


Abstract
Repair of DSB induced by IR is primarily carried out by Non-Homologous End Joining (NHEJ), a pathway in which 53BP1 plays a key role. We have discovered that the EMT-inducing transcriptional repressor ZEB1 (i) interacts with 53BP1 and that this interaction occurs rapidly and is significantly amplified following exposure of cells to IR; (ii) is required for the localization of 53BP1 to a subset of double-stranded breaks, and for physiological DSB repair; (iii) co-localizes with 53BP1 at IR-induced foci (IRIF); (iv) promotes NHEJ and inhibits Homologous Recombination (HR); (v) depletion increases resection at DSBs and (vi) confers PARP inhibitor (PARPi) sensitivity on BRCA1-deficient cells. Lastly, ZEB1’s effects on repair pathway choice, resection, and PARPi sensitivity all rely on its homeodomain. In contrast to the well-characterized therapeutic resistance of high ZEB1-expressing cancer cells, the novel ZEB1-53BP1-shieldin resection axis described here exposes a therapeutic vulnerability: ZEB1 levels in BRCA1-deficient tumors may serve as a predictive biomarker of response to PARPis.
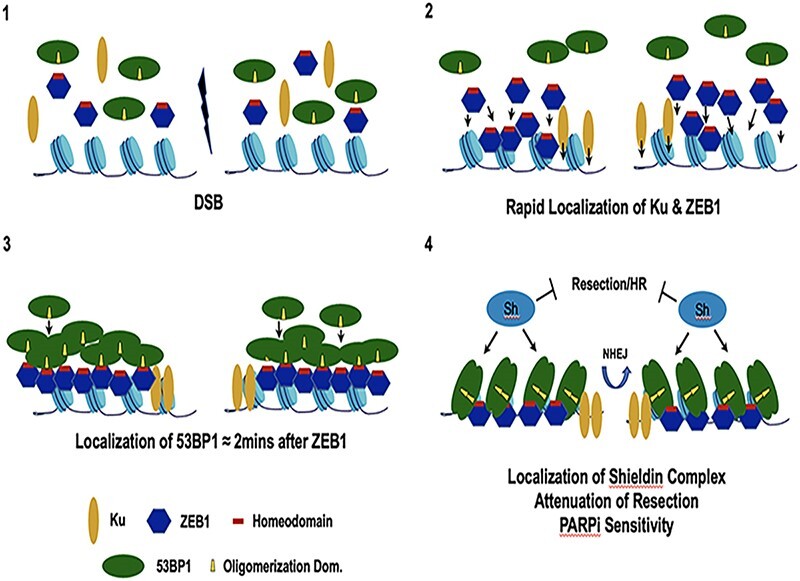
Graphical Abstract
Graphical Abstract.
INTRODUCTION
DNA double strand breaks (DSB) induced by ionizing radiation (IR), chemo-therapeutic agents, or produced as by-products of physiological processes such as replication stress, transcriptional block or aberrant somatic recombination pose an existential threat to survival (1–3). Ionizing radiation-induced DSBs are mostly repaired through-out the cell cycle by Non-Homologous End Joining (NHEJ), which is rapid, but somewhat error-prone. With the appearance of newly replicated DNA in S phase and throughout G2, the opportunity to carry out templated repair via strand invasion between sister chromatids is afforded through the alternate end-joining, single-strand annealing (both error prone) and homologous repair (virtually error-free; HR) pathways (4,5). The details of how chromatin context, cell-cycle phase, cell type and differentiation state influence the DDR, regulating (among other things), the extent of strand resection, and length of the repair process are far from understood.
The Zinc finger E-box-Binding protein ZEB1 (also known as delta-EF1) has a well-documented role as a transcriptional regulator. ZEB1 and ZEB2 (SIP1) are the sole members of the zinc-finger-homeodomain family of transcription factors. Each has two clusters—amino and a carboxy-terminal—of C2H2-type zinc finger domains that can bind CANNT(G) elements (E-boxes) at promoters and enhancers and a central POU-like homeodomain, the function of which (in any context) has not been determined (6). Although initially described as transcriptional repressors through their interaction with the CtBP1/2 corepressors, ZEB1 can also activate transcription, through its interaction with coactivators (e.g. p300 and YAP (7,8)).
Through its activity at a number of genetic targets, notably the E-cadherin gene promoter, and at specific microRNA cluster loci, ZEB1 is one of a family of factors (incl. Snail, SLUG, TWIST, etc.) required for the initiation and maintenance of the epithelial-to-mesenchymal transition (EMT) during both development and tumor progression (9,10). ZEB1 has been implicated in virtually all aspects of tumor biology, including progression, metabolism, metastasis, cancer stem cell (CSC) maintenance (including induction of CSC-specific surface markers) and plasticity, and induces both chemo- and radio-resistance (11–18). Consistently, ZEB1 is highly expressed in both the interior of tumor masses, mirroring levels observed in stem cell (SC) compartments (as observed during normal development, tissue regeneration and tumor progression), and at the leading edge of invasive tumors (11,13–15,19). In the context of normal mammary duct development, high ZEB1 expression in epithelial stem cells is further implicated in suppressing oncogenic-induced genomic instability by up-regulating the methionine sulfoxide reductase anti-oxidant pathway (16). This affect appears to be tissue/tumor-type-specific, however, as ZEB1-mediated transcriptional repression of N-methyl-purine glycosylase, a key enzyme initiating base-excision repair, contributes to inflammation-driven colorectal cancer (17). With respect to DSBs, recent work has shown that exclusive of its role as an inducer of the EMT, IR stabilizes ZEB1 which facilitates repair by recruiting the deubiquitinase USP7 to, in turn, stabilize CHK1 (20). Furthermore, it has been reported that ZEB1 transcriptionally regulates several DDR-related genes (21), including the Pol Q promoter, the effect of which may promote genome stability (22). Despite its potential importance to the DDR, however, essentially nothing is known about whether ZEB1 functions directly at DSBs.
Initially isolated based on its physical interaction with p53, 53BP1 is a key player in the DDR. 53BP1 rapidly localizes to DSBs, via recognition of specific ubiquitinated and methylated histone marks, to promote NHEJ by nucleating the anti-resection complex shieldin (23–28). 53BP1 is required for Class Switch Recombination (CSR), mid-range V[D]J recombination, and fusion of de-protected/dysfunctional telomeres (29). Depletion of 53BP1 radio-sensitizes normal as well as tumor cells in culture and in xenografts, dramatically increases the number and size of insertions and deletions (indels) at repair junctions, and increases chromosomal aberrations (30–34). While largely dispensable for cNHEJ in the context of therapeutically induced DSBs, there is extensive evidence that 53BP1, via the shieldin complex, inhibits more error-prone NHEJ pathways (i.e. alternative end-joining and single-strand annealing) that rely on increasing levels of resection. A recent report describes a requirement for the transactivator SP1 for localizing 53BP1 to DSBs during G1, though whether these two factors physically interact or arrive at a DSB coincidentally is not known (35).
We present data showing that ZEB1 promotes 53BP1-mediated effects on DSB repair. Our results demonstrate that ZEB1 (i) concentrates very rapidly at LASER-induced lesions, (ii) physically interacts with (through it's homeodomain region), and recruits 53BP1 at DSBs; (iii) promotes NHEJ and PARPi sensitivity, while attenuating HR and resection through the homeodomain region; (iv) is required for physiological distal NHEJ-mediated DSB end-joining. These data establish a novel link between ZEB1, a key mediator of the EMT, and 53BP1, a critical determinant of DSB repair by NHEJ.
MATERIALS AND METHODS
Cell culture
HEK293T, and U2OS cell lines were obtained from ATCC. The U2OS-based DIvA cell line was a generous gift from G. Legube (CNRS, Toulouse). The U2OS-based EJ-DR cell line was a generous gift from S. Powell and R. Bindra (Sloan-Kettering). HEK 293T, I-SceI Knock-In 293T, 293T ZEB1 KO, U2OS and U2OS ZEB1 KO cells were cultured in DMEM (+4.5 g/l glucose)/10% FCS/1× Penn-Strep. DIvA and ZEB1 KO DIvA cells were cultured in DMEM (+4.5 g/l glucose)/10% FCS/1 mM NaPyruvate/1× Penn-Strep. EJ-DR cells were cultured in DMEM (+4.5 g/l glucose)/10% Tet-minus FCS/1× Penn-Strep. The human breast cancer cell lines HCC-1937, MDA-MB-231, MDA-MB-361 and MDA-MB-436 were obtained from ATCC. The HCC-1937 cells were cultured in RPMI (+4.5 g/l glucose)/10% FCS/1× Penn-Strep, while the latter three were cultured in DMEM (+4.5 g/l glucose)/10% FCS/1× Penn-Strep. The CH12.F3 mouse B lymphocyte cell line (a generous gift from T. Honjo (Kyoto Univ.)) was cultured in RPMI/10% FCS/5% NCTC-109 (Invitrogen)/1× Penn-Strep/50 μM βMeOH. All cells were cultured in a humidified atmosphere of 5% CO2 at 37°C and were verified mycoplasma negative. After reviving from liquid nitrogen storage, cells were routinely passaged no more than 5–6 times before using in an experiment.
Generation of stable cell lines
U2OS cells obtained from ATCC were expanded upon receipt and 4th passage aliquots stored in liquid N2. A fresh aliquot of these cells was used in subsequent experiments, always between passage three and seven. For transient transfection, cells were thawed, passaged three-to-four times, and then transfected (3:1 ratio of PEI to total DNA) with expression constructs for either flZEB1, the ΔHD, PLDLS or ΔNH3 mutant cDNAs. Two days later cells were subjected to polyclonal selection with 800 μg/ml G418. Low passage HCC-1937 breast cancer cells were transfected as above with either fl human ZEB1 cDNA in pcDNA3 alone, fl human BRCA1 cDNA in p3PA-Puro-iTk alone or both together. Two days after transfection, stable cell lines were generated using 350 μg/ml G418 (ZEB1 alone), 0.25 μg/ml Puromycin (BRCA1 alone) or both drugs for the combination. Selection media was changed every 2–3 days to generate polyclonal stable cell lines.
Plasmid constructs
Full-length (fl) ZEB1 cDNA, (variant 2, NCBI Ref. Seq.: NM_030751.5) was amplified (see table for primers), with Bam HI (5′) and Not I (3′) overhangs and cloned via Gibson Assembly (GA) into the CMV-promoter driven expression vector pcDNA3.1, yielding pcDNA3.1flZEB1. The pcDNA3.1ΔNH3 construct, which removes 160aa from the Amino terminal end of ZEB1 was generated in this expression vector using Q5 Polymerase deletion mutagenesis (NEB Q5 Site-Directed Mutagenesis Kit), and the primer pair listed in the ‘Cloning Primers’ Table in the Supplemental Methods and Materials. A 183 bp region encompassing the homeodomain was deleted using Q5 deletion mutagenesis (see table for primer sequences), yielding pcDNA3.1ΔHD. flZEB1 was amplified with Hind III (5′) and Bam HI (3′) overhangs (see table for primer sequences) and fused in frame at its carboxyl end via GA to eGFP in the vector eGFP-N3 (CLONTECH), to generate flZEB1-GFP. 53BP1-RFP was gift from S. Jackson, (Cambridge, UK).
