


Abstract
The SARS-CoV-2 Nsp8 protein is a critical component of the RNA replicase, as its N-terminal domain (NTD) anchors Nsp12, the RNA, and Nsp13. Whereas its C-terminal domain (CTD) structure is well resolved, there is an open debate regarding the conformation adopted by the NTD as it is predicted as disordered but found in a variety of complex-dependent conformations or missing from many other structures. Using NMR spectroscopy, we show that the SARS CoV-2 Nsp8 NTD features both well folded secondary structure and disordered segments. Our results suggest that while part of this domain corresponding to two long α-helices forms autonomously, the folding of other segments would require interaction with other replicase components. When isolated, the α-helix population progressively declines towards the C-termini but surprisingly binds dsRNA while preserving structural disorder.
Graphical Abstract
Graphical Abstract.

INTRODUCTION
The COVID-19 pandemic has currently (10 March 2023) affected over 675 million persons and caused over 6.8 million deaths worldwide (https://coronavirus.jhu.edu/map.html). The virus responsible for the disease, SARS-CoV-2, is a coronavirus whose unusually long (30 kB) RNA genome is replicated by a rather sophisticated RNA polymerase. This polymerase complex is composed of several non-structural proteins (Nsp) including the RNA-dependent RNA polymerase, Nsp12, as well as Nsp7 and Nsp8, which embrace the RNA to promote progressivity (1). Nsp8 serves as the platform onto which Nsp7 and Nsp12 bind, and cryo-EM studies have shown that it also contains binding regions to recruit the helicase Nsp13 within the replicase-transcription complex (RTC) (2,3).
Due to this important role, a score of cryo-EM and crystallographic studies, detailed in Supplementary Table S1, have been applied to resolve the structure of Nsp8 in combination with Nsp7 (PDBs: 2AHM (1), 6YHU (4), 6M5I (unpublished), 6WQD (5), 6XIP (5), 7JLT (6)) or Nsp7 plus Nsp12 (PDBs: 6M71 (7), 7BTF (7), 6XQB (unpublished)) and a model RNA template/primer (PDBs: 6YYT (2), 7AAP (8), 7BW4 (9), 7B3B (10), 7B3C (10), 7B3D (10), 7BV1 (11), 7BV2 (11), 7BZF (12), 7C2K(12)) and Nsp13 (PDBs: 6XEZ (3), 7CXM (13), 7CXN (13), 7CYQ (14)) among others (see Supplementary Table S1 for a complete list). In all these, the Nsp8 CTD appears well structured, adopting the same globular fold which interacts with Nsp7. Conversely, the NTD of Nsp8 shows a high degree of plasticity, with different conformations present in distinct contexts. Interestingly enough, bioinformatic analyses predict disorder in half of the Nsp8 NTD (15) and residues 1–15 and 50–84 are predicted with lower confidence by AlphaFold2 (16), which is a hallmark of disorder (17) and which may correlate with the absence of large portions of NTD residues in many crystallographic and cryo-EM structures (Supplementary Table S1). This, together with the number of distinct non-structural proteins that anchor to Nsp8 to create the replicase, suggest that some regions of Nsp8 behave like intrinsically disordered segments that fold upon complexation. Indeed, an early crystallographic study of SARS Nsp8, whose NTD is identical to SARS CoV-2 Nsp8 except for the conservative Y15→F substitution, showed that the NTD can adopt two strikingly different conformations in combination with a Nsp7 to form a hollow PCNA-like complex (1). Building on the critical observation that the Nsp8 NTD integrates Nsp12, Nsp13 and RNA (11), and that its predicted intrinsic disorder and observed conformational plasticity prevent direct observation by crystallography or cryo-EM, we sought to characterize its conformation, dynamics and dsRNA binding using solution NMR spectroscopy.
MATERIALS AND METHODS
Sample production and isotopic labeling
The gene coding for the Nsp8 NTD, whose sequence is: G0 AIASE5 FSSLP10 SYAAF15 ATAQE20 AYEQA25 VANGD30 SEVVL35 KKLKK40 SLNVA45 KSEFD50 RDAAM55 QRKLE60 KMADQ65 AMTQM70 YKQAR75 SEDKR80 AKVT84, was purchased from Genscript (New Jersey, NJ) with codons optimized for E. coli expression and subcloned in a pET28 derived vector containing in 5′ position an encoding sequence for thioredoxin followed by a hexa-histidine tag and a TEV-protease cleavage site. This construct was cloned in BL21star (DE3) cells. Afterwards, cells were grown in LB at 37°C until OD600 = 0.8, then the temperature was decreased to 25°C, and 0.5 mM IPTG was added to induce the expression overnight. Uniformly isotopically labeled samples (13C + 15N) were obtained using a protocol derived from those described by Marley et al. (18) and Sivashanmugan et al. (19). Briefly, transformed cells were grown in 2 L LB media until OD600 = 0.6- 0.8, then they were centrifuged, and the pellet resuspended in 0.5 L minimal medium containing 13C-glucose and 15NH4Cl. Cells were incubated at 37°C for 1.5 h to enhance labeling. Next, the temperature was decreased to 25°C before overnight induction with 0.5 mM IPTG.
