


Abstract
Eukaryotic transcription is dependent on specific histone modifications. Their recognition by chromatin readers triggers complex processes relying on the coordinated association of transcription regulatory factors. Although various modification states of a particular histone residue often lead to differential outcomes, it is not entirely clear how they are discriminated. Moreover, the contribution of intrinsically disordered regions outside of the specialized reader domains to nucleosome binding remains unexplored. Here, we report the structures of a PWWP domain from transcriptional coactivator LEDGF in complex with the H3K36 di- and trimethylated nucleosome, indicating that both methylation marks are recognized by PWWP in a highly conserved manner. We identify a unique secondary interaction site for the PWWP domain at the interface between the acidic patch and nucleosomal DNA that might contribute to an H3K36-methylation independent role of LEDGF. We reveal DNA interacting motifs in the intrinsically disordered region of LEDGF that discriminate between the intra- or extranucleosomal DNA but remain dynamic in the context of dinucleosomes. The interplay between the LEDGF H3K36-methylation reader and protein binding module mediated by multivalent interactions of the intrinsically disordered linker with chromatin might help direct the elongation machinery to the vicinity of RNA polymerase II, thereby facilitating productive elongation.
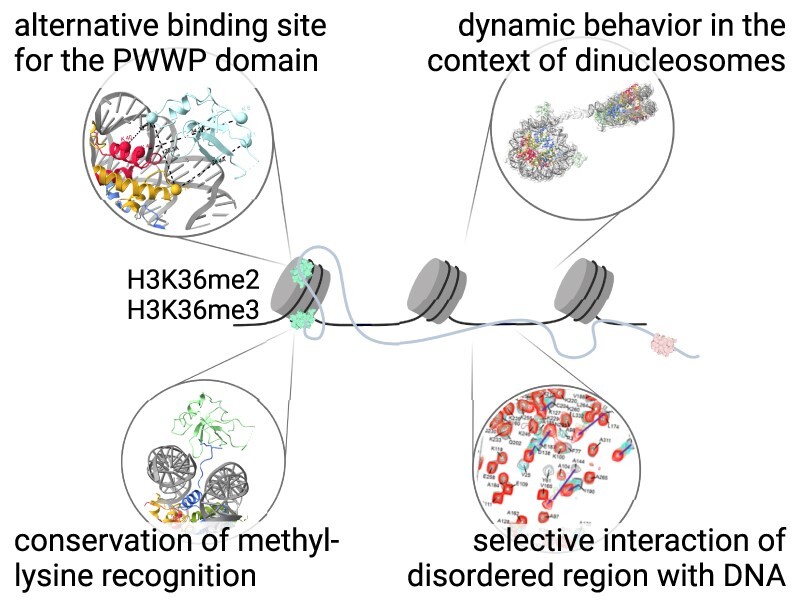
Graphical Abstract
Graphical Abstract.
INTRODUCTION
Nucleosomes, basic units of chromatin assembled from eight histones and DNA, are subjected to various covalent modifications of specific amino-acids within histone flexible tails that define distinct chromatin states. These modified histone side chains serve as docking sites for many regulatory proteins. Representing one type of these modifications, methylation marks are implicated in both repression and activation of transcription, depending on which residue is deposited. The chromatin-interacting proteins then utilize specific domains that recognize various methylated histone residues and recruit transcription regulation machinery (1). Methylated lysine 36 on histone H3, known as a marker of active transcription, is recognized by proteins containing a PWWP (Pro-Trp-Trp-Pro) domain (2), such as de novo DNA methyltransferases (DNMT3A and DNMT3B), subunits of acetyltransferase complexes (BRPF1–3), transcriptional repressors (HDGF and ZMYND11), histone lysine methyltransferases (NSD1–3), or hepatoma-derived growth factor-related protein family transcriptional coactivators (LEDGF/p75 and HRP2). Interestingly, the PWWP domain is often accompanied within these proteins by other chromatin mark reading/modifying domains, therefore contributing to a more complex recognition or definition of chromatin states (3).
The histone H3 lysine 36 side-chain can be mono-, di- or trimethylated (H3K36me1, me2 and me3). While the H3K36me1-specific reader and function remain unknown (4), the H3K36me2 and H3K36me3 marks play a role in a multitude of eukaryotic processes: transcriptional activation, DNA repair and recombination, alternative splicing, dosage compensation, and transcription repression in certain cases (5). There are differences in the genomic distribution of di- and trimethylated marks. H3K36me2 exhibits a more disperse pattern in regions near the transcription start sites (TSS) up to the first intron as well as the intergenic regions, whereas H3K36me3 is enriched throughout the gene bodies (2,6). Although numerous studies strived to clarify the in vivo preference of various PWWP-containing proteins towards either me2 or me3, they highlighted the physiological importance of the H3K36me2 (6–12). In higher eukaryotes, me2 and me3 methylation marks are deposited by distinct methyltransferases with different pathological outcomes linked to their enzymatic activity affecting mutations (2), suggesting that each mark might play diverse roles.
Lens epithelium-derived growth factor (LEDGF/p75 or PSIP1) belongs to the PWWP family of chromatin readers. It is implicated in the tethering of lentiviral integrases to actively transcribed genes (13,14) through its prominent protein-protein interaction module, an integrase-binding domain (IBD) that adopts a fold characteristic for the TFIIS N-terminal domains (TNDs) (15). We recently showed that LEDGF/p75 plays an essential role in the transcription elongation machinery (16), where TNDs coordinate assembly of the network of elongation factors through recognition of unstructured TND-interaction motifs (TIMs). Additional functional roles of LEDGF/p75, relying on an interplay between the PWWP and IBD domain interactions, include transcription coactivation (17), regulation of developmental genes (18), acute leukemia development (dependent on MLL1 fusion proteins) (19), DNA homologous recombination-mediated repair (20) and overcoming the nucleosomal barrier to transcription in cells lacking the FACT (Facilitates Chromatin Transcription) complex (7). Although LEDGF/p75 was originally thought to be functionally coupled only to the H3K36me3 mark (9,21–23), it is now also linked with the H3K36me2 mark (7,8,10).
A prominent feature of the PWWP domain is an extensive basic patch on its surface that can bind to DNA in a non-specific manner and with a micromolar affinity. In addition, the domain contains a deep hydrophobic cavity responsible for recognizing the methyl-lysine side-chain at position 20 of histone H4, and 36 or 79 of histone H3 with a low millimolar affinity (24). A combination of these two binding modes results in a nanomolar affinity of PWWP towards methyl-marked nucleosomes (21,23,25,26). The recently published cryo-electron microscopy (cryo-EM) structure of the LEDGF/p75 PWWP bound to the H3K36me3 nucleosome revealed that the interaction is maintained by a cooperative interaction of the PWWP domain with the trimethylated lysine 36 side-chain and both DNA gyres (27). Despite the fact that the complex included the full-length LEDGF/p75 molecule and was stabilized by chemical crosslinking, only the PWWP domain bound to the nucleosome was resolved. The IBD as well as the central and C-terminal intrinsically disordered regions remained undetected in cryo-EM maps.
Here, we show that LEDGF/p75 binds to the H3K36me3 and H3K36me2 nucleosomes with comparable affinity and that both the H3K36me3 and H3K36me2 nucleosomes structurally engage with the PWWP domain of LEDGF/p75 in a highly similar manner. Unexpectedly, we revealed that PWWP also binds to a less populated non-canonical site at the interface between nucleosomal DNA and the histone H2A-H2B acidic patch. Although we attempted to stabilize the C-terminal part of LEDGF/p75 by an additional interaction with a 64 kDa dimeric protein, all LEDGF/p75 regions except the PWWP domain remained unresolved. Therefore, we used NMR spectroscopy to explore additional interacting parts, and we experimentally identified three nucleosomal DNA interacting hotspots in the central LEDGF region that outline the protein's modularity in the context of methylated nucleosomes.
MATERIALS AND METHODS
Protein expression and purification
The expression plasmids coding for LEDGF/p52, LEDGF/p75 and PogZ(1117–1410) were derived from a pMCSG7 vector (T7 driven, ampicillin resistance). While LEDGF/p52 and LEDGF/p75 contained no tag, the protein coding sequence for PogZ(1117–1410) was preceded by an N-terminal His6 affinity tag and a tobacco etch virus (TEV) protease recognition site. As a result of TEV protease cleavage, five additional amino acid residues (SNAAS) were retained at the N-terminus of the natural protein sequence as a cloning artifact. Proteins were overexpressed in Escherichia coli BL21 (DE3) cells using LB broth (Sigma) supplemented with 100 μg/ml ampicillin and 0.5% glycerol, with the exception of 15N/13C-labeled LEDGF/p52, which was produced in minimal medium containing 15N-ammonium sulphate and 13C-glucose as the sole nitrogen and carbon sources and 100 μg/ml ampicillin. All cultures were grown at 37 °C until induction of protein expression at OD600 ∼ 0.8 with 200 μM IPTG (isopropyl-β-d-thiogalactopyranoside). Protein expression was continued overnight at 18 °C. Cells were harvested by centrifugation, resuspended in lysis buffer (30 mM Tris–HCl, 100 mM NaCl, 0.05% β-mercaptoethanol, pH 7.0 for LEDGF/p52, 15N/13C-LEDGF/p52 and LEDGF/p75; 25 mM Tris–HCl, 1 M NaCl, 0.05% β-mercaptoethanol, 10 μM EDTA, pH 7.5 for PogZ(1117–1410)) containing protease inhibitors (cOmplete™, EDTA-free, Roche) and sonicated.