RNA interference (si-RNA)
Knock-down of factors in Figure 5A was carried out using 100 pmol Smart-pool SiRNAs (53BP1, BRCA1, NT control, Dharmacon); or, in the case of ZEB1, 25 pmol siRNA targeting the ZEB1 3′ UTR (IDT; see Supplemental Materials and Methods for list of sequences). One million cells were electroporated (Nucleofector, Lonza), and after 48h, ZEB1 KD EJ-DR cells were rescued via transfection (3:1 ratio of PEI to total DNA) with either empty vector or indicated expression constructs in Figure 3A, and cultured for an additional 48h before induction of DSBs. KD of ZEB1 in the mouse B lymphoma cell line CH12.F3 (Figure 8A) was carried out using 25 pmol siRNA targeting the ZEB1 3′ UTR, as above.
Figure 5.

ZEB1 promotes NHEJ to a similar extent as 53BP1 and attenuates HR. (A) Representative WBs showing the effect of the NT, anti-BRCA1, anti-53BP1 and anti-ZEB1 siRNAs. (B) Top, schematic indicating major functional domains of ZEB1: ZF, Zinc Finger Cluster; SID, SMAD-Interacting Domain; HD, Homeodomain; CID, Co-Repressor-Interacting Domain. Bottom, the EJ-DR U2OS-based cell line with stably integrated, I-Sce I-based reporters for both NHEJ (RFP) and HR (GFP), were transfected with either NT or indicated SiRNAs, and 48 h later, DSBs were induced (see Materials and Methods). Cells were subjected to FLOW analysis 72 h after DSB induction. The anti-ZEB1 siRNA targeted its 3′UTR, allowing rescue by either full-length (fl) or a homeodomain-deleted (ΔHD) cDNAs. ZEB1 KD, similar to the effect of 53BP1 KD, results in a 20% reduction in NHEJ repair, with a more modest effect on HR. This effect is rescued, with a potentiation of NHEJ and a concomitant reduction in HR upon transfection of the fl ZEB1 expression vector, while transfection of the ΔHD construct had the opposite effect. Results are normalized to induced (IND) control; UN Cont., un-induced/No SiRNA; IND. Cont., induced/No SiRNA. error bars = S.E.M. Results are the avg. of three independent expts. with S.E.M. P > 0.05; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005.
Figure 3.

ZEB1 is required for localization of 53BP1 to a subset of DSBs. (A) Top: Time course of 53BP1 foci formation and resolution, ZEB1-proficient or -knockout U2OS cells (see Supplementary Figure S4 for analysis of protein levels) grown on coverslips were subjected to 2 Gy, immediately returned to the incubator, and after the indicated times fixed and processed for immunostaining for 53BP1. Nuclei are visualized with Hoechst 33342. Shown are representative images derived from one of three experimental replications (done on different days), Below: At least 80 cells per condition were scored for 53BP1-positive foci in a blinded fashion and the mean number of 53BP1-positive foci are plotted as a function of time. (B) Loss of ZEB1 dramatically reduces localization of 53BP1 800 bp from AsiSI-induced DSBs. ChIP/qPCR analysis of 53BP1 binding near AsiSI-generated DSBs on either chromosome 18 or 21, in wt or ZEB1 KO DIvA U2OS cells (see Supplementary Figure S4 for analysis of protein levels). IgG neg. control was carried out using the chr. 18 primer set. (C) Spatial distribution of 53BP1 chromatin localization from 80 to approximately 106 bp from the AsiSI-induced DSB is lost in the absence of ZEB1. In panels B and C, bar graphs depict relative ChIP/qPCR results, adjusted for background (ChIP with non-specific IgG) and normalized to Input; representative plots from three separate experiments. qPCR results in all ChIP expts were corrected for minor differences in cutting efficiency between wt and KO conditions (see Supplementary Table S1 and Material and Methods). IF staining analysis of ZEB1 effects on IRIF and all ChIP/qPCR experiments were carried out at least three times. Statistical analysis employed a two-tailed unpaired Student's t-test, Error bars are S.E.M. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005.
Figure 8.

ZEB1 is required for physiological DSB repair. (A) Class Switch recombination: the murine B lymphocyte cell line CH12.F3 was electroporated with either non-targeting (NT) or anti-ZEB1 SiRNA (ZEB1 KD) and 48h later stimulated to undergo CSR. After 72 h cells were stained and gated live cells (7AAD negative) analyzed by FLOW for % of surface IgA expression. ZEB1 KD resulted in greater than 50% reduction in IgA-positive cells. (B) V[D]J recombination: wt or ZEB1 KO 293T cells (see Figure 4, bottom for WB) were transfected with the RAG-responsive GFPi (inverted, non-expressed GFP cDNA) reporter and CMV-driven expression vectors for RAG1 and RAG2 and gated live cells (7AAD negative) scored 48 h later for percentage of GFP-positive signal (inside an RFP + gate—see MAterials and Methods). Loss of ZEB1 resulted in >50% reduction in recombination-mediated inversion of the GFP cDNA to the expressed orientation; shown are representative results from experiments performed a minimum of three times; data are the average ± S.E.M.; ***P ≤ 0.005.
CRISPR/cas KO of ZEB1
293T, U2OS and DIvA-AID cells were each transiently co-transfected (3 μl linear PEI to 1 μg total DNA) with the Cas9 expression vector Sp5 (Sant Cruz Biotech.) and an sgRNA-expression vector harboring a ZEB1-targeting sequence (see table). After 5h media was replaced and 48 h later cells were subjected to Puromycin selection (1 μg/ml for 293T, 1.5 μg/ml for U2OS and DIvA cells) for an additional 48 h. Cells were then detached and serially diluted in regular media (w/o puromycin) onto 15 cm culture dishes to achieve well-separated single-cell-derived colonies. Colonies were picked, expanded and screened for disruption of the ZEB1 gene via PCR (see table in Supplemental Materials and Methods for primer sequences) as well as for protein levels via WB.
Endogenous co-IP
U2OS cells subjected to 8Gy or no IR control were allowed to recover for 30′ at 37°C, placed on ice and washed 1X with ice-cold PBS. Cells were scraped, pelleted at 4°C, transferred to microfuge tubes and lysed in Buffer A 20 mM HEPES (7.4), 150 mM NaCl2, 0.2% NP-40, 0.2 mM EDTA, 5 mM MgCl2, 1 mM DTT, 50 U Benzonase/ml, 10% glycerol, protease and phosphatase inhibitors (Roche), 1 mM PMSF, for 30 min at 4°C. After sonication (30″on/30″off, 10′ total time) in a Bioruptor (Diagenode) to disrupt aggregates, lysate was spun at 20 000×g @4°C, 15′, and protein concentration was measured in the saved supernatant (BCA assay, Pierce). After pre-clearing the lysate with protein G Sepharose for 2 h at 4°C, 500 μg of protein was subjected to IP o/n at 4°C with rotation using 5 μg of specified antibody, followed by protein G Sepharose for an additional 2 h. Beads were washed 3× with 1ml Buffer A, then 2× in Buffer A with 300 mM NaCl. A subset of these IP experiments (Supplementary Figure S2A) included 200 μg/ml EtBr in the tissue culture media just prior to irradiating the cells as well as in the buffers at all subsequent steps from the initial washing of the cells at the time of harvest. Protein complexes were eluted in 2× SDS sample buffer and subjected to standard WB on nitrocellulose membranes using the indicated antibodies (listed in Table in Supplemental Materials and Methods).
Chromatin immunoprecipitation
For the U2OS-based DIvA system, the protocol of Nelson, et.al. (36) was followed with minor variations. After fixing in 1% formaldehyde for 10′ and glycine quenching, cells were rinsed with ice-cold PBS, scraped, pelleted, and resuspended at 50 μl/106 cells in Lysis buffer (50 mM Tris pH 8.0,150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.1% Na deoxycholate, 0.5% SDS, protease and phosphatase inhibitors (Roche), 1 mM PMSF). Chromatin was sonicated to an average size of 250–750 bp and 5% of the cleared lysate was removed for input. The remainder was diluted 5-fold in Chip dilution Buffer (50 mM Tris–Cl pH 8.0,150 mM NaCl, 2 mM EDTA, 1.2% Triton X-100, protease inhibitors and phosphatase inhibitors, 1 mM PMSF). After addition of Abs (or isotype-specific IgG; see table in Supplemental Materials and Methods), samples were rotated at 4°C o/n, followed by addition of biotinylated secondary Ab, rotation for 1 h, 4°C, and then with pre-blocked streptavidin-coupled magnetic beads, and further rotation for 2 h, 4°C. Beads were washed 3× with ice-cold low-salt Wash Buffer (20 mM Tris–Cl pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100) and then once with ice-cold high-salt Wash Buffer (20 mM Tris–Cl pH 8.0, 500 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100). Following the Chelex-100 isolation step, DNA was purified using a standard PCR-clean-up column (Qiagen). 2 μl of purified ChIPed DNA was used in the subsequent qPCR reaction.
Quantitative PCR (qPCR)
Real-time qPCR analysis was carried out using iTaq Universal SYBR Green Supermix (Bio-Rad) for 39 cycles on a CFX96RealTime PCR machine (Bio-Rad). Data were calculated using the ΔCt method and presented as percent of input. All data shown are the results of at least three independent ChIP experiments with qPCR performed in triplicate and results averaged with SEM. Primer pairs for ChIP analysis are listed in Supplemental Materials and Methods. Percent AsiSI cutting efficiencies (listed in Table I in the Supplemental Materials and Methods) between wt and ZEB1 KO cells were determined as described in (37), using flanking primers across the sites in Chrom 18 and Chrom 21 (listed in Supplemental Materials and Methods) to determine Ct values before and after administration of 4-OHT.
Immunofluorescence
Co-immunofluorescence staining of 53BP1 and ZEB1 (Figure 1B) was carried out as described (38). Immuno-staining for γH2AX and RAD51 was carried out as follows. Wild-type or ZEB1 KO 293T cells were grown on poly-lysine-coated coverslips, and, at given time points, IR-treated (or no IR control) cells were gently rinsed in 1× PBS, fixed in freshly-made 4% paraformaldehyde for 15′, permeabilized in 0.1% Triton X-100/PBS, 5′, and blocked for 1h in 1% BSA/0.05% Triton X-100/PBS (all at RT), then stained o/n at 4°C with an anti-γH2A.X-Alexafluor-488 Ab diluted 1:2500 in blocking buffer. For Rad51-Alexafluor-568 staining (1:250 in blocking buffer), an extraction step (CSX buffer for 5′ at RT) was included after the first PBS wash. After washing 3×, 5′ in 0.05%Triton-X100/PBS at RT, with gentle rocking, coverslips were mounted and imaged on a Zeiss LSM-980 inverted microscope using a 20× objective.
