

Abstract
The effect of coadministration of ritonavir and zidovudine (ZDV) on the pharmacokinetics of these drugs was investigated in a three-period, multidose, crossover study. Eighteen asymptomatic, human immunodeficiency virus-positive men were assigned randomly to six different sequences of the following three regimens: ZDV (200 mg every 8 h [q8h]) alone for 4 days, ritonavir (300 mg q6h) alone for 4 days, and ZDV with ritonavir for 4 days. Ritonavir pharmacokinetics were unaffected by coadministration with ZDV. However, ZDV exposure was reduced by about 26% (P < 0.05) in the presence of ritonavir. The maximum concentration in (Cmax) of ZDV plasma decreased from 748 ± 375 (mean ± standard deviation) to 546 ± 296, and area under the concentration-time curve from 0 to 24 h (AUC0–24) decreased from 3,052 ± 1,007 to 2,261 ± 715 when coadministered with ritonavir. In contrast, the ZDV elimination rate constant was unaffected by ritonavir, suggesting that there was no change in ZDV systemic metabolism. Correspondingly, differences in ZDV-glucuronide Cmax and AUC were not statistically significantly different between regimens (P > 0.31). Also, there were no apparent differences in the formation of 3′-amino-3′-deoxythymidine or in the adverse event profiles between the regimens. The lack of change in ritonavir pharmacokinetics suggests that dosage adjustment of ritonavir is unnecessary when it is administered concurrently with ZDV. The clinical relevance of a 26% reduction in ZDV exposure when ZDV is administered with ritonavir is unknown. In addition to other multidrug regimens, the long-term safety and efficacy of coadministration of ritonavir and ZDV is being investigated.
Ritonavir is a highly potent human immunodeficiency virus (HIV) protease inhibitor (15) that leads to exponential decreases in plasma viral RNA within a few days after administration (9, 13, 22). The HIV reverse transcriptase inhibitor zidovudine (ZDV) also delays the progression of disease in patients infected with HIV (11). The development of viral resistance to antiretroviral drugs is more likely to occur when a single antiretroviral drug is administered alone (19, 21, 28, 29). Thus, in an attempt to reduce the incidence of the development of HIV variants with resistance and improve the safety and efficacy profile, a combination of drugs of different classes (e.g., ritonavir and ZDV) and possibly other drugs within a class (i.e., triple-drug therapy) may be administered concurrently.
Treatment of HIV infection with a multidrug regimen introduces the possibility of drug-drug interactions. Ritonavir is metabolized extensively by the cytochrome P450 (CYP) system, particularly CYP3A, and ritonavir is a potent inhibitor of CYP3A (17). In addition, there is evidence that ritonavir induces glucuronidation in rats (18) and humans (25). ZDV is excreted to a minor extent (mean of 14%) as unchanged drug in the urine, while on average, 75% of a dose is excreted in the urine as the 5′ glucuronide metabolite (ZDV-G) (1). A small portion of ZDV is metabolized to 3′-amino-3′-deoxythymidine (AMT), which is more toxic than ZDV (8). Both cytochrome P450 reductase and cytochrome P450 are involved in the reduction of ZDV to AMT (5, 8, 26), and the formation of AMT is inhibited by ketoconazole (10), a potent inhibitor of CYP3A (24). Because of the potential ritonavir-induced changes in cytochrome P450 activity and glucuronidation, the pharmacokinetics of both ritonavir and ZDV were investigated when the drugs were administered alone and concurrently.
(This study was presented in part at the 35th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, Calif., 17 to 20 September 1995.)
MATERIALS AND METHODS
Patients.
The 18 patients enrolled in this study were asymptomatic, HIV-positive men. Patients were otherwise healthy based upon the results of a medical history, physical exam, ophthalmologic exam, laboratory profile, electrocardiogram, and a negative hepatitis B antigen test and had no recent history of drug or alcohol abuse. Patients were excluded from study participation if they had a CD4 lymphocyte count of <300/mm3, had received any investigational drug within the 4-week period before the initial drug administration in this study, or were using any other drug that could not be discontinued, including over-the-counter medications, at least 2 weeks before the initial drug dose was given in the present study. All patients gave written, informed consent in compliance with Food and Drug Administration regulations, and approval was obtained from the institutional review board.
Study design.
This was a single-center, multiple-dose, open-label, three-period complete crossover study. Patients were assigned randomly in equal numbers to six different sequences of the following three regimens: ZDV (200 mg every 8 h [q8h]) alone for 4 days, ritonavir (300 mg q6h) alone for 4 days, and ZDV (200 mg q8h) with ritonavir (300 mg q6h) for 4 days. Patients received doses on days 1 to 5 of each period, with the morning ritonavir and ZDV doses administered at approximately 6:30 and 8:30 a.m., respectively. The final dose on day 5 was administered at approximately 12:30 a.m. A solution formulation of ritonavir was used for this study because the capsule formulation had not been developed. For ZDV, Retrovir capsules (100 mg/capsule; Glaxo Wellcome Company) were administered. Ritonavir doses were administered within 10 min after a meal or snack; ZDV was administered between meals. All doses were taken with approximately 200 ml of water. The study was conducted during three confinement periods, each lasting approximately 6 days and separated by a washout interval of at least 9 days during which neither drug was administered. Blood samples (5 ml each) were collected beginning on day 4 at approximately 6:30 a.m. (within 5 min prior to the morning ritonavir dose [time 0] and/or 2 h prior to the morning ZDV dose) and 1, 2, 2.5, 3, 4, 5, 6, 7, 8, 9, 10, 10.5, 11, 12, 13, 14, 15, 16, 18, 19, 20, 21, 22, and 24 h later. Administration of ritonavir or ZDV continued during the sample collection period (Table 1). Samples were obtained immediately prior to dosing when sampling and dosing times coincided. The plasma was harvested and stored frozen at −20°C or colder in appropriately labeled tubes until assayed for ritonavir or ZDV and ZDV-G. Additionally, AMT plasma concentrations of selected samples were measured.