LEDGF/p52, 15N/13C-LEDGF/p52 and LEDGF/p75 bacterial lysates were purified on an ÄKTA™ pure system using a HiTrap® Heparin HP 5 ml colum (GE Healthcare), a Superdex® 200 Increase 10/300 GL column (for LEDGF/p52, 15N/13C-LEDGF/p52; GE Healthcare) and a HiLoad® 16/600 Superdex® 200 pg column (for LEDGF/p75; GE Healthcare) using the aforementioned lysis buffer. The final purification step for all three proteins was carried out on a MonoS® 5/50 GL column (GE Healthcare) using 50 mM HEPES, 1 M NaCl, 0.05% β-mercaptoethanol, pH 7.6 as an elution buffer. PogZ(1117–1410) bacterial lysate was loaded on HIS-Select® Nickel Affinity Gel resin (Sigma), bound fractions were eluted with the appropriate lysis buffer containing 300 mM imidazole and subjected to overnight His6-tag cleavage by TEV protease during dialysis. The successfully cleaved His6-tag was then removed with a second round of Ni-NTA affinity chromatography, and the pure protein was concentrated in Amicon® Ultracel® (Merck Milipore) and further purified with a Superdex® 200 10/300 GL column (GE Healthcare) using a GF buffer (25 mM Tris–HCl, 200 mM NaCl, 0.05% β-mercaptoethanol, pH 7.5). All proteins were stored at −80 °C.
Unmethylated and methylated nucleosome preparation
Xenopus laevis histone proteins (H2A, H2B, H3, H4) and a modified H3, which has Cys111 mutated to Ala and Lys36 mutated to Cys (H3KC36), were expressed in E. coli BL21 RIL cells using a YT medium as an insoluble fraction, as described earlier (28,29). To briefly summarize: After extraction from the inclusion bodies, histones were purified on an anion exchange column (HiPrep® Q FF 16/10, GE Healthcare) and a cation exchange column (HiPrep® SP FF 16/10, GE Healthcare) using anion (7 M deionized urea, 20 mM Tris–HCl, 1 M NaCl, 1 mM EDTA, 5 mM β-mercaptoethanol, pH 7.5) and cation (7 M deionized urea, 20 mM Na-acetate, 1 M NaCl, 1 mM EDTA, 5 mM β-mercaptoethanol, pH 5.2) exchange buffers. Pure fractions were pooled and dialyzed against 2 mM β-mercaptoethanol in MilliQ water, flash-frozen, lyophilized and stored at −80 °C. Dimethylation and trimethylation of histone H3KC36 was carried out exactly as described elsewhere (30,31), and the efficacy was confirmed by MALDI-TOF MS.
Widom 601 147 bp and 166 bp dsDNA for nucleosome reconstitution was obtained via PCR with 5′ Cy5-labeled forward primer (Integrated DNA Technologies) and purified using a MonoQ® 5/50 GL column (GE Healthcare) followed by ethanol precipitation, or via restriction digest of high copy number plasmid. 314 bp dsDNA for dinucleosomes (Widom 601–20 bp linker sequence-Widom 601) was obtained via restriction digest of high copy number plasmid. Plasmids for DNA production (except for the one with 166 bp long sequence) were a generous gift from prof. Danesh Moazed (Harvard Medical School, Boston, MA).
Histone octamers were refolded as described in (28,29); equimolar amounts of individual histones were resuspended in unfolding buffer (6 M GuHCl, 20 mM Tris–HCl, 5 mM DTT, pH 7.5), mixed in final concentration of 1 mg/ml and refolded during dialysis into 10 mM Tris–HCl, 2 M NaCl, 1 mM EDTA, 5 mM DTT, pH 7.5. The sample was concentrated and applied to a Superdex® 200 Increase 10/300 GL column (GE Healthcare) that was equilibrated in the aforementioned buffer. Histone octamer-containing fractions were pooled, concentrated and stored at 4 °C until further usage.
To produce nucleosomes, appropriately methylated histone H3-containing octamers (4–4.4 μM) were mixed with DNA (4 μM) in high salt buffer (10 mM Tris–HCl, 2 M KCl, 1 mM EDTA, 1 mM DTT, pH 7.5), and the mixture was dialyzed for ∼18 h against a gradually decreasing salt concentration, as described previously (28,29). The nucleosomes used for cryo-EM experiments were purified using preparative native PAGE (Model 491 Prep Cell, BioRad) with a 7.5 cm long 5% 59:1 acrylamide:bisacrylamide TBE gel running in 0.2× TBE buffer. All nucleosomes were assessed using native PAGE (5% 59:1 acrylamide:bisacrylamide TBE gel running in 0.5× TBE buffer at 150 V, stained with GelRed® (Biotinum)), concentrated in Amicon® Ultracel® (Merck Milipore) to 4–50 μM and stored at 4 °C until further usage.
Electrophoretic mobility shift assay (EMSA)
0.25 μM nucleosomes with 5′ Cy5-labeled 147 bp Widom DNA (Supplementary Figure 6A) and 1 μM respective proteins or protein complexes were incubated in a total volume of 10 μl in TCS buffer (20 mM Tris–HCl, 1 mM EDTA, 1 mM DTT, pH 7.5) for 30 minutes on ice. Samples containing 0.25 pmol of nucleosome and 0.25–7 pmols of binding partner were analyzed on native PAGE (5% 59:1 acrylamide:bisacrylamide TBE gel running in 0.5× TBE buffer at 150 V) and imaged using Azure 600 (Azure Biosystems). The amount of unbound nucleosomes was quantified as a band intensity of unshifted nucleosome band using gel analysis tool in ImageJ (NIH) and visualized in GraphPad Prism.
Micro-scale thermophoresis
All MST data were measured on a NanoTemper Monolith NT.LabelFree instrument at 24 °C. LEDGF/p52 or LEDGF/p75 was kept constant at 1 μM concentration, as it gave a reasonable fluorescent intensity. Nucleosomes (147 bp or 166 bp H3KC36me0/2/3) were added in a 14-step dilution series, ranging from 1.2 nM to 20 μM in MST buffer (20 mM HEPES, 50 mM NaCl, 1 mM TCEP, 0.05% Tween-20, pH 7.5). Each dilution step was centrifuged and incubated at room temperature for 15 minutes in a final sample volume of 30 μl. Samples were loaded into the Monolith NT.LabelFree capillaries, and measurement was performed with 60% LED Power and 40% MST Power. All measurements were performed in triplicates. Values were fitted in the GraphPad Prism using a built-in equation for specific binding with Hill slope.
Cryo-electron microscopy
To prepare the LEDGF/p75-PogZ(1117–1410) complex, PogZ(1117–1410) was mixed with LEDGF/p75 in a molar ratio of 1.5:1 (75:50 μM) in suitable binding buffer (20 mM HEPES, 200 mM NaCl, 1 mM DTT, pH 7.5). The mixture was centrifuged, incubated for 30 minutes on ice, and applied to a Superdex® 200 Increase 10/300 GL column (GE Healthcare) that was equilibrated in the aforementioned buffer. Fractions containing the protein complex were pooled, concentrated in 10 MWCO Amicon® Ultracel® (Merck Milipore), flash frozen and stored at −80 °C. For mononucleosome samples, the LEDGF/p75-PogZ(1117–1410) complex was mixed with H3KC36me2 or H3KC36me3 nucleosome in a molar ratio of 1:2 nuc:protein (2:4 μM) in 30 μl and incubated on ice. After 30 mins, the sample was diluted with an extra 10 μl of TCS buffer and centrifuged. Complex samples (3.5 μl, ∼1.3 μM) were applied to freshly glow-discharged TEM grids (Quantifoil®, Cu, 300 mesh, R1.2/1.3) and vitrified in liquid ethane using ThermoScientific™ Vitrobot Mark IV (4 °C, 100% rel. humidity, 20 s waiting time, 4.5 s blotting time, blot force 5). For H3KC36me3 dinucleosome sample, the LEDGF/p75-PogZ(1117–1410) complex was mixed with dinucleosome in a molar ratio of 1:2 dinuc:protein (4:8 μM), incubated on ice and centrifuged. Complex samples (3.5 μl, ∼2 μM) were applied to freshly glow-discharged TEM grids (Quantifoil®, Cu, 200 mesh, R2/1) and vitrified in liquid ethane using ThermoScientific™ Vitrobot Mark IV (4 °C, 100% rel. humidity, 30 s waiting time, 5 s blotting time, blot force 5). All the grids were subsequently mounted onto the Autogrid cartridges and loaded to Titan Krios (ThermoScientific™) transmission electron microscope for data acquisition.
