEC: uterine corpus endometrial carcinoma. Statistical significance was estimated using non-parametric paired Wilcoxon signed-rank test, with FDR adjusted using Benjamini-Hochberg procedure. Box center line: median; box limits: upper and lower quantiles; box whiskers: 1.5×IQR. (D) Lowess smooth curves showing positive correlations between TMT scores and CCS in selected cancer types. (E) Association of TMT score with patient overall survival based on both univariate and multivariate Cox proportional hazards models in different cancer types. CESC: cervical squamous cell carcinoma and endocervical adenocarcinoma, COAD: colon adenocarcinoma, GBM: glioblastoma multiforme, LGG: lower-grade glioma, OV: ovarian serous cystadenocarcinoma, PAAD: pancreatic adenocarcinoma, PCPG: pheochromocytoma and paraganglioma, READ: rectum adenocarcinoma, SARC: sarcoma, SKCM: skin cutaneous melanoma, TGCT: testicular germ cell tumors. We applied two multivariate Cox models: one with just well-known clinical confounders as covariates and the other with clinical confounders plus CCS as a covariate. Size denotes statistical significance at the cutoff of FDR=0.1; color denotes the hazard ratio. (F-K) Kaplan-Meier estimates of overall survival, according to TMT score calculated from RNA-seq data of BRCA (F), HNSC (G), LGG) (H), LIHC (I), LUAD (J) and PAAD (K). The patients were stratified into two groups (TMT high and low) based on median value of TMT scores. Statistical significance was evaluated using log-rank test. See also Figure S6 and Table S6.
To further investigate the functional relevance of MT transfer in cancer, we estimated the TMT score of 10,628 human cancer or adjacent normal samples in The Cancer Genome Atlas (TCGA) database. To ensure statistical power, we analyzed the 22 major cancer types with over 100 patients. In 9 out of the 12 cancers with at least 10 adjacent normal samples, we observed significantly higher TMT scores in the tumors than in the matched normal tissues (Figure 6C), consistent with the enhanced ability of mitochondrial capture in cancer cells29,42,44. Further, as MT transfer is reported to promote recipient cell proliferation and survival through improved energy production45,46, we assessed the association of TMT score with cell cycle activity47. To get an unbiased estimation, we applied partial spearman correlation corrected for tumor purity for the related variables. Consistently, the TMT score is positively correlated with cell cycle score (CCS) in almost all cancer types (Figure S6D). In 12 out of 22 cancers, the TMT scores are highly correlated with CCS (partial Spearman ρ>0.2, FDR<0.05, Figure 6D), suggesting increased cancer cell proliferation induced by MT trafficking.
Hypoxia is a common signature of cancer48, and therefore, oxygen deficiency in the tumor microenvironment could influence the activity of MT transfer. We next investigated this potential connection using a recently published estimation of hypoxia status in the TCGA samples49. Interestingly, in non-small cell lung and pancreatic cancers, tumor hypoxia level is significantly correlated with the TMT score (partial Spearman ρ>0.2 adjusted for tumor purity, FDR<0.05, Figures S6E and S6F). One possible explanation to this result is the previously reported mechanism that limited oxygen supply promotes cancer cells to hijack foreign mitochondria to keep up with energy production50.
Finally, to investigate the potential role of MT transfer in prognosis, we performed univariate and multivariate Cox regression with TMT score as the primary predictor, while controlled for potential clinical confounders including age, tumor grade, clinical stage, tumor purity, etc. We observed that high TMT scores were consistently associated with poor survival in multiple cancer types (Figure 6E), including breast, head and neck, brain, liver, lung and pancreatic cancers (Figures 6F–6K). Given the high correlation between TMT score and cell cycle activity, we speculated that TMT score’s impact over patient outcome might be through CCS, which has also been reported to influence survival47. Interestingly, when adding CCS as a covariate in Cox model, the hazard ratio of TMT score became non-significant in brain (LGG) and pancreatic (PAAD) cancers, while the other cancer types remain unaffected (Figure 6E). These results provided evidence that MT acquisition by cancer cells might promote proliferation, which ultimately caused shortened survival in selected cancers. Meanwhile, TMT score still serve as an independent prognostic predictor for other cancer types, which is in line with the fact that mitochondrial transfer metabolically empowers the aggressive behavior of cancer cells and causes immune cell dysfunction26.
Discussion
In this work, we described MERCI for tracing MT transfer from T cells to cancer through combined use of MT mutation and gene expression profiles from single cell RNA-seq samples. MERCI was validated and benchmarked using experimentally generated ground-truth data, with demonstrated utilities in the real-world settings. Application of MERCI to the human cancer samples uncovered a previously unreported cancer phenotype related to MT transfer. Analysis of large human patient cohorts through TMT score further revealed that MT transfer might negatively influence patient outcome, partially through promoting cancer cell proliferation.
Nuclear mitochondrial DNA segments (NUMT)51,52 generated via the translocation of mtDNA fragments into the nuclear genome has been presented as a confounder in studies using MT variants as cell lineage markers. This is because variants derived from the NUMT regions usually satisfy the inclusion criteria designed for lineage tracing, such as high cellular heteroplasmy. NUMT were not explicitly handled our study, though, as they usually occur in all cell types and will unlikely contribute T cell enriched variants for the downstream inference in MERCI.
Horizontal MT transfer between mammalian cells is mediated by tunneling nanotubes (TNTs), gap junctions, microvesicles or direct extrusion and internalization24,53,54, with TNT formation being the most prevalent mechanism for intercellular mitochondrial transport20,29. TNT-mediated MT hijack from T cells might serve as another immune evasive approach in vivo26. Thus, selectively blocking the formation of TNTs can be a promising antitumor therapeutic opportunity. However, due to the lack of specific TNT-markers, the current pan-inhibitors (such as farnesyltransferase inhibitors and cytochalasin) only partially prohibit TNT formation26,55, leading to limited tumor growth control. We discovered that MT transfer is highly associated with many putative regulators of the actin filament network (Table S3–4), such as PIM1, MYO1B, PFN1 and ABI1, which might be candidates for TNT disruption24,56,57. Another interesting target is cell adhesion molecule PVRL2 (encoding Nectin-2), a top predicted marker of MT transfer (Figure 5D). PVRL2 might participate in MT transport, as its disruption results in morphologically aberrant spermatozoa of mice with defects in mitochondrial localization58.
The dysfunction of tumor infiltrating lymphocytes (TILs) is one of the main reasons for uncontrolled tumor progression59,60. In parallel to the heavily-investigated immune checkpoint molecules (such as PD-1, LAG3, TIM-3 and CTLA-4), the newly discovered mitochondrial drainage by cancer cells provided another possible mechanism for the metabolic dysfunction and exhaustion of TILs. As reported in previous work, partial loss of mitochondria in T cells may lead to shortage of energy production and dysfunction61,62. Capturing the signatures of T cell exhaustion resulted from MT transfer may reveal novel genes or pathways for T cell revitalization. Although MERCI was optimized to detect gain-of-MT signals in the cancer cells, its framework could be reconfigured to detect donor T cells with proper benchmark experiments and training data. We thus anticipate a better understanding of tumor-immune interactions to unfold with the future investigation of the donor T cells in the process of MT transfer. Related to this point, in fact, both this study and Saha et al.26 observed a small fraction (3–5%) of T cells that received some mitochondria from cancer cells. This signal is much weaker compared to the transfer from T cells to cancer cells, both in receiver cell percentage as well as the amount of mitochondrial translocation. Although weak, this effect might be useful to detect antigen-specific T cells closely attached to the cancer cells.
Limitations of the Study
While our analyses provided more insights into this process, there are several important aspects that remain unknown. First, what is the driving mechanism that regulates mitochondria uptake by the cancer cells? GTPase Miro1 has been reported to mediate mitochondrial trafficking63,64, yet it did not emerge as a top hit in the unbiased genomic analysis. Our data suggests that MT receivers upregulated multiple genes involved in cell junction and cytoskeleton remodeling pathways (ACTB, GBP1/2, PIM1, PFN1, etc) that might directly or indirectly influence MT trafficking65,66. Selective knockout of these top targets will be useful to illustrate their roles in mitochondrial transfer. Second, can non-malignant cells acquire mitochondria from T cells under physiological conditions? Mitochondrial transfer to damaged or stressed epithelial cells has been repeatedly observed in previous studies as a way of quick tissue repair67,68. So far, mitochondrial drainage targeting T cells from any non-malignant cell types has not been reported. We posit that due to their small size and relatively smaller mitochondrial reservoir, T cells are not ideal donors of mitochondria in damaged tissues. Last, why different cancers (lung, breast, pancreatic, skin, etc) all demonstrated the same phenotype? Despite high-level of heterogeneity, cancer cells exhibited common hallmarks, such as sustained growth signaling, metastasis, immune evasion, etc69. It is possible that drainage of mitochondria from tumor-infiltrating T cells delivers selective advantages that caused the convergent evolution of cancer cells with diverse genetic backgrounds. While immune evasion is an apparent mechanism for survival benefit70, further investigations will be needed to confirm: 1) if tumor-associated T cells are preferentially targeted in this process and 2) if and how T cells become dysfunctional after mitochondrial transfer.
