

Abstract
Background
DJ‐1 is a causative gene for Parkinson's disease. DJ‐1‐deficient mice develop gait‐associated progressive behavioural abnormalities and hypoactive forearm grip strength. However, underlying activity mechanisms are not fully explored.
Methods
Western blotting and quantitative real‐time polymerase chain reaction approaches were adopted to analyse DJ‐1 expression in skeletal muscle from aged humans or mice and compared with young subjects. Skeletal muscle‐specific‐DJ‐1 knockout (MDKO) mice were generated, followed by an assessment of the physical activity phenotypes (grip strength, maximal load capacity, and hanging, rotarod, and exercise capacity tests) of the MDKO and control mice on the chow diet. Muscular atrophy phenotypes (cross‐sectional area and fibre types) were determined by imaging and quantitative real‐time polymerase chain reaction. Mitochondrial function and skeletal muscle morphology were evaluated by oxygen consumption rate and electron microscopy, respectively. Tail suspension was applied to address disuse atrophy. RNA‐seq analysis was performed to indicate molecular changes in muscles with DJ‐1 ablation. Dual‐luciferase reporter assays were employed to identify the promoter region of Trim63 and Fbxo32 genes, which were indirectly regulated by DJ‐1 via the FoxO1 pathway. Cytoplasmic and nuclear fractions of DJ‐1‐deleted muscle cells were analysed by western blotting. Compound 23 was administered into the gastrocnemius muscle to mimic the of DJ‐1 deletion effects.
Results
DJ‐1 expression decreased in atrophied muscles of aged human (young men, n = 2; old with aged men, n = 2; young women, n = 2; old with aged women, n = 2) and immobilization mice (n = 6, P < 0.01). MDKO mice exhibited no body weight difference compared with control mice on the chow diet (Flox, n = 8; MDKO, n = 9). DJ‐1‐deficient muscles were slightly dystrophic (Flox, n = 7; MDKO, n = 8; P < 0.05), with impaired physical activities and oxidative capacity (n = 8, P < 0.01). In disuse‐atrophic conditions, MDKO mice showed smaller cross‐sectional area (n = 5, P < 0.01) and more central nuclei than control mice (Flox, n = 7; MDKO, n = 6; P < 0.05), without alteration in muscle fibre types (Flox, n = 6; MDKO, n = 7). Biochemical analysis indicated that reduced mitochondrial function and upregulated of atrogenes induced these changes. Furthermore, RNA‐seq analysis revealed enhanced activity of the FoxO1 signalling pathway in DJ‐1‐ablated muscles, which was responsible for the induction of atrogenes. Finally, compound 23 (an inhibitor of DJ‐1) could mimic the effects of DJ‐1 ablation in vivo.
Conclusions
Our results illuminate the crucial of skeletal muscle DJ‐1 in the regulation of catabolic signals from mechanical stimulation, providing a therapeutic target for muscle wasting diseases.
Keywords: Atrogenes, Atrophy, DJ‐1, Skeletal muscle
Introduction
Muscle wasting or atrophy can occur systemically under various conditions, including aging, immobilization, cancer, and metabolic and neurodegenerative diseases. 1 , 2 Although multiple proteolytic pathways are involved in muscle protein breakdown, degradation primarily through the ubiquitin‐proteasome system activation may account for up to 80% of proteolysis during skeletal muscle loss. 3 Polyubiquitination of protein substrates is required for ubiquitin‐proteasome system activation, and E3 ligases can tag ubiquitin to specific proteins. For example, the muscle RING finger 1 (MuRF1, encoded by Trim63) and Atrogin‐1 (encoded by Fbxo32) E3 ligases are proposed as key regulators in the atrophy process. MuRF1 has been shown to play a direct role in myosin heavy chain ubiquitination and degradation, 4 while Atrogin‐1 can control the processes associated with protein synthesis. 5 Considering the crucial involvement of Atrogin‐1 and MuRF1 in the progression of typical muscle wasting, studies have emphasized understanding the upstream regulation of these primary atrophy markers.
The Forkhead box O (FoxO) transcription factor family, including FoxO1, FoxO3a, and FoxO4, is critical for controlling muscle degradation through regulating atrogenes expression. Under catabolic conditions, the phosphoinositide 3‐kinase (PI3K)‐AKT pathway is suppressed and hypophosphorylated FoxO translocates into the nucleus, causing MuRF‐1 and Atrogin‐1 induction leading to muscle atrophy. 6 , 7 In line with this, FoxO1 has been shown to manipulate MuRF‐1 and Atrogin‐1 transcription. 8 , 9 In another report, FoxO4 was linked to tumour necrosis factor α‐induced expression of Atrogin‐1 in the C2C12 myotubes. 10 These findings indicate that different members of the FoxO family respond to various catabolic conditions by promoting MuRF‐1 and/or Atrogin‐1 expression.
DJ‐1 was first characterized as a causative gene for Parkinson's disease. 11 In addition, several subsequent studies have suggested that inactivation and/or excess activation of DJ‐1 functions leads to the onset of oxidative stress‐related diseases, including type 2 diabetes mellitus, 12 male infertility, 13 and various neurodegenerative diseases such as Alzheimer's 14 and amyotrophic lateral sclerosis. 15 Further studies have reported that DJ‐1 knockout mice are hypoactive on the chow diet compared with Flox mice. 16 , 17 Notably, Seyfarth et al. showed the reduced activity of DJ‐1−/− mice, accompanied by a decreased lean mass. 16 However, additional clarification is needed to specify whether the reduced muscle mass is causative or a consequence of hypoactivity.
To elucidate the role of DJ‐1 in controlling muscle mass, we generated DJ‐1 conditional knockout mice by crossing the myosin light polypeptide 1 (Myl1)‐cre mice with DJ‐1 Flox mice. Although the skeletal muscle‐specific DJ‐1 knockout (MDKO) mice exhibited normal metabolic phenotypes on the chow diet, their activities were impaired with mildly decreased lean mass. Moreover, under atrophic conditions (immobilization), DJ‐1 ablated muscles became severely dystrophic, with a smaller cross‐sectional area (CSA) and more central nuclei than Flox mice. Mechanistically, we observed that DJ‐1 exerted its effects via the FoxO1 signalling pathway. In the present study, we have demonstrated the novel role of DJ‐1 in muscular atrophy development, providing a potential therapeutic target for muscle wasting conditions.
Methods
Human subjects
A total of four men and four women from Huashan Hospital and Zhongshan Hospital, Fudan University were included in this study. All subjects underwent routine physical and laboratory examinations. The subjects were divided into men and women groups and then were further divided into young and old groups based on age. Skeletal muscle tissue near knee joint was collected with use of arthroscopy during operation. All studies were in accordance with the 1975 Declaration of Helsinki and approved by the ethics committee of Huashan Hospital and Zhongshan Hospital, Fudan University. Each subject provided written informed consent.
Animals
All animal experiments were conducted in accordance with the guidelines of Science Research Ethics Committee at the Shanghai University of Sport (No. 102772022DW016) and approved by the Animal Care and Use Committee at the Shanghai University of Sports. Male and female C57BL/6J wild‐type mice were from GemPharmatech Co., Ltd (Jiangsu, China). DJ‐1flox/flox mice were generated using CRISPR/Cas9 technology. DJ‐1 gene has 10 transcripts. According to the structure of DJ‐1 gene, exon3‐exon5 of DJ‐1 (ENSMUST00000030805.13) was set as the knockout region. To delete DJ1 expression in myocyte, we bred the DJ‐1Flox/Flox mice with the Myl1‐Cre transgenic mice and obtained the Myl1‐Cre; DJ‐1Flox/Flox (referred to as MDKO). The Cre‐negative Floxed DJ‐1 mice (i.e., DJ‐1Flox/Flox) were used as controls (referred to as Flox) in this study.
Immobilization protocol
For immobilization‐induced muscle atrophy, a tail ring was formed with a 2‐0 sterile surgical steel wire. Mouse tail were inserted with the tail ring, then fixed and hung. All mice were sacrificed to collect samples for further experiments after 14 days suspension.
Compound 23 treatment
Compound 23 was dissolved in corn oil at a concentration of 2 mg/kg and treated during immobilization. Mice were immobilized as previously described. Compound 23 and corn oil were injected into gastrocnemius (GAS) of left and right leg, respectively, at 3rd day of immobilization. All mice were killed after 7 days immobilization.
Autophagy flux protocol
Autophagy flux mice model was constructed using colchicine. 18 Briefly, mice were injected with either 0.4 mg/kg colchicine or a control (vehicle) solution and then starved for 15 h. After that, mice were treated with colchicine or the control solution repeatedly and starved again. Finally, 15 h later, muscle tissues of the mice were harvested to examine the autophagy flux.
In vivo muscle contractility analysis
TA muscles contractility analysis was conducted by 1300 A 3‐in‐1 whole animal system (Aurora Scientific). Mice were anaesthetised with isopentane (0.8 g/kg body weight) and warmed by heat lamp. The hair of the hind legs was shaved, and the legs were fixed and immobilized in a frame without affecting blood flow. Tie around the patellar ligament of the distal TA muscle. Expose the sciatic nerve and tie another knot around the proximal end of the sciatic nerve. The distal suture ring of TA tendon was connected to the lever arm hook of the instrument to measure the contractile force. Results were analysed by DMA software (Aurora Scientific). The measurement of muscle contractility was based on an average of five tests.