Data acquisition: Cryo-EM data were collected using a Titan Krios transmission electron microscope operated at 300 kV using SerialEM software (32). The data were acquired on the K2 or K3 (Gatan) direct electron detector operating in electron counting mode and positioned behind the Gatan Imaging Filter (GIF). The energy selecting slit was set to 20 and 10 eV for the K2 and K3 detector, respectively. Micrographs were collected at the calibrated pixel size of 0.822 Å/px (K2) or 0.8336 Å/px (K3) as a set of 40 frames. Data collection details are listed in Supplementary Table S1.
Data analysis: All movie frames were aligned using MotionCor2 (33). Thon rings from summed power spectra of every 4 e−/Å2 were used for contrast transfer function parameter calculation with CTFFIND 4.1. (34). Particles were selected with TOPAZ (35) using trained picking models for each individual dataset. Further 2D and 3D cryo-EM image processing was performed in Relion3.1 or 4.0 (36), as illustrated in Supplementary Figure S3.
In case of the mononucleosome complex sample datasets, particles with defined features were pooled and subjected to 3D focus-classification on either the PWWP pose 1 or pose 2 region. Particles with the most defined features for PWWP domain pose 1 or pose 2 region were 3D refined separately.
In case of the dinucleosome complex dataset, particles with defined features were pooled and subjected to 3D focus-classification on the more defined nucleosome. Particles from the most defined class were selected, duplicate particles were removed in 200 Å range, and remaining particles (154,299) were re-extracted to 1.656 Å/pixel. A global 3D refinement was performed as a basis for 3D focus-refinement on the more defined nucleosome with respective masking. The final 3D reconstruction of the defined nucleosome was estimated at 3.34 Å resolution which corresponds to the Nyquist frequency of the extracted particles. The signal for the defined nucleosome was subtracted from the experimental particle images, and the resulting images were globally 3D refined with restricted angular assignment to 3.7 degrees based on the reconstruction of the defined nucleosome. The resultant 3D reconstruction was further 3D focus-classified and particles from the most defined class of the second nucleosome were selected (49,681) and further 3D focus-refined. To verify a signal presence for both nucleosome moieties, the selected particles were reextracted without signal subtraction and 3D refined with restricted angular assignment to 1.8 degrees based on the reconstruction of the second nucleosome.
To define the mutual movement of the defined and second nucleosome we performed a multi-body refinement in RELION3.1 of the 154,299 particles selected after the 3D focus-classification on the more defined nucleosome (37). We defined two bodies as the two nucleosomes with respective masking and used the principal component analysis to visualize the mutual nucleosome movement.
Model building: The starting model for H3KC36me3 pose 1 mononucleosome consisted of the protein components of PDB entry 6S01 (38) and the DNA strands from PDB entry 7OHC (39) rigid body docked into the cryo-EM map using ChimeraX 1.5 (40). The DNA duplex was extended on each end by one base pair to cover the nucleotide sequence between positions −73 and 73, and the terminal regions were mutated to match the DNA sequence used for the nucleosome reconstitution (Supplementary Figure S6B). The model was then subjected to iterative cycles of real-space refinement (41) in Phenix Version 1.20.1–4487 (42) against the post-processed mrc map, combined with manual adjustments in Coot 0.9.8.7 (43) based on the blurred mtz map (blurring B = 50) generated in CCP-EM 1.5.0 (44,45). External DNA restraints, automatically generated by the program libG (46) run under Refmac 5 (44) in the CCP4Interface 8.0.010 (47), were manually edited and used during DNA rebuilding in Coot. In later stages of model refinement, self-restraints were used in Coot, and starting model was used as a reference for model restraints in Phenix. The refined model (Supplementary Table S1) was validated with MolProbity (48) and the wwPDB (49) validation server.
The model described above was rigid-body docked into the cryo-EM map for the H3KC36me2 pose 1 mononucleosome using ChimeraX 1.5 (40) and adjusted in the same way as the H3KC36me3 pose 1 mononucleosome model. The trimethylated S-aminoethyl-L-cysteine residue was manually replaced with a dimethylated analog in the structure and the sidechain was refined against the post-processed mrc map in Coot 0.9.8.7 (43), using ligand restraints generated by the eLBOW program (50) from the Phenix suite Version 1.20.1-4487 (42).
The two generated H3KC36me3 and H3KC36me2 pose 1 mononucleosome models were rigid body docked to cryo-EM maps for the H3KC36me3 and H3KC36me2 pose 2 mononucleosome, respectively. The PWWP domains for pose 2 were rigid body docked using JiggleFit tool within Coot 0.9.8.7 (43) into the blurred mtz map (blurring B = 150). The final models (Supplementary Table S1) were real-space refined in Phenix using self-restraints for the input model. All final mononucleosome cryo-EM maps were reference-based local amplitude scaled in LocScale (51). PDB model 6S01 was rigid body docked to the cryo-EM maps of the H3KC36me3 dinucleosome using MolRep within CCP-EM 1.5.0 (44,45).
NMR spectroscopy
All NMR data were acquired at 298 K on an 850 MHz Bruker Avance spectrometer, equipped with a triple-resonance (15N/13C/1H) cryoprobe. The sample volume was either 0.16 ml in 3 mm or 0.35 ml in 5 mm Shigemi tube in TRIS (25 mM Tris-d11, 200 mM NaCl, 1 mM TCEP, 10 μM EDTA, 5% D2O, 0.02% sodium azide, pH 7.5) or HEPES buffer (30 mM HEPES, 200 mM NaCl, 1 mM TCEP, 5% D2O, pH 7.0). The binding of peptides and DNA oligonucleotides was monitored using 100 μM 15N/13C-LEDGF/p52 and a variable concentration of histone H3 peptides (0–12 mM) SAPATGGVKKPHRY, SAPATGGVKme1KPHRY, SAPATGGVKme2KPHRY, SAPATGGVKme3KPHRY and dsDNA oligomers derived from a 601 Widom sequence (0–0.8 mM) 5′-CTGGAGAATCCCGGTGC-3′ (17 bp), 5′-GTGTCAGATATATACATC-3′ (18 bp), 5′-GTCGTAGACAGCTCTAGCAC-3′ (20 bp). Peptides were synthesized by solid phase synthesis using Fmoc-protected un-, mono-, di- and trimethylated lysines at the Institute of Organic Chemistry and Biochemistry of the CAS, v.v.i., and their 40 mM stock solution in the TRIS buffer was used for titrations. ssDNA oligomers were purchased from Sigma, annealed (95 °C for 5 min with subsequent cooling to room temperature) in the TRIS buffer and their 5 mM stock solution was used for titrations. The interaction of 15N/13C-LEDGF/p52 with 147 bp nucleosomal DNA or the nucleosome was monitored in a sample containing 30 μM nucleosomal DNA or nucleosome and 60 μM 15N/13C-LEDGF/p52, in the HEPES buffer. A series of double- and triple-resonance experiments (52,53) were recorded to assign 15N/13C-LEDGF/p52 backbone signals using NMRFAM Sparky (54). Chemical shift perturbation (CSP) values for each assigned resonance in either the 2D 15N/1H HSQC or 3D HNCO spectra of the protein was calculated as a geometrical distance in ppm to the peak in the corresponding type of 2D or 3D spectra acquired in the absence or presence of unlabeled interaction molecule using the formula 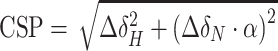 or
or 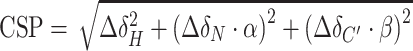 , where α and β are weighing factors of 0.2 and 0.1 used to account for differences in the backbone proton and nitrogen or carbonyl spectral widths (55). From CSPs, the dissociation constant KD was determined by a nonlinear least-squares analysis using a MestReNova.
, where α and β are weighing factors of 0.2 and 0.1 used to account for differences in the backbone proton and nitrogen or carbonyl spectral widths (55). From CSPs, the dissociation constant KD was determined by a nonlinear least-squares analysis using a MestReNova.