Low detection sensitivity is a potential limitation of MERCI because per cell MT read coverage could be low for some 10x libraries, leading to compromised detection power of MT mutations. Using libraries sequenced at 100K reads per cell (2.3K reads in the MT region), MERCI reached 56.3% sensitivity in our benchmark. Further increase of coverage of the mitochondrial genome is expected to improve its performance. Recently, Tyler et al. introduced the MAESTER scRNA-seq platform, which combined standard 10x protocol with mitochondrial transcriptome enrichment to increase per site and per allele MT coverage for each cell, enabling high-confidence MT variant detection in scRNA-seq dataset33. mtscATAC-seq implemented in this study is another example of successful mtDNA enrichment for single cell genomic analysis. Adaptation of these new technologies for future data generation will likely increase the prediction accuracy of MERCI and broaden its application. In addition, although we defined TMT score to investigate MT transfer in the bulk tumor samples, it should be noted that this score is derived from a composite gene signature that might not be exclusive to this biological process. Conclusions regarding this analysis require future validations with tailored clinical samples.
STAR ★ METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources, reagents and codes should be directed to and will be fulfilled by the Lead Contact, Dr. Bo Li (lib3@chop.edu).
Materials availability
This study did not generate any new unique reagents or models.
Data and code availability
scRNA-seq datasets generated in this study, as well as the MT variants and coverage information are available at Zenodo https://doi.org/10.5281/zenodo.8065206, and Gene Expression Omnibus (GEO) database with accession number GSE235675. The other scRNA-seq datasets of human cancer are available under the GEO accession codes provided in Table S1 and Key Resources Table. The TCGA data is available at https://portal.gdc.cancer.gov/. The source codes of MERCI-mtSNP and MERCI R package, and the codes to run example data are available at: https://github.com/shyhihihi/MERCI. Any additional information is also available from the corresponding author (Dr. Bo Li) upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor® 700 anti-mCD8α (clone53–6.7) | Biolegend | Cat# 100730 |
| Ultra-LEAF™ Purified anti-mouse CD3 Antibody | Biolegend | Cat# 100239 |
| Ultra-LEAF™ Purified anti-mouse CD28 Antibody | Biolegend | Cat# 102115 |
| Chemicals, peptides, and recombinant proteins | ||
| MitoTracker™ green FM | ThermoFisher | Cat# M7514 |
| MitoTracker™ red CMXRos | ThermoFisher | Cat# M7512 |
| PEI MAX | Polysciences | Cat# 24765–1 |
| Fixable viability Dye eFluor 506 | eBioscience | Cat# 65–0866-18 |
| Critical commercial assays | ||
| CD8+ T cell isolation kit | Stem Cell Technologies | Cat# 19853 |
| Chromium Next GEM Single Cell ATAC Kit v2 | 10x Genomics | PN-1000406 |
| Chromium Single Cell V(D)J Reagent Kit v1.1 | 10x Genomics | PN-1000167 |
| Chromium Next GEM Chip H Single Cell Kit | 10x Genomics | PN-1000162 |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | Cat# 5067–4626 |
| Mitochondria Isolation Kit for Cultured Cells | Thermo Fisher Scientific | Cat# 89874 |
| Deposited data | ||
| Raw and processed data | This paper | GEO: GSE235675 |
| Processed data | This paper | Zenodo:https://doi.org/10.5281/zenodo.8065206 |
| Human reference genome UCSC, GRCh38; Murine reference genome UCSC, mm10 | University of California Santa Cruz | https://genome.ucsc.edu/cgi-bin/hgGateway |
| Full list of public single cell datasets used in this study | N/A | Table S1 |
| scRNA-seq data for BCC samples | Yost et al.37 | GEO: GSE123814 |
| scRNA-seq data for ESCC samples | Zhang et al.38 | GEO: GSE160269 |
| scRNA-seq data for NSCLC samples | Zeng et al.41 | EBI: E-MTAB-6149 |
| scRNA-seq data for CRC samples | Zeng et al.41 | EBI: E-MTAB-8410 |
| scRNA-seq data for MCC samples | Zeng et al.41 | GEO: GSE117988 |
| scRNA-seq data for MCC samples | Zeng et al.41 | GEO: GSE118056 |
| scRNA-seq data for LUAD samples | Zeng et al.41 | GEO: GSE123904 |
| scRNA-seq data for STAD samples | Zeng et al.41 | GEO: GSE134520 |
| scRNA-seq data for NSCLC samples | Zeng et al.41 | GEO: GSE143423 |
| scRNA-seq data for TNBC samples | Zeng et al.41 | GEO: GSE148673 |
| scRNA-seq data for MIUBC samples | Zeng et al.41 | GEO: GSE145137 |
| scRNA-seq data for PDAC samples | Zeng et al.41 | GSA: PRJCA001063 |
| bulk RNA sequencing data for cancer samples | TCGA | https://portal.gdc.cancer.gov/ |
| Experimental models: Cell lines | ||
| KrasG12D/p53ko (KP) lung cancer cells | Dr. Esra Akbay | N/A |
| KP-mito-DsRed | This paper | N/A |
| MC38 colon cancer cells | ATCC | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jackson Laboratory | Cat# 000664 |
| Recombinant DNA | ||
| pDsRed2-Mito | Takara | Cat# 632421 |
| Software and algorithms | ||
| MERCI-mtSNP and MERCI R package code | This paper | https://github.com/shyhihihi/MERCI |
| CytExpert | Beckman Coulter, Inc | https://www.beckman.com/coulter-flow-cytometers/cytoflex/cytexpert |
| FlowJo | Tree Star Inc. | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism software 7.0 | GraphPad Software, Inc. | https://graphpad.com/scientific-software/prism/ |
| Python version 3.8 | Python Software Foundation | https://www.python.org/downloads/ |
| R version 4.0.2 | The R Foundation | https://www.r-project.org/ |
| Cell Ranger 3.1.0 | 10x Genomics | https://10xgenomics.com/ |
| Cell Ranger ATAC 2.0.0 | 10x Genomics | https://10xgenomics.com/ |
| Seurat 3.1.2 | Seurat developers | https://satijalab.org/seurat/articles/get_started.html |
| pysam | pysam developers | https://github.com/pysam-developers/pysam |
| e1071(version 1.7.4) | N/A | https://cran.r-project.org/web/packages/e1071/index.html |
| fgesa(version 1.16.0) | N/A | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| msigdbr (version 7.2.1) | N/A | https://cran.r-project.org/web/packages/msigdbr/index.html |
| simplifyEnrichment (version 1.0.0) | N/A | https://bioconductor.org/packages/release/bioc/html/simplifyEnrichment.html |
| Cytoscape (version 3.9.1) | Cytoscape developers | https://cytoscape.org/ |
| GSVA | Hänzelmann et al.43 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| BioRender | biorender.com | https://www.biorender.com/ |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cancer cell line and culture
The KP lung cancer cells were provided initially by Dr. Esra Akbay. The KP-mito-DsRed stable cell line was generated in the lab with pDsRed2-Mito vector (Takara, Cat# 632421) transfection using PEI MAX® (Polysciences, Cat# 24765–1) with long-term selection using G418 following manufacture’s protocol. KP cell lines were grown in 5% FBS (ThermoFisher Scientific, Cat# 26140079) supplemented RPMI 1640 (Millipore Sigma, Cat# R8758). Cell lines were routinely tested using a mycoplasma contamination kit (E-myco, Cat# 25233, Bulldog Bio) and cultured under 5% CO2 at 37 °C. The MC38 cell lines were purchased from the American Type Culture Collection and cultured in DMEM supplemented with 10% heat-inactivated FBS, 100 U ml−1 penicillin and 100 U ml−1 streptomycin under 5% CO2 at 37 °C.
