

Abstract
Cruzain, an important drug target for Chagas disease, is a member of clan CA of the cysteine proteases. Understanding the catalytic mechanism of cruzain is vital to the design of new inhibitors. To this end, we have determined pH−rate profiles for substrates and affinity agents and solvent kinetic isotope effects in presteady-state and steady-state modes using three substrates: Cbz-Phe-Arg-AMC, Cbz-Arg-Arg-AMC, and Cbz-Arg-Ala-AMC. The pH−rate profile of for Cbz-Arg-Arg-AMC indicated (unprotonated) and (protonated) groups were required for catalysis. The temperature dependence of the group exhibited a value of , typical of histidine. The pH−rate profile of inactivation by iodoacetamide confirmed that the catalytic cysteine possesses a of 9.8. Normal solvent kinetic isotope effects were observed for both and for all three substrates. Pre-steady-state kinetics revealed exponential bursts of AMC production for Cbz-Phe-Arg-AMC and Cbz-Arg-Arg-AMC, but not for Cbz-Arg-Ala-AMC. The overall solvent isotope effect on can be attributed to the solvent isotope effect on the deacylation step. Our results suggest that cruzain is unique among papain-like cysteine proteases in that the catalytic cysteine and histidine have neutral charges in the free enzyme. The generation of the active thiolate of the catalytic cysteine is likely preceded (and possibly triggered) by a ligand-induced conformational change, which could bring the catalytic dyad into the proximity to effect proton transfer.
Graphical Abstract
Cruzain is the major cysteine protease expressed by Trypanosoma cruzi, the causative agent of Chagas disease.1 With Chagas disease categorized as one of the most important neglected tropical diseases (NTDs), it is estimated that almost 8–9 million people are currently affected by this disease, with 50000 new cases annually reported in Latin America and the Caribbean.2 Present treatment options of Chagas disease are limited to benznidazole and nifurtimox, both of which have severe side effects and are inefficacious against long-term infections.3 The viability of cruzain as a drug target for new trypanocidal agents was demonstrated both in cell culture4,5 and in animal models,6,7 although its exact physiological role remains unclear. Recent studies have suggested that cruzain is potentially involved in immune evasion through its proteolysis of NF- P65,8 and the activation of latent TGF- that induces increased infectivity of T. cruzi.9
Extensive efforts over the past three decades to develop inhibitors or covalent inactivators of cruzain have encompassed structure-based design, high-throughput screening, and virtual screening.10 The most successful inhibitor developed so far is K11777, which is comprised of a phenylalanyl-homophenylalanyl dipeptide “scaffold” linked to an electrophilic vinyl sulfone warhead that forms an irreversible Michael adduct with the eponymous active-site cysteine (Figure 1).11 K11777 cures Chagas disease in mouse models and protects dogs from cardiac damage during T. cruzi infection.12 However, preclinical studies of K11777 were halted because of its poor tolerability in primates and dogs even at low doses, which may be attributable to its irreversible mode of inactivation.
Figure 1.
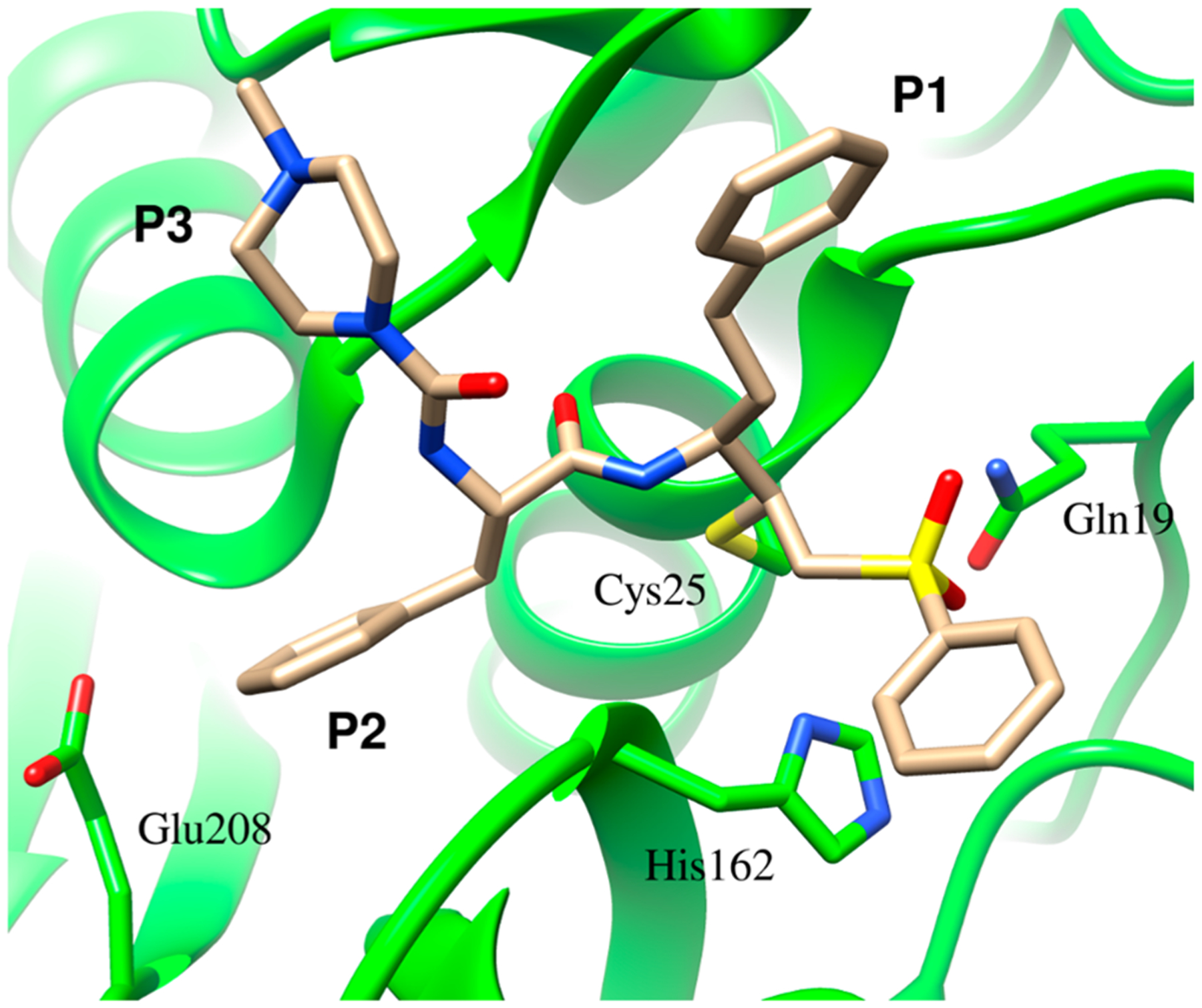
X-ray crystal structure of cruzain complexed with the irreversible inhibitor K11777, demonstrating the formation of a covalent bond between the active-site and the -carbon of the vinyl sulfone warhead.11 Protein Data Bank entry 2OZ2.
While the development of new cruzain inhibitors has been extensive, no detailed studies of the catalytic mechanism of cruzain exist. Sequence analysis and substrate profiling studies indicate that cruzain is a member of the cathepsin L-like papain family and belongs to clan CA of the cysteine proteases.13,14 The active sites of cysteine proteases contain a catalytic cysteine-histidine “dyad” that exists as either a neutral Cys-SH:His or a thiolate-imidazolium ion pair Cys–S−:H-His+ species in the free enzyme. The initial protonation state of this catalytic dyad has been the subject of considerable debate.15–18 Only recently has the catalytic mechanism of cruzain been investigated by molecular dynamics. In that study, an imidazolium-thiolate catalytic dyad exists in ligand-free cruzain, and the imidazolium ion of active-site protonates the amide nitrogen prior to attack of the thiolate on the amide carbonyl group.19 The catalytic mechanism of cysteine proteases involves two discrete half-reactions: the acylation of the active-site cysteine by the scissile carbonyl of the substrate with release of the amine product and the subsequent hydrolysis of the enzyme−peptide thioester (deacylation) to produce the carboxylate product. During acylation, the imidazolium ion of the active-site histidine provides the proton to the leaving amine (Scheme 1). Kinetic and structural analysis of papain is consistent with a Cys-S−:H-His+ ion pair in the free enzyme.20–23 Kinetic studies by Schneck et al. demonstrated that for human cathepsin , like papain, the thiolate-imidazolium ion pair predominates in the active site (Scheme 1) in which the values for the Cys (4.3) and His (6.5) conform to “reverse protonation” of these residues, and the observed inverse solvent kinetic isotope effect for is consistent with enrichment of the thiolate ion in in the free enzyme.24 However, in other cysteine proteases/hydrolases, such as ubiquitin-specific protease 1 (USP1), a neutral Cys-SH:His dyad exists in the free enzyme, as indicated by normal sKIEs, and the binding of its protein substrate induces apparent proton transfer from Cys-SH to the His to initiate catalysis.25 Here we investigate in detail the catalytic mechanism of cruzain by pH−rate profiles and the measurement of solvent kinetic isotope effects on steady-state and pre-steady-state kinetics of the proteolytic reaction of different dipeptide substrates.
Scheme 1.
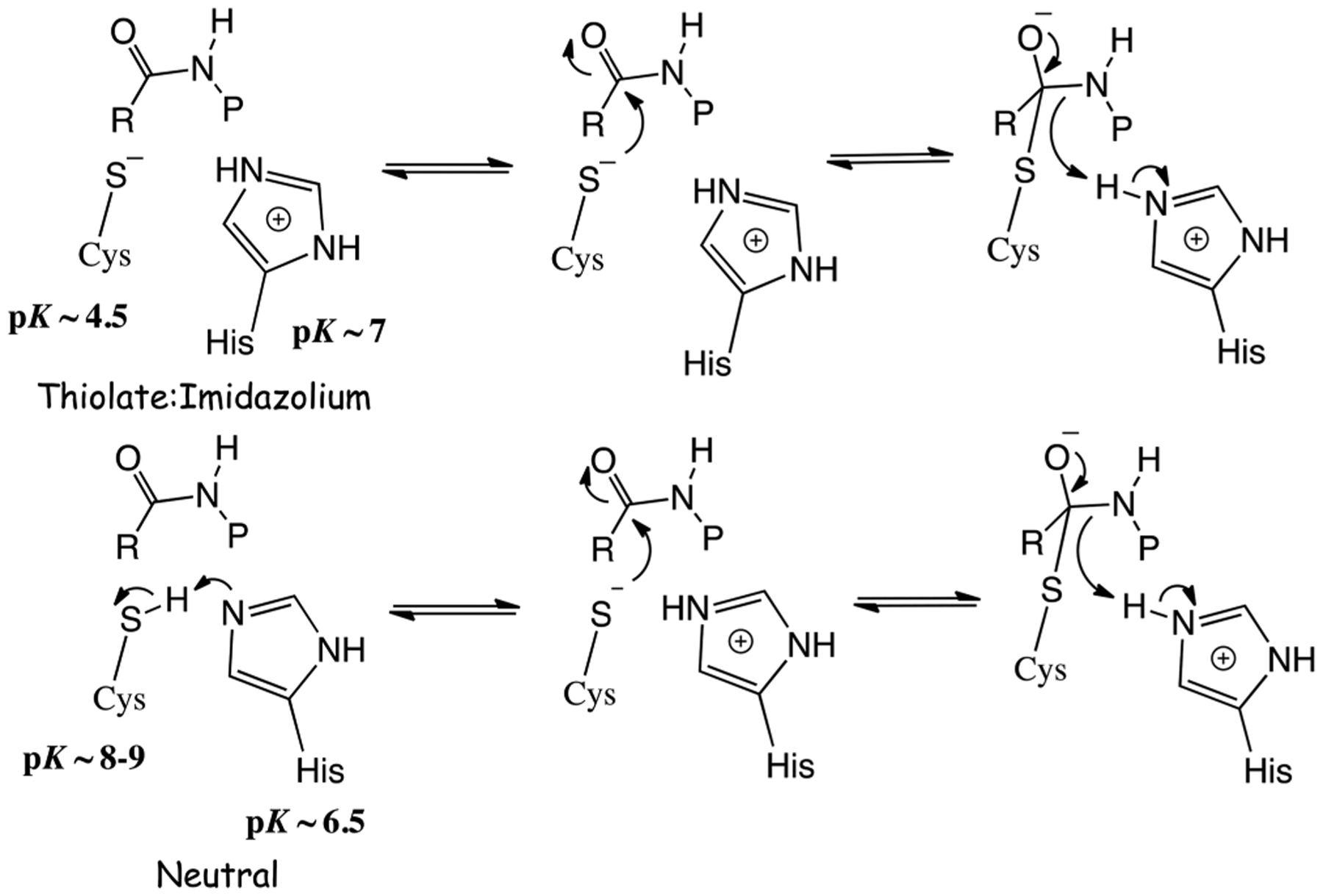
Potential Chemical Mechanisms for Cruzain of Attack of Thiolate on a Peptide Substrate Involving a Thiolate-Imidazolium or Neutral Cysteine-Histidine Dyad
EXPERIMENTAL PROCEDURES
Materials.
HisPur Ni-NTA Superflow Agarose was purchased from ThermoFisher. HiTrap Q Sepharose FF columns were purchased from GE Healthcare. Deuterium oxide (≥99.9% gram atom D) was purchased from Cambridge Isotope Laboratories. Deuterium chloride [35% (w/w), 99% D], sodium acetate, sodium deuterium oxide [40% (w/w), 99% D], TAPSO, TEA, DEA, DEPC, MES, MMTS, disodium EDTA, glycerol, and sucrose were purchased from Sigma-Aldrich. DNase I was purchased from Roche. Z-FR-AMC was purchased from Biotium. Z-RR-AMC and Z-RA-AMC were purchased from Bachem or Enzo. All other chemicals were reagent grade or better and used without further purification.
Enzyme Preparations.
General procedures for cruzain expression, purification, and activation were performed according to published protocols26 with some modifications.
Protein Expression.
The plasmid encoding the C-terminally truncated procruzain (Δc; GenBank entry M84342.1) was a generous gift from C. S. Craik. The primer 5’-GCCTTGTCAAGGAGGCGGCGAGCTCCGCGGTGGTCGG-3’ was used to generate the G208A mutant of cruzain. The sequence of this mutant plasmid was confirmed by Eton Bioscience. ArcticExpress (DE3) cells were used as the expression strain. After inoculation with frozen glycerol stocks of transformed ArcticExpress cells, small cultures (5 mL) of Luria broth containing carbenicillin (100 μg/mL) and gentamycin (20 μg/mL) were grown overnight at 37 °C to saturation. Five milliliters of this starting culture was added to 1 L of Terrific Broth medium containing the same amount of carbenicillin and gentamycin. After being grown for ~6 h (OD600 = 0.6), cultures were moved to 18 °C, and protein expression was initiated by adding 0.4 mM isopropyl -d-1-thiogalactopyranoside. Cells were harvested after overnight expression, and the cell pellets were stored at −20 °C.
Protein Purification and Activation of Cruzain from Procruzain.
Frozen cell pellets were thawed and resuspended at a concentration of 50 mL/L of growth medium in lysis buffer [50 mM Tris (pH 10), 300 mM NaCl (buffer A), containing 10 mM imidazole, 1 mM CaCl2, 1 mM MgSO4, 1 μM DNase I, 1 mM phenylmethanesulfonyl fluoride (PMSF), and 2 mM methylmethanethiosulfonate (MMTS)]. After resuspension, samples were lysed via sonication (ThermoFisher model FB505 sonicator) with 20 s pulses (50% amplitude) followed by 59 s intervals at 50% amplitude for a 10 min total period of sonication. The resulting lysate was centrifuged at 17000g for 1 h. The supernatant was passed through a 0.2 μm filter prior to being loaded onto a HisPur Ni-NTA column (ThermoFisher Scientific) equilibrated with buffer A. After being extensively washed with buffer A containing 50 mM imidazole, bound procruzain was eluted with buffer A containing 500 mM imidazole. MMTS at a final concentration of 5 mM was added to each of the fractions containing His6-tagged procruzain upon elution.
Fractions containing procruzain were combined and dialyzed overnight at 4 °C against 2 L of activation buffer [50 mM sodium acetate (pH 5.0), 100 mM NaCl, and 0.1 mM disodium EDTA] in the absence of reducing agents. The resulting cloudy solution was transferred into 50 mL conical centrifuge tubes (1 L growth medium per 50 mL activation sample). Autoproteolysis was initiated by addition of 10 mM DTT, followed by incubation at 37 °C for 1–3 h until the dialysate solution became completely clear. The clarified solution was then dialyzed against 50 mM Tris (pH 8.0) to remove salts, and this dialysate was loaded onto two 5 mL HiTrap Q FF columns connected in a series. Cruzain was eluted with 500 mL of 50 mM Tris (pH 8.0) buffer containing a gradient of 50–400 mM NaCl. The resulting cruzain was flash-frozen and stored at −80 °C in the eluant containing 20% glycerol, at a protein concentration of 5 mg/mL. The protein was judged to be homogeneous with >95% purity based on sodium dodecyl sulfate−polyacrylamide gel electrophoresis. The N-terminus of the protein (H2N-APAAVD) was confirmed by Edman sequencing performed by the Protein Chemistry Laboratory at Texas A&M University. Activation of cruzain fractions required removal of MMTS. This was accomplished by five rounds of spin dialysis versus assay buffer containing 5 mM DTT using a 10000 molecular weight cutoff Amicon centrifuge tube. The final enzyme concentration was determined by absorbance at 280 nm using an ε280 value of 60430 M−1 cm−1 (oxidized Cys) or 59930 M−1 cm−1 (reduced Cys) calculated from the ProtParam tool available on the Expasy server.27
Enzyme Assays.
Unless otherwise noted, all enzyme assays were performed at 25 °C. Initial rates of the peptidolytic reaction catalyzed by cruzain were measured by monitoring the fluorescence generated by cleavage of the dipeptide−AMC bond. Assays were conducted in 96-well plates (Greiner, flat-bottom, clear black plates) in a total volume of 250 μL, containing either 50 mM sodium acetate, 50 mM MES, and 100 mM TEA (pH 3.5–8.0) or 50 mM MES, 50 mM TAPSO, and 100 mM DEA (pH 5.5–9.8), each mixed with 1 mM CHAPS, 1 mM Na2EDTA (Overlap Buffers), 5 mM DTT, and 2 or 10% (v/v) DMSO. Substrates were dissolved in 100% DMSO and then diluted 10-fold such that when they were added to reaction mixtures, the final DMSO concentration was either 2% (v/v) (as used in the pH studies such that high levels of DMSO would not obscure the acid/base dissociation constants) or 10%. Reactions were initiated via the addition of 5 μL of cruzain (final concentrations of 0.017–3.0 nM). Fluorescence was measured on either a SpectraMax M5 (Molecular Devices) or a Synergy HTX (Biotek, Wisnooki, VT) microplate reader . Initial rates were determined from continuous kinetic time courses and calculated from the earliest time points, typically in <10 min.
pH−rate Profiles.
The pH dependence of and for cruzain-catalyzed peptidolysis of Z-FR-AMC, Z-RR-AMC, and Z-RA-AMC was investigated at pH 3.5–10 with two different three-component mixed buffer systems. Cruzain was preincubated for 30 min at each pH, after which the pH was readjusted to 7.0 to determine the stability of the enzyme over the experimental pH range. Cruzain activity was unchanged from pH 4.5 to 9.5, affording a wide range for the evaluation of pH−rate profiles. To maintain a constant ionic strength of 0.1, 50 mM sodium acetate/MES and 100 mM TEA were used for pH 3.5–8.0 and 50 mM MES/TAPSO and 100 mM DEA were used for pH 5.5–10.0 (Overlap Buffers);28 the pH was adjusted with small aliquots of concentrated HCl or NaOH. There were no significant differences in values of and at the overlap pH points for the two buffers. Typical enzyme concentrations (wild type) used in kinetic measurements were 17 pM, 0.17–1.7 nM, and 1–70 nM for Z-FR-AMC, Z-RR-AMC, and Z-RA-AMC, respectively. Concentrations for the E208A mutant of cruzain used in kinetic measurements were 17 pM, 3.4–34 nM, and 20–1400 nM for Z-FR-AMC, Z-RR-AMC, and Z-RA-AMC, respectively. The temperature dependence of the of wild-type (WT) cruzain was determined in a single mixed buffer of 50 mM MES/TAPSO, 100 mM DEA, 1 mM Na2EDTA, and 1 mM CHAPS (pH 4.5–7.5) at 25–40 °C. The apparent pH for the buffer was determined at each temperature used, and data for versus pH were corrected for a diminution in pH of 0.012 unit/°C.
Solvent Kinetic Isotope Effects.
To determine the solvent kinetic isotope effects of cruzain substrates, Overlap Buffers were prepared in 100% and adjusted to the desired pD using either sodium deuteroxide or deuterium chloride (pD 3.5–10). The calculated final percentage of deuterium in these buffers was 93%, based on precedent.24 pD values were determined as the measured pH value + 0.4.29 Values of and for Z-FR-AMC, Z-RR-AMC, and Z-RA-AMC were acquired from initial rate studies in deuterated Overlap Buffers also containing 5 mM DTT and 2% DMSO. Solvent kinetic isotope effects were then ascertained from comparison of the pH- and pD-independent values of these kinetic parameters as described in Data Analysis. To rule out any viscosity effect of on the initial rate, the kinetic parameters and were measured with 0.5–100 μM Z-FR-AMC in Overlap Buffers containing 5 mM DTT, 10% DMSO, and either 0–25% (w/v) sucrose or 0–12% (w/v) glycerol.
Inactivation of Cruzain by lodoacetamide and Diethylpyrocarbonate.
WT cruzain (1–3 nM) was preincubated for 0–80 min with 0–1 mM iodoacetamide at pH 5.5–10 in 50 mM MES, 50 mM TAPSO, 100 mM DEA, and 10% DMSO. Because DTT reacts with iodoacetamide and potentially with DEPC, it was omitted from the preincubation and assay mixtures. Likewise, 1–3 nM WT cruzain was preincubated for 0–5 min with 0–100 μM DEPC (dissolved in either 100% DMSO or ice-cold ethanol) at pH 5.5–10 in 50 mM MES, 50 mM TAPSO, 100 mM DEA, and 10% DMSO. Aliquots (5 μL) of both types of preincubation mixtures were diluted into 250 μL assay mixtures containing Overlap Buffers (pH 7.5) and 10 μM Z-FR-AMC [10% (v/v) DMSO], and residual cruzain activity was measured.
Pre-Steady-State Kinetics.
Pre-steady-state kinetics for cruzain-catalyzed reactions of 0.5–10 μM Z-FR-AMC and 1.6–60 μM Z-RR-AMC were conducted at 25 °C and pH(D) 7.5. Time courses (0.002–0.2 s) of AMC production were measured using a Kintek AutoSF-120 stopped-flow fluorimeter (Kintek, Snow Shoe, PA), where fluorescence readings of AMC (excitation at 348 nm and emission at 438 nm with a 400 nm cutoff filter) were collected and analyzed by Kintek software. Reaction mixtures contained 50 mM MES, 50 mM TAPSO, 100 mM DEA, 1 mM CHAPS, and 1 mM Na2EDTA and were prepared in either or . Solutions of WT cruzain (0.5 μM) and the substrate were loaded into separated syringes that each contained buffer, 10% DMSO, and 5 mM DTT. Fluorescence time courses were comprised of 1000 time points for each fixed substrate concentration, and time courses of AMC formation (millivolts) were converted to nanomolar concentrations of product formed following calibration of the system with known concentrations of AMC. Calibration of the stopped-flow fluorimeter was ascertained by measurement of the concentration of free AMC found in each solution of substrate, and two kinetic traces were generated: one background trace measuring the signal of the contaminating free AMC present in the substrate and one reaction trace monitoring the newly generated AMC from the cleavage of the peptide substrate by cruzain. After the background had been subtracted, the voltage signals were converted into concentration according to a calibration curve generated from solutions containing known concentrations of AMC. Separate experiments were conducted to determine the binding constant for both substrates in and using a larger range of substrate concentrations (0–80 μM).
Data Analysis.
Values of and for cruzain-catalyzed reactions of the fluorogenic peptide substrates were determined by nonlinear least-squares fits of the initial velocity data to eq 1 using GraphPad Prism 6.0 or SigmaPlot 12.0 (Systat, Inc.). For eq 1, is the turnover number, is the concentration of active sites of cruzain, and is the Michaelis constant for substrate A.
| (1) |
In enzyme inactivation studies using affinity agents, residual cruzain activity plotted against time (eq 2) was used to obtain pseudo-first-order rate constants of inactivation at each inactivator concentration . The second-order rate constants, , for inactivation of cruzain by iodoacetamide were determined from the slopes of the replots of the pseudo-first-order rate constants versus iodoacetamide concentration, as fitted to eq 3a. Similarly, for DEPC, values of versus time were fitted to eq 3b, and for both equations, and are the maximal rate of inactivation and the concentration of inactivator at which the rate of inactivation is half that of , respectively.
| (2) |
| (3a) |
| (3b) |
Data for all pH profiles were fitted as semilogarithmic plots to eq 4
| (4) |
where is the observed kinetic parameter or is the pH-independent value of , and and are the apparent acid and base dissociation constants, respectively. For plots of versus pH(D) in which a “hollow” was observed, rather than apparent slopes of 1 and −1 characteristic of “bell-shaped” curves (eq 4), data were fitted to eq 5
| (5) |
where is the pH-independent value of , where is a collection of rate constants, is the apparent acid dissociation constant, and is a collection of rate constants that comprise the “stickiness ratio” for the substrate.30
The solvent kinetic isotope effects (sKIEs, ) were calculated using eq 6
| (6) |
where and are pH(D)-independent values of or and is the fraction of used in the studies. Error propagation on calculated values of the sKIEs was determined from the ratios of the experimental values in and , by the use of eq 7
| (7) |
where , and are experimental standard deviations of , and , respectively, and , and are the respective partial derivatives of eq 6.
Pre-steady-state data were fitted to a single-exponential function (eq 8) for each fixed concentration of Z-FR-AMC and Z-RR-AMC. In eq 8, is product AMC formed (bound or free, micromolar), is the apparent steady-state rate, is the time in seconds, is the apparent burst amplitude, is the apparent rate of the exponential phase, and is a constant of calibration.24 Steady-state rates, burst amplitudes, and transient rate constants were replotted at each concentration of substrate ([A]) using eqs 1,9, and 10, respectively, for which is the burst amplitude (micromolar), is the transient rate constant (inverse seconds), is the steady-state turnover number (inverse seconds), and are the apparent rate constants of the acylation and deacylation half-reactions, respectively (inverse seconds), is the cruzain concentration (micro-molar), and and are the Michaelis and dissociation constants of the substrate, respectively.
| (8) |
| (9) |
| (10) |
| (11) |
Additionally, pre-steady-state data conforming to a singleexponential function were fitted globally to eq 11, in which more than 10000 data points representing pre-steady-state time courses for all fixed concentrations of Z-FR-AMC or Z-RR-AMC were simultaneously fitted. In all cases, the most appropriate fit for each equation was determined by an F-test analysis of the results of nonlinear regression.
Nomenclature.
Isotope effects are expressed using the notation of Cook, Cleland, and Northrop.31,32 Solvent kinetic and equilibrium isotope effects measured on and other kinetic parameters are notated as leading superscripts, designated as “D” for the sake of simplicity, with the variable substrate as the right subscript.
RESULTS AND DISCUSSION
Steady-State Kinetics and Substrate Specificity.
Initial velocity data for the three fluorogenic dipeptide substrates are summarized in Table 1. The value of is largest for Z-FR-AMC (17 s−1), which is more than twice than that of Z-RR-AMC, suggesting a better accommodation for a Phe residue at the P233 position than for Arg. The possibility that cruzain may accommodate both Phe and Arg at the P2 position of dipeptide substrates has been attributed to the role of Glu208 found in the S2 binding subsite (Figure 1). It was shown from structural studies that Glu208 points toward a P2 Arg residue on the substrate but away from Phe in the P2 position of the substrate, as depicted in Figure 1, for which the irreversible inactivator K11777 (N-methyl-piperazine-Phe-homoPhe-vinylsulfone-phenyl) contains a phenylalanine as its P2 residue.11 The of Z-RA-AMC (0.89 s−1) is nearly 20-fold lower than that of Z-FR-AMC, indicating that a larger residue at the position is preferred for catalysis, while a hydrophobic residue at is more favorable than a charged one. Values of for the three substrates range from 104 to 107 M−1 s−1, wherein for Z-FR-AMC is 3 orders of magnitude larger than the lowest value of for Z-RA-AMC. On the basis of the rank order of values for the three different substrates, cruzain clearly prefers substrates with a large, charged residue at the P1 position and a hydrophobic residue in the P2 position, consistent with findings from a comprehensive analysis of peptide substrate specificity.14
Table 1.
Steady-state Kinetic Parameters for Cruzain-catalyzed Proteolytic Reactions of Z-FR-AMC, Z-RR-AMC and Z-RA-AMC.a
| Wildtypeb | E208Ac | |||||
|---|---|---|---|---|---|---|
| Substrate | ||||||
| Z-FR-AMC | 17 ± 2 | 0.62 ± 0.02 | (2.7 ± 0.4) × 107 | 21 ± 1 | 1.6 ± 0.1 | (1.3 ± 0.1) × 107 |
| Z-RR-AMC | 7.2 ± 0.1 | 3.7 ± 0.4 | (1.9 ± 0.2) × 106 | 0.27 ± 0.01 | 45 ± 4 | (6.0 ± 0.1) × 103 |
| Z-RA-AMC | 0.89 ± 0.06 | 38 ± 2 | (2.3 ± 0.3) × 104 | 0.017 ± 0.001 | 163 ± 3 | (1.0 ± 0.1) × 102 |
Under assay conditions of 50 mM MES, 50 mM TAPSO, 100 mM DEA, 1 mM CHAPS, 1 mM EDTA, 5 mM DTT, 2% (v/v) DMSO, pH 7.5 and 25 °C.
The errors for wildtype-catalyzed reactions were determined from the average of kinetic parameters determined in duplicates.
The errors for E208A cruzain mutant-catalyzed reactions are the standard deviations determined from the nonlinear least squares fits of the kinetic data.
Mutagenesis of Glu208.
We prepared the E208A mutant of cruzain to investigate the role of this residue in recognition of the P2 substituent of substrates. For Z-FR-AMC, for WT and E208A cruzain is equivalent, is increased 2.5-fold, and is halved for the mutant enzyme (Table 1). Accordingly, while the E208A mutant “recognizes” Z-FR-AMC less effectively than wild-type cruzain does, turnover of the substrate is comparable, suggesting that once bound, mutant cruzain processes Z-FR-AMC at rates similar to that of the wild-type enzyme. However, mutation of Glu208 exhibited profound effects on all three kinetic parameters of substrates bearing an Arg residue at the P2 position. Diminutions by 300- and 200-fold in the values of Z-RR-AMC and Z-RA-AMC, respectively, were observed for the E208A mutant cruzain compared to that of the wild type, suggesting that ion pairing between Glu208 and P2 Arg is essential for recognition of these substrates. This is also reflected in values of , which are increased for Z-RR-AMC (10-fold) and Z-RA-AMC (4-fold). Values of for mutant cruzain, unlike Z-FR-AMC, are greatly diminished for the substrates containing Arg at P2: Z-RR-AMC (26-fold lower) and Z-RA-AMC (50-fold lower).
pH−rate Profiles.
The pH–rate profiles for Z-FR-AMC, Z-RR-AMC, and Z-RA-AMC with wild-type cruzain over a pH range of 3.5–10.0 are shown in Figure 2 (blue plots) with results summarized in Table 2. For this pH range, there are no prototropic groups on any of the substrates studied. The plot of versus pH (3.5–10.0) for Z-FR-AMC was apparently invariant with pH, while those of Z-RR-AMC and Z-RA-AMC decreased with apparent slopes of +1 and −1 at low and high pH, respectively. When fitting to eq 4 was performed, decreased upon protonation of an enzymatic residue with values of 5.5 and 6.1, for Z-RR-AMC and Z-RA-AMC, respectively, and upon deprotonation of an enzymatic residue with a of ≥ 10. For Z-FR-AMC, the plot of versus pH was characterized by a “hollow” as pH decreased, with an apparent slope of less than +1. This plot indicates that Z-FR-AMC is a “sticky” substrate;30 that is, the substrate desorbs from its Michaelis complex at a rate equal to or less than that of its progression through catalysis.
Figure 2.
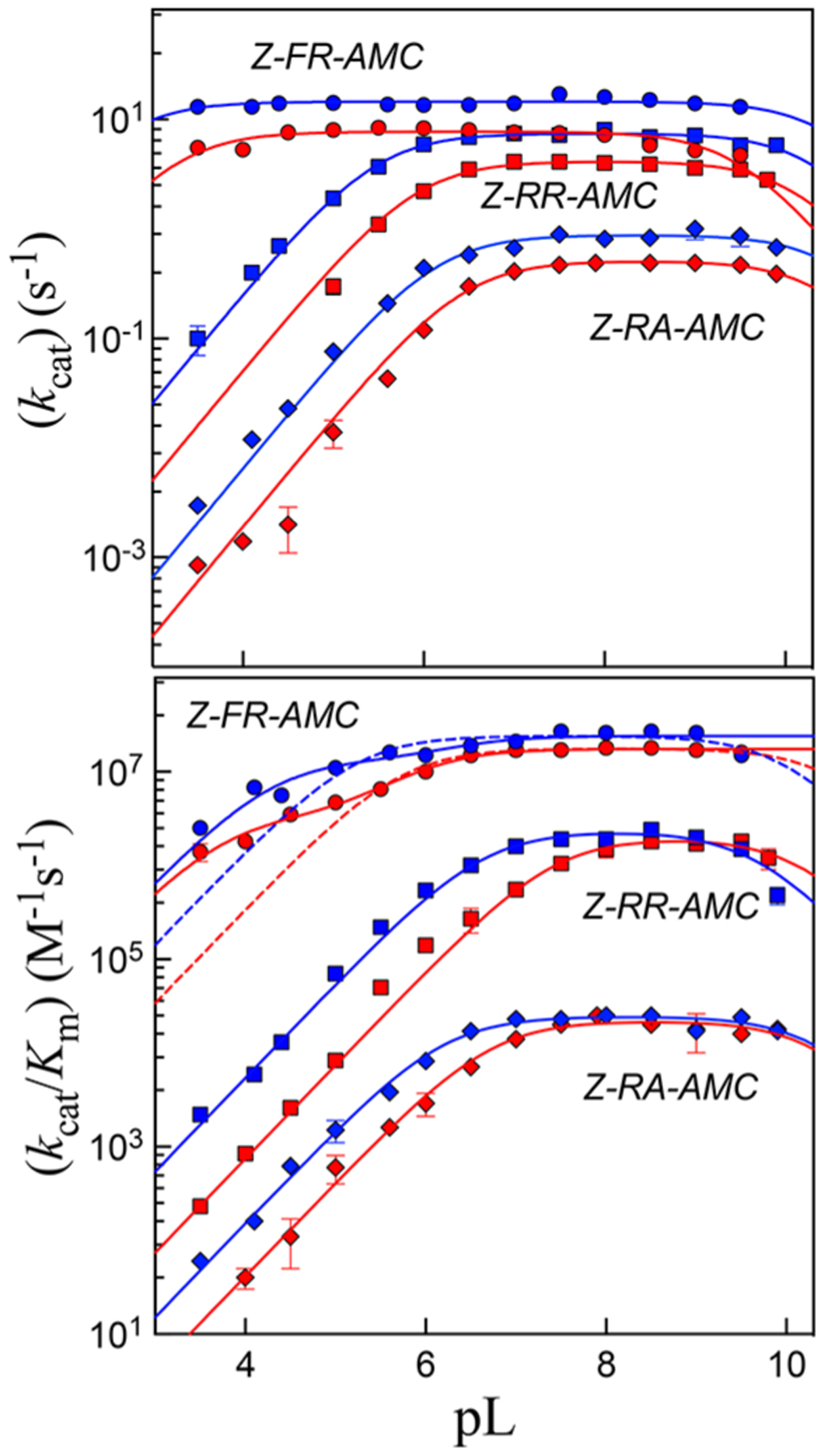
Plots of pH(D) (pL) rate profiles of (top) and (bottom) for cruzain-catalyzed hydrolysis of Z-FR-AMC (circles), Z-RR-AMC (squares), and Z-RA-AMC (diamonds). Blue and red symbols are for data obtained in and , respectively. Lines drawn through the experimental data of plots of are from fitting to eq 4, while lines drawn through the experimental data of plots of vs are from fitting to eq 4 (solid line, dashed line for Z-FR-AMC) or eq 5 (solid line for Z-FR-AMC). Error bars represent standard deviations from two independent measurements.
Table 2.
pH-rate Profiles and Solvent Kinetic Isotope Effects from Steady-state Kinetics of Wild-Type Cruzain.a
| b | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrate | Solvent | Eq. | c | Eq. | |||||||||
| Z-FR-AMC | H2O | n.a. e | n.d. d | n.d. | 14.5 ± 0.4 | 4 | 6.4 ± 0.7 | n.d. | n.d. | 24 ± 2 | 1.88 ± 0.06 | 1.4 ± 0.1 | |
| 5 | 6.2 ± 0.4 | n.d. | 5.8 ± 0.4 | 26 ± 2 | 70 ± 50 | ||||||||
| D2O | n.a. | n.d. | n.d. | 7.7 ± 0.1 | 4 | 6.0 ± 0.1 | n.d. | n.d. | 17 ± 1 | ||||
| 5 | 6.0 ± 0.1 | n.d. | 5.2 ± 0.2 | 17.4 ± 0.7 | 34 ± 8 | ||||||||
| Z-RR-AMC | H2O | 4 | 5.5 ± 0.1 | ≥ 10 | 7.4 ± 0.2 | 4 | 6.6 ± 0.1 | 9.6 ± 0.1 | n.a. | 0.23 ± 0.01 | n.a. | 1.76 ± 0.06 | 1.15 ± 0.08 |
| D2O | 4 | 5.9 ± 0.1 | ≥ 10 | 4.2 ± 0.1 | 4 | 7.4 ± 0.1 | ~ 10 | n.a. | 0.2 ± 0.01 | n.a. | |||
| Z-RA-AMC | H2O | 4 | 6.1 ± 0.1 | ≥ 10 | 0.88 ± 0.03 | 4 | 6.2 ± 0.1 | ≥ 10 | n.a. | 0.024 ± 0.001 | n.a. | 1.73 ± 0.07 | 1.09 ± 0.07 |
| D2O | 4 | 6.4 ± 0.1 | ≥ 10 | 0.51 ± 0.01 | 4 | 6.7 ± 0.1 | ≥ 10 | n.a. | 0.022 ± 0.001 | n.a. | |||
This result is unsurprising given that the value of for Z-FR-AMC is near the limit of diffusion control (2.4 × 107 M−1s−1), and the pH independence of the values of . For sticky substrates, apparent values of are perturbed in plots of versus pH, resulting in “hollows” in the plots, and do not reflect the true acid or base dissociation constants of the enzymatic residues involved in binding and catalysis.30 However, fitting of these data to eq 5 uncovered a value of 6.2 associated with an enzymatic group that must be unprotonated for substrate binding. In contrast, the plots of versus pH for Z-RR-AMC and Z-RA-AMC exhibited apparent slopes of +1 and −1 at low and high pH, respectively. Fitting of plots of versus pH to eq 4 resulted in values of 6.6 and 6.2 for Z-RR-AMC and Z-RA-AMC, respectively. The basic dissociation constant, , while poorly determined at these extreme values of pH, equaled ~10 for both substrates. The current pH profiles are similar to those previously reported for cruzain in which pH–rate profiles were also substrate-dependent.34
Scheme 2 describes a model for the pH−rate profiles for wild-type cruzain. HE is the monoprotonated form of cruzain to which all three substrates bind to form the competent Michaelis complex HEA, which proceeds to form the thioester form of cruzain (F), followed by deacylation to regenerate the free enzyme. Plots of versus pH report on the rate of the acylation half-reaction, measure the enzymatic rate at substrate concentrations approaching zero, and describe the protonation state of free enzyme HE. Here, protonation of an enzymatic group with a of ~6 or deprotonation of the enzymatic group with a of ~10 produces or , respectively, each of which is incapable of catalysis. However, a sticky substrate (A) may also bind to , and with loss of a proton from the group of , the resulting HEA complex is catalytically competent.30,35 In addition, for a sticky substrate, the proton of the HEA complex may not be subject to removal at high pH such that is apparently independent of pH. The “hollow” observed for the plot of versus pH arises from protonation of the HEA complex , and the flattening of the curve is due to perturbation of the value of where . 30,35 Fitting of the plot of versus pH using eq 5 provided an estimate of the stickiness of the substrate from the parameter , indicating that Z-FR-AMC proceeds to form acylated cruzain ~70 times faster than it desorbs from the HEA complex.
Scheme 2.
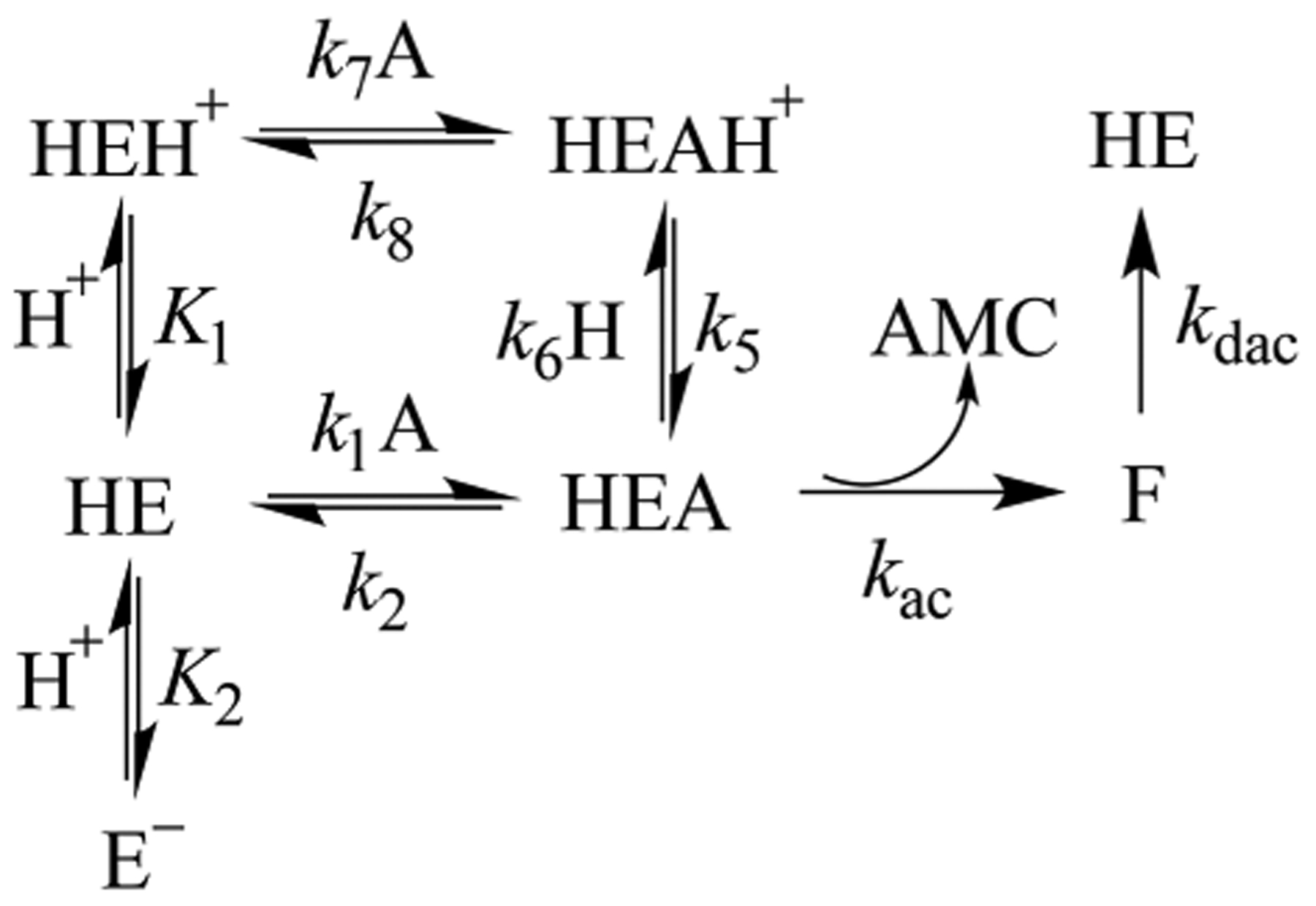
Kinetic Model for pH−Rate Profiles for Cruzain
For cruzain, the essential basic group with a of 6.2–6.6 could be the active-site histidine, or given the salience of an enzymatic group of in the pH–rate profiles of substrates bearing an Arg at the P2 position, this could reflect a carboxylate group such as that of Glu208. We therefore investigated the pH dependence of the E208A mutant of cruzain for Z-FR-AMC, Z-RR-AMC, and Z-RA-AMC (Figures S1–S5). The semilogarithmic profile of versus pH for E208A cruzain was similar to that of the wild type except that the curve exhibited less of a “hollow” than WT cruzain did, suggesting that Z-FR-AMC is a less sticky substrate with the mutant enzyme (Figure S1). For Z-RR-AMC and Z-RA-AMC, the pH−rate profiles indicated that a residue with a pK of ~5.3 and ~6.1, respectively, must be unprotonated for catalysis, and that the pH-independent values of and are almost 20-fold lower than that of wild-type cruzain (Figures S2 and S3). These results rule out Glu208 as the residue corresponding to .
Solvent Kinetic Isotope Effects under Steady-State Conditions.
Solvent kinetic isotope effects from the kinetic parameters and were acquired from pD–rate profiles generated under conditions identical to those described above using Overlap Buffers prepared in . Plots of and versus pD (Figure 2, red points) were identical in form to their counterparts in water, and each plot had been apparently right-shifted by approximately 0.5–0.8 unit for , due to the solvent equilibrium isotope effect29 on enzymatic residues (Table 2). A value of 0.5 is typical for all enzymatic residues except cysteine.29,36 The values in both the and profiles were poorly determined because of the lack of data beyond pD = 10. As in for Z-FR-AMC, was pD-independent, while versus pD was best fitted to eq 5 because of the presence of a “hollow” in the profile. In , the stickiness ratio was half that obtained in , indicating that a solvent isotope effect on slowed catalysis with respect to substrate desorption. Solvent kinetic isotope effects were ascertained from the pH(D)-independent values of and . Normal sKIEs were found for with the following values: for Z-FR-AMC, for Z-RR-AMC, and for Z-RA-AMC. The sKIEs for were normal and only slightly greater than unity (1.1–1.4) for all three substrates (Table 2). These results differ from the sKIEs observed on cathepsin C24 and papain,20–22 both of which exhibited and (discussed below). The kinetic parameters and were unchanged in the presence of either 0–25% (w/v) sucrose or 0–12% (w/v) glycerol, indicating that the experimental normal sKIEs were not a result of the increased viscosity of , which is comparable to the viscosity of an aqueous solution of 9% (w/v) glycerol.37
Temperature Dependence of .
The temperature dependence of acid dissociation constants provides an additional method for identification of an enzymatic group due to its distinctive values of enthalpy of ionization. Because the dissociation constant for the enzyme side chain with a of 6 could be observed in the plot of versus pH at 25 °C, we investigated the temperature dependence of from plots of versus pH obtained at 25–40 °C. The resulting average values (n ≥ 2) of decreased from 6.2 ± 0.1 at 25 °C to 5.9 ± 0.1 at 40 °C (corrections for the temperature dependence of pH for the buffer used were applied at each temperature). The plot of versus was linear (Figure 3). The temperature dependence of acid dissociation constant was fitted to eq 12
| (12) |
where is the temperature in Kelvin, is the universal gas constant, and and are the values of enthalpy and entropy of ionization, respectively.
Figure 3.
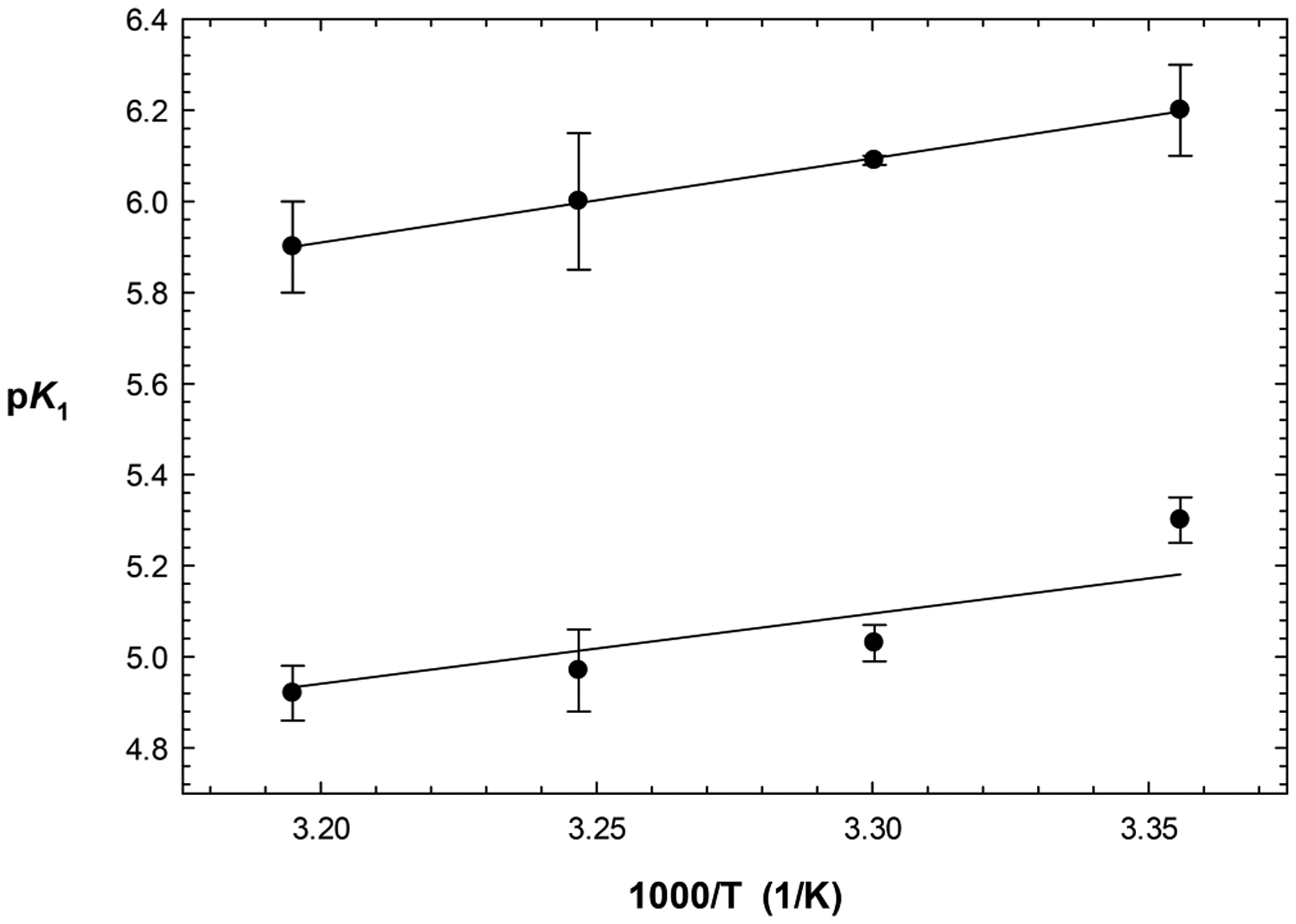
Effect of temperature on the acid dissociation constant from plots of (top line) and (bottom line) vs pH. Values of vs (Kelvin) were fitted to eq 12, from which values of 8.4 ± 0.2 and and a of were obtained.
The fitting of the temperature-dependent data resulted in values of and for and respectively. This value of is consistent with either a cysteine or histidine active-site residue, but not a carboxylic acid. From the plots of versus pH for Z-RR-AMC, we in kind examined the temperature dependence of (5.3–5.5). The resulting average values (n ≥ 2) of decreased from 5.34 ± 0.06 at 25 °C to 4.92 ± 0.06 at 40 °C, and again, fitting of versus (Kelvin) was linear (Figure 3). Fitting to eq 12 resulted in and values of and , respectively. The residue involved in the profile is also likely to be a basic histidine.
Affinity Labeling with Iodoacetamide and Diethyl Pyrocarbonate.
We employed affinity labeling studies of cruzain using the thiol-specific reagent, iodoacetamide. Preincubation of iodoacetamide (0–1 mM) with cruzain, followed by its dilution and analysis of residual enzymatic activity, resulted in irreversible, time-dependent inactivation. The observed first-order rate constants of inactivation, , obtained from fitting of residual enzyme activity versus time (eq 2) demonstrated higher rates of inactivation with increasing concentrations of iodoacetamide (Figure 4A). The inclusion of increasing, fixed concentrations of Z-FR-AMC in the reaction mixtures ablated the inactivation, indicating that iodoacetamide reacted with catalytic Cys25 (data not shown). A replot of the first-order rate constant of inactivation, , versus iodoacetamide concentration demonstrated that inactivation of cruzain by iodoacetamide was not saturable (Figure 4A), which is typical for affinity labeling agents. The second-order rate constant for inactivation, , was obtained from the slopes of replots of versus iodoacetamide concentration by fitting to eq 3a (Figure 4A, inset). At pH 8.0, .
Figure 4.
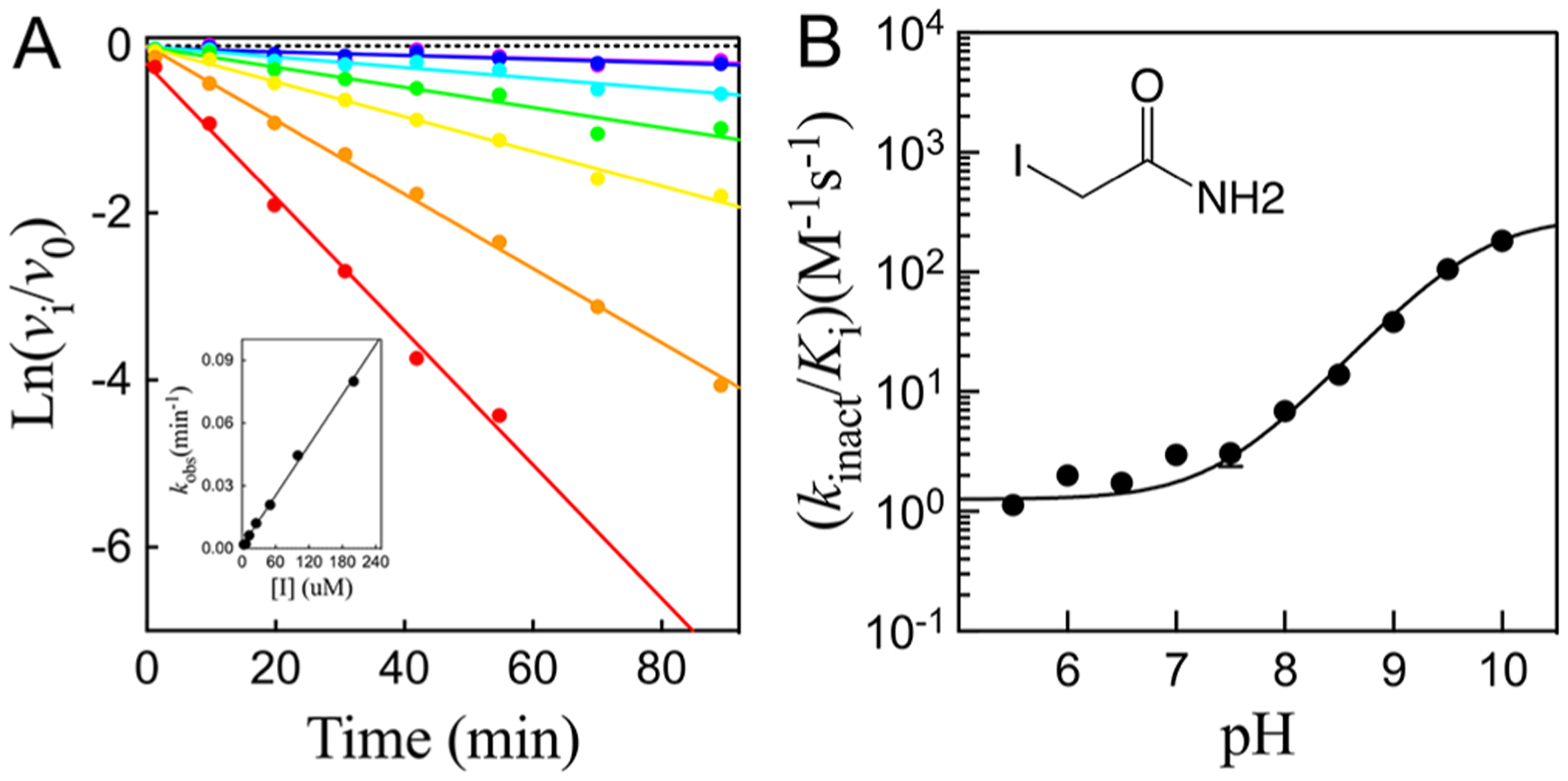
(A) Representative data of the time-dependent inactivation of cruzain by iodoacetamide at pH 8.0. Lines drawn through the experimental data were from fitting to eq 2. The inset displays a replot of the inactivation rate vs iodoacetamide concentration, and the line drawn through the experimental data points was obtained from fitting to eq 3a. (B) pH dependence of the second-order rate constant for cruzain inactivation by iodoacetamide (0–1 mM). The line drawn through the experimental data was obtained by fitting of the data to eq 13.
We determined the pH dependence (Ph 5.5–10.0) of cruzain inactivation by iodoacetamide (Figure 4B). The resulting plot of versus pH exhibited a “wave” in which the level of enzyme inactivation increased from an invariant value at neutral pH to a higher value of 180 ± 20 M−1 s−1 at pH 10. The pH dependence of the second-order rate constant of inactivation of cruzain by iodoacetamide can be fitted to eq 13
| (13) |
where and are values of at low and high pH, respectively, and is the acid dissociation constant for an enzymatic group. Fitting of the data indicated that deprotonation of an enzymatic group with a of 9.76 ± 0.04, likely to be the thiol of active-site Cys25, resulted in the increased rate of inactivation at high pH.
Cruzain can also be inactivated by the histidine-specific affinity agent, diethyl pyrocarbonate (DEPC),38 in a time-dependent manner. At pH 7.2, preincubation of 1 nM cruzain with 1.6–104 μM DEPC for 40–340 s led to an apparent biphasic inactivation of cruzain (Figure 5A). The initial phase occurred too rapidly to be measured; at the shortest preincubation time of 40 s, 11–50% of cruzain was already inactivated by respective DEPC concentrations of 6.5–104 μM. The slower second phase of inactivation was observed at 40–340 s. Replotting the (from fitting to eq 2) versus [DEPC] resulted in a hyperbolic plot (Figure 5B), from which fitting of these data to eq 3b provided the following values: , and . Cruzain is therefore exceptionally sensitive to inactivation by DEPC, especially considering the unmeasurable, rapid phase of inactivation (for comparison, for phosphotriesterase39). The biphasic kinetics of DEPC inactivation suggested that either more than a single histidine had been carboethoxylated or a single histidine had been labeled on both of its nitrogens. No pH dependence of the second phase of DEPC inactivation was observed (pH 5–8) Increasing concentrations of Z-FR-AMC in preincubation mixtures containing cruzain and DEPC led to protection from inactivated by DEPC, which indicated that inactivation occurs via covalent modification of the active site of cruzain.
Figure 5.
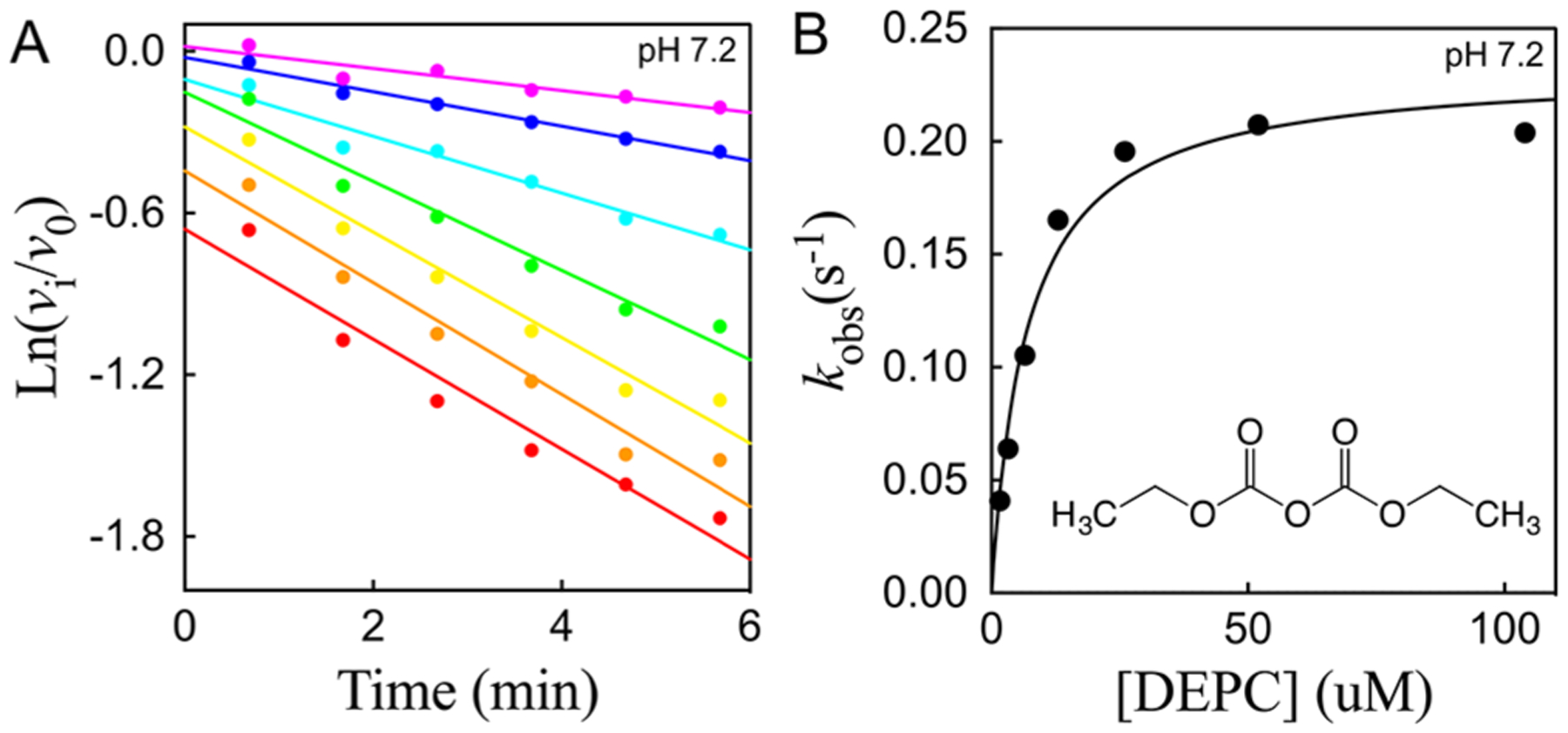
(A) Time-dependent inactivation of cruzain by 1.6–104 μM DEPC at pH 7.2. The lines drawn through the experimental data points were from fitting of the data to eq 2. (B) Replot of [DEPC]. Fitting of the data to eq 3b results in the following values: , and .
Protonation States of the Catalytic Cysteine and Histidine Groups.
Cysteine proteases/hydrolases universally possess a conserved catalytic Cys-His dyad, for which the catalytic cysteine serves as a nucleophile to effect acyl transfer, while the histidine facilitates the deprotonation of the thiol of the cysteine and the lytic water and provides a proton for the amine product. The active, monoprotonated forms of cysteine proteases that exist at the maxima of bell-shaped pH−rate profiles (HE in Scheme 2) could result from two tautomeric species of the catalytic dyad: a neutral thiol-imidazole (Cys-SH/His) or an ionic thiolate-imidazolium species (Cys-S−/HisH+). Bednar has measured the thiolate:thiol nucleophilicity ratio to be 1010:1.40 Polgar showed via pH−rate profiles of the inactivation of papain by haloacetamide that the values of the active-site cysteine (4.0) and histidine (8.4) comprised a case of “reverse protonation”,20 consistent with the sKIE studies of Creighton et al.21,22 Solvent kinetic isotope effects on in these studies were inverse, supporting an enrichment of the thiolate species in arising from a fractionation factor for thiol of 0.45.24,36 Lewis et al. demonstrated via nuclear magnetic resonance studies of papain that the imidazoliumthiolate form was the predominant species in the unbound enzyme.23 In kind, detailed sKIE studies of human cathepsin C were consistent with that of papain; that is, the thiolateimidazolium species of the free enzyme predominates in catalysis: , and .
In contrast, the pH−rate profiles of for all three cruzain substrates were characterized by essential unprotonated and protonated catalytic groups with and , respectively, consistent with neutral forms of the His and Cys species at the pH optimum of cruzain. Other studies described herein confirm the assignment of these . (a) Mutagenic replacement of Glu208 does not eliminate cruzain activity or the observed in the pH−rate profile of Z-RR-AMC. (b) Values of obtained from temperature variation studies for a of 6.2 are in agreement with that of a histidine group. (c) The pH dependence of inactivation of cruzain by the thiol-specific agent iodoacetamide increased upon deprotonation of a residue with a of 9.76 ± 0.04. (d) An absence of an inverse sKIE on indicates that Cys25 is protonated in the free enzyme. (e) For Z-RR-AMC and Z-RA-AMC, the of 0.5–0.8 is consistent with a histidine group, but not a cysteine.35,36 Also, from the temperature dependence of a of 5.3 from vs pH, the obtained value of was also consistent with the involvement of a basic histidine despite the low value of its . This value of 5.3, for which cruzain is saturated with substrate, is identical to the value of 5.2–5.3 for His162 obtained from nuclear magnetic resonance analysis of cruzain covalently complexed to dipeptide analogue K11777.26
The finding of a neutral Cys-His catalytic dyad for free cruzain has several implications. In free cruzain, it is likely that the respective sulfur and nitrogen atoms of Cys25 and His162 are not sufficiently proximal to allow the formation of the thiolateimidazolium dyad seen in papain and cathepsin C. In addition, the neutral Cys-His dyad of cruzain would be a far poorer catalyst for hydrolysis than the thiolate-imidazolium dyad, and removal of the proton from would be required upon substrate binding. As stated above, USP1 hydrolase also possesses a neutral Cys-His dyad for which proton transfer apparently occurs upon binding of the large ubiquitinated protein substrate.25 In the cases of the cysteine hydrolases dimethylarginine dimethylaminohydrolase (DDAH)41 and protein arginine deiminases 2 (PAD2),42 it was similarly proposed that the binding of the positively charged substrates facilitates catalysis by depressing the of the catalytic cysteine to facilitate its deprotonation. Close examination of the crystal structure of PAD2 revealed that Cys645 and His471 are further separated than the catalytic dyad is in PAD4, a closely related isozyme found to have the thiolate form of the activesite cysteine irrespective of substrate binding. Cruzain may be a member of an apparent subclass of cysteine hydrolases that, possibly because of active-site geometry, has a neutral Cys-His dyad in their ligand-free forms that requires substrate binding to assist proton transfer from His to Cys, as suggested for USP1.25
Pre-Steady-State Kinetics and Solvent Isotope Effects.
Pre-steady-state kinetics for the cruzain-catalyzed hydrolysis of Z-FR-AMC and Z-RR-AMC in both and were generated at pH(D) 7.5 in a stopped-low fluorimeter by measurement of product AMC. While solvent kinetic isotope effects should be determined over a range of pH(D) values to eliminate the possibility that observed sKIEs are due to the equilibrium isotope effects that perturb values of active-site residues, the lack of pH(D) dependence of either substrate between pH(D) 7.0 and 8.0 allows us to assess the sKIEs at pH(D) 7.5. For each substrate concentration in either solvent, all time courses exhibited an initial, rapid burst of AMC, followed by a linear, steady-state rate of AMC product formation (Figure 6, Figure S4, and Table 3). Burst amplitudes increased with increasing fixed concentrations of substrates to apparent saturation. In contrast, no transient burst was observed for Z-RA-AMC. These results are very similar to those of cathepsin C, in which dipeptide-AMC substrates with high values are characterized by a burst phase in which the transient:steady-state rate ratio was ~ 20:1, while poor substrates displayed no transient bursts.24,43 In these previous studies, the rate constant of the transient phase could be attributed to the acylation half-reaction that encompasses the catalytic steps described by , while the steady-state rate constant was equivalent to the rate constant of the deacylation half-reaction . Likewise, for Z-FR-AMC and Z-RR-AMC, the observed transient rates demonstrated that the rate of transfer of an acyl group to cruzain exceeded the steadystate rate of deacylation, while for the poor substrate Z-RA-AMC, and are comparable, for which no pre-steady-state burst would be observed. For the pre-steady-state kinetics of Z-FR-AMC (Figure 6) and Z-RR-AMC (Figure S4) performed in , transient bursts were also observed, albeit with slower transient and steady-state rates (data for the substrate Z-RR-AMC are found in the Supporting Information).
Figure 6.
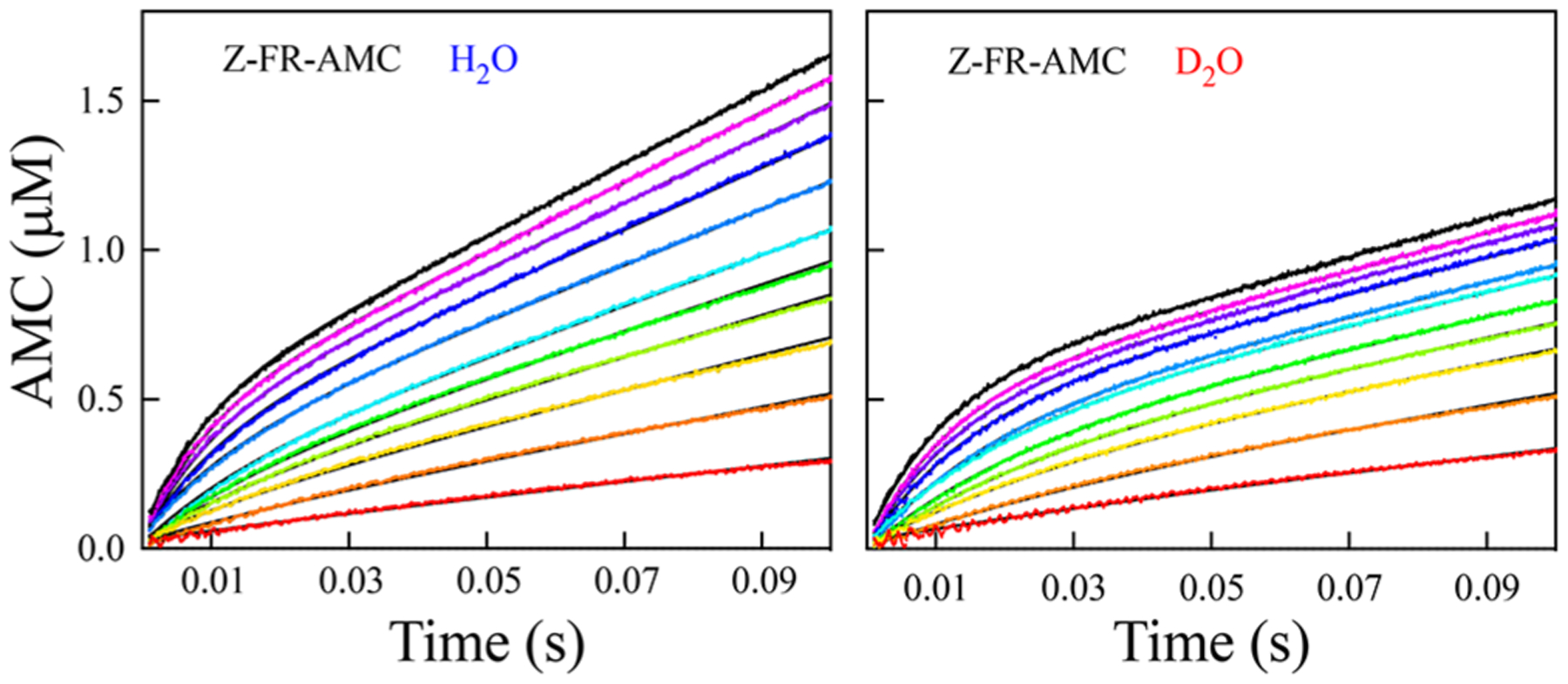
Pre-steady-state time courses of cruzain-catalyzed hydrolysis of Z-FR-AMC [0.5 (red), 1.0, 1.5, 2.0, 2.5, 3.0, 3.75, 5.0. 6.25, 7.5, and 8.75 (black) μM] in and at 0.002–0.1 s, with the product formation shown on the same scale. The black lines drawn through the experimental data represent fitting to eq 11.
Table 3.
Pre-Steady-State Kinetic Parameters of Cruzain-Catalyzed Hydrolysis of Z-FR-AMC and Z-RR-AMC in and a
| substrate | solvent | b | c | |||
|---|---|---|---|---|---|---|
| Z-FR-AMC | H2O | 30.5 ± 0.1 | 239 ± 6 | 35 ± 1 | 2.3 ± 0.1 | 9.8 ± 0.6 |
| D2O | 14.7 ± 0.1 | 179 ± 2 | 16 ± 1 | 0.86 ± 0.01 | 9.0 ± 0.7 | |
| sKIEd | ||||||
| Z-RR-AMC | H2O | 15.2 ± 0.1 | 64.5 ± 0.2 | 19.9 ± 0.1 | 7.0 ± 0.1 | 14.0 ± 3.0 |
| D2O | 9.5 ± 0.1 | 46.1 ± 0.1 | 11.9 ± 0.1 | 5.5 ± 0.1 | 15.0 ± 3.0 | |
| sKIEd |
Data were obtained as described in Experimental Procedures at pH(D) 7.5 in 10% (v/v) DMSO. The kinetic parameters were determined from global fitting according to eq 11.
was calculated from and , and values of in and for both substrates were determined separately as described. The errors for all the kinetic parameters are the standard deviations determined from the nonlinear least-squares fitting of the data.
Determined from fitting the transient burst rate vs substrate concentration according to eq 11.
Solvent kinetic isotope effects as determined from the ratio of the two sets of kinetic parameters in and . The errors were calculated on the basis of the propagation of fractional errors.
For both substrates, the time courses obtained at each fixed concentration of substrate in either solvent were best fitted to a single-exponential function at all concentrations (eq 8), and values for the resulting steady-state rate constant , burst amplitude , and transient rate constant were replotted separately versus substrate concentration and fitted to eqs 1,9, or 10 (Figure 7 and Figure S5). For both solvents, we determined the transient rate constant over a larger range of fixed concentrations of both substrates (≤80 μM) to observe saturation (Figure 7C). The steady-state rate is diminished in , but more so at higher concentrations of Z-FR-AMC in agreement with kinetics obtained in the steady state (Figure 7A; from fitting to eq 1), as expected, because the steady-state sKIE is larger on than on . Interestingly, the apparent effect of on the burst amplitude is to make it larger than that observed in , which is due to the large differences in fitted values of . Replotting of the transient rate constant versus [Z-FR-AMC] provided the following values upon fitting to eq 10: and . The latter value is identical to that which is obtained for the substrate Ser-Tyr-AMC with human cathepsin C.24
Figure 7.
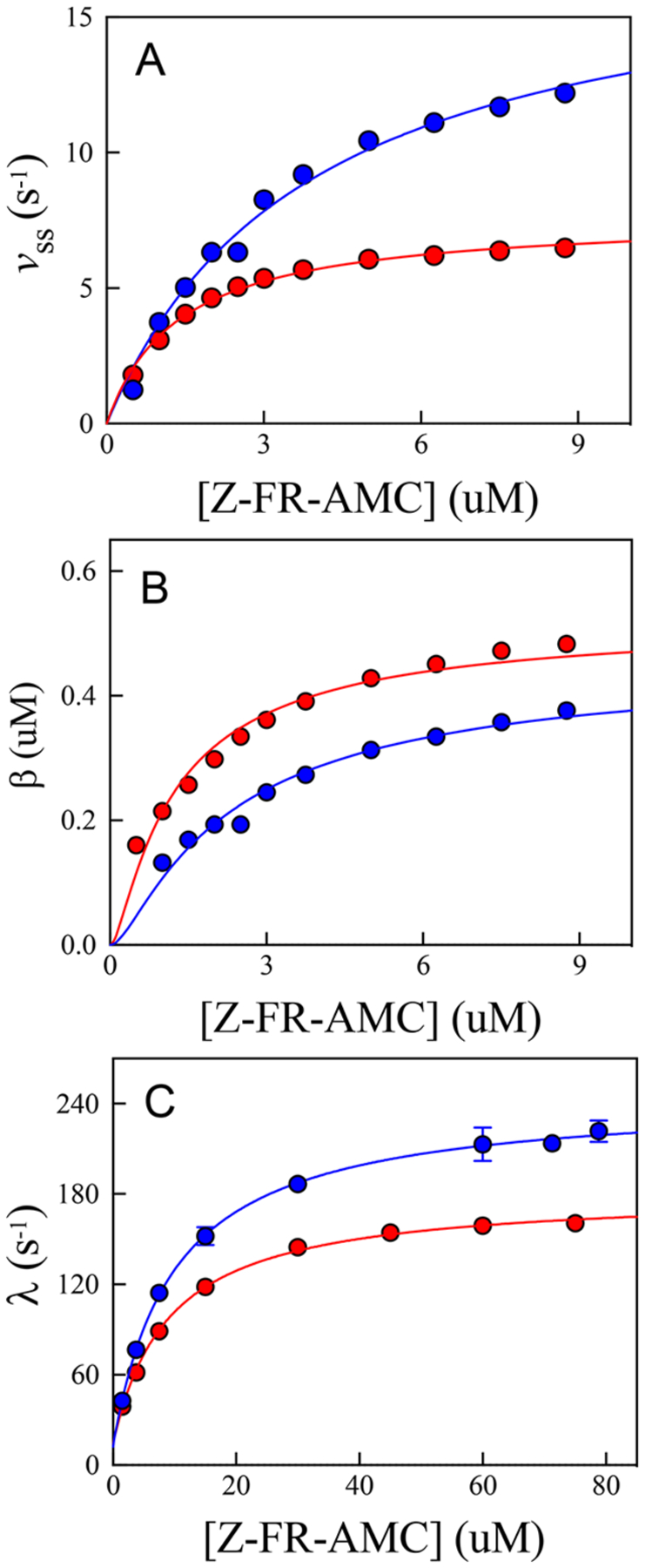
Replots of the kinetic parameters obtained from fitting individual time courses at each substrate concentration to eqs 8–10 in (blue) and (red). Substrate concentrations used to determine the transient rate constants were 1.5–79 μM, compared to substrate concentrations used for the steady-state rate and burst amplitudes (0.5–8.75 μM). (a) Fitting of the steady-state rate vs [Z-FR-AMC] (eq 1) resulted in the following values: and in , and and in . (b) Fitting of the burst amplitude vs [Z-FR-AMC] (eq 9) resulted in the following values: , and , and , and . (c) Fitting of the transient rate constant vs [Z-FR-AMC] (eq 10) resulted in the following values: , and , and , and .
Kinetic parameters for Z-FR-AMC and Z-RR-AMC, including sKIEs on , and as obtained from global fitting using eq 11 of pre-steady-state data are listed in Table 3. This full set of kinetic parameters was well-determined, as opposed to those obtained from replots as depicted in Figure 7. For Z-FR-AMC, the rate constant of cruzain acylation was 8-fold higher than the turnover number and the equivalent rate constant of deacylation . For Z-RR-AMC, these rate constants were lower: , which was 4-fold higher than the turnover number and its similar rate constant of deacylation . The normal sKIEs for for the two substrates [ (Z-FR-AMC), and (Z-RR-AMC)] are comparable to the pL-independent values obtained in the steady-state studies. For both substrates, normal sKIEs were obtained for the rate constants of acylation and deacylation: and (Z-FR-AMC), and and (Z-RR-AMC). The fact that for both substrates provides further evidence that the rate constants of enzyme deacylation determine the turnover numbers. We attribute the differences in measured values of and under steady-state and presteady-state conditions to the increased concentration of DMSO in the latter experiments, as borne out by additional studies in the steady state where these parameters were obtained in both 2% (v/v) and 10% (v/v) DMSO (data not shown).
Catalytic Mechanism of Cruzain.
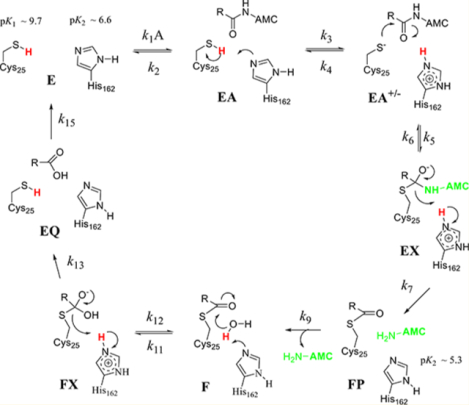
Similar to the mechanism of human cathepsin , the acylation half-reaction of cruzain is significantly faster than deacylation for those substrates with the highest values of .43 Where the two enzymes differ is that for cruzain, all solvent kinetic isotope effects are normal, indicating no enrichment of the Cys-S−:His-H+ tautomer of the ligand-free enzyme species in , so the Cys-His catalytic dyad is neutral at the initiation of catalysis. Accordingly, we may consider two catalytic mechanisms for cruzain for which Cys25-SH and His162 are both neutral in free enzyme E (Figure 8). Binding of substrate to form EA facilitates proton transfer from to , at which point the two mechanisms diverge. In mechanism 1 (Figure 8, green box), conformational isomerization of complex EA to EA’ positions , and the substrate for a concerted reaction of general base-catalyzed proton transfer and attack of the incipient thiolate of on the scissile amide carbonyl. In mechanism 2 (Figure 8, magenta box), thiolation of the amide carbonyl occurs in two discrete steps. (1) General basecatalyzed proton transfer to establishes the imidazoliumthiolate dyad , and (2) the resulting thiolate attacks the amide carbonyl to form tetrahedral intermediate EX. One expects that the equilibrium isotope effect (EIE) for the formation of the thiolate in EA+/− from EA would be inverse ( as in the case of cathepsin C),24 which could allow discrimination of these two mechanisms. The acylation half-reaction is then completed upon the collapse of the EX intermediate with proton transfer from the imidazolium group to the departing AMC, which completes the transient phase seen in pre-steady-state kinetics. Discrimination of these two potential mechanisms is discussed below. The slower, deacylation half-reaction is the same for both mechanisms: Neutral in the F complex deprotonates the lytic water leading to the second tetrahedral intermediate, FX, which then collapses to elaborate the carboxylate product and restores and to their neutral forms.
Figure 8.
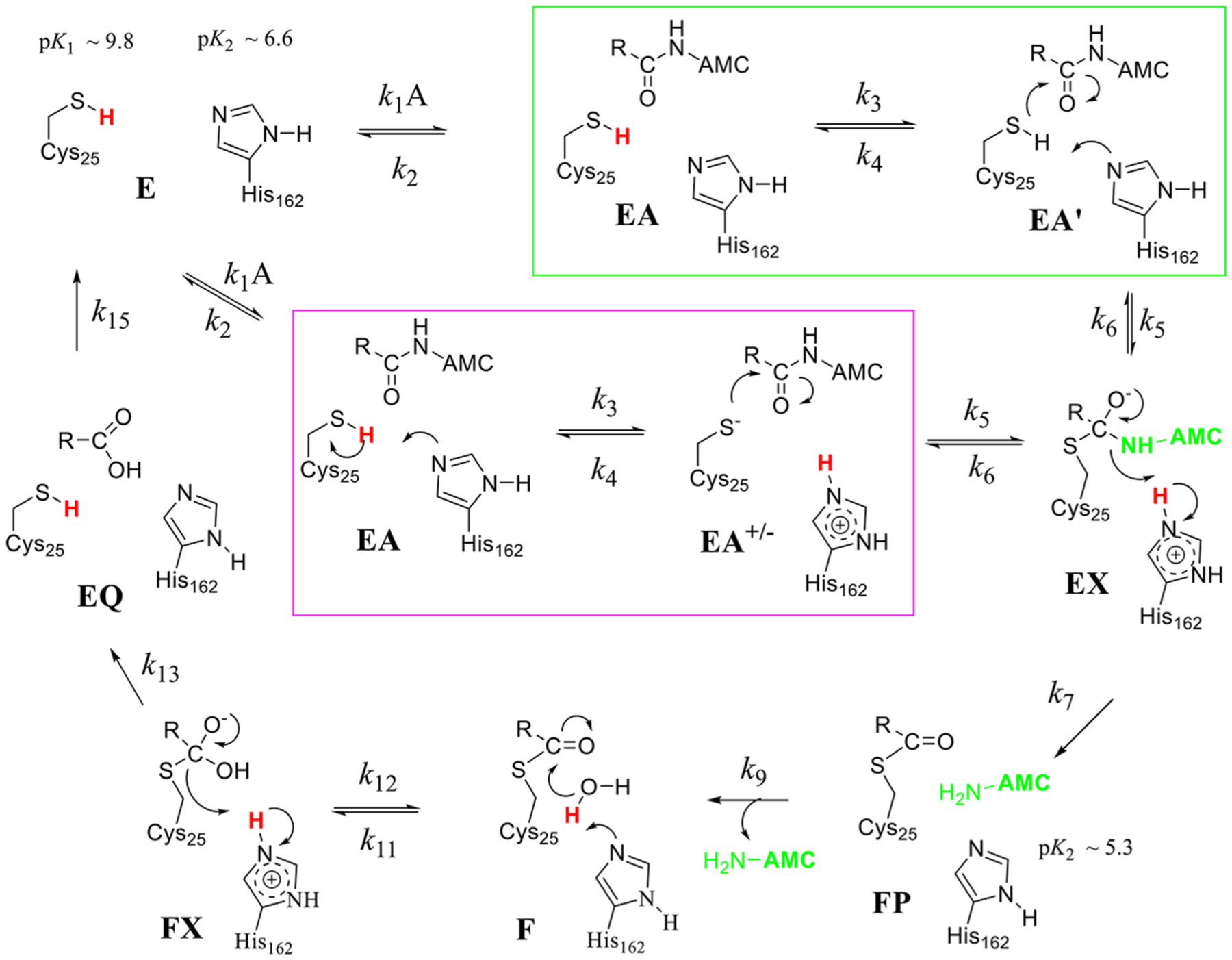
Proposed catalytic mechanisms for cruzain each involving acylation and deacylation half-reactions. Ligand-free cruzain contains neutral forms of and that are not positioned for proton transfer until substrate is bound. In mechanism 1 (green box), proton transfer from to is concerted with thiolate attack on the amide carbonyl to form tetrahedral intermediate EX. This occurs in two discrete steps in mechanism 2 (magenta box) in which the thiolate of attacks the amide. Steps in the deacylation half-reaction are the same for both mechanisms: thio-ester form undergoes attack by a hydroxide ion to form a second tetrahedral intermediate FX, which collapses to form the carboxylate product. Protons involved in primary solvent kinetic isotope effects are colored red. Free AMC is colored light green where it is expected to show full fluorescence.
Estimation of the Individual Kinetic Constants and Intrinsic Isotope Effects.
We sought to discriminate between the two proposed catalytic mechanisms by ascertaining values of the individual rate constants and intrinsic isotope effects on the catalytic steps. Expressions for the eight experimental kinetic parameters were derived in the steady state by the method of net rate constants for both mechanisms (eqs 14−21), for which , and represent commitment factors.44 Equations 18a and 20a are for mechanism 1, and eqs 18b and 20b are for mechanism 2.
| (14) |
| (15) |
| (16) |
| (17) |
| (18a) |
| (18b) |
| (19) |
| (20a) |
| (20b) |
| (21) |
To generate estimates of microscopic rate constants and intrinsic isotope effects (, etc.) found in the eight measured kinetic parameters (eqs 14−21) acquired for cruzain under presteady-state and steady-state conditions (Tables 2 and 3), we utilized the dynamic simulation capability of Kintek Explorer45 to iteratively search for and optimize values of for the two mechanisms in Figure 8 until we generated simulated time courses that superimposed with the experimental data in for both Z-RR-AMC and Z-FR-AMC. We conducted a similar simulation for the time courses performed in , and the intrinsic kinetic isotope effects for each catalytic step were then calculated by dividing each individual rate constant obtained in by its counterpart obtained in ; for example, . The simulated time courses generated from these “candidate” rate constants are in good agreement with the experimental time courses for both Z-FR-AMC and Z-RR-AMC in as shown in Figure 9. By calculating the corresponding rate constants in by division of each rate constant by its intrinsic isotope effect , we likewise obtained simulated time courses that can be superimposed on the experimental ones obtained in (Figure 9). The rate constants and intrinsic kinetic isotope effects , and in Table 4 comprise reasonable values for mechanism 2. To differentiate between mechanisms 1 and 2 in Figure 8, we reinserted the determined intrinsic values and individual rate constants into eqs 14−21 for both mechanisms 1 and 2 and calculated values for the eight kinetic parameters (Table 4, calculated kinetic parameters). The calculated kinetic parameters listed in Table 4 are for mechanism 2, where eqs 18b and 20b were used.
Figure 9.
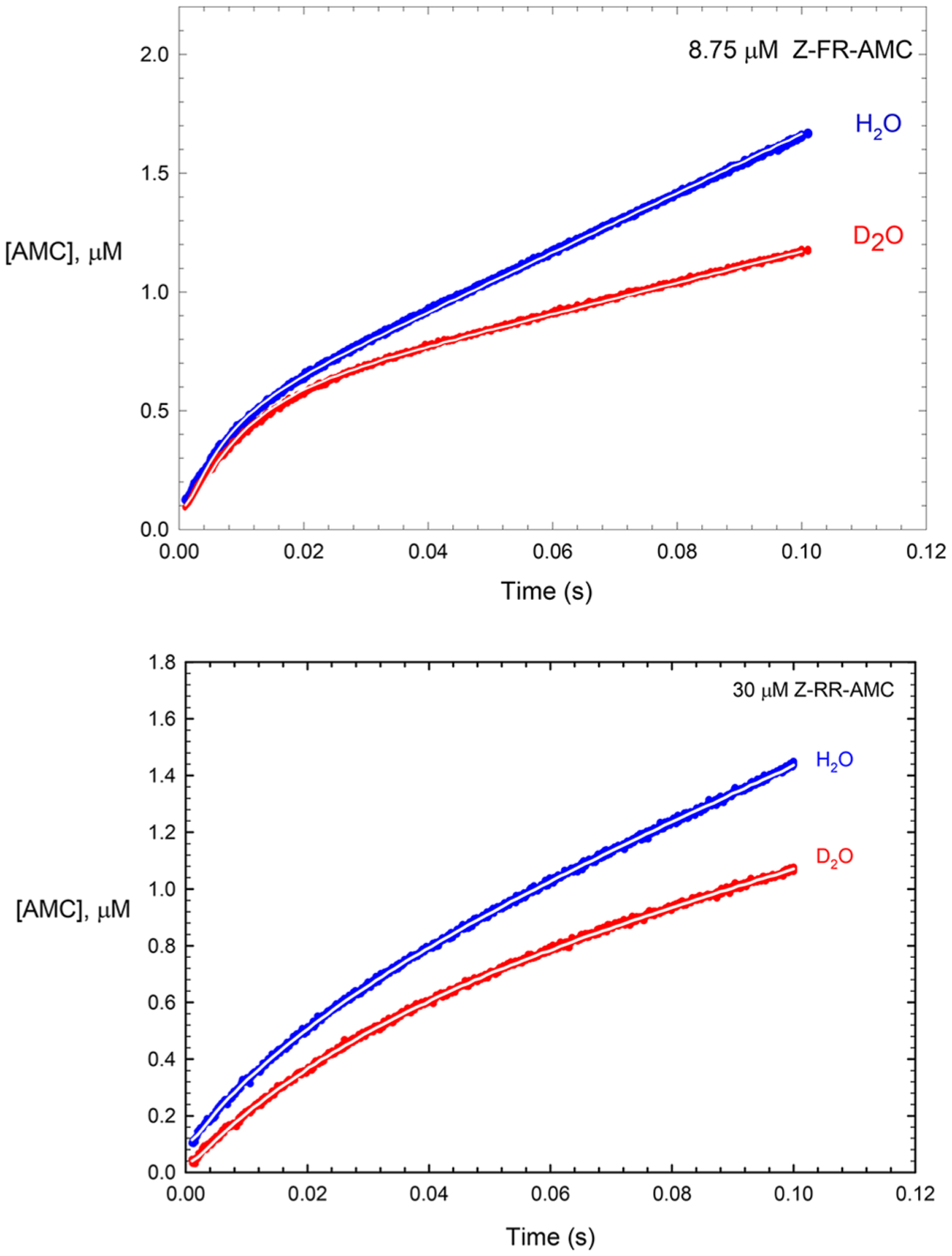
Experimental pre-steady-state time courses of cruzaincatalyzed hydrolysis of 8.75 μM Z-FR-AMC and 30 μM Z-RR-AMC in (blue) and (red) from data found in Figure 6 and Figure S4. The white lines drawn through all curves were produced by dynamic simulation using Kintek Explorer; here the enzyme species EX, FP, and P (P = AMC) and product carboxylic acid were assigned as the observables (generation of a fluorescence signal). For each experimental time course, values of rate constants were iteratively selected until the simulated curve could be superimposed upon the experimental time course for both substrates, as shown here. The rate constants from this approach led to the “candidate values” listed in Table 4. Values of the intrinsic isotope effects in Table 4 were obtained by dividing each individual rate constant optimized in by that optimized in ; for example, . The four simulated curves were generated with Z-RR-AMC and Z-FR-AMC: = 50 and and and 300 and and and and and and and 1.6, and and 1.9.
Table 4.
Experimental and Calculated Kinetic Parameters for the Catalytic Mechanism of Cruzain.a
| Substrate | Cbz-Arg-Arg-AMC | Cbz-Phe-Arg-AMC | ||||
|---|---|---|---|---|---|---|
| Kinetic Parameter | Experimental Value | Calculated Value | Candidate Value | Experimental Value | Calculated Value | Candidate Value |
| 1.15 ± 0.08 | 1.14 | 1.4 ± 0.1 | 1.4 | |||
| 64.5± 0.2 s−1 | 65 s−1 | 239 ± 6 s−1 | 239 s−1 | |||
| 1.41 ± 0.01 | 1.2 | 1.34 ± 0.01 | 1.2 | |||
| 19.9 ± 0.1 s−1 | 20 s−1 | 35 ± 1 s−1 | 35 s−1 | |||
| 1.67± 0.02 | 1.6 | 2.2 ± 0.02 | 2.3 | |||
| 15.2 ± 0.1 s−1 | 15 s−1 | 30 ± 1 s−1 | 30 s−1 | |||
| 1.61 ± 0.02 | 1.5 | 2.07 ± 0.02 | 2.08 | |||
| 4 | 1.2 | |||||
| 1.6 | 1.8 | 2.2 | 2.1 | |||
| 4 | 4 | 70 ± 50 | 9 | |||
Experimental kinetic parameters are those reported in Tables 2 and 3. Solvent equilibrium isotope effects, , and , intrinsic isotope effects on each step , and microscopic rate constants were obtained by dynamic simulation of the experimental pre-steady-state kinetic data using Kintek Explorer.47 Candidate values obtained from these fittings were then inserted into eqs 14–21 to produce the calculated values in the table. Values for and presented here resulted from extrapolation to an infinite concentration of the variable substrates to produce limiting values and .
From this and for both substrates, the “candidate” microscopic rate constants were more in accord with mechanism 2 than mechanism 1. Experimental and calculated values for both substrates (mechanism 2) agreed within a variation of ≤ 10%, except for for Z-RR-AMC, indicating that otherwise these candidate values, while not necessarily unique, are reasonable. By calculating kinetic parameters for both substrates for mechanism 1 using the values listed in Table 4 (eqs 18a and 20a; ) we obtained and , which exhibited poorer agreement with experimental results than with the kinetic parameters calculated for mechanism 2. One would expect the conformational change in the step of mechanism 1 to be faster than the subsequent chemical step and to exhibit no solvent KIE. For Z-FR-AMC under mechanism 2, proton transfer in the step is 4 times faster than that of Z-RR-AMC compared to ), suggesting that the Cbz-Phe-Arg dipeptide scaffold is more favorable for initiating proton transfer from Cys25 to His162 than Cbz-Arg-Arg is, and this difference in rate constants accounts for the 4-fold difference observed for . The stickiness of Z-FR-AMC is reflected in the calculated ratio of 9, while it is lower than the value determined from the plot of versus pH. This indicates that upon binding of Z-FR-AMC and proton transfer from Cys25 to His162, the enzyme–substrate complex EA +/− is highly committed to further catalytic steps , and accordingly, the subsequent attack by the resulting thiolate on the substrate carbonyl carbon to form EX is rapid ). Collapse of tetrahedral intermediate EX to form the enzyme–dipeptide thioester FP is very rapid with a modest intrinsic isotope effect on te proton transfer from to the departing amine (AMC) .
For Z-RR-AMC, the catalytic steps for are unsurprisingly slower than those of Z-FR-AMC, possibly because of a favorable ionic interaction of the Arg group at with , while Z-RR-AMC has a lower stickiness ratio of 4. As with Z-FR-AMC, the step (100 s−1) is the slowest in the acylation half-reaction, and the intrinsic KIE of accounts for . The steps are similar, if not identical, in rate to that of Z-FR-AMC. From the simulation, for both substrates, calculated values for of 0.6–0.7 (deprotonation of cysteine) and for of 1.3 (hydroxide attack on the thioester) were obtained, where the former value is inverse because . These equilibrium isotope effects compare favorably with values calculated from fractionation factors and .30,36
In the deacylation half-reaction, the determined rate constants indicated that the rates of formation of the tetrahedral complex for both substrates in FX and are similar, followed by rate-determining collapse of this intermediate to form the cruzain−carboxylate (EQ) complex ( and , for which the intrinsic kinetic isotope effects of and 1.9 for Z-RR-AMC and Z-FR-AMC, respectively, account for the measured values of and .