Muscle‐grip strength test
Mice were measured for four limbs grip strength with a grip strength meter. Mice were gently pulled away by the tail until the mice released themselves from a metal grid with forepaws. The measurement of grip strength was based on an average of three tests. Mice were weighed and acclimated to the grip strength meter for 2 days prior to the first test.
Maximal load capacity test
The maximal load capacity was tested in male and female MDKO mice and age‐matched Flox mice. Before the test, mice were acclimated to the ladder for 2 days. Acclimation consisted of 10–12 repetitions per day with a 2 min rest in between. During the test, loads were increased from 30% body weight followed by a 4 g increased every repetition. A 2 min rest was set between repetitions, and the maximal load was analysed.
Hanging test
Mice were measured for four limbs and forelimbs strength. For the four limbs test, mice were placed on the grid of the big cage so that they were able to grasp it with all four paws. By inverting the grid, the mice hung, and the timer started immediately. The test ended for mice that were able to hang for 600 s. Mice that fall before this limit are given a maximum of two more tries. Analysing the maximum hanging time. For forelimbs test, mice were placed on a 2 mm thick metallic wire. Then let the mice grasping the wire with the only two forepaws, and lowered their hindlimbs so that the mice only hold onto the wire with two forepaws. The timer started immediately when the mice were released. The test ended for mice that were able to hang for 600 s. We analysed the maximum hanging time.
Rotarod
Mice were assessed for motor function using a rotarod test, conducted on a rotating rod (Ugo Basile) that accelerated from 4 to 40 rpm during the course of 5 min. For aged mice, the rotated rod accelerated from 0 to 20 rpm. A record of the time spent on the rod (before falling) was kept for each trial. We analysed the longest duration time.
Treadmill performance test
Mice were acclimated to the treadmill for 2 days before the test. During the performance test, mice were warmed up for 5 min at 10° and 10 m/min, and the speed was then increased by 2 m/min every 5 min until exhaustion.
Histology and immunofluorescence
GAS and soleus (SOL) muscles were collected on a piece of cork material, immobilized with tragacanth gum, then frozen in liquid nitrogen‐cooled 2‐methylbutane. For haematoxylin–eosin staining, cross‐sections with 10 μm thickness were cut and stained with haematoxylin–eosin (H&E) (BBI, E607317, and E607321). For immunostaining, sections with 20 μm thick. Primary antibodies against MyHC‐I (BA‐D5), MyHC‐IIa (SC‐71), and MyHC‐IIb (BF‐F3) were from Development Studies Hybridoma Bank (DSHB, Iowa, USA). The secondary antibodies against Alexa Fluor 350 anti‐mouse IgG2b (A‐21140), Alexa Fluor 488 anti‐mouse IgG1 (A‐21121), and Alexa Fluor 555 anti‐mouse IgM (A‐21426) were from Thermo Fisher. Photographs were obtained by confocal microscope (Zeiss), and quantifications were performed by Fiji.
Transmission electron microscopy
GAS and SOL muscles were collected, cut into pieces and pre‐fixed in 2.5% (vol/vol) glutaraldehyde overnight at 4°C (Solarbio, P1126). Then the muscles were washed in cold PBS three times and incubated in 1% OSO4 on ice for 1 h. Using en bloc stained with 2% Uranyl acetate on ice overnight, dehydrated in ethanol of different gradients, and infiltrated and embedded in SPON of different gradients. The grids were imaged with a JEM‐2100 transmission electron microscope (Hitachi HT7700, Japan).
Nuclear and cytoplasmic protein extraction
Nuclear and cytosolic fractions were conducted using Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, P0028) according to the instructions.
Muscle fibres permeabilization and mitochondrial respiration studies
GAS muscles were excised, cut into 15–20 mg weight and put into ice cold BIOPS (10 mM Ca‐EGTA, 0.1 μM free calcium, 20 mM imidazole, 20 mM taurine, 50 mM K‐MES, 0.5 mM DTT, 6.56 mM MgCl2, 5.77 mM ATP, and 15 mM phosphocreatine). Then fibre bundles were dissected with sharp forceps, incubated with ice cold BIOPS and saponin, and shaken for 30 min at 140 rpm. Then the fibres were gently and quickly transferred into MIRO5 (0.5 mM EGTA, 3 mM MgCl2, 60 mM lactobionic acid, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, and 110 mM d‐sucrose) at 140 rpm for 5 min. After permeabilization, a total of 2–3 mg fibres were added into the O2K chambers; 5 mM pyruvate, 2 mM malate, 10 mM glutamate, and 2.5 mM ADP were used to measure the maximal respiration flux; 0.5 μM Rotenone was added for respiration through complex I; 1 M succinate was added for complex II; 0.2 M N,N,N′,N′‐tetramethyl‐p‐phenylenediamine dihydrochloride, and 0.8 M ascorbate were for complex IV.
RNA sequencing studies
GAS tissues from immobilized MDKO mice and Flox mice were extracted for RNA sequencing. The sequencing was conducted using Illumina BGIseq500 platform (BGI‐Shenzhen, China). For signalling pathway analysis, the filtered datasets were uploaded into DAVID Bioinformatics Resources for reviewing biological pathways using the Functional Categories database. KEGG pathway analysis was used to interpret data and pathways were ranked by P value. 19
Cell lines, cell culture, C2C12 myotube differentiation, and transfection
HEK293 T cell and C2C12 cells were obtained from (national collection of authenticated cell cultures) and cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) at 37°C with 5% CO2. To induced myotube differentiation, the C2C12 cells were incubated with differentiation medium (DMEM containing 2% horse serum) when cell confluence to 85% for 6 days. Human shDj‐1 were cloned into pLKO.1‐puro vector to produce pLKO.1 + Dj‐1 (sh Dj‐1). Mice trim63 promoter, Trim63 mut (−192/−188 mutated) promoter, Fbxo32 promoter, and Fbxo32 mut (−38/−31 mutated) were inserted into pGL3‐basic vector. HEK293 T cells were transfected with indicated plasmids using polyethylenimine (PEI, PolySciences, 23966‐100) according to the instructions.
qRT‐PCR and western blot analysis
Total RNA of muscles was extracted using RNeasy Mini Kit. Then the RNA was converted into complementary deoxyribonucleic acid (cDNA) using High‐Capacity cDNA Reverse Transcription Kit (AB Applied Biosystems) according to the manufacturer's instructions. Quantitative real‐time PCR (qRT‐PCR) was conducted by QuantStudio 7 Flex Read Time PCR System (Life Technologies) using SYBR Green PCR Master Mix (QIANGEN). Table S1 lists the sequences of the primers used in this study. Cells were lysed in RIPA buffer, and tissue lysates were prepared as previously described. Protein concentrations were determined using the BCA Protein Assay Kit (P0010S, Beyotime, Shanghai, China). For western blot analysis, the protein solution was diluted by 5× loading buffer and heated at 95°C for 5 min. Protein lysates were loaded by SDS‐PAGE, transferred to a PVDF membrane, blocked in fat‐free milk for 1 h at room temperature, and incubated with the indicated primary antibodies overnight at 4°C and their corresponding secondary antibodies. Blots were developed with ECL. Antibodies information are listed in Table S2. For western blotting analyses for expression of total and phospho‐proteins, because both proteins migrate with a similar molecular mass, we run duplicate or multiple gels with same amounts of proteins (i.e., 20 μg per lane). For these experiments, only one loading control from one gel blot is provided. Signal intensities were quantified by ImageJ software.
Reactive oxygen species staining
Reactive oxygen species levels were determined in vivo in immobilized mice GAS muscles through DHE staining. The frozen GAS slides were incubated with DHE (50 μmol/L in PBS) 18 at 37°C. Thirty minutes later, the slides were washed with PBS for 5 min and three times. The slides were imaged by confocal microscope (Zeiss).
Luciferase reporter assay
HEK293 T cells were seeded into 24‐well plates in DMEM containing 10% FBS medium. Twenty‐four hours later, the cells were changed into DMEM without FBS medium for 2 h and then transfected with indicated plasmids (a total of 1 μg DNA per well) for 48 h. Cells were lysed and measured using the Dual‐luciferase reporter assay system (Promega).
Statistical analyses
Data were analysed using GraphPad Prism 9. Unpaired two‐tailed Student's t‐tests were set as statistical methods between two groups. All data were shown as mean ± standard error of the mean.