TABLE 1.
Dose administration and pharmacokinetic sample collection schedule for one of six potential sequences of administration of ZDV alone, ritonavir alone, or ZDV with ritonavir
| Regimen | Study days | Doses administereda |
|---|---|---|
| ZDV alone (samples collected) |
1–5 | Yes |
| 4–5 | Yes | |
| Washout | 5–13 | No |
| Ritonavir alone (samples collected) | 14–18 | Yes |
| 17–18 | Yes | |
| Washout | 18–26 | No |
| ZDV with ritonavir (samples collected) | 27–31 | Yes |
| 30–31 | Yes |
The first dose was administered the morning of day 1, and the final dose of each period was administered at 12:30 a.m. on days 5, 18, and 31.
Drug analyses.
Ritonavir samples were analyzed by a validated high-pressure liquid chromatography (HPLC) method (23). Briefly, samples were extracted by liquid-liquid extraction with ethyl acetate:hexane (9:1 [vol/vol]), the organic phase was evaporated to dryness, and the sample was reconstituted and washed twice with hexane. An aliquot of the reconstituted extract was then analyzed by reverse-phase HPLC with UV detection at 205 nm. Calibration standards (10 to 15,000 ng/ml) and quality control samples (150, 7,500, and 12,000 ng/ml) had coefficients of variation of 14.1% or lower and ranged in accuracy from 91.7 to 104%. The lower limit of quantification (LLQ) was 10 ng/ml. Samples with ritonavir concentrations greater than 15,000 ng/ml were diluted and reanalyzed. Samples for ZDV and ZDV-G concentrations were analyzed with a validated competitive direct equilibrium radioimmunoassay with the ZDV-Trac 125I radioimmunoassay kit (Incstar Corporation, Stillwater, Minn.). Calibration standards ranged from 15 to 3,000 ng/ml for ZDV and 25 to 2,000 ng/ml for ZDV-G. Quality control samples (15, 25, 500, and 2,000 ng/ml for ZDV; 25, 70, 500, and 2,000 ng/ml for ZDV-G) had coefficients of variation of 15.3% or lower and ranged in accuracy from 85.9 to 102%. ZDV concentrations after hydrolysis with β-glucuronidase (ZDVh) were used to estimate ZDV-G concentrations according to the following equation:
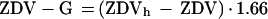 |
where the multiplier converts the molecular weight of ZDV to that of ZDV-G. The LLQs were 15 ng/ml for ZDV and 25 ng/ml for ZDV-G. Samples for AMT concentrations were analyzed by a previously published HPLC assay with fluorescence detection (32). AMT calibration standards ranged from 3 to 75 or 3 to 100 ng/ml. Estimated concentrations of quality control samples were similar to theoretical values of 7 and 30 ng/ml, ranging from 7.1 to 7.8 and 27.1 to 29.6 ng/ml, respectively. The LLQ and limit of detection were 3.0 and 0.5 ng/ml, respectively.
Pharmacokinetic and statistical analyses.
All pharmacokinetic parameters were calculated by noncompartmental methods for a 24-h period beginning at approximately 6:30 a.m. on the morning of day 4 of each period. The time to reach the observed maximum concentration (Tmax) for individual dose intervals was obtained directly from the plasma concentration-time data. For the 24-h period, Tmax was calculated as the mean Tmax of individual dose intervals. Maximum and minimum observed concentrations (Cmax and Cmin, respectively) were obtained directly from the plasma concentration-time data for the 0- to 24-h interval and for each individual dose interval. Area under the plasma concentration-time curve (AUC) was calculated by the linear trapezoidal method. For ritonavir, ZDV, and ZDV-G, the AUC for the 24-h interval (AUC0–24) was calculated beginning from time 0 (immediately prior to the morning ritonavir dose). For each dose interval completely sampled (all intervals except the last ZDV/ZDV-G interval), the concentration-time data used to calculate AUC ranged from the values obtained immediately prior to the dose to those obtained immediately prior to the subsequent dose. Assuming ZDV and ZDV-G had reached steady-state concentrations by the fourth day of dosing, the AUC of the last ZDV and ZDV-G dose interval was calculated as AUC18–24 + AUC0–2. The apparent oral clearance (CL/F) for individual dose intervals was estimated as the quotient of dose and AUC. The average concentration for the 24-h interval (Cavg) and the 24-h CL/F were estimated as the quotient of AUC0–24 and 24 h and the quotient of the total daily dose and AUC0–24, respectively. For ZDV, β (elimination phase) was estimated for each dose of the 24-h interval as the negative of the slope of the straight line obtained by regression of the logarithms of the concentrations versus time in the log-linear terminal phase of the curve. All regressions were based on the measurable concentrations from Cmax to the sample collected immediately prior to the subsequent dose. For ritonavir, no log-linear terminal phase could be observed with this dosage regimen. ZDV half-life (t1/2) was calculated as ln(2)/β, and the harmonic mean and pseudo standard deviation t1/2 values are reported.
