

Abstract

Nucleic acids and proteins possess encoded “languages” that can be used for information storage or to direct function. However, each biopolymer is limited to encoding its respective “language.” Using a peptide nucleic acid (PNA) scaffold, nucleobase and amino acid residues can be installed on a singular backbone, enabling a single biopolymer to encode both languages. Our laboratory previously reported the development of a “bilingual” PNA biopolymer that incorporates a sequence-specific nucleic acid code interspersed with hydrophobic (alanine) and hydrophilic (lysine) amino acid residues at defined positions to produce amphiphilic character. We observed the amphiphilic amino acid residues directing the biopolymer to undergo self-assembly into micelle-like structures, while the nucleic acid recognition was harnessed for disassembly. Herein, we report a series of bilingual PNA sequences having amino acid residues with varying lengths, functional group charges, hydrophobicities, and spacings to elucidate the effect of these parameters on micelle assembly and nucleic acid recognition. Negative charges in the hydrophilic block or increased bulkiness of the hydrophobic side chains led to assembly into similarly sized micelles; however, the negative charge additionally led to increased critical micelle concentration. Upon PNA sequence truncation to decrease the spacing between side chains, the biopolymers remained capable of self-assembling but formed smaller structures. Characterization of disassembly revealed that each variant retained sequence recognition capabilities and stimuli-responsive disassembly. Together, these data show that the amino acid and nucleic acid sequences of amphiphilic bilingual biopolymers can be customized to finely tune the assembly and disassembly properties, which has implications for applications such as the encapsulation and delivery of cargo for therapeutics.
Introduction
Nature primarily uses two biopolymer structures—nucleic acid and protein—to encode and transmit information into structure and function. Nucleic acids are adept at storing genetic information and performing sequence recognition to regulate biological function. While this system is privileged by predictable molecular recognition properties in the form of Watson–Crick–Franklin base pairing, the relative lack of chemical diversity in backbone and nucleobase components limits its potential functions.1–3 In contrast, peptides and proteins are decorated with amino acid side chains, which possess a diverse range of chemical structures and functionalities, leading to greater diversity of function but making folding and molecular recognition less predictable. Due to their uniquely adaptable capabilities, both biopolymers show great promise in applications outside their canonical biological roles.4–6 We hypothesized that a “bilingual” biopolymer capable of integrating both the sequence-recognition properties of nucleic acids and the structure- and function-defining properties of peptides in a cohesive manner would open new avenues for creating programmable materials for applications such as drug delivery and development of diagnostic probes.
Researchers have described chimeric polymers containing both nucleic acid and peptide blocks, but the separation of these domains limits the overall function of the polymers.3,7,8 We envisioned that peptide nucleic acid (PNA), with its modifiable pseudopeptide backbone, would provide an ideal scaffold for simultaneously and precisely encoding nucleic acid and protein sequences.9,10 PNA confers additional advantages such as increased duplex stability and resistance to enzymatic degradation.3,11–14 Previously, we designed and synthesized a “bilingual” PNA biopolymer by incorporating amino acid residues at the γ-position along the PNA backbone to encode for amphiphilicity.9 Specifically, we incorporated hydrophilic (lysine) and hydrophobic (alanine) residues to generate an amphiphilic sequence capable of self-assembly into spherical micelle-like structures. Upon the introduction of a complementary nucleic acid sequence, recognition via hybridization leads to a change in the electrostatic properties and induces stimuli-responsive disassembly. Through this design, we harnessed the peptide code for assembly and the nucleic acid code for disassembly. Further exploration of this system allowed us to design a masking system that renders the PNA amphiphile in an initially disassembled state, enabling stimuli-responsive assembly in the presence of a target nucleic acid.10 Recognizing the potential use of these amphiphilic bilingual biopolymers for applications such as drug delivery and biosensing, we were curious to explore the design rules for assembly and function with the goal of being able to tune the physical properties of the assembled structures.15
Herein, we describe the design, synthesis, and characterization of a series of amphiphilic PNA sequences that vary in amino acid side chain identity and placement, enabling us to explore the parameters of charge, hydrophobicity, and sequence length on assembly properties (Figure 1A). We demonstrate that altering the structure of either the hydrophilic or hydrophobic residues alters the micellar architecture, as observed by transmission electron microscopy (TEM) and dynamic light scattering (DLS) (Figure 1B). Variation in the sequence length of the oligomer plays a role in determining the size of the assemblies, as shorter sequences produced smaller assemblies (Figure 1C). Finally, our studies confirmed that the bilingual biopolymer sequences remained capable of undergoing stimuli-responsive disassembly upon the introduction of a complementary nucleic acid sequence irrespective of micellar size and charge of corona. Taken together, these results demonstrate our ability to manipulate the physicochemical properties of bilingual PNA amphiphiles through defined amino acid codes and oligomer length, thus advancing our understanding of this new class of materials for use in diverse biological and nanotechnology applications.
Figure 1.