Results
DJ‐1 expression is positively correlated with muscle mass in humans and mice
Loss of muscle mass and strength is a part of the natural aging process 20 ; therefore, DJ‐1 expression was reduced in aged human muscles (Figures 1A and S1A). We examined muscle gene expression in a published transcriptome dataset indicating that the DJ‐1 transcripts were also decreased in muscle biopsy from healthy aged humans (Figure 1B). 21 Likewise, the mice proteomics analysis showed a 50% reduction in DJ‐1 protein in aged muscles (Figure 1C). 22 Additionally, DJ‐1 protein was observed to be 30% lower in the muscle of 24‐month‐old mice than in the 7‐month‐old mice (Figure 1D). Furthermore, the single nuclei RNA sequence (snRNA seq) analysis showed an approximate 50% decline in DJ‐1 expression from mature muscle fibres of aged mice compared with that of young mice (Figure S1C). 23 To explore the relationship between DJ‐1 and muscular atrophy, DJ‐1 expression was assessed in muscle wasting mice models with unloaded hindlimbs, showing a decline in expression levels (Figure 1E, F). On the contrary, DJ‐1 expression in human skeletal muscles was slightly but significantly induced during rehabilitation intervention after cast immobilization (Figure S1B). 24 These results indicated that DJ‐1 expression positively correlated with muscle mass in both humans and mice.
Figure 1.
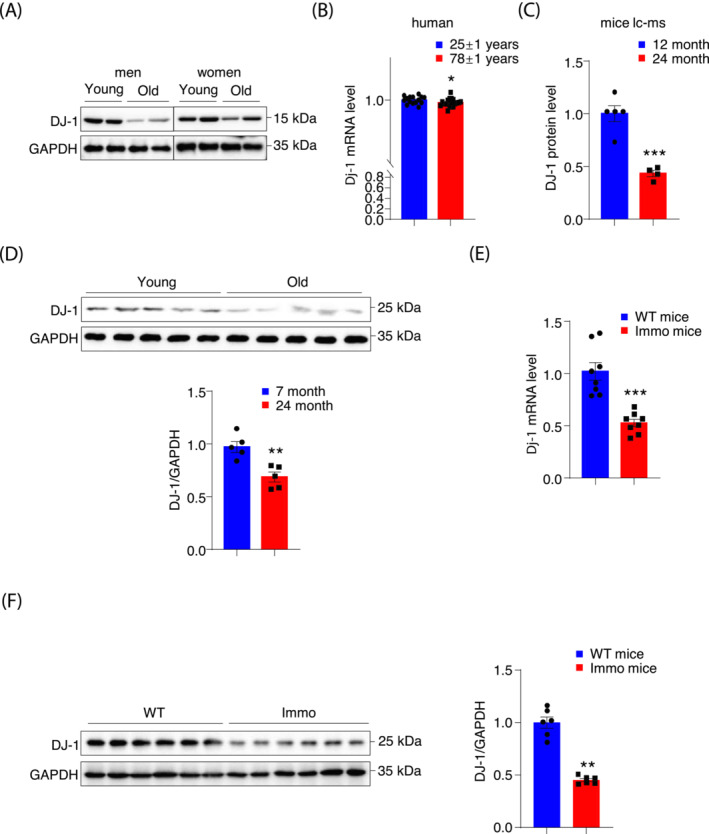
DJ‐1 expression is decreased in skeletal muscle from human and mice with muscle atrophy. (A) DJ‐1 immunoblots of young and old aged men (young, 28 and 29 years; old, 65 and 88 years) and women (young, 15 and 26 years; old, 58 and 69 years) muscles lysates. (B) DJ‐1 RNA‐seq expression data from young (n = 15, 25 ± 1 years) and old (n = 21, 78 ± 1 years) human muscles in GSE25941 datasets. (C) The fold change of DJ‐1 protein expression in young (n = 5, 12 months) and old (n = 4, 24 months) male mice muscles. (D) DJ‐1 immunoblots of young (n = 5, 7 months) and old (n = 5, 24 months) male mice muscles lysates and the quantification of blots. (E) DJ‐1 expression in GAS muscle of Flox male mice and immobilized Flox male mice (n = 8). (F) DJ‐1 immunoblots of GAS from mice without/with immobilization and the quantification of blots. Data represented the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, a two‐tailed Student's t‐test was used for statistical analysis.
Mice with skeletal muscle‐specific DJ‐1 ablation have slightly decreased lean mass and impaired exercise performance
To investigate the role of DJ‐1 in muscle atrophy, we constructed skeletal MDKO mice by crossing DJ‐1flox/flox mice with Myl1‐cre mice (Figure S2A). Protein and mRNA levels of DJ‐1 were dramatically reduced in skeletal muscles, but not in other tissues of MDKO mice (Figures 2A, B and S2B–E). The body weight of MDKO mice showed no difference compared with Flox mice on the chow diet (Figures 2C and S3A); however, the fat mass was marginally increased while lean mass was moderately decreased in MDKO mice than in controls at the age of 5 months (Figures 2D, S2F‐a‐d, S3B‐C, and S3D‐a‐e). Furthermore, muscle contractility was measured in situ; the specific tetanic force of MDKO mice was significantly weaker than that of Flox mice, and the time to relaxation was longer for MDKO mice (Figure 2E). Next, exercise performance was evaluated, revealing weaker grip strength of MDKO mice on a horizontal grid than Flox mice (Figures 2F‐a and S3E‐a). For resistance exercise, mice climbed a vertical ladder with weights affixed to their tail base. The maximal weight‐carrying capacity of MDKO mice was 23% lower than that of Flox mice (Figures 2F‐b and S3E‐b). The muscle coordination and strength were assessed by employing two distinctive hanging tests with a wire or grid using either the two forelimbs or all four limbs, respectively. MDKO mice exhibited shorter suspension time than control mice in both hanging tests (Figures 2F‐c‐d and S3E‐c‐d). In addition, muscle strength was also monitored by the rotarod test, with a 76% reduction in the maximum running time of MDKO mice (Figures 2F‐e and S3E‐e). Consistent with the ability to perform anaerobic exercise, the maximal running capacity of MDKO mice was also diminished (Figures 2F‐f and S3E‐f). To avoid muscle growth that may contribute to lean mass loss, additional body composition experiments were conducted using 18‐month‐old mice. MDKO mice had lower lean mass than controls, while no difference in body weight was observed (Figure S4A‐B). Moreover, the exercise performances were also impaired in MDKO mice (Figure S4C–E). These results indicated that DJ‐1 deletion in skeletal muscles mildly decreased muscle mass and impaired exercise performance.
Figure 2.
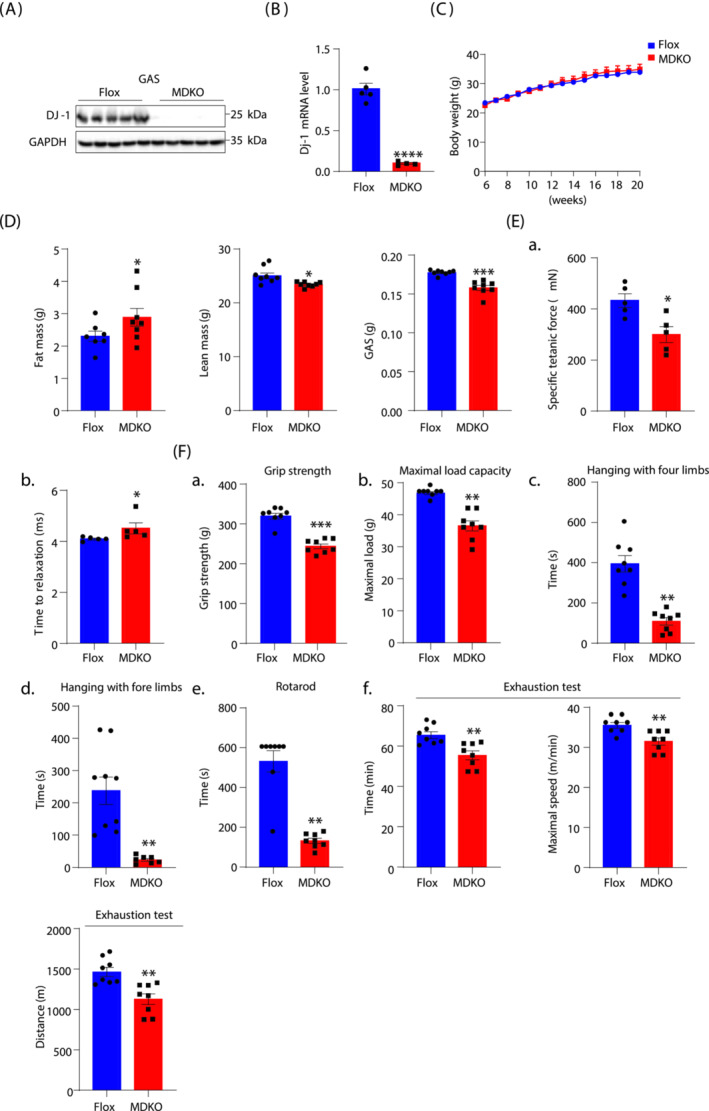
Mice with DJ‐1‐specific knockout in skeletal muscle show decreased muscle mass and impaired exercise performance. (A) Western blot analysis of DJ‐1 in GAS of muscle‐specific DJ‐1 knockout (MDKO) and Flox mice. (B) DJ‐1 expression level in GAS by qPCR. (C) Body weight of male MDKO and Flox mice fed on chow diet (n = 8–9). (D) Body composition and muscle weight of male MDKO and Flox mice at the age of 18 weeks (n = 7–8). Fat mass, lean mass, GAS weight. (E) Tibialis anterior (TA) muscle force analysis of male mice at the age of 18 weeks (n = 5). Specific titanic force of TA muscles (A), relaxation time (B). (F) Exercise performance tests. Grip strength of mice at the age of 13 weeks (n = 8) (A). Maximum load capacity of mice in a resistance test at the age of 14 weeks (n = 8) (B). Maximum hanging time with four or fore limbs of mice at the age of 15 weeks (n = 8) (C, D). Rotarod performance test of mice at the age of 16 weeks (n = 8) (E). Total time, maximum speed, and total distance of mice in exhaustion test at the age of 17 weeks (n = 8) (F). Data represented the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, a two‐tailed Student's t‐test was used for statistical analysis.