Preliminary analyses of variance (ANOVAs) were performed to detect possible unequal carryover effects or other causes of period-regimen interaction for ritonavir, ZDV, and ZDV-G pharmacokinetic parameters. The sources of variation included in the model were patient, period, regimen, and period by regimen interaction, with patients viewed as a random sample. Because there was little evidence of regimen by period interaction in any of these analyses (all P values exceeded 0.10), the ANOVA models were simplified by omitting this term. Parameters for the individual dose intervals also were analyzed with this model. Because most ZDV and ZDV-G Cmin values were below the LLQ, they were not analyzed statistically. In addition, although Cavg was not analyzed statistically, results of the analyses of AUC0–24 apply to Cavg.
RESULTS
The mean ± standard deviation (range) age and body weight of the 18 men enrolled were 37.4 ± 6.8 (28 to 49) years and 77.8 ± 11.1 (61.2 to 109.8) kg, respectively. One patient was removed from the study because of noncompliance (unable to eat several meals). Six patients were withdrawn from the study during the ritonavir alone or combination regimens due to various adverse events, most commonly nausea (n = 3) and paresthesia (n = 2). Three additional patients had documented or apparent dosage interruptions that affected the ZDV and ZDV-G concentration-time profiles during the ZDV alone or combination regimen. For ritonavir, only one patient clearly had a dosage interruption, and this patient was excluded from analysis of the ritonavir 24-h interval and the third and fourth dose intervals. The concentration-time profiles of two other patients (very low concentrations) suggested that some doses were missed during both periods that ritonavir was administered. However, because dosage interruptions were not as demonstrable for these two patients (i.e., no clear log-linear decline in concentrations over an entire dose interval), they were not excluded from analysis. Thus, ritonavir comparisons are based on the data for 10 or 11 patients, and ZDV/ZDV-G comparisons are based on the data for 9 or 11 patients (depending on the dose interval).
Ritonavir mean 24-h pharmacokinetic parameters (Table 2) and plasma concentration-time profiles of ritonavir (Fig. 1) reflect minimal differences in concentrations between regimens. Ritonavir Cmax, Cmin, and AUC mean values were higher for the first dose interval than for those of other intervals, although the actual difference in any parameter between dose intervals was relatively minor (Table 3). Similarly, differences in mean Cmax and AUC due to the addition of ZDV to the regimen of ritonavir alone were 8% or less for all dose intervals (Cmax increases, 5, 5, 8, and 6% and AUC differences, 0, −2, 8, and 5% for the four dose intervals). Differences in ritonavir Tmax, Cmin, Cmax and AUC with coadministration compared to administration of ritonavir alone were not statistically significant (P > 0.09). Thus, ritonavir pharmacokinetics were unaffected by coadministration with ZDV.
TABLE 2.
Ritonavir 24-h pharmacokinetics after multiple doses when the drug was administered alone or with ZDV
| Regimen | Parameter (mean ± SD) (n = 10)
|
|||||
|---|---|---|---|---|---|---|
| Avg Tmax (h) | 0- to 24-h Cmax (μg/ml) | 0- to 24-h Cmin (μg/ml) | AUC0–24 (μg · h/ml) | CL/F (liter/h) | Cavg (μg/liter) | |
| Ritonavir alone | 3.8 ± 0.5 | 12.31 ± 5.45 | 4.93 ± 2.65 | 188.5 ± 83.4 | 10.1 ± 10.3 | 7.9 ± 3.5 |
| Ritonavir and ZDV | 3.8 ± 1.0 | 12.34 ± 5.32 | 5.21 ± 2.88 | 192.7 ± 93.4 | 13.8 ± 22.9 | 8.0 ± 3.9 |
FIG. 1.

Mean ritonavir plasma concentrations after administration of ritonavir (300 mg q6h) alone (circles) or in combination (squares) with ZDV (200 mg q8h).
TABLE 3.