Figure 1.

Endogenous interaction between ZEB1 and 53BP1 is amplified post-IR. (A) Representative WB of reciprocal co-IP, shows that in U2OS cells, ZEB1 and 53BP1 each co-IP the other, an interaction that is amplified within minutes after exposure to 4 Gy IR, peaking 1 h later and returning to near base-line levels at the 24 h time point. (B) Top, indicated expression constructs of deletion mutants encoding FLAG-tagged ZEB1 or HA-tagged 53BP1 were introduced into 293T cells, and 36 h later, complexes were immunoprecipitated using anti-FLAG-coupled beads. Bottom, representative WBs of the physical interaction between ZEB1 and 53BP1 depends on a 147aa peptide of ZEB1 that includes the homeodomain (left panel) and the 57aa oligomerization domain of 53BP1 (right panel). Both endogenous and over-expression IPs were carried out a minimum of three times.
Live cell imaging
U2OS cells were plated onto 35 mm glass bottomed dishes (Ibidi) and 24 h later transfected with the indicated fluorescence reporter constructs (ratio of 3:1 linear PEI to DNA), and cells were imaged 48 h later. Initial plating numbers were calculated so that cells were growing exponentially on day of imaging. One hour before imaging, media was exchanged for its identical phenol red-minus counterpart (DMEM). Laser ablation and live cell imaging were carried out on a Zeiss LSM 780 laser scanning confocal microscope with an integrated IR Chameleon Vision-II, ultrafast Ti:Sapphire laser (see Supplementary Table S2 for detailed microscope settings). Dishes were imaged on a heated stage under sparged, 5% CO2. To minimize confounding cellular damage, the focal plane was positioned at the center of each nucleus and a thin rectangular region of interest was drawn that traversed the nucleus while avoiding the nuclear membrane. Images were recorded through a 40× oil immersion objective using continuous capture as follows. Two images (1.94″ total time per image to capture both color channels) were recorded and the laser (tuned to 780 nm, 10% power, for three iterations that combined totaled 12 μs) was triggered at the beginning of the third image capture.
EJ-DR-based DSB repair pathway choice assay
I-SceI-mediated DSBs were induced in the EJ-DR cell line as described (40,41). Briefly, at the appropriate time after administration of RNAi and rescue constructs, cells were rinsed 1× in 37°C PBS, and replenished with complete media (with 10% Tet-minus FCS) containing 1 μM Sheild1 (Aobious) and 0.1 μM triamcinolone acteonide (Cayman). After 24 h, cells were again rinsed with PBS, replenished with complete media and incubated at 37°C for an additional 96 h to allow expression and maturation of fluorescence proteins. Cells were dissociated from the culture dish using Accutase (Innovative Cell Technologies), diluted directly in PBS/1% FCS and analyzed by FLOW.
Sample preparation for imaging flow cytometry (IFC)
U2OS cells were subjected to either 4Gy IR or mock irradiated, then returned to the incubator for 30′. Preparation of cells for IFC was carried out essentially as in Forment et al. (39). Following trypsinization and washing 1× in PBS and counting, 2 × 106 cells/sample in microfuge tubes were pre-extracted in 100 μl PBS/0.2% Triton X-100 for 5′ on ice. After washing in 0.5 ml ice-cold Buffer A (PBS/1 mg/ml BSA), samples were pelleted (10 K rpm, 10″, 4°C), and resuspended in 50 μl Buffer A. An equal volume of 4% paraformaldehyde/PBS was added and incubated at RT for 20′. Fixed cells were diluted with addition of 0.5 ml 1× permeabilization/wash buffer (PW buffer, Becton/Dickenson), quickly pelleted (10 K rpm, 10″, RT), then resuspended in 100 μl P/W buffer with Alexa-fluor-conjugated Ab and incubated at 4°C, o/n. The following day, cells were washed in 0.5 ml P/W buffer, pelleted and resuspended in 75 μl Buffer A, with 250 μg/ml RNase A and 0.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI), then incubated at 4°C overnight in the dark, and then analyzed. Samples stained individually with each fluorophore, including DAPI alone, were included in every analysis for calibration of the ImageStream FLOW cytometer. The anti-RPA2 antibody (1:750) was conjugated to Alexa 568 (Lightning-Link kit, Abcam).
Imaging flow cytometry (IFC) analysis
Combining the statistical power of flow cytometry with fluorescence imaging to capture distinct, prominent morphological parameters, this technique allows the number of RPA2 foci/cell to be scored—the ‘spot count’–in each phase of the cell-cycle (proportional to DAPI staining). The intensity of a given spot/focus is proportional to extent of RPA decoration (the ‘bright detail Intensity’, itself proportional to the extent of resection). During post-flow analysis, acquired scored data (spot counts and bright detail intensity) for a given cell can be matched to an individual captured image of that cell (a representative panel of such images is presented in Supplementary Figure S5). IFC associates the scored events/parameters of a given cell (such as fluorescence intensity, side scatter, etc. each represented via familiar graphical scatter/dot-plots, including gates, etc.) with a captured image of that cell; every single plotted event can be matched to a corresponding image and vice-versa (40). Analysis was carried out on an IDEAS software-driven Amnis ImageStreamX Mark II instrument, using DAPI and Alexa 568 excitation channels and extended depth of field (EDF). The EDF optical focus, analogous to confocal imaging, dramatically increases the focusing capability through the depth of the cell, projecting structures such as foci onto a single clear plane (see Supplementary Figure S5). We arbitrarily chose to score cells with five or more foci (‘High Spot Count’) in our analysis. ‘Bright Detail Intensity’, is a relative measure of the fluorescence intensity of a given spot, graphically represented as the sum of the scored cells for a given condition. At least 20 000 cells were analyzed per condition. Cell cycle phase is scored as in regular flow cytometry via measurement of DNA content through DAPI staining. Cells were gated on Gradient RMS versus Normalized Frequency to eliminate unfocused cells, and then gated by plotting the area and aspect ratio of the brightfield (M01). To produce spot count data sets (a direct measurement of the number of RPA2-staining foci), IDEAS Spot Count Wizard was implemented and groups of high count and low count (∼50 cells each) were manually assigned. Further post-acquisition analysis was performed in FCS Express 7, where cell-cycle specific data was acquired by dot-plotting DAPI intensity versus Brightfield aspect ratio and gating for G1, S and G2. The aforementioned gates were then applied to histograms plotting Spot Count datasets as acquired in IDEAS software. Bright detail intensity (directly proportional to the extent of resection, (40)) was plotted in an identical fashion.
DNA end-resection assays
Isolation of genomic DNA, o/n digestion with indicated restriction enzymes, and subsequent CyberGreen-based qPCR analysis was performed using standard procedures. The extent of resection at an AsiSI-induced DSB was quantitated in asynchronous or synchronized DIvA cells as previously described (37). Synchronization was achieved using double thymidine block as follows. For synchronization in G1 and G2, cells were treated with 4-OHT (or vehicle) 11 and 6 h, respectively after release from the second block, and collected 4 h later (41). After o/n digestion with Bsr GI, genomic DNA was purified and ssDNA near the AsiSI-cut site on Chromosome 1 measured by qPCR, using CyberGreen fluorescence. All qPCR reactions were carried out in triplicate at least three times. Primer pairs used for normalization of DNA levels (flanking a region of Chromosome 22 devoid of an AsiSI site) and for determining cutting efficiency before and after administration of 4-OHT at the AsiSI site on Chrom 1 were from ref (37). Percent ss DNA at a given Bsr GI site was calculated as: %ssDNA = 1/(2(ΔCt – 1) + 0.5) × 100. This result was then divided by the cutting efficiency to give the final value plotted in Figure 6B. Results using the primer pairs targeting Chrom 22 (a region devoid of an AsisI site) were used to normalize the amount of genomic DNA between experimental conditions in the qPCR reactions.
Figure 6.

ZEB1 Inhibits resection. (A) Imaging FLOW cytometry shows that, in response to IR, ZEB1 inhibits number and intensity of RPA2-labled foci. Top left panel, in either wt or ZEB1 KO U2OS cells, in the absence of IR, there are virtually no detectable RPA2-positive foci; Bottom left panel, blue bars, 30 min after 4 Gy, wt cells with RPA2-positive foci appear in all three CC phases, with cells in S having the highest percentage of cells with greater than 5 foci per cell (high spot count); their ZEB1 KO counterparts, depicted with red bars, show a significant increase in high spot count in all three phases. Upper right panel, ZEB1 KO cells stably expressing the indicated constructs were subjected to 4 Gy and processed for Imaging FLOW cytometry as above; compared to vector control (red bars, top row), expression of flZEB1 cDNA dramatically reduces RPA2-positive foci, while the homeodomain deletion (ΔHD) mutant has a minimal effect (bottom row). A minimum of 20000 cells were analyzed per condition. Data shown in this panel are representative of three independently performed analyses (carried out on separate days). (B) ZEB1 inhibits resection at an enzymatically-induced DSB. An AsiSI-generated DSB was induced in synchronized DIvA cells (see Supplementary Figure S7A for WB of these ZEB1 KO U2OS cells), genomic DNA isolated, digested with restriction enzyme BsrGI and subjected to qPCR (in triplicate) using PCR primers that flank BsrGI sites at 335, 1618 or 3500 bp downstream of an AsiSI cut site on chromosome I. Resection, yielding increased levels of SS DNA and subsequent PCR product, is increased in asynchronized cells with ZEB1 KD, a result amplified in G2. Results, normalized to AsiSI cutting efficiency (see Supplementary Table S1 and Materials and Methods), are the avg. of three different expts. with S.E.M. * P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005.
Analysis of PARP inhibitor sensitivity
One hundred thousand cells/well were plated in a 12-well plate and the following day, transfected with either 5 nM [final] anti-BRCA1 or non-targeting control SiRNA using RNAiMax (Invitrogen), according to the manufacturer's instructions. Twenty-four hrs later, cells were re-plated at 500 cells/well in 96-well plates, with five experimental replicates/condition, and, after an additional 24 h, PARPi (or Optimem vehicle) was added to 0, 5, 15 or 30 nM final concentration for each condition. Plates were then returned to the incubator for 5 additional days, after which proliferation, as a function of metabolic activity, was analyzed using the MTT assay (Cyquant, Invitrogen), according to the manufacturer's instructions.
Analysis of CSR in vitro
Wild-type or ZEB KD CH12.F3 B lymphoma cells induced to undergo CSR from IgM to IgA as described (60). Cells were split at a density of 5 × 105/ml and the next day, stimulated to undergo CSR as described, by addition of 1 μg/ml anti-murine CD40 Ab; BD Biosciences), 5 ng/ml recombinant murine IL-4 (R&D Systems), and 5 ng/ml recombinant human TGF-β (R&D Systems). After 72 h, cells were collected and stained on ice with Alexa-fluor 488-coupled anti-IgA Ab (Southern Biotech) and 7AAD (Roche) then analyzed by FLOW.
Analysis of V[D]J recombination in vitro
The GFPi reporter and CMV-RAG1, CMV-RAG2 expression constructs were a generous gift of J. Demengeot (Inst. G. de Ciencia, Portugal). Recombination Activation Gene (RAG)-mediated induction of DSBs, re-orientation and subsequent expression of GFP (GFPi) cDNA was carried out as described (61). 70% confluent wt or ZEB1 KO 293T cells were co-transfected (linear PEI) with the GFPi reporter alone (background) or in combination with CMV-driven RAG1 and RAG2 expression constructs, at a ratio of 1 μg GFPi:0.32 μg RAG1:0.28 μg RAG2. The following day, media was changed and after an additional 48 h, cells were harvested and analyzed by FLOW.
Statistical analysis
Bar graphs represent the mean ± S.E.M. of at least three independent experiments. The unpaired two-tailed Student's t test was used to calculate statistical significance, and a P < 0.05 value was considered significant.
RESULTS
The physical interaction between ZEB1 and 53BP1 is potentiated by ionizing radiation and they co-localize at DSBs
In its role as a transcription factor, ZEB1 has been shown to interact with a number of chromatin-modifying and transcriptional-regulating factors (6,8–10,42). To identify interacting partners with which ZEB1 promotes radio-therapeutic resistance we performed a yeast two-hybrid screen using full length human ZEB1 as the bait. Screening a HeLa cDNA library, 53BP1 was a prominent positively-interacting candidate. Reversing the configuration of the assay, with fl53BP1 as the bait, like-wise, resulted in colony growth (Supplementary Figure S1).
The association between ZEB1-53BP1 was confirmed in resting mammalian U2OS cells via reciprocal endogenous co-immunoprecipitation. Exposure of these cells to 4 Gy ionizing radiation amplified this interaction very rapidly–within 1–2 min of treatment–peaking at about 1h post-IR and declined thereafter. Twenty-four hours after treatment, this association had returned to baseline levels (see Discussion), (Figure 1A). To limit (but not eliminate) the possibility that the amplified interaction between these two proteins required DNA, we added the DNA intercalating agent Ethidium Bromide in the culture media just prior to irradiation and also during the harvesting, processing and immunoprecipitation steps. We observed no effect on the rapid–within at least 2 mins of IR administration–amplification of the ZEB1-53BP1 interaction (Supplementary Figure S2A). In addition, western blot analysis shows that in the 2 min time frame that this amplification occurs, steady-state protein levels of ZEB1 and 53BP1 remain unchanged (Supplementary Figure S2B).
As 53BP1 is a well-established marker of DSBs, we next asked whether ZEB1 localizes to IR-induced lesions. Double immunofluorescence staining of U2OS cells 30 min after exposure to 4 Gy showed co-localization of endogenous ZEB1 and 53BP1 at ionizing radiation-induced foci (IRIF; Supplementary Figure S3).
Using over-expressed epitope-tagged cDNAs in U2OS cells, we narrowed the interaction between ZEB1 and 53BP1 to a 147aa region that includes the homeodomain in the former and to the 57aa oligomerization domain (OD) in the latter (Figure 1B). Interestingly, deletion of 305aa from the amino terminus of ZEB1 (which includes the amino-terminal zinc-fingers and SMAD interaction domain (9), left panel of Figure 1B) resulted in loss of 53BP1 binding, whereas the interaction was robustly regained after a further 225 deletion (530aa total from the amnio terminus; see discussion).
ZEB1 is rapidly recruited to LASER-induced lesions
As localization of 53BP1 to DSBs occurs within 2 min of a LASER-induced lesion (43), we next sought to determine the timing of ZEB1 localization relative to this benchmark. Using continuous-capture, live cell imaging and near-infrared LASER ablation, we introduced a complex lesion (including DSBs, with a minimum of pyrimidine dimers, (44)) into nuclei of U2OS cells co-expressing ZEB1-GFP and 53BP1-RFP. Concentration of ZEB1-GFP fluorescence at the lesion was very rapid—within at least 2 s—and precedes 53BP1 (Figure 2A), which, consistent with previous findings (43), arrives at the lesion after about 90 s. This speed of recruitment—faster than the ability of our system to record it as a live-cell image–led us to compare ZEB1 to that of PARP1, one of the earliest factors known to localize—within seconds–to such lesions (45–47). To the extent that we can accurately measure, ZEB1-GFP and PARP1-mcherry arrive simultaneously to IF-mediated LASER stripes (Figure 2B). Quantification of the kinetics of these fluorescence readouts is presented below these images. Virtually identical results were obtained–simultaneous rapid co-localization within the 2 s timeframe required to capture both color channels–when ZEB1-GFP was co-transfected with the cNHEJ repair factor Ku80 fused to mCherry (Supplementary Figure S13). The speed of this co-localization to the LASER lesion is clearly seen in time-lapse capture of a second ZEB1-GFP/Ku80-mCherry co-transfected cell subjected to the same IFR LASER ablation protocol (Supplementary Figure S14).
Figure 2.

ZEB1 localization to LASER-induced Lesion is rapid and precedes 53BP1. (A) U2OS cells were transfected with the indicated fluorescence-tagged cDNAs and 48 h later, subjected to 780 nM near-infrared LASER ablation in the stripes defined by the yellow rectangles (regions of Interest, ROIs). Frames were continuously captured (2 s total time for both channels) and images extracted. ZEB1-GFP concentrates at the lesion within 2 s; 53BP1-RFP co-localizes about 1.5 mins later. (B) Identical experimental parameters as in panel A, with co-transfection of ZEB1-GFP and PARP1-mCherry. In the composite image, the lower yellow ROI marks a region not subjected to LASER ablation (to serve as background fluorescence), while the upper yellow ROI marks the site of LASER targeting (at this magnification the position of the cell had drifted slightly over the course of the imagining period, several minutes). Fluorescence intensity for each channel in each frame was measured (subtracting background fluorescence of nuclear region not subjected to ablation) after images had been captured by refreshing the ROI around the site of the lesion (red rectangle in panel B) to generate the graphs below the images, plotting fluorescence intensity versus time. The arrow in each graph indicates the moment–the beginning of the fourth imaging capture–when the LASER was fired (a 12 μs burst). Captured images are representative of at least six separate transfections/analyses (done on different days). Kinetics localization plots are the mean of each time point derived from at least three separate experiments; error bars are S.E.M. 40× oil objective; detailed microscope parameters provided in Supplementary Table S2.
ZEB1 depletion impairs 53BP1 recruitment to both IR- and enzymatically-induced DSBs
Given the relative timing of localization of these two factors to a LASER-mediated lesion (Figure 2A), we asked whether, following induction of DSBs, ZEB1 is required for formation of at least a subset of 53BP1 foci, over a 24 h period. Wild-type or CRISPR/Cas9-mediated ZEB1 KO U2OS cells (see Supplementary Figure S4 for analysis of protein levels in these cells) were irradiated with 2 Gy, and various time points, fixed and stained for endogenous 53BP1 via IF. Compared with wt, ZEB1 KO cells not only display approximately half the 53BP1 foci (Figure 3A; quantified below). Interestingly, at 24 h-post treatment, this ratio is reversed, suggesting that DSB resolution is delayed in ZEB1-deficient cells (an effect mirrored in our analysis of γH2A.X resolution (Figure 4, see below). A similar effect–irradiated ZEB1-deficient cells had half the number of 53BP1-positive foci as their wt counterparts–was observed in 293T cells 20 min after IR (data not shown). ZEB1-KO U2OS cells displayed no significant difference in cell cycle phase distribution from their wt counterparts (Supplementary Figure S15).
Figure 4.

In response to IR, ZEB1 KO delays resolution of γH2AX foci and increases the average number of cells with high levels of RAD51 foci. (A) Top: IF staining time course of appearance, post-1Gy, and resolution of γH2AX foci, in either wt, or ZEB1-depleted (via CRISPR KO) 293T cells; Bottom, left: quantitation of staining results (200 cells from each condition were scored in a blinded fashion). Bottom right: WB shows virtual absence of ZEB1 protein in the KO line. (B) wt or ZEB1 KO U2OS cells were subjected to either Mock (Control) or 2 Gy, and, 2 h later stained for RAD51 (see Materials and Methods); below, at least 200 cells from each condition were scored (blinded) for presence of RAD51 foci and resulting percentages categorized as shown. Data shown in all panels is a representative example of three independent experiments. Asterisks indicate statistically significant differences by Student's t test, Error bars are S.E.M.; ns, not significant: P > 0.05; *P ≤ 0.05, **P ≤ 0.01,***P ≤ 0.005.
Next, using the U2OS (p53 wt)-based DIvA (Double-stranded breaks Induced via AsiSI) cell line, a 4-OHT-inducible system that allows for the reliable interrogation of approximately 180 AsiSI endonuclease-targeted sites dispersed throughout the human genome (virtually all of these reside in euchromatin, previously mapped and characterized (48,49). This number of DSBs provides a reasonable, enzymatically-generated analogue of a therapeutic dose of IR (48,50,51), the consequences of which can be interrogated biochemically. As above, we generated a ZEB1 KO derivative of this line (for WB analysis, see Supplementary Figure S4) and, following induction of DSBs, determined via ChIP/qPCR the extent of 53BP1 localization to two sites, comparing the parent line to ZEB1 KO. Localization of 53BP1 was dramatically reduced—approximately 5-fold—at a site in Chr 18 and 9-fold at one in Chr 21 (normalized to the minor effect that ZEB1 depletion had on AsiSi cutting efficiency in ZEB1 KO cells versus wt—see Supplementary Table S1; Figure 3B). Furthermore, ChIP/qPCR analysis of the spatial distribution of 53BP1 and ZEB1 at these two DSBs showed that ZEB1 is required for 53BP1 localization up to one megabase from either of these AsiSI-generated cuts, and it associates with chromatin to an extent that mirrors the pattern seen for 53BP1 (Figure 3C). In the context of these results, it is important to note that depletion of ZEB1 had no effect on 53BP1 protein levels in both 293T (HEK) and in DiVA (U2OS) cells (Supplementary Figure S4); and had only negligible effects on AsiSI enzymatic cutting efficiency (Supplementary Table S1).
Loss of ZEB1 delays resolution of γH2AX foci and increases the number of RAD51 foci
Wild-type or Crisper/Cas9-generated ZEB1 KO U2OS cells were subjected to 1 Gy, and appearance and resolution of γH2AX foci, a surrogated marker of DSB, was monitored over the next 24 h. Residual γH2AX foci were significantly greater in the ZEB1 KO cells compared to control (Figure 4, quantified in lower panel), consistent with the results seen with 53BP1 foci observed 24 h post-irradiation (Figure 3). As above, there was no discernible effect of ZEB1 depletion in these cells on 53BP1 levels (Supplementary Figure S4). As ZEB1 is required for localizing a key regulator of resection and promoter of NHEJ, 53BP1, to a subset of DSBs, we then asked whether it affects a classic hallmark of HR, the Rad51 recombinase (52). Wild-type and ZEB1KO U2OS cells were either mock-treated or subjected to 2 Gy, and 2 h later were processed for IF staining for the presence of Rad51 foci. Both unirradiated and irradiated ZEB1 KO cells showed clear evidence of greater RAD51 foci than their ZEB1-proficient counterparts (Figure 4B, top; quantitation is shown below the images). Collectively, these results suggest that loss of ZEB1 delays the repair of DSBs and may be accompanied by enhanced HR repair.
ZEB1 promotes NHEJ-mediated DSB repair and inhibits resection through its homeodomain
To determine whether ZEB1 influences the repair of DSBs via NHEJ or HR pathways (or both), we utilized the EJ-DR U2OS cell line (a gift from S. Powell and R. Bindra), which has three stably integrated constructs: reporters measuring either NHEJ (RFP) or HR (GFP) mediated repair, and a cDNA encoding an inducible I-SceI enzyme (53,54). These cells were treated with siRNAs targeting ZEB1, 53BP1, BRCA1 or non-targeting control, and, following induction of DSBs, the repair outcomes were analyzed by flow cytometry (54). The results show that ZEB1 promotes NHEJ to an extent similar to 53BP1 (Figure 5A; representative WBs showing KD specificity are shown in Figure 5B).
Depletion of ZEB1 via SiRNA-targeting of its 3′UTR in the EJ-DR reporter cell line (see representative WB, Figure 5B, Bottom) reduced NHEJ-mediated fluorescence and up-regulated the HR fluorescence output (Figure 5A). Re-introduction of a full-length ZEB1 cDNA, restored NHEJ/HR fluorescence to normal levels, supporting a role for ZEB1 in the promotion of NHEJ and inhibition of HR. Interestingly, introducing a ZEB1 cDNA with only the homeodomain region deleted (ΔHD, Figure 5A; see schematic) failed to rescue this repair switch phenotype.
Using high-throughput imaging flow cytometry (IFC;(39,40) to score the number and intensity of IR-induced RPA2-positive foci (a well-established marker for ssDNA—see Discussion (55)), in wt versus ZEB1 KO U2OS cells, we further found that ZEB1 promotes NHEJ, at least in part, through the attenuation of resection. In the absence of ZEB1, the number of IR-induced RPA2-positve foci increased throughout the cell cycle, with the greatest effect–3.9-fold–in G2 (Figure 6A, bottom, left). ZEB1-depleted cells (which showed no discernable difference in the rate of proliferation from wt cells, Supplementary Figure S7), like-wise, showed a comparable increase in the extent of resection, as reflected in significantly greater bright detail Intensity throughout the cell cycle (Supplementary Figure S6). Additionally, mirroring the results seen in the EJ-DR fluorescent reporter system (Figure 5A), rescue of the U2OS ZEB1 KO cells by stably expressing either wt flZEB1 cDNA or the HD-deletion mutant resulted in the attenuation or promotion of resection, respectively (Figure 6, top, right). Cell cycle phase profiles–derived from intensity of DAPI-staining for DNA content as part of the IFC analysis—remained relatively unchanged across these conditions (Supplementary Figure S7). Statistical analysis of cell cycle phase distribution from asynchronously dividing wt versus KO cells that are derived from these Imaging Flow Cytometry data is presented in Supplementary Figure S15. Protein levels of these stably expressed cDNAs were verified via western blot (Supplementary Figure S8A).
These results were corroborated in the context of enzymatically-induced ‘clean’ DSBs using the DIvA system, and a qPCR-based resection assay (37). Loss of ZEB1 significantly increased the appearance of ssDNA near the break, an effect virtually all confined to the G2 phase as shown in double-thymidine-blocked cells (Figure 6B; see discussion). Cell cycle profiles post-thymidine release were similar in ZEB1-proficient and their knock-out counterparts (Supplementary Figure S16). The effect of ZEB1 depletion on AsiSI cutting efficiency at this target site was negligible (Supplementary Table S1) and consistent with that observed with 53BP1 KD (37). As ZEB1 is required for localization of 53BP1 to a subset of IR-, as well as to enzymatically-induced DSBs (Figure 3), this result is consistent with the hypothesis that it promotes the 53BP1/shieldin-mediated-attenuation of resection activity (23–28). Depletion of ZEB1 significantly increased that activity up to at least 3500 bases from the AsiSI-mediated DSB (Figure 6B).
ZEB1 confers sensitivity to PARP inhibitors through its homeodomain
Micro-Homology-Mediated End Joining (MMEJ, also called Theta-Mediated End Joining/TMEJ), is an error-prone, PARP1/Pol Theta-reliant repair pathway that is up-regulated in resection-deficient genetic backgrounds (49–51). Given that the resection-regulating 53BP1/Shieldin complex promotes this pathway in BRCA1-deficient backgrounds, and that ZEB1 is required both for 53BP1 localization to a subset of DSBs (Figure 3), and attenuation of resection (Figure 6), we tested whether ZEB1 impacts the sensitivity of cancer cells to PARP inhibitors. Wild-type and ZEB1 KO U2OS cells, depleted of BRCA1 via siRNA KD (or control si-RNA) were subjected to increasing concentrations the PARP1/2 inhibitor Olaparib (or DMSO vehicle) for 5 days. Analysis of cell growth at the end of this period showed that, in the context of BRCA1-depletion, ZEB1-deficincy conferred significantly more resistance to PARP inhibition compared to ZEB1-proficinert cells (Figure 7A, summarized in Figure 7B), an effect phenocopying that of 53BP1 KO (J. Hurwitz, personal communication). Loss of ZEB1 in these cells had no significant effect on the cell cycle (Supplementary Figures S7, S9, S15 and S16). Similar results were obtained using the PARP inhibitor Veliparib (Supplementary Figure S10). PARPi sensitivity could be restored in ZEB1 KO U2OS cells after stable transfection of full-length ZEB1 cDNA (but not vector control, Figure 7C, verification of protein expression in Supplementary Figure S8A).
Figure 7.

ZEB1 confers PARPi sensitivity. Upper panel: (A) loss of ZEB1 in U2OS cells significantly increases PARP inhibitor resistance in BRCA1-deficient cells, as quantified by cell proliferation (MTT assay). (B) Referring to panel A, at 30 nM concentration of Olaparib, ZEB1-positive cells are significantly more sensitive in a BRCA1-deficient background. (C) Rescue of fl ZEB1 in ZEB1-KO U2OS cells enhances PARP inhibitor sensitivity in BRCA1-deficient cells. (D) Over-expression of the ZEB1 transcriptional repression minus mutant ‘PLDLS’ still confers PARPi sensitivity, while a the homeodomain deletion mutant ‘Delta’ does not rescue this effect. (E) In tumor-derived breast cancer cell lines, ZEB1+, BRCA1-deficient cells (MDA-MB-436) are sensitive to PARP inhibition as quantified by cell proliferation (MTT assay), an effect that is lost in BRCA1-proficient cells (MDA-MB-231). (F) Stable cell lines derived from the ZEB1-deficient/BRCA1 deficient HCC-1937 cells over-expressing either ZEB1 alone, BRCA1 alone, or both, were subjected to the same PARPi experimental protocol. As in the previous examples, cells expressing ZEB1 alone (BRCA1-deficient) were sensitive to PARPi. WB showing expression of rescue cDNAs in U2OS cells is shown in Supplementary Figure S7A; WB demonstrating the expected expression profiles for ZEB1 and BRCA1 in the four cell lines employed in panel E and in the rescued HCC1937 cells are presented in Supplementary Figure S11. All graphs depict representative results and all experiments were performed a minimum of three times; data shown are average values ± S.E.M. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005, ****P ≤ 0.001.
Given the results above, we predicted that the homeodomain-deletion mutant of ZEB1, which fails to interact with 53BP1 [and, by extension, will fail, in certain contexts, to localize the anti-resection Shieldin complex, (23–26)], should allow resection in a BRCA1-deficient background, conferring PARPi resistance. This was, indeed the case, as shown if Figure 7D (verification of protein expression in Supplementary Figure S8A).
We next asked whether a key, well described cellular function of ZEB1–as a nucleator of chromatin modifying activities that collectively either repress or activate transcription (6–8) would play role in the response to PARPi. ZEB1 KO U2OS cells rescued with either a transcription repression-deficient (Figure 7D; see Supplementary Figure S8A for protein expression and Supplementary Figure S8B for verification of the functional activity of this cDNA (56,57)) or transcription activation-deficient mutant cDNA (58) (Supplementary Figure S11) were as sensitive to olaparib as the wt ZEB1 cDNA. These data establish that ZEB1’s ability to promote NHEJ and suppress HR is independent of its canonical transcriptional activity.
Lastly, to obtain a more clinically-relevant understanding of the relationship between ZEB1 and the cellular response to PARPi, we examined the sensitivity of a panel of breast cancer cell lines harboring all four combinations of ZEB1/BRCA1 genotypes to PARPi. Recapitulating the results in U2OS cells, BRCA1-deficient cells with high ZEB1 expression (MDA-MB-436) were significantly more sensitive to olaparib treatment than cells with low ZEB1 expression (Figure 7E). To control for the genetic variability of these distinct patient-derived lines, we introduced into the ZEB1-null/BRCA1-null HCC-1937 cells either flZEB1, flBRCA1 or both cDNAs, generating isogenic polyclonal stable cell lines expressing these constructs, and subjected these, in turn, to PARP inhibitor treatment. The results recapitulated those we observed in U2OS cells and the four patient-derived lines (Figure 7F). Protein expression analysis for all of these cell lines is shown in Supplementary Figure S12.
ZEB1 is required for the repair of physiological DSBs
Extensive characterization of Class Switch Recombination (CSR) and Variable [Diversity] Joining (V[D]J) recombination have demonstrated an absolute requirement for 53BP1 in the former (59–61) and for a subset of recombination events in the latter (62). A putative role for ZEB1 in V[D]J recombination is further supported by a phenotypic outcome in a ZEB1 loss-of-function mutant mouse, which harbors a 99% reduction in the total thymic T cell population (63). We therefore asked, as is the case for non-physiological DSBs, whether ZEB1 plays a role in somatic recombination. To investigate CSR, we employed the standard CH12.F3 mouse B cell line, which can be triggered (via Activation-Induced Deaminase) to carry out a distal NHEJ reaction in the constant region of the IgH gene, resulting in up-regulated expression of cell surface IgA isotype, detectable by FLOW (64). Depleting ZEB1 using specific si-RNA resulted in a greater than 50% reduction in cell-surface expression of IgA (Figure 8A).
To investigate the role of ZEB1 in V[D]J recombination (in the context of 53BP1 involvement, the result of long-range distal end-joining of two enzymatically-induced DSBs (62)), we made use of a straight-forward fluorescent reporter-based test system. Co-transfection with cDNAs expressing the Recombination Activation Genes, RAG1 and RAG2, results in either an NHEJ-mediated inversion of a GFP cDNA into the correct orientation for expression downstream of a constitutively acting promoter or as an excised, non-expressing fused ring (65). When we introduced these three constructs into ZEB1 KO 293T cells, we observed, via FLOW, an approximately 40% reduction in the establishment of productive, GFP-expressing cells compared to wt controls (Figure 8B). Taking into account that others have shown the distance between the distal DSBs is determinative for the requirement for 53BP1 in V[D]J (62) this result suggests that ZEB1 is required for at least a subset of physiological distal NHEJ repair.
DISCUSSION
ZEB1 is known for promoting the EMT in both development and in metastasis, as well as for having pleiotropic effects that collectively potentiate tumor progression and therapeutic resistance (10,19). In addition, recent studies have implicated ZEB1 in regulating the DDR, potentially contributing to its role in therapeutic resistance (20,22,66). Most pertinent to this study, Zhang et al. have shown that, in response to IR, ZEB1 activates the G2/M checkpoint independent of its role in the EMT. Mechanistically (in a radio-resistant derivative of the SUM159 human breast cancer cell line), ZEB1, through ATM-targeting (at S585), is stabilized and recruits the deubiquitinase USP7 to, in turn, stabilize CHK1, allowing for repair (20).
Our data demonstrate a novel role for ZEB1, independent of ATM or checkpoint activation, directly in repair of DSBs. We show here, initially through a classic yeast two-hybrid genetic screen (Supplementary Figure S1) and, in endogenous Co-IP and in over-expression analysis (Figure 1), that ZEB1 physically interacts, through its homeodomain, with 53BP1, a central player in the regulation of resection and the promotion of NHEJ.
We have demonstrated that ZEB1 is required for localizing 53BP1 to a subset of DSBs, promoting NHEJ and inhibiting HR, attenuating resection, and conferring PARPi sensitivity–each of which can be attributed to ZEB1s homeodomain region. As loss of ZEB1 had no significant effect on proliferation rates or cell cycle distribution (Supplementary Figures S7 and S9, S15 and S16), and, as loss of ZEB1 increased resection throughout the cell cycle (Figure 6), these data strongly support a direct role for ZEB1 in DSB repair, per se. Together with our observations on the effects of ZEB1 depletion on experimental analogues of physiological DSB repair (V[D]J and CSR), these findings establish that ZEB1 plays a direct, non-transcriptional role in cis at DSBs, promoting NHEJ at the expense of HR (48,59,60,62,67,68).
ZEB1’s rapid appearance—within at least two seconds (Figure 2)—to DSBs, and its requirement for the subsequent localization of the 53BP1/shieldin complex mirrors it's well-established function as a transcription factor, nucleating chromatin modifying/re-modeling factors/activities at promoters and enhancers (69,70). The influences that determine where (e.g. chromatin context, PIKK apical kinase activity) and when (e.g. CC phase, differentiation state, tumor stage) ZEB1 localizes 53BP1 to DSBs are currently under investigation. We note that 53BP1 recruitment to IR-induced DSBs is not entirely eliminated in the absence of ZEB1 (Figure 3A), suggesting that ZEB1 recruits 53BP1 to only a subset of DSBs, presumably influenced by certain chromatin context. This conclusion is supported by the loss of 53BP1 recruitment at enzymatically-induced DSBs in ZEB1-deficient DiVA cells (Figure 3A), where AsiSI activity is limited almost exclusively to euchromatic sites (48,49). We speculate that ZEB1 acts by establishing a 53BP1-permissive chromatin environment at certain DSBs (29,71,72), by promoting the functional dimerization of 53BP1 at these sites (73,74), by nucleating the 53BP1/shieldin complex via homeodomain-mediated protein contacts (23,24,26,75), or through a combination of these. Another (non-mutually-exclusive) possibility is that 53BP1 may be recruited to complex DSBs or clustered DSBs (such as those induced by IR) through an alternate mechanism that does not require ZEB1. Further investigation is required for elucidating the mechanism for this differential requirement of ZEB1 for recruiting 53BP1 at DSBs.
Interestingly, another well-studied transcription factor, SP1, has recently been shown to be required for G1-specific localization of 53BP1 to DSBs (and the subsequent promotion of NHEJ (35)). Although ZEB1 deficiency results in an increase in resection throughout interphase (Figure 6A), it remains to be determined whether ZEB1-mediated localization of 53BP1 is confined to a specific cell cycle phase. With respect to timing, our data from live cell LASER ablation studies shows that ZEB1 localization to that lesion occurs at least as rapidly as PARP1 (46). Whether SP1 is upstream, downstream, coincident with or independent of ZEB1 remains to be investigated. Likewise, it remains to be determined if, like ZEB1, SP1 physically interacts with 53BP1. Although ZEB1 and SP1 both contain Cys2/His2-type zinc fingers, ZEB1 uniquely harbors an homeodomain (HD) with a classic helix-turn-helix structure, and, as discussed above, we have shown that this 144aa domain is required for ZEB1s effects in DSB repair.
A central finding of this work, that ZEB1 attenuates resection, is consistent with the recent expansion of the mechanistic role for 53BP1 in pro-NHEJ activity. A number of recent reports show that 53BP1 localizes both the shieldin protein complex, which directly inhibits exo-nucleolytic activities and long-range resection (23–27), as well as the resection-countering fill-in Polymerase-α (28). Consistent with our data demonstrating that 53BP1 localization at a subset of DSBs requires ZEB1, reduction or loss of ZEB1 results in an increase in HR-promoting resection (Figure 6). The resection phenotype we observed here is initiated through the interaction between the HD of ZEB1 the oligomerization domain (OD) of 53BP1 (Figures 1B, 5A6A and 7D), required components of the ZEB-53BP1 axis. HD-containing proteins play well-established key roles as transcription factors during embryonic structural patterning and organogenesis (76,77), but can also function non-transcriptionally in translation (78), protein stabilization (79) and in export of mRNA (80). In this report, we find that the 147aa region incorporating the ZEB1 homeodomain is required for (i) physical interaction with 53BP1 (Figure 1B; whether this is direct is currently under investigation); (ii) promoting NHEJ (Figure 5); (iii) attenuating resection (Figures 6, Supplementary Figure S6) and (iv) PARPi sensitivity (Figure 7).
ZEB1’s effect on resection was measured two ways, by scoring RPA2-positive foci via imaging flow cytometry (IFC) and quantifying the presence of ssDNA at defined distances from known AsiSI cut sites via qPCR (Figure 6). With regard to the former method, hyperphosphorylation of the RPA2 subunit (Ser4/8, Ser32 and Thr21), increases during resection, but not at replication forks. Accordingly, this marker has been used to distinguish between these two events using immunofluorescence of fixed cells (81–83). Despite considerable effort, we could not find an IFC-compatible anti-phospho-RPA2 antibody to distinguish between these two ssDNA targets. The fact that ZEB1-deficiency causes an increase in both the number and intensity of RPA2-positive foci throughout interphase (Figures 6, Supplementary Figure S6), with no apparent effect on the length of S-phase (Supplementary Figure S7), makes this a suitable readout for resection. If, in post-IR ZEB1 KO cells, an increase in the number of cells with a High Spot Count/Bright Detail Intensity occurred only in S phase, it might, for example, suggest a replication stalling effect. In both of these readouts, however, we observed that ZEB1-deficient cells show an increase in all three phases.
In contrast to the Image Stream analysis, the qPCR analysis of DNA upstream of an enzymatically-derived DSB showed virtually no increase in resection in G1 in ZEB1 KO cells (Figure 6B). A reasonable explanation for this discrepancy is that the former analysis involved subjecting cells IR-mediated DNA damage (4Gy), resulting in a complex mixture of lesions (including DSBs, a large number of SS breaks, multiple adducts, etc.) requiring extensive processing, including resection. A ‘clean’ enzymatically-induced DSB, however, can be rapidly repaired through cNHEJ with minimal processing, and, as resection is down-regulated in G1, this pathway will predominate.
One unresolved question raised by this study is whether the interaction between ZEB1 and 53BP1 is direct or indirect and, in the subset of DSBs where it occurs, whether it requires DNA/chromatin. The increase in the association between endogenous ZEB1 and 53BP1 proteins occurs within 2 min after exposure of U2OS to therapeutic levels of IR (4 Gy; Figure 1A), a timing consistent with kinetics of localization we observed in our live-cell imaging analysis (Figure 2). While we cannot rule out a role for DNA/chromatin in the initiation of this amplification, during post-treatment processing, addition of an agent known to reduce conformational flexibility of DNA–ethidium bromide (84)—had no apparent effect on the temporal stability of this association, suggesting that chromatin, per se, may not be required for its persistence (Supplementary Figure S2A).
Another critical question relates to the chromatin context in which 53BP1 requires ZEB1 for localization. Our Co-IP analysis also identifies the 47aa OD of 53BP1 as being required for this interaction (Figure 1B). Not only is this domain responsible for oligomerization/multimerization of this factor at DSBs (29,74), but has very recently been shown to mediate liquid-liquid phase separation (LLPS; (85)) through interaction with the core Heterochromatin Protein 1α (HP1 α) and the repressive epigenetic modification H3K9me3 (86). In addition, a very recent report from the Soutoglou group has shown that such 53BP1-sponsored LLPS repair compartments are restricted in scope again through the oligomerization domain–in G1 by the scaffold protein AHNAK. They demonstrated that loss of AHNAK results in increased foci condensation and droplet fusion with a concomitant increase in p53 accumulation/activation and downstream consequences, such as apoptosis and senescence (87). As ZEB1 and 53BP1 co-localize at IRIF (Supplementary Figure S3), but ZEB1 is required for 53BP1 localization to only a subset of these sites (Figure 3A), our current efforts to characterize the specific chromatin environment to which ZEB1 localizes at a DSB is being informed by, among other things, the homeodomain–OD interaction.
ZEB1 plays a broad pleiotropic role in cancer biology, particularly in metastatic compartments of solid tumors (15,18) and this applies, as well, to ZEB1’s role in the DDR. Recent data from the Positgo, Ma and Tissier laboratories show that ZEB1 regulates base excision repair (88), CHK1 stability (20) and MMEJ/TMEJ (22), respectively. In the case of the last study, the authors provide convincing evidence, including from patient samples, that the inverse correlation between ZEB1 and Pol Theta in aggressive, claudin low, triple negative breast cancers is due to direct transcriptional repression of the PolQ gene by ZEB1. The consequent reduction in TMEJ may play a role in reducing genomic instability in at least a subset of these triple negative tumor cells, conferring a selective advantage over their ZEB1-deficient counterparts (22). In light of these data, a functional role for ZEB1 as a regulator of resection (via localization of the 53BP1/shieldin complex) will likely depend on a number of factors beyond chromatin context, including cell cycle phase, complexity of the DSB, and tumor stage.
The clinical relevance of the ZEB1–53BP1 axis described here, however, is centered largely on BRCA-deficient tumors, which rely on TMEJ as a back-up repair pathway in S/G2—one basis for the synthetic lethality of PARP inhibition. Critically in this regard, ZEB1KO/BRCA1 KD cells over-expressing either the ZEB1 transcriptional repression mutant or wt cDNA were equally sensitive to PARPi (Figure 7). Whether the down-regulation of PolQ by ZEB1 is confined to G1–inhibiting the error-prone TMEJ pathway in a CC phase dependent on the less error-prone rapid cNHEJ pathway (which is clearly potentiated by the ZEB1–53BP1 axis), remains to be elucidated. Consistent with this reasoning, in their report, Prudhomme et al. speculate that the down-regulation of TMEJ by ZEB1 may result in a parallel induction of cNEHJ, an outcome supported by the present study (22).
In contrast to the well-characterized therapeutic resistance of high ZEB1-expressing cancer cells, the ZEB1→53BP1→shieldin –| resection axis described here exposes a potential novel vulnerability: ZEB1-expressing/BRCA1/2-deficient cells are sensitive to PARP inhibitors. ZEB1 levels in BRCA-deficient patients and/or tumors may, therefore serve as a biomarker to predict response to PARP inhibition, and we are currently exploring this possibility.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank S. Jackson, G. Legube, T. Honjo, F. Alt, M. Gellert, J. Demengeot, R. Bindra, S. Powell, G. Chinnadurai and G.M. Anderson, for cell lines and plasmid constructs; and M. Solga and T. Harper for help with ImageStream Analysis. We thank R. Cao, S. Hao and A. Periasamy for help with live cell and IF image capture, which were acquired using the UVA Keck Center Zeiss 780 Confocal and 980 microscopy systems, respectively (NIH OD016446).
Contributor Information
Thomas L Genetta, Dept. of Radiation Oncology, University of Virginia School of Medicine, PO Box 800383, Charlottesville, VA 22908, USA.
Joshua C Hurwitz, Dept. of Radiation Oncology, University of Virginia School of Medicine, PO Box 800383, Charlottesville, VA 22908, USA.
Evan A Clark, Dept. of Radiation Oncology, University of Virginia School of Medicine, PO Box 800383, Charlottesville, VA 22908, USA.
Benjamin T Herold, Dept. of Radiation Oncology, University of Virginia School of Medicine, PO Box 800383, Charlottesville, VA 22908, USA.
Shadi Khalil, Dept. of Radiation Oncology, University of Virginia School of Medicine, PO Box 800383, Charlottesville, VA 22908, USA.
Tarek Abbas, Dept. of Radiation Oncology, University of Virginia School of Medicine, PO Box 800383, Charlottesville, VA 22908, USA; Dept. of Biochemistry and Molecular Genetics University of Virginia School of Medicine, Charlottesville, VA 22908, USA.
James M Larner, Dept. of Radiation Oncology, University of Virginia School of Medicine, PO Box 800383, Charlottesville, VA 22908, USA.
DATA AVAILABILITY
The data underlying this article will be shared on reasonable request to the corresponding author.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [1R01CA259054-01A1]; Charles Burnett Fund. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Jackson S.P., Bartek J.. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sirbu B.M., Cortez D. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb. Perspect. Biol. 2013; 5:a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Her J., Bunting S.F.. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018; 293:10502–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scully R., Panday A., Elango R., Willis N.A.. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019; 20:698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ceccaldi R., Rondinelli B., D’Andrea A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016; 26:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gheldof A., Hulpiau P., van Roy F., De Craene B., Berx G.. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell. Mol. Life Sci. 2012; 69:2527–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sanchez-Tillo E., de Barrios O., Valls E., Darling D.S., Castells A., Postigo A.. ZEB1 and TCF4 reciprocally modulate their transcriptional activities to regulate wnt target gene expression. Oncogene. 2015; 34:5760–5770. [DOI] [PubMed] [Google Scholar]
- 8. Lehmann W., Mossmann D., Kleemann J., Mock K., Meisinger C., Brummer T., Herr R., Brabletz S., Stemmler M.P., Brabletz T.. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat. Commun. 2016; 7:10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nieto M.A., Huang R.Y., Jackson R.A., Thiery J.P.. Emt: 2016. Cell. 2016; 166:21–45. [DOI] [PubMed] [Google Scholar]
- 10. Stemmler M.P., Eccles R.L., Brabletz S., Brabletz T.. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019; 21:102–112. [DOI] [PubMed] [Google Scholar]
- 11. Wellner U., Schubert J., Burk U.C., Schmalhofer O., Zhu F., Sonntag A., Waldvogel B., Vannier C., Darling D., zur Hausen A.et al.. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009; 11:1487–1495. [DOI] [PubMed] [Google Scholar]
- 12. Chaffer C.L., Marjanovic N.D., Lee T., Bell G., Kleer C.G., Reinhardt F., D’Alessio A.C., Young R.A., Weinberg R.A.. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013; 154:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Siebzehnrubl F.A., Silver D.J., Tugertimur B., Deleyrolle L.P., Siebzehnrubl D., Sarkisian M.R., Devers K.G., Yachnis A.T., Kupper M.D., Neal D.et al.. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013; 5:1196–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Preca B.T., Bajdak K., Mock K., Sundararajan V., Pfannstiel J., Maurer J., Wellner U., Hopt U.T., Brummer T., Brabletz S.et al.. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int. J. Cancer. 2015; 137:2566–2577. [DOI] [PubMed] [Google Scholar]
- 15. Zhang P., Sun Y., Ma L.. ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle. 2015; 14:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morel A.P., Ginestier C., Pommier R.M., Cabaud O., Ruiz E., Wicinski J., Devouassoux-Shisheboran M., Combaret V., Finetti P., Chassot C.et al.. A stemness-related ZEB1-MSRB3 axis governs cellular pliancy and breast cancer genome stability. Nat. Med. 2017; 23:568–578. [DOI] [PubMed] [Google Scholar]
- 17. de Barrios O., Sanchez-Moral L., Cortes M., Ninfali C., Profitos-Peleja N., Martinez-Campanario M.C., Siles L., Del Campo R., Fernandez-Acenero M.J., Darling D.S.et al.. ZEB1 promotes inflammation and progression towards inflammation-driven carcinoma through repression of the DNA repair glycosylase MPG in epithelial cells. Gut. 2019; 68:2129–2141. [DOI] [PubMed] [Google Scholar]
- 18. Chen L., Gibbons D.L., Goswami S., Cortez M.A., Ahn Y.H., Byers L.A., Zhang X., Yi X., Dwyer D., Lin W.et al.. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014; 5:5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Caramel J., Ligier M., Puisieux A.. Pleiotropic roles for ZEB1 in cancer. Cancer Res. 2018; 78:30–35. [DOI] [PubMed] [Google Scholar]
- 20. Zhang P., Wei Y., Wang L., Debeb B.G., Yuan Y., Zhang J., Yuan J., Wang M., Chen D., Sun Y.et al.. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014; 16:864–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang M., He S.F., Liu L.L., Sun X.X., Yang F., Ge Q., Wong W.K., Meng J.Y.. Potential role of ZEB1 as a DNA repair regulator in colorectal cancer cells revealed by cancer-associated promoter profiling. Oncol. Rep. 2017; 38:1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Prodhomme M.K., Pommier R.M., Franchet C., Fauvet F., Bergoglio V., Brousset P., Morel A.P., Brunac A.C., Devouassoux-Shisheboran M., Petrilli V.et al.. EMT transcription factor ZEB1 represses the mutagenic poltheta-mediated end-joining pathway in breast cancers. Cancer Res. 2021; 81:1595–1606. [DOI] [PubMed] [Google Scholar]
- 23. Dev H., Chiang T.W., Lescale C., de Krijger I., Martin A.G., Pilger D., Coates J., Sczaniecka-Clift M., Wei W., Ostermaier M.et al.. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018; 20:954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gupta R., Somyajit K., Narita T., Maskey E., Stanlie A., Kremer M., Typas D., Lammers M., Mailand N., Nussenzweig A.et al.. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018; 173:972–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mirman Z., Lottersberger F., Takai H., Kibe T., Gong Y., Takai K., Bianchi A., Zimmermann M., Durocher D., de Lange T.. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature. 2018; 560:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noordermeer S.M., Adam S., Setiaputra D., Barazas M., Pettitt S.J., Ling A.K., Olivieri M., Alvarez-Quilon A., Moatti N., Zimmermann M.et al.. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018; 560:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tomida J., Takata K.I., Bhetawal S., Person M.D., Chao H.P., Tang D.G., Wood R.D.. FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO J. 2018; 37:e99543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barazas M., Annunziato S., Pettitt S.J., de Krijger I., Ghezraoui H., Roobol S.J., Lutz C., Frankum J., Song F.F., Brough R.et al.. The CST complex mediates end protection at double-strand breaks and promotes PARP inhibitor sensitivity in BRCA1-deficient cells. Cell Rep. 2018; 23:2107–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mirman Z., de Lange T.. 53BP1 a DSB Escort. Genes & Dev. 2020; 34:7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Panier S., Boulton S.J.. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014; 15:7–18. [DOI] [PubMed] [Google Scholar]
- 31. Gupta A., Hunt C.R., Chakraborty S., Pandita R.K., Yordy J., Ramnarain D.B., Horikoshi N., Pandita T.K.. Role of 53BP1 in the regulation of DNA double-strand break repair pathway choice. Radiat. Res. 2014; 181:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zimmermann M., de Lange T.. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014; 24:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Squatrito M., Vanoli F., Schultz N., Jasin M., Holland E.C.. 53BP1 is a haploinsufficient tumor suppressor and protects cells from radiation response in glioma. Cancer Res. 2012; 72:5250–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ward I.M., Minn K., van Deursen J., Chen J.. Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 2003; 23:2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Swift M.L., Beishline K., Flashner S., Azizkhan-Clifford J.. DSB repair pathway choice is regulated by recruitment of 53BP1 through cell cycle-dependent regulation of Sp1. Cell Rep. 2021; 34:108840. [DOI] [PubMed] [Google Scholar]