The domain was purified by three chromatographic steps. Briefly, after lysis by sonication, the obtained supernatant was purified on a HisTrap FF crude 5 mL column (Cytiva, Marlborough, MA) and eluted using an imidazole gradient whose initial and final concentrations were 10 and 500 mM, respectively. The eluted protein fusion was cleaved overnight at room temperature with TEV protease and dialyzed to eliminate imidazole. The cleaved sample was reloaded on the same column and the flowthrough was collected and applied to a Bio-Scale™ Mini Bio-Gel®P-6 Desalting Cartridges (Bio-rad, Hercules, CA) for desalting. Finally, the sample was loaded on a 5 ml HiTrap Q HP anion exchange column (Cytiva, Marlborough, MA) at pH 8 and eluted, collecting the non-retained fraction. Homogeneity following purification was confirmed by gradient (4–20% acrylamide) SDS PAGE and NMR spectroscopy. Prior to NMR spectroscopy, the sample was transferred into ‘Buffer A’ consisting of 90% H2O mQ, 10% D2O, 50 mM NaCl, 10 mM KH2PO4, pH 6.1. The spectra were referenced using DSS as the internal chemical shift standard. Spectra were recorded at 5.0°C, except 2D 1H–15N HSQC and 3D HNCA spectra registered at 25 and 37°C. Essentially the same procedures were used to express and purify a shorter construct containing residues 1 - 64 of Nsp8 (Supp. Methods Text).
An RNA duplex with the sequence:
5′GAUGCUACGCGGUAGUA3’
3′CUACGAUGCGCCAUCAUG5’,
which is almost identical to part of the dsRNA in a recent study (14), was purchased from Integrated DNA Technologies (IDT). Its purity and the presence of duplex structure were corroborated by 1D 1H NMR spectroscopy. In particular, the observation of 1HN imine signals in the 12.3–14.5 ppm chemical shift ranges are indicative of Watson–Crick base pairing. A 510 μM stock solution of dsRNA for addition to 15N-labeled Nsp8 NTD was prepared by dissolving 51.3 nmol of dsRNA in 100 μl of buffer A. Alternatively, a 100 μM sample of dsRNA was prepared in buffer A, and titrated the unlabeled protein to monitor the interaction from the perspective of the nucleic acid. For comparison, a dsDNA with the sequence 5′GATGCTACGCGGTAGTA3 and 3′CTACGATGCGCCATCATG5’purchased from IDT was also titrated with Nsp8 in buffer A. Its purity and duplex structure were also confirmed by 1D 1H NMR.
Circular dichroism spectroscopy
To corroborate the α-helical secondary structure of Nsp8 NTD, circular dichroism (CD) spectra of Nsp8 NTD were recorded on a Jasco J810 spectropolarimeter equipped with a Peltier module for fine temperature control. The buffer reference spectrum was subtracted from the spectrum of the protein. Far UV CD spectra were recorded from 193–260 nm at a scan rate of 50 nm/min and using a bandwidth of 1 nm at 5°C. Four scans were averaged per spectrum. The CD signal at 206 nm was used to monitor the thermal denaturation of a 15 μM sample of Nsp8 NTD in buffer A employing a heating rate of 60°C/h.
NMR spectroscopy
A series of 2D 1H–1H TOCSY, 1H–1H NOESY (mixing time = 80 ms), 1H–15N HSQC, 1H–13C HSQC and 3D HNCO, HNCACO, HNCA, CBCAcoNH, CBCANH as well as 3D hNcocaNH and HncocaNH (20) spectra were recorded on a Bruker Avance Neo 800 MHz (1H) spectrometer fitted with a cryoprobe and z-gradients. These experiments provide the connectivities between the distinct spin systems. Spectral parameters are listed in Supplementary Table S2. Additional experiments for side chain assignments including HcccoNH, hCCcoNH, HCCH-TOCSY, 1H–15N HSQC-NOESY, and HBHAcoNH were also acquired. An 3D HNHA experiment was recorded to obtain 3JHNHA coupling constants. All spectra were transformed with Topspin 4.0.8 (Bruker Biospin) and were assigned by two independent operators using the aid of the program Sparky (21). These experiments were collected at 5°C, and additional HNCA and 1H–15N HSQC were collected at 25°C and 37°C to corroborate that the structure is preserved at physiological relevant temperatures.
