

Abstract
Stress response pathways (SRPs) mitigate the cellular effects of chemicals, but excessive perturbation can lead to adverse outcomes. Here, we investigated a computational approach to evaluate SRP activity from transcriptomic data using gene set enrichment analysis (GSEA). We extracted published gene signatures for DNA damage response (DDR), unfolded protein response (UPR), heat shock response (HSR), response to hypoxia (HPX), metal-associated response (MTL), and oxidative stress response (OSR) from the Molecular Signatures Database (MSigDB). Next, we used a gene-frequency approach to build consensus SRP signatures of varying lengths from 50 to 477 genes. We then prepared a reference dataset from perturbagens associated with SRPs from the literature with their transcriptomic profiles retrieved from public repositories. Lastly, we used receiver-operator characteristic analysis to evaluate the GSEA scores from matching transcriptomic reference profiles to SRP signatures. Our consensus signatures performed better than or as well as published signatures for 4 out of the 6 SRPs, with the best consensus signature area under the curve (% performance relative to median of published signatures) of 1.00 for DDR (109%), 0.86 for UPR (169%), 0.99 for HTS (103%), 1.00 for HPX (104%), 0.74 for MTL (150%) and 0.83 for OSR (148%). The best matches between transcriptomic profiles and SRP signatures correctly classified perturbagens in 78% and 88% of the cases by first and second rank, respectively. We believe this approach can characterize SRP activity for new chemicals using transcriptomics with further evaluation.
Keywords: stress response pathways, transcriptomics, gene signatures, receiver operating characteristic, computational toxicology
1. Introduction
Modern high-throughput screening technologies (HTS) (Houck, et al. 2013) promise chemical hazard and risk assessment without using whole animals (ICCVAM 2018; Thomas, et al. 2019). Known as new approach methodologies (NAMs), they include any technology, methodology, approach, or combination of methods to provide information about chemical hazards and risks (USEPA 2018a - Strategic Plan to Promote the Development and Implementation of Alternative Test Methods Within the TSCA Program). Adverse outcome pathways (AOPs), a central tenet of NAMs, assume that stressors act through molecular initiating events (MIEs), ultimately resulting in an adverse outcome (Ankley, et al. 2010). Using MIEs as surrogates for the adverse outcomes, and measuring MIE activation using HTS, thousands of environmental chemicals can be prioritized for targeted testing (Thomas, et al. 2019). However, the problem is that many environmental chemicals are promiscuous and do not specifically activate a single MIE (Escher, et al. 2020; Judson, et al. 2016). Instead, they disrupt cellular homeostasis, resulting in activation of adaptive stress response pathways (SRPs) to recover from chemical perturbation (Simmons, et al. 2009). Here we explore a NAM to help characterize such non-specific chemicals by elucidating their activation of SRPs using transcriptomic data.
Non-specifically acting chemicals are hypothesized to perturb the normal cellular state resulting in DNA damage, misfolded proteins, deoxygenation, and shifts in cytosolic reductive potential (Judson, et al. 2016; Simmons, et al. 2009). These perturbations are detected by sensors that activate transcription factors (TFs), inducing the expression of effector genes responsible for cellular recovery (Welch 1992). If the magnitude or duration of perturbation exceeds specific thresholds, it can lead to autophagy (Hiemstra, et al. 2017; Mazure, et al. 2010), senescence, or apoptotic cell death (Muñoz-Pinedo, et al. 2014). The sensor, TF, and effector architecture of SRPs are conserved across cells and species in canonical modules described as DNA damage response (DDR), unfolded protein response (UPR), heat shock response (HSR), hypoxic response (HPX), response to metals (MTL) and oxidative stress response (OSR) (Simmons, et al. 2009). Not surprisingly, SRPs have been linked to drug-induced liver injury (Podtelezhnikov, et al. 2020), the critical stages of diabetes (Ozcan, et al. 2004), neurodegenerative diseases (Lindholm, et al. 2006; Wang, et al. 2015), and cancers (Spaan, et al. 2019).
Multiple methodologies have been used to measure SRP activity in vitro. Wink, et al. (2014) developed a novel SRP reporter assay based on high-content imaging (HCI) to measure TF activity in a platform focused on drug-induced liver injury. Using multiplexed time-course HCI data, Shah et al. developed a technique to estimate chemical concentrations for in vitro tipping points associated with SRPs (Shah, et al. 2016). Recently, Hatherell et al. used an HCI-based SRP panel to differentiate high-risk from low-risk chemicals (Hatherell, et al. 2020). The activity of SRPs has also been characterized by the expression of effector genes using reporter assays to confirm OSR following reactive oxygen species exposure (Plusquin, et al. 2012) and real-time quantitative PCR (qPCR) based quantification for both the activation and downstream markers of UPR (Oslowski, et al. 2011). An example of the use of a diverse set of stress assays can be found in Escher, et al. (2014). A more comprehensive evaluation of SRP activity is now possible using transcriptomics based on the expression of effector genes quantified with gene set enrichment analysis (GSEA) (Subramanian, et al. 2005).
Quantifying SRP activity using GSEA requires a signature, a set of genes whose activation is putatively associated with a biological state, namely, a disease state, pathway perturbation, or TF activation (Maleki, et al. 2020). Signatures can be derived from transcriptomic data. Researchers have successfully identified SRP signatures from transcriptomic data for DDR (Corton, et al. 2018; Li, et al. 2017), HSR, MTL (Jackson, et al. 2020), and OSR (Rooney, et al. 2020). SRP signatures have also been inferred from promoter-TF associations (Chen, et al. 2013; Chorley, et al. 2012) or based on biological pathways curated from the literature (e.g., the Gene Ontology biological process “Response to DNA Damage Stimulus”, GO:0034984), (Ashburner, et al. 2000). The Molecular Signatures Database (MSigDB) is a central repository of many gene sets derived from transcriptomic studies, literature-curated gene lists, or biological pathway annotations (Liberzon, et al. 2015; Liberzon, et al. 2011), and we make extensive use of this resource to identify published SRP gene signatures.
One of the challenges in developing accurate signatures is ensuring their sensitivity and specificity (Liu, et al. 2020; Maleki, et al. 2020); this is of particular concern for SRP signatures. Because of overlapping signaling and regulatory networks, perturbing distinct SRPs can produce similar patterns of effector gene expression (Schlage, et al. 2011), which limits their specificity. For instance, in a recent study by Gong et al., a protective isoform of p53, a key TF in DDR, is activated via a HIF1-dependent mechanism (Gong, et al. 2020), directly linking DDR and HPX stress systems. Metal stress TFs, MTF1, and MTF2 can activate the OSR-associated TF NFE2L2 after chelation, linking MTL and OSR (Jennings, et al. 2013). Further still, UPR can be activated alongside OSR via misfolding due to covalent bonding with redox species (Cao, et al. 2014), linking UPR and OSR. Using SRPs to assay chemical hazards in a NAM will require disentangling key genes associated with each pathway.
Our work attempts to address this issue in two novel ways. First, we use a “crowd-sourcing” approach to develop representative consensus gene signatures for SRPs from published gene sets by giving less importance to genes frequently involved in multiple SRPs. Second, we systematically evaluate all signatures’ cooperative performance to correctly categorize perturbagens as a synergistic unit. To our knowledge, a “crowd-sourcing” approach to develop consensus signatures and a cooperative performance evaluation have not been used to investigate SRPs before.