Mice
6–8 weeks old female C57BL/6J mice were purchased from Jackson Laboratory and housed at UTSW Animal Resource Center. All mice were maintained at 68–79°F, 30–70% humidity, in individually ventilated cages, with no more than five mice per cage on 12-hour on:off light:dark cycles. Mice had unrestricted access to RO chlorinated water and irradiated 2916 Teklab global diet (Envigo, Cat# 2916). The mice were housed at least for a week before starting the downstream process. Animal care and experiments were performed to comply with institutional and National Institutes of Health protocols and guidelines. This study has been approved by the Institutional Animal Care and Use Committee of University of Texas Southwestern Medical Center.
METHOD DETAILS
T cell isolation, activation and mitochondria labeling
Splenocytes were isolated by harvesting the spleen from 6–8 weeks old mice following red blood cell lysis with ACK buffer. CD8+ T cells were isolated by immunomagnetic negative selection using EasySep™ mouse CD8+ T cell isolation kit (Stem Cell Technologies, Cat# 19853). Isolated T cells were activated with 1ug/mL anti-CD3/CD28 (Biolegend, Cat#100239 and Cat#102115) antibodies for 24h. Activated CD8+ T cells were then stained with 100 nM MitoTracker™ green FM (ThermoFisher, Cat# M7514) at 37°C for 30 min following the manufacturer’s instruction. MitoTracker™ green FM (ThermoFisher, Cat# M7514) at indicated concentration would not stain all T cell mitochondria positive, to avoid non-mitochondrial labeling. Stained CD8+ T cells were then washed with medium, centrifuged down, and then resuspended to remove exceeded dye in the medium. Isolated CD8+ T cells were maintained in 10% heat deactivated FBS supplemented RPMI 1640 (Millipore Sigma, Cat# R8758). In the control experiment, KP-mito-DsRed cancer cells were labeled with MitoTracker™ red CMXRos (ThermoFisher, Cat# M7512) using the same procedure.
Coculture assay and flow cytometry analysis
The KP-mito-DsRed cancer cells were plated at 2×106 cells/well of 6 well plate overnight to allow cell attachment before co-culturing. MitoTracker™ green FM stained CD8+ T cells were then added to the tumor cells at 2×105 cells/well with a final volume of 2ml of RPMI 1640 supplemented with 10% heat deactivated FBS in each well. Cancer cells and CD8+ T cells were then cocultured under 5% CO2 at 37 °C for 24h before mitochondria horizontal transfer evaluation. Co-cultured supernatant containing CD8+ T cells were then collected, as well as 0.25% trypsinized co-cultured KP-mito-DsRed cells. All the cells were spined down, and a single cell suspension was used to perform staining and analysis. Samples were incubated with anti-FcγIII/II receptor (clone 2.4G2) for 15 min at 4 °C to block potential non-specific binding of conjugated antibodies. Anti-mCD8α (Biolegend, Cat# 100730) were incubated with single-cell suspension for 30 min at 4 °C. Fixable viability Dye eFluor 506 (eBioscience, Cat# 65–0866-18) was used to exclude the dead cells. Subsequently, the dissociated cells were filtered through a 70 μM cell strainer and resuspended in FACS buffer before analysis. Data were collected and analyzed on CytoFlex flow cytometer (Beckman Coulter, Inc). To control the effect of different co-culture ratios on mitochondrial transfer. A wide range of co-culture ratios (10:1, 5:1 and 2:1) were also tested in our control experiments for KP and MC38 cancer cells.
Single cell suspension preparation and flow sorting
CD8+ T cells successfully labeled with MitoTracker™ were sorted as FITC positive through BD FACSMelody™ sorter. Single-cell suspension of co-cultured KP cells (either DsRed or mitoTracker Red labeled cells) and CD8+ T cells were prepared and stained as described during flow cytometry analysis. KP cells containing mitochondria derived from T cells (CD8 negative, FITC and DsRed/MitoTracker Red positive) and CD8+ T cells were sorted with BD Melody flow sorter. Monocultured cancer cells and CD8+ T cells were prepared as single-cell suspension simultaneously following the same protocol as the cocultured cancer cells.
mtscATAC-seq libraries preparation and sequencing
Mitochondrial single-cell assay for transposase-accessible chromatin with sequencing (mtscATAC-seq) technique was performed strictly following the revised 10x scATAC-seq protocol developed by Lareau et al.10,71. Briefly, after washing, the sorted cells were fixed in 1% formaldehyde (FA; ThermoFisher #28906) in PBS for 10 min at RT, quenched with glycine solution to a final concentration of 0.125 M before washing cells twice in PBS via centrifugation at 400 g, 5 min, 4°C. Cells were subsequently treated with lysis buffer (10mM Tris-HCL pH 7.4, 10mM NaCl, 3mM MgCl2, 0.1% NP40, 1% BSA) for 3 min for CD8+ T cells and 5 min for KP cells on ice, followed by adding 1 ml of chilled wash buffer and inversion (10mM Tris-HCL pH 7.4, 10mM NaCl, 3mM MgCl2, 1% BSA) before centrifugation at 500 g, 5 min, 4°C. The supernatant was discarded, and cells were diluted in 1x Diluted Nuclei buffer (10x Genomics) before counting using Trypan Blue and a Countess II FL Automated Cell Counter (Invitrogen). mtscATAC-seq libraries were generated using the 10x Chromium Controller and the Chromium Next GEM Single Cell ATAC Kit v2 (PN-1000406) according to the manufacturer’s instructions (CG000496 Rev-B), and sequencing was run on Illumina NovaSeq 6000 using paired-end sequencing of 150-bp read length, aiming to allocate 35,000 paired-end reads per cell in a mtscATAC-seq library.
scRNA-seq libraries preparation and sequencing
scRNA-seq was performed by the 10x Genomics single-cell 5′ V(D)J library platform. Single-cell suspension of negative controls and sorted cells were counted manually under a microscope stained with trypan blue. The concentration of single-cell suspensions was adjusted to 900–1000 live cell/μl. Cells were loaded at 10,000 cells/chip position. Single-cell libraries were generated with Chromium Single Cell V(D)J Reagent Kit (10x Genomics, PN-10000006, PN-10000009) following per manufacturer’s instruction. Purified libraries were analyzed by Illumina NovaSeq with 150-bp paired-end reads at a targeted median read depth of 100,000 reads per cell.
Data processing and read alignment
For each scRNA-seq library used in this study, fastq files were processed using the Cell Ranger (v6.0.0) Software Suite. Cellranger count was used to align reads to mouse mm10 or human GRCh38 reference genome, as well as to filter and quantify. The generated files of Cell Ranger for each library included a cell barcode file, a feature file and a gene expression matrix (in unique molecular identifier (UMI) count). Seurat pipeline (v3.1.2) was applied to the raw UMI count table, following log-normalization. A normalized gene expression matrix was generated for each sample. The cell barcode file was used for subsequent mitochondrial variant calling, and only MT reads associated with cell barcodes listed in this file were retained. The raw data from mtscATAC-seq libraries underwent preprocessing with the Cell Ranger ATAC pipeline (version 2.0.0) and alignment with the mm10 reference via the cellranger-atac count command. The cell barcodes listed in ‘filtered_peak_bc_matrix’ of the output files were utilized for subsequent mitochondrial variants calling.
Analysis of mouse scRNA-seq data
All normalized expression matrices were merged into one file by the standard Seurat v3 integration workflow72 to remove potential technical variabilities. Since our purpose is to analyze the mitochondrial genomic feature of every single cell, we performed an additional filtering operation to remove those cells with low read depths in the mitochondrial genome region. The aligned and processed .bam files generated from Cell Ranger were further processed using customized Python scripts. Cells with at least 1,000 reads uniquely mapping to the mitochondrial genome were considered for downstream analysis.
UMAP projection using mitochondrial reads
To visualize the mitochondrial transcriptional data in a 2-dimensional space, we performed a uniform manifold approximation and projection (UMAP) embedding of the cells by their mitochondrial read coverage. First, we extracted all the high-quality reads (mapping score=255) that uniquely mapped to the mitochondrial genome, and calculated the basewise coverage for each cell. When counting the coverage for each base, we only considered the reads with base quality > 15. The 16.3-kb long genome of mouse MT provided a read-depth matrix, with 16.3 k rows and columns corresponding to each cell. Raw read counts were then log-normalized for each cell using Seurat. The ScaleData function was used to calculate the scaled z-scores for mitochondrial genome sites, which were used as input of the principal component analysis (PCA). With the first 10 principal components as features, the UMAP projections were generated using the RunUMAP function.