A series of 2D 1H–15N HSQC-based experiments were recorded to determine the {1H}–15N NOE, R1 and R1ρ relaxation rates in order to assess dynamics on the ps/ns timescale. These spectra were collected at 5 and 25°C, recorded without non-uniform sampling and were transformed without applying linear prediction. For the {1H}–15N NOE ratios, peak integrals were measured using Topspin 4.0.8 and the uncertainties were calculated as the ratio of the noise (estimated as the standard deviation of the integrals of several peakless spectral regions) times 21/2 to the peak integral measured without applying the NOE. R1 and R1ρ relaxation rates were obtained by using Bruker's Dynamics Center program (version 2.6.3). For R1, uncertainties reported here are those obtained from the least squares fit. Transverse relaxation rates (R2) were calculated from R1 and R1ρ and the uncertainties in R2 were obtained by error propagation. The NMR assignments, {1H}–15N NOE ratios, 3JHNHA coupling constants and R1 and R1ρ relaxation rates have been deposited in the Biological Magnetic Resonance Data Bank under accession code 50788.
NMR structure determination
Conformational chemical shifts were calculated using the statistical coil values calculated for this sequence, pH and temperature using the parameters reported by Poulsen and coworkers (22,23). For the structure calculation of the Nsp8 NTD a 2D 1H–1H NOESY spectrum was recorded (mixing time 80 ms). Experimental NOE-derived distance constraints and Talos+-derived dihedral constraints (24) were input to the program Cyana v3.98.13 (25) for the standard 7-cycle iterative process plus a final annealing step using the list of restraints obtained in the last cycle. One hundred structures were generated. Constraint files included 630 upper distance restraints for protons (360 intra, 131 medium-range, 122 long-range), complemented with 163 φ and ψ restrictions. The 20 conformers with the lowest target function values were selected for further refinement and energy minimization with the Amber9 software (26) using the Gibbs-Boltzmann continuum solvation model. The structural ensembles were visualized and examined using MolMol (27) and Pymol (28). PROCHECK-NMR (29) version 3.4.4 was used to analyze the quality of the refined structures, which have been deposited in the Protein Data Bank under the accession number 7YWR. Statistics of the calculation are summarized in Supplementary Table S3.
Interaction with nucleic acid
The interaction between Nsp8 and dsRNA was followed by adding small aliquots of dsRNA to the 15N-labeled protein and following signal changes in 2D 1H–15N HSQC spectra. In addition, a 100 μM solution of dsRNA was titrated with unlabeled Nsp8 NTD and their union was detected by changes in the imine region of 1D 1H spectra (imine 1H) and phosphodiester signals of 1D 31P NMR spectra using a Bruker 600 MHz Avance Neo spectrometer equipped with a Z-gradients and a cryoprobe with channels for 1H, 13C, 15N and 31P.
Molecular complex modeling
Molecular docking of Nsp8 with an RNA duplex was done using HADDOCK (30) as implemented in the HADDOCK 2.4 webserver (31,32), using the lowest-energy Nsp8 structure from the NMR ensemble as described above. Active residues were defined from titrations of Nsp8 with dsRNA, and default parameters were used to drive the docking, except the number of structures during all three stages it0, it1, and itw, were increased to 10 000, 400 and 400, respectively, and random removal of ambiguous restraints was turned off. The two top-scored clusters were retained as plausible Nsp8 NTD-RNA complexes.
RESULTS
SARS CoV-2 nsp8 NTD production and purification
Following purification by Ni++-NTA affinity, His tag cleavage and polishing with anion exchange chromatography, sample homogeneity was confirmed by SDS PAGE (Supplementary Figure S1). The average yields were 5.0 mg/l from LB broth and 2.8 mg/L from minimal media for labeled samples.
Nsp8 NTD solution NMR structure
The 2D 1H–15N HSQC spectrum of the Nsp8 NTD shows the excellent dispersion that is a hallmark of well-folded proteins for most signals (Figure 1A). Closer inspection reveals a subset of narrower, more intense resonances corresponding to residues at the N- and C-termini. Spectral analysis led to the assignment of over 95% of the backbone 13CO, 13Cα, 1Hα, 15N, with 1H–15N assignments for all non-proline residues except Q56. The 13Cα conformational chemical shifts (Δδ) reveal two highly populated α-helical structures spanning residues 11–27 and 32–50 (Figure 1B). Following residue 50, the helix continues but its population gradually declines, with the last stretch of residues (74–84) being chiefly disordered. The first ten residues are also mostly disordered according to the 13Cα conformational shifts. The position and high population of the two α-helical segments was corroborated by 1Hα and 13CO and conformational chemical shifts and 1HN–1Hα coupling constants (3JHNHA) (Supplementary Figure S2), and further corroborated by 13Cα conformational shifts using a distinct reference data set (Supplementary Figure S3). This secondary structure, observed at 5°C and quasi physiological pH, is maintained at 25°C and 37°C according to 13Cα conformational shifts (Supplementary Figure S4). A substantial population of α-helical structure and a thermal unfolding midpoint of 44°C were revealed by CD spectroscopy (Supplementary Figure S5). Regarding the residues linking the two long α-helices, N28-G29-D30-S31, their 13Cα,13Cβ, 13CO, 1HN and 1Hα chemical shifts suggest that they adopt a type II′ tight turn (33,34). Using a natural abundance sample to record a 2D 1H-1H NOESY, we obtained 630 distance restraints from ca. 950 carefully picked cross-peaks. This information was input along with 163 protein dihedral angles obtained from TALOS + to CYANA to determine the solution NMR structure of the Nsp8 NTD. Figure 1C shows the refined ensemble of the resulting 20 lowest-energy conformers, which recapitulates all the features extracted from the secondary structure analyses. In particular, whereas the two α-helices 11–37 and 32–50 pack with the same conformations as seen in the replicase-transcription complex, there are distinctive aspects in the non-complexed state, namely, a 310 helical turn embedded within the disordered N-terminal 1–10 stretch (residues 5–8), and the gradual loss in secondary structure from residue 50 towards the C-terminus. AlphaFold2 was used to predict the NTD Nsp8 structure and cross-validate our NMR results (Supplementary Figure 6). Although the helix-turn-helix motif up to ca. residue 55 was correctly predicted, it is intriguing that residues 55–84 were also modeled with low confidence as an extension of the second α-helix as opposed to our NMR data that clearly indicate disorder. Remarkably, the short N-terminal helix is also predicted by AlphaFold2, but is mispositioned at residues I2-F6, instead of F6-S8 as detected by NMR. The disordered behavior of this segment seems to be maintained in full-length Nsp8 (Supplementary Figure S7), which is not required for the autonomous folding of residues 1–55 (Supplementary Figure S8).