Ritonavir pharmacokinetics for each dose interval after multiple doses administered alone or with ZDV
| Regimen and dose intervala | Parameter (mean ± SD)
|
|||
|---|---|---|---|---|
| Tmax (h) | Cmax (μg/ml) | Cmin (μg/ml) | AUC (μg · h/ml) | |
| Ritonavir alone | ||||
| 1 | 3.5 ± 2.0 | 11.51 ± 5.21 | 7.21 ± 3.38 | 56.4 ± 24.7 |
| 2 | 4.4 ± 1.7 | 8.85 ± 3.28 | 5.83 ± 2.52 | 45.0 ± 18.8 |
| 3 | 3.0 ± 1.8 | 8.63 ± 3.97 | 5.45 ± 2.87 | 42.2 ± 19.7 |
| 4 | 3.8 ± 1.9 | 10.05 ± 4.43 | 5.51 ± 3.00 | 49.8 ± 22.1 |
| Ritonavir and ZDV | ||||
| 1 | 3.5 ± 2.0 | 12.05 ± 6.17 | 7.10 ± 3.34 | 56.6 ± 26.3 |
| 2 | 4.1 ± 1.8 | 9.28 ± 3.88 | 5.72 ± 2.77 | 44.1 ± 18.9 |
| 3 | 4.1 ± 2.5 | 9.32 ± 4.42 | 6.31 ± 3.70 | 45.4 ± 23.5 |
| 4 | 3.1 ± 1.9 | 10.70 ± 5.61 | 6.89 ± 4.04 | 52.3 ± 27.9 |
For dose intervals 1 to 4, n = 12, 11, 10, and 10, respectively.
For ZDV, mean 24-h pharmacokinetic parameters (Table 4) and plasma concentration-time profiles of ZDV (Fig. 2) indicate small but statistically significant differences in concentrations between regimens. For individual dose intervals, there was a trend of decreasing Cmax and AUC mean values within each regimen (Table 5). However, changes in ZDV Cmax and AUC due to the addition of ritonavir (decreases of 22 to 33%) compared to ZDV administered alone were not obviously related to the dose interval. Differences between regimens in ZDV 0- to 24-h Cmax and AUC0–24 were similar to those of the individual dose intervals, approximately a 26% reduction in parameter mean values with coadministration compared to ZDV alone (P < 0.03). Because of the minimal amount of AUC contained in the 6- to 8-h postdose portion of the dosage interval, the method of calculation of AUC0–24 had no effect on the estimates; AUC18–24 + AUC0–2 values were similar to those calculated as AUC18–24 + AUC24–26, with AUC24–26 obtained by extrapolation with β. The 24-h AUC was the primary statistical parameter; thus, the AUC based on the actual data collected was reported. ZDV AUC and Cmax were statistically significantly different between regimens for all dose intervals (P < 0.05). There were no statistically significant differences between regimens in ZDV mean Tmax (P = 0.19) or in Tmax of the first and third dose intervals (P > 0.42), but there was a statistically significant difference in Tmax of the second dose interval (P = 0.047). Coadministration with ritonavir had no effect on ZDV β (P > 0.55 for each dose interval).
TABLE 4.
ZDV and ZDV-G 24-h pharmacokinetics after multiple doses administered alone or with ritonavir
| Regimen | Parameter (mean ± SD) (n = 9)a
|
||||||
|---|---|---|---|---|---|---|---|
| Avg Tmax (h) | 0- to 24-h Cmax (ng/ml) | AUC0–24 (ng · h/ml) | Cavg (ng/ml) | CL/F (liter/h) | β (h−1) | t1/2 (h) | |
| ZDV alone | |||||||
| ZDV value | 1.2 ± 0.6 | 748 ± 375 | 3,052 ± 1,007 | 127 ± 42 | 213 ± 58 | 0.645 ± 0.12 | 1.08 ± 0.21 |
| ZDV-G value | 1.3 ± 0.4 | 4,964 ± 1,626 | 20,448 ± 3,362 | 852 ± 140 | 0.694 ± 0.13 | 1.00 ± 0.18 | |
| ZDV and ritonavir | |||||||
| ZDV value | 1.4 ± 0.7 | 546 ± 296* | 2,261 ± 715* | 94 ± 30 | 285 ± 73 | 0.657 ± 0.16 | 1.06 ± 0.26 |
| ZDV-G value | 1.5 ± 0.4 | 4,046 ± 1,418 | 20,446 ± 5,322 | 852 ± 222 | 0.747 ± 0.19 | 0.93 ± 0.23 | |
∗, statistically significant difference between regimens (ANOVA, P < 0.030).
FIG. 2.

Mean ZDV plasma concentrations after administration of ZDV (200 mg q8h) alone (circles) or in combination (squares) with ritonavir (300 mg q6h).
TABLE 5.