Overview of MERCI
Mitochondrial variant calling by MERCI-mtSNP
To efficiently retrieve the cell-level mitochondrial variants from single-cell sequencing data, we developed a mutation calling pipeline called MERCI-mtSNP (Figures S2B). MERCI-mtSNP was designed to call single-nucleotide variants (SNV) in MT genomics data generated from popular bulk or single-cell sequencing technologies, such as 10x Genomics scRNA-seq, smart-seq2 and ATAC-seq. More specifically, the aligned bam file was used as the input of MERCI-mtSNP, and all reads uniquely aligned to the mitochondrial genome were extracted. For 10x scRNA-seq and mtscATAC-seq data, the reads were further separated by cell barcodes. This step generated a new mitochondrial bam (MT bam) file only containing those extracted mitochondrial reads. We performed variant calling for each cell independently, keeping cells with at least K mitochondrial reads for the scRNA-seq (K=1,000) and mtscATAC-seq (K=2,000) data. The imported MT bam file using the pysam module determined per base and per allele read counts. We removed those reads marked with ‘duplicate’, ‘secondary’, ‘supplementary’ or ‘ummaped’, and only kept the reads with the high mapping score (255 for scRNA-seq, >=5 for mtscATAC-seq). In mtscATAC-seq data, we observed that some regions in the mtDNA regions are similar to the mouse genome, resulting in multiple mapping locations low mapping score. To include reads in these regions, we lowered the mapping quality cutoff while restricted the primary mapping location to mtDNA. Per-based coverage statistics were then calculated using the remaining mitochondrial reads. For each altered base at a given locus, the variant allele frequency (VAF) was computed by using the number of the supporting reads divided by the total read depth. To get high-quality variants, only the aligned reads with base-quality score >15 (>25 for mtscATAC-seq) were used to calculate the VAF values. In the scRNA-seq data, we noticed the existence of ‘mutation clusters’, which are consecutive variants occurring within a small genomic region, which are likely to be some false positives due to technical artifact. Therefore, variants observed in the clustered genome loci (within 5 bp) with only supported by one altered read were removed. A csv file and txt file would be generated as the output of MERCI-mtSNP, representing the information of MT coverage and MT variants, respectively.
Since MERCI-mtSNP ignores the mutations in the nuclear genome, it has a favorable computational efficiency with 52s for running out a single cell smart-seq2 data (450,358 MT reads) or 2.5h for a normal-sized10x scRNA-seq data (8,721 cells, 1,147 MT reads per cell) with 256G memory and 12 CPU nodes.
Binomial model-based filtering of MT variants
Assuming a mitochondrial single nucleotide variant (SNV) with a true heteroplasmy level , the statistical power for detecting this mutation depends on the local sequencing coverage at the site of this variant. It should be noted that heteroplasmy indicates the proportion of mitochondrial genomes containing a specific mutation, which is different with observed VAF. VAF is usually determined by multiple factors including heteroplasmy, sequencing error and read depth. Assuming the sequencing error to be ), the probability of observing at least one alternate read can be written as:
in which
Where and represent the probabilities of a read being altered and wild type, respectively. The probability of detecting mutation at a heteroplasmy of 0.2 is greater than 60% when read coverage is 5 or higher (Figures S2F). Therefore, only variants with at least 5 total reads (wild-type and variant) reads were kept. For a given cell and a given variant, if the coverage at the locus is >= 5, we define this variant “observable” in this cell. Variants observable in less than 10% of cells were removed. Unobservable positions are represented as missing values. We implemented additional filtering for mtscATAC-seq data because its comparatively higher coverage per site per cell can result in more sequencing errors. Specifically, the probability of observing at least identical alternate reads due to sequencing error can be expressed as follows:
The minimum number of alternate reads supporting that is less than a given false-positive rate (FPR) is:
With FPR set to 5×10−4, we can compute a value for each variant. All variants with supporting reads fewer than were considered as potential sequencing errors (FPR > 5×10−4), and removed from the mtSNV profile. Finally, we obtained a matrix of VAF with rows being SNPs and columns being cells, which was further transformed into a binary matrix: with 0 being VAF=0 and 1 VAF>0.
Defining T cell enriched/depleted MT variants
The monocultured cancer cells were randomly split into a training and a validation set. T cell enriched mtSNVs were defined as those more frequently occurring in T cells than in cancer cells of the training set (FDR<0.1, OR>1). Similarly, T cell depleted variants represented the mutations less observed in T cells (FDR<0.1, OR<1). Statistical significance was estimated using Chi-square test followed by FDR correction with the Benjamini–Hochberg (BH) procedure.
‘Effective count statistic’ of T cell enriched mtSNV
The observation of T cell enriched mtSNVs in cancer cells provides key evidence of MT transfer. In this study, we consider each cancer cell as a potential receiver of mitochondria from T cells. Let denote the set of mutations that are enriched in the T cell population. For a given cancer cell and mutation in , we observe the coverage of this site and the count of mutated reads ). Although the ratio is an unbiased estimation of the heteroplasmy of the mutation , given the relatively shallow per-cell coverage of the single cell RNA-seq data, this estimate is noisy and can be inaccurate. Therefore, we used the following hierarchical Bayesian approach to determine if mutation is resulted from MT transfer from T cells or otherwise. We define a weight associated with mutation i as the ratio of marginal likelihood:
| (1) |
where
Intuitively, if MT transfer occurs, we anticipate for mutation . Next, we calculate the likelihood given the probability of observing a mutated read:
| (2) |
Under the null scenario, i.e. variant coming from either sequencing error or as a result of the background population heteroplasmy :
| (3) |
The variable in equation (3) can be estimated by pooling all the single cells from the given cell population. Assuming that a total of altered reads support the mutation , with sequencing depth , then . According to the Central Limited Theorem, when is a sufficiently large number, is asymptotically Normal with estimated deviation:
Then the standard deviation of should be:
We hypothesized that the condition of with or without MT transfer will alter the range of the true heteroplasmy of the variant allele in the expressions of and in equation (1). Specifically, when there is no MT transfer, it is reasonable to believe that the mutation allele frequency is unlikely to take a value that is ‘a lot’ higher than , with ‘a lot’ quantified by . Given the property of Normal distribution, we set the upper limit of in the case of no MT transfer to be such that will be smaller than this limit 97.5% of the chance. As it is not possible to learn which T cells had been in contact with the cancer cell of interest, when MT transfer occurs, we assume that the heteroplasmy of the variant in the T cell population can uniformly take value somewhere between to 1. Considering the statistical uncertainty, we anticipate to be higher than . Accordingly,
The integral can be written as a form of incomplete Beta function to get the expression of :
| (4) |
where
Therefore, the final expression of is:
| (5) |
All the variables in the equation (5) can be estimated from the single cell RNA-seq data. Based on the value of , we define the ‘effective count statistic’ () of each cancer cell as follows:
in which is indicator function:
Effective count is an estimate of MT transfer occurrence for each cancer cell of interest based on T cell enriched MT mutations, while controlled for sequencing error and read depth. We use in MERCI as the DNA source of information, which is further taken the rank transformation to reduce the impact of outliers.
Deconvolution model with support vector regression
The mixed mitochondrial transcriptome expressed in receiver cells allowed us to propose a deconvolution model on mitochondrial mRNA mixture for imputing an unknown fraction of mitochondrial composition for each cell34,73,74. The mitochondrial genome contains 13 protein-coding genes (MT genes) that provide essential functions for making enzymes involved in oxidative phosphorylation. The mRNA expression of each MT gene in a receiver cell is expected to be contributed by both intrinsic (endogenous) MT and donor-transferred (exogeneous) MT. Thus, the deconvolution problem can be represented by the equation:
Where denotes a vector of the mitochondrial fraction of different source of origins in the mixture and is the signature matrix of MT gene expression profiles (MGEP) in purified donor and receiver cell types. is the random noise following a normal distribution with zero mean. In this work, MERCI adopted epsilon-support vector regression (ε-SVR) with linear kernel application35 to estimate the fraction of external mitochondria by decomposing the relative RNA transcripts of MT genes. -SVR is an extended use of support vector machine (SVM) in regression problems. Unlike ordinary linear regression, the objective of -SVR is to find an appropriate line (or hyperplane in higher dimensions) to fit the data with acceptable error (ε) instead of minimizing the squared error sum. Therefore, -SVR can largely reduce overfitting and be robust to noisy data, making it suitable for deconvolving the mixture profile of gene expression34,75.
For a given receiver cell, the regression line of ε-SVR can be written in the equation:
| (6) |
Where denotes the weights of the exogenous and endogenous MT within the cytoplasm of this cell, and is the vectorized reference MGEP of potential donor cells and receiver cells. This formula represents the linear mixture of different cell types’ gene expression pattern with trainable weights. The goal is to minimize the difference between the mixed expression pattern and the observed data at given constraints. Let denote the observed expression of a MT gene. To solve the weights, the following objective function of ε-SVR (-norm penalty function) and constraints are used:
| (7) |
With Constraints:
| (8) |
| (9) |
where is the number of data points, is the input penalty coefficient and , are positive slack variables ensuring equality. is set as 0.1 in this study.