Ablation of DJ‐1 decreases fibre CSA without affecting fibre types
To examine the physiological consequences of DJ‐1 deletion in skeletal muscle, imaging analysis using haematoxylin and eosin (H&E) staining, immune histology fluorescent staining, and electron microscopy (EM) were carried out. Despite the overall similar architecture of GAS and SOL muscles in MDKO mice, the fibre size appeared smaller in H&E staining (Figures 3A‐a and S5A‐a), which was further confirmed by CSA determination. DJ‐1 depletion caused a 20% reduction in the median CSA than the control fibres (Figures 3A‐b‐c and S5A‐b‐c). Different muscle fibres have been reported to have variable fibre size. 25 Accordingly, we verified the fibre type switch as the probable reason for decreased CSA. Surprisingly, no difference in slow or fast twitch fibre number was observed between MDKO and Flox mice (Figures 3B‐a‐b and S5B‐a‐b). However, each fibre type CSA was smaller in MDKO mice than in control mice (Figures 3B‐c and S5B‐c), and the key genes related to muscle fibre type I, IIA, and IIX were down‐regulated in DJ‐1‐depleted skeletal muscles (Figure 3C). In summary, our results suggested that the ablation of DJ‐1 results in muscular atrophy development.
Figure 3.
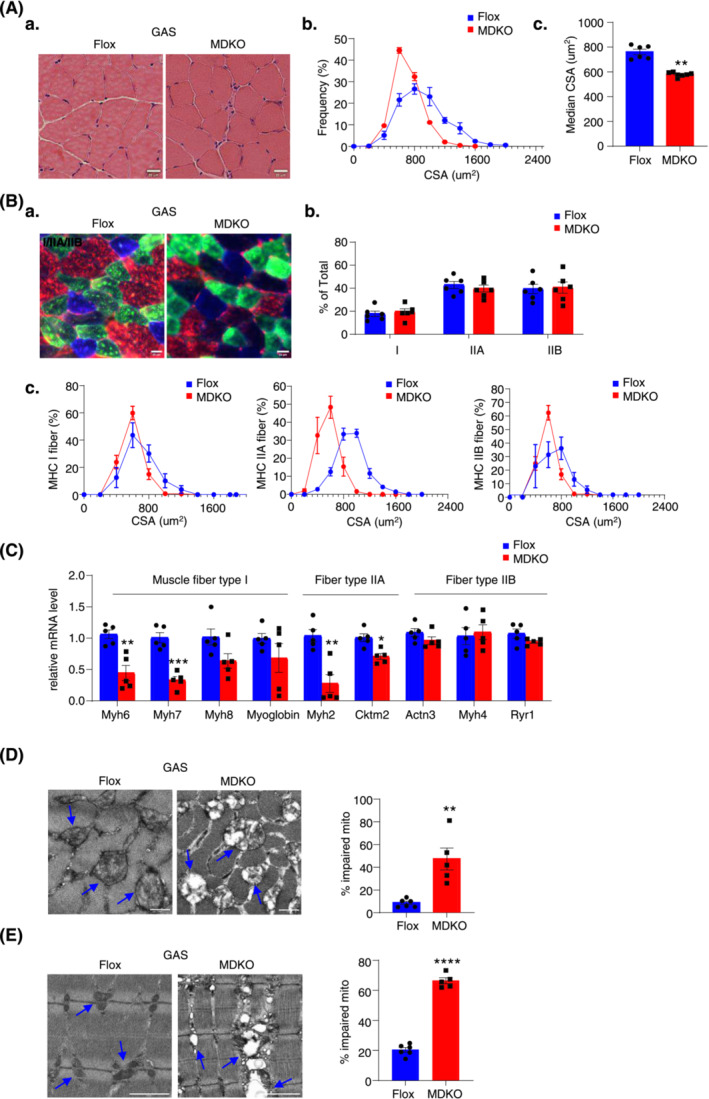
DJ‐1 ablation in skeletal muscle decreases fibre cross‐sectional area and impairs mitochondrial function. (A) Physiological consequences of GAS H&E staining in male mice at the age of 18 weeks (scale bars, 20 μm). Representative H&E staining of GAS (A). Fibre cross‐sectional area (CSA) distribution and median CSA in GAS of male mice (n = 6–7) (B, C). (B) Immunofluorescence analysis of fibre type composition in GAS. The different myosin heavy chain isoforms were stained in blue (MyHC‐I), green (MyHC‐IIa) or red (MyHC‐IIb) (scale bars, 20 μm). Representative immunofluorescence staining of GAS (A). The ratio of muscle fibre type (n = 6) (B). Fibre cross‐sectional area (CSA) distribution in different muscle fibres (n = 6) (C). (C) Expression of muscle fibre type related genes measured by qPCR (n = 5). (D) Representative electron micrographs of cross and the ratio of impaired mitochondria in GAS of male mice (scale bars, 0.5 μm). (E) Representative electron micrographs and quantification of abnormal mitochondria of longitudinal section. Blue arrows point to the healthy mitochondria in Flox mice and the impaired mitochondria in MDKO mice (scale bars, 0.5 μm). Data represented the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, a two‐tailed Student's t‐test was used for statistical analysis.
Mitochondrial function is impaired in muscles with DJ‐1 deletion
DJ‐1 has been reported to act as an antioxidant signal molecule, and mutations in the DJ‐1 gene are associated with oxidative stress, resulting in mitochondrial dysfunction. 26 EM imaging revealed abnormal mitochondria structure, with a significant loss of cristae and numerous enlarged mitochondria (Figures 3D, E and S5C‐D). However, the mitochondrial DNA (mtDNA) number and expression of some OXPHOS genes in DJ‐1 deleted muscles were similar to that in controls (Figure S5E‐a‐c). Likewise, the expression level of a master gene, PGC‐1α, for mitochondrial biogenesis was also not altered in MDKO mice muscles compared with Flox mice muscles (Figure S5E‐d).
MDKO mice show aggravated muscle dystrophy during immobilization
To further decipher the role of DJ‐1 in muscle atrophy, a muscle disuse model was adopted with hindlimb suspension of 10‐week‐old male mice for 14 days. The body weight of MDKO mice was significantly dropped after suspension compared with that of Flox mice showing no difference (Figure 4A). Similarly, lean mass was lower in MDKO mice than in Flox mice (Figure 4B), without changes in other tissues (fat tissues, heart, and liver) (Figure S6A‐a‐e). In addition, MDKO mice muscles showed signs of progressive dystrophy, including decreased CSA and increased centralized nuclei, which were indicative of ongoing de‐ and regeneration (Figure 4C, Figure S6B‐a‐c). Moreover, the area of each muscle fibre type in MDKO mice was smaller than that in control mice, without alteration in the number of fibre types (Figures 4D and S6C, D‐a‐c). Furthermore, the muscle fibre type I (Myh6, Myh7, Myh8, troponin, and myoglobin) and type IIa (Myh2 and Cktm2) genes expression was decreased; however, the type IIb genes (Myh4 and Actn3) expression levels were increased in muscles with DJ‐1 disruption (Figure 4E).
Figure 4.