ZDV and ZDV-G pharmacokinetics for each dose interval after multiple doses administered alone or with ritonavir
| Regimen and dose intervala | Parameter (mean ± SD)b
|
|||||
|---|---|---|---|---|---|---|
| Tmax (h) | Cmax (ng/ml) | Cmin (ng/ml) | AUC (ng · h/ml) | β (h−1) | t1/2 (h) | |
| ZDV alone | ||||||
| ZDV value | ||||||
| 1 | 0.9 ± 0.5 | 714 ± 331 | 3.0 ± 6.7 | 1,111 ± 465 | 0.682 ± 0.094 | 1.02 ± 0.14 |
| 2 | 0.9 ± 0.4 | 594 ± 399 | 1.6 ± 5.4 | 991 ± 354 | 0.637 ± 0.159 | 1.09 ± 0.27 |
| 3 | 1.8 ± 1.5 | 394 ± 135 | 6.5 ± 10.5 | 896 ± 223 | 0.607 ± 0.105 | 1.14 ± 0.20 |
| ZDV-G value | ||||||
| 1 | 1.0 ± 0.4 | 4,655 ± 1,577 | 12.4 ± 27.9 | 7,760 ± 1,551 | 0.736 ± 0.074 | 0.94 ± 0.09 |
| 2 | 1.2 ± 0.5 | 3,346 ± 1,050 | 5.3 ± 17.6 | 6,734 ± 1,486 | 0.703 ± 0.178 | 0.99 ± 0.25 |
| 3 | 1.7 ± 0.9 | 2,508 ± 651 | 25.4 ± 41.6 | 6,666 ± 942 | 0.632 ± 0.073 | 1.10 ± 0.13 |
| ZDV and ritonavir | ||||||
| ZDV value | ||||||
| 1 | 1.0 ± 0.5 | 495 ± 280* | 1.8 ± 5.9 | 744 ± 331* | 0.707 ± 0.137 | 0.98 ± 0.19 |
| 2 | 1.2 ± 0.6* | 423 ± 231* | 1.5 ± 5.0 | 713 ± 250* | 0.664 ± 0.162 | 1.04 ± 0.25 |
| 3 | 2.2 ± 1.6 | 291 ± 125* | 3.8 ± 8.0 | 701 ± 200* | 0.587 ± 0.191 | 1.18 ± 0.39 |
| ZDV-G value | ||||||
| 1 | 1.2 ± 0.6 | 4,267 ± 1,448 | 9.8 ± 32.6 | 7,441 ± 1,909 | 0.813 ± 0.175 | 0.85 ± 0.19 |
| 2 | 1.3 ± 0.5 | 3,412 ± 1,411 | 3.6 ± 12.1 | 6,730 ± 1,797 | 0.724 ± 0.179 | 0.96 ± 0.24 |
| 3 | 2.2 ± 0.9 | 2,689 ± 899 | 17.9 ± 44.4 | 6,999 ± 2,185 | 0.695 ± 0.211 | 1.00 ± 0.32 |
For dose intervals 1 and 2, n = 11; for dose interval 3, n = 10.
∗, statistically significant difference between regimens (ANOVA, P < 0.05).
Similar to ZDV Cmax and AUC, there was a trend of decreasing ZDV-G Cmax and AUC mean values of individual dose intervals within each regimen, and differences between regimens were not obviously related to the dose interval (Table 5). In contrast to the statistically significant changes in ZDV AUC0–24, ZDV-G AUC0–24 with the combination was similar to that for ZDV administration alone (P = 0.64). In fact, there were no statistically significant differences between regimens for any ZDV-G pharmacokinetic parameters (P > 0.13; Tables 4 and 5), and plasma concentration-time profiles of ZDV-G were similar between regimens (Fig. 3).
FIG. 3.

Mean ZDV-G plasma concentrations after administration of ZDV (200 mg q8h) alone (circles) or in combination (squares) with ritonavir (300 mg q6h).
Of the samples assayed for AMT, there were no apparent differences in AMT concentrations between the two regimens (data not shown). Of the 47 samples analyzed for AMT, 39 had AMT concentrations below the LLQ, with the majority of these below the lower limit of detection, regardless of whether ZDV was dosed alone or in combination with ritonavir. The highest concentration of AMT was 7.5 ng/ml for ZDV administered alone and 7.6 ng/ml for the combination regimen. Because most of the samples had AMT concentrations below the LLQ, pharmacokinetic and statistical analyses of AMT concentrations could not be performed.
DISCUSSION
Although ZDV therapy prolongs survival, there is a higher incidence of the development of viral resistance when the drug is administered alone (19, 21, 27–30) through a variety of potential mechanisms (12). Thus, the prevention of the development of resistant viruses by the use of combination regimens of drugs of different classes (e.g., reverse transcriptase inhibitors and protease inhibitors) is paramount. However, with multidrug regimens, there is an increase in the potential for drug-drug interactions causing altered pharmacokinetics of either drug, possibly leading to an increase in adverse events or a reduction in efficacy.
Of the 18 patients enrolled in the present study, 6 did not complete at least one regimen because of adverse events, and several others had dosage interruptions during at least one sample collection period. It is possible that the adverse events that caused five patients to be unable to complete the combination regimen were due to a pharmacokinetic interaction. However, of the adverse effects experienced by the six patients who did not complete the study, those associated with the combination regimen do not seem to have differed much from those associated with the administration of ritonavir or ZDV alone. Thus, it is unlikely that these patients were unable to complete the study due to a drug-drug interaction leading to high concentrations of ZDV or ritonavir.
The addition of ZDV to the regimen of ritonavir alone had little if any effect on ritonavir pharmacokinetics. Ritonavir Cmax and AUC for each dosage interval differed by 8% or less between regimens. Ritonavir Tmax also was unaffected by the addition of ZDV to the regimen and was approximately 4 h for both regimens, similar to that of recent studies (2, 3). Also consistent with recent studies (2, 3), the maximum values of Cmax and AUC generally occurred after the morning dose regardless of regimen, suggesting that absorption of ritonavir may proceed more efficiently during the morning dose interval (i.e., diurnal variation of absorption).