Chemical crosslinking and mass spectrometry
LEDGF/p75-PogZ(1117–1410)-H3KC36me3 ternary complex was loaded on the Superdex® 200 Increase 10/300 GL column (GE Healthcare). The fraction of the ternary complex was crosslinked with a 500 molar excess of DSBU (urea crosslinker–C4-arm, NHS ester). Cysteine residues of crosslinked complex were reduced by 10 mM TCEP (10 min at 37 °C) and alkylated by 20 mM IAA (20 min, room temperature, in dark). Complex was subsequently two times diluted by 100 mM ethylmorpholine buffer (pH 8.5) and digested by benzonase (10 U per 1 ug of complex; 30 min at 37 °C) and subsequently by trypsin (ratio complex:protease 10:1; 8 hours at 37 °C). 1 μg of peptide mixture was injected onto an analytical reversed phase column (Luna® Omega Polar C18, 3 μm, 100 Å, 0.3 × 150 mm heated to 50 °C) with a trap pre-column (Luna® Omega Polar C18 5 μm, 100 Å, 0.3 × 30 mm) and separated using a 5%–40% acetonitrile gradient over 35 minutes. Throughout chromatography, 0.1% formic acid was used as an ion-pairing agent. The HPLC system was directly coupled with the ESI source of an FT-ICR mass spectrometer solariX XR equipped with a 15 T superconducting magnet (Bruker Daltonics, Billerica, MA, USA). The mass spectrometer was operated in data independent mode and positive broadband mode over a 250–2500 m/z range with 1M data point acquisition. Fragmentation data was acquired with 16 eV collision voltage, 0.2 s MS/MS ion accumulation and 2 scans were accumulated per spectra. Data processing was performed using Data Analysis 5.2 (Bruker Daltonics, Billerica, MA, USA), and crosslinks were assigned within 1 ppm mass accuracy for precursor and 2 ppm for fragments using MeroX (56) and LinX (57) software. All crosslinks identified in triplicate were manually checked in raw spectra.
RESULTS
LEDGF/p75 binds the H3K36 di- and trimethylated nucleosomes with a similar affinity
To date, several studies reported preferential binding of the LEDGF PWWP domain to a H3-derived peptide trimethylated at lysine 36 (H3K36me3) with KD values ranging between 2.7 and 17 mM (10,21,23). However, there is also evidence for considerable binding of LEDGF to H3K36me2 nucleosomes (7,8,10). In order to substantiate the binding preference of LEDGF to various H3K36 methylation marks in vitro, we obtained dissociation constants for LEDGF complexes with differentially methylated peptides as well as nucleosomes. First, we used NMR to monitor the binding between the shorter 15N/13C-labeled LEDGF/p52 splice variant (Figure 1A) and a panel of H3-derived peptides (unmodified or mono-, di- and trimethylated at lysine 36). The chemical shift perturbations of amide group signals were monitored in 15N/1H HSQC spectra obtained at variable peptide concentrations. As expected, binding of all peptide variants affected only residues in the PWWP domain of LEDGF (Supplementary Figure S1A–D), which suggests that the central region of LEDGF is not involved in the histone H3 N-terminal tail binding. Subsequent analysis of the NMR binding profiles yielded KD values of 6.0/5.4 mM for H3K36me0/H3K36me1 and 2.4/2.7 mM for H3K36me2/H3K36me3 peptides, respectively (Figure 1B, H). 15N/13C-labeled LEDGF/p52 was also titrated by short Widom sequence-derived dsDNA oligonucleotides to mimic a nucleosomal DNA binding, and KD values were determined as 23–32 μM (Figure 1C, H). Next, we explored both the LEDGF/p52 and LEDGF/p75 interaction with analogously methylated nucleosomes using label-free microscale thermophoresis (MST). The nucleosomes were reconstituted using either wild-type H3 or its KC36 di- or trimethylated variants (Supplementary Figure S2A), the 147 bp or a 166 bp Widom sequence (Supplementary Figure S2B), and incubated with LEDGF/p52 or LEDGF/p75. The MST data revealed that both LEDGF/p52 and LEDGF/p75 bind to the H3KC36me2 or me3 nucleosomes with a similar ∼200 nM affinity, while the affinity of the interaction with unmodified nucleosomes is in a μM range (Figure 1D, E, H). Moreover, according to the Hill slope coefficient h > 1 obtained from data fitting for the H3KC36me2/3 nucleosomes, their interaction is maintained through multiple binding sites with moderate positive cooperativity (58), consistent with nucleosomes carrying two copies of methylated histone H3 being able to accommodate two LEDGF molecules. Since there are also regions on LEDGF/p75 that bind DNA, we compared LEDGF binding to simple 147 bp NCP and to NCP with a 20 bp DNA extension on one side (166 bp in total). There was no significant change in affinity for the nucleosomes with longer DNA, as well as no difference between the dimethylated and trimethylated state (Figure 1F, H). To increase the likelihood of resolving the portions of LEDGF bound to nucleosomes downstream of the PWWP domain in cryo-EM density maps, we assembled the nucleosome-LEDGF/p75 complex in the presence of a PogZ domain that is known to interact with the C-terminally positioned LEDGF/p75 IBD domain (Supplementary Figure S2C–G) (59,60). However, the effect of PogZ on LEDGF interaction with nucleosomes could not be monitored by MST because the presence of PogZ interfered with the fluorescence signal. Therefore, we followed the binding of purified LEDGF/p75-PogZ(1117–1410) binary complex and LEDGF/p75 alone to H3KC36me2 or H3KC36me3 nucleosomes using the electromobility shift assay (EMSA). We observed no significant preference of LEDGF/p75 for individual methylation states, which is in good agreement with the MST analysis. However, the presence of PogZ(1117–1410) led to reduced binding of LEDGF/p75 to di- and trimethylated nucleosomes (Figure 1G and Supplementary Figure S2H, I). The predicted stabilizing effect of PogZ did not lead to increased LEDGF/p75 affinity to methylated nucleosomes.
Figure 1.

Binding of LEDGF to unmodified or methylated histone H3-derived peptides, oligonucleotides and NCPs. (A) Domain architecture of the LEDGF splice variants p75 and p52. (B) KD fits from NMR titrations obtained using 15N-labeled LEDGF and the unlabeled short H3-derived peptides (unmodified, mono-, di- and trimethylated at K36). Dissociation constants were determined by following the chemical shift perturbations (CSP) of the LEDGF/p52 backbone amide signals induced upon titration with H3 peptides. Error bars represent the error of the fit for most perturbed residues (n = 10). (C) KD fits from NMR titrations obtained using 15N-labeled LEDGF and three 601-derived dsDNA oligonucleotides (17 bp–cyan, 18 bp – blue and 20 bp – red). Dissociation constants were determined by following the chemical shift perturbations (CSP) of the LEDGF/p52 backbone amide signals induced upon titration with oligonucleotides. Error bars represent the error of the fit for most perturbed residues (n = 10). (D, E) Equilibrium binding between the LEDGF/p52 or p75 splice variants and unmodified, di- or trimethylated nucleosomes, respectively, determined using microscale thermophoresis (MST). Error bars represent the standard deviation from the mean values obtained from n = 3 experiments. (F) Equilibrium binding between the full length LEDGF/p75 splice variant and di- or trimethylated nucleosomes reconstituted with 147 or 166 bp DNA. (G) EMSA showing the effect of the presence of PogZ(1117–1410) on the binding of LEDGF/p75 to di- or trimethylated nucleosomes. Error bars represent the standard deviation from the mean values obtained from n = 3 experiments. (H) Table summarizing the dissociation constants and hill coefficients for figures B-F.
H3K36me3 and H3K36me2 nucleosomes are recognized by the PWWP domain through a conserved mechanism
To prepare the cryo-EM samples, the LEDGF/p75-PogZ(1117–1410) binary complex was incubated with freshly reconstituted H3KC36me2 or H3KC36me3 147 bp nucleosomes and vitrified. After the initial 2D classification, there was an electron density identified for the PWWP domain region right next to both exit channels of the H3 N-terminal tail from the nucleosome (Supplementary Figure S3A, E). The published structure of the PWWP/H3KC36me3 nucleosome core particle (NCP) is in a good agreement with our experimental maps obtained for different methylation states (38). The structural conservation of the di- and trimethylation mark recognition by PWWP was recently shown for the ternary complex of the isolated PWWP domain from HRP3 (HDGF-related protein 3), 10 bp dsDNA and either H3K36me3- and H3K36me2-containing short peptides (26). The interaction between the PWWP aromatic cage and both the tri- and dimethylated Lys36 side-chains is maintained by cation–π interactions. The only difference is in additional stabilization of the dimethylated side-chain by a salt bridge to a side-chain from conserved Glu53 (Glu49 in LEDGF). The highly overlapping experimental electron density maps obtained for both modifications (Figure 2A, B) confirm the structural conservation of the recognition of both marks by PWWP in the nucleosomal context.
Figure 2.

Conservation of H3K36 di- and trimethylated nucleosome recognition by PWWP domain. (A) Structure of the PWWP domain from LEDGF/p75 bound to dimethylated H3KC36 mononucleosome, viewed from the nucleosome dyad. A close-up is showing densities for dimethylated S-aminoethyl-L-cysteine on histone H3 (ML2) and surrounding residues from PWWP (M15, Y18, W21, F44, T47). (B) Structure of the PWWP domain from LEDGF/p75 bound to trimethylated H3KC36 mononucleosome, viewed from the nucleosome dyad. A close-up is showing densities for trimethylated S-aminoethyl-L-cysteine on histone H3 (ML3) and surrounding residues from PWWP (M15, Y18, W21, F44, T47).