CONCLUSIONS
Similar to human cathepsin C, the most efficient substrates of cruzain undergo rapid acylation, which precedes rate-limiting deacylation. However, cruzain differs from papain, cathepsin C, and other cysteine proteases, in that its ligand-free form houses the less reactive, neutral Cys-SH/His dyad, implicit in both its pH–rate profiles and sKIEs of >1. This neutral dyad apparently undergoes proton transfer from the thiol of only upon substrate binding, which most likely occurs in a stepwise fashion (mechanism 2). That general base deprotonation of by elaborates the fact that the reactive thiolate comprises unusual chemistry for cruzain catalysis. Leinhard and Jencks demonstrated more than 50 years ago that general base deprotonation of thiols was not a feature of their reactions with the carbonyl groups of aldehydes and ketones but rather the thiolate fraction present was the sole reactant.46 Why then would unliganded cruzain adopt the unreactive Cys-SH:His tautomer and rely on this general base catalytic step to unmask the catalytic upon substrate binding? One is tempted to speculate that this is a feature of stringent substrate specificity in that only those bound substrates that affect the proton transfer step ( of mechanism 2) proceed to peptidolysis.
The rate-limiting steps for these two substrates occur during deacylation of these substrates exhibiting normal sKIEs. The less efficient substrate Z-RA-AMC is devoid of a pre-steadystate burst, suggesting that its diminished ability to induce this proton transfer renders . In terms of the future design of peptidomimetic covalent inactivators of cruzain such as K11777, the choice of amino acid residues at the and residues may determine whether the dipeptide “scaffold” binds in a fashion that promotes the transfer of the proton on to to elaborate the more nucleophilic thiolate species, which would then progress to alkylation of the activesite catalytic cysteine by its vinyl sulfone warhead. Our results suggest that a peptidomimetic inactivator bearing a Phe-Arg dipeptide may be more favorable than that with an Arg-Arg or Arg-Ala dipeptide.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Professor Charles S. Craik for providing a construct for the expression of cruzain and Dr. Larry Dangott for providing protein sequencing. The authors also thank Professor Ken Johnson for assistance with the use of Kintek Explorer.
Funding
The authors thank Texas A&M AgriLife Research for financial support of this work. This work was also supported by National Institutes of Health Grant R21AI127634.
ABBREVIATIONS
- MMTS
methylmethanethiosulfonate
- DEPC
diethylpyrocarbonate
- DTT
dithiothreitol
- DMSO
dimethyl sulfoxide
- AMC
7-amino-4-methylcoumarin
- Z-FR-AMC
carboxybenzyl-L-phenylalanine-L-arginine-7-amino-4-methylcoumarin
- Z-RR-AMC
carboxybenzyl-L-arginine-L-arginine-7-amino-4-methylcoumarin
- Z-RA-AMC
carboxybenzyl-L-arginine-L-alanine-7-amino-4-methylcoumarin
- EDTA
ethylenediaminetetraacetic acid
- CHAPS
3-[3-(cholamidopropyl)dimethylammonio]-1-propanesulfonate
- TAPSO
3-{[1,3-dihydroxy-2-(hydroxymethyl)-propan-2-yl] amino}-2-hydroxypropane-1-sulfonic acid
- MES
2-(N-morpholino)ethanesulfonic acid
- TEA
triethanolamine
- DEA
diethanolamine
- sKIE
solvent kinetic isotope effect
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b01250.
Steady-state kinetic data for wild-type cruzain-catalyzed reactions of Z-FR-AMC, Z-RR-AMC, and Z-RA-AMC in and and E208A mutant cruzain-catalyzed reactions of Z-FR-AMC, Z-RR-AMC, and Z-RA-AMC in values of E208A cruzain; pH−rate profiles of the E208A mutant-catalyzed reactions of Z-FR-AMC, Z-RR-AMC, and Z-RA-AMC in ; and pre-steady-state data and replots of pre-steady-state kinetic constants for the wild-type cruzain-catalyzed reaction of Z-RR-AMC in and (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Coura JR, and Vinas PA (2010) Chagas disease: a new worldwide challenge. Nature 465, S6–S7. [DOI] [PubMed] [Google Scholar]
- (2).Hotez PJ, Bottazzi ME, Franco-Paredes C, Ault SK, and Periago MR (2008) The neglected tropical diseases of Latin America and the Caribbean: a review of disease burden and distribution and a roadmap for control and elimination. PLoS Neglected Trop. Dis 2, e300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wilkinson SR, and Kelly JM (2009) Trypanocidal drugs: mechanisms, resistance and new targets. Expert Rev. Mol. Med 11, e31. [DOI] [PubMed] [Google Scholar]
- (4).Meirelles MN, Juliano L, Carmona E, Silva SG, Costa EM, Murta AC, and Scharfstein J (1992) Inhibitors of the major cysteinyl proteinase (GP57/51) impair host cell invasion and arrest the intracellular development of Trypanosoma cruzi in vitro. Mol. Biochem. Parasitol 52, 175–184. [DOI] [PubMed] [Google Scholar]
- (5).Harth G, Andrews N, Mills AA, Engel JC, Smith R, and McKerrow JH (1993) Peptide-fluoromethyl ketones arrest intracellular replication and intercellular transmission of Trypanosoma cruzi. Mol. Biochem. Parasitol 58, 17–24. [DOI] [PubMed] [Google Scholar]
- (6).Engel JC, Doyle PS, Hsieh I, and McKerrow JH (1998) Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J. Exp. Med 188, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Doyle PS, Zhou YM, Engel JC, and McKerrow JH (2007) A cysteine protease inhibitor cures Chagas’ disease in an immunodeficient-mouse model of infection,. Antimicrob. Agents Chemother 51, 3932–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Doyle PS, Zhou YM, Hsieh I, Greenbaum DC, McKerrow JH, and Engel JC (2011) The Trypanosoma cruzi protease cruzain mediates immune evasion,. PLoS Pathog. 7, e1002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ferrao PM, d’Avila-Levy CM, Araujo-Jorge TC, Degrave WM, Gonçalves A. d. S., Garzoni LR, Lima AP, Feige JJ, Bailly S, Mendonca-Lima L, and Waghabi MC (2015) Cruzipain Activates Latent TGF-beta from Host Cells during T. cruzi Invasion. PLoS One 10, e0124832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Martinez-Mayorga K, Byler KG, Ramirez-Hernandez AI, and Terrazas-Alvares DE (2015) Cruzain inhibitors: efforts made, current leads and a structural outlook of new hits. Drug Discovery Today 20, 890–898. [DOI] [PubMed] [Google Scholar]
- (11).Kerr ID, Lee JH, Farady CJ, Marion R, Rickert M, Sajid M, Pandey KC, Caffrey CR, Legac J, Hansell E, McKerrow JH, Craik CS, Rosenthal PJ, and Brinen LS (2009) Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem 284, 25697–25703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Barr SC, Warner KL, Kornreic BG, Piscitelli J, Wolfe A, Benet L, and McKerrow JH (2005) A cysteine protease inhibitor protects dogs from cardiac damage during infection by Trypanosoma cruzi. Antimicrob. Agents Chemother 49, 5160–5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lecaille F, Kaleta J, and Bromme D (2002) Human and parasitic papain-like cysteine proteases: their role in physiology and pathology and recent developments in inhibitor design. Chem. Rev 102,4459–4488. [DOI] [PubMed] [Google Scholar]
- (14).Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, Ellman JA, and Craik CS (2006) Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem 281, 1282412832. [DOI] [PubMed] [Google Scholar]
- (15).Storer AC, and Menard R (1994) Catalytic mechanism in papain family of cysteine peptidases. Methods Enzymol. 244, 486–500. [DOI] [PubMed] [Google Scholar]
- (16).Shokhen M, Khazanov N, and Albeck A (2009) Challenging a paradigm: theoretical calculations of the protonation state of the Cys25-His 159 catalytic diad in free papain. Proteins: Struct., Funct., Genet 77, 916–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Solowiej J, Thomson JA, Ryan K, Luo C, He M, Lou J, and Murray BW (2008) Steady-state and pre-steady-state kinetic evaluation of severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro cysteine protease: development of an ion-pair model for catalysis. Biochemistry 47, 2617–2630. [DOI] [PubMed] [Google Scholar]
- (18).Paasche A, Zipper A, Schafer S, Ziebuhr J, Schirmeister T, and Engels B (2014) Evidence for substrate binding-induced zwitterion formation in the catalytic Cys-His dyad of the SARS-CoV main protease. Biochemistry 53, 5930–5946. [DOI] [PubMed] [Google Scholar]
- (19).Arafet K, Ferrer S, and Moliner V (2017) Computational Study of the Catalytic Mechanism of the Cruzain Cysteine Protease. ACS Catal. 7, 1207–1215. [Google Scholar]
- (20).Polgar L (1974) Mercaptide-imidazolium ion-pair: the reactive nucleophile in papain catalysis. FEBS Lett. 47, 15–18. [DOI] [PubMed] [Google Scholar]
- (21).Creighton DJ, Gessouroun MS, and Heapes JM (1980) Is the thiolate−imidazolium ion pair the catalytically important form of papain? FEBS Lett. 110, 319–322. [DOI] [PubMed] [Google Scholar]
- (22).Creighton DJ, and Schamp DJ (1980) Solvent isotope effects on tautomerization equilibria of papain and model thiolamines. FEBS Lett. 110, 313–318. [DOI] [PubMed] [Google Scholar]
- (23).Lewis SD, Johnson FA, and Shafer JA (1976) Potentiometric determination of ionizations at the active site of papain. Biochemistry 15, 5009–5017. [DOI] [PubMed] [Google Scholar]
- (24).Schneck JL, Villa JP, McDevitt P, McQueney MS, Thrall SH, and Meek TD (2008) Chemical mechanism of a cysteine protease, cathepsin C, as revealed by integration of both steady-state and pre-steady-state solvent kinetic isotope effects. Biochemistry 47, 8697–8710 [DOI] [PubMed] [Google Scholar]
- (25).Villamil MA, Chen J, Liang Q, and Zhuang Z (2012) A noncanonical cysteine protease USP1 is activated through active site modulation by USP1-associated factor 1,. Biochemistry 51, 2829–2839. [DOI] [PubMed] [Google Scholar]
- (26).Lee GM, Balouch E, Goetz DH, Lazic A, McKerrow JH, and Craik CS (2012) Mapping inhibitor binding modes on an active cysteine protease via nuclear magnetic resonance spectroscopy. Biochemistry 51, 10087–10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, and Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. Proteomics Protocols Handbook, 571–607. [Google Scholar]
- (28).Ellis KJ, and Morrison JF (1982) Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol. 87, 405–426. [DOI] [PubMed] [Google Scholar]
- (29).Schowen KB, and Schowen RL (1982) Solvent isotope effects of enzyme systems. Methods Enzymol. 87, 551–606. [PubMed] [Google Scholar]
- (30).Cleland WW (1977) Determining the chemical mechanisms of enzyme-catalyzed reactions by kinetic studies. Adv. Enzymol. Relat. Areas Mol. Biol 45, 273–387. [DOI] [PubMed] [Google Scholar]
- (31).Cook PF, and Cleland WW (1981) Mechanistic deductions from isotope effects in multireactant enzyme mechanisms. Biochemistry 20, 1790–1796 [DOI] [PubMed] [Google Scholar]
- (32).Northrop DB (1981) The expression of isotope effects on enzyme-catalyzed reactions,. Annu. Rev. Biochem 50, 103–131. [DOI] [PubMed] [Google Scholar]
- (33).Schechter I, and Berger A (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun 27, 157–162. [DOI] [PubMed] [Google Scholar]
- (34).Gillmor SA, Craik CS, and Fletterick RJ (1997) Structural determinants of specificity in the cysteine protease cruzain. Protein Sci. 6, 1603–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cleland WW (1982) The use of pH studies to determine chemical mechanisms of enzyme-catalyzed reactions,. Methods Enzymol. 87, 390–405. [DOI] [PubMed] [Google Scholar]
- (36).Quinn DM, and Sutton LD (1991) Theoretical basis and mechanistic utility of solvent isotope effects. In Enzyme Mechanism from Isotope Effects (Cook PF, Ed.) p 73, CRC Press, Inc., Boca Raton, FL. [Google Scholar]
- (37).Karsten WE, Lai C-J, and Cook PF (1995) Inverse solvent isotope effects in the NAD-malic enzyme reaction are the result of the viscosity difference between and : implications for solvent isotope effect studies. J. Am. Chem. Soc 117, 5914–5918. [Google Scholar]
- (38).Miles EW (1977) Modification of histidyl residues in proteins by diethylpyrocarbonate. Methods Enzymol. 47, 431–442. [DOI] [PubMed] [Google Scholar]
- (39).Dumas DP, and Raushel FM (1990) Chemical and kinetic evidence for an essential histidine in the phosphotriesterase from Pseudomonas diminuta. J. Biol. Chem 265, 21498–21503. [PubMed] [Google Scholar]
- (40).Bednar RA (1990) Reactivity and pH dependence of thiol conjugation to -ethylmaleimide: detection of a conformational change in chalcone isomerase. Biochemistry 29, 3684–3690. [DOI] [PubMed] [Google Scholar]
- (41).Stone EM, Costello AL, Tierney DL, and Fast W (2006) Substrate-assisted cysteine deprotonation in the mechanism of dimethylargininase (DDAH) from Pseudomonas aeruginosa. Biochemistry 45, 5618–5630. [DOI] [PubMed] [Google Scholar]
- (42).Dreyton CJ, Knuckley B, Jones JE, Lewallen DM, and Thompson PR (2014) Mechanistic studies of protein arginine deiminase 2: evidence for a substrate-assisted mechanism. Biochemistry 53, 4426–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rubach JK, Cui G, Schneck JL, Taylor AN, Zhao B, Smallwood A, Nevins N, Wisnoski D, Thrall SH, and Meek TD (2012) The amino-acid substituents of dipeptide substrates of cathepsin C can determine the rate-limiting steps of catalysis,. Biochemistry 51, 7551–7568. [DOI] [PubMed] [Google Scholar]
- (44).Cleland WW (1975) Partition analysis and concept of net rate constants as tools in enzyme kinetics. Biochemistry 14, 3220–3224. [DOI] [PubMed] [Google Scholar]
- (45).Johnson KA, Simpson ZB, and Blom T (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem 387, 20–29. [DOI] [PubMed] [Google Scholar]
- (46).Lienhard GE, and Jencks WP (1966) Thiol addition to the carbonyl group: equilibria and kinetics. J. Am. Chem. Soc 88, 3982–3995. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.