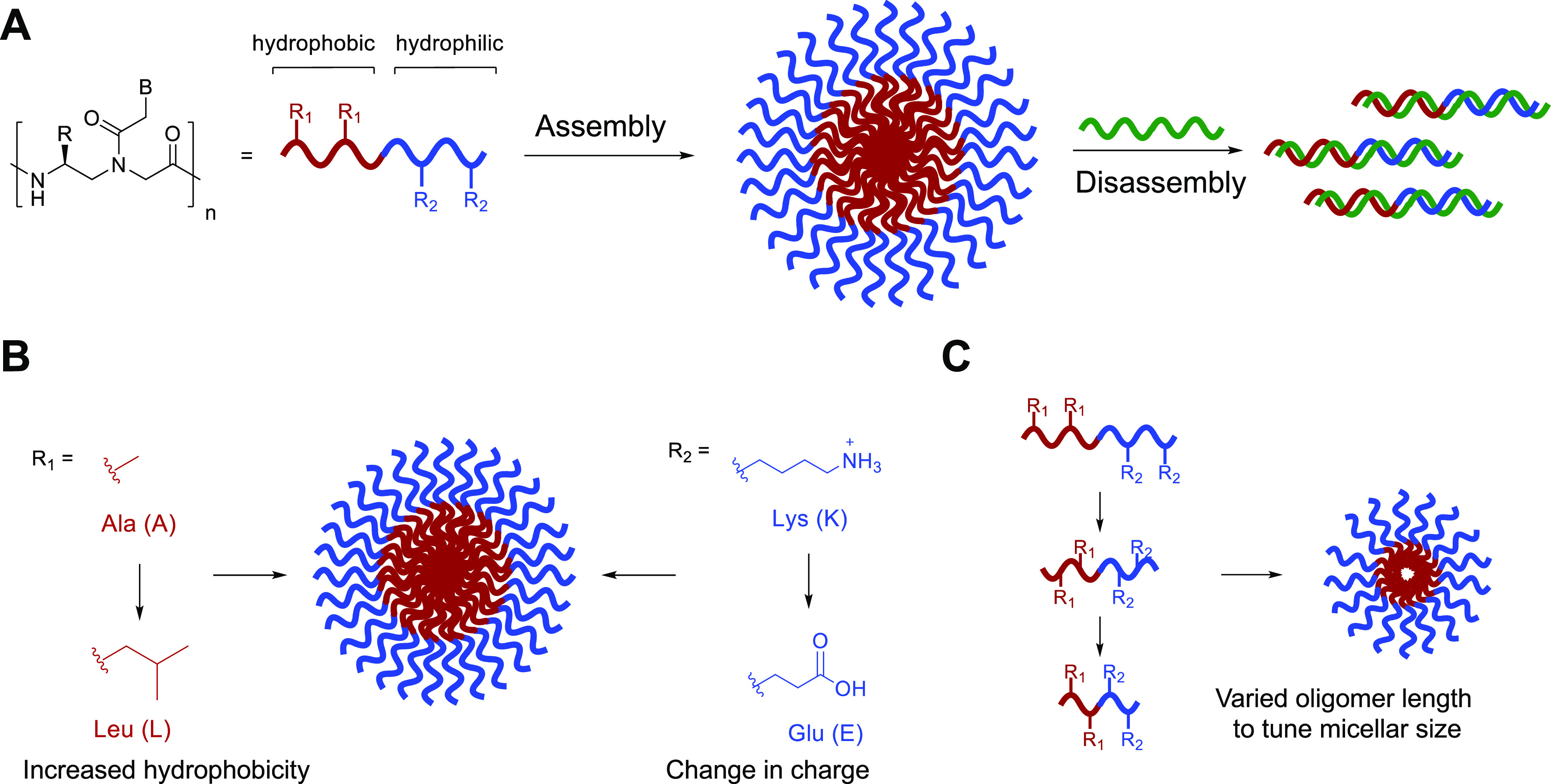
(A) Bilingual PNA biopolymers contain hydrophobic and hydrophilic amino acid residues at the γ-position to direct self-assembly. Upon the introduction of a complementary sequence, nucleobase recognition induces stimuli-responsive disassembly. (B) Changes to the amino acid code (R1, R2) do not significantly impact assembly. (C) Nucleic acid sequence truncation allows for control over assembly size.
Results and Discussion
Design and Synthesis of Amphiphilic PNA Sequences
Previously, our lab designed and characterized PNA-A, a dodecameric PNA sequence that contains two alanine and two lysine residues that create the hydrophobic and hydrophilic blocks, respectively (Table 1).9,10 We recognized that the application of amphiphilic bilingual biopolymers would be advanced by elucidating the impact of the amino acid side chain identity and spacing on the assembly and disassembly properties. To test the effect of a larger hydrophobic side chain, we chose to replace the methyl group with an isobutane side chain derived from leucine to produce PNA-B-A.16–18 To determine the influence of charge in the hydrophilic block, we replaced the lysine side chain in PNA-A with glutamic acid in PNA-N-A to switch from positive to negative charge while maintaining similar, but not identical side chain length to lysine.19,20 Finally, to test the influence of the side chain spacing and nucleic acid length on the biopolymer assembly characteristics, we explored octamer (PNA-8-A) and decamer (PNA-10-A) sequences containing the same number and identity of amino acid side chains as first generation PNA-A but in a shorter polymer (Figure 1C).21,22 Sequence truncation impacted residue spacing with the amino acid side chains present on every other monomer instead of on every third monomer, altering the general structure of the hydrophilic and hydrophobic block. As a facile method to evaluate assembly and determine critical micelle concentration (CMC), we also incorporated a fluorogenic 4-DMN PNA monomer into the hydrophobic block as previously reported.9
Table 1. Oligonucleotide Sequences Used to Explore the Impact of Structure on Self-Assembly and Stimuli-Responsive Disassemblya.

“D” denotes the 4-DMN dye monomer. * Denotes acetyl N terminal. COOH denotes carboxylic acid at C-terminus. C denotes amide at C-terminus. Subscript denotes the amino acid residue single letter code.
To generate the small library of sequences, we first synthesized the γ-modified PNA monomers as previously reported.23,24 Starting with Fmoc-protected l-amino acids, the PNA backbones containing either lysine, alanine, leucine, or glutamic acid, were synthesized followed by coupling to protected nucleobase acetic acids using HATU in the presence of N,N-diisopropylethylamine to produce the desired monomers. Synthesis of the 4-DMN PNA monomer was completed as previously reported.9 Following monomer synthesis, the amphiphilic and control PNA sequences were synthesized in solid-phase, purified, and characterized via mass spectrometry and high-performance liquid chromatography (HPLC). The C-to-N terminal peptide synthesis produces oligomers having an N-terminal positive charge, which is ideal for amphiphiles having a positively charged hydrophilic block but could interfere with our design of amphiphiles having a negatively charged hydrophilic block. To account for the charge distribution for the case of PNA-N-A, we chose to reverse the ordering of the hydrophobic and hydrophilic residues, with the 4-DMN being placed at the N-terminus and an acetyl group used to cap the free amine.25 Additionally, for positively charged amphiphiles, we used rink amide MBHA resin to produce an uncharged C-terminal carboxyamide. Alternatively, for negatively charged PNA-N-A, we used Wang resin to result in a C-terminal carboxylic acid, thus providing a terminal negative charge in the hydrophilic block analogous to the terminal positive charge present in PNA-A.
After obtaining the PNA sequences, UV melting temperature studies were performed to analyze hybridization to complementary DNA target sequences. To measure melting temperature, equimolar amounts of PNA and DNA were combined in 1× phosphate buffered saline (1× PBS) at final concentrations of 3 μM, and a heat–cool cycle was performed to anneal the two strands. We previously observed that amphiphilic PNA sequences have higher melting temperatures than their control strand counterparts, which lack γ-modifications, as the introduction of γ-modifications is known to increase hybridization stability.26,27 Of note, in the case of PNA-N-A, we observed a decrease in melting temperature from 58.1 to 55.7 °C as a result of adding the amino acid side chains. This may be due to the swapping of the hydrophilic and hydrophobic blocks in the sequence, but more likely results from the presence of the additional glutamic acid at the N-terminus and addition of a glycine at the C-terminus, which may reduce preorganization, as observed for peptides.27 While electrostatic repulsion between the negative side chains and the negatively charged DNA backbone may also play a role, previous studies have found that at physiological ionic strength, the polarity of the side chains has little impact on hybridization strength with DNA and RNA.24
Characterization of PNA Assembly
In parallel, we determined the effect of each structural change on the self-assembly properties of the bilingual biopolymers. In our previous study, we had strategically placed the 4-DMN dye such that it would become sequestered in a hydrophobic environment upon assembly, leading to increased fluorescence enabling us to measure CMC.9,28 By measuring the relative fluorescence of each amphiphilic sequence and unbound dye monomer in solution, we were able to determine the CMC value for each PNA amphiphile (Figure 2A). Similar CMC values were found for PNA-B-A, PNA-10-A, and PNA-8-A as compared to the previously reported PNA-A (Table 2).9 This is likely due to the similarity in the peptide sequence that drives assembly, having two hydrophobic and two positively charged residues. In the case of PNA-B-A, this was somewhat surprising as we anticipated that the increased hydrophobicity of the leucine side chain would increase the driving force for assembly. However, any gain from increased hydrophobicity could be offset by greater steric hindrance in the close packing of PNA strands. Alternatively, it is possible that the positively charged block plays a more significant role in determining the thermodynamics of assembly. In comparison, PNA-N-A containing glutamic acid had a significantly higher CMC, indicating that it has a higher solubility and reduced driving force for assembly. Importantly, these experiments demonstrate our ability to tune the assembly strength of the micelles by altering the side chain identity, enabling these materials to be tuned for varying biological applications, including drug delivery and biosensors. Experiments using the control sequences resulted in lower and delayed increases in fluorescence, similar to the previously reported PNA-C,9 indicating that the amino acid residues are driving self-assembly.
Figure 2.

(A) Activation of solvatochromic 4-DMN dye upon PNA assembly. (B) CMC measurement using the fluorescence ratio. The fluorescence ratio represents the PNA fluorescence relative to dye monomer fluorescence. Samples tested in water. The blue line represents the baseline of the curve. The red line represents the linear increase upon assembly. CMC is determined by the intersection of these two linear trends. (C) Normalized size distribution of PNA assemblies measured by DLS. Samples tested at 200 μM in 1× PBS.
Table 2. Tabulated CMC Value.
| Strand | CMC |
|---|---|
| PNA-A[9] | 317 nM |
| PNA-B-A | 860 nM |
| PNA-N-A | 3.58 μM |
| PNA-10-A | 548 nM |
| PNA-8-A | 523 nM |
Next, we evaluated the size of the assemblies in solution using DLS experiments compared to the previously reported PNA-A (110.2 ± 31.9 nm). Samples were prepared using 200 μM PNA and their size distribution was measured (Figure 2C). Each amphiphile sequence produced assemblies having a measurable hydrodynamic diameter, and PNA-B-A and PNA-N-A were found to be of similar sizes at 107.8 ± 47.0 and 161.7 ± 44.4 nm, respectively. The truncated sequences PNA-10-A and PNA-8-A were found to form smaller assemblies, measuring 58.9 ± 17.8 and 70 ± 43.1 nm, respectively (Table 3). This suggests that the length of the oligomer determines the size of the assembly, while the peptide sequence determines the assembly strength, measured as decreasing CMC values. DLS analysis of the control sequences revealed only objects below 5 nm in size, which corresponds approximately to the size of single-stranded PNA and, thus, indicates the lack of measurable aggregation.
Table 3. Average Size of PNA Assembly Particles Using DLS.
| Strand | Diameter (nm) |
|---|---|
| PNA-A[9] | 110.2 ± 31.9 |
| PNA-B-A | 107.8 ± 47.0 |
| PNA-N-C | 1.3 ± 0.1 |
| PNA-N-A | 161.7 ± 44.4 |
| PNA-10-C | 1.6 ± 0.3 |
| PNA-10-A | 58.9 ± 17.8 |
| PNA-8-C | 1.5 ± 0.3 |
| PNA-8-A | 70.0 ± 43.1 |
Finally, we used TEM to visualize and characterize the assemblies. All sequences were prepared in ultrapure water and diluted to 100 μM. The assemblies were spotted onto the grid and stained with uranyl acetate to image. We observed that all of the amphiphilic sequences were capable of forming uniform, micelle-like assemblies. The dodecameric sequences PNA-B-A and PNA-N-A were found to be 30.3 ± 7.6 and 29.1 ± 14.3 nm, respectively, which are similar in size to the parent PNA-A (36.9 ± 9.9 nm). These assembly sizes were expected, as the DLS measurements of PNA-N-A and PNA-B-A similarly matched those of PNA-A.9 The shorter sequences PNA-10-A and PNA-8-A were found to be smaller in size, measuring 17.7 ± 4.12 and 18.6 ± 3.1 nm, respectively, again in alignment with the DLS data (Figure 3 and Table 4). The control PNA sequences were also analyzed, and we did not observe the spherical assemblies as seen for the amphiphiles (Figure S2). Rather, the only observable objects were amorphous in nature and likely due to uncontrolled PNA aggregation at high concentrations. Overall, these results demonstrate that PNA bilingual biopolymers undergo self-assembly and retain uniform shape, and their size is determined by the length of the oligomer.
Figure 3.

TEM images of PNA-B-A, PNA-N-A, PNA-10-A, and PNA-8-A assembled in water at 100 μM. (A) Scale bar = 200 nm. (B) Scale bar = 50 nm.
Table 4. Average Size of PNA Assemblies As Measured by TEM.
| Strand | Diameter (nm) |
|---|---|
| PNA-A[9] | 36.9 ± 9.9 |
| PNA-B-A | 30.3 ± 7.6 |
| PNA-N-A | 29.1 ± 14.3 |
| PNA-10-A | 17.7 ± 4.12 |
| PNA-8-A | 18.6 ± 3.1 |
Characterization of Stimuli-Responsive PNA Disassembly
While the amino acid code of our bilingual biopolymers drives assembly, the nucleic acid code is responsible for stimuli-responsive disassembly as hybridization to a highly anionic DNA or RNA strand shields the hydrophilic block and diminishes the amphiphilicity. Thus, we were curious to study the effect of changing the sequence length and amino acid residues on stimuli-responsive disassembly. To test this, we used a 22 bp DNA sequence that would be fully complementary to all the PNA sequences. Using circular dichroism (CD), we collected spectra for each PNA and PNA/DNA duplex, as well as a DNA/DNA control (Figure 4).29 All solutions were prepared in 1× PBS at a 100 μM final concentration of each oligonucleotide. As previously reported, the unmodified PNA sequences imparted no chirality, while the γ-modified sequences produced a small chiral signal, as the backbone modifications orient the single stranded oligomer into a helical structure.27,30 With the exception of PNA-B-A and PNA-8-A, all the sequences demonstrated a shift in CD signal to maxima of 220 and 263 nm and minima of 200 and 244 nm, indicative of formation of a PNA/DNA duplex.12,27,29 We also tested complementary DNA alone and DNA hybridized to a fully complementary DNA sequence (CS-DNA), which enabled us to distinguish between the ssDNA, PNA/DNA, and DNA/DNA CD signals. While PNA-B-A/DNA and PNA-8-A/DNA did not display the expected changes in the CD signal for PNA-DNA duplex formation, the decrease in the CD signal intensity is likely indicative of a change in the hybridization state. For PNA-B-A, this unexpected result could be due to the change in the amino acid bulk, which may result in a tighter helix confirmation, thus reducing the CD signal, as observed in helical peptides.16,31,32 For PNA-8-A, this result is likely due to the change in the proximal nucleobases adjacent to the γ-modified PNA monomers, as previously reported.27 These hypotheses are also consistent with our Tm data that support the hybridization of these PNA sequences with their complementary DNA counterparts. The control sequences were also tested under similar conditions and were found to be capable of binding to DNA, inducing a similar change in CD signal.
Figure 4.

(A) PNA assemblies undergoing stimuli-responsive disassembly in the presence of a complementary sequence. CD spectra of (B) PNA-B-A, (C) PNA-N-A, (D) PNA-10-A, and (E) PNA-8-A in 1× PBS at 100 μM to demonstrate the change in maxima and minima on the addition of DNA.
Finally, we used TEM to image the changes in aggregation patterns upon the addition of the complementary DNA sequence to confirm that the bilingual biopolymers can undergo stimuli-responsive disassembly. Samples containing a final concentration of 100 μM each of PNA and DNA in 1× PBS was prepared. As previously reported, we observe the appearance of dark precipitates in all samples containing DNA, likely due to the negative stain undergoing a nonspecific interaction with the phosphate backbone in DNA (Figure 5).9,10 As expected, we noted the disappearance of the previously observed spherical assemblies, indicating disassembly through hybridization. Thus, through CD and TEM, we were able to establish the binding of complementary DNA sequences to the PNA amphiphiles and the corresponding disassembly of the micellar assemblies, demonstrating all of the bilingual biopolymers remain capable of nucleic acid molecular recognition, irrespective of amino acid identity and PNA sequence length.
Figure 5.

TEM images of PNA-B-A, PNA-N-A, PNA-10-A, and PNA-8-A disassembled through hybridization to DNA in 1× PBS at 100 μM. (A) Scale bar = 200 nm. (B) Scale bar = 50 nm.
Conclusions
By synthesizing and characterizing a series of novel bilingual PNA biopolymers having variations in both the amino acid and nucleic acid codes, we were able to understand the impact of these parameters on assembly and stimuli-responsive disassembly. Our studies demonstrate that varying the identity and spacing of the amphiphilic side chains can modulate assembly strength but does not significantly alter the overall morphology. Somewhat predictably, we found that the assembly size can be tuned by altering the sequence length. Finally, all of the sequences tested were capable of undergoing stimuli-responsive disassembly upon hybridization to a complementary DNA sequence. This further suggests that the amino acid residues direct assembly but do not interfere with nucleic acid sequence recognition.
In Nature, the languages of nucleic acids and proteins encode information and impart function. Our previous work brought these two separate information systems together to produce a bilingual biopolymer that incorporates both amino acid and nucleobase sequences to direct assembly and stimuli-responsive disassembly. In our initial studies, we focused on a single sequence, and here, we explore the impact of amino acid side chains and nucleic acid sequence length. The ability to modulate assembly and disassembly properties, by altering structure enables the generation of materials having tunable properties, providing insight into the assembly process and, in turn, increasing utility for biological and nanotechnology applications. As such, the scope of this work further expands the capabilities of bilingual PNA biopolymers by informing future applications, such as drug delivery in nanomedicine and biomedical applications. We envision future studies involving the incorporation of unnatural amino acids and variations in amino acid patterning to increase the structural and functional repertoire of this class of biomaterials.
Experimental Procedures
PNA Monomer Synthesis
Unmodified PNA monomers were purchased from PolyOrg, Inc. Thymine acetic acid was purchased from Sigma-Aldrich. The protected nucleobases adenine and cytosine and Fmoc protected γ-methyl, γ-lysyl, γ-glutamyl, and γ-leucyl modified PNA backbones were synthesized following previously published procedures.24,33–36 The Fmoc-protected l-amino acids and nucleobase starting materials were purchased from Chem-Impex Inc. All other reagents were purchased from Fisher Scientific, Chem-Impex, and Sigma-Aldrich and used without additional purification unless otherwise stated. Merck silica gel 60 F254 was used to monitor the small molecule synthesis through thin layer chromatography with UV light for visualization. Synthesized compounds were purified using flash chromatography with SiliaFlash F60 grade silica purchased from SiliCycle Inc. Characterization of compounds was completed using proton (1H) and carbon (13C) NMR using a Varian Inova 400 MHz spectrometer. The spectra were analyzed by using MestReNova Software. The mass of the compounds was confirmed using an Agilent 6230 electrospray ionization–time-of-flight (ESI–TOF) mass spectrometer. PNA monomer synthesis was completed as described previously.9
PNA Oligomer Synthesis
PNA oligomers were synthesized as previously described using a Biotage SP Wave semiautomatic peptide synthesizer using rink amide MBHA and Wang resins, respectively.9 Monomer coupling efficiencies were monitored using a Nanodrop 2000 spectrophotometer to measure the dibenzofulvene-piperidine adduct absorbance at 301 nm. Upon cleavage, sequences were purified through reverse-phase HPLC using an Agilent Eclipse XDB-C18 5 μm, 9.4 × 250 mm column at 60 °C with a flow rate of 2 mL/min, monitoring 260 and 495 nm using a linear gradient (10–40%) of 0.1% TFA/ACN in 0.1% TFA/H2O. The sequences were confirmed by ESI–TOF mass spectrometry.
Melting Temperature Analysis of PNA Strands
All DNA sequences were purchased from the University of Utah DNA/Peptide Synthesis Core Facility or from Integrated DNA Technologies. Samples containing 3 μM complementary nucleic acids were prepared using stock solutions in 1× PBS. Samples were then added to an 8-cell quartz microcuvette with a path length of 1 cm path length. A Shimadzu UV-1800 spectrophotometer equipped with a temperature controller and Julabo Corio CD water circulator was used for measurement of absorbance at 260 nm while heating from 25 to 95 °C at a rate of 0.5 °C/min. The melting temperatures were calculated using a first derivative method, and experiments were carried out in triplicate.
Preparation of PNA Amphiphile Assemblies
To prepare the amphiphile assembly, samples of PNA and PNA/DNA were dissolved in water or 1× PBS and were annealed as previously described. All samples were allowed to incubate at room temperature for 1 h.
Determination of CMC Using Fluorescence
The CMC of each sequence was determined by using the 4-DMN monomer fluorescence response. PNA samples and fully deprotected 4-DMN ranging from 32 to 0.061 μM in 1% dimethyl sulfoxide (DMSO)/H2O were prepared in stock solutions. 4-DMN was initially dissolved in DMSO, followed by dilution with water to a final solution containing 1% DMSO/H2O. Each sample was prepared using the amphiphile assembly protocol as previously described. Samples were transferred to a 384-well clear-bottom plate for analysis on a Biotek Cytation5 plate reader (higher concentrations) or to a 50 μL quartz cuvette for analysis by a Horiba Scientific Dual-FL fluorometer (lower concentrations). Fluorescence measurements at 550 nm were collected by using an excitation of 458 nm. Absorbance was also measured at 458 nm to correct for any changes in the concentration between PNA samples and the monomer. A plot of the fluorescence enhancement of the PNA samples over the monomer as a function of concentration was created by using GraphPad Prism software. A semilogarithmic fit to the data provided equations for the upper and lower portions of the curve, which were then compared to determine the CMC value.
Determination of Size Using DLS
Samples of PNA and PNA/DNA were prepared in 1× PBS at 200 μM from stock solutions and were annealed through heating and slow cooling as described above. Samples were then transferred to a quartz cuvette and analyzed using a Particular Systems NanoPlus DLS nanoparticle analyzer, and data were analyzed using NanoPlus Software.
Confirmation of DNA Hybridizing to PNA by CD
CD was used to confirm the hybridization of PNA/DNA. Individual samples of PNA and DNA were prepared at 200 μM in 1× PBS. Thirty μL portion of PNA was combined with 30 μL of DNA to form the final 100 μM in 1× PBS solutions. Samples were incubated for 1 h, after which they were analyzed using a JASCO J-1500 CD spectrometer. Data points from 190 to 300 nm were collected at 1 nm intervals by using a continuous scanning mode of 200 nm/min at 23.5 °C. A sample with PNA alone was used as a control and had the signal overlapped using JASCO software.
Characterization of PNA Assemblies by TEM
Samples of PNA were prepared at 100 μM in water from stock solutions. Using a 200-mesh Formvar/carbon- coated copper grid, 3.5 μL of sample was spotted for 2 min and was wicked away with filter paper. Then, 3.5 μL of 1% uranyl acetate stain solution was spotted for 30 s before wicking. Grids were dried at room temperature for 30 min prior to imaging using a JEOL JEM-1400 transmission electron microscope.
Characterization of PNA-A Disassembled Using Masking Sequence DNA by TEM
Samples of PNA/DNA at 100 μM were prepared in 1× PBS and annealed through heating and slow cooling as described above. The duplex sample was then spotted and prepared for TEM imaging, as described above.
Acknowledgments
This work was supported by the National Science Foundation (DMR 2003987 to J.M.H.). The authors also acknowledge the Robert P. Apkarian Integrated Electron Microscopy Core and NMR Research Center at Emory University for instrument access and technical assistance. They would also like to thank Mike Hanson and the oligonucleotide and peptide synthesis facility and the University of Utah for oligonucleotide materials.
Glossary
Abbreviations
- Fmoc
fluorenylmethyloxycarbonyl
- FAM
fluorescein
- ACN
acetonitrile
- H2O
water
- DIPEA
diisopropylethylamine
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- TFA
trifluoroacetic acid
- DCM
dichloromethane
- Boc
tert-butyloxycarbonyl
- NMP
N-methyl-2-pyrrolidone
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c05528.
Characterization of PNA sequences, Tm data, TEM images, and additional displacement studies (PDF)
Author Present Address
‡ Department of Chemistry, Washington University in St. Louis, St. Louis, Missouri 63130, United States
The authors declare no competing financial interest.
Special Issue
Published as part of ACS Omegavirtual special issue “Nucleic Acids: A 70th Anniversary Celebration of DNA”.
Supplementary Material
References
- Tan X.; Jia F.; Wang P.; Zhang K. Nucleic acid-based drug delivery strategies. J. Controlled Release 2020, 323, 240–252. 10.1016/j.jconrel.2020.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Gong C.; Yuan X.; Wei G. Controlling the self-assembly of biomolecules into functional nanomaterials through internal interactions and external stimulations: A review. Nanomaterials 2019, 9 (2), 285. 10.3390/nano9020285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson A. M.; Heemstra J. M. Controlling self-assembly of DNA-polymer conjugates for applications in imaging and drug delivery. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2015, 7 (3), 282–297. 10.1002/wnan.1309. [DOI] [PubMed] [Google Scholar]
- Seeman N. C.; Sleiman H. F. DNA nanotechnology. Nat. Rev. Mater. 2017, 3 (1), 17068. 10.1038/natrevmats.2017.68. [DOI] [Google Scholar]
- Sato K.; Hendricks M. P.; Palmer L. C.; Stupp S. I. Peptide supramolecular materials for therapeutics. Chem. Soc. Rev. 2018, 47 (20), 7539–7551. 10.1039/C7CS00735C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi N.; Kamaly N.; Memic A.; Shafiee H. Self-assembled peptide-based nanostructures: Smart nanomaterials toward targeted drug delivery. Nano today 2016, 11 (1), 41–60. 10.1016/j.nantod.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak M.; Herrmann A. Nucleic acid amphiphiles: synthesis and self-assembled nanostructures. Chem. Soc. Rev. 2011, 40 (12), 5745–5755. 10.1039/c1cs15138j. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Nangreave J.; Liu Y.; Yan H. Structural DNA nanotechnology: state of the art and future perspective. J. Am. Chem. Soc. 2014, 136 (32), 11198–11211. 10.1021/ja505101a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson C. S.; Velusamy A.; Argueta-Gonzalez H. S.; Heemstra J. M. Bilingual peptide nucleic acids: encoding the languages of nucleic acids and proteins in a single self-assembling biopolymer. J. Am. Chem. Soc. 2019, 141 (48), 19038–19047. 10.1021/jacs.9b09146. [DOI] [PubMed] [Google Scholar]
- Argueta-Gonzalez H. S.; Swenson C. S.; Song G.; Heemstra J. M. Stimuli-responsive assembly of bilingual peptide nucleic acids. RSC Chem. Biol. 2022, 3, 1035–1043. 10.1039/d2cb00020b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egholm M.; Buchardt O.; Nielsen P. E.; Berg R. H. Peptide nucleic acids (PNA). Oligonucleotide analogs with an achiral peptide backbone. J. Am. Chem. Soc. 1992, 114 (5), 1895–1897. 10.1021/ja00031a062. [DOI] [Google Scholar]
- Moccia M.; Adamo M. F.; Saviano M. Insights on chiral, backbone modified peptide nucleic acids: properties and biological activity. Artif. DNA PNA XNA 2014, 5 (3), e1107176 10.1080/1949095X.2015.1107176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakeel S.; Karim S.; Ali A. Peptide nucleic acid (PNA)—a review. J. Appl. Chem. Biotechnol. 2006, 81 (6), 892–899. 10.1002/jctb.1505. [DOI] [Google Scholar]
- Egholm M.; Buchardt O.; Christensen L.; Behrens C.; Freier S. M.; Driver D. A.; Berg R. H.; Kim S. K.; Norden B.; Nielsen P. E. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 1993, 365 (6446), 566–568. 10.1038/365566a0. [DOI] [PubMed] [Google Scholar]
- Peng L.; Wu C. S.; You M.; Han D.; Chen Y.; Fu T.; Ye M.; Tan W. Engineering and applications of DNA-grafted polymer materials. Chem. Sci. 2013, 4 (5), 1928–1938. 10.1039/c2sc21198j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurezka R.; Laage R.; Brosig B.; Langosch D. A heptad motif of leucine residues found in membrane proteins can drive self-assembly of artificial transmembrane segments. J. Biol. Chem. 1999, 274 (14), 9265–9270. 10.1074/jbc.274.14.9265. [DOI] [PubMed] [Google Scholar]
- Wang K.; Keasling J. D.; Muller S. J. Effects of the sequence and size of non-polar residues on self-assembly of amphiphilic peptides. Int. J. Biol. Macromol. 2005, 36 (4), 232–240. 10.1016/j.ijbiomac.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Klein T.; Ulrich H. F.; Gruschwitz F. V.; Kuchenbrod M. T.; Takahashi R.; Fujii S.; Hoeppener S.; Nischang I.; Sakurai K.; Brendel J. C. Impact of amino acids on the aqueous self-assembly of benzenetrispeptides into supramolecular polymer bottlebrushes. Polym. Chem. 2020, 11 (42), 6763–6771. 10.1039/D0PY01185A. [DOI] [Google Scholar]
- Koshti B.; Kshtriya V.; Singh R.; Walia S.; Bhatia D.; Joshi K. B.; Gour N. Unusual Aggregates Formed by the Self-Assembly of Proline, Hydroxyproline, and Lysine. ACS Chem. Neurosci. 2021, 12 (17), 3237–3249. 10.1021/acschemneuro.1c00427. [DOI] [PubMed] [Google Scholar]
- Dzwolak W.; Marszalek P. E. Zipper-like properties of [poly (L-lysine)+ poly (L-glutamic acid)] β-pleated molecular self-assembly. Chem. Commun. 2005, (44), 5557–5559. 10.1039/b510150f. [DOI] [PubMed] [Google Scholar]
- Mendes A. C.; Baran E. T.; Reis R. L.; Azevedo H. S. Self-assembly in nature: using the principles of nature to create complex nanobiomaterials. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2013, 5 (6), 582–612. 10.1002/wnan.1238. [DOI] [PubMed] [Google Scholar]
- Edwards-Gayle C. J.; Hamley I. W. Self-assembly of bioactive peptides, peptide conjugates, and peptide mimetic materials. Org. Biomol. Chem. 2017, 15 (28), 5867–5876. 10.1039/C7OB01092C. [DOI] [PubMed] [Google Scholar]
- Nielsen P. E.; Haaima G.; Lohse A.; Buchardt O. Peptide nucleic acids (PNAs) containing thymine monomers derived from chiral amino acids: hybridization and solubility properties of D-lysine PNA. Angew Chem. Int. Ed. Engl. 1996, 35 (17), 1939–1942. 10.1002/anie.199619391. [DOI] [Google Scholar]
- De Costa N. T. S.; Heemstra J. M. Evaluating the effect of ionic strength on duplex stability for PNA having negatively or positively charged side chains. PLoS One 2013, 8 (3), e58670 10.1371/journal.pone.0058670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallaro V.; Thompson P.; Hearn M. Solid-phase synthesis of a dendritic peptide related to a retinoblastoma protein fragment utilizing a combined boc-and fmoc-chemistry approach. J. Pept. Sci. 2001, 7 (5), 262–269. 10.1002/psc.314. [DOI] [PubMed] [Google Scholar]
- Sforza S.; et al. Induction of Helical Handedness and DNA Binding Properties of Peptide Nucleic Acids (PNAs) with Two Stereogenic Centres; Wiley Online Library, 2007. [Google Scholar]
- Dragulescu-Andrasi A.; Rapireddy S.; Frezza B. M.; Gayathri C.; Gil R. R.; Ly D. H. A simple γ-backbone modification preorganizes peptide nucleic acid into a helical structure. J. Am. Chem. Soc. 2006, 128 (31), 10258–10267. 10.1021/ja0625576. [DOI] [PubMed] [Google Scholar]
- Loving G.; Imperiali B. A versatile amino acid analogue of the solvatochromic fluorophore 4-N, N-dimethylamino-1, 8-naphthalimide: a powerful tool for the study of dynamic protein interactions. J. Am. Chem. Soc. 2008, 130 (41), 13630–13638. 10.1021/ja804754y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccini A.; Tortori A.; Tedeschi T.; Sforza S.; Tonelli R.; Pession A.; Corradini R.; Marchelli R. Circular dichroism study of DNA binding by a potential anticancer peptide nucleic acid targeted against the MYCN oncogene. Chirality Pharmacol. Biologicaland Chem. Consequences Mol. Asymmetry 2008, 20 (3–4), 494–500. 10.1002/chir.20489. [DOI] [PubMed] [Google Scholar]
- Rapireddy S.; He G.; Roy S.; Armitage B. A.; Ly D. H. Strand invasion of mixed-sequence B-DNA by acridine-linked, γ-peptide nucleic acid (γ-PNA). J. Am. Chem. Soc. 2007, 129 (50), 15596–15600. 10.1021/ja074886j. [DOI] [PubMed] [Google Scholar]
- Xu H.; Wang J.; Han S.; Wang J.; Yu D.; Zhang H.; Xia D.; Zhao X.; Waigh T. A.; Lu J. R. Hydrophobic-region-induced transitions in self-assembled peptide nanostructures. Langmuir 2009, 25 (7), 4115–4123. 10.1021/la802499n. [DOI] [PubMed] [Google Scholar]
- Xu C.; Liu R.; Mehta A. K.; Guerrero-Ferreira R. C.; Wright E. R.; Dunin-Horkawicz S.; Morris K.; Serpell L. C.; Zuo X.; Wall J. S.; et al. Rational design of helical nanotubes from self-assembly of coiled-coil lock washers. J. Am. Chem. Soc. 2013, 135 (41), 15565–15578. 10.1021/ja4074529. [DOI] [PubMed] [Google Scholar]
- Kleiner R. E.; Brudno Y.; Birnbaum M. E.; Liu D. R. DNA-Templated Polymerization of Side-Chain-Functionalized Peptide Nucleic Acid Aldehydes. J. Am. Chem. Soc. 2008, 130, 4646–4659. Copyright (C) 2011 American Chemical Society (ACS). All Rights Reserved. 10.1021/ja0753997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna A.; Rapireddy S.; Sureshkumar G.; Ly D. H. Synthesis of optically pure γPNA monomers: a comparative study. Tetrahedron 2015, 71 (21), 3507–3514. 10.1016/j.tet.2015.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcheddu A.; Giacomelli G.; Piredda I.; Carta M.; Nieddu G. A practical and efficient approach to PNA monomers compatible with Fmoc-mediated solid-phase synthesis protocols. Eur. J. Org Chem. 2008, 2008, 5786–5797. Copyright (C) 2011 American Chemical Society (ACS). All Rights Reserved. 10.1002/ejoc.200800891. [DOI] [Google Scholar]
- Wu Y.; Xu J.-C. Synthesis of chiral peptide nucleic acids using Fmoc chemistry. Tetrahedron 2001, 57 (38), 8107–8113. 10.1016/S0040-4020(01)00789-X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.