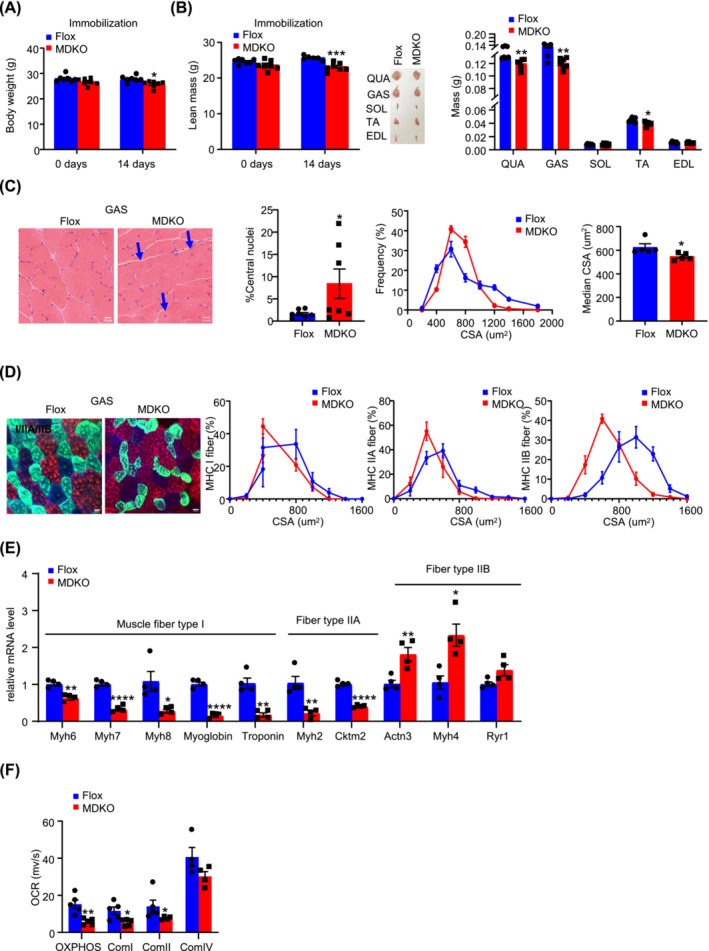
DJ‐1 deletion in skeletal muscle aggravates atrophy during immobilization. (A) Body weight of male mice during immobilization at the age of 10‐week‐old (n = 6–7). (B) Lean mass, representative morphology and weight of muscles male mice during immobilization at the age of 10‐week‐old (n = 6–7). Lean mass. Representative morphology of quadriceps (QUA), GAS, soleus (SOL), TA, and extensor digitalis anterior (EDL) after immobilization. Weight of QUA, GAS, SOL, TA, and EDL after immobilization (n = 6–7). (C) Physiological consequences of GAS H&E staining in immobilized mice. Representative H&E staining of GAS (scale bars, 10 μm), the percentage of central nuclei after immobilization, CSA distribution, and median CSA in GAS of immobilized mice (n = 5). Blue arrows point to the nuclei inward migration in MDKO mice. (D) Physiological consequences of immunofluorescence staining in GAS. The different myosin heavy chain isoforms were stained in blue (MyHC‐I), green (MyHC‐IIa), and red (MyHC‐IIb) (scale bars, 20 μm). Representative immunofluorescence of muscle fibre type composition in GAS of immobilized mice and CSA distribution in GAS of immobilized mice (n = 6–7). (E) Expression of muscle fibre type related genes of GAS (n = 4). (F) Oroboros O2k respirometer oxygen flux analysis of permeabilized immobilized GAS (n = 5). Data represented the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, a two‐tailed Student's t‐test was used for statistical analysis.
Next, to demonstrate modifications in mitochondrial function, O2K analysis was performed, which showed an obviously impaired oxidative phosphorylation, a diminished activity of mitochondrial complex I and II in the GAS muscle of MDKO mice, while a trend of decrease in the complex IV activity was observed (Figure 4F). Determination of reactive oxygen species activity with DHE staining showed no obvious differences between MDKO mice and Flox mice (Figure S6E‐a‐b). Besides, the mtDNA number and protein levels of OXPHOS genes (SDHB and NDUFB8) were reduced in MDKO mice muscles (Figure S6F‐a‐c). Taken together, these data demonstrated that DJ‐1 loss in skeletal muscle promoted muscle wasting and mitochondria dysfunction during immobilization.
MDKO mice muscles show molecular characteristics specific to atrophy
To elucidate the underlying molecular mechanisms for atrophy induced by DJ‐1 deletion, we performed RNA‐seq of GAS muscle from immobilization models. The principal component analysis showed two obvious groups between MDKO and Flox mice (Figure 5A), with 1113 differentially regulated genes; of the 763 down‐regulated genes, most were related to muscle structure and function (Figure 5B). GO analysis revealed up‐regulation of carbohydrate catabolism, down‐regulation of electron transport chain, and aerobic respiration pathways (Figure 5C). We next analysed the muscle fibre type genes. In line with this, oxidative muscle fibre (type I and IIa) genes were decreased, whereas glycolic fibre (type IIb) genes were increased in DJ‐1 ablated muscles (Figure 5D). Nonetheless, the muscle satellite cell‐specific gene markers (MyoD, Myf5, Pax7, and Myh3) were not altered (Figure S7A‐a). In addition, the in vitro experiments also showed that no modifications in myogenic proteins expression (MyoD) and myogenin (MyoG) between DJ‐1 knockdown and controls muscle cells (Figure S7A‐b).
Figure 5.
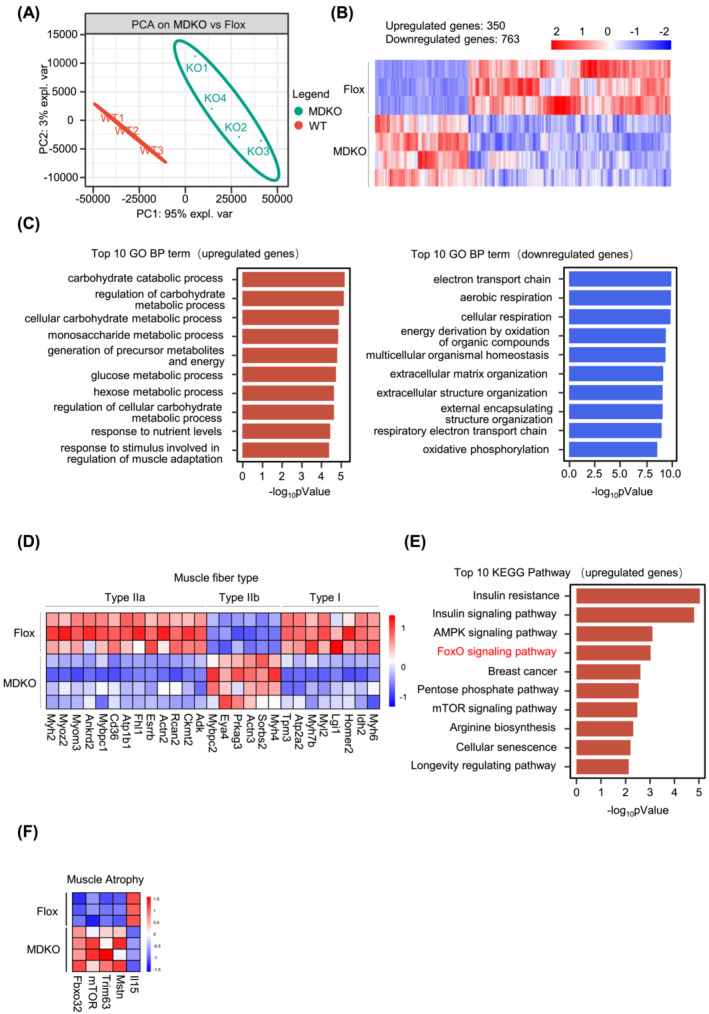
DJ‐1 knockout in skeletal muscle shows molecular signs of atrophy. (A) Principal component analysis (PCA) was analysed from RNA‐seq data of GAS in immobilized mice (n = 3–4). (B) Heatmap of 1113 differential genes of GAS in immobilized mice (n = 3–4). (C) GO enrichment analysis based on differential expression genes. Top 10 GO BP term of upregulated genes (left). Top 10 GO BP term down‐regulated genes (right). (D) Expression of muscle fibre type related genes from RNA‐seq data of immobilization models (n = 3–4). (E) KEGG enrichment analysis based on upregulated genes. (F) Atrogenes expression from RNA‐seq data (n = 3–4). Data represented the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, a two‐tailed Student's t‐test was used for statistical analysis. UBI, ubiquitination; Immo model, immobilized model.
According to the KEGG pathway analysis, the top 10 up‐regulated genes list included two muscle atrophy‐related pathways: FoxO and mTOR signalling pathway (Figure 5E). Western blotting illustrated enhanced mTOR phosphorylation in MDKO mice muscles (Figure S7B). Enhanced activation of the mTOR signalling has been reported to increase protein synthesis, which might subsequently increase muscle mass. Thus, in the present study, the has been elevated activity of the mTOR signalling pathway might compensate for muscle wasting in MDKO mice. Next, analysis of the FoxO signalling pathway, playing a vital role in muscle atrophy, indicated a notable increase in two important atrophy genes (Trim63 and Fbxo32) among the FoxO regulated genes 18 in DJ‐1 deleted muscles (Figure S7C‐a). These results were further validated by qPCR and western blotting (Figure S7C‐c‐f). Because Trim63 and Fbxo32 are key ubiquitin‐protein ligases (E3), their ubiquitination levels were increased in DJ‐1 ablated muscles (Figure S7D). Moreover, myostatin (Mstn) levels, an inhibitor of muscle growth reportedly regulated by FoxO1, 27 were also increased in MDKO mice muscles after suspension (Figure 5F). On the contrary, a drop in interleukin‐15 (IL‐15, an anabolic factor for skeletal muscle which inhibits muscle wasting) 28 was observed in DJ‐1 knockout mice muscles (Figure 5F). We also checked the normal condition models. Under normal conditions, only Trim63 levels were significantly elevated, while Fbxo32 and Mstn showed a moderate trending rise (Figure S7E‐a‐b). These data suggested that the FoxO signalling pathway might account for DJ‐1‐induced muscle loss.