To solve this constrained optimization problem, Lagrange multipliers and are introduced76,77 and the Lagrangian function can be obtained to be:
| (10) |
By using the Lagrange multiplier techniques, the optimization problem in support vector regression is transformed to:
| (11) |
This leads to the following dual optimization problem35.
| (12) |
Using the condition that the partial derivatives of with respect to the primal variables will be zero at optimality. The dual problem can be written as following77:
Maximize
| (13) |
s.t.
in the primal formulation can expressed easily in terms of and .
| (14) |
Thus, the linear -SVR hyperplane can also be represented by
| (15) |
where is computed using the fact that (8) becomes an equality with if and (9) becomes an equality with if . The current implementation of -SVR in MERCI is based on the R package e1071. The estimated regression coefficients ( and ) in MERCI denote the relative, not absolute, quantity of intrinsic and foreign mitochondria in a receiver cell.
Rank score calculation
MERCI generated two rank scores (DNA rank, RNA rank) for each input candidate cell. Based on the statistic of donor cell (T cell in this study) enriched MT variants , the DNA rank score is calculated as the descending order of . The RNA rank score is obtained from decreasingly ranking the estimated abundance of donor cell derived MT calculated from the equation (14) . In Figures S3C–S3D, the combined rank score of each cell was computed by averaging the DNA rank score and RNA rank score. MERCI considers both DNA and RNA rank scores to predict receiver cells using an AND gate. A candidate cell will be assigned as a receiver only if its DNA and RNA rank scores are both within the user-defined threshold :
Depth-normalized counts of T cell enriched mtSNVs
The probabilities of observing a mtSNV are influenced by the read depth (Figure S2F). Therefore, we compared the observed counts of T cell enriched/depleted mtSNVs between CC cells and MC cells only within the same read depth range. We explored different ranges (1000–2000, 2000–3000, 3000–4000, 4000–5000) and showed the comparison using cells with 1000–2000 MT reads (Figure 1F) because this range covered the most of cells. We did not consider cells with MT reads > 5000 because this range contains very few (<100) MC cells.
MERCI LOO strategy for real-world application
In case the reference data of true non-receivers is not available, we used a leave-one-out strategy to get the reference MGEP and donor-enriched MT variants. When MERCI calculates the rank scores for a given cell, we left this cell in the input cell set and used remaining cells as reference non-receiver cells. The reference MGEP was then obtained by averaging the expression of MT genes in these cells. Donor cell enriched MT variants are derived from comparing the mitochondrial mutation profile of these non-reference receiver cells to the potential donor cells. The other steps are the same as the regular MERCI pipeline.
Significance estimation
Bona fide mitochondrial receivers are expected to have higher DNA and RNA rank scores. This effect causes an association between the two scores, enabled us to test if the sample contains true receivers. For example, if no receiver cell is included, the two rank scores are expected to be independent. When we set the rank threshold at top rank 10%, theoretically ~1% of input cells will be randomly captured By MERCI as the intersection of two cell sets: one with top 10% DNA rank scores and the other with top 10% RNA rank scores. However, if the input cells include a certain proportion of true receivers, the fraction of captured cells will be significantly higher than 1%. Thus, we used randomly permutated ranks to produce a null distribution of captured cell numbers. At a given rank cutoff, MERCI predicts receivers based on two lists of random rank scores. This process is repeated 10,000 times to generate the empirical estimate of (with a list of , or ). Let denotes count of predicted receivers using the original ranks, we define a statistic as below:
We used a strict criterion to determine whether the receiver cells are included in the input cells or not. The captured cell number is considered significant only if , with empirical p value < 0.0001, i.e. all the predicted cell counts by random permutations are smaller than the real value.
Down-sampling analysis
In the ground-truth data, the fraction of cocultured cancer cell accounts for 43% of total input cancer cells. To analyze the dependence of the number of MERCI captured cells on the faction of true receiver cells in the input cell set, down-sampling was performed to test the influence of signal/noise ratio on . Down-sampling of cocultured cancer cells generated simulated data sets with different fraction (from 0.5%~20%) of true receivers (Figures 3D–3F). There were 100 simulated data sets produced for each fixed fraction. Significance estimation and calculation was then repeated for these subsets of cells from ground-truth data, which enabled us to determine whether the number of captured cells is stably significant when analyzing each subset with smaller positive signal. We then calculated the values for each simulation to determine the power of MERCI at lower receiver cell percentage.
Analysis of BCC scRNA-seq data
We downloaded the expression data of 11 patients with advanced basal cell carcinoma (BCC) from the GEO database (GSE123814)37. Of these patients, only three of them, ‘su005’, ‘su006’ and ‘su008’ contained both T cells and tumor cells, with each subset comprising more than 500 cells, and were kept for the downstream analysis. We accessed the raw fastq files of these patients from the SRA database (PRJNA509907), which were further processed with Cell Ranger and Seurat as described above. We directly used the cell type labels provided by the cell information table37. Different subpopulations of T cells such as CD8+ memory, CD8+ active and CD4+ T cells were merged into one T cell cluster. After obtaining the MT mutation profile for each cell, MERCI significance estimation was then performed for each patient. A significant signal was observed for the tumor cells from samples of patient ‘su006’. Thus, the MERCI LOO pipeline was applied to predict the mitochondrial receiver tumor cells and quantify the MT composition.
Analysis of DEGs and MT transfer related genes
The DEGs of the predicted receiver cells in BCC sample ‘su006’ were identified using the FindMarkers function with a Wilcoxon rank-sum test. A total of 608 DEGs were called with an adjusted FDR < 0.05 (Benjamini–Hochberg method) (Table S3). Candidate genes of MT transfer were defined as those with a significantly positive correlation with the estimated abundance of exogenous MT across cancer cells (Spearman ρ > 0, FDR<0.05, two-sided Spearman’s correlation coefficient test following BH correction). Totally, 398 and 1,678 MT transfer related genes were obtained for murine CC cells and BCC tumor cells, respectively. The two lists overlapped by 95 genes, which were defined as MT transfer related genes.
Analysis of ESCC scRNA-seq data.
The scRNA-seq data sets of 58 patients with esophageal squamous-cell carcinoma (ESCC) were downloaded from the GEO (GSE160269) and SRA (PRJNA672851) databases38. Of them, we only analyzed the raw fastq files for twenty-seven patients because they contained both sufficient T cells and tumor cells (n>500). The preprocess of single cell data is the same as the BCC data. Using T cells as potential donors, we calculated statistics for tumor cells (epithelial cells) of all samples. Significant signals of receiver existence were detected in samples of three patients ‘P16T’, ‘P52T’ and ‘P39T’. Next, we used MERCI LOO pipeline to predict the MT receivers from tumor cells of these three patients using the rank parameter cutoff 50%.
Analysis of other single cell transcriptome data
We investigated the single cell RNA-seq datasets in the CancerSCEM database41 and downloaded the scRNA-seq data of 156 samples with both cancer and T cells reported in the original publications. We used the same data processing workflow as described in CancerSCEM to process the downloaded data and reproduced the T cell and cancer clusters for each sample. We then applied MERCI LOO pipeline to the 61 samples that contained sufficient number (>100) of T cells and cancer cells. Subsequently, we calculated Pearson’s correlation coefficient between the estimated fraction of T cell-derived mitochondria and two UMAP coordinates (umap1, umap2) for each sample to measure the phenotypic impact of MT transfer (Table S5). We further evaluated if a gene is associated with MT transfer by calculating the Spearman’s correlation coefficient between the estimated T cell mitochondrial fraction and the gene expression level and corrected the FDR using the Benjamini-Hochberg method.
Gene set enrichment analysis
The R packages fgesa (v1.16.0) and msigdbr (v7.2.1) were used to perform the gene ontology and pathway enrichment analysis. The pathway gene sets used in this work were extracted from the online databases Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology, BioCarta, Reactome and curated Hallmark gene sets78. Function terms or pathways were considered significant if Benjamini–Hochberg adjusted FDR<0.05. To investigate the programs associated with MT transfer, we extracted the common GO biological processes enriched by the candidate genes of MT transfer using the benchmark data and human BCC sample, respectively. An enrichment map was produced based on these ontologies using simplifyEnrichment (v1.0.0) and Cytoscape (v3.9.1).
Analysis of MT transfer score for TCGA samples
From 95 MT transfer related genes, we selected a 17 gene signatures (ABI1, ARF6, F11R, BAIAP2L1, MYADM, ACTR3, ARHGEF5, SRC, CAPZA2, GOLPH3, RAB10, ARF4, CAP1, PSEN1, YWHAG, YWHAZ, HSPA4) that are involved in pathways of mitochondrial transportation or nanotube formation (Table S6). We excluded genes with direct association with oxygen-dependent metabolism. The tumor MT transfer (TMT) score for each cell or tumor sample was calculated by using gene set variation analysis43 based on the mRNA expression of these 17 genes. The Wilcoxon paired signed-rank test was used to assess the statistical difference between TMT scores of tumoral and match normal samples in different cancer types. The cell cycle scores (CCS) and hypoxia scores of TCGA cancer samples were obtained from Lundberg et al.47 and Ye et al.49. Partial Spearman’s rank correlation coefficient was used to assess the correlation of TMT scores with CCS and hypoxia scores in TCGA cancer types by adjusting for tumor purity.
QUANTIFICATION AND STATISTICAL ANALYSIS
All sequencing data were analyzed using R v4.0.2 and Python v3.8. The statistical analyses in this work, including two-sided Wilcoxon rank-sum or signed-rank, chi-squared, odds ratio, correlation tests and downsampling analysis were implemented with the R programming language. All P values from multiple tests were corrected using the Benjamini–Hochberg method and p.adjust function. Survival analyses were conducted with Kaplan–Meier and Cox proportional hazards regression methods using R package survival v3.3.1. Cox regression was adjusted for age, race, gender (excluding BRCA), tumor purity, molecular subtypes (for BRCA), clinical stage (for CESC and HNSC), histologic diagnosis (for CESC, HNSC, LGG, LIHC and PAAD), tobacco smoking history (for CESC, HNSC, LUAD and PAAD), tumor grade (for HNSC, LGG, LIHC and PAAD) and clinical TNM stage subgroups (for HNSC) as clinical covariates. Survival curves were compared across groups with log-rank test. The other method details (such as sequencing data processing and gene set enrichment analysis etc.) and the related software or packages could be found in METHOD DETAILS.
Supplementary Material
Table S1. Sample and sequence information used in this study. Related to Figure 4.
Table S2. The list of T cell-enriched mtSNVs. Related to Figure 4.
Table S3. The differential expressed genes (DEGs) of predicted receivers vs non-receivers for scRNA-seq data of BCC patient ‘su006’. Related to Figure 5 and STAR Methods.
Table S4. The enriched pathways of 608 DEGs. Related to Figure 5.
Table S5. Associations of MT transfer and UMAP phenotype in human cancer samples. Related to Figure 5 and STAR Methods.
Table S6. The involved pathways of 17 gene signatures. Related to Figure 6 and STAR Methods.
Highlights:
Cancer cells ‘hijack’ mitochondria from nearby T cells
Development of MERCI for tracing intercellular mitochondrial (MT) transfer
Genomic inference of receiver cancer cells at single cell resolution using MERCI
Receiver cancer cells exhibit a distinct phenotype from the rest
Acknowledgments
This work is supported by NCI 1R01CA245318 (B.L.), 1R01CA258524 (B.L.), and NCI P50 CA070907 (J.M., H.L.).
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andreyev AY, Kushnareva YE, and Starkov AA (2005). Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 70, 200–214. 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 2.Rizzuto R, De Stefani D, Raffaello A, and Mammucari C (2012). Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13, 566–578. 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 3.Wang C, and Youle RJ (2009). The role of mitochondria in apoptosis*. Annu Rev Genet 43, 95–118. 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang E, Wang X, Tippner-Hedges R, Ma H, Folmes CD, Gutierrez NM, Lee Y, Van Dyken C, Ahmed R, Li Y, et al. (2016). Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs. Cell Stem Cell 18, 625–636. 10.1016/j.stem.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Sigurgardottir S, Helgason A, Gulcher JR, Stefansson K, and Donnelly P (2000). The mutation rate in the human mtDNA control region. Am J Hum Genet 66, 1599–1609. 10.1086/302902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stewart JB, and Chinnery PF (2015). The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet 16, 530–542. 10.1038/nrg3966. [DOI] [PubMed] [Google Scholar]
- 7.Fazzini F, Schopf B, Blatzer M, Coassin S, Hicks AA, Kronenberg F, and Fendt L (2018). Plasmid-normalized quantification of relative mitochondrial DNA copy number. Sci Rep 8, 15347. 10.1038/s41598-018-33684-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace DC, and Chalkia D (2013). Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol 5, a021220. 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenuth JP, Peterson AC, Fu K, and Shoubridge EA (1996). Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet 14, 146–151. 10.1038/ng1096-146. [DOI] [PubMed] [Google Scholar]
- 10.Lareau CA, Ludwig LS, Muus C, Gohil SH, Zhao T, Chiang Z, Pelka K, Verboon JM, Luo W, Christian E, et al. (2021). Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat Biotechnol 39, 451–461. 10.1038/s41587-020-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ludwig LS, Lareau CA, Ulirsch JC, Christian E, Muus C, Li LH, Pelka K, Ge W, Oren Y, Brack A, et al. (2019). Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176, 1325–1339 e1322. 10.1016/j.cell.2019.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Nuno K, Litzenburger UM, Qi Y, Corces MR, Majeti R, and Chang HY (2019). Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. Elife 8. 10.7554/eLife.45105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwok AWC, Qiao C, Huang R, Sham MH, Ho JWK, and Huang Y (2022). MQuad enables clonal substructure discovery using single cell mitochondrial variants. Nat Commun 13, 1205. 10.1038/s41467-022-28845-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin L, Zhang Y, Qian W, Liu Y, Zhang Y, Lin F, Liu C, Lu G, Sun D, Guo X, et al. (2022). LINEAGE: Label-free identification of endogenous informative single-cell mitochondrial RNA mutation for lineage analysis. Proc Natl Acad Sci U S A 119. 10.1073/pnas.2119767119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park S, Mali NM, Kim R, Choi JW, Lee J, Lim J, Park JM, Park JW, Kim D, Kim T, et al. (2021). Clonal dynamics in early human embryogenesis inferred from somatic mutation. Nature 597, 393–397. 10.1038/s41586-021-03786-8. [DOI] [PubMed] [Google Scholar]
- 16.Coorens THH, Moore L, Robinson PS, Sanghvi R, Christopher J, Hewinson J, Przybilla MJ, Lawson ARJ, Spencer Chapman M, Cagan A, et al. (2021). Extensive phylogenies of human development inferred from somatic mutations. Nature 597, 387–392. 10.1038/s41586-021-03790-y. [DOI] [PubMed] [Google Scholar]
- 17.Wei W, Gaffney DJ, and Chinnery PF (2021). Cell reprogramming shapes the mitochondrial DNA landscape. Nat Commun 12, 5241. 10.1038/s41467-021-25482-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore L, Cagan A, Coorens THH, Neville MDC, Sanghvi R, Sanders MA, Oliver TRW, Leongamornlert D, Ellis P, Noorani A, et al. (2021). The mutational landscape of human somatic and germline cells. Nature 597, 381–386. 10.1038/s41586-021-03822-7. [DOI] [PubMed] [Google Scholar]
- 19.Berridge MV, McConnell MJ, Grasso C, Bajzikova M, Kovarova J, and Neuzil J (2016). Horizontal transfer of mitochondria between mammalian cells: beyond co-culture approaches. Curr Opin Genet Dev 38, 75–82. 10.1016/j.gde.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Liu D, Gao Y, Liu J, Huang Y, Yin J, Feng Y, Shi L, Meloni BP, Zhang C, Zheng M, and Gao J (2021). Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct Target Ther 6, 65. 10.1038/s41392-020-00440-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torralba D, Baixauli F, and Sanchez-Madrid F (2016). Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front Cell Dev Biol 4, 107. 10.3389/fcell.2016.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, and Bhattacharya J (2012). Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18, 759–765. 10.1038/nm.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spees JL, Olson SD, Whitney MJ, and Prockop DJ (2006). Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A 103, 1283–1288. 10.1073/pnas.0510511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, and Lo EH (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. 10.1038/nature18928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho YM, Kim JH, Kim M, Park SJ, Koh SH, Ahn HS, Kang GH, Lee JB, Park KS, and Lee HK (2012). Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS One 7, e32778. 10.1371/journal.pone.0032778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saha T, Dash C, Jayabalan R, Khiste S, Kulkarni A, Kurmi K, Mondal J, Majumder PK, Bardia A, Jang HL, and Sengupta S (2022). Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat Nanotechnol 17, 98–106. 10.1038/s41565-021-01000-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desdin-Mico G, Soto-Heredero G, and Mittelbrunn M (2018). Mitochondrial activity in T cells. Mitochondrion 41, 51–57. 10.1016/j.mito.2017.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Lim AR, Rathmell WK, and Rathmell JC (2020). The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 9. 10.7554/eLife.55185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zampieri LX, Silva-Almeida C, Rondeau JD, and Sonveaux P (2021). Mitochondrial Transfer in Cancer: A Comprehensive Review. Int J Mol Sci 22. 10.3390/ijms22063245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pham AH, McCaffery JM, and Chan DC (2012). Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis 50, 833–843. 10.1002/dvg.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Liu Z, Liu L, Zhang H, Han C, Girard L, Park H, Zhang A, Dong C, Ye J, et al. (2022). AXL targeting restores PD-1 blockade sensitivity of STK11/LKB1 mutant NSCLC through expansion of TCF1(+) CD8 T cells. Cell Rep Med 3, 100554. 10.1016/j.xcrm.2022.100554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 33.Miller TE, Lareau CA, Verga JA, DePasquale EAK, Liu V, Ssozi D, Sandor K, Yin Y, Ludwig LS, El Farran CA, et al. (2022). Mitochondrial variant enrichment from high-throughput single-cell RNA sequencing resolves clonal populations. Nat Biotechnol. 10.1038/s41587-022-01210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457. 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scholkopf B, Smola AJ, Williamson RC, and Bartlett PL (2000). New support vector algorithms. Neural Comput 12, 1207–1245. 10.1162/089976600300015565. [DOI] [PubMed] [Google Scholar]
- 36.Petti AA, Williams SR, Miller CA, Fiddes IT, Srivatsan SN, Chen DY, Fronick CC, Fulton RS, Church DM, and Ley TJ (2019). A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat Commun 10, 3660. 10.1038/s41467-019-11591-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259. 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Peng L, Luo Y, Zhang S, Pu Y, Chen Y, Guo W, Yao J, Shao M, Fan W, et al. (2021). Dissecting esophageal squamous-cell carcinoma ecosystem by single-cell transcriptomic analysis. Nat Commun 12, 5291. 10.1038/s41467-021-25539-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ranzinger J, Rustom A, Abel M, Leyh J, Kihm L, Witkowski M, Scheurich P, Zeier M, and Schwenger V (2011). Nanotube action between human mesothelial cells reveals novel aspects of inflammatory responses. PLoS One 6, e29537. 10.1371/journal.pone.0029537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarma V, Wolf FW, Marks RM, Shows TB, and Dixit VM (1992). Cloning of a novel tumor necrosis factor-alpha-inducible primary response gene that is differentially expressed in development and capillary tube-like formation in vitro. J Immunol 148, 3302–3312. [PubMed] [Google Scholar]
- 41.Zeng J, Zhang Y, Shang Y, Mai J, Shi S, Lu M, Bu C, Zhang Z, Zhang Z, Li Y, et al. (2022). CancerSCEM: a database of single-cell expression map across various human cancers. Nucleic Acids Res 50, D1147–D1155. 10.1093/nar/gkab905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jang M, Kim SS, and Lee J (2013). Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med 45, e45. 10.1038/emm.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caicedo A, Fritz V, Brondello JM, Ayala M, Dennemont I, Abdellaoui N, de Fraipont F, Moisan A, Prouteau CA, Boukhaddaoui H, et al. (2015). MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci Rep 5, 9073. 10.1038/srep09073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marlein CR, Piddock RE, Mistry JJ, Zaitseva L, Hellmich C, Horton RH, Zhou Z, Auger MJ, Bowles KM, and Rushworth SA (2019). CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res 79, 2285–2297. 10.1158/0008-5472.CAN-18-0773. [DOI] [PubMed] [Google Scholar]
- 47.Lundberg A, Lindstrom LS, Parker JS, Loverli E, Perou CM, Bergh J, and Tobin NP (2020). A pan-cancer analysis of the frequency of DNA alterations across cell cycle activity levels. Oncogene 39, 5430–5440. 10.1038/s41388-020-1367-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hockel M, and Vaupel P (2001). Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 93, 266–276. 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 49.Ye Y, Hu Q, Chen H, Liang K, Yuan Y, Xiang Y, Ruan H, Zhang Z, Song A, Zhang H, et al. (2019). Characterization of Hypoxia-associated Molecular Features to Aid Hypoxia-Targeted Therapy. Nat Metab 1, 431–444. 10.1038/s42255-019-0045-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snyder CM, and Chandel NS (2009). Mitochondrial regulation of cell survival and death during low-oxygen conditions. Antioxid Redox Signal 11, 2673–2683. 10.1089/ARS.2009.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cihlar JC, Strobl C, Lagace R, Muenzler M, Parson W, and Budowle B (2020). Distinguishing mitochondrial DNA and NUMT sequences amplified with the precision ID mtDNA whole genome panel. Mitochondrion 55, 122–133. 10.1016/j.mito.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Wei W, Pagnamenta AT, Gleadall N, Sanchis-Juan A, Stephens J, Broxholme J, Tuna S, Odhams CA, Genomics England Research C, BioResource N, et al. (2020). Nuclear-mitochondrial DNA segments resemble paternally inherited mitochondrial DNA in humans. Nat Commun 11, 1740. 10.1038/s41467-020-15336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rustom A, Saffrich R, Markovic I, Walther P, and Gerdes HH (2004). Nanotubular highways for intercellular organelle transport. Science 303, 1007–1010. 10.1126/science.1093133. [DOI] [PubMed] [Google Scholar]
- 54.Nakajima A, Kurihara H, Yagita H, Okumura K, and Nakano H (2008). Mitochondrial Extrusion through the cytoplasmic vacuoles during cell death. J Biol Chem 283, 24128–24135. 10.1074/jbc.M802996200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bukoreshtliev NV, Wang X, Hodneland E, Gurke S, Barroso JF, and Gerdes HH (2009). Selective block of tunneling nanotube (TNT) formation inhibits intercellular organelle transfer between PC12 cells. FEBS Lett 583, 1481–1488. 10.1016/j.febslet.2009.03.065. [DOI] [PubMed] [Google Scholar]
- 56.Liu K, Ji K, Guo L, Wu W, Lu H, Shan P, and Yan C (2014). Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res 92, 10–18. 10.1016/j.mvr.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 57.Onfelt B, Nedvetzki S, Benninger RK, Purbhoo MA, Sowinski S, Hume AN, Seabra MC, Neil MA, French PM, and Davis DM (2006). Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J Immunol 177, 8476–8483. 10.4049/jimmunol.177.12.8476. [DOI] [PubMed] [Google Scholar]
- 58.Bouchard MJ, Dong Y, McDermott BM Jr., Lam DH, Brown KR, Shelanski M, Bellve AR, and Racaniello VR (2000). Defects in nuclear and cytoskeletal morphology and mitochondrial localization in spermatozoa of mice lacking nectin-2, a component of cell-cell adherens junctions. Mol Cell Biol 20, 2865–2873. 10.1128/MCB.20.8.2865-2873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Philip M, and Schietinger A (2022). CD8(+) T cell differentiation and dysfunction in cancer. Nat Rev Immunol 22, 209–223. 10.1038/s41577-021-00574-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hellstrom KE, and Hellstrom I (2019). From the Hellstrom paradox toward cancer cure. Prog Mol Biol Transl Sci 164, 1–24. 10.1016/bs.pmbts.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 61.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016). The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 374–388. 10.1016/j.immuni.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng Y, Delgoffe GM, Meyer CF, Chan W, and Powell JD (2009). Anergic T cells are metabolically anergic. J Immunol 183, 6095–6101. 10.4049/jimmunol.0803510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen TT, Oh SS, Weaver D, Lewandowska A, Maxfield D, Schuler MH, Smith NK, Macfarlane J, Saunders G, Palmer CA, et al. (2014). Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc Natl Acad Sci U S A 111, E3631–3640. 10.1073/pnas.1402449111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zinsmaier KE (2021). Mitochondrial Miro GTPases coordinate mitochondrial and peroxisomal dynamics. Small GTPases 12, 372–398. 10.1080/21541248.2020.1843957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borillo GA, Mason M, Quijada P, Volkers M, Cottage C, McGregor M, Din S, Fischer K, Gude N, Avitabile D, et al. (2010). Pim-1 kinase protects mitochondrial integrity in cardiomyocytes. Circ Res 106, 1265–1274. 10.1161/CIRCRESAHA.109.212035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paszek E, Zajdel W, Rajs T, Zmudka K, Legutko J, and Kleczynski P (2021). Profilin 1 and Mitochondria-Partners in the Pathogenesis of Coronary Artery Disease? Int J Mol Sci 22. 10.3390/ijms22031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, et al. (2014). Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J 33, 994–1010. 10.1002/embj.201386030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, et al. (2015). Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 21, 81–94. 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 69.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 70.Zhang L, Zhang W, Li Z, Lin S, Zheng T, Hao B, Hou Y, Zhang Y, Wang K, Qin C, et al. (2022). Mitochondria dysfunction in CD8+ T cells as an important contributing factor for cancer development and a potential target for cancer treatment: a review. J Exp Clin Cancer Res 41, 227. 10.1186/s13046-022-02439-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lareau CA, Liu VC, Muus C, Praktiknjo SD, Nitsch L, Kautz P, Sandor K, Yin YJ, Gutierrez JC, Pelka K, et al. (2023). Mitochondrial single-cell ATAC-seq for high-throughput multi-omic detection of mitochondrial genotypes and chromatin accessibility. Nat Protoc 18, 1416–1440. 10.1038/s41596-022-00795-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gong T, Hartmann N, Kohane IS, Brinkmann V, Staedtler F, Letzkus M, Bongiovanni S, and Szustakowski JD (2011). Optimal deconvolution of transcriptional profiling data using quadratic programming with application to complex clinical blood samples. PLoS One 6, e27156. 10.1371/journal.pone.0027156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liebner DA, Huang K, and Parvin JD (2014). MMAD: microarray microdissection with analysis of differences is a computational tool for deconvoluting cell type-specific contributions from tissue samples. Bioinformatics 30, 682–689. 10.1093/bioinformatics/btt566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cherkassky V, and Ma Y (2004). Practical selection of SVM parameters and noise estimation for SVM regression. Neural Netw 17, 113–126. 10.1016/S0893-6080(03)00169-2. [DOI] [PubMed] [Google Scholar]
- 76.Balasundaram S, Gupta D, and Kapil (2014). Lagrangian support vector regression via unconstrained convex minimization. Neural Netw 51, 67–79. 10.1016/j.neunet.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 77.Musicant DR, and Feinberg A (2004). Active set support vector regression. IEEE Trans Neural Netw 15, 268–275. 10.1109/TNN.2004.824259. [DOI] [PubMed] [Google Scholar]
- 78.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sample and sequence information used in this study. Related to Figure 4.
Table S2. The list of T cell-enriched mtSNVs. Related to Figure 4.
Table S3. The differential expressed genes (DEGs) of predicted receivers vs non-receivers for scRNA-seq data of BCC patient ‘su006’. Related to Figure 5 and STAR Methods.
Table S4. The enriched pathways of 608 DEGs. Related to Figure 5.
Table S5. Associations of MT transfer and UMAP phenotype in human cancer samples. Related to Figure 5 and STAR Methods.
Table S6. The involved pathways of 17 gene signatures. Related to Figure 6 and STAR Methods.
Data Availability Statement
scRNA-seq datasets generated in this study, as well as the MT variants and coverage information are available at Zenodo https://doi.org/10.5281/zenodo.8065206, and Gene Expression Omnibus (GEO) database with accession number GSE235675. The other scRNA-seq datasets of human cancer are available under the GEO accession codes provided in Table S1 and Key Resources Table. The TCGA data is available at https://portal.gdc.cancer.gov/. The source codes of MERCI-mtSNP and MERCI R package, and the codes to run example data are available at: https://github.com/shyhihihi/MERCI. Any additional information is also available from the corresponding author (Dr. Bo Li) upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor® 700 anti-mCD8α (clone53–6.7) | Biolegend | Cat# 100730 |
| Ultra-LEAF™ Purified anti-mouse CD3 Antibody | Biolegend | Cat# 100239 |
| Ultra-LEAF™ Purified anti-mouse CD28 Antibody | Biolegend | Cat# 102115 |
| Chemicals, peptides, and recombinant proteins | ||
| MitoTracker™ green FM | ThermoFisher | Cat# M7514 |
| MitoTracker™ red CMXRos | ThermoFisher | Cat# M7512 |
| PEI MAX | Polysciences | Cat# 24765–1 |
| Fixable viability Dye eFluor 506 | eBioscience | Cat# 65–0866-18 |
| Critical commercial assays | ||
| CD8+ T cell isolation kit | Stem Cell Technologies | Cat# 19853 |
| Chromium Next GEM Single Cell ATAC Kit v2 | 10x Genomics | PN-1000406 |
| Chromium Single Cell V(D)J Reagent Kit v1.1 | 10x Genomics | PN-1000167 |
| Chromium Next GEM Chip H Single Cell Kit | 10x Genomics | PN-1000162 |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | Cat# 5067–4626 |
| Mitochondria Isolation Kit for Cultured Cells | Thermo Fisher Scientific | Cat# 89874 |
| Deposited data | ||
| Raw and processed data | This paper | GEO: GSE235675 |
| Processed data | This paper | Zenodo:https://doi.org/10.5281/zenodo.8065206 |
| Human reference genome UCSC, GRCh38; Murine reference genome UCSC, mm10 | University of California Santa Cruz | https://genome.ucsc.edu/cgi-bin/hgGateway |
| Full list of public single cell datasets used in this study | N/A | Table S1 |
| scRNA-seq data for BCC samples | Yost et al.37 | GEO: GSE123814 |
| scRNA-seq data for ESCC samples | Zhang et al.38 | GEO: GSE160269 |
| scRNA-seq data for NSCLC samples | Zeng et al.41 | EBI: E-MTAB-6149 |
| scRNA-seq data for CRC samples | Zeng et al.41 | EBI: E-MTAB-8410 |
| scRNA-seq data for MCC samples | Zeng et al.41 | GEO: GSE117988 |
| scRNA-seq data for MCC samples | Zeng et al.41 | GEO: GSE118056 |
| scRNA-seq data for LUAD samples | Zeng et al.41 | GEO: GSE123904 |
| scRNA-seq data for STAD samples | Zeng et al.41 | GEO: GSE134520 |
| scRNA-seq data for NSCLC samples | Zeng et al.41 | GEO: GSE143423 |
| scRNA-seq data for TNBC samples | Zeng et al.41 | GEO: GSE148673 |
| scRNA-seq data for MIUBC samples | Zeng et al.41 | GEO: GSE145137 |
| scRNA-seq data for PDAC samples | Zeng et al.41 | GSA: PRJCA001063 |
| bulk RNA sequencing data for cancer samples | TCGA | https://portal.gdc.cancer.gov/ |
| Experimental models: Cell lines | ||
| KrasG12D/p53ko (KP) lung cancer cells | Dr. Esra Akbay | N/A |
| KP-mito-DsRed | This paper | N/A |
| MC38 colon cancer cells | ATCC | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jackson Laboratory | Cat# 000664 |
| Recombinant DNA | ||
| pDsRed2-Mito | Takara | Cat# 632421 |
| Software and algorithms | ||
| MERCI-mtSNP and MERCI R package code | This paper | https://github.com/shyhihihi/MERCI |
| CytExpert | Beckman Coulter, Inc | https://www.beckman.com/coulter-flow-cytometers/cytoflex/cytexpert |
| FlowJo | Tree Star Inc. | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism software 7.0 | GraphPad Software, Inc. | https://graphpad.com/scientific-software/prism/ |
| Python version 3.8 | Python Software Foundation | https://www.python.org/downloads/ |
| R version 4.0.2 | The R Foundation | https://www.r-project.org/ |
| Cell Ranger 3.1.0 | 10x Genomics | https://10xgenomics.com/ |
| Cell Ranger ATAC 2.0.0 | 10x Genomics | https://10xgenomics.com/ |
| Seurat 3.1.2 | Seurat developers | https://satijalab.org/seurat/articles/get_started.html |
| pysam | pysam developers | https://github.com/pysam-developers/pysam |
| e1071(version 1.7.4) | N/A | https://cran.r-project.org/web/packages/e1071/index.html |
| fgesa(version 1.16.0) | N/A | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| msigdbr (version 7.2.1) | N/A | https://cran.r-project.org/web/packages/msigdbr/index.html |
| simplifyEnrichment (version 1.0.0) | N/A | https://bioconductor.org/packages/release/bioc/html/simplifyEnrichment.html |
| Cytoscape (version 3.9.1) | Cytoscape developers | https://cytoscape.org/ |
| GSVA | Hänzelmann et al.43 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| BioRender | biorender.com | https://www.biorender.com/ |