Figure 1.

Isolated Nsp8 NTD adopts two central α-helices bordered by flexible termini. (A) 2D 1H–15N HSQC spectrum of Nsp8 NTD recorded at 5.0°C, in 50 mM NaCl, 10 mM KH2PO4, pH 6.1. Assigned backbone 1H–15N resonances are labeled. A set of sharper, more intense peaks with less 1H dispersion corresponding to N- and C-terminal residues are found amid a larger set of resonances with the wide range of 1HN chemical shifts characteristic of well folded proteins. (B) 13Cα (blue bars) conformational chemical shifts (Δδ) reveal two highly populated α-helical structures spanning residues 11–27 and 32–50 (gray shading), as judged by the Δδ 13Cα value expected for 100% α-helix. Following residue 50, the helix continues, but its population declines in intensity (light gray shading). The thin line at 3.1 ppm marks the average Δδ13Cα value seen for fully populated α-helices. (C) Superposition of the family of 20 conformers of the Nsp8 NTD monomer with side chains colored black and mainchains colored blue. No side chains are shown for the terminal disordered zones. (D) The heteronuclear {1H}–15N NOE. High ratios approaching the value of 0.86 (thin line) expected for full rigidity on ns/ps timescales are observed for the two main helical regions. (E) Longitudinal (R1) and (F) transverse (R2) relaxation rates gauge μs/ms timescale dynamics. Their relatively low (R1) and high (R2) values reflect stiffness.
Nsp8 NTD solution dynamics
The per-residue {1H}–15N NOE ratios, which are sensitive to dynamics on fast ps/ns timescales, are plotted in Figure 1D. The residues composing the two α-helical segments show relatively high values averaging about 0.75. This indicates considerable stiffness, but is significantly below the theoretical ratio of 0.86 expected for complete rigidity, which is often observed in well-folded proteins. Ratios are lower in the first ten residues and decrease progressively beyond residue 60, reflecting increased mobility. The values for the residues 60–80 are in the neighborhood of 0.5, meaning that this segment is moderately mobile but significantly more rigid than a statistical coil which typically has values close to zero or even negative (35).
In line with the {1H}–15N NOE analyses, residues 11–55 also show reduced longitudinal (R1) and elevated transverse (R2) relaxation rates, reflecting dampened mobility due to slowed reorientational motions of the 1H–15N bond vectors as expected for folded regions, and the opposite trends for residues 56–84 in agreement with their disordered nature (Figure 1E and F). For this relatively rigid region including the two helices, a correlation time (τc) of 6.24 ns can be estimated, which corresponds to an approximate molecular weight of 10.4 kDa in solution, assuming the protein behaves as an isotropic sphere (36,37). These results, which indicate that the Nsp8 NTD behaves as a monomer under the conditions of this study, were further corroborated by equilibrium sedimentation (Supplementary Figure S9).