The region of the histone octamer acidic patch at the DNA interface forms an additional PWWP interaction site
One 3D class obtained for each methylation state comprised an additional density in proximity to the octamer acidic patch besides the density representing the PWWP bound to the exit channel of the N-terminal H3 tail (Figure 3A, B and Supplementary Figure S3A, E). We expected that this site might be occupied by the 80 kDa 2:2 IBD-PogZ(1117–1410) complex, but the size and shape of this additional density resembled the density observed for the canonical position of the PWWP domain, which suggests an alternative binding site for PWWP (Figure 3A). Next, we used chemical crosslinking coupled with mass spectrometry to confirm the density as PWWP. We used an urea-based 12.5 Å long crosslinker for crosslinking the ternary complex of the LEDGF-PogZ(1117–1410)-H3KC36me3 nucleosome. Subsequent proteolysis and mass spectrometry analysis revealed a number of crosslinked intermolecular lysine pairs that suggests a spatial proximity of specific LEDGF regions to core histones H2A and H2B (Figure 3C and Supplementary Figure S4). In particular, there were four crosslinks identified between the PWWP domain and H2A-H2B dimer which support the assignment of the additional density to the alternatively bound PWWP domain. To investigate the ability of the PWWP domain to interact with H2A-H2B in the absence of DNA, we titrated the 15N-labeled LEDGF/p52 with up to 16-fold excess of the unlabeled H2A-H2B dimer by NMR spectroscopy, but there were no changes in amide backbone signal positions (data not shown), further confirming that the interaction at this site is primarily maintained by contacts with nucleosomal DNA. We fitted the density with a PWWP orientation, based on the X-ray structure of the HRP3 PWWP domain bound in the minor groove of a short dsDNA (26). The obtained model is supported by a charge complementarity between the DNA phosphate backbone and the positively charged loops that are implicated in DNA contacts within the canonical PWWP pose (Figure 3C).
Figure 3.

Structure of the PWWP domain from LEDGF bound to di- or trimethylated H3KC36 nucleosome. (A) Trimethylated nucleosome with both the canonical and the alternative PWWP domain position density, with superhelical locations (SHL) indicated. (B) The position of the alternative PWWP binding site relatively to the acidic patch formed by the H2A-H2B dimeric interface. (C) The pose was validated by chemical crosslinks (dashed black lines) between H2A, H2B and the PWWP domain.
Specific sites within the intrinsically disordered central region of LEDGF selectively interact with nucleosomal or extranucleosomal DNA
We used NMR spectroscopy to provide more insight into the dynamic nature of the LEDGF association with nucleosomes. The central region of LEDGF that is N-terminally flanked by the PWWP domain and by the IBD domain at the C-terminus is predicted to be highly disordered (IDRs) (Figure 4A). We analyzed the chemical shift values obtained for the backbone resonances of the LEDGF/p52 splice variant and revealed that the central region is almost entirely dynamic, with the exception of a short α-helical segment between residues 310 and 316 at the very end of LEDGF/p52 (Figure 4A, B). Interestingly, amide group NMR signals from the three short putative DNA interaction regions, nuclear localization signal (NLS) and a pair of canonical AT-hooks, were not present in the triple-resonance NMR spectra, perhaps due to their unfavorable chemical exchange properties.
Figure 4.

NMR analysis of LEDGF binding to DNA and H3K36me3-modified nucleosome. (A) Disorder probability and NMR-derived order parameter S2 calculated for LEDGF/p52 residues. (B) Distribution of regular secondary structure elements in LEDGF/p52. (C) DNA binding of LEDGF/p52 monitored in the 2D 15N/1H HSQC spectra. Overlaid LEDGF backbone signals reveal the effect of an increasing DNA concentration (stoichiometry protein:DNA 1:0.5, 1, 2, 4, 8 and 16). (D) Chemical shift perturbations per LEDGF/p52 residues induced by DNA binding at the 16fold excess of DNA. The positions of the PWWP domain and DNA binding modules (NLS – nuclear localization signal and AT-hooks) are highlighted above the graph. (E) An overlay of the 3D HNCO LEDGF/p52 spectra acquired in the absence (grey) and presence of the 0.5 molar equivalent of Widom 147 bp DNA (red), H3KC36me3-modified (orange) and native nucleosomes (cyan). Changes in signal positions are highlighted by black arrows. (F–H) Chemical shift perturbations induced by an interaction with the H3KC36me3, native nucleosomes and nucleosomal DNA.
Titration of 15N/13C-labeled LEDGF/p52 with short Widom DNA-derived oligonucleotides (Figure 4C) was in addition to determining KD values (Figure 1C) used to obtain information on the DNA binding properties of LEDGF. Chemical shift perturbation (CSP) analysis (Figure 4D) revealed that multiple regions of LEDGF interact with a linear DNA duplex. As expected, the DNA binds to the extensive positively charged patch on the PWWP domain that is involved in the direct contact with both the di- and trimethylated H3KC36 nucleosome. In addition, DNA binding affects three regions in the central IDR of LEDGF—residues 134–145 adjacent to the NLS, residues 186–204, which include a pair of putative AT-hooks, and residues 286–290, which precede the more rigid α-helical region.
A similar experimental setup was used to investigate the binding of LEDGF to nucleosomes by comparison of the LEDGF/p52 NMR spectra obtained in the presence and absence of native nucleosomes, H3KC36me3 nucleosomes or 147 bp Widom DNA (Figure 4E). In all three cases, the NMR signals from the PWWP domain disappeared after binding due to line broadening associated with the domain stably interacting with relatively much larger molecules while accepting their slower tumbling rates (Figure 4F–H). The NMR signals from the central region of LEDGF remained detectable in the spectra which is suggesting that this region remains relatively dynamic upon binding. Overall, the CSP profiles resembled that observed for the short DNA interaction (Figure 4D). Signals in the first affected region (residues 134–145) showed an extensive change in position, indicating a substantial conformational rearrangement of these residues upon binding to the native or H3KC36me3 nucleosomes and short or 147 bp Widom DNA. However, the CSPs observed for the residues in the two remaining regions (186–204 and 286–290) for both unmethylated and K36-methylated nucleosomes (Figure 4F, G) were significantly lower than those obtained for short or Widom DNA interactions (Figure 4E,H). This indicates that the short regions between residues 186–204 and 286–290 have a clear preference for extranucleosomal DNA, while the region encompassing residues from 134 to 145 does not distinguish between the intra- and extranucleosomal DNA. The absence of trimethylated mark of H3K36 had no effect on the CSP profiles (Figure 4F, G), most likely due to the concentration of interacting molecules being at least 30-fold higher than the dissociation constant of the interaction (Figure 1H).
LEDGF complex with H3K36-trimethylated dinucleosome
Based on the preferential binding of the two regions within the central LEDGF IDR to extranucleosomal DNA, we attempted to resolve the central region of LEDGF in the context of the H3KC36me3 dinucleosome. In order to minimize the flexibility of the system, we reconstituted the dinucleosomes using a short 20 bp DNA linker joining two 147 bp Widom DNA sequences. Similar to the mononucleosome samples, we incubated H3KC36me3 dinucleosomes with the LEDGF/p75-PogZ(1117–1410) binary complex before preparing cryo-EM samples. Despite the short DNA linker, the 2D classes showed densities for one defined and for one blurred nucleosome (referred as ‘second nucleosome’) (Supplementary Figure S3I), which is a consequence of the relative motions between NCP subunits and did not allow selection of a subpopulation of particles that would provide a single rigid, high-resolution structure of the dinucleosome. Therefore, we first performed 3D focus-classification and refinement on the defined nucleosome, followed by signal subtraction, 3D classification and refinement of the second nucleosome. This procedure yielded two independent 3D reconstructions with a global resolution of 3.34 Å for the defined half and 4.0 Å for the second half of the dinucleosome (Figure 5 and Supplementary Figure S3I–K). Next, we determined the relative motion between two nucleosomes using principal component analysis in a multi-body refinement approach (37) (Figure 5A). The two independent maps were rigid body fitted by individual NCPs, two PWWP domains bound at canonical H3KC36me3 sites of the defined nucleosome and one PWWP domain at the DNA linker proximal H3KC36me3 site of the second nucleosome (Figure 5B). The placement of PWWP domains on the defined NCP corresponds to distinct densities in the 2D classification. There was no additional density resolved in the DNA linker region or at the interface between the acidic patch and nucleosomal DNA occupied by a second PWWP domain in the complexes of LEDGF with di- or trimethylated mononucleosomes. The data suggest that the IBD of LEDGF/p75 bound to the PogZ(1117–1410) is not tightly associated with the dinucleosome and the central LEDGF IDR does not adopt a single distinct conformation upon binding to the internucleosomal DNA, but retains its dynamic properties.
Figure 5.

Structure of the PWWP domain from LEDGF bound to trimethylated H3KC36 dinucleosome. (A) Mutual nucleosome movements illustrated by the extremities of their relative orientation and visualized by three principal component analysis. (B) The densities with rigid body fitted nucleosomes and PWWP domains bound at three out of the four available canonical H3KC36me3 sites.