The autophagy‐lysosomal protein degradation was also determined; the protein levels of two autophagy marker genes (LC3 and P62) were neither altered in immobilized samples nor during autophagic flux measurements (Figure S7F‐a‐b). Likewise, the mRNA levels of autophagic genes (lc3, p62, and bnip3) did not differ between immobilized Flox mice and MDKO mice (Figure S7E‐c).
FoxO1 pathway is responsible for DJ‐1 ablation‐induced muscle atrophy
To identify the specific FoxO family member involved in DJ‐1‐mediated atrophy, the phosphorylation of FoxO1, FoxO3A, and FoxO4 proteins was examined. FoxO1 phosphorylation was reduced in MDKO mice muscles, while no change in the other two proteins was observed (Figures 6A and S8A). Similarly, C2C12 myotubes with DJ‐1 knockdown showed decreased FoxO1 phosphorylation (Figure S8B). FoxO1 has been reported phosphorylated by AKT, 29 and DJ‐1 regulates AKT phosphorylation in cancer cells. 30 As anticipated, AKT phosphorylation was attenuated in MDKO mice compared with Flox mice (Figure 6A), suggesting that DJ‐1 could regulate FoxO1 phosphorylation through the AKT pathway in skeletal muscles.
Figure 6.
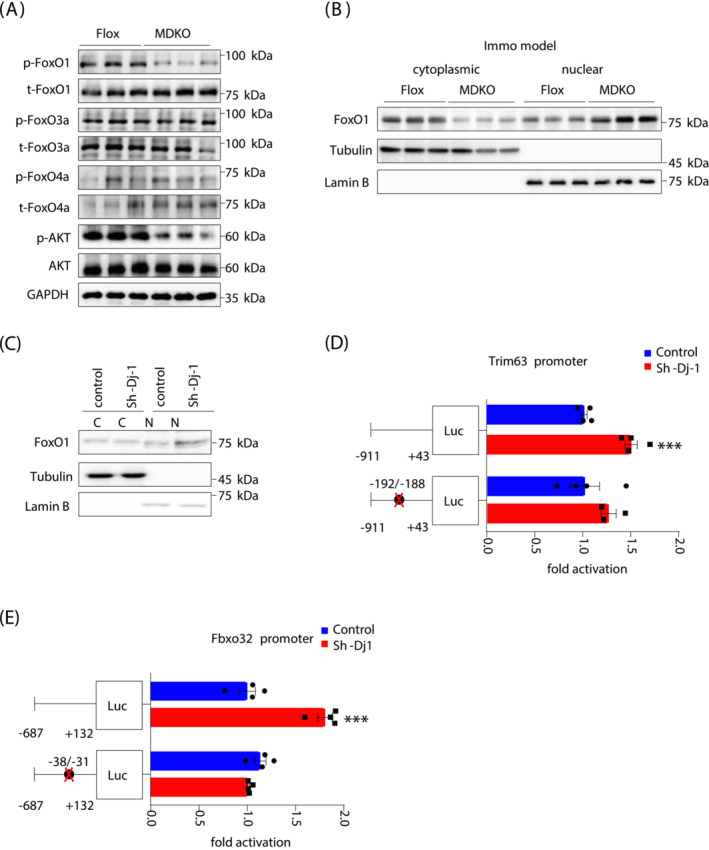
FoxO1 pathway is responsible for DJ‐1 ablation‐induced muscle atrophy. (A) Immunoblot analysis of FoxO family and AKT of GAS in immobilization models. (B) Protein level of FoxO1 in cytoplasmic and nuclear fractions of GAS in immobilization mice. (C) Protein level of FoxO1 in cytoplasmic and nuclear fractions of C2C12 myotubes with/without DJ‐1 knockdown. (D, E) Dual‐luciferase assays of Trim63 and Fbxo32 promoter. Luciferase activity was corrected for Renilla luciferase activity and normalized to control group. The putative FoxO site was noted by black circles. Binding site for FoxO1 in the Trim63 promoter was identified at −192/−188. Binding site for FoxO1 in the Fbxo32 promoter was identified at −38/−31. Data represented the mean ± SEM. *P < 0.05, ***P < 0.001, a two‐tailed Student's t‐test was used for statistical analysis. Immo model, immobilized model; Luc, luciferase.
FoxO1 can regulate nuclear genes; however, it is phosphorylated by AKT before translocation to the nuclear. 29 Therefore, the FoxO1 level was measured in the nucleus and cytoplasm, showing a lessened cytoplasmic and elevated nuclear expression in DJ‐1 ablated muscles (Figures 6B and S8C). This was consistent with the FoxO1 phosphorylation levels (Figure 6A). Likewise, DJ‐1 knockdown increased FoxO1 protein levels in the nucleus of C2C12 myotubes (Figure 6C), suggesting a cell‐autonomous effect of DJ‐1 in myotubes.
The 5′ flanking regions of Trim63 and Fbxo32 genes were also examined to determine DJ‐1 mediated regulation of MuRF‐1/Atrogin‐1 expression through FoxO1. Trim63/Fbxo32 promoter‐luciferase constructs, transfected into 293 cells, were activated by DJ‐1 knockdown (Figures 6D, E and S8D). FoxO1‐assisted transcriptional regulation of Trim63 and Fbxo32 via forkhead‐binding element located at −195/−189 and −38/−31 positions within the promoter, respectively, was evaluated. 7 , 29 Mutation in forkhead‐binding element abolished the DJ‐1 knockdown‐inducted luciferase expression (Figure 6D, E), indicating that FoxO1 serves as a transcriptional regulator for DJ‐1′‐mediated muscle atrophy.
Mice treated with compound 23, a DJ‐1 antagonist, exhibited progressive muscular atrophy symptoms during immobilization
Compound 23 (comp‐23) has been reported to interact with endogenous DJ‐1 and protect against oxidative stress‐induced dopaminergic neuronal death in Parkinson's disease and ischaemia models. 31 , 32 Therefore, comp‐23 was directly injected into the GAS muscle, resulting in reduced DJ‐1 expression in the treated leg (Figure 7A). In addition, H&E staining revealed an apparently smaller CSA in the comp‐23 treated leg than in the control (Figures 7B and S8E). To further confirm that comp‐23 exerts its action depending on DJ‐1, comp‐23 was injected into MDKO mice. However, no aggravated atrophy symptoms in comp‐23 treated DJ‐1 deleted muscle were observed (Figures 7C and S8F). Besides, Trim63 levels were increased in either DJ‐1 knockout or comp‐23 treated muscle but did not rise synergistically in comp‐23 treated DJ‐1‐ablated muscle (Figure 7D). Expression levels of Fbxo32 and Mstn were increased only in DJ‐1‐1deleted muscle (Figure 7D). Similar results were obtained during the expression of muscle fibre type genes (Figure 7E). These data suggested that the effects of comp‐23 in the skeletal muscle were DJ‐1 dependent.
Figure 7.
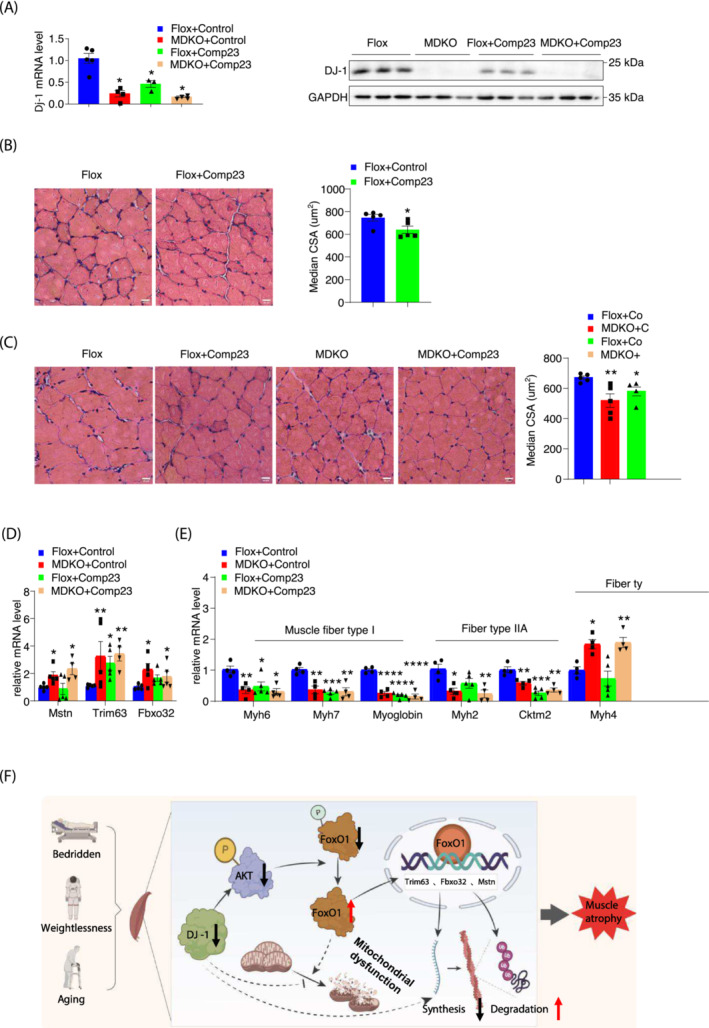
Compound 23 mimics the effects of skeletal muscle DJ‐1 ablation in vivo. (A) DJ‐1 mRNA level and protein level expression of GAS from indicated mice after immobilization (n = 3–5). (B, C) Physiological consequences of GAS in comp23‐treated mice. Representative H&E staining of GAS in comp23‐treated mice (scale bars, 20 μm) (left). The median of fibre CSA in GAS from a (n = 5) (right). (D) Expression of GAS atrogenes (n = 4–5). (E) Expression of muscle fibre type related genes of GAS. (F) Working models. Data represented the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, a two‐tailed Student's t‐test was used for statistical analysis. comp23, Compound 23.