Nsp8 NTD can bind an RNA duplex
Having established that the Nsp8 NTD autonomously folds into a helix-turn-helix motif followed by a disordered segment, the latter unseen in the complexed state, we next wondered whether this species retains the ability to interact with RNA when isolated from the rest of the replicase complex. The interaction of the Nsp8 NTD with a model dsRNA duplex was studied by NMR affording insight into the binding stoichiometry, strength, kinetics and impact on each macromolecule's conformation. Overall, when mixed with Nsp8 NTD, RNA 1HN imine signals continue to resonate at 12.2–14.5 ppm, which means that the RNA duplex structure is not significantly affected by the protein's presence (Supplementary Figure S10). However, these RNA peaks do progressively decrease in intensity as protein is added, which indicates interaction (Supplementary Figure S10). In addition, intensity changes and chemical shift alterations are detectable in the 31P signals of the phosphodiester backbone, which corroborates protein binding (Supplementary Figure S11). Likewise, the 1H–15N HSQC spectra of Nsp8 NTD continued to show excellent chemical shift dispersion upon adding dsRNA (Figure 2A). While this is consistent with the protein's 310helix-α-helix-turn-α-helix structure remaining folded in the presence of dsRNA, residues 55–84 stay disordered yet undergo a moderate stiffening. Many Nsp8 NTD 1H–15N signals do show significant intensity changes or chemical shift differences upon addition of dsRNA (Figure 2A) indicating that binding occurs. In particular, residues 12–58 and 60–77 show different behaviors upon titration with dsRNA (Figure 2A, B). The former show loss of signal intensity, which is indicative of intermediate exchange, while the latter show progressive chemical shift changes, a hallmark of fast exchange. These two distinct dsRNA binding regimes correspond with the folded and less ordered segments of the Nsp8 NTD. Whereas the disappearance of signals undergoing intermediate exchange makes it difficult to quantify their binding, based on residues featuring fast exchange, a KD in the range of 10–20 μM can be estimated.
Figure 2.

Nsp8 NTD binds a dsRNA duplex. (A) Region of 2D 1H–15N HSQC spectra recorded at 5°C of 100 μM 15N-labeled Nsp8 NTD with 0 (blue contours), 2.2 μM (green), 6.6 μM (gold) and 10.9 μM (red) of unlabeled dsRNA. Residues with large 1HN and 15N chemical shifts displacements are labeled red and residues whose signal intensity drops significantly are labeled magenta. (B) Per residue 1H·15N CPS values for 15N-Nsp8 NTD in the presence of 10.9 μM dsRNA at 5°C. The solid line represents the mean CSP value and the dotted line is the mean plus one standard deviation. The main chain 1H–15N signals with significantly high CSP values (> μ + σ) are E60, M67, R75, S76, E77, K79. The side chain 1H15Nϵ signal of Arg51 (not shown) also shows a high CSP value. This is indicative of binding in the fast regime. The signals of residues Y12, K36, L38, K39, L42, K46, D50, R51, D52, A53, A54, M55 and K58 had weakened to the point that they are undetectable at this RNA concentration and this is evidence for slow/intermediate regime binding. The approximate position of these residues is marked with magenta asterisks. (C) Representative structures from the two top-scored HADDOCK clusters calculated to model the Nsp8 NTD dsRNA complex. Each cluster is composed of five structures. Interacting residues in the protein were defined according to the titration analysis (magenta for residues undergoing intermediate exchange and red for residues undergoing fast exchange), while for the RNA the entire molecule was considered active for the interaction. The Nsp8 NTDs are shown in blue and purple and dsRNA is represented in orange and green/blue for the backbone and bases, respectively.
Considering the identity of the residues whose chemical shifts are most affected by dsRNA binding, an atomistic model for the Nsp8 NTD – dsRNA complex was prepared using HADDOCK and is shown in Figure 2C. Interestingly, facing residues from the long 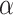 -helix and the disordered segment are seen to interact with the major groove and phosphodiester backbone to capture the dsRNA.
-helix and the disordered segment are seen to interact with the major groove and phosphodiester backbone to capture the dsRNA.
DISCUSSION
Nsp8 is an essential anchoring scaffold for the replication-transcription complex (RTC) of the SARS-CoV-2, whose NTD binds with the other critical components, particularly the RNA-dependent RNA polymerase Nsp12, the helicase Nsp13 and RNA. In these assemblies, the Nsp8 NTD adopts well-folded structures. This contrasts with bioinformatics analyses that predict this domain to be largely disordered (15). In this regard, in other many crystallographic or cryo-EM structures, large portions of the NTD are missing (Supplementary Table S1) or adopt dramatically distinct conformations (Figure 3), which evinces structural plasticity and correlates well with a low confidence of the AF2 structure predictions for the C-terminus of Nsp8 NTD. In particular, the Nsp8 NTD of SARS-CoV, which is almost identical to SARS-CoV-2 Nsp8 NTD, adopts two strikingly different conformations when eight Nsp8 monomers combine with eight Nsp7 monomers to form a hexadecameric ring (1) (Figure 3A). In one conformation, most of the Nsp8 NTD is absent whereas in the other, three α-helices are adopted (Figure 3A). By contrast, when combined with Nsp7, Nsp12 and RNA (with or without Nsp9 and Nsp13), the NTD of Nsp8 forms two α-helices which extend out like arms from the body of the RTC to embrace the RNA (2,3,7–13) (Figure 3B). Two Nsp8 subunits are present per RTC, but they are not in contact. There is variation in the helix length, disorder and the position of the connecting turn among the reported structures. So what is the conformation of Nsp8’s NTD in isolation and what can it tell us about RTC assembly? One possibility is a concomitant structure formation upon binding event wherein an isolated, disordered Nsp8 NTD folds upon complexation. In line with this scenario, the Nsp8 forms distinct dimers with Nsp7 wherein the Nsp8 NTD folds in one type of dimer (1) (Figure 3A). In this structure and larger complexes, Nsp7 interacts with the CTD of Nsp8 (2,3,7–13) (Figure 3B), which might suggest that the Nsp8 CTD then coaxes its NTD to fold. By contrast, here we show by NMR spectroscopy that the Nsp8 NTD, in the absence of other subunits and its CTD, exists as a 310-helix-α-helix-turn-α-helix structure followed by an intrinsically disordered C-terminus that intriguingly binds dsRNA in its disordered state. The two rather long, rigid α-helices spanning residues 11–50 might fold first and thus provide a foundation for anchoring Nsp7, Nsp12, Nsp13 and RNA to build up the RTC (Figure 3C). Within this working model, the 51–84 residue segment could become fully helical as the complex grows (Figure 3B), while the two monomers would reorganize to accommodate themselves within the large complex.