DISCUSSION
Covalent modifications of histones represent an essential mechanism for expression regulation of specific genes in living organisms. Ectopic changes in this highly regulated system often lead to the development of human diseases. Unfortunately, the molecular mechanisms of the recognition of various states of particular marks are not entirely understood. Here, we employed cryo-EM coupled with NMR spectroscopy and micro-scale thermophoresis to understand the basis of the H3K36 di- and trimethylation mark recognition in the nucleosomal context.
The recently published cryo-EM structure obtained for LEDGF/p75 bound to the H3KC36me3 nucleosome showed only the PWWP domain, while the IDR as well as the IBD domain remained unresolved (38). Most particles contained a single resolved PWWP at one of the exit channels of the H3 N-terminus. However, a significant population of particles showed a less resolved density at the second exit channel, revealing that the H3KC36me3 NCP can bind two LEDGF/p75 molecules simultaneously. In addition, the study required a longer DNA and chemical crosslinking in order to produce a stable LEDGF-H3KC36me3 NCP complex that yielded high-resolution cryo-EM data. Our binding data suggested that the 2:1 complex is formed in vitro for both methylation states (Figure 1H). Recently, we uncovered the mechanism of LEDGF dimerization through its C-terminal domain (61). We hypothesized that the constitutive dimer stitched by the dimeric C-terminal domain of chromatin regulator PogZ might facilitate the formation of a 2:1 LEDGF-NCP due to an increased avidity that would not dissociate upon vitrification during cryo-EM sample preparation. In addition, the presence of a large dimeric 64 kDa assembly might help resolve the LEDGF IBD in cryo-EM data. However, we observed reduced binding of LEDGF/p75 to methylated nucleosomes in the presence of PogZ (Figure 1G), therefore the predicted stabilizing effect was not manifested by increased affinity of LEDGF/p75 towards NCPs.
We obtained cryo-EM datasets for two ternary complexes of LEDGF-PogZ-NCP carrying di- or trimethylation at H3KC36 (Figures 2 and 3; Supplementary Figures S2 and S3). The 3D classification for both complexes yielded similar classes, and the best resolved ones were subjected to full 3D refinement with an overall resolution of 2.69 Å for di- and 3.02 Å for trimethylated complex, respectively. The experimental maps revealed similar positioning of PWWP to that observed previously (38), with one of the PWWP poses significantly enriched. This confirms the capacity of methylated nucleosomes to accommodate two LEDGF molecules with some cooperativity, as suggested by the analysis of the MST binding data (Figure 1D–F). The published structural model could be satisfactorily fitted in the maps obtained for both modifications, which suggests that the di- or trimethylation state of KC36 does not affect the orientation of the domain relative to the nucleosome. The LEDGF regions downstream of the PWWP domain again remain unresolved despite the presence of PogZ dimer, which suggests that the PWWP remains flexibly connected to the rest of the complex via the central intrinsically disordered LEDGF region. Our data demonstrate that the distinct transcriptional programs triggered by H3K36 di- and trimethylation marks are independent of the actual recognition mode by a cognate reader domain and might require an orthogonal regulatory mechanism, such as a specific recognition of unstructured linear motifs in relevant transcription elongation factors by the LEDGF protein interaction module (16,62).
A pair of the less populated 3D classes obtained for di- and trimethylated LEDGF-PogZ-NCP complexes revealed a novel LEDGF/p75 interaction site at the interface between the acidic patch and nucleosomal DNA (Figure 3). The relatively lower local resolution of the maps due to a limited number of particles did not allow for the building of a high-resolution model for this PWWP pose. Using NMR, we confirmed that this molecular arrangement cannot be sustained in the absence of nucleosomal DNA. This is analogous to the interaction of PWWP at the canonical site, where a disproportionately larger interaction energy contribution comes from the DNA (Figure 1B, C). As it was shown by LeRoy et al. (7) before, LEDGF and its ortholog HRP2 can overcome the nucleosomal barrier to transcription for RNA polymerase II in differentiated cells. Therefore, it is tempting to speculate that the alternative binding site for PWWP, together with LEDGF IDR, provide a molecular basis for this LEDGF role, as two FACT subunit regions bind to the same H2A-H2B dimeric interface as PWWP. In the case of FACT, however, the affinity of the electrostatically driven interaction is in the nanomolar range and does not require DNA (63). Still, the new PWWP pose might play a role in nucleosome reorganization. Interestingly, as we did not identify particles with a solely non-canonical PWWP pose (Figure 3), there might be a positive cooperativity between the two PWWP domains that could be linked through IBD-mediated dimerization, or even stapled by the interacting PogZ dimer.
The multivalent nature of binding of chromatin interacting proteins to multiple nucleosome components is relatively common (64). There are high-resolution structural data for several proteins that bind to the H2A-H2B dimer interface and DNA adjacent to the non-canonical PWWP binding site (summarized in Supplementary Figure S5). These include Regulator of Chromosome Condensation (RCC1) (65), cyclic GMP-AMP synthase (cGAS) (66–68), transcription factors OCT4 and SOX2 (69), AEBP2—a cofactor of the polycomb repressive complex 2; (70) and Set1—histone methyltransferase from the COMPASS complex (71). In addition, this region is targeted by a number of histone chaperones, such as Swc5 (72), that utilize the DEF/Y motifs for binding to the H2A–H2B dimer interface but require nucleosomal DNA displacement. However, none of these structurally resolved complexes reveal significantly overlapping binding interfaces with the non-canonical PWWP interface, which suggests that PWWP might interact with a unique binding site on the nucleosome. Of note, the structures of chromatin remodeling complexes bound to the nucleosome, such as the human BAF complex (73), leave this surface unoccupied despite extensive contact with neighboring nucleosomal surfaces.
The modular organization of LEDGF/p75, including the N-terminal chromatin recognition domain attached to the C-terminal protein interaction platform through a 200-residue long flexible chain, allows for a large action radius of the protein that could ‘crosslink’ several spatially neighbouring nucleosomes (Figure 6A,B). Using NMR spectroscopy, we probed the free as well as nucleosomal DNA interaction properties of the central LEDGF flexible region that consist of several DNA binding elements, e.g. AT-hooks. The NMR titration experiments revealed a preference of the two specific regions towards extranucleosomal DNA, while the DNA interaction properties of the third, more PWWP-proximal region are not influenced by DNA being part of the fully assembled nucleosome (Figure 4). Subsequent cryo-EM structural characterization of the ternary complex between LEDGF-PogZ and H3KC36-trimethylated dinucleosome did not resolve a stably located central region of LEDGF bound to the DNA linker (Figure 5), which probably remains conformationally heterogeneous.
Figure 6.

The chromatin interaction landscape of LEDGF. (A) Spatial considerations for LEDGF binding to chromatin. Approximate dimensions of nucleosomes and LEDGF molecule in extended or compacted form. (B) Possible distribution for LEDGF domains across a mononucleosome or nucleosomal array. (C) Hypothetical role of the central region of LEDGF in the positioning of elongation factors near RNA polymerase II at actively transcribed gene bodies marked by a H3K36 methylation. The PWWP domain is highlighted in green and the IBD in pink.
Multiple interactions of LEDGF with intra- or extranucleosomal DNA might have functional consequences: (i) increased affinity of LEDGF for chromatin, (ii) restriction of the space accessed by complexes formed between LEDGF and its cellular partners (59,60) or (iii) cooperation with chromatin binding activities of larger multi-subunit complexes involved in RNA polymerase II-dependent transcription elongation (62). The capacity of multivalent interactions with nucleosomes, extranucleosomal DNA and other proteins further highlights the role of LEDGF as an important component of the transcriptional machinery. The interactions of the LEDGF IBD (TND) domain have been recently shown to be finely orchestrated in the context of transcription elongation, where their selective disruption has caused elongation defects in gene expression (16). The interplay between the chromatin recognition module (PWWP) and the protein interaction hub (IBD) controlled by the central IDR DNA interactions might be therefore essential for the appropriate positioning of the LEDGF-bound factors near the elongating RNA polymerase II (Figure 6C).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Michal Svoboda, Anatolij Filimonenko (both IOCB Prague) and Ladislav Bumba (Institute of Microbiology, Prague) for help with data acquisition and discussion.
Author contributions: E.K., V.L. and V.V. designed research; E.K., V.L., T.K., J.Š., J.N., P.S., R.H., H.Š., Z.K., P.N., M.F., S.P. and V.V. performed research and analyzed data; E.K. and V.V. wrote the paper and all authors commented on the manuscript.
Contributor Information
Eliška Koutná, Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague 160 00, Czech Republic; Department of Cell Biology, Faculty of Science, Charles University, Prague 128 00, Czech Republic.
Vanda Lux, Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague 160 00, Czech Republic.
Tomáš Kouba, Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague 160 00, Czech Republic.
Jana Škerlová, Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague 160 00, Czech Republic.
Jiří Nováček, CEITEC, Brno 625 00, Czech Republic.
Pavel Srb, Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague 160 00, Czech Republic.
Rozálie Hexnerová, Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague 160 00, Czech Republic.
Hana Šváchová, Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague 160 00, Czech Republic.
Zdeněk Kukačka, Institute of Microbiology of the Czech Academy of Sciences, Prague 142 20, Czech Republic.
Petr Novák, Institute of Microbiology of the Czech Academy of Sciences, Prague 142 20, Czech Republic.
Milan Fábry, Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague 160 00, Czech Republic.
Simon Poepsel, Center for Molecular Medicine Cologne (CMMC), Faculty of Medicine and University Hospital Cologne, Cologne 509 31, Germany; Cologne Excellence Cluster for Cellular Stress Responses in Ageing-Associated Diseases (CECAD), University of Cologne, Cologne 509 31, Germany.
Václav Veverka, Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague 160 00, Czech Republic; Department of Cell Biology, Faculty of Science, Charles University, Prague 128 00, Czech Republic.
DATA AVAILABILITY
Analyses were performed using publicly or commercially available software. Figure 6 and the graphical abstract were created with BioRender.com. The structures were deposited in the Protein Data Bank (PDB accession codes: 8PC5, 8PC6, 8PEP, 8PEO, 8CBN and 8CBQ) and the electron microscopy densities were deposited in the Electron Microscopy Data Bank (EMDB, accession codes: EMD-17594, EMD-17595, EMD-17634, EMD-17633, EMD-16546 and EMD-16549). The assigned chemical shift values for the LEDGF/p52 backbone resonances were deposited in the Biological Magnetic Resonance Bank (BMRB accession code: 51794).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Czech Science Foundation (GACR) [22-03028S to V.V., 22-27695S to P.N.]; Ministry of Education of the Czech Republic [LO1304]; European Regional Development Fund; OP RDE; project Chemical Biology for Drugging Undruggable Targets (ChemBioDrug) [CZ.02.1.01/0.0/0.0/16_019/0000729 to V.V.]; Charles University [GA UK No. 1180119 to E.K.]; Center of Molecular Structure Core Facility at BIOCEV and the Cryo-EM Core Facility of CIISB, Instruct-CZ Centre, supported by the Ministry of Education of the Czech Republic [LM2018127, LM2023042]; European Regional Development Fund-Project ‘UP CIISB’ [CZ.02.1.01/0.0/0.0/18_046/0015974]; molecular graphics and analyses were performed with UCSF ChimeraX (developed by the Resource for Biocomputing, Visualization and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases). Funding for open access charge: GACR [22-03028S].
Conflict of interest statement. None declared.
REFERENCES
- 1. Biswas S., Rao C.M.. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018; 837:8–24. [DOI] [PubMed] [Google Scholar]
- 2. Huang C., Zhu B.. Roles of H3K36-specific histone methyltransferases in transcription: antagonizing silencing and safeguarding transcription fidelity. Biophysics Reports. 2018; 4:170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Qin S., Min J.. Structure and function of the nucleosome-binding PWWP domain. Trends Biochem. Sci. 2014; 39:536–547. [DOI] [PubMed] [Google Scholar]
- 4. DiFiore J.V., Ptacek T.S., Wang Y., Li B., Simon J.M., Strahl B.D.. Unique and Shared Roles for Histone H3K36 Methylation States in Transcription Regulation Functions. Cell Rep. 2020; 31:107751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wagner E.J., Carpenter P.B.. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012; 13:115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weinberg D.N., Papillon-Cavanagh S., Chen H., Yue Y., Chen X., Rajagopalan K.N., Horth C., McGuire J.T., Xu X., Nikbakht H.et al.. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature. 2019; 573:281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. LeRoy G., Oksuz O., Descostes N., Aoi Y., Ganai R.A., Kara H.O., Yu J.R., Lee C.H., Stafford J., Shilatifard A.et al.. LEDGF and HDGF2 relieve the nucleosome-induced barrier to transcription in differentiated cells. Sci. Adv. 2019; 5:eaay3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Okuda H., Kawaguchi M., Kanai A., Matsui H., Kawamura T., Inaba T., Kitabayashi I., Yokoyama A.. MLL fusion proteins link transcriptional coactivators to previously active CpG-rich promoters. Nucleic Acids Res. 2014; 42:4241–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sankaran S.M., Wilkinson A.W., Elias J.E., Gozani O.. A PWWP Domain of histone-lysine N-methyltransferase NSD2 binds to dimethylated lys-36 of histone H3 and regulates NSD2 function at chromatin. J. Biol. Chem. 2016; 291:8465–8474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu L., Li Q., Wong S.H.K., Huang M., Klein B.J., Shen J., Ikenouye L., Onishi M., Schneidawind D., Buechele C.et al.. ASH1L links histone H3 lysine 36 dimethylation to MLL leukemia. Cancer Discov. 2016; 6:770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu X., Lan B., Yi X., He C., Dang L., Zhou X., Lu Y., Sun Y., Liu Z., Bai X.et al.. HRP2–DPF3a–BAF complex coordinates histone modification and chromatin remodeling to regulate myogenic gene transcription. Nucleic Acids Res. 2020; 48:6563–6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shirane K., Miura F., Ito T., Lorincz M.C.. NSD1-deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb-associated silencing. Nat. Genet. 2020; 52:1088–1098. [DOI] [PubMed] [Google Scholar]
- 13. Ciuffi A., Llano M., Poeschla E., Hoffmann C., Leipzig J., Shinn P., Ecker J.R., Bushman F.. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005; 11:1287–1289. [DOI] [PubMed] [Google Scholar]
- 14. Maertens G., Cherepanov P., Pluymers W., Busschots K., De Clercq E., Debyser Z., Engelborghs Y.. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 2003; 278:33528–33539. [DOI] [PubMed] [Google Scholar]
- 15. Booth V., Koth C.M., Edwards A.M., Arrowsmith C.H.. Structure of a conserved domain common to the transcription factors TFIIS, elongin A, and CRSP70. J. Biol. Chem. 2000; 275:31266–31268. [DOI] [PubMed] [Google Scholar]
- 16. Cermakova K., Demeulemeester J., Lux V., Nedomova M., Goldman S.R., Smith E.A., Srb P., Hexnerova R., Fabry M., Madlikova M.et al.. A ubiquitous disordered protein interaction module orchestrates transcription elongation. Science. 2021; 374:1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ge H., Si Y., Roeder R.G.. Isolation of cDNAs encoding novel transcription coactivators p52 and p75 reveals an alternate regulatory mechanism of transcriptional activation. EMBO J. 1998; 17:6723–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sutherland H.G., Newton K., Brownstein D.G., Holmes M.C., Kress C., Semple C.A., Bickmore W.A.. Disruption of Ledgf/Psip1 Results in Perinatal Mortality and HomeoticSkeletal Transformations. Mol. Cell. Biol. 2006; 26:7201–7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Méreau H., De Rijck J., Čermáková K., Kutz A., Juge S., Demeulemeester J., Gijsbers R., Christ F., Debyser Z., Schwaller J.. Impairing MLL-fusion gene-mediated transformation by dissecting critical interactions with the lens epithelium-derived growth factor (LEDGF/p75). Leukemia. 2013; 27:1245–1253. [DOI] [PubMed] [Google Scholar]
- 20. Daugaard M., Baude A., Fugger K., Povlsen L.K., Beck H., Sørensen C.S., Petersen N.H.T., Sorensen P.H.B., Lukas C., Bartek J.et al.. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012; 19:803–810. [DOI] [PubMed] [Google Scholar]
- 21. Eidahl J.O., Crowe B.L., North J.A., McKee C.J., Shkriabai N., Feng L., Plumb M., Graham R.L., Gorelick R.J., Hess S.et al.. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013; 41:3924–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pradeepa M.M., Sutherland H.G., Ule J., Grimes G.R., Bickmore W.A.. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012; 8:e1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Nuland R., Van Schaik F.M.A., Simonis M., Van Heesch S., Cuppen E., Boelens R., Timmers H.T.M., Van Ingen H.. Nucleosomal DNA binding drives the recognition of H3K36-methylated nucleosomes by the PSIP1-PWWP domain. Epigenetics Chromatin. 2013; 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rona G.B., Eleutherio E.C.A., Pinheiro A.S.. PWWP domains and their modes of sensing DNA and histone methylated lysines. Biophys. Rev. 2016; 8:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qiu Y., Zhang W., Zhao C., Wang Y., Wang W., Zhang J., Zhang Z., Li G., Shi Y., Tu X.et al.. Solution structure of the Pdp1 PWWP domain reveals its unique binding sites for methylated H4K20 and DNA. Biochem. J. 2011; 442:527–538. [DOI] [PubMed] [Google Scholar]
- 26. Tian W., Yan P., Xu N., Chakravorty A., Liefke R., Xi Q., Wang Z.. The HRP3 PWWP domain recognizes the minor groove of double-stranded DNA and recruits HRP3 to chromatin. Nucleic Acids Res. 2019; 47:5436–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang H., Farnung L., Dienemann C., Cramer P.. Structure of H3K36-methylated nucleosome-PWWP complex reveals multivalent cross-gyre binding. Nat. Struct. Mol. Biol. 2020; 27:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luger K., Rechsteiner T.J., Richmond T.J.. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 1999; 119:1–16. [DOI] [PubMed] [Google Scholar]
- 29. Dyer P.N., Edayathumangalam R.S., White C.L., Bao Y., Chakravarthy S., Muthurajan U.M., Luger K.. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004; 375:23–44. [DOI] [PubMed] [Google Scholar]
- 30. Margueron R., Justin N., Ohno K., Sharpe M.L., Son J., Drury W.J., Voigt P., Martin S.R., Taylor W.R., De Marco V.et al.. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009; 461:762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Simon M.D., Chu F., Racki L.R., de la Cruz C.C., Burlingame A.L., Panning B., Narlikar G.J., Shokat K.M.. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007; 128:1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mastronarde D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005; 152:36–51. [DOI] [PubMed] [Google Scholar]
- 33. Zheng S.Q., Palovcak E., Armache J.P., Verba K.A., Cheng Y., Agard D.A.. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017; 14:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 2016; 193:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bepler T., Morin A., Rapp M., Brasch J., Shapiro L., Noble A.J., Berger B.. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods. 2019; 16:1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zivanov J., Nakane T., Scheres S.H.W.. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ. 2020; 7:253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakane T., Kimanius D., Lindahl E., Scheres S.H.. Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. Elife. 2018; 7:e36861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang H., Farnung L., Dienemann C., Cramer P.. Structure of H3K36-methylated nucleosome–PWWP complex reveals multivalent cross-gyre binding. Nat. Struct. Mol. Biol. 2019; 27:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang H., Xiong L., Cramer P.. Structures and implications of TBP-nucleosome complexes. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2108859118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.H., Ferrin T.E.. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021; 30:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Afonine P.V., Poon B.K., Read R.J., Sobolev O.V., Terwilliger T.C., Urzhumtsev A., Adams P.D.. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018; 74:531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liebschner D., Afonine P.V., Baker M.L., Bunkoczi G., Chen V.B., Croll T.I., Hintze B., Hung L.W., Jain S., McCoy A.J.et al.. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct. Biol. 2019; 75:861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 44. Kovalevskiy O., Nicholls R.A., Long F., Carlon A., Murshudov G.N.. Overview of refinement procedures within REFMAC5: utilizing data from different sources. Acta Crystallogr D Struct. Biol. 2018; 74:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wood C., Burnley T., Patwardhan A., Scheres S., Topf M., Roseman A., Winn M.. Collaborative computational project for electron cryo-microscopy. Acta. Crystallogr. D Biol. Crystallogr. 2015; 71:123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brown A., Long F., Nicholls R.A., Toots J., Emsley P., Murshudov G.. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta. Crystallogr. D Biol. Crystallogr. 2015; 71:136–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A.et al.. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B.et al.. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018; 27:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Berman H., Henrick K., Nakamura H.. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003; 10:980–980. [DOI] [PubMed] [Google Scholar]
- 50. Moriarty N.W., Grosse-Kunstleve R.W., Adams P.D.. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta. Crystallogr. D Biol. Crystallogr. 2009; 65:1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jakobi A.J., Wilmanns M., Sachse C.. Model-based local density sharpening of cryo-EM maps. Elife. 2017; 6:e27131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Renshaw P.S., Veverka V., Kelly G., Frenkiel T.A., Williamson R.A., Gordon S.V., Hewinson R.G., Carr M.D.. Sequence-specific assignment and secondary structure determination of the 195-residue complex formed by the Mycobacterium tuberculosis proteins CFP-10 and ESAT-6. J. Biomol. NMR. 2004; 30:225–226. [DOI] [PubMed] [Google Scholar]
- 53. Veverka V., Lennie G., Crabbe T., Bird I., Taylor R.J., Carr M.D.. NMR assignment of the mTOR domain responsible for rapamycin binding. J. Biomol. NMR. 2006; 36:3. [DOI] [PubMed] [Google Scholar]
- 54. Lee W., Tonelli M., Markley J.L.. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015; 31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Veverka V., Crabbe T., Bird I., Lennie G., Muskett F.W., Taylor R.J., Carr M.D.. Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor: compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene. 2008; 27:585–595. [DOI] [PubMed] [Google Scholar]
- 56. Gotze M., Pettelkau J., Fritzsche R., Ihling C.H., Schafer M., Sinz A.. Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J. Am. Soc. Mass. Spectrom. 2015; 26:83–97. [DOI] [PubMed] [Google Scholar]
- 57. Kukacka Z., Rosulek M., Jelinek J., Slavata L., Kavan D., Novak P.. LinX: a software tool for uncommon cross-linking chemistry. J. Proteome Res. 2021; 20:2021–2027. [DOI] [PubMed] [Google Scholar]
- 58. Abeliovich H. An empirical extremum principle for the Hill coefficient in ligand-protein interactions showing negative cooperativity. Biophys. J. 2005; 89:76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sharma S., Čermáková K., De Rijck J., Demeulemeester J., Fábry M., El Ashkar S., Van Belle S., Lepšík M., Tesina P., Duchoslav V.et al.. Affinity switching of the LEDGF/p75 IBD interactome is governed by kinase-dependent phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E7053–E7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tesina P., Cermáková K., Horejší M., Procházková K., Fábry M., Sharma S., Christ F., Demeulemeester J., Debyser Z., Rijck J.D.et al.. Multiple cellular proteins interact with LEDGF/p75 through a conserved unstructured consensus motif. Nat. Commun. 2015; 6:7968. [DOI] [PubMed] [Google Scholar]
- 61. Lux V., Brouns T., Cermakova K., Srb P., Fabry M., Madlikova M., Horejsi M., Kukacka Z., Novak P., Kugler M.et al.. Molecular Mechanism of LEDGF/p75 Dimerization. Structure. 2020; 28:1288–1299. [DOI] [PubMed] [Google Scholar]
- 62. Cermakova K., Veverka V., Hodges H.C.. The TFIIS N-terminal domain (TND): a transcription assembly module at the interface of order and disorder. Biochem. Soc. Trans. 2023; 51:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kemble D.J., McCullough L.L., Whitby F.G., Formosa T., Hill C.P.. FACT disrupts nucleosome structure by binding H2A-H2B with conserved peptide motifs. Mol. Cell. 2015; 60:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McGinty R.K., Tan S.. Recognition of the nucleosome by chromatin factors and enzymes. Curr. Opin. Struct. Biol. 2016; 37:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Makde R.D., England J.R., Yennawar H.P., Tan S.. Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature. 2010; 467:562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cao D., Han X., Fan X., Xu R.M., Zhang X.. Structural basis for nucleosome-mediated inhibition of cGAS activity. Cell Res. 2020; 30:1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Boyer J.A., Spangler C.J., Strauss J.D., Cesmat A.P., Liu P., McGinty R.K., Zhang Q.. Structural basis of nucleosome-dependent cGAS inhibition. Science. 2020; 370:450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pathare G.R., Decout A., Gluck S., Cavadini S., Makasheva K., Hovius R., Kempf G., Weiss J., Kozicka Z., Guey B.et al.. Structural mechanism of cGAS inhibition by the nucleosome. Nature. 2020; 587:668–672. [DOI] [PubMed] [Google Scholar]
- 69. Michael A.K., Grand R.S., Isbel L., Cavadini S., Kozicka Z., Kempf G., Bunker R.D., Schenk A.D., Graff-Meyer A., Pathare G.R.et al.. Mechanisms of OCT4-SOX2 motif readout on nucleosomes. Science. 2020; 368:1460–1465. [DOI] [PubMed] [Google Scholar]
- 70. Kasinath V., Beck C., Sauer P., Poepsel S., Kosmatka J., Faini M., Toso D., Aebersold R., Nogales E.. JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science. 2021; 371:eabc3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hsu P.L., Shi H., Leonen C., Kang J., Chatterjee C., Zheng N.. Structural basis of H2B ubiquitination-dependent H3K4 methylation by COMPASS. Mol. Cell. 2019; 76:712–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Huang Y., Sun L., Pierrakeas L., Dai L., Pan L., Luk E., Zhou Z.. Role of a DEF/Y motif in histone H2A-H2B recognition and nucleosome editing. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:3543–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. He S., Wu Z., Tian Y., Yu Z., Yu J., Wang X., Li J., Liu B., Xu Y.. Structure of nucleosome-bound human BAF complex. Science. 2020; 367:875–881. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Analyses were performed using publicly or commercially available software. Figure 6 and the graphical abstract were created with BioRender.com. The structures were deposited in the Protein Data Bank (PDB accession codes: 8PC5, 8PC6, 8PEP, 8PEO, 8CBN and 8CBQ) and the electron microscopy densities were deposited in the Electron Microscopy Data Bank (EMDB, accession codes: EMD-17594, EMD-17595, EMD-17634, EMD-17633, EMD-16546 and EMD-16549). The assigned chemical shift values for the LEDGF/p52 backbone resonances were deposited in the Biological Magnetic Resonance Bank (BMRB accession code: 51794).