Overall, the results suggested that DJ‐1 manipulates FoxO1 phosphorylation for its translocation into the nucleus to transcriptionally regulate atrogenes expression, the latter eventually ubiquitinates proteins involved in sarcomere assemble and protein synthesis (Figure 7F).
Discussion
Skeletal muscle is a dynamic tissue that has the capacity to continuously regulate its size in response to various external cues, such as mechanical load, neural activity, hormones/growth factors, stress, and nutritional status. 7 In the present study, DJ‐1 was observed to be down‐regulated in muscle wasting mice and human models. Although skeletal muscle‐specific DJ‐1 knockout mice did not show significant body weight difference on the chow diet, the body mass was slightly changed with: higher fat mass and lower lean mass than the control mice. In addition, hindlimb suspension of MDKO mice led to the development of progressive muscular dystrophy. Biochemical analysis suggested the course of events for atrophy induction in MDKO mice involving the enhanced activity of the FoxO1 signalling pathway, following up‐regulation of MuRF1 and Atrogin‐1, and subsequent sarcomere disassembly through the myosin stabilizing protein ubiquitination. This is consistent with a previous study reporting that DJ‐1 protects against protein degradation by inhibiting 20 s proteasome activity. 33
The production of new myofibrils and degradation of existing proteins is a delicate balance, which can promote muscle growth or loss. The PI3K/AKT signalling cascade is critical for regulating diverse cellular functions including metabolism, growth, proliferation, survival, and protein synthesis. 34 DJ‐1 can inhibit PTEN (phosphatase and tensin homologue) to activate PI3K/AKT pathway in neuron and cancer cells. 35 Consistent with this, AKT phosphorylation was reduced in muscles with DJ‐1 deletion, indicating the involvement of the AKT pathway in DJ‐1 exerted function in skeletal muscle. However, the AKT pathway has two major downstream branches; the FoxO pathway blocked by AKT and the mTOR pathway activated by AKT. 36 FoxO pathway activity, controlling protein degradation, was enhanced in DJ‐1 deletion‐induced atrophic muscles. Contrastingly, mTOR phosphorylation was also increased, which might be stimulated by amino acids from protein breakdown. The activated mTOR pathway may increase protein synthesis to compensate for protein degradation partially. Although the contribution of cellular turnover to the homeostasis of adult fibres and its role in hypertrophy has been recently debated, 37 the population of satellite cells in MDKO mice was not affected based on the RNA‐seq data. Additionally, the expression of a few essential satellite cell genes (Pax7, MyoD, and MyoG) was not altered in MDKO mice muscles or DJ‐1 knockdown C2C12 cells. However, the concentration of myostatin, a master negative regulator of muscle growth, was increased in DJ‐1 ablated muscles.
The FoxO family in skeletal muscle comprises of three isoforms: FoxO1, FoxO3a, and FoxO4. AKT phosphorylates FoxOs to promote their export from the nucleus to the cytoplasm. The three isoforms have overlapping and different functions. For example, all three are required for the up‐regulation of MuRF1 and Atrogin‐1 in response to catabolic stimuli. 29 However, under specific conditions, such as glucocorticoid treatment (Dex), FoxO1‐mediated overactivity of the MuRF1 promoter is synergized by the concomitant binding of the glucocorticoid receptor. FoxO3a and FoxO4 can neither activate the MuRF1 promoter nor potentiate glucocorticoid receptor upon Dex treatment. 7 A concomitant reduction in the AKT pathway activity observed in DJ‐1‐depleted muscle led to decreased cytoplasmic levels of phosphorylated FoxO1 and a marked increase in nuclear FoxO1 protein. Nevertheless, organ‐ and cell‐specific deletion of FoxO family members combined with DJ‐1 ablation will provide comprehensive insights into the tissue‐ and cell‐specific role of DJ‐1 in skeletal muscle.
Numerous transcription factors are important in regulating the cytoarchitecture. 38 In the present study, the RNA‐seq analysis revealed altered expression of some transcription factors, such as serum response factor (Srf) and nuclear factor kappa B (NFκB) subunits, except FoxO1, in DJ‐1‐deleted skeletal muscle. The embryonic and perinatal sarcomere defects in Srf knockout studies indicated the essential role of Srf in the formation and maintenance of the myofibrillar structure. 38 , 39 , 40 However, DJ‐1 deletion‐induced Srf expression, suggesting that Srf may not contribute to the atrophy phenotype. Additionally, DJ‐1 can activate NFκB, a transcriptional repressor of muscle structural genes, 41 by activating IκB kinase. 34 Consequently, the expression of two subunits of NFκB, relA and relB, was increased in the DJ‐1 ablated muscle. Therefore, the NFκB signalling pathway may also account for the decline muscle mass in MDKO mice.
Collectively, we have proposed a DJ‐1‐regulated AKT‐FoxO1 signalling pathway to control muscle mass during disuse‐induced atrophy. Enhancing or maintaining DJ‐1 levels represents a novel therapeutic target for developing drugs that can ameliorate muscle wasting accompanied by several systemic diseases.
Conflict of interest
The authors declare no competing interests.
Supporting information
Figure S1. DJ‐1 is increased in skeletal muscle during rehabilitation in human. (A) The information of muscles sample donors. (B) Dj‐1 Genome‐wide profiling expression data from immobilized human muscles during the 0‐day, 4‐week, 6‐week and 4‐month rehabilitation (n = 16–19). (C) The expression of Dj‐1 in mature myotubes of mice with 5‐month, 24‐month and 30‐month‐old. FBG: Fasting blood glucose, **p < 0.01, ***p < 0.001, a two‐tailed Student t test was used for statistical analysis.
Figure S2. Generation and verification of skeletal muscle specific DJ‐1 knockout mice. (A) Schematic of Flox targeting‐construct for the Dj‐1 locus. (B‐E) Western blot analysis of DJ‐1 in different muscle depots and heart of MDKO and Flox mice. (F) Muscles weight of male mice at the age of 18 weeks (n = 8). QUA, quadriceps (a). SOL, soleus (b). TA, tibialis anterior (c). EDL, extensor digitorum longus (d). Data represented the mean±s.e.m.. *p < 0.05, **p < 0.01, a two‐tailed Student t test was used for statistical analysis.
Figure S3. DJ‐1 knockout in skeletal muscle decreases muscle mass and impairs exercise performance in female mice. (A) Body weight of female mice fed on chow diet at 18‐week‐old (n = 8). (B‐C) Body composition of female mice at the age of 18 weeks (n = 8). Fat mass (B). Lean mass (C). (D) Muscles weight of female mice at the age of 20 weeks (n = 4). QUA (a). GAS (b). SOL (c). TA (d). EDL (e). (E) Exercise performance tests. Grip strength of female mice at the age of 13 weeks (n = 7–8) (a). Maximum load capacity of female mice in a resistance test at the age of 14 weeks (n = 7–8) (b). Maximum hanging time with four or fore limbs of mice at the age of 15 weeks (n = 7) (c‐d). Rotarod performance test of female mice at the age of 16 weeks (n = 8) (e). Total time, maximum speed and total distance of female mice in exhaustion test at the age of 17 weeks (n = 7) (f). Data represented the mean ± s.e.m.. *p < 0.05, ***p < 0.001, ****p < 0.0001, a two‐tailed Student t test was used for statistical analysis.
Figure S4. DJ‐1 knockout in skeletal muscle decreases muscle mass and impairs exercise performance in aged male mice. (A) Body weight of aged male mice fed on chow diet at 18‐month‐old (n = 5). (B) Lean mass of aged male mice at the age of 18 months (n = 5). (C) Grip strength of aged male mice at the age of 18 months (n = 5). (D) Maximum load capacity of aged male mice in a resistance test at the age of 18 months (n = 5). (E) Rotarod performance test of aged male at the age of 18 months (n = 5). Data represented the mean ± s.e.m.. *p < 0.05, ***p < 0.001, ****p < 0.0001, a two‐tailed Student t test was used for statistical analysis.