Figure 3.

Conformational plasticity of the Nsp8 NTD. (A) Two strikingly different conformations are adopted in Nsp8 NTD dimers in the hexadecameric ring adopted by combination of Nsp7 and Nsp8 dimers in SARS-CoV (PDB 2AHM) (1). The zoomed box illustrates that one of the two monomers in the Nsp8 NTD dimers contains three helices, while in the second monomer part of the NTD is missing (see Supporting Table S1) but the remaining C-terminal part of the NTD preserves the helix. (B) Two different structures of the RTC of SARS-CoV-2. The NTD of Nsp8 harbors interaction sites to bind RNA, Nsp12 (left, PDB 6YYT (2) and Nsp13 (right, PDB 6XEZ (3)), and adopts a helix-loop-helix conformation (zoomed regions). The bottom box illustrates an NMR-based 2D representation of the NTD in the absence of RNA, Nsp12 and Nsp13. Based on data shown in Figure 1, a small yet rigid helix-loop-helix core forms autonomously, and is flanked by two N- and C-terminal segments. Whereas the N-terminal stretch is fully disordered, the C-terminal part possesses both nascent helical structure and a disordered segment. Nsp12 and Nsp13 bind to these C-terminal regions of the NTD, which become fully helical upon complexation. (C) When isolated from other SARS CoV-2 replicase subunits, the Nsp8 NTD spontaneously folds into an autonomous helical structure that retains some disorder beyond residue 55(Left). This disordered region can bind an RNA duplex without folding on a fast submillisecond NMR timescale, representing one of the few examples to date of stable interactions between protein disordered regions and RNA duplexes, whereas the helical section binds and interacts on slow/intermediate timescales (>ms). AlphaFold2 incorrectly predicts this RNA binding region as fully helical and not disordered (right).
One puzzling detail is the oligomerization state of the Nsp8 NTD of SARS CoV, which could contribute to the formation of a hexadecameric ring structure (1) (Figure 3A). However, in recently reported assemblies of the RTC from SARS CoV-2, there are two Nsp8 NTD that do not contact each other (2,3,7–13) (Figure 3B) but bind to opposite sides of the same dsRNA and to Nsp7 and Nsp12 Our NMR data are in line with the Nsp8 NTD subunits acting independently to bind dsRNA. This is broadly consistent with a recent study modeling SARS-CoV-2 replicase assembly (38). Furthermore, the ability of the Nsp8 NTD to autonomously fold and readily bind dsRNA also suggests that it may not just nucleate the formation of the protein components of the replicase, but also be involved in accommodating nascent dsRNA.
The NMR data generated in this study represent valuable tools to test these scenarios by following changes in the assigned 1H–15N HSQC spectrum of Nsp8 NTD upon titration with other RTC components. As Nsp8 is the foundation of the replicase, these assignments shall also be key for identifying drug-like inhibitors for developing improved therapeutics by blocking Nsp8 NTD associations with other replicase subunits. Beyond the replicase, these data can be used to map interactions with Orf6, a known partner (39), which plays a key role in blocking the interferon response. Finally, the interaction between Nsp8 NTD and dsRNA represents one of few examples of interaction between a duplex RNA and an intrinsically disordered protein region.
Supplementary Material
ACKNOWLEDGEMENTS
MM acknowledges the Ramón y Cajal program (grant RYC2019-026574-I) of the Spanish AEI-Ministry of Science and Innovation MCIN/AEI/10.13039/501100011033 and ‘ESF Investing in your future’, and the Junior Leader Program (LCR/BQ/PR19/11700003) from “La Caixa" Foundation (ID 100010434).
Contributor Information
Miguel Á Treviño, “Blas Cabrera” Institute for Physical Chemistry, Spanish National Research Council, Serrano 119, Madrid 28006, Spain.
David Pantoja-Uceda, “Blas Cabrera” Institute for Physical Chemistry, Spanish National Research Council, Serrano 119, Madrid 28006, Spain.
Douglas V Laurents, “Blas Cabrera” Institute for Physical Chemistry, Spanish National Research Council, Serrano 119, Madrid 28006, Spain.
