

Abstract
PURPOSE
To determine prognostic and predictive clinical outcomes in metastatic hormone-sensitive prostate cancer (mHSPC) and metastatic castrate-resistant prostate cancer (mCRPC) on the basis of a combination of plasma-derived genomic alterations and lipid features in a longitudinal cohort of patients with advanced prostate cancer.
METHODS
A multifeature classifier was constructed to predict clinical outcomes using plasma-based genomic alterations detected in 120 genes and 772 lipidomic species as informative features in a cohort of 71 patients with mHSPC and 144 patients with mCRPC. Outcomes of interest were collected over 11 years of follow-up. These included in mHSPC state early failure of androgen-deprivation therapy (ADT) and exceptional responders to ADT; early death (poor prognosis) and long-term survivors in mCRPC state. The approach was to build binary classification models that identified discriminative candidates with optimal weights to predict outcomes. To achieve this, we built multi-omic feature-based classifiers using traditional machine learning (ML) methods, including logistic regression with sparse regularization, multi-kernel Gaussian process regression, and support vector machines.
RESULTS
The levels of specific ceramides (d18:1/14:0 and d18:1/17:0), and the presence of CHEK2 mutations, AR amplification, and RB1 deletion were identified as the most crucial factors associated with clinical outcomes. Using ML models, the optimal multi-omics feature combination determined resulted in AUC scores of 0.751 for predicting mHSPC survival and 0.638 for predicting ADT failure; and in mCRPC state, 0.687 for prognostication and 0.727 for exceptional survival. The models were observed to be superior than using a limited candidate number of features for developing multi-omic prognostic and predictive signatures.
CONCLUSION
Using a ML approach that incorporates multiple omic features improves the prediction accuracy for metastatic prostate cancer outcomes significantly. Validation of these models will be needed in independent data sets in future.
Multi-omic blood-based biomarkers identify prognostic patients in metastatic prostate cancer.
INTRODUCTION
The complexity of tumorigenesis and clonal heterogeneity has been rationalized in several hallmarks of cancer.1 Although not all accumulating somatic alterations during clonal expansion with cancer initiation and progression contribute to cancer mutagenesis or affect cancer outcomes, several will affect clinical outcomes. It has been estimated that clonal evolutionary processes after tumorigenesis occur on an average of 1-10 mutations per cell division2 and many of these can affect disease outcomes after cancer initiation, including the natural history of progression, response to treatments, and resistance to interventions. To identify key biological drivers of cancer outcomes, bioinformatic methods have largely focused on single omic platform (either DNA or RNA) biomarker probing. A more comprehensive approach to biomarkers that integrate gene, transcriptome, and protein/lipid products in the context of a stage of progression and with treatment interactions is typically lacking for several reasons. Limited availability of clinically annotated data sets and databases that can provide such multi-omic sequencing/profiles in patients with cancer with longitudinal outcomes and a paucity of robust computational neural network methodologic approaches remain challenges to achieve the desired outcome.
CONTEXT
Key Objective
To identify multi-omic, machine learning (ML) classifiers of clinical outcomes in metastatic prostate cancer (mPC), using measurements of a variety of plasma lipid species combined with gene alterations detected in circulating tumor DNA.
Knowledge Generated
Logistic regression with elastic net regularization ML approach was observed to be the most optimal of all ML methods in generating a lipid molecule combined with gene alteration–based predictive algorithm that identified short- and long-term responses to androgen-deprivation therapy (ADT), and survival in metastatic hormone-sensitive prostate cancer and metastatic castrate-resistant prostate cancer states. This ML classifier was observed to be superior to conventional biostatistical approaches that use a limited number of biological candidates for identifying prognostic and predictive outcomes in mPC.
Relevance
A blood-based multi-omic classifier in different states of mPC progression could potentially be used to define cohorts of mPC patients destined to have different clinical outcomes.
Prostate cancer is a leading cause of cancer death in males in the Western world.3 Metastatic prostate cancer (mPC) is subdivided into an earlier metastatic hormone-sensitive prostate cancer (mHSPC) state and then a more progressed metastatic castrate-resistant prostate cancer (mCRPC) state after failure of androgen-deprivation therapy (ADT), which is the prime way to treat mHSPC. Both mHSPC and mCRPC are heterogeneous in clinical behavior, and patients in these states can either quickly progress or have slowly progressing disease. Response to treatments given in these states can similarly be short- or long-lasting responses. An increasing number of treatment options for managing mHSPC4 and mCRPC5 states are now available, while there are no specific molecular multi-omic alterations identified in these states predictive for response to ADT or prognostic for survival. We have previously reported plasma-based cell-free DNA (cfDNA) genomic alterations using next-generation sequencing (NGS)6-8 and liquid chromatography-mass spectrometry (LC-MS)–based lipid profiles9 in a mPC cohort.
In this study, we hypothesized that a multi-omic molecular classifier on the basis of stage-specific genetic and lipidomic alterations detected in plasma can predict treatment and survival outcomes in mPC. We combined blood-based profiling of genomic alterations in cfDNA for detecting somatic alterations and mass spectrometry–generated lipidomic features in plasma in a cohort of patients with mPC and then applied a machine learning (ML) multi-omic platform approach to build multi-omic classifiers of outcomes. To achieve this, we applied a ML approach that combined the two platforms for identifying a multi-omic classifier that may predict clinical outcomes in mHSPC and mCRPC states.
METHODS
Patient Cohort
A real-world prospectively collected and retrospectively analyzed hospital-based cohort of patients with mPC, in mHSPC and mCRPC states were considered for this study. Previously, this cohort's plasma cfDNA alterations using NGS,6-8 plasma-based LC-MS–generated lipid species,9 and a combined candidate gene with a candidate 3-lipid signature10 for predicting clinical outcomes have been reported. The current study attempted to integrate beyond limited number of candidates, all 722 lipid species, and genomic alteration features for building multi-omic classifiers by applying ML approaches.
The mPC cohort was enrolled between September 2009 and January 2013 after obtaining a written consent and institutional review board approval (Mayo IRB 09-001889). Details of NGS methods and LC-MS–generated lipid profiles have been previously described.6,9 Clinical follow-up for this cohort in the current study was extended from October 20, 2018, in the cutoff date for analysis in previous reports6,9,10 to March 1, 2022. Progression on ADT, labeled as ADT failure for patients with mHSPC, was defined as the time from initiating hormone therapy for metastatic state to development of castrate resistance, and survival was calculated as the time from diagnosis of mHSPC state to death at the date of cutoff for analysis (March 1, 2022). Patients with mHSPC who did not have a death event at the time of data analysis were censored. In the mHSPC cohort, we labeled patients who failed ADT within 6 months after initiation as early failure. The survival time of patients with mCRPC was determined from the date of castrate resistance to the date of death at the time of analysis (March 1, 2022). The range of survival time of the patients with mCRPC was divided into tertiles with the top 33% and bottom 33% being selected. Thus, patients in the bottom 33% of survival time who died within 20.6 months after initial progression to mCRPC were classified as belonging to the poor-prognosis group, while those who were still alive in the top 33% of the survival range at or after 50 months of initial progression to mCRPC were grouped as exceptional survivors.
Four multi-omic classifier-building tasks were pursued using machine learning methods, two in mHSPC state and two in mCRPC state. For patients with mHSPC, we included ML classifier model that predicted mHSPC survival and a machine learning–based classifier that predicted patients who experienced early failure of ADT. In the mCRPC state, we developed a multi-omic classifier model that predicted patients with poor prognosis, as defined by death within 20.6 months of turning castration-resistant. We also developed ML classifiers that predict exceptional survival in mCRPC state, as defined by long-term survival of 50 month or more.
Feature Selection Process
The outline for our overall methodology workflow is illustrated in Figure 1. We used the human-in-the-loop principle to identify the most informative features from previously published results. To start, we considered 120 ctDNA-based gene alterations and 722 plasma-measured lipidomic species from our published platforms.6,9 On the basis of previous results that confirm specific cfDNA-based signatures in these 120 genes to have predictive and prognostic value in mPC,6,11,12 we adopted these alterations to proceed to the next step. Previously, we have reported extensively6 on both, the frequencies of alterations and the relevance of individual alterations to state-specific clinical outcomes including survival and ADT-related treatment outcomes. In patients with untreated mHSPC at the individual-gene level, alterations in TP53 and ATM were significantly associated with shorter overall survival. Collectively, patients with untreated mHSPC with somatic alterations detected in multiple DNA repair genes (ATM, BRCA1, BRCA2, and CHEK2) were also observed to have had significantly short overall survival and a shorter time to failure with ADT even after adjusting for clinical prognostic factors. For the mCRPC groups, RB1 deletions had the most significant prognostic value for poor outcomes in multivariate analyses after adjusting for Gleason score and alkaline phosphate levels. Somatic perturbations detected in other genes in multivariable analyses that had prognostic significance included AR, TP53, and BRCA2. Based both on the survival impact and the state-specific frequency of alterations, we selected mutations in ATM, BRCA1, BRCA2, and CHEK2 for patients in the mHSPC state, and mutations in TP53 and copy number alterations in RB1 loss, and AR amplification for patients in the mCRPC state, for integration with lipidomic features as potential classifiers.
FIG 1.

Workflow of integrating genomic and lipidomic features and building machine learning pipelines. Number of individual lipid species included in analyses for each lipid type indicated in parentheses. Acy, acylcarnitine; Cer, ceramide; ctDNA, circulating tumor DNA; DG, diacylglycerol; LCA, latent class analysis; LC-MS, liquid chromatography-mass spectrometry; ML, machine learning; Sph, sphingolipid; TG, triacylglycerol.
For the 772 lipidomic features, we applied Cox regression and latent component analysis (LCA) as previously described9 to identify candidate lipid species in the multi-omic model build. This resulted in the selection of 61 ceramide (Cer) species, 25 diacylglycerol (DG) species, and 142 triacylglycerol (TG) species for patients in the mHSPC state and 61 ceramide (Cer) species, 3 sphingosine (Sph) species, and 14 acylcarnitine (Acy) species for patients in the mCRPC state.
For feature processing, genomic alterations were transformed into binary variables (present/absent). Lipidomic species' measurements being continuous features were binarized using a novel approach called bin-split binarization. This is different from the traditional mean-split binarization methods13-15 that categorize continuous features on the basis of the mean/median levels. The bin-split binarization, inspired by the k-group split,14 maps each numerical lipid feature to two separate binary features, high-level lipid and low-level lipid, allowing for a finer-grained analysis of the effects of different lipid levels. Use of this approach was performed to improve robustness and interpretability of the model. More details on bin-split binarization approach are included in the Data Supplement ([Supplementary Methods]).
Machine Learning Methods
To identify the most optimal data-driven algorithm that can determine mHSPC and mCRPC clinical outcomes on the basis of the selected features, we evaluated several traditional machine learning techniques, including Logistic regression16 with and without elastic net regularization,17 kernel support vector machines (kernel-SVM), and Gaussian process regression (GPR) with multiple kernel configurations. After identifying the optimal model and to mitigate overfitting, we trained and evaluated the model using a standard five-fold cross-validation approach.18 All patients with mHSPC and mCRPC were divided into five equal groups, with one group used to test the model, while the other four were used to train the model. This process was repeated five times to ensure each group was included in the model evaluation. To create multi-omics classifiers for each of the four clinical outcomes of interest, we used different genomic and lipidomic features feature sets, as well as combinations of both. The performance of each classifier was evaluated using five metrics: AUC, calculated as concordance index or C-index, accuracy, precision, recall, and sensitivity. Using Monte-Carlo five-fold cross-validation, we report the mean and a 0.95 CI for each metric.18,19 The risk threshold was established on the basis of established clinical and data-driven approaches.20,21 Further details for metrics' definition and test-score computation in the machine learning pipeline are included in the Data Supplement ([Supplementary Methods]).
Methods for Determination of Feature Effects
We analyzed the top 20 features that were observed to have high positive or negative weights for each clinical outcome and then calculated their relative ratios on the basis of the raw weight values over the feature groups yielding the most optimal performance. This was performed to get an insight into a feature's predictive or protective contributory role for each clinical outcome. Details on the approach used for calculating the relative effect ratio computations in our feature analysis are included in the Data Supplement ([Supplementary Methods]).
Next, we adopted the survival prediction task for patients with mHSPC and mCRPC to compute the test accuracy of the multi-omic model that included both genomic and lipidomic features, and then compared our method with the previously published candidate-based, three-lipid-signature approach,9 which was used for predicting clinical outcomes. Computation details for this comparative approach are detailed in the Data Supplement ([Supplementary Methods]).
Using the Multi-Omic Model Predicting Outcomes in Future Patients
Finally, we explored the construction of a predictive model for future patients using the combined multi-omic features identified in the current study. For this, we selected the top number of multi-omic features with the most significant effects and evaluated the following equation to generate a probability score:
where N is the number of used features, are the top n-th feature's weight from the trained models, and are evaluation values of the future patients. The number of features used can be a relatively small number compared with the total number of feature candidates and still yield accurate predictions. Details for generating a predictive model probability score are detailed in the Data Supplement ([Supplementary Methods]).
RESULTS
Briefly, for the patients with mHSPC, sample draw was performed before any initiation of ADT treatments and for patients with mCRPC, sample blood draw was performed after ADT failure and before initiation of mCRPC therapies.6 Patient baseline and follow-up characteristics for both mHSPC and mCRPC cohorts are available in the Data Supplement ([Patient Demographics Table P1]). We observed the predictive performance of logistic regression with elastic net regularization using various feature sets for four target tasks to be superior than all other ML approaches. Results for the classical machine learning techniques, including logistic regression without elastic net regularization, kernel-SVM, and GPR with multiple kernel configurations, are summarized in the Data Supplement ([Tables S1-4]). The logistic regression with elastic net regularization approach was observed to enable examination of the impact of features since the model weights reflect feature effects after training.
Tables 1-4 demonstrate the prediction results using logistic regression with elastic net regularization. Names of the specific genes and lipid species for each table/task have been detailed in the Legends section. In addition, effect ratios of the top 20 weighted features with an individual positive or negative weight in predicting outcomes are presented in Figure 2A-2D. The full list of all features analyzed with their raw weights and effect ratio is listed in the Data Supplement ([Tables S6A-S6H]).
TABLE 1.
AUC, Accuracy, Precision, Recall, and Sensitivity of Different Gene Alteration-Lipid Feature Combinations' Association With Survival in Metastatic Hormone-Sensitive Prostate Cancer State

TABLE 4.
AUC, Accuracy, Precision, Recall, and Sensitivity of Different Feature Combinations Associated With Exceptional Survival in Metastatic Castrate-Resistant Prostate Cancer State

FIG 2.
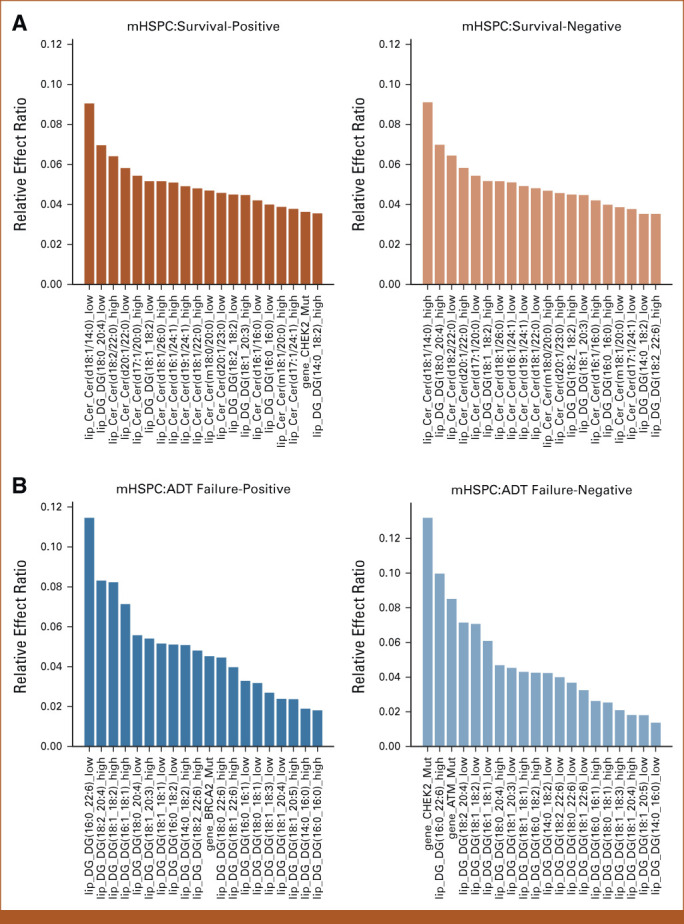
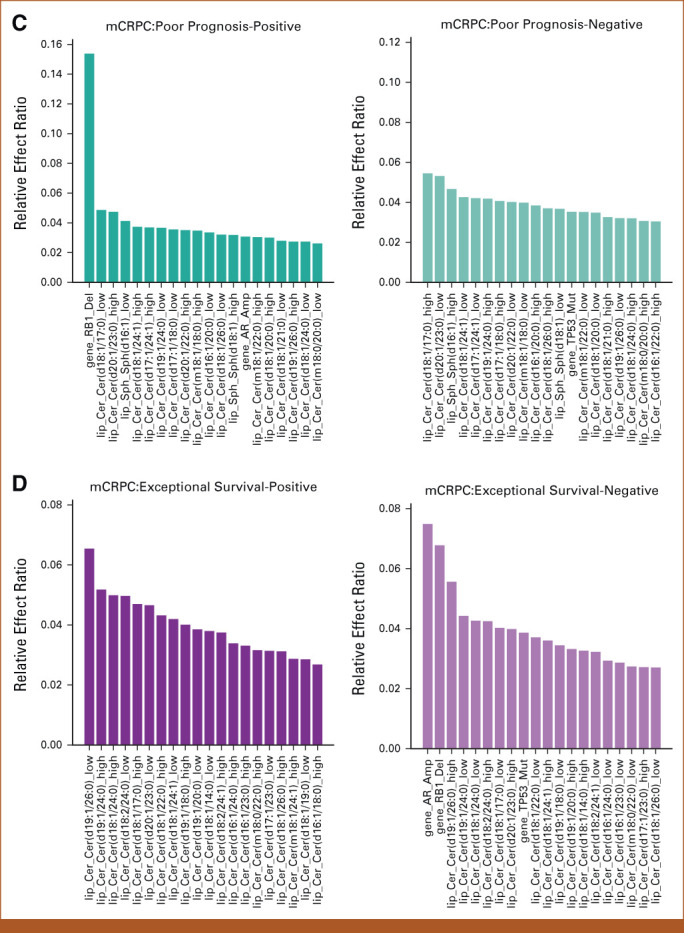
Results of feature effects analysis for association of multi-omic classifier with clinical outcomes. (A-D) For each target task, the top 20 genomic and lipidomic features with most positive and negative effects along with their relative effect ratios are shown respectively. Machine learning was used to compute feature weights. The relative effect ratio computations are in the Data Supplement ([Supplementary Methods]). (A) Top 20 multi-omic features with most positive and negative effects associated with survival prediction in mHSPC state. (B) Top 20 multi-omic features with most positive and negative effects on ADT-failure prediction in mHSPC state. (C) Top 20 multi-omics features with most positive and negative effects associated with poor prognosis prediction in mCRPC state. (D) Top 20 multi-omics features with most positive and negative effects on exceptional survival prediction in mCRPC state. Acy, acylcarnitine; ADT, androgen-deprivation therapy; Cer, ceramide; DG, diacylglycerol; Gene, genomic feature sets; mCRPC, metastatic castrate-resistant prostate cancer; mHSPC, metastatic hormone-sensitive prostate cancer; Sph, sphingolipid; TG, triacylglycerol.
Table 1 shows that the best performance in predicting mHSPC patient survival, which is achieved through the combination of gene features (mutations in ATM, BRCA1, BRCA2, and CHEK2) and diacylglycerol (DG) and triacylglycerol (TG) lipids, with an AUC of 0.754. Figure 2A demonstrates that the lipid feature Cer (d18:1/14:0) at a low level, with the largest effect ratios among all negative-weights-value features, has the most significant predictive effect for survival prediction. Conversely, at a high level, this lipid feature has the most substantial protective effect for the task, also with the largest effect ratios among all negative-weights-value features. Table 2 shows the highest AUC of 0.638 for predicting ADT failure in patients with mHSPC achieved by combining gene features (mutations in ATM, BRCA1, BRCA2, and CHEK2) and diacylglycerol (DG) levels. Figure 2B shows the most positive effect of low level of DG (16:0/22:6) and the negative effect of CHEK2 alteration. Table 3 displays the highest AUC of 0.687 for predicting poor prognosis in mCRPC state achieved by combining gene features (mutations in TP53 along with RB1 loss and AR amplification) and sphingosine (Sph) levels. Figure 2C shows the significance predictive effect of the RB1 gene deletion on poor prognosis, as it has the largest effect ratios among all negative-weights-value features.
TABLE 2.
AUC, Accuracy, Precision, Recall, and Sensitivity of Different Feature Combinations for Association With Androgen-Deprivation Therapy Failure in Metastatic Hormone-Sensitive Prostate Cancer State

TABLE 3.
AUC, Accuracy, Precision, Recall, and Sensitivity of Different Feature Combinations for Association With Early Death in Metastatic Castrate-Resistant Prostate Cancer State

Table 4 presents the optimal AUC of 0.727 for predicting exceptional survivors in mCRPC, achieved through combining gene features (mutations in TP53 along with RB1 loss and AR amplification) with Cer, Sph, and Acy levels. Figure 2D highlights the negative effect of AR amplification and RB1 gene deletion, and the positive effect of low level of ceramide levels (19:1/26:1) in exceptional survival prediction.
In comparing logistic regression with elastic net regularization to the previously published limited candidate three-lipid signature-based derivatives,9,10 we observe greater test performance for the current setting. Detailed results are listed in the Data Supplement ([Table S5, Fig S1]).
Results for using the top number features to predict the probability scores in future patients results in a more efficient and rapid modeling process, with no significant reduction in performance. The number of features selected can be substantially smaller than the total number of features, for instance, only 50% of the features can be used. A detailed analysis of the trade-off for using lesser number of features for predicting outcomes is presented in the Data Supplement ([Fig S2-S5]).
DISCUSSION
We observed that the results of logistic regression with elastic net regularization in this study obtained greater performance metrics for identifying the performance of multi-omic classifier models with different combinations of lipid species and gene alterations in different stages of mPC progression predictive of prespecified treatment outcomes. For patients with mHSPC, we achieved optimal 0.751 AUC for survival prediction and 0.638 for ADT failure prediction; and in mCRPC state, we got 0.687 for prognostication and 0.727 for exceptional survival. It is not surprising that a set of different species and combinations of genomic and lipid molecules may affect clinical outcomes in different stages of cancer progression. In fact, the phylogenetic evolutionary tree of most cancers appears to be characterized by early mutations in a constrained set of driver genes and then is followed by the continuous diversification of the mutational spectrum leading to increased genomic instability in later cancer stages with clonal evolutionary complexities after cancer treatments that result in treatment-induced lineage plasticity (TILP). Evaluating the composite outcomes of host-treatment interactions along with clonal diversification is critical for enhancing precision clinical medicine applications as the therapeutic landscapes for treating metastatic stage disease across all tumor types have increased considerably in recent years with the approval of several novel drugs. The Pan Cancer Analysis of the Whole Genome (PCAWG) Consortium and The Cancer Genome Atlas (TCGA) reconstructed the life history and evolution of driver mutational sequences in 2,778 cancers from 38 tumor types,22 but efforts in these large tissue-based data sets did not account for the impact of treatments and stage progression on tumor biology after diagnosis. Additionally, it is also not possible to obtain serial tissue biopsies in metastatic stages to identify multi-omic alterations, which potentially is possible to characterize using blood of other easily obtained biofluid specimens.
The limitation of using biofluid and blood samples, however, is that creating robust multi-omic clinical models with many noisy features is challenging. Classic biostatistical methods use manual variable selection and simple models, but struggle with numerous multi-omics features. Data-driven machine learning models possibly offer a more promised solution by incorporating many features into a best-fit classifier for clinical outcome prediction. In this study, we evaluated this using multiple mainstream machine learning models, and observed logistic regression with elastic net regularization to be the most satisfactory approach with a balance between model complexity and overfitting in this data set, which had a limited number of patients and a large number of features. During the feature processing and model building stage, we combined all features for each patient into a comprehensive vector, serving as the input for the models. To address the challenge of relying solely on data-driven machine learning models, which could result in overfitting and unstable models, we leveraged human knowledge from previous research findings to construct candidate feature groups and enhance the stability of the models, while increasing the total number of features than have been previously included. Merging domain knowledge at the feature selection stage addresses some of the challenges posed by a limited number of samples and a large number of features. The issue of noisy data still persists and can negatively affect the robustness of the model, especially when dealing with continuous numerical lipidomic features. To address this, we used further binarization techniques to enhance the robustness of the model and observed acceptable test accuracies for prediction of all outcomes.
Our results also compared the study methods with the limited candidate three species-level lipid features (ceramide [d18:1/24:1], sphingosine [d18:2/16:0], phosphatidylcholine [16:0/16:0]) previously reported on the basis of a linear logistic model for reaching significant associations with outcomes. Our data-driven machine learning approach incorporating a greater number of genomic and lipidomic features and guided by elastic net regularization showed better test accuracies (Data Supplement [Supplementary Results]). Finally, we were able to construct a probability model that can accommodate multiple features and using a specific feature to predict the outcome in future patients.
One limitation of our study is that the final model with optimal performances is a linear machine learning method with limited capacity, and we believe that if we had access to a larger patient data set, we can train a more powerful and robust nonlinear model, leading to better performance. Nevertheless, encompassing tumor heterogeneity into a classifier model that is based on measuring biomarkers from different pathways for prediction of clinical outcomes is likely more robust than a single-molecule, single-pathway–based classifier. In this study, our attempt was to perform this deterministic evaluation of multi-omic classifiers. Independent validation of this preliminary model approach, however, will need to be tested in future patients and larger cohorts.
Hui-Ming Lin
Patents, Royalties, Other Intellectual Property: Provisional patent for PCPro
Arun A. Azad
Honoraria: Janssen, Astellas Pharma, Novartis, Tolmar, Amgen, Pfizer, Bayer, Telix Pharmaceuticals, Bristol Myers Squibb, Merck Serono, AstraZeneca, Sanofi, Ipsen, Merck Sharp & Dohme, Noxopharm, Aculeus Therapeutics
Consulting or Advisory Role: Astellas Pharma, Novartis, Janssen, Sanofi, AstraZeneca, Pfizer, Bristol Myers Squibb, Tolmar, Telix Pharmaceuticals, Merck Sharp & Dohme, Bayer, Ipsen, Merck Serono, Amgen, Noxopharm, Aculeus Therapeutics
Speakers' Bureau: Astellas Pharma, Novartis, Amgen, Bayer, Janssen, Ipsen, Bristol Myers Squibb, Merck Serono
Research Funding: Astellas Pharma, Merck Serono (Inst), Novartis (Inst), Pfizer (Inst), Bristol Myers Squibb (Inst), Sanofi (Inst), AstraZeneca (Inst), GlaxoSmithKline (Inst), Aptevo Therapeutics (Inst), MedImmune (Inst), Bionomics (Inst), Synthorx (Inst), AstraZeneca, Astellas Pharma (Inst), Ipsen (Inst), Merck Serono, Lilly (Inst), Gilead Sciences (Inst), Janssen (Inst), Exelixis (Inst), MSD (Inst), Hinova Pharmaceuticals (Inst)
Travel, Accommodations, Expenses: Astellas Pharma, Sanofi, Merck Serono, Amgen, Janssen, Tolmar, Pfizer
Edmond M. Kwan
Honoraria: Janssen, Ipsen, Astellas Pharma, Research Review
Consulting or Advisory Role: Astellas Pharma, Janssen, Ipsen
Research Funding: Astellas Pharma (Inst), AstraZeneca (Inst)
Travel, Accommodations, Expenses: Astellas Pharma, Pfizer, Ipsen, Roche
Lisa Horvath
Employment: Connected Medical Solutions
Leadership: Connected Medical Solutions
Stock and Other Ownership Interests: Connected Medical Solutions, Imagion Biosystems
Honoraria: Janssen, Astellas Pharma
Consulting or Advisory Role: Imagion Biosystems, Bayer
Research Funding: Astellas Pharma
Patents, Royalties, Other Intellectual Property: Provisional patent Australian No. 2022902527 Prognostic Markers (plasma lipid prognostic signature in metastatic prostate cancer). Inventors: Horvath L, Meikle P, Scheinberg S, Lin HM, Sullivan D The patent is owned by the Chris O'Brien Lifehouse (my institition) (Inst)
Travel, Accommodations, Expenses: Astellas Pharma, Janssen-Cilag, Pfizer, AstraZeneca, MSD Oncology
Pan Du
Employment: Predicine
Leadership: Predicine
Stock and Other Ownership Interests: Predicine
Shidong Jia
Employment: Predicine, Genentech
Leadership: Predicine
Stock and Other Ownership Interests: Predicine
Patents, Royalties, Other Intellectual Property: Liquid biopsy in cancer detection, therapy monitoring, MRD and early cancer detection
Manish Kohli
Employment: nference
Honoraria: Advanced Accelerator Applications
Consulting or Advisory Role: Bristol Myers Squibb/Celgene, Genapsys, Tempus
Patents, Royalties, Other Intellectual Property: Patent number: 10982286. Algorithmic approach for determining the plasma genome abnormality PGA and the urine genome abnormality UGA scores based on cell free cfDNA copy number variations in plasma and urine
Travel, Accommodations, Expenses: Celgene
No other potential conflicts of interest were reported.
SUPPORT
Supported in part by the Computational Oncology Research Initiative at the Huntsman Cancer Institute, Salt Lake City, UT.
*M.K. and R.M.K. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Shikai Fang, Shandian Zhe, Robert M. Kirby, Manish Kohli
Provision of study materials or patients: Arun A. Azad, Shidong Jia
Collection and assembly of data: Shikai Fang, Arun A. Azad, Heidi Fettke, Lisa Horvath, Blossom Mak, Edmond Kwan, Manish Kohli
Data analysis and interpretation: Shikai Fang, Shandian Zhe, Hui-Ming Lin, Tiantian Zheng, Pan Du, Shidong Jia, Robert M. Kirby, Manish Kohli
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/cci/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Hui-Ming Lin
Patents, Royalties, Other Intellectual Property: Provisional patent for PCPro
Arun A. Azad
Honoraria: Janssen, Astellas Pharma, Novartis, Tolmar, Amgen, Pfizer, Bayer, Telix Pharmaceuticals, Bristol Myers Squibb, Merck Serono, AstraZeneca, Sanofi, Ipsen, Merck Sharp & Dohme, Noxopharm, Aculeus Therapeutics
Consulting or Advisory Role: Astellas Pharma, Novartis, Janssen, Sanofi, AstraZeneca, Pfizer, Bristol Myers Squibb, Tolmar, Telix Pharmaceuticals, Merck Sharp & Dohme, Bayer, Ipsen, Merck Serono, Amgen, Noxopharm, Aculeus Therapeutics
Speakers' Bureau: Astellas Pharma, Novartis, Amgen, Bayer, Janssen, Ipsen, Bristol Myers Squibb, Merck Serono
Research Funding: Astellas Pharma, Merck Serono (Inst), Novartis (Inst), Pfizer (Inst), Bristol Myers Squibb (Inst), Sanofi (Inst), AstraZeneca (Inst), GlaxoSmithKline (Inst), Aptevo Therapeutics (Inst), MedImmune (Inst), Bionomics (Inst), Synthorx (Inst), AstraZeneca, Astellas Pharma (Inst), Ipsen (Inst), Merck Serono, Lilly (Inst), Gilead Sciences (Inst), Janssen (Inst), Exelixis (Inst), MSD (Inst), Hinova Pharmaceuticals (Inst)
Travel, Accommodations, Expenses: Astellas Pharma, Sanofi, Merck Serono, Amgen, Janssen, Tolmar, Pfizer
Edmond M. Kwan
Honoraria: Janssen, Ipsen, Astellas Pharma, Research Review
Consulting or Advisory Role: Astellas Pharma, Janssen, Ipsen
Research Funding: Astellas Pharma (Inst), AstraZeneca (Inst)
Travel, Accommodations, Expenses: Astellas Pharma, Pfizer, Ipsen, Roche
Lisa Horvath
Employment: Connected Medical Solutions
Leadership: Connected Medical Solutions
Stock and Other Ownership Interests: Connected Medical Solutions, Imagion Biosystems
Honoraria: Janssen, Astellas Pharma
Consulting or Advisory Role: Imagion Biosystems, Bayer
Research Funding: Astellas Pharma
Patents, Royalties, Other Intellectual Property: Provisional patent Australian No. 2022902527 Prognostic Markers (plasma lipid prognostic signature in metastatic prostate cancer). Inventors: Horvath L, Meikle P, Scheinberg S, Lin HM, Sullivan D The patent is owned by the Chris O'Brien Lifehouse (my institition) (Inst)
Travel, Accommodations, Expenses: Astellas Pharma, Janssen-Cilag, Pfizer, AstraZeneca, MSD Oncology
Pan Du
Employment: Predicine
Leadership: Predicine
Stock and Other Ownership Interests: Predicine
Shidong Jia
Employment: Predicine, Genentech
Leadership: Predicine
Stock and Other Ownership Interests: Predicine
Patents, Royalties, Other Intellectual Property: Liquid biopsy in cancer detection, therapy monitoring, MRD and early cancer detection
Manish Kohli
Employment: nference
Honoraria: Advanced Accelerator Applications
Consulting or Advisory Role: Bristol Myers Squibb/Celgene, Genapsys, Tempus
Patents, Royalties, Other Intellectual Property: Patent number: 10982286. Algorithmic approach for determining the plasma genome abnormality PGA and the urine genome abnormality UGA scores based on cell free cfDNA copy number variations in plasma and urine
Travel, Accommodations, Expenses: Celgene
No other potential conflicts of interest were reported.
REFERENCES
- 1.Hanahan D, Weinberg RA: Hallmarks of cancer: The next generation. Cell 144:646-674, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Martincorena I, Campbell PJ: Somatic mutation in cancer and normal cells. Science 349:1483-1489, 2015 [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A: Cancer statistics, 2016. CA Cancer J Clin 66:7-30, 2016 [DOI] [PubMed] [Google Scholar]
- 4.Fallara G, Robesti D, Nocera L, et al. : Chemotherapy and advanced androgen blockage, alone or combined, for metastatic hormone-sensitive prostate cancer a systematic review and meta-analysis. Cancer Treat Rev 110:102441, 2022 [DOI] [PubMed] [Google Scholar]
- 5.Ingrosso G, Bottero M, Becherini C, et al. : A systematic review and meta-analysis on non-metastatic castration resistant prostate cancer: The radiation oncologist's perspective. Semin Oncol 49:409-418, 2022 [DOI] [PubMed] [Google Scholar]
- 6.Kohli M, Tan W, Zheng T, et al. : Clinical and genomic insights into circulating tumor DNA-based alterations across the spectrum of metastatic hormone-sensitive and castrate-resistant prostate cancer. EBioMedicine 54:102728, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwan EM, Dai C, Fettke H, et al. : Plasma cell-free DNA profiling of PTEN-PI3K-AKT pathway aberrations in metastatic castration-resistant prostate cancer. JCO Precis Oncol 5:622-637, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fettke H, Kwan EM, Docanto MM, et al. : Combined cell-free DNA and RNA profiling of the androgen receptor: Clinical utility of a novel multianalyte liquid biopsy assay for metastatic prostate cancer. Eur Urol 78:173-180, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin HM, Huynh K, Kohli M, et al. : Aberrations in circulating ceramide levels are associated with poor clinical outcomes across localised and metastatic prostate cancer. Prostate Cancer Prostatic Dis 24:860-870, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mak B, Lin HM, Kwan EM, et al. : Combined impact of lipidomic and genetic aberrations on clinical outcomes in metastatic castration-resistant prostate cancer. BMC Med 20:112, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nava Rodrigues D, Casiraghi N, Romanel A, et al. : RB1 heterogeneity in advanced metastatic castration-resistant prostate cancer. Clin Cancer Res 25:687-697, 2019 [DOI] [PubMed] [Google Scholar]
- 12.Hamid AA, Gray KP, Shaw G, et al. : Compound genomic alterations of TP53, PTEN, and RB1 tumor suppressors in localized and metastatic prostate cancer. Eur Urol 76:89-97, 2019 [DOI] [PubMed] [Google Scholar]
- 13.Iacobucci D, Posavac SS, Kardes FR, et al. : The median split: Robust, refined, and revived. J Consumer Psychol 25:690-704, 2015 [Google Scholar]
- 14.Knüppel L, Hermsen O: Median split, k-group split, and optimality in continuous populations. AStA Adv Stat Anal 94:53-74, 2010 [Google Scholar]
- 15.Maxwell SE, Delaney HD: Bivariate median splits and spurious statistical significance. Psychol Bull 113:181-190, 1993 [Google Scholar]
- 16.Wright RE: Logistic regression. 1995 [Google Scholar]
- 17.Zou H, Hastie T: Regularization and variable selection via the elastic net. J R Stat Soc Stat Methodol 67:301-320, 2005 [Google Scholar]
- 18.Refaeilzadeh P, Tang L, Liu H: Cross-validation. Encyclopedia of database Systems 5:532-538, 2009 [Google Scholar]
- 19.Nicholls A: Confidence limits, error bars and method comparison in molecular modeling. Part 2: Comparing methods. J Comput Aided Mol Des 30:103-126, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chalkidis G, McPherson J, Beck A, et al. : Development of a machine learning model using limited features to predict 6-month mortality at treatment decision points for patients with advanced solid tumors. JCO Clin Cancer Inform 6:e2100163, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Royston P, Altman DG, Sauerbrei W: Dichotomizing continuous predictors in multiple regression: A bad idea. Stat Med 25:127-141, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Gerstung M, Jolly C, Leshchiner I, et al. : The evolutionary history of 2,658 cancers. Nature 578:122-128, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]