In contrast to the lack of effect of ZDV on ritonavir pharmacokinetics, ZDV Cmax and AUC decreased by about 26% in the presence of ritonavir. The small differences in ZDV-G Cmax and AUC were not statistically significant. In general, ZDV is rapidly and completely absorbed, but first-pass metabolism reduces the absolute bioavailability to about 60%. Maximum plasma concentrations of ZDV are achieved within about 1 h, and elimination is rapid, with a mean half-life of approximately 1.1 to 1.5 h (1, 4, 16, 20), similar to the mean of 1.1 h in the present study. Plasma concentrations of ZDV-G often exceed those of ZDV, and parallel decline of plasma concentration-time curves for ZDV and ZDV-G suggest formation-rate limited elimination of ZDV-G.
Although about 90% of a dose of ZDV is excreted in the urine as unchanged drug or ZDV-G, a second metabolite, AMT, has been identified in studies with human liver (8), human gastrointestinal bacteria (14), and rat liver microsomes and hepatocytes (7, 8) and in the plasma of rhesus monkeys (6). In addition, AMT has been detected in the plasma (but not bile) of patients receiving 2.5 mg of ZDV per kg by a 1-h intravenous infusion (31). There is evidence of the involvement of both cytochrome P450 reductase and cytochrome P450 in the reduction of ZDV to AMT (5, 8, 10, 26). A wide range of substrates and inhibitors of different cytochrome P450 isozymes were investigated in human liver microsomes. The most marked inhibition of the reduction of ZDV to AMT occurred with ketoconazole (10), a potent inhibitor of CYP3A (24). Induction in rats of CYPs 2B, 3A, and 4A resulted in increased hepatic microsomal formation of AMT and increases in the intrinsic clearance of approximately 1.6- to 3-fold (10). Although AMT is a relatively minor metabolite of ZDV, AMT is five- to sevenfold more toxic than ZDV in several in vitro tests (8), suggesting that alterations in the formation of AMT could affect the adverse event profile of ZDV.
The combination of ZDV and ritonavir caused a decrease in ZDV Cmax and AUC compared to those for ZDV administered alone, suggesting that ritonavir induced the metabolism of ZDV. Although there was minimal change in ZDV-G pharmacokinetics, and ritonavir is a potent CYP3A inhibitor, the effect on the formation of AMT was unknown. Therefore, several samples for AMT analysis were selected in an attempt to provide a representative subset of all samples collected during the study (i.e., samples from each dose interval, samples with typical or high ZDV concentrations, and samples during the time of peak ZDV concentrations for both regimens).
AMT mean peak plasma concentrations of approximately 160 ng/ml (n = 6) were detected in the plasma of patients receiving 2.5 mg of ZDV per kg by intravenous infusion over 1 h (31) (a dose resulting in approximately 50% greater exposure of ZDV than the dose used in the present study). Assuming absolute bioavailability of about 60%, AMT peak concentrations of approximately 60 ng/ml could occur with the dosage regimen used in the present study. However, for both regimens, AMT concentrations were quite low. Most of the samples tested had AMT concentrations below the LLQ, with the majority of these below the lower limit of detection, regardless of whether ZDV was dosed alone or in combination with ritonavir. Thus, ritonavir had essentially no effect on the conversion of ZDV to AMT by CYP3A, and toxicity associated with high plasma concentrations of AMT did not occur in the presence of ritonavir.
The decrease in ZDV AUC by approximately 25% with no apparent change in ZDV-G AUC or AMT concentrations is not easily explained. A decrease in absorption could account for the change in ZDV, but a corresponding decrease in metabolite AUC would be expected. Rather than changes in absorption, enhanced ZDV metabolism by induction of glucuronyl transferase could have caused lower ZDV concentrations and relatively little change in metabolite AUC, as slightly more ZDV would be converted to ZDV-G rather than eliminated unchanged in the urine. However, enhanced ZDV metabolism would be expected to have a corresponding effect on ZDV β, which was not observed. Thus the reduction in ZDV concentrations when administered concurrently with ritonavir are not completely explained by either reduced absorption (ZDV-G concentrations were unchanged) or by increased metabolism (ZDV β was unchanged). Also, if glucuronidation of ZDV was induced by ritonavir, the full extent of induction may not have been reached after 4 days of dosing and the effect on ZDV concentrations may have been underestimated in this study. Regardless of the actual mechanism(s) involved, plasma concentrations of ZDV were reduced by about 25% when administered concurrently with ritonavir, but ZDV-G, AMT, and ritonavir concentrations were similar between regimens.
In conclusion, ritonavir pharmacokinetics were not influenced by the addition of ZDV to the regimen of ritonavir alone, and dosage adjustment of ritonavir is unnecessary when it is administered concurrently with ZDV. In contrast, ZDV exposure apparently was reduced when it was coadministered with ritonavir; ZDV 0- to 24-h Cmax and AUC0–24 decreased 27 and 26%, respectively, with coadministration. However, there was little or no apparent difference in ZDV-G or AMT plasma concentrations between regimens. The mechanism of the interaction between ritonavir and ZDV is unclear. No differences in adverse events between the combination and single-drug regimens were apparent. The clinical relevance of a 26% reduction in ZDV exposure when the drug is administered with ritonavir is unknown; however, the long-term safety and efficacy of coadministration of ZDV and ritonavir continues to be evaluated clinically.
REFERENCES
- 1.Blum, M. R., S. H. T. Liao, S. S. Good, and P. De Miranda. 1988. Pharmacokinetics and bioavailability of zidovudine in humans. Am. J. Med. 85(Suppl. 2A):189–194. [PubMed]
- 2.Cato A, Hsu A, Granneman R, Leonard J, Carlson G, Carothers L, Cao G. Program and abstracts of the 35th Interscience Conference on Antimicrobial Agents and Chemotherapy. Washington, D.C: American Society for Microbiology; 1995. Assessment of the pharmacokinetic interaction between the HIV-1 protease inhibitor ABT-538 and fluconazole, abstr. I33; p. 211. [Google Scholar]
- 3.Cato A, Qian J, Carothers L, Carlson G, Locke C, Hsu A, Granneman R, Leonard J. Evaluation of the pharmacokinetic interaction between ritonavir and didanosine. Clin Pharmacol Ther. 1996;59:144. [Google Scholar]
- 4.Collins J M, Unadkat J D. Clinical pharmacokinetics of zidovudine: an overview of current data. Clin Pharmacokinet. 1989;17:1–9. doi: 10.2165/00003088-198917010-00001. [DOI] [PubMed] [Google Scholar]
- 5.Cretton E M, Placidi L, Sommadossi J P. Abstracts of the 94th Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics. 1993. Conversion of 3′-azido-3′-deoxythymidine (AZT) to its toxic metabolite 3′-amino-3′-deoxythymidine (AMT) is mediated by cytochrome P450 and NADPH-cytochrome C reductase in liver microsomes, abstr. PII-78; p. 189. [Google Scholar]
- 6.Cretton E M, Schinazi R F, McClure H M, Anderson D C, Sommadossi J P. Pharmacokinetics of 3′-azido-3′-deoxythymidine and its catabolites and interactions with probenecid in rhesus monkeys. Antimicrob Agents Chemother. 1991;35:801–807. doi: 10.1128/aac.35.5.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cretton E M, Sommadossi J P. Modulation of 3′-azido-3′-deoxythymidine catabolism by probenecid and acetaminophen in freshly isolated rat hepatocytes. Biochem Pharmacol. 1991;42:1475–1480. doi: 10.1016/0006-2952(91)90461-d. [DOI] [PubMed] [Google Scholar]
- 8.Cretton E M, Xie M Y, Bevan R J, Goudgaon N M, Schinazi R F, Sommadossi J P. Catabolism of 3′-azido-3′-deoxythymidine in hepatocytes and liver microsomes, with evidence of formation of 3′-amino-3′-deoxythymidine, a highly toxic catabolite for human bone marrow cells. Mol Pharmacol. 1991;39:258–266. [PubMed] [Google Scholar]
- 9.Danner S A, Carr A, Leonard J M, Lehman L M, Gudiol F, Gonzales J, Raventos A, Rubio R, Bouza E, Pintado V, Aguado A G, De Lomas J G, Delgado R, Borleffs J C C, Hsu A, Valdes J M, Boucher C A B, Cooper D A. A short-term study of the safety, pharmacokinetics, and efficacy of ritonavir, an inhibitor of HIV-1 protease. N Engl J Med. 1995;333:1528–1533. doi: 10.1056/NEJM199512073332303. [DOI] [PubMed] [Google Scholar]
- 10.Eagling V A, Howe J L, Barry M J, Back D J. The metabolism of zidovudine by human liver microsomes in vitro: formation of 3′-amino-3′-deoxythymidine. Biochem Pharmacol. 1994;48:267–276. doi: 10.1016/0006-2952(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 11.Fischl M A, Richman D D, Causey D M, Grieco M H, et al. Prolonged zidovudine therapy in patients with AIDS and advanced AIDS-related complex. JAMA. 1989;262:2405–2410. [PubMed] [Google Scholar]
- 12.Hirsch M S, D’Aquila R T. Therapy for human immunodeficiency virus infection. N Engl J Med. 1993;328:1686–1695. doi: 10.1056/NEJM199306103282307. [DOI] [PubMed] [Google Scholar]
- 13.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 14.Howe J L, Back D J, Colbert J. Extrahepatic metabolism of zidovudine. Br J Clin Pharmacol. 1992;33:190–192. doi: 10.1111/j.1365-2125.1992.tb04024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kempf D J, Marsh K C, Denissen J F, McDonald E, Vasavanonda S, Flentge C A, Green B E, Fino L, Park C H, Kong X P, Wideburg N E, Saldivar A, Ruiz L, Kati W M, Sham H L, Robins T, Stewart K D, Hsu A, Plattner J J, Leonard J M, Norbeck D W. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc Natl Acad Sci USA. 1995;92:2484–2488. doi: 10.1073/pnas.92.7.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klecker R W, Collins J M, Yarchoan R, Thomas R, Jenkins J F, Broder S, Myers C E. Plasma and cerebrospinal fluid pharmacokinetics of 3′-azido-3′-deoxythymidine: a novel pyrimidine analog with potential application for the treatment of patients with AIDS and related diseases. Clin Pharmacol Ther. 1987;41:407–412. doi: 10.1038/clpt.1987.49. [DOI] [PubMed] [Google Scholar]
- 17.Kumar G N, Rodrigues A D, Buko A M, Denissen J F. Cytochrome P450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J Pharmacol Exp Ther. 1996;277:423–431. [PubMed] [Google Scholar]
- 18.Kumar G N, Grabowski B, Lee R, Denissen J F. Hepatic drug-metabolizing activities in rats after 14 days of oral administration of the human immunodeficiency virus-type 1 protease inhibitor ritonavir (ABT-538) Drug Metab Dispos. 1996;24:615–617. [PubMed] [Google Scholar]
- 19.Land S, Treloar G, McPhee D, Birch C, Doherty R, Cooper D, Gust I. Decreased in vitro susceptibility to zidovudine of HIV isolates obtained from patients with AIDS. J Infect Dis. 1990;161:326–329. doi: 10.1093/infdis/161.2.326. [DOI] [PubMed] [Google Scholar]
- 20.Langtry H D, Campoli-Richards D M. Zidovudine: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs. 1989;37:408–450. doi: 10.2165/00003495-198937040-00003. [DOI] [PubMed] [Google Scholar]
- 21.Larder B A, Darby G, Richman D D. HIV with reduced sensitivity to zidovudine (AZT) isolated during prolonged therapy. Science. 1989;243:1731–1734. doi: 10.1126/science.2467383. [DOI] [PubMed] [Google Scholar]
- 22.Markowitz M, Saag M, Powderly W G, Hurley A M, Hsu A, Valdes J M, Henry D, Sattler F, La Marca A, Leonard J M, Ho D D. A preliminary study of ritonavir, an inhibitor of HIV-1 protease, to treat HIV-1 infection. N Engl J Med. 1995;333:1534–1539. doi: 10.1056/NEJM199512073332204. [DOI] [PubMed] [Google Scholar]
- 23.Marsh K C, Eiden E, McDonald E. Determination of ritonavir, a new HIV protease inhibitor, in biological samples using reversed-phase high-performance liquid chromatography. J Chromatogr B. 1997;704:307–313. doi: 10.1016/s0378-4347(97)00454-4. [DOI] [PubMed] [Google Scholar]
- 24.Maurice M, Pichard L, Daujat M, Fabre I, Joyeux H, Domergue J, Maurel P. Effects of imidazole derivatives on cytochromes P450 from human hepatocytes in primary culture. FASEB J. 1992;6:752–758. doi: 10.1096/fasebj.6.2.1371482. [DOI] [PubMed] [Google Scholar]
- 25.Ouellet D, Hsu A, Qian J, Cavanaugh J, Leonard J. Abstracts of the XI International Conference on AIDS. 1996. Effect of ritonavir on the pharmacokinetics of ethinyl estradiol in healthy female volunteers, abstr. Mo.B.1198; p. 88. [Google Scholar]
- 26.Placidi L, Cretton E M, Placidi M, Sommadossi J P. Reduction of 3′-azido-3′-deoxythymidine to 3′-amino-3′-deoxythymidine in human liver microsomes and its relationship to cytochrome P450. Clin Pharmacol Ther. 1993;54:168–176. doi: 10.1038/clpt.1993.128. [DOI] [PubMed] [Google Scholar]
- 27.Ragni M V, Kingsley L A, Zhou S J. The effect of antiviral therapy on the natural history of human immunodeficiency virus infection in a cohort of hemophiliacs. J Acquired Immune Defic Syndr. 1992;5:120–126. [PubMed] [Google Scholar]
- 28.Richman, D. D. 1990. Zidovudine resistance of human immunodeficiency virus. Rev. Infect. Dis. 12(Suppl. 5):S507–S510. [DOI] [PubMed]
- 29.Rooke R, Tremblay M, Soudeyns H, DeStephano L, Yao X-J, Fanning M, Montaner J S G, O’Shaughnessy M, Gelmon K, Tsoukas C, Gill J, Ruedy J, Wainberg M A. Isolation of drug-resistant variants of HIV-1 from patients on long-term zidovudine therapy. AIDS. 1989;3:411–415. doi: 10.1097/00002030-198907000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Shirasaka T, Kavlick M F, Ueno T, Gao W Y, Kojima E, Alcaide M L, Chokekijchai S, Roy B M, Arnold E, Yarchoan R, Mitsuya H. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc Natl Acad Sci USA. 1995;92:2398–2402. doi: 10.1073/pnas.92.6.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stagg M P, Cretton E M, Kidd L, Diasio R B, Sommadossi J P. Clinical pharmacokinetics of 3′-azido-3′-deoxythymidine (zidovudine) and catabolites with formation of a toxic catabolite, 3′-amino-3′-deoxythymidine. Clin Pharmacol Ther. 1992;51:668–676. doi: 10.1038/clpt.1992.79. [DOI] [PubMed] [Google Scholar]
- 32.Zhou X J, Sommadossi J P. Quantification of 3′-amino-3′-deoxythymidine, a toxic catabolite of 3′-azido-3′-deoxythymidine (zidovudine) in human plasma by high-performance liquid chromatography using precolumn derivatization with fluorescamine and fluorescence detection. J Chromatogr. 1994;656:389–396. doi: 10.1016/0378-4347(94)00118-9. [DOI] [PubMed] [Google Scholar]