2. Methods
2.1. Workflow overview
The construction of SRP consensus signatures sets was completed in three steps. First, we constructed consensus signatures by merging and pruning relevant gene sets from the MSigDB v7.1 (Liberzon, et al. 2015). Second, we developed an independent gene expression validation set by identifying reference perturbagens from the literature and curating their transcriptomic profiles from publicly available sources. Third, we used gene set enrichment analysis (GSEA) (Barbie, et al. 2009; Subramanian, et al. 2005) to score matches between signatures and transcriptomic profiles. Lastly, we evaluated the performance of GSEA scores as classifiers of SRP activity within reference perturbagen transcriptomic profiles using ROC AUC analysis. The entire workflow and associated data are available from the authors upon request.
2.2. Obtaining published stress response signatures
An “essential” gene list was compiled for each stress response system investigated in this study to capture the genes most commonly associated with each pathway in the literature (Gunther, et al. 2012; Ma 2013; Radons 2016; Simmons, et al. 2009; Tu, et al. 2004). For each SRP, we identified sensors, TFs, and effectors; a table of these genes and references may be found in Supplemental Material 1. We next supplied a subset of the essential gene lists as queries to MSigDB and identified relevant signatures for each SRP. The list of all queries used for each SRP is provided in Supplemental Material 2. Initial queries resulted in 261 signatures comprised of 113, 14, 21, 52, 51, and 10 signatures for SRPs DDR, UPR, HSR, HPX, MTL, and OSR, respectively. The resulting signatures were not always relevant so they were filtered by keywords (Supplemental Material 2) to reduce each result to 4–17 signatures per SRP. For example, signatures in MSigDB returned for UPR essential genes were refined by matching the description field with the terms: “protein” and “endoplasmic reticulum.” In all, we identified 17, 9, 9, 5, 6, and 4 signatures from which we constructed consensus signatures DDR, UPR, HSR, HPX, MTL, OSR, respectively. The queries, filters, and resulting signatures from MSigDB, which were the source of our consensus signatures, are provided in Supplemental Material 2. Only three duplicate published signatures were present in all collected published signatures. These were shared between UPR and HSR, two closely related SRPs (Copple, et al. 2019; Simmons, et al. 2009).
2.3. Developing consensus stress response signatures
The collection of 46 published signatures from MSigDB identified in the previous step included 4660 genes and an average of 776.67 (253 SD) genes per SRP. There were 1,207, 639, 477, 647, 842, and 848 unique genes representing the SRPs DDR, UPR, HSR, HPX, MTL, and OSR, respectively. Despite our best attempt at selecting representative signatures from MSigDB, we found that 889/4660 (19%) of genes were present in multiple SRPs to the extent that key TFs overlapped between SRPs in these published signatures. For example, the central UPR TF ATF4 was associated with UPR, MTL, and OSR. Additionally, critical effectors of OSR such as heme oxygenase 1, HMOX1, and superoxide dismutase, SOD1, were present in published signatures for DNA, HPX, and OSR, among others. Therefore, to find unique consensus signatures, we developed a “crowd-sourcing” strategy in which the MSigDB signatures represented a “crowd” of published signatures from which we derived the consensus gene set most associated with one SRP. This crowd-sourcing strategy was based on calculating a normalized occurrence frequency (NOF) for weighing the membership for each of the 4660 genes in one of the six SRPs. The NOF of each gene for an SRP is based on three factors: (a) the intra-SRP frequency, which is the frequency of the gene across all signatures in the SRP (), and (b) the inter-SRP frequency, which is the frequency of the gene across the other signatures (). We defined the NOF of a gene for an SRP as the difference of the intra- and inter-group frequencies normalized by their sum (denoted as, ) and we found −1<NOF<1. A NOF=1 means that a gene is ideal for inclusion in the consensus signature for an SRP, while NOF=−1 means that it should be excluded altogether. Next, we sorted all genes for an SRP by NOF in descending order and constructed consensus signatures based on the top N genes (where N= 50, 100, 200, 300, 400, and 477, limited by total HPX genes identified). We produced consensus gene signatures by labeling them with the SRP name and the size restriction. Where mentioned in the text, consensus signatures restricted by a NOF cutoff are denoted by an SRP abbreviation linked to the cutoff. For example, HPX-300 is the hypoxia consensus signature restricted to the NOF rank-ordered first 300 genes. The overlap between all NOF restricted signature sets was analyzed and is discussed further in Supplemental Material 3. An overlap minimum was found for all NOF signatures restricted to the first 200 NOF ranked genes and was therefore selected as a starting point for analysis.
2.4. Identifying Reference Perturbagens
We define a reference perturbagen as a chemical or a treatment that has been experimentally shown to activate a particular SRP with high confidence. Reference perturbagens were identified using three independent methods. First, we searched the literature to identify perturbagens associated with the six SRPs. This approach captured well-known reference perturbagens, e.g., hydrogen peroxide for OSR and heat for HSP. Additional reference perturbagens were identified by searching databases for chemicals associated with the activation of essential genes in each SRP. Querying the Library of Integrated Network-based Cellular Signatures (LINCS, March 2020 build) (Stathias, et al. 2020) database with the essential genes yielded chemicals associated with each stress response pathway ranked by median tau score, an enrichment score that measures the average expression of the query genes across thousands of gene expression profiles (Pilarczyk, et al. 2020). We filtered the high-scoring chemicals by searching activity descriptions in LINCS with terms associated with SRPs, e.g., heat shock and HSP70 activator. We also searched for chemicals that activate the essential genes using the Comparative Toxicogenomic Database (CTD) (Davis, et al. 2020). We identified 49 perturbagens among the three approaches, with the majority, 45 of 49, identified in the literature. Perturbagens identified in CTD matched with literature searches, while LINCS provided 2 additional HIF1A inducers (Table1). The list of perturbagens and search phrases used to identify relevant transcriptomic profiles is provided in Supplemental Material 4.
Table 1.
Summary of reference perturbagens.
| Assigned Stress Category1 | Chemical | Perturbance (dose) | Time | Cell | GSE2 | Reference |
|---|---|---|---|---|---|---|
| DDR | Benzo(a)pyrene | 5uM | 24h | HepaRG | 146549 | (Hockley, Arlt et al. 2007) |
| Glycidamide | 2mM | 1h | HT1080 | 74725 | (McMullen, Pendse et al. 2016) | |
| Lasicoarpine | 1uM | 24h | HepaRG | 146549 | (Chen, Ning et al. 2018) | |
| Methylmethanesulfonate | 200uM | 1h | HT1080 | 74725 | (McMullen, Pendse et al. 2016) | |
|
| ||||||
| UPR | Brefeldin A | 10uM | 24h | MCF7 | LINCS3 | (Oslowski and Urano 2011) |
| Brefeldin A | 10uM | 6h | MCF7 | LINCS | (Oslowski and Urano 2011) | |
| Thapsigargin | 7.6uM | 24h | LBD | 31447 | (Oslowski and Urano 2011) | |
| Thapsigargin | 7.6uM | 3h | LBD | 31447 | (Oslowski and Urano 2011) | |
| Thapsigargin | 500mM | 2h | LBD | 19519 | (Oslowski and Urano 2011) | |
| Tunicamycin | 10uM | 24h | MCF7 | LINCS | (Wang, Wang et al. 2015) | |
| Tunicamycin | 10uM | 6h | MCF7 | LINCS | (Wang, Wang et al. 2015) | |
| Tunicamycin | 4.8uM | 8h | LBD | 19519 | (Wang, Wang et al. 2015) | |
|
| ||||||
| HSR | Geldanamycin | 0.2uM | 6h | MCF7 | LINCS | (West, Wang et al. 2012) |
| Geldanamycin | 0.37uM | 3h | MCF7 | LINCS | (West, Wang et al. 2012) | |
| Geldanamycin | 1.11uM | 3h | MCF7 | LINCS | (West, Wang et al. 2012) | |
| Geldanamycin | 10uM | 6h | HepG2 | LINCS | (West, Wang et al. 2012) | |
| Heat with recovery | 43degree C | 4h | THP.1 | 9916 | (Simmons, Fan et al. 2009) | |
| Radicicol | 10uM | 24h | MCF7 | LINCS | (West, Wang et al. 2012) | |
| Radicicol | 10uM | 6h | MCF7 | LINCS | (West, Wang et al. 2012) | |
|
| ||||||
| HPX | Oxygen | 0.1% | 24h | HeLa | 3051 | (U. R. Jewel, I. Kvietikova et al. 2001) |
| Oxygen | 0.1% | 6h | P493.6 | 4086 | (U. R. Jewel, I. Kvietikova et al. 2001) | |
| VU-0418946-1 | 10uM | 24h | MCF7 | LINCS | (Hu, Jin et al. 2018) | |
| VU-0418946-1 | 10uM | 6h | MCF7 | LINCS | (Hu, Jin et al. 2018) | |
| VU-0418946-2 | 10uM | 24h | MCF7 | LINCS | (Hu, Jin et al. 2018) | |
| VU-0418946-2 | 10uM | 6h | MCF7 | LINCS | (Hu, Jin et al. 2018) | |
|
| ||||||
| MTL | Silver Nitrate | 2.9uM | 24h | CaCo | 62253 | (Lichtlen and Schaffner 2001) |
| Zinc | 50uM | 24h | LBD | 2964 | (Lichtlen and Schaffner 2001) | |
| Zinc | 25uM | 24h | LBD | 2964 | (Lichtlen and Schaffner 2001) | |
|
| ||||||
| OSR | Hydrogen peroxide | 2.5uM | 4h | Thyroid | 39156 | (McMullen, Pendse et al. 2016) |
| Hydrogen peroxide | 300uM | 4h | Thyroid | 39156 | (McMullen, Pendse et al. 2016) | |
| Hydrogen peroxide | 50uM | 24h | HepG2 | 39291 | (McMullen, Pendse et al. 2016) | |
| Tert-butyl hydrogen peroxide | 200uM | 24h | HepG2 | 39291 | (Masaki, Kyle et al. 1989) | |
Abbreviations:
Assigned Stress Categories: DDR: DNA Damage Response; UPR: Unfolded Protein Response; HPX: Hypoxia; MTL: Metals stress response; OSR: Oxidative stress response.
GSE: geo series identifier.
LINCS: Library of Integrated Cellular Signatures
2.5. Curating Transcriptomic Profiles
Transcriptomic data for perturbagens were obtained from the NCBI gene expression omnibus (GEO, accessed March 2020) (Barrett, et al. 2013). For 43 of the 49-potential reference perturbagens, transcriptomic datasets were searched in GEO using the GEOparse python package alongside queries used to identify the perturbagens. This query resulted in 2000 GEO data sets (GEO series, GSE), consisting of multiple transcriptomic profiles related to perturbagens and other potential stress inducers. We filtered the data sets to include human studies focusing on stress response and using the Affymetrix Human Genome U133 Plus 2.0 Array (GEO platform GPL570). Each GEO series was manually inspected to confirm that the phenotype data were extracted correctly. GEO title and description fields were filtered to include “stress response” and associated publications were inspected to validate that the SRP in question was studied and to ensure that the stress response was likely active. Only 11 series were determined to have an adequate title and description to ensure stress response pathway activation and accurately reconstruct sample treatment conditions. These series contain data for perturbagens: glycidamide, heat, hydrogen peroxide, methyl methanesulfonate, silver nitrate, oxygen deprivation, tert-butyl hydrogen peroxide, thapsigargin, tunicamycin, and zinc (Table 1). Transcriptomic data for these perturbagens were prepared from normalized intensity data from which log2 fold changes were calculated using the relevant controls. Samples not pertinent to the study were discarded, e.g. mixtures with a non-stress-related chemical. Transcriptomic data for benzo(a)pyrene and lasiocarpine were obtained from GSE 146549 (Kreuzer, et al. 2020), which is an RNAseq data set. We extracted the normalized count data, processed with DESeq2 from Love, et al. (2014), to estimate differential expression. Moderated z-score LINCS data, termed level 5 data, were obtained using cmapR (Natoli 2020). These data were restricted to the MCF7 cell line. In total, we compiled a data set containing 4, 8, 7, 6, 3, and 4 transcriptomic profiles for DDR, UPR, HSR, HPX, MTL, and OSR, respectively (summarized in Table 1). Additional details about the perturbagens, SRP class and source are provided as Supplemental Material 5.
2.6. Scoring gene signatures and transcriptomic profiles
We used gene set variance analysis (GSVA) to score the enrichment of SRP signatures in transcriptomic profiles (Hanzelmann, et al. 2013). Single sample GSVA uses sample level differential expression data to score gene ranks independently, therefore the log2 fold changes, and moderated z-scores were not normalized. GSEA scores were calculated for each signature from a differential expression matrix (with gene-level log2-fold changes or moderated z-scores); matrix rows corresponded to gene expression, and columns corresponded to perturbagen treatment. The GSEA scores for all reference transcriptomic profiles and SRP signatures are provided as Supplemental Material 6.
2.7. Evaluating performance
The performance of gene signatures to classify SRP activation in reference profiles was evaluated by receiver operating characteristic (ROC) area under the curve (AUC). As an example, the ROC AUC analysis for OSR was conducted as follows. First, reference profiles for OSR perturbagens were assigned a positive class label, and all other profiles were assigned a negative class label. We next calculated the GSEA scores between the test OSR signature (e.g., OSR-100) and all reference profiles. Lastly, the GSEA scores and the class labels for the profiles were used to systematically analyze the sensitivity and specificity for all score thresholds.
The ROC of each signature was characterized as the sensitivity, defined as the true positive rate, vs. specificity, defined as the true negative rate, for each signature’s GSEA scores relative to the designated SRP class. The overall performance was quantified by the AUC enclosed by the boundaries of the ROC curve, with AUC = 1 representing perfect performance. A total of six evaluations were performed, corresponding to the SRPs analyzed in this study. Signatures ranked by AUCs enumerated the top-performing signatures for each SRP. The ROC was calculated with an implementation of the R package, pROC (Robin, et al. 2011).
In addition to evaluating the performance of signatures against a reference data set, their sensitivity was also evaluated against a random dataset. The random dataset was constructed by permuting the log2 fold change values for all genes of all reference perturbagen in the expression matrix without replacement. Permuted data was scored with the best performing signatures ranked by ROC-AUC using GSEA to generate a background with which evaluated signature sensitivity.
3. Results
3.1. Results overview
Comparative analysis of consensus signatures relative to published signatures was completed in three steps. First, we examined the performance of the consensus signatures based on the first 200 genes, ranked by NOF. Second, we assessed specificity and sensitivity using ROC-AUC analysis of our consensus signatures alongside respective published signatures. Third, we identified an optimized subset of signatures composed of four consensus signatures and two published signatures. Lastly, we evaluated the performance of this optimized set against a randomized null data set.
3.2. Performance of Consensus Signatures with 200 genes
We used consensus signatures restricted to the top 200 NOF-ranked genes because they represented the largest set of unique genes for each SRP (Figure 1; Supplemental Material 7). This subset of the consensus signatures produced the highest-scoring match with the perturbagen profiles in the same stress category in 47% of the cases (Figure 1B). Including the first and the second highest-scoring profile matches for each consensus SRP signature in the evaluation increased performance to 75% (or 91% if we included the top three matches; Figure 1B). Only the DDR signature responded perfectly from the top 200 occuring genes (Figure 1C). DDR-200 was the most accurate signature with 75% of reference profiles assigned correctly. All remaining signatures performed with less than 70% accuracy. The MTL-200 signature classified only 33% of chemicals that cause metal stress, and the HSR-200 signature classified only 14% of assigned reference profiles. The HSR related signature, UPR-200, classified 63% of assignments matching top-scoring GSEA values. In the SRP-200 scored set, many HSR profiles showed activity in UPR-200 (Figure-1A). MTL-200 and HPX-200 also have similar levels of cross-induction (Figure-1A); MTL-200 appears to indicate hypoxia. The MTL-200 AUC for HPX is greater than for MTL (AUC = 0.84 and 0.66, respectively).
Figure 1.

Stress gene activity and accuracy for the 200 genes most frequently associated with canonical stress pathways. A) Z-transformed gene set enrichment scores for consensus signatures filtered to the top 200 genes occurring in associated published signature sets. Enrichment scores were computed using a single sample approach based on a Zhang C statistic (Zhang 2002). B) Classification accuracy for consensus signatures filtered to the top 200 most associated genes. Accuracy is calculated as the total percent of reference profiles stress assignments cumulatively matching the largest enrichment score by rank depth (e.g., by the 3rd highest enrichment score). C) Area under the receiver operating characteristic curve of each top 200 genes consensus signatures for all stress response pathways. The area under the curve was calculated as sensitivity vs specificity.
3.3. Evaluate all signatures with ROC-AUC to find the best performing for classification of each SRP
Next, we systematically evaluated the performance of all consensus signatures and all MSigDB published source signatures for classifying the perturbagen profiles using ROC-AUC analysis, and the results are shown in Figure 2. In general, consensus signature sets performed better than almost all published signature sets for almost all SRPs. Consensus signatures DDR, UPR, MTL, and OSR, performed better than published MSigDB signatures. In contrast, HSR and HPX performed similarly to respective published signatures in classifying assigned profiles. The DDR-050, DDR-100, and DDR-200 signatures performed perfectly (AUC=1.00) along with three of the 17 published signatures sets and reflected the larger DDR consensus signatures (Figure 2A). Shorter UPR consensus signatures UPR-050 and UPR-100 had AUCs of 0.83 and 0.86, respectively, and were among the best-performing signatures (Figure 2B). Larger UPR signatures performed with lower accuracy. Although the Winter HPX signature (Winter, et al. 2007) performed perfectly, HPX-400 and HPX-477 signatures performed almost equally as well with AUCs of 0.99 (Figure 2D). Shorter HPX consensus signatures interestingly performed worse with AUC<0.92. The OSR-200 and OSR-050 signatures were the two best-performing signatures for OSR assigned profiles with AUCs of 0.83 and 0.79, respectively; no published OSR signature performed better than an AUC of 0.76 (Figure 2F). HSR and MTL were the weakest ranked performing consensus signatures (Figure 2C and Figure 2E, respectively). The HSR published signature, GO de novo protein folding (GO:0006458) performed the best with an AUC of 0.99, while the HSR-477 signature was the best performing HSR consensus signature, with an AUC of 0.91. The MTL-100 signature was the best MTL performing signature (AUC=0.74); however, MTL assigned profiles were more associated with OSR consensus and DDR published signatures.
Figure 2.

Area under receiver operator characteristic curve for consensus and contributing published signature sets for each canonical stress response pathway (A-F). The receiver operator characteristic was calculated as sensitivity vs. specificity. The top nine signatures for each stress system are listed in tables next to each boxplot. Consensus signatures are shown in bold and consensus signatures relevant to the identified stress response pathway are underlined.
The best overall performing signatures were selected by AUC rank. Here all signatures, both published and consensus, were ranked by AUC and the top signatures were selected for a subsequent accuracy analysis. The consensus signatures, OSR-200 was the best OSR signature (AUC=0.83), and UPR-100 was the best UPR signature (AUC=0.86). The published signatures WINTER_HYPOXIA and GO_DE_NOVO_PROTEIN_FOLDING, were the best performing signatures for HPX (AUC=1.00) and HSR (AUC=0.99), respectively. If more than one signature was tied by AUC (e.g., DDR) then a consensus signature was selected. Here, consensus signatures DDR-050, DDR-100, DDR-200, and published signatures GO DNA Repair (GO:0006281), GO Double Strand Repair (GO:0006302), and Reactome DNA repair (R-HSA-73894) all had AUC=1.00; as such, DDR-200 was chosen as the representative signature. Only in the case of MTL were consensus signatures and published signatures that contributed to the formulation of the consensus signatures outranked by other SRP signatures. In this case, the highest-scoring MTL signature, MTL-100 (AUC=0.74), was selected.
3.4. Classify perturbagens by SRP using best-performing signatures
The resulting accuracy improved to 78% of profiles being accurately classified by the selected group of top-performing signatures and 88% by the second-ranked GSEA score with all but three profiles successfully classified (Figure 3A and B). The OSR signature was the least accurate in classifying assigned reference profiles, with only one out of the four reference profiles successfully classified despite good overall sensitivity within SRP during ROC analysis (0.83 AUC; Figure 3B). The hallmark OSR stress inducer, hydrogen peroxide, was interestingly varied in its responses, inducing UPR and HSR in addition to OSR. Hydrogen peroxide treated Thyroid derived cells, GSE39156, had a z-score of 0.72 for UPR-100 at the lowest dose and a z-score of 1.1 for HSR at the highest dose, 0.3 mM. This possibly indicates a protein misfolding effect resulting from exposure to an oxidative environment or a cell type dependent response. OSR stress agents (hydrogen peroxide and tert-butyl hydroperoxide) both resulted in some activation of DDR SRPs, indicating that, despite our best efforts, the selection of reference profiles might still show signs of off-SRP activity. The Winter hypoxia set (HPX-WINT) scored perfectly.
Figure 3.
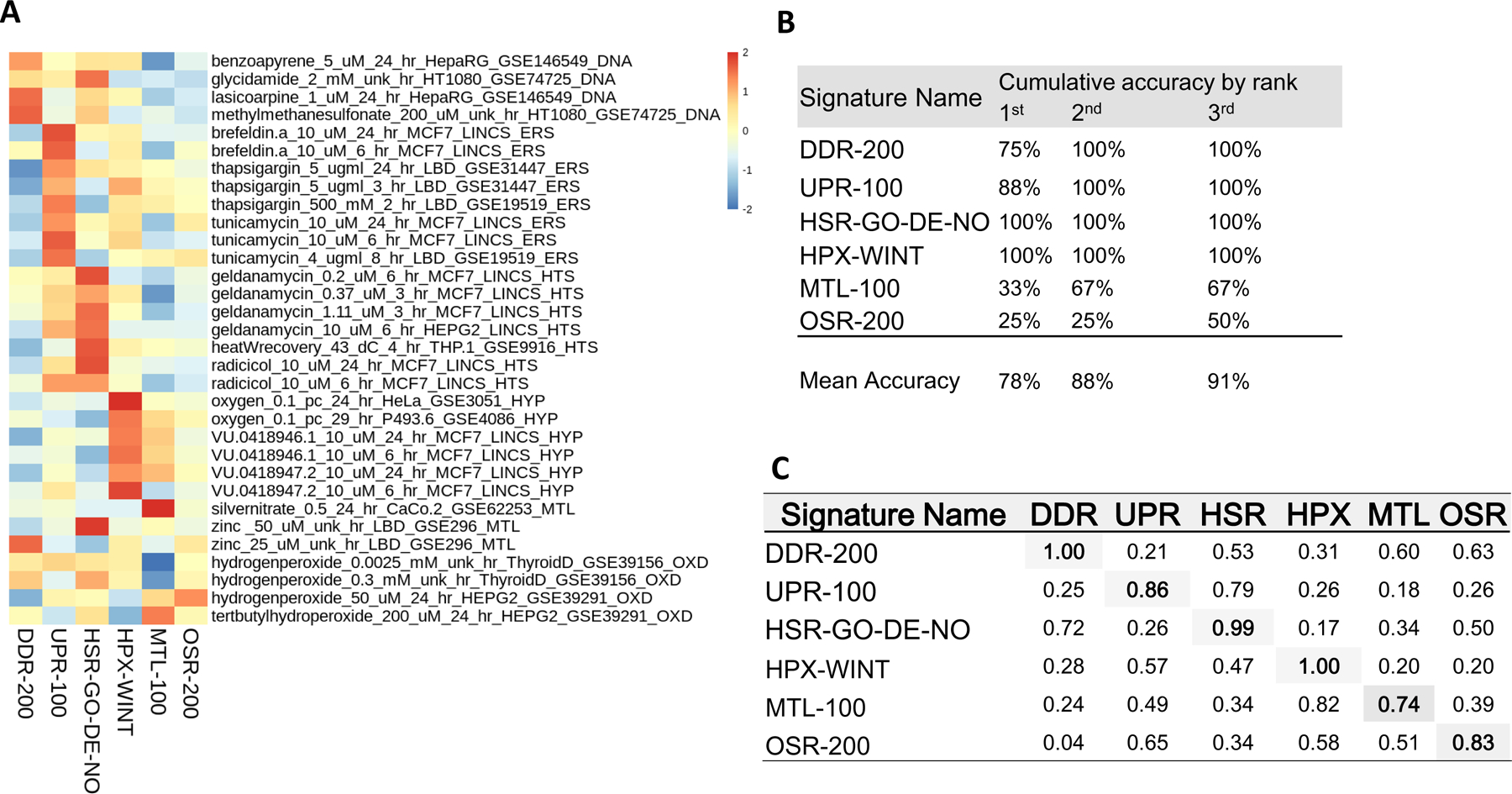
Stress gene activity and accuracy for the best-performing signatures. A) Z-transformed gene set enrichment scores for the highest-ranked area under the receiver operating characteristic curve signatures for each stress response pathway. Enrichment scores were computed using a single sample approach based on a Zhang C statistic (Zhang 2002) B) Classification accuracy for best-performing signatures. Accuracy is calculated as the total percent of reference profiles stress assignments cumulatively matching the largest enrichment score by rank depth (e.g., by the 3rd highest enrichment score). C) Area under the receiver operating characteristic curve of the best-performing signatures for all stress response pathways. The area under the curve was calculated as specificity vs selectivity.
3.5. Performance of selected signatures against a randomly permuted background dataset
The perturbagen signature classification was challenged further by permuting differential expression values for each gene across all perturbagens to create a random dataset from which to render a background distribution of scores. The permuted data were scored with the same signatures listed in Figure 3C using GSEA (henceforth, referred to as the “background”). These results were compared against negative profiles representing all reference perturbagens not assigned to a respective SRP, and positive profiles, representing all profiles assigned to a respective SRP. The data were 0-centered to the background for each SRP and z-scored (Figure 4). All signatures have median GSEA scores separated at least one standard deviation from their median background score. DDR and HSR were the best performing signatures and were 3 and 4 standard deviations from their respective median background scores and negative profile scores (Figure 4A and C). Consensus signatures for UPR, HPX, MTL, and OSR were at least one standard deviation from their respective background scores (Figure 4B, D, E, and F), however, both UPR and HPX (Figure 4B and D) had good separation from respective negative profile scores, at approximately 2 standard deviations. The two least specific consensus signatures were MTL and OSR and had the least separation between respective negative profile scores and positive profile scores (Figure 4E and F).
Figure 4.
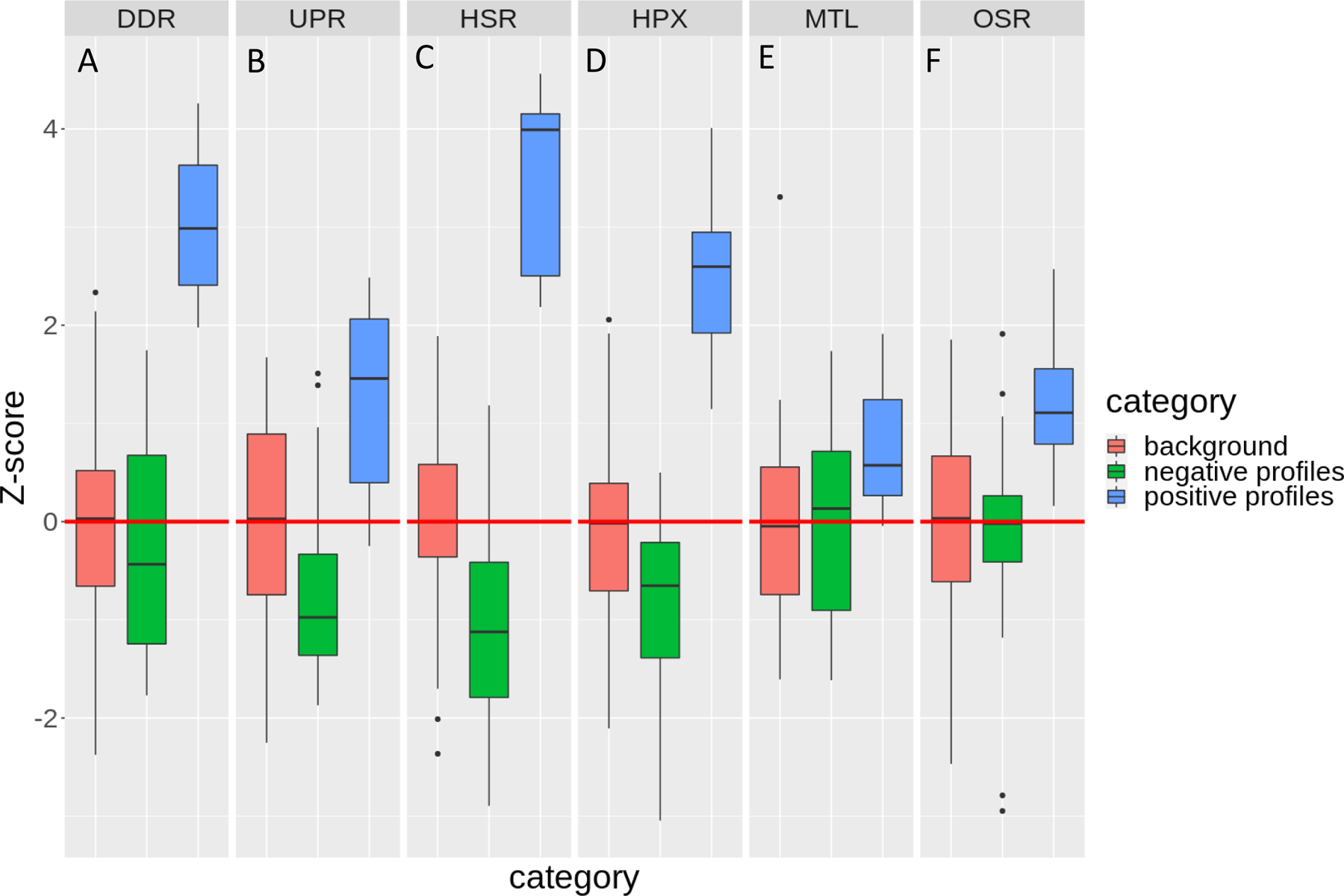
Z- background transcriptomic data set compared against reference profiles scored by best-performing signatures. Best performing signature gene set enrichment scores were zero-centered and z-score transformed for all reference profiles assigned to the tested stress response pathway (denoted by dark gray bar; termed positive profiles), all reference profiles not assigned to the tested stress response pathway (termed negative profiles) relative to an expressional data set permuted by gene from the reference profile set (termed background).
4. Discussion
The use of SRPs to characterize disease states and adverse outcomes (Copple, et al. 2019; Hatherell, et al. 2020; Podtelezhnikov, et al. 2020) hint at the potential for an SRP-based NAM with which to evaluate the effects of non-specific chemicals using transcriptomics. However, at large, the current approaches rely on SRP signatures that have been constructed independently, while SRPs often share a common set of effector genes (Schlage, et al. 2011) due to substantial crosstalk between TFs and signaling pathways (Simmons, et al. 2009). Therefore, we believe that SRP signatures should be constructed to minimize overlap between shared genes to improve the selectivity and sensitivity of NAMs. In this study, we laid the foundation for such a method by binning genes based upon their occurrence within published SRPs as the basis of a “wisdom of the crowd” approach to defining consensus signatures. We have developed a simple data-driven and automated approach to building consensus signatures and evaluated their performance with transcriptomic profiles for a small pilot set of reference perturbagens. We believe that appropriately combining published signatures leverages the valuable contributions others have made. Also, our findings set a performance baseline for published signatures, our proposed consensus signatures or for SRP signatures built de novo.
Our data-driven approach produced an optimized set of signatures to assess SRP activity with approximately 78% accuracy for the best-performing signature and 88% by the second-ranked GSEA score. Consensus signature sets were better or equal to published signature sets’ performance in four of six SRPs, namely DDR, UPR, MTL, and OSR (Figure 2A-F). The abundance of consensus signatures ranking among the best-performing signatures classifying SRPs highlights the usefulness of a tunable data-driven approach for signature development. Our results show that some published signatures performed well, but others were not very specific. For example, many published DDR signatures also matched profiles for perturbagens that cause OSR and MTL due to the number of shared effector genes. This underscores the need for tailoring signatures for cooperative use (that is, characterizing multiple SRPs simultaneously) by optimizing performance based on sensitivity and specificity. Moreover, the consensus signatures produced using this approach will incrementally improve as new relevant signatures are added. To our knowledge, no cooperative and tunable signature sets for SRPs have been published previously, and this report is the first, however small, step in this direction.
The usefulness of SRPs to assess the bioactivity of chemicals is limited by signature specificity in a highly interconnected system. This is because crosstalk between SRPs can mask the mechanisms of action, decreasing the diagnostic usefulness of gene signatures. In the present study, the best-performing signatures had sufficient discriminatory potential to classify DDR, UPR, HSR, and HPX reference perturbagens but not MTL and OSR (Figure 2E and F, Figure 3C). We ascribe this performance limitation to the centrality of the OSR evidenced by the overlap between the genes in this stress response system and all other SRPs (Supplemental Materials 7, A, and C) (Fulda, et al. 2010). Critical OSR effectors HMOX1, SOD1, and SOD2 are shared between published signatures for HPX and MTL and OSR. Therefore, the abundance of OSR genes in other published SRP signatures could exclude critical genes from the OSR signatures. Oxidative stress also induces DDR (Coluzzi, et al. 2014), UPR/HSR (Adams, et al. 2019), and MSR (Lichtlen, et al. 2001). Because OSR mechanistically precedes the SRPs as mentioned earlier, it can be challenging to assign OSR effector genes using differential expression analysis (Fulda, et al. 2010). An independent second source of gene-SRP relationships would be helpful when constructing consensus signatures to reduce such conflict in gene assignment. For example, mapping genes in published SRP signatures onto a protein-protein interaction network before assignment to a consensus set could potentially resolve the attribution of genes to SRPs by considering the underlying causal pathways.
Our approach succeeds in constructing consensus signatures that perform better than their constituent signatures, on average. However, improvements in gene-SRP assignments and signature scoring could enhance performance by increasing sensitivity and specificity. Specifying directionality can provide additional information by which to tune signature scoring thereby increasing sensitivity (Varemo, et al. 2013). As of now, many published signatures do not contain this information. Improving gene-SRP assignment is also critical, exemplified by OSR. Network-based consensus signature construction can increase the accuracy of gene-SRP assignments by incorporating additional TF-effector information (Frohlich 2011). Signature scoring methods beyond GSEA (Boufea, et al. 2020; Maleki, et al. 2020; Maleki, et al. 2020) can also impact performance. Lastly, these improvements must be paired with an expanded reference chemical catalog. However, curating an accurate database of accurately identified positive and negative reference chemicals is perhaps the most difficult measure to improve because chemicals can induce multiple SRPs. For example, a classic DDR activator, benzo(a)pyrene damages DNA by oxidative stress and could activate both DDR and OSR (Park, et al. 2006). Therefore, creating a curated catalog of reference chemicals for SRPs requires substantial domain expertise and literature review. Existing concentration-time rich transcriptomic databases like LINCS can provide a valuable resource for curating more reference profiles and improving this work.
A comprehensive set of SRP classifiers paired with rich transcriptomic data could efficiently screen environmental chemicals for non-specific effects (Hatherell, et al. 2020) and potential drugs for off-target effects (Hatherell, et al. 2020; He, et al. 2019). Although there are multiple areas for improvement, we believe this work takes an essential first step in demonstrating the utility of a new resource for environmental chemical screening: a cooperative SRP consensus signature set as the basis of a NAM for identifying non-specific chemicals from transcriptomic data.
5. Conclusion
Efficiently evaluating and characterizing chemicals is challenging given the multitude of indirect and non-specific routes by which chemicals induce toxicity. A key question is understanding the role that adaptive stress responses play in mitigating the effects of non-specifically acting chemicals. The SRP signatures developed in this work contribute to our understanding of the transcriptional bioactivity of adaptive stress responses. Also, they start to address the issue of crosstalk between SRPs by using a data-driven approach to resolve the contributions of effector genes to signatures. With further evaluation, we believe that these signatures could form the basis of NAMs that can efficiently characterize non-specific bioactivity of thousands of untested chemicals.
Supplementary Material
Funding
Funding for this work was provided by the US EPA Office of Research and Development and the Oak Ridge Institute for Science and Education.
List of abbreviations
- AOP
adverse outcome pathway
- AUC
area under the curve
- CTD
Comparative Toxicogenomic Database
- DDR
DNA damage response
- GEO
Gene Expression Omnibus
- GO
Gene Ontology
- GSE
GEO Series
- GSEA
Gene Set Enrichment Analysis
- HPX
hypoxic stress response
- HSR
heat shock response
- HTS
high throughput sequencing
- LINCS
Library of Integrated Network-Based Cellular Signatures
- MIE
molecular initiating event
- MeSH
medical subject heading
- MSigDB
the Molecular Signatures Database
- MTL
metals stress response
- NAM
new approach methodology
- NOF
normalized occurrence frequency
- OSR
oxidative stress response
- qPCR
quantitative real-time polymerase chain reaction
- ROC
receiver operating characteristic
- SRP
stress response pathway
- TF
transcription factor
- UPR
unfolded protein response
Footnotes
Competing financial interests: The authors declare they have no actual or potential competing financial interests
Disclaimer: The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency.
Conflict of Interest
The authors declare no conflict of interest.
References
- Adams CJ, Kopp MC, Larburu N, Nowak PR and Ali MMU Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Frontiers in Molecular Biosciences 2019;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, … Villeneuve DL Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem 2010;29(3):730–741. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, … Sherlock G Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25(1):25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, … Hahn WC Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462(7269):108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, … Soboleva A NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res 2013;41(Database issue):D991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boufea K, Seth S and Batada NN scID Uses Discriminant Analysis to Identify Transcriptionally Equivalent Cell Types across Single-Cell RNA-Seq Data with Batch Effect. iScience 2020;23(3):100914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao SS and Kaufman RJ Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 2014;21(3):396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Hu Z, Phatak M, Reichard J, Freudenberg JM, Sivaganesan S and Medvedovic M Genome-wide signatures of transcription factor activity: connecting transcription factors, disease, and small molecules. PLoS Comput Biol 2013;9(9):e1003198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, … Bell DA Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res 2012;40(15):7416–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluzzi E, Colamartino M, Cozzi R, Leone S, Meneghini C, O’Callaghan N and Sgura A Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS One 2014;9(10):e110963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copple IM, den Hollander W, Callegaro G, Mutter FE, Maggs JL, Schofield AL, … Park BK Characterisation of the NRF2 transcriptional network and its response to chemical insult in primary human hepatocytes: implications for prediction of drug-induced liver injury. Arch Toxicol 2019;93(2):385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JC, Williams A and Yauk CL Using a gene expression biomarker to identify DNA damage-inducing agents in microarray profiles. Environmental and Molecular Mutagenesis 2018;59(9):772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AP, Grondin CJ, Johnson RJ, Sciaky D, Wiegers J, Wiegers TC and Mattingly CJ Comparative Toxicogenomics Database (CTD): update 2021. Nucleic Acids Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA US Strategic Plan to Promote the Development and Implementation of Alternative Test Methods Within the TSCA Program. In: Prevention, O.o.C.S.a.P, editor. United States: Environmental Protection Agency; 2018. [Google Scholar]
- Escher BI, Allinson M, Altenburger R, Bain PA, Balaguer P, Busch W, … Leusch FD Benchmarking organic micropollutants in wastewater, recycled water and drinking water with in vitro bioassays. Environ Sci Technol 2014;48(3):1940–1956. [DOI] [PubMed] [Google Scholar]
- Escher BI, Henneberger L, Konig M, Schlichting R and Fischer FC Cytotoxicity Burst? Differentiating Specific from Nonspecific Effects in Tox21 in Vitro Reporter Gene Assays. Environ Health Perspect 2020;128(7):77007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich H Network based consensus gene signatures for biomarker discovery in breast cancer. PLoS One 2011;6(10):e25364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Gorman AM, Hori O and Samali A Cellular stress responses: cell survival and cell death. Int J Cell Biol 2010;2010:214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Pan X, Abali GK, Little JB and Yuan Z-M Functional interplay between p53 and Δ133p53 in adaptive stress response. Cell Death & Differentiation 2020;27(5):1618–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther V, Lindert U and Schaffner W The taste of heavy metals: gene regulation by MTF-1. Biochim Biophys Acta 2012;1823(9):1416–1425. [DOI] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R and Guinney J GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatherell S, Baltazar MT, Reynolds J, Carmichael PL, Dent M, Li H, … Middleton AM Identifying and Characterizing Stress Pathways of Concern for Consumer Safety in Next-Generation Risk Assessment. Toxicol Sci 2020;176(1):11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Su J, Lan B, Gao Y and Zhao J Targeting off-target effects: endoplasmic reticulum stress and autophagy as effective strategies to enhance temozolomide treatment. Onco Targets Ther 2019;12:1857–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiemstra S, Niemeijer M, Koedoot E, Wink S, Pip JE, Vlasveld M, … van de Water B Comprehensive Landscape of Nrf2 and p53 Pathway Activation Dynamics by Oxidative Stress and DNA Damage. Chemical Research in Toxicology 2017;30(4):923–933. [DOI] [PubMed] [Google Scholar]
- Houck KA, Richard AM, Judson RS, Martin MT, Reif DM and Shah I ToxCast: Predicting Toxicity Potential Through High-Throughput Bioactivity Profiling. In, High-Throughput Screening Methods in Toxicity Testing 2013. p. 1–31. [Google Scholar]
- ICCVAM. A Strategic Roadmap for Establishing New Approaches to Evaluate the Safety of Chemicals and Medical Products in the United States. In.: Interagency Coordintating Committee on the Validation of Alternative Methods; 2018. [Google Scholar]
- Jackson AC, Liu J, Vallanat B, Jones C, Nelms MD, Patlewicz G and Corton JC Identification of novel activators of the metal responsive transcription factor (MTF-1) using a gene expression biomarker in a microarray compendium. Metallomics 2020;12(9):1400–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings P, Limonciel A, Felice L and Leonard MO An overview of transcriptional regulation in response to toxicological insult. Arch Toxicol 2013;87(1):49–72. [DOI] [PubMed] [Google Scholar]
- Judson R, Houck K, Martin M, Richard AM, Knudsen TB, Shah I, … Thomas RS Editor’s Highlight: Analysis of the Effects of Cell Stress and Cytotoxicity on In Vitro Assay Activity Across a Diverse Chemical and Assay Space. Toxicol Sci 2016;152(2):323–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzer K, Bohmert L, Alhalabi D, Buhrke T, Lampen A and Braeuning A Identification of a transcriptomic signature of food-relevant genotoxins in human HepaRG hepatocarcinoma cells. Food Chem Toxicol 2020;140:111297. [DOI] [PubMed] [Google Scholar]
- Li HH, Chen R, Hyduke DR, Williams A, Frotschl R, Ellinger-Ziegelbauer H, … Fornace AJ Jr. Development and validation of a high-throughput transcriptomic biomarker to address 21st century genetic toxicology needs. Proc Natl Acad Sci U S A 2017;114(51):E10881–E10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP and Tamayo P The Molecular Signatures Database Hallmark Gene Set Collection. Cell Systems 2015;1(6):417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P and Mesirov JP Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011;27(12):1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtlen P and Schaffner W Putting its fingers on stressful situations: the heavy metal-regulatory transcription factor MTF-1. Bioessays 2001;23(11):1010–1017. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Wootz H and Korhonen L ER stress and neurodegenerative diseases. Cell Death & Differentiation 2006;13(3):385–392. [DOI] [PubMed] [Google Scholar]
- Liu B, Lindner P, Jirmo AC, Maus U, Illig T and DeLuca DS A comparison of curated gene sets versus transcriptomics-derived gene signatures for detecting pathway activation in immune cells. BMC Bioinformatics 2020;21(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W and Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q Role of Nrf2 in Oxidative Stress and Toxicity. Annual Review of Pharmacology and Toxicology, Vol 53, 2013. 2013;53:401-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki F and Kusalik AJ The Effect of Gene Set Overlap on Specificity of Over-representation Analysis 2020. [Google Scholar]
- Maleki F, Ovens K, Hogan DJ and Kusalik AJ Gene Set Analysis: Challenges, Opportunities, and Future Research. Front Genet 2020;11:654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki N, Kyle ME and Farber JL tert-Butyl hydroperoxide kills cultured hepatocytes by peroxidizing membrane lipids. Archives of Biochemistry and Biophysics 1989;269(2):390–399. [DOI] [PubMed] [Google Scholar]
- Mazure NM and Pouysségur J Hypoxia-induced autophagy: cell death or cell survival? Current Opinion in Cell Biology 2010;22(2):177–180. [DOI] [PubMed] [Google Scholar]
- Muñoz-Pinedo C and Martin SJ Autosis: a new addition to the cell death tower of babel. Cell Death & Disease 2014;5(7):e1319–e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli T cmapR: CMap Tools in R. R package version 1.2.1, https://github.com/cmap/cmapR. In.; 2020. [Google Scholar]
- Oslowski CM and Urano F Chapter Four - Measuring ER Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. In: Conn PM, editor, Methods in Enzymology Academic Press; 2011. p. 71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, … Hotamisligil GS Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004;306(5695):457–461. [DOI] [PubMed] [Google Scholar]
- Park SY, Lee SM, Ye SK, Yoon SH, Chung MH and Choi J Benzo[a]pyrene-induced DNA damage and p53 modulation in human hepatoma HepG2 cells for the identification of potential biomarkers for PAH monitoring and risk assessment. Toxicol Lett 2006;167(1):27–33. [DOI] [PubMed] [Google Scholar]
- Pilarczyk M, Kouril M, Shamsaei B, Vasiliauskas J, Niu W, Mahi N, … Medvedovic M Connecting omics signatures of diseases, drugs, and mechanisms of actions with iLINCS. Preprint 2020. [Google Scholar]
- Plusquin M, Stevens AS, Van Belleghem F, Degheselle O, Van Roten A, Vroonen J, … Smeets K Physiological and molecular characterisation of cadmium stress in Schmidtea mediterranea. Int J Dev Biol 2012;56(1–3):183–191. [DOI] [PubMed] [Google Scholar]
- Podtelezhnikov AA, Monroe JJ, Aslamkhan AG, Pearson K, Qin CH, Tamburino AM, … Tanis KQ Quantitative Transcriptional Biomarkers of Xenobiotic Receptor Activation in Rat Liver for the Early Assessment of Drug Safety Liabilities. Toxicological Sciences 2020;175(1):98–112. [DOI] [PubMed] [Google Scholar]
- Radons J The human HSP70 family of chaperones: where do we stand? Cell Stress and Chaperones 2016;21(3):379–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC and Muller M pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12(12):77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JP, Chorley B, Hiemstra S, Wink S, Wang X, Bell DA, … Corton JC Mining a human transcriptome database for chemical modulators of NRF2. PLoS One 2020;15(9):e0239367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlage WK, Westra JW, Gebel S, Catlett NL, Mathis C, Frushour BP, … Deehan R A computable cellular stress network model for non-diseased pulmonary and cardiovascular tissue. BMC Syst Biol 2011;5:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah I, Setzer RW, Jack J, Houck KA, Judson RS, Knudsen TB, … Kavlock RJ Using ToxCast Data to Reconstruct Dynamic Cell State Trajectories and Estimate Toxicological Points of Departure. Environ Health Perspect 2016;124(7):910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons SO, Fan CY and Ramabhadran R Cellular stress response pathway system as a sentinel ensemble in toxicological screening. Toxicol Sci 2009;111(2):202–225. [DOI] [PubMed] [Google Scholar]
- Spaan CN, Smit WL, van Lidth de Jeude JF, Meijer BJ, Muncan V, van den Brink GR and Heijmans J Expression of UPR effector proteins ATF6 and XBP1 reduce colorectal cancer cell proliferation and stemness by activating PERK signaling. Cell Death & Disease 2019;10(7):490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathias V, Turner J, Koleti A, Vidovic D, Cooper D, Fazel-Najafabadi M, … Schürer SC LINCS Data Portal 2.0: next generation access point for perturbation-response signatures. Nucleic Acids Res 2020;48(D1):D431–d439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, … Mesirov JP Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 2005;102(43):15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RS, Bahadori T, Buckley TJ, Cowden J, Deisenroth C, Dionisio KL, … Williams AJ The Next Generation Blueprint of Computational Toxicology at the U.S. Environmental Protection Agency. Toxicological Sciences 2019;169(2):317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP and Weissman JS Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 2004;164(3):341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varemo L, Nielsen J and Nookaew I Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res 2013;41(8):4378–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang X, Ke ZJ, Comer AL, Xu M, Frank JA, … Luo J Tunicamycin-induced unfolded protein response in the developing mouse brain. Toxicol Appl Pharmacol 2015;283(3):157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch WJ Mammalian stress response: cell physiology, structure/function of stress proteins, and implications for medicine and disease. Physiological Reviews 1992;72(4):1063–1081. [DOI] [PubMed] [Google Scholar]
- Wink S, Hiemstra S, Huppelschoten S, Danen E, Niemeijer M, Hendriks G, … van de Water B Quantitative High Content Imaging of Cellular Adaptive Stress Response Pathways in Toxicity for Chemical Safety Assessment. Chemical Research in Toxicology 2014;27(3):338–355. [DOI] [PubMed] [Google Scholar]
- Winter SC, Buffa FM, Silva P, Miller C, Valentine HR, Turley H, … Harris AL Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Research 2007;67(7):3441–3449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.