- 36. Nelson J.D., Denisenko O., Bomsztyk K.. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 2006; 1:179–185. [DOI] [PubMed] [Google Scholar]
- 37. Zhou Y., Caron P., Legube G., Paull T.T.. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic. Acids. Res. 2014; 42:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chakraborty S., Pandita R.K., Hambarde S., Mattoo A.R., Charaka V., Ahmed K.M., Iyer S.P., Hunt C.R., Pandita T.K.. SMARCAD1 Phosphorylation and ubiquitination are required for resection during DNA double-strand break repair. iScience. 2018; 2:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Forment J.V., Walker R.V., Jackson S.P.. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry Part A. 2012; 81A:922–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee Y., Wang Q., Shuryak I., Brenner D.J., Turner H.C.. Development of a high-throughput gamma-H2AX assay based on imaging flow cytometry. Radiat. Oncol. 2019; 14:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cohen S., Puget N., Lin Y.L., Clouaire T., Aguirrebengoa M., Rocher V., Pasero P., Canitrot Y., Legube G.. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018; 9:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sanchez-Tillo E., Lazaro A., Torrent R., Cuatrecasas M., Vaquero E.C., Castells A., Engel P., Postigo A.. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010; 29:3490–3500. [DOI] [PubMed] [Google Scholar]
- 43. Altmeyer M., Neelsen K.J., Teloni F., Pozdnyakova I., Pellegrino S., Grofte M., Rask M.D., Streicher W., Jungmichel S., Nielsen M.L.et al.. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 2015; 6:8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reynolds P., Botchway S.W., Parker A.W., O’Neill P. Spatiotemporal dynamics of DNA repair proteins following laser microbeam induced DNA damage - when is a DSB not a DSB?. Mutat. Res. 2013; 756:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mortusewicz O., Ame J.C., Schreiber V., Leonhardt H.. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007; 35:7665–7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haince J.F., McDonald D., Rodrigue A., Dery U., Masson J.Y., Hendzel M.J., Poirier G.G.. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008; 283:1197–1208. [DOI] [PubMed] [Google Scholar]
- 47. Mahadevan J., Rudolph J., Jha A., Tay J.W., Dragavon J., Grumstrup E.M., Luger K.. Q-FADD: a mechanistic approach for modeling the accumulation of proteins at sites of DNA damage. Biophys. J. 2019; 116:2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aymard F., Bugler B., Schmidt C.K., Guillou E., Caron P., Briois S., Iacovoni J.S., Daburon V., Miller K.M., Jackson S.P.et al.. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014; 21:366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iacovoni J.S., Caron P., Lassadi I., Nicolas E., Massip L., Trouche D., Legube G.. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010; 29:1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Powell S.N. DNA damage and repair following treatment with ionizing radiation. Radiother. Oncol. 1990; 19:95–108. [DOI] [PubMed] [Google Scholar]
- 51. Ward J.F. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988; 35:95–125. [DOI] [PubMed] [Google Scholar]
- 52. Wassing I.E., Esashi F.. RAD51: beyond the break. Semin. Cell Dev. Biol. 2021; 113:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bindra R.S., Goglia A.G., Jasin M., Powell S.N.. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 2013; 41:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goglia A.G., Delsite R., Luz A.N., Shahbazian D., Salem A.F., Sundaram R.K., Chiaravalli J., Hendrikx P.J., Wilshire J.A., Jasin M.et al.. Identification of novel radiosensitizers in a high-throughput, cell-based screen for DSB repair inhibitors. Mol. Cancer Ther. 2015; 14:326–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shibata A., Conrad S., Birraux J., Geuting V., Barton O., Ismail A., Kakarougkas A., Meek K., Taucher-Scholz G., Löbrich M.et al.. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011; 30:1079–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao L.J., Kuppuswamy M., Vijayalingam S., Chinnadurai G.. Interaction of ZEB and histone deacetylase with the PLDLS-binding cleft region of monomeric C-terminal binding protein 2. BMC Mol. Biol. 2009; 10:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Postigo A.A., Dean D.C.. Independent repressor domains in ZEB regulate muscle and T-cell differentiation. Mole. Cell Biol. 1999; 19:7961–7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Postigo A.D., Taylor J.J., Kroll K.L.. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003; 22:2453–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bothmer A., Robbiani D.F., Di Virgilio M., Bunting S.F., Klein I.A., Feldhahn N., Barlow J., Chen H.T., Bosque D., Callen E.et al.. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol. Cell. 2011; 42:319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ward I.M., Reina-San-Martin B., Olaru A., Minn K., Tamada K., Lau J.S., Cascalho M., Chen L., Nussenzweig A., Livak F.et al.. 53BP1 is required for class switch recombination. J. Cell Biol. 2004; 165:459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stavnezer J., Guikema J.E., Schrader C.E.. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 2008; 26:261–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Difilippantonio S., Gapud E., Wong N., Huang C.Y., Mahowald G., Chen H.T., Kruhlak M.J., Callen E., Livak F., Nussenzweig M.C.et al.. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008; 456:529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Higashi Y., Moribe H., Takagi T., Sekido R., Kawakami K., Kikutani H., Kondoh H.. Impairment of T cell development in δ EF1 mutant mice. J. Exp. Med. 1997; 185:1467–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nakamura M., Kondo S., Sugai M., Nazarea M., Imamura S., Honjo T.. High frequency class switching of an lgM+ B lymphoma clone CH12F3 to lgA+ cells. Int. Immunol. 1996; 8:193–201. [DOI] [PubMed] [Google Scholar]
- 65. Trancoso I., Bonnet M., Gardner R., Carneiro J., Barreto V.M., Demengeot J., Sarmento L.M.. A novel quantitative fluorescent reporter assay for RAG targets and RAG activity. Front. Immunol. 2013; 4:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schuhwerk H., Kleemann J., Gupta P., van Roey R., Armstark I., Kreileder M., Feldker N., Ramesh V., Hajjaj Y., Fuchs K.. The EMT transcription factor ZEB1 governs a fitness-promoting but vulnerable DNA replication stress response. Cell Rep. 2022; 41. [DOI] [PubMed] [Google Scholar]
- 67. Kumar V., Alt F.W.. Encyclop. Immunobiol. 2016; 107–114. [Google Scholar]
- 68. Crowe J.L., Shao Z., Wang X.S., Wei P.C., Jiang W., Lee B.J., Estes V.M., Alt F.W., Zha S.. Kinase-dependent structural role of DNA-pkcs during immunoglobulin class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:8615–8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Aghdassi A., Sendler M., Guenther A., Mayerle J., Behn C.O., Heidecke C.D., Friess H., Buchler M., Evert M., Lerch M.M.et al.. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012; 61:439–448. [DOI] [PubMed] [Google Scholar]
- 70. Wang J., Scully K., Zhu X., Cai L., Zhang J., Prefontaine G.G., Krones A., Ohgi K.A., Zhu P., Garcia-Bassets I.et al.. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007; 446:882–887. [DOI] [PubMed] [Google Scholar]
- 71. Fradet-Turcotte A., Canny M.D., Escribano-Díaz C., Orthwein A., Leung C.C., Huang H., Landry M.-C., Kitevski-LeBlanc J., Noordermeer S.M., Sicheri F.. 53BP1 is a reader of the DNA-damage-induced H2A lys 15 ubiquitin mark. Nature. 2013; 499:50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tang J., Cho N.W., Cui G., Manion E.M., Shanbhag N.M., Botuyan M.V., Mer G., Greenberg R.A.. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013; 20:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lou J., Priest D.G., Solano A., Kerjouan A., Hinde E.. Spatiotemporal dynamics of 53BP1 dimer recruitment to a DNA double strand break. Nat. Commun. 2020; 11:5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lottersberger F., Bothmer A., Robbiani D.F., Nussenzweig M.C., de Lange T.. Role of 53BP1 oligomerization in regulating double-strand break repair. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:2146–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Setiaputra D., Durocher D. Shieldin–the protector of DNA ends. EMBO Rep. 2019; 20:e47560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. McGinnis W., Krumlauf R.. Homeobox genes and axial patterning. Cell. 1992; 68:283–302. [DOI] [PubMed] [Google Scholar]
- 77. Panganiban G.R., Rubenstein J.L.. Developmental functions of the Distal-less/Dlx homeobox genes. Development. 2002; 129:4371–4386. [DOI] [PubMed] [Google Scholar]
- 78. Dubnau J., Struhl G.. RNA recognition and translational regulation by a homeodomain protein. Nature. 1996; 379:694–699. [DOI] [PubMed] [Google Scholar]
- 79. Aoki K., Kakizaki F., Sakashita H., Manabe T., Aoki M., Taketo M.M.. Suppression of colonic polyposis by homeoprotein CDX2 through its nontranscriptional function that stabilizes p27Kip1. Cancer Res. 2011; 71:593–602. [DOI] [PubMed] [Google Scholar]
- 80. Topisirovic I., Kentsis A., Perez J.M., Guzman M.L., Jordan C.T., Borden K.L.. Eukaryotic translation initiation factor 4E activity is modulated by HOXA9 at multiple levels. Mol. Cell. Biol. 2005; 25:1100–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vassin V.M., Wold M.S., Borowiec J.A.. Replication protein A (RPA) phosphorylation prevents RPA association with replication centers. Mol. Cell. Biol. 2004; 24:1930–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Soniat M.M., Myler L.R., Kuo H.C., Paull T.T., Finkelstein I.J.. RPA phosphorylation inhibits DNA resection. Mol. Cell. 2019; 75:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Françon P., Lemaître J.-M., Dreyer C., Maiorano D., Cuvier O., Méchali M.. A hypophosphorylated form of RPA34 is a specific component of pre-replication centers. J. Cell Sci. 2004; 117:4909–4920. [DOI] [PubMed] [Google Scholar]
- 84. Lai J.S., Herr W.. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:6958–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kilic S., Lezaja A., Gatti M., Bianco E., Michelena J., Imhof R., Altmeyer M.. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 2019; 38:e101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhang L., Geng X., Wang F., Tang J., Ichida Y., Sharma A., Jin S., Chen M., Tang M., Pozo F.M.et al.. 53BP1 regulates heterochromatin through liquid phase separation. Nat. Commun. 2022; 13:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ghodke I., Remisova M., Furst A., Kilic S., Reina-San-Martin B., Poetsch A.R., Altmeyer M., Soutoglou E.. AHNAK controls 53BP1-mediated p53 response by restraining 53BP1 oligomerization and phase separation. Mol. Cell. 2021; 81:2596–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. de Barrios O., Sanchez-Moral L., Cortés M., Ninfali C., Profitós-Pelejà N., Martínez-Campanario M., Siles L., del Campo R., Fernández-Aceñero M.J., Darling D.S.et al.. ZEB1 promotes inflammation and progression towards inflammation-driven carcinoma through repression of the DNA repair glycosylase MPG in epithelial cells. Gut. 2019; 68:2129–2141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.