Figure S5. DJ‐1 ablation decreases CSA of soleus, but has no effects in mitochondrial DNA number or OXPHOS genes expression. (A) Physiological consequences of GAS in male mice fed on chow diet at the age of 18 weeks. Representative H&E staining of SOL (scale bars, 20 μm) (a). CSA distribution and median in SOL of male mice (n = 4–6) (b‐c). (B) Physiological consequences of SOL immunofluorescence staining in male mice. Representative immunofluorescence analysis of muscle fibre type in SOL (a). The different myosin heavy chain isoforms were stained in blue (MyHC‐I) or green (MyHC‐IIa) (scale bars, 20 μm). The ratio of muscle fibre type (n = 4–6) (b). CSA distribution in SOL (n = 4–6) (c). (C‐D) Representative electron micrographs of cross or longitudinal section in SOL of male mice (scale bars, 1 μm). Cross section (C), Longitudinal section (D). Blue arrows point to the healthy mitochondria in Flox mice and the impaired mitochondria in MDKO mice. (E). Mitochondria function in GAS of male mice. Mitochondrial DNA number (n = 4) (a). Expression of mitochondria complex related genes and proteins measured by qPCR (b) and western blots (c). Immunoblot analysis of PGC1α protein in GAS muscle of male KO and Flox mice (d). Data represented the mean±s.e.m.. *p < 0.05, a two‐tailed Student t test was used for statistical analysis.
Figure S6. DJ‐1 deletion shows progressive atrophy in SOL of immobilization models. (A) The weight of tissues of male mice at the age of 10 weeks in immobilization models (n = 6–7). BAT (a). iWAT (b). eWAT (c). Heart (d). Liver (e). (B). Physiological consequences of GAS H&E staining in immobilization models. Representative H&E staining of SOL (scale bars, 10 μm) (a). CSA distribution (b) and median (c) of SOL in immobilization models. (C) The ratio of muscle fibre type of GAS in immobilization models. (D) Physiological consequences of SOL immunofluorescence staining in immobilization models. Representative immunofluorescence analysis of muscle fibre type of SOL in immobilization models (a). The different myosin heavy chain isoforms were stained in blue (MyHC‐I) and green (MyHC‐IIa) (scale bars, 50 μm). The ratio of muscle fibre type (b) and CSA distribution (c) of SOL in immobilization models (n = 6–8). Data represented the mean±s.e.m.. ***p < 0.001, a two‐tailed Student t test was used for statistical analysis. (E) ROS was assessed by DHE staining in GAS (scale bars, 20 μm). Representative DHE staining images (a), DHE intensity analysis by Image J (b). (F) Mitochondria function was estimated in immobilized mice. Mitochondrial DNA number in GAS of immobilized mice (n = 3) (a). Expression of mitochondria complex related genes measured by western blots (b) and qPCR (c).
Figure S7. Myogenesis genes and autophagy related genes are not altered in DJ‐1 disrupted myotubes, but atrogenes are increased in GAS muscles. (A) Expression of myogenesis genes and proteins. Expression of muscle satellite cell related genes of GAS in immobilization models (n = 3–5) (a), Immunoblot analysis of MyoD and MyoG protein level in C2C12 myotubes (b). (B) Immunoblot analysis of phosphorylation and total mTOR of GAS in immobilization models. (C) Expression of GAS FoxO regulated genes, atrogenes, proteins and some transcript factors from mice on immobilization model (n = 5). Genes expression by RNA‐SEQ analysis (a). Atrogenes expression determined by qPCR of GAS (n = 3) (b). Immunoblot analysis of MSTN, MuRF1 and Atrogin‐1 protein level of GAS in immobilization models (n = 3) (c). Proteasome related genes (d), FoxOs regulated genes (e), other pathways related genes(f). (D) Ubiquitination expression level in GAS of immobilized mice (c). (E‐a) Expression of GAS atrogenes from mice on chow diet (n = 3–5). (E‐b) Immunoblot analysis of MuRF1 protein level in GAS of mice on chow diet. (F) Expression of GAS autophagy pathway proteins and genes in Immobilization model and autophagy flux model mice (n = 3–5). P62 and LC3 proteins level in GAS of immobilized mice and autophagy flux mice (a), Expression of P62 and LC3 proteins in autophagy flux model mice (b). (c) P62 and Lc3 genes level in GAS of immobilized mice. Data represented the mean±s.e.m.. *p < 0.05, a two‐tailed Student t test was used for statistical analysis. Ctsl, Cathepsin l; Gab, Gabarapl1; 4EPB, Eif4ebp1; Ncl, nucleolin.
Figure S8. FoxO1 pathway is responsible for DJ‐1‐ablation induced muscle atrophy. (A) Western blots analysis of phosphorylation and total FoxO1 protein level in GAS of mice fed on chow diet. (B) Immunoblot analysis of phosphorylation and total FoxO1 in C2C12 myotubes. (C) Protein level of FoxO1 in cytoplasmic fractions and nuclear fractions of GAS from mice fed on chow diet. (D) The effects of DJ‐1 knockdown or overexpression in 293 T cells. (D‐E) The distribution of fibre CSA in comp23‐treated mice. comp23, Compound23.
Table S1. Primer sequences.
Table S2. Antibody list.
Acknowledgements
The authors gratefully acknowledge Electron Microscopy core of NIBS for performing electron microscopy. This work was funded by the National Natural Science Foundation of China (92157203) to X.K., the National Key R&D Program of China (2019YFA0801900 and 2018YFA0800300), the National Natural Science Foundation of China (92249302, 31971074, and 32150610475) to T.L., Natural Science Foundation of Shanghai Science and Technology Commission, General Project (21ZR1413200) to H.Y., National Natural Science Foundation of China (32011540004, 31900823, and 20220508080RC) to X.L., and NIH NIDDK (DK125354) to Z.Z. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 42
Zhang S., Yan H., Ding J., Wang R., Feng Y., Zhang X., et al (2023) Skeletal muscle‐specific DJ‐1 ablation‐induced atrogenes expression and mitochondrial dysfunction contributing to muscular atrophy, Journal of Cachexia, Sarcopenia and Muscle, 14, 2126–2142, 10.1002/jcsm.13290
Shuang Zhang, Hongmei Yan, Jiyang Ding, and Ruwen Wang contributed equally to this work.
Contributor Information
Ru Wang, Email: wangru@sus.edu.cn.
Peijie Chen, Email: chenpeijie@sus.edu.cn.
Tiemin Liu, Email: tiemin_liu@fudan.edu.cn.
Xingxing Kong, Email: kongxingxing@fudan.edu.cn.
References
- 1. Hasselgren PO, Fischer JE. Muscle cachexia: current concepts of intracellular mechanisms and molecular regulation. Ann Surg 2001;233:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mitch WE, Goldberg AL. Mechanisms of muscle wasting. The role of the ubiquitin‐proteasome pathway. N Engl J Med 1996;335:1897–1905. [DOI] [PubMed] [Google Scholar]
- 3. Tawa NE Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest 1997;100:197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone‐treated skeletal muscle. Cell Metab 2007;6:376–385. [DOI] [PubMed] [Google Scholar]
- 5. Attaix D, Baracos VE. MAFbx/Atrogin‐1 expression is a poor index of muscle proteolysis. Curr Opin Clin Nutr Metab Care 2010;13:223–224. [DOI] [PubMed] [Google Scholar]
- 6. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- 7. Waddell DS, Baehr LM, van den Brandt J, Johnsen SA, Reichardt HM, Furlow JD, et al. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy‐associated MuRF1 gene. Am J Physiol Endocrinol Metab 2008;295:E785–E797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al. The IGF‐1/PI3K/Akt pathway prevents expression of muscle atrophy‐induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 2004;14:395–403. [DOI] [PubMed] [Google Scholar]
- 9. Smith IJ, Alamdari N, O'Neal P, Gonnella P, Aversa Z, Hasselgren PO. Sepsis increases the expression and activity of the transcription factor Forkhead Box O 1 (FOXO1) in skeletal muscle by a glucocorticoid‐dependent mechanism. Int J Biochem Cell Biol 2010;42:701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moylan JS, Smith JD, Chambers MA, McLoughlin TJ, Reid MB. TNF induction of atrogin‐1/MAFbx mRNA depends on Foxo4 expression but not AKT‐Foxo1/3 signaling. Am J Physiol Cell Physiol 2008;295:C986–C993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bandopadhyay R, Kingsbury AE, Cookson MR, Reid AR, Evans IM, Hope AD, et al. The expression of DJ‐1 (PARK7) in normal human CNS and idiopathic Parkinson's disease. Brain 2004;127:420–430. [DOI] [PubMed] [Google Scholar]
- 12. Jain D, Jain R, Eberhard D, Eglinger J, Bugliani M, Piemonti L, et al. Age‐ and diet‐dependent requirement of DJ‐1 for glucose homeostasis in mice with implications for human type 2 diabetes. J Mol Cell Biol 2012;4:221–230. [DOI] [PubMed] [Google Scholar]
- 13. An CN, Jiang H, Wang Q, Yuan RP, Liu JM, Shi WL, et al. Down‐regulation of DJ‐1 protein in the ejaculated spermatozoa from Chinese asthenozoospermia patients. Fertil Steril 2011;96:19–23 e2. [DOI] [PubMed] [Google Scholar]
- 14. Choi J, Sullards MC, Olzmann JA, Rees HD, Weintraub ST, Bