

Abstract
Researchers can often successfully generate antibodies to predicted epitopes. Especially when the epitopes are on the surface of a protein or in a hydrophilic loop. But it is difficult to direct recombinant antibodies to bind either to- or near a specific amino acid on a protein or peptide. We have developed a unique immune-targeting strategy, that we call “Epivolve,” that enables us to make site-specific antibodies (Abs). Epivolve technology leverages a highly immunogenic modified amino acid that acts as a “pseudo-hapten” immunotarget and takes advantage of Ab affinity maturation technologies to make high-affinity site-specific antibodies. Epivolve functions by the evolution of an Ab paratope to either synonymous or especially non-synonymous amino acid (aa) binding. Here we describe the use of Epivolve technology in phage display and the protocols for developing site-specific antibodies.
Keywords: Phage display, Antibody, Site-specific, Site-directed, Biopanning, Epivolve
1. Introduction
A systematic and targeted means for isolating antibodies (Abs) against a targeted amino acid sequence within the core of- or on the exposed surface of a protein is needed. We invented a unique technology, termed “‘Epivolve” (short for Epitope Evolution), to develop site-specific Abs, with as low as (<10 nM) KD binding affinities (not published data) to solvent-exposed residues that incorporate the adjacent “context” sequences. A principle of Epivolve (Fig. 1) is to use a synthetic modified amino acid residue to replace a targeted specific native amino acid residue in a peptide or protein. This replacement can be either synonymous or non-synonymous amino acid. This modified residue is termed “mod1.” The mod1 in the antigen (Ag) functions as a “pseudo-hapten” or “immuno-attractant.” It also serves to enrich for and differentiate between modification-specific affinity reagents. The mod1-specific Abs are then evolved to recognize the native sequence Ag, which is termed “NAT,” using directed evolution by affinity maturation processes. Epivolve technology can be used in in vitro by phage display technology described here and in vivo using animal immunization in our recent publication [1].
Fig. 1.
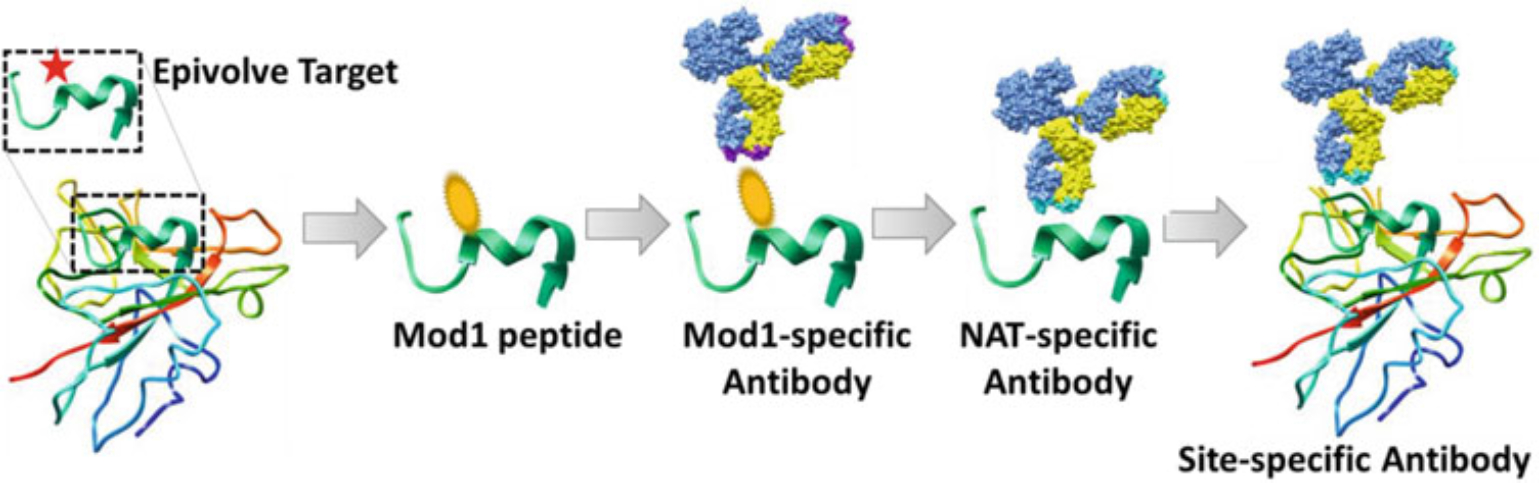
Illustration the principles of Epivolve technology. An important residue (the Epivolve targeted site) on the target protein is chosen. A 10–15-mer peptide of the target sequence will then be synthesized with the modification on the Epivolve targeted amino acid, named mod1-peptide. This mod1-peptide is biopanned using a naïve phage display library to generate a mod1-specific Ab. The identified mod1-specific Abs are then mutagenized using error-prone PCR to generate a Discovery Maturation (DisMat) library. The DisMat library is then biopanned against the NAT peptide (or full-length protein if available) to identify anti-NAT (or anti-full-length protein) clonotypes. The resulting anti-NAT Abs can be further engineered for increased binding affinities using Affinity Maturation (AffMat)
In a typical Epivolve in vitro phage display workflow (Fig. 2), 3 rounds of phage display biopanning are performed in 3 phases. Phase I is to discover the mod1-specific antibodies using a discovery phage library against mod1-Biotin-peptide antigens. In Phase II, the “Discover Maturation” (DisMat) round, it is used to convert the mod1-specific scFv Abs into anti-NAT Abs by using errorprone PCR-generated mutagenesis phage libraries and biopanning against NAT peptides antigens. Phase III is optional, which can be used to mature the NAT-specific Abs to increase the binding affinity and binding specificities against NAT peptides and/or NAT full-length proteins. For clarity, we define the following terms used throughout this protocol: (1) “mod1”: a modified amino acid that acts as an “immune-attractant” and is emplaced at the targeted site on a protein, (2) “NAT”: the non-synonymous naturally-occurring amino acid at the targeted site that was replaced by mod1, (3) “Site-specific Abs”: Abs recognizing the targeted peptide sequence wherein the mod1 is replaced by NAT.
Fig. 2.
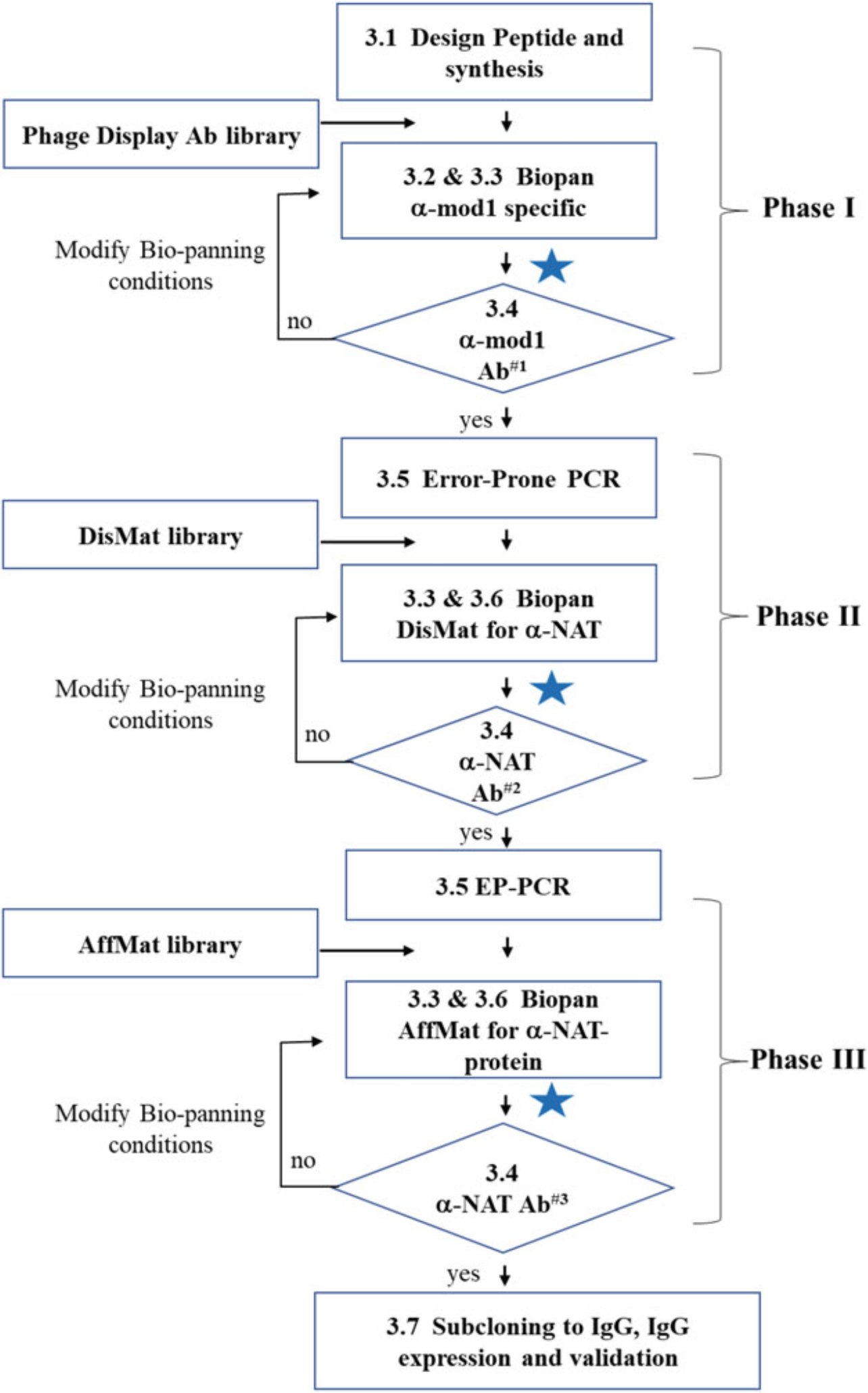
In vitro Epivolve workflow Developing site-specific Abs using Epivolve technology by in vitro phage display can be illuminated as 2 phases (Discovery and DisMat) and an optional third phase (AffMat). In Phase I, discovery biopanning against a mod1-peptide is used to identify mod1-specific scFvs. In phase II (DisMat), anti-mod1 specific scFvs from phase I are used as the template for Error-Prone PCR to make DisMat libraries. The DisMat library is then biopanned against a NAT peptide or full-length protein to identify anti-NAT clones. In the optional Phase III (AffMat), anti-NAT clones are subjected to a second round of Error-Prone PCR to make AffMat libraries. These libraries are then used to develop high-affinity and high-specificity anti-NAT peptide or an-NAT full-length protein Abs, which will be validated by the appropriate applications such as by MILKSHAKE Western-blot [25], ELISA, and other functional assays. The blue star indicates where there are several steps in between the two steps shown, which can include phage supernatant ELISA, scFv protein expression and purification with details we have published [2, 19]. Typical result of Ab#1 is shown in Fig. 3a. Typical results of Ab#2 and Ab#3 are shown in Fig. 3b and Fig. 3c
The advantages of Epivolve technology are: (a) avoiding in vivo immune tolerance, (b) precisely targeting any antigenic epitope regardless of its immunogenicity, and (c) taking advantage of in vitro mutagenesis to generate affinity reagents with different clonotypes against one target residue.
With Epivolve technology, a researcher can generate antibodies showing unique specificities: (1) “Pan-variant specificity”: Epivolve generated Abs can bind two or more different NAT amino acids at the mod site of the targeted epitope; (2) “Polymorphism-specific Abs”: Epivolve derived Abs can differentiate between two or more variants at the mod1 site of the targeted epitope by specifically recognize point-mutated residue; (3) “Conformational Abs”: Epivolve derived Ab can bind Ag proteins at its certain structural conformation; (4) Post-Translational Modification (PTM) specificity: Abs can specifically recognize target’s post-translational modification status: either requiring PTM for binding (“PTM-specific”), absence of binding if the site is PTM (“NAT-specific”), or binding regardless of PTM status (“PTM-status independent”).
Here we describe the protocols for developing site-specific antibodies by Epivolve technology using phage display in vitro. In the following protocols, we note that some of the techniques can be used with reagents and methods developed by other means. For example, we typically use a constant framework scFv phage library in our research which was built using our published protocols [2, 3]. But the protocols here can be adapted for use with any other display library. The same applies to other well-characterized standard methods such as methods for mutagenesis, cloning, and protein expression and purification. Although not tested by any of us, presumably other in vitro display methods, such as yeast display [4] and ribosomal display [5] can be adapted to Epivolve.
2. Materials, Reagents, and Equipment
Bacterial Strains
CJ236 strain (Genotype: 0046Δ(HindIII)::cat (Tra+ Pil+ CamR)/ung-1 relA1 dut-1 thi-1 spoT1 mcrA) can be purchased from New England BioLabs (Waverly, MA). This strain lacks functional dUTPase and uracil N-glycosylase, and yields uracilated, single-stranded DNA template when infected with M13 bacteriophage.
TG1 E. coli strain (F’ (traD36 proAB+ lacIq lacZΔM15) supE thi-1 Δ(lac-proAB) Δ(mcrB-hsdSM)5, (rK-mK-) is available from Lucigen (Middleton, WI) and several other suppliers. It is important to note that the M13 phage attachment site is the F pilus of E. coli. The bacterium does not produce the pilus if grown at less than 30 °C. TG1 cells are F+ and therefore they cannot be infected or transduced if grown at a temperature less than 30 °C or stored on ice or room temperature [6].
Equipment and Facilities:
Specific instrumentation for the protocols in this chapter include the following. Many equipment can be substituted with other instruments having equivalent specifications.
37 °C and 30 °C incubator: Thermo Scientific, Catalog number 3950 or equivalent.
Microplate shaker variable to 600 rpm: Lab Line Model 4625 or equivalent.
Spectrophotometer capable of measuring OD600: Thermo Fisher, Genesys 20 or equivalent.
Table top centrifuge: Sorvall model ST 40R or equivalent.
Nutator: Clay Adams model number 421105 or equivalent.
Muti-channel electronic pipette: Sartorius model Picus 735491 or equivalent.
Microplate Plate washer: Biotek, model EL406 or equivalent.
Plate reader capable of reading OD450: Perkin Elmer Envision 2150 plate reader or equivalent.
Media and Solutions: (see Note 1)
Luria Broth (LB) medium: 10 g/L of tryptone, 5 g/L of yeast extract, 10 g/L of NaCl. To prepare the LB medium, combine the above reagents, and shake until the solutes have dissolved. Adjust the pH to 8.0 with 5 N NaOH. Adjust the volume of the solution to 1 L with deionized H2O.
1× phosphate-buffered saline (PBS) buffer: 137 mM Sodium Chloride, 2.7 mM Potassium Chloride, and 10 mM Na2HPO4, 1.8 mM KH2PO4 adjusted to pH 7.4.
1× PBST: 1× PBS with final concentration of 0.1% Tween 20.
2% MPBS: 2 g non-fat dry milk dissolved in 100 mL of 1× PBS.
10× TM buffer: 200 μL 0.5 M MgCl2 (final concentration 0.1 M), 500 μL 1 M Tris pH 7.5 (final concentration 0.5 M), 300 μL sterile ddH20. Must be made fresh each time.
2YT medium: measure around 900 mL of distilled water, add 16 g tryptone, 10 g yeast extract, 5 g NaCl, combine the reagents and shake until the solutes have dissolved. Adjust pH to 7.0 with 5 N NaOH. Adjust to 1 L with distilled water. Sterilize by autoclaving.
10× mutagenic PCR buffer: 70 mM MgCl2, 500 mM KCl, 100 mM Tris (pH 8.3), 0.1% (wt/vol) gelatin) is combined with 10 μL of 10× dNTP mix (2 mM dGTP, 2 mM dATP, 10 mM dCTP, 10 mM dTTP).
10× Trypsin: Dissolve 0.5 g of trypsin into ddH2O, add 2.5 mL of 1 M Tris–HCl, add 50 μL of 1 M CaCl2, adjust volume to 50 mL ddH2O. Store at −20 °C for up to one year.
3. Methods
3.1. Peptide Design
For each targeted site, 4 peptides are needed, the 5th and 6th peptides are optional: (i) “NAT-peptide,” composed of 4–6 amino acids on either side of the intended target site, (ii) “NAT-biotin peptide,” composed of the NAT peptide with N-terminal biotin conjugation, (iii) “mod1-peptide,” same sequence as the NAT-peptide except that the central, intended target site is substituted ideally with a modified amino acid with a highly-charged side chain, as one example, phosphate (as examples, modified serine, threonine, tyrosine or histidine), and (iv) “mod1-biotin peptide,” composed of the mod1-peptide with N-terminal biotin conjugation. Two additional peptides are used as controls but they are optional and they are somewhat application-specific: (v) “scrambled-NAT peptide,” composed of a central target site from native sequence and the adjacent amino acids in randomized sequence, and (vi) “scrambled-mod1 peptide,” composed of a central mod1 target site and the adjacent amino acids in randomized sequence.
3.2. Phage Display Library
Generally, phage display Ab libraries are engineered to display the variable, Ag-binding domains of Abs, either as single chains (scFv) [7] or as heavy and light chain Ag binding fragments (Fabs) [8]. Several methods have been used to generate Ab diversity in vitro. Ab variable sequences can be amplified from the natural immunoglobulin gene repertoire in human B-cells [9–12]. Alternatively, recombinant Ab libraries can be built using synthetic diversity [13–15] in which Ab complementarity determining region (CDR) gene fragments are generated with mixed-nucleotide synthesis [16]. A semi-synthetic approach can also be used to further diversify native heavy and light chain genes by synthetically randomizing the CDRs [17]. Many types of libraries can be used with these protocols, but the diversity should typically be >1011 with phage Ab libraries. Our protocols for producing such libraries have been published [3, 18].
3.3. Isolation of mod1-specific scFvs by Phage Display Biopanning.
Standard phage display screening conditions can be used and have been previously described [3, 18, 19]. For each phage library biopanning in each of the 3 phases, we perform 3–5 rounds of bio-screenings. We usually use subtraction and/or competition using counter peptides such as non-biotinylated mod1-peptides, non-biotinylated NAT-peptide (non-modified peptide), and native protein (if available) to increase the stringency during the bio-screening. For example, in phase I stage, in the phage display discovery screening to discover mod1-specific Ag, 10-fold excess of NAT-peptide competitor is added during positive phage selection against the biotinylated mod1-peptide coated to wells in round 2 and round 3 of bio-screenings described below.
Coat 96-well Immuno-plates (Thermo Fisher, Nunc Maxisorp) with NeutrAvidin with 10ug mL−1 concentration overnight at 4 °C. Afterwards, wash the plates with excess PBS, then block plates with 250 μL per well of MPBS for 1 h at room temperature or at 4 °C overnight.
After an additional PBS wash, biotinylated peptide Ags are coated onto plates at a concentration of 10 μg mL−1 for 1 h at room temperature. Excess PBS is again used to wash the plates followed by blocking the plates with MPBS.
Prepare 1 mL of the discovery phage library with titer of >1 × 1012, diluted 1:10 in 2% MPBS for each Ag being screened. 100 μL of diluted library is added to each well of the plate and incubated for 1 h at room temperature with shaking at ~600 rpm on a microplate shaker. If the phagemid contains a trypsin cleavage site between the scFv and phage coat III protein gpIII, then after rigorous washing (10 times) with PBST, bound phage can be eluted by an addition of 100 μL per well of 1× trypsin in 1× PBS buffer and incubating with shaking at ~600 rpm on a microplate shaker at room temperature for 10 min. (see Note 2)
Transfer the eluate in a new 96-well deep-well plate containing 200 μL per well exponentially growing E. coli TG1 (OD600 of 0.4–0.6). Transduce for 30 min at 37 °C without shaking. (see Note 3)
Pellet the transduced cells by spinning the 96-well deep-well plate at 3000 rpm in a Sorvall RT-7 with RTH-750 rotor for 10 min. Pour off excess supernatant gently so as to not disturb the pellet.
Resuspend the pellet in each well with 600 μL 2YT medium supplemented with ampicillin (100 g μL−1 ) and 1% glucose. Cover the 96-well deep-well block with a plastic plate seal and grow with shaking at ~600 rpm on a microplate shaker at 30 o C overnight.
The next day: for each well, dilute 5 μL of the overnight cultures into 200 μL fresh 2YT medium containing 100 μg μL−1 ampicillin and 1% glucose in a new 96-well deep-well plate. Cover the plate with a plastic plate seal and incubated with shaking at 37 °C until the culture OD600 reaches 0.4. (see Note 4)
When OD600 reaches 0.4, 16 μL KM13 helper phage (Multiplicity of Infection (MOI) of 20) or hyperphage (Progen, PRHYPE, MOI of 10) is added to each well and then incubated at 37 °C for 30 min without shaking.
Transduced cells are then pelleted by spinning at 3000 rpm for 10 min.
Pour off excess supernatant gently so as to not disturb the pellet.
Resuspend the pellets using 600 μL per well of 2YT media supplemented with ampicillin (100 ug μL−1), kanamycin (50 ug μL−1 ), and 0.1% glucose. Cover the plate with a plastic plate seal.
Incubate the plate with shaking at ~600 rpm on a microplate shaker at 30 °C overnight.
Apply the resulting phage supernatant to another Ag-coated Maxisorp microplate.
This is the beginning of 2nd round bio-screening process. This whole process from step 1 to step 14 will be repeated for total of 3–5 rounds.
At the end of final biopanning round, we plate bio-screened phages on agar plates to pick single clones: At the end of the final round, we elute the bound phage from the biopanning plate by adding 100 μL per well with 1× trypsin in 1× PBS.
Incubate the plate with shaking at ~600 rpm on a microplate shaker at room temperature for 10 min.
Transfer the eluents from the biopanning plate to a 96-well deep-well plate.
Transduce TG1 cells with eluted phage by adding 200 μL of mid-log phase (OD600 of 0.4–0.6) TG1 cells to the 100 μL of eluate already in the deep-well plate. Incubate without shaking at 37 °C for 30 min.
Pool the transduced cells from each well in a column on the plate (corresponding to each Ag being screened on the plate). For example, for the first Ag being screened, pool column 1 (wells A1-H1).
Take 1 mL of pooled cells to make a serial dilution of by diluting 100-fold down to 1:1,000,000 (10−6 ). Plate 250 μL of each dilution (10−2, 10−4, 10−6 ) onto large TYE plates (150 mm diameter) supplemented with ampicillin (final concentration of 100 μg μL−1) and glucose (final concentration of 1%) individually. (see Note 5). Store the remaining pooled transduced eluates at 4 °C overnight in case this step needs to be repeated.
Incubate overnight at 37 °C.
The next day, prepare 2YT media supplemented with 100 μg mL−1 ampicillin and 1% glucose and transfer 200 μL per well into 96-well deep-well plates. One plate will be required for each Ag being screened.
For each Ag target, from the agar plates of transduced eluates, pick 88 single colonies into columns 1–11 of a deep-well plate containing the prepared media. Leave the last column (12) free of single colonies for ELISA controls. Pick a single colony from positive control plate (separately prepared) into wells A12 through D12, leaving wells E12 through H12 as medium-only negative control wells. Cover the plate with a plastic plate seal.
Incubate the deep-well plate with shaking at ~250 rpm at 37 °C overnight.
These single clones will be analyzed by scFv expression and single-point ELISA testing to identify the best hits (See Note 6).
Once the mod1-specific hits are identified, scaled-up soluble scFv proteins from those hits are then expressed by inducing with IPTG and purified with standard HisPur Cobalt Resin column purification.
Perform standard titration ELISA to analyze purified scFv proteins (Subheading 3.4). Typical positive clones from phase I will be mod1-peptide specific antibodies as shown in Fig. 3a.
Best mod1-specific antibodies will be moved forward to DisMat or AffMat to in vitro envolve to NAT-specific antibodies. Alternatively, to generate a final model1-specific antibody, the positive scFv can be subcloned into IgG vectors and retested using titration ELISA following standard protocols (Subheading 3.4) and validated by applications.
Fig. 3.
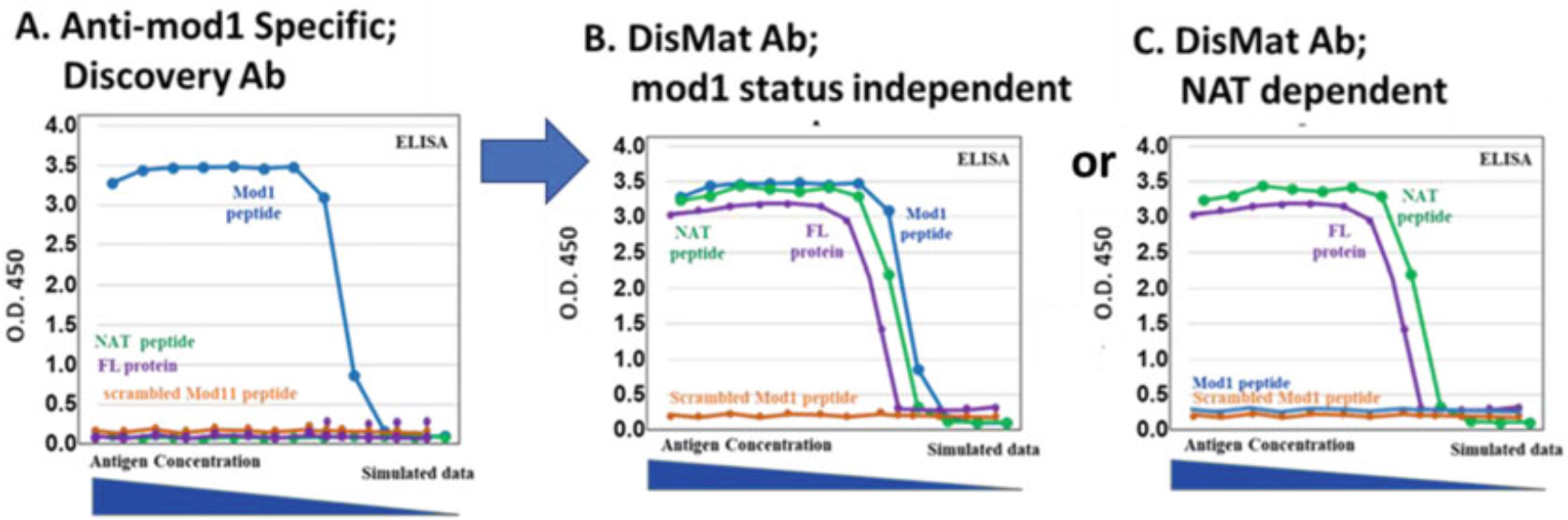
Simulated examples of scFv protein or full IgG protein titration ELISA results of site-specific antibodies discovered by Epivolve (a) Mod1-specific scFv or IgG protein titration ELISA results from anti-mod1 discovery phage display biopanning in phase I. A typical anti-Mod1 Ab will demonstrate specific binding to the mod1 peptide, and absence of binding to the NAT-biotin-peptide, or NAT-full-length protein, or scrambled mod1 peptide. (b) and (c) are scFv or IgG protein titration ELISA results of two different clonotypes of final anti-NAT Abs: (b) a mod1-independent Ab which demonstrates binding to mod1-peptide, NAT-peptide, NAT-full-length protein, and absence of binding to scrambled Mod1 peptide; (c) a NAT-specific Ab which demonstrates binding to NAT-peptide, NAT-full-length protein, and absence of binding to mod1-peptide and scrambled Mod1 peptide
3.4. Site-Specific scFv Protein Titration
ELISA Coat the Maxisorp 96-well plates with 100 μL per well of NeutrAvidin at a final concentration of 10 μL mL−1. Incubate at 4 °C overnight.
Wash NeutrAvidin-coated plates three times with 250 μL per well of 1× PBS using a handheld electronic multichannel pipette or plate washer. (see Note 7)
Discard the last wash and gently tap the plates on a dry paper towel to remove residual PBS. Add 250 μL per well of 3% BSA in 1× PBS to block each plate using a handheld multichannel electronic pipette. Incubate the plates at room temperature for 1 h.
-
Prepare the Ag dilutions in an untreated 96-well plate.
Dilute each target Ag to 10 μg mL−1 in 1× PBS and add 300 μL to Row A of each corresponding column of the 96-well plate.
Add 150 μL of 1× PBS to Rows B-H of the 96-well plate.
Move 150 μL from Row A to Row B (1:2 dilution) using a multichannel pipette. Be sure to mix each well significantly by pipetting up and down several times.
Continue the 1:2 serial dilutions until Row G. Final Ag concentrations will be per mL: 10 μg, 5 μg, 2.5 μg, 1.25 μg, 0.62 μg, 0.31 μg, 0.16 μg. It is important to be sure to use new pipette tips for each serial dilution.
DO NOT add Ag to Row H. Leave row H as 150 μL 1× PBS. It is the negative control.
Wash the NeutrAvidin-coated plates three times with 250 μL per well of 1× PBS using BioTek plate washer.
Using a hand-held multichannel pipette, transfer 100 μL of the serially diluted Ags from the untreated 96-well plate to the NeutrAvidin-coated 96-well plate. In the last column of the plate, add 100 μL of each Ag at starting concentration of 10 μg mL−1 to one well for the secondary only negative control. Incubate the plates at room temperature for 1 h.
Wash the plates three times with 250 μL per well of 1× PBS on the BioTek plate washer.
Using a hand-held multichannel electronic pipette, add 250 μL per well of 3% BSA/in 1× PBS block solution to the washed wells of the plates. Incubate the plates at room temperature for 1 h.
While the plates are in block, prepare the scFv dilutions in 1.7 mL microcentrifuge tubes: add 1 μg of scFv protein to 1 mL of 3% BSA in 1× PBS block solution for a final concentration of 1 μg mL−1 and mix well.
Wash the Ag-coated plates three times with 250 μL per well of 1× PBS on the BioTek plate washer.
Using an electronic multichannel pipette, add 100 μL of scFv (at 1 μg mL−1) to Rows A-H of the corresponding column on the plate. Add 3% BSA in 1× PBS block only (no scFv) to the last column for secondary only control. Incubate the plates at room temperature for 1 h.
Wash the plates four times with 250 μL per well of 1× PBSTon plate washer. Discard the last wash and gently tap the plates on a dry paper towel to remove residual 1× PBST.
Add 100 μL per well of the appropriate secondary Ab diluted 1: 5000 in 3% BSA in 1× PBS per well to all ELISA plates, including the secondary only column. Incubate the plates at room temperature for 1 h.
Wash the plates three times with 250 μL per well of 1× PBST on the BioTek plate washer.
Add 100 μL per well of Ultra TMB developing reagent (ThermoFisher, Catalog number 34028) that has come to room temperature.
To stop the developing reaction, add 50 μL per well of 2 M H2SO4 stop solution.
Place plates on Perkin Elmer Envision plate reader (or equivalent) and measure the absorbance at 450 nm. A typical result of mod1-specific Abs discovered from phase I biopanning is shown in Fig. 3a. Typical results of final NAT-specific Abs discovered from phase II and phase III biopanning are shown in Fig. 3b, c.
3.5. Error-Prone PCR
DisMat (Discovery Maturation) and AffMat (Affinity Maturation) phage libraries are used for directed evolution from mod1-specificic antibodies to either mod1-independent, or NAT-specific site-specific antibodies. Several methods for the manufacturing of directed evolution mutagenesis libraries are known. Two of the methods are described below.
The first is AXM mutagenesis method which is we have previously developed [2]. This “AXM mutagenesis” method relies on the ability of T7 exonuclease to sequentially hydrolyze DNA in the 5′ → 3′ direction and its inability to hydrolyze DNA that contains several phosphorothioate groups at its 5′ terminus [20]. A large (i.e., 800 nt long), mutated DNA fragment is produced using polymerase chain reaction (PCR) conditions that promote nucleotide misincorporation into newly synthesized DNA. In the PCR reaction, one of the primers contains phosphorothioate linkages at its 5′ end. Treatment of the error-prone generated PCR product with T7 exonuclease is used to preferentially remove the strand synthesized with the non-modified primer, resulting in a single-stranded DNA segment, or “megaprimer.” The use of this megaprimer in a Kunkel-like mutagenesis reaction takes advantage of the E. coli DNA base excision repair pathway to bias nucleotide base-changes between the megaprimer and a complementary uracilated DNA sequence in favor of the in vitro synthesized megaprimer [21–23]. A second method is the Error-Prone PCR developed by Cadwell and Joyce [24]. The protocol is as follows.
10 μL of 10× mutagenic PCR buffer combined with 10 μL of 10× dNTP mix (2 mM dGTP, 2 mM dATP, 10 mM dCTP, 10 mM dTTP), 20 fmole of input DNA, 30 pmole each of forward and reverse primers, and H2O for a final volume of 88 μL.
10 μL of 5 mM MnCl2 is added to the reaction, mixed well, and the absence of any precipitate need to be visually verified. 2 μL of Taq DNA polymerase is added to bring the final volume to 100 μL. Cycling conditions consist of 30 cycles of: 94 °C 1 min, 55 °C 1 min, and 72 °C 1 min. It is important to use standard Taq polymerase. It is important NOT to use a proof-reading polymerase.
After the PCR is complete, cleanup is performed using the Qiagen QIAquick PCR Purification Kit (Qiagen, catalog number 28104) according to the manufacturer’s protocol. The purified DNA is eluted in 40 μL of Buffer EB. For scFvs template DNA, we expect a PCR amplification product of approximately 800 base pairs.
The EP-PCR product is then cloned into the appropriate expression vector using standard conditions. (see Note 8)
3.6. Discovery Maturation (DisMat) and Affinity Maturation (AffMat) Biopanning
Both the DisMat and AffMat biopanning can be performed using the protocol previously described (Subheading 3.3) with the following modifications.
3.6.1. DisMat Modifications
DisMat is used to convert mod1-specific Abs to anti-NAT Abs. Typically, we choose 6–10 positive mod1-specific hits to move forward to Discovery Maturation (DisMat). Three rounds of positive selecting bio-screenings are used against the NAT-biotin-peptide. And in the 2nd and 3rd round, we add a 10× concentration of the non-biotinylated NAT-peptide during the bio-screening as competitors.
3.6.2. AffMat Modifications
AffMat is used to evolve the binding affinities of DisMatted Abs to NAT peptides and NAT full-length proteins. Typically, we will pick 2–3 best NAT-specific hits to move forward to AffMat. Three rounds of positive selecting bio-screenings are used against the NAT full-length proteins (if not available, then use NAT-biotin-peptides). And in the 2nd round and 3rd round, we successively reduce 10-fold screening Ag concentration while coating the plates. Additionally, we use a range of 10-fold to 100-fold excess of the non-biotinylated NAT-peptides as competitors.
3.7. Converting scFv Hits into Full-Length IgG Proteins and Final Antibody Validation.
Final validated positive scFv hits should be sequenced and assembled into suitable plasmid expression vectors for IgG expression and purification. Final IgGs can be validated by our innovative Western-Blot based MILKSHAKE and Sundae methods published recently [25] and also described in a separate chapter in this book [26]. Final validation of the Epivolve-derived Abs will depend on the desired specific applications.
4. Notes
Prepare all solutions using ultrapure water (prepared by purifying deionized water to attain a sensitivity of 18 MΩ-cm at 25 °C). Prepare and store all reagents at room temperature unless indicated otherwise. Follow all waste disposal regulations when disposing of waste materials
It is important to note that trypsin elution method will only work with phage libraries constructed with trypsin-cleavage phagemid vectors, which contains a trypsin cleavage site between the scFv and phage coat III protein gpIII, such as pIT-2 based vectors. If another type of elution condition is needed, use the recommended elution condition for that specific library in use
The eluted selected phage can be stored at 4 °C for no longer than 6 h if needed.
Make a duplicate 96-well deep-well plate which grows the same cells at the same timeline as the experimental plate. Use this plate to monitor the cell growth by measuring OD600 to avoid any contamination concerns to the experimental plate
If autoclaved glass beads are available, use autoclaved glass beads to spread the culture thoroughly over the agar plate. Allow the plate to dry with beads on the agar at 37 °C for around 20 min before inverting the plates to incubate overnight. Alternatively, cell spreaders can be used for plating cells on agar plates
Selection criteria of the best hits to move forward from phage-induced cell supernatant ELISA: we export the ELISA data at OD450 absorbance into Excel file. We calculate the ratio of absorbance of each well over the absorbance from background well (medium-only control wells of E12 through H12). We consider a positive hit only when all the following 3 criteria will be met: (1) the Ab can detect the Ag coated on plate at 2.5 μg mL−1 concentration; (2) the absorbance OD450 must be ≥0.2; (3) The binding absorbance signal must be ≥2-fold over absorbance on background wells
For consistency between rounds and different experiments, it is recommended that electronic washing conditions such as Biotek plate washer will be used. Manual pipetting may deliver variable results
We typically use heteroduplex formation cloning (Kunkel mutagenesis) after T7 exonuclease treatment to make the mutagenesis libraries [2, 3]. But standard enzymatic digestion followed by standard ligase ligation to a vector cloning will also work for making the mutagenesis libraries
Acknowledgments
The authors would like to thank Dr. Brian K. Kay for insightful comments and discussion. This research was funded by NIH Appl. ID 1R43GM146473–01 and NIH Appl. ID 1R44AI177126–01. Patent protection for the Epivolve technology has been submitted for Abbratech Inc.
References
- 1.Fuller EP, O’Neill RJ, Weiner MP (2022) Derivation of splice junction-specific antibodies using a unique hapten targeting strategy and directed evolution. New Biotechnol 71:1–10. 10.1016/j.nbt.2022.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holland EG, Buhr DL, Acca FE et al. (2013) AXM mutagenesis: an efficient means for the production of libraries for directed evolution of proteins. J Immunol Methods 394(1–2): 55–61. 10.1016/j.jim.2013.05.003 [DOI] [PubMed] [Google Scholar]
- 3.Batonick M, Holland EG, Busygina V et al. (2016) Platform for high-throughput antibody selection using synthetically-designed antibody libraries. New Biotechnol 33(5 Pt A):565–573. 10.1016/j.nbt.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Deventer JA, Wittrup KD (2014) Yeast surface display for antibody isolation: library construction, library screening, and affinity maturation. Methods Mol Biol (Clifton, N.J.) 1131:151–181. 10.1007/978-1-62703-992-5_10 [DOI] [PubMed] [Google Scholar]
- 5.Zahnd C, Amstutz P, Plückthun A (2007) Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat Methods 4(3):269–279. 10.1038/nmeth1003 [DOI] [PubMed] [Google Scholar]
- 6.Novotny CP, Lavin K (1971) Some effects of temperature on the growth of F pili. J Bacteriol 107(3):671–682. 10.1128/jb.107.3.671-682.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marks JD, Hoogenboom HR, Bonnert TP et al. (1991) By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol 222(3):581–597. 10.1016/0022-2836(91)90498-u [DOI] [PubMed] [Google Scholar]
- 8.Hoogenboom HR, Griffiths AD, Johnson KS et al. (1991) Multi-subunit proteins on the surface of filamentous phage: methodologies for displaying antibody (Fab) heavy and light chains. Nucleic Acids Res 19(15):4133–4137. 10.1093/nar/19.15.4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheets MD, Amersdorfer P, Finnern R et al. (1998) Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc Natl Acad Sci U S A 95(11):6157–6162. 10.1073/pnas.95.11.6157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaughan TJ, Williams AJ, Pritchard K et al. (1996) Human antibodies with sub-nan-omolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol 14(3):309–314. 10.1038/nbt0396-309 [DOI] [PubMed] [Google Scholar]
- 11.de Haard HJ, van Neer N, Reurs A et al. (1999) A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J Biol Chem 274(26):18218–18230. 10.1074/jbc.274.26.18218 [DOI] [PubMed] [Google Scholar]
- 12.Haidaris CG, Malone J, Sherrill LA et al. (2001) Recombinant human antibody single chain variable fragments reactive with Candida albicans surface antigens. J Immunol Methods 257(1–2):185–202. 10.1016/s0022-1759(01)00463-x [DOI] [PubMed] [Google Scholar]
- 13.Knappik A, Ge L, Honegger A et al. (2000) Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol 296(1):57–86. 10.1006/jmbi.1999.3444 [DOI] [PubMed] [Google Scholar]
- 14.Sidhu SS, Li B, Chen Y et al. (2004) Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol 338(2):299–310. 10.1016/j.jmb.2004.02.050 [DOI] [PubMed] [Google Scholar]
- 15.Rauchenberger R, Borges E, Thomassen-Wolf E et al. (2003) Human combinatorial Fab library yielding specific and functional antibodies against the human fibroblast growth factor receptor 3. J Biol Chem 278(40): 38194–38205. 10.1074/jbc.M303164200 [DOI] [PubMed] [Google Scholar]
- 16.Nelson B, Sidhu SS (2012) Synthetic antibody libraries. Methods Mol Biol (Clifton, N.J.) 899:27–41. 10.1007/978-1-61779-921-1_2 [DOI] [PubMed] [Google Scholar]
- 17.Strachan G, McElhiney J, Drever MR et al. (2002) Rapid selection of anti-hapten antibodies isolated from synthetic and semi-synthetic antibody phage display libraries expressed in Escherichia coli. FEMS Microbiol Lett 210(2):257–261. 10.1111/j.1574-6968.2002.tb11190.x [DOI] [PubMed] [Google Scholar]
- 18.Zhao Q, Buhr D, Gunter C et al. (2018) Rational library design by functional CDR resampling. New Biotechnol 45:89–97. 10.1016/j.nbt.2017.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiss MM, Babineau EG, Bonatsakis M et al. (2011) Phage ESCape: an emulsion-based approach for the selection of recombinant phage display antibodies. J Immunol Methods 367(1–2):17–26. 10.1016/j.jim.2010.09.034 [DOI] [PubMed] [Google Scholar]
- 20.Nikiforov TT, Rendle RB, Kotewicz ML et al. (1994) The use of phosphorothioate primers and exonuclease hydrolysis for the preparation of single-stranded PCR products and their detection by solid-phase hybridization. PCR Methods Appl 3(5):285–291. 10.1101/gr.3.5.285 [DOI] [PubMed] [Google Scholar]
- 21.Kunkel TA (1985) Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A 82(2):488–492. 10.1073/pnas.82.2.488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scholle MD, Kehoe JW, Kay BK (2005) Efficient construction of a large collection of phage-displayed combinatorial peptide libraries. Comb Chem High Throughput Screen 8(6):545–551. 10.2174/1386207054867337 [DOI] [PubMed] [Google Scholar]
- 23.Huang R, Fang P, Kay BK (2012) Improvements to the Kunkel mutagenesis protocol for constructing primary and secondary phage-display libraries. Methods (San Diego, Calif.) 58(1):10–17. 10.1016/j.ymeth.2012.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cadwell RC, Joyce GF (1994) Mutagenic PCR. PCR Methods Appl 3(6):S136–S140. 10.1101/gr.3.6.s136 [DOI] [PubMed] [Google Scholar]
- 25.Jones KS, Chapman AE, Driscoll HA et al. (2022) MILKSHAKE: novel validation method for antibodies to post-translationally modified targets by surrogate Western blot. BioTechniques 72(1):11–20. 10.2144/btn-2021-0078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferguson FM, Mendez MQ, Acca EF et al. (this volume) Validation and the determination of antibody bioactivity using MILKSHAKE and sundae protocols. In: Phage display: methods and protocols. Springer, New York: [DOI] [PMC free article] [PubMed] [Google Scholar]