Miguel Mompeán, “Blas Cabrera” Institute for Physical Chemistry, Spanish National Research Council, Serrano 119, Madrid 28006, Spain.
DATA AVAILABILITY
The refined NMR ensemble for the SARS-CoV-2 NTD NSP8 monomer has been deposited in the Protein Data Bank (https://www.rcsb.org) under accession code 7YWR.
Chemical shifts, 15N relaxation data and 3JHNHA coupling constants are deposited in the Biological Magnetic Resonance Databank (https://bmrb.io) under accession code 51325. Molecular models derived by HADDOCK using NMR information are available as supplementary material.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Carlos III Institute of Health and the Spanish Ministry of Science and Innovation [COV20/00764 to M.M. and D.V.L.]; MCIN/AEI/10.13039/501100011033 [PID2020-113907RA-I00 to M.M.]; cofunded by the European Union (ERC) [101042403-BiFOLDOME]; Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council; Neither the European Union nor the granting authority can be held responsible for them; NMR experiments were performed in the ‘Manuel Rico’ NMR Laboratory (LMR) of the Spanish National Research Council (CSIC), a node of the Spanish Large-Scale National Facility for Biomolecular NMR (ICTS R-LRB). Funding for open access charge: Ministerio de Ciencia e InnovaciónInstituto de Salud Carlos III [COV20/00764].
Conflict of interest statement. None declared.
REFERENCES
- 1. Zhai Y., Sun F., Li X., Pang H., Xu X., Bartlam M., Rao Z.. Insights into SARS-CoV transcription and replication from the structure of the Nsp7-Nsp8 hexadecamer. Nat. Struct. Mol. Biol. 2005; 12:980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hillen H.S., Kokic G., Farnung L., Dienemann C., Tegunov D., Cramer P.. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020; 584:154–156. [DOI] [PubMed] [Google Scholar]
- 3. Chen J., Malone B., Llewellyn E., Grasso M., Shelton P.M.M., Olinares P.D.B., Maruthi K., Eng E.T., Vatandaslar H., Chait B.T.et al.. Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell. 2020; 182:1560–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Konkolova E., Klima M., Nencka R., Boura E.. Structural analysis of the putative SARS-CoV-2 primase complex. J. Struct. Biol. 2020; 211:107548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilamowski M., Hammel M., Leite W., Zhang Q., Kim Y., Weiss K.L., Jedrzejczak R., Rosenberg D.J., Fan Y., Wower J.et al.. Transient and stabilized complexes of Nsp7, Nsp8, and Nsp12 in SARS-CoV-2 replication. Biophys. J. 2021; 120:3152–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biswal M., Diggs S., Xu D., Khudaverdyan N., Lu J., Fang J., Blaha G., Hai R., Song J.. Two conserved oligomer interfaces of NSP7 and NSP8 underpin the dynamic assembly of SARS-CoV-2 RdRP. Nucleic Acids Res. 2021; 49:5956–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L.et al.. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020; 368:779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Naydenova K., Muir K.W., Wu L.F., Zhang Z., Coscia F., Peet M.J., Castro-Hartmann P., Qian P., Sader K., Dent K.et al.. J. Structure ofthe SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2021946118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peng Q., Peng R., Yuan B., Zhao J., Wang M., Wang X., Wang Q., Sun Y., Fan Z., Qi J.et al.. Structural and biochemical characterization of the Nsp12-Nsp7-Nsp8 core polymerase complex from SARS-CoV-2. Cell Rep. 2020; 31:107774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kokic G., Hillen H.S., Tegunov D., Dienemann C., Seitz F., Schmitzova J., Farnung L., Siewert A., Höbartner C., Cramer P.. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021; 12:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yin W., Mao C., Luan X., Shen D.-D., Shen Q., Su H., Wang X., Zhou F., Zhao W., Gao M.et al.. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020; 368:1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Q., Wu J., Wang H., Gao Y., Liu Q., Mu A., Ji W., Yan L., Zhu Y., Zhu C.et al.. Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell. 2020; 182:417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yan L., Zhang Y., Ge J., Zheng L., Gao Y., Wang T., Jia Z., Wang H., Huang Y., Li M.et al.. Architecture of a SARS-CoV-2 mini replication and transcription complex. Nat. Commun. 2020; 11:5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yan L., Ge J., Zheng L., Zhang L., Gao Y., Wang T., Huang Y., Yang Y., Gao S., Li M.et al.. Cryo-EM structure of an extended SARS-CoV-2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell. 2021; 184:184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giri R., Ghardwaj T., Shegane M., Gehi B.R., Kumar P., Gadhave K., Oldfield C.J., Uversky V.N.. Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronavirus. Cell. Mol. Life Sci. 2021; 78:1655–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ruff K.M., Pappu R.V.. AlphaFold and implications for intrinsically disordered proteins. J. Mol. Biol. 2021; 433:167208. [DOI] [PubMed] [Google Scholar]
- 18. Marley J., Lu M., Bracken C.A.. Method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR. 2001; 20:71–75. [DOI] [PubMed] [Google Scholar]
- 19. Sivashanmugam A., Murray V., Cui C., Zhang Y., Wang J., Li Q.. Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Sci. Publ. Protein Soc. 2009; 18:936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pantoja-Uceda D., Santoro J.. Aliasing in reduced dimensionality NMR spectra: (3,2)D HNHA and (4,2)D HN(COCA)NH experiments as examples. J. Magn. Reson. 2009; 45:351–356. [DOI] [PubMed] [Google Scholar]
- 21. Lee W., Tonelli M., Markley J.L.. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinforma. Oxf. Engl. 2015; 31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen Y., Delaglio F., Cornilescu G., Bax A.. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009; 44:213–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Güntert P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004; 278:353–378. [DOI] [PubMed] [Google Scholar]
- 24. Case D., Cheatham T.E. 3rd, Darden T., Gohlke H., Luo R., Merz K.J., Onufriev A., Simmerling C., Wang B., Woodsk R.J.. The Amber biomolecular simulation programs. J. Comput. Chem. 2005; 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koradi R., Billeter M., Wüthrich K.. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996; 14:51–55. [DOI] [PubMed] [Google Scholar]
- 26. Schrodinger L.L.C. 2015; The PyMOL Molecular Graphics System. Version 1.8.
- 27. Laskowski R.A., Rullmannn J.A., MacArthur M.W., Kaptein R., Thornton J.M.. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996; 8:477–486. [DOI] [PubMed] [Google Scholar]
- 28. Dominguez C., Boelens R.M.J.J., Bonvin A.M.J.J.. HADDOCK: a protein-protein docking approach based on biochemical and/or biophysical information. J. Am. Chem. Soc. 2003; 125:1731–1737. [DOI] [PubMed] [Google Scholar]
- 29. Kjaergaard M., Brander S., Poulsen F.M.. Random coil chemical shift for intrinsically disordered proteins: effects of temperature and pH. J. Biomol. NMR. 2011; 49:139–149. [DOI] [PubMed] [Google Scholar]
- 30. Kjaergaard M., Poulsen F.M.. Sequence correction of random coil chemical shifts: correlation between neighbor correction factors and changes in the Ramachandran distribution. J. Biomol. NMR. 2011; 50:157–165. [DOI] [PubMed] [Google Scholar]
- 31. van Zundert G.C.P., Rodrigues J.P.G.L.M., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S.J., van Dijk M., de Vries S.J., Bonvin A.M.J.J.. The HADDOCK2.2 webserver: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016; 428:720–725. [DOI] [PubMed] [Google Scholar]
- 32. Honorato R.V., Koukos P.I., Jimenez-Garcia B., Tsaregorodtsev A., Verlato M., Giachetti A., Rosato A., Bonvin A.M.J.J. Structural biology in the clouds: the WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021; 8:729513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang C.-C., Lai W.-C., Chuang W.-J.. Type I and II β-turns prediction using NMR chemical shifts. J. Biomol. NMR. 2014; 59:175–184. [DOI] [PubMed] [Google Scholar]
- 34. Barbato G., Ikura M., Kay L.E., Pastor R.W., Bax A.. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry. 1992; 31:5269–5278. [DOI] [PubMed] [Google Scholar]
- 35. Farrow N.A., Zhang O., Forman-Kay J.D., Kay L.E.. Comparison of the backbone dynamics of a folded and an unfolded SH3 domain existing in equilibrium in aqueous buffer. Biochemistry. 1995; 34:868–878. [DOI] [PubMed] [Google Scholar]
- 36. Su X.-C., Jergic S., Ozawa K., Burns N.D., Dixon N.E., Otting G.. Measurement of dissociation constants of high-molecular weight protein-protein complexes by transferred 15N-relaxation. J. Biomol. NMR. 2007; 38:65–72. [DOI] [PubMed] [Google Scholar]
- 37. Bae S.-H., Dyson H.J., Wright P.E.. Prediction of the rotational tumbling time for proteins with disordered segments. J. Am. Chem. Soc. 2009; 131:6814–6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perry J.K., Appleby T.C., Bilello J.P., Feng J.Y., Schmitz U., Campbell E.A.. An atomistic model of the coronavirus replication-transcription complex as a hexamer assembled around nsp15. J. Biol. Chem. 2021; 297:101218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumar P., Gunalan V., Liu B., Chow V.T.K., Druce J., Birch C., Catton M., Fielding B.C., Tan Y.-J., Lal S.K.. The nonstructural protein 8 (Nsp8) of the SARS coronavirus interacts with Its ORF6 accessory protein. Virology. 2007; 366:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The refined NMR ensemble for the SARS-CoV-2 NTD NSP8 monomer has been deposited in the Protein Data Bank (https://www.rcsb.org) under accession code 7YWR.
Chemical shifts, 15N relaxation data and 3JHNHA coupling constants are deposited in the Biological Magnetic Resonance Databank (https://bmrb.io) under accession code 51325. Molecular models derived by HADDOCK using NMR information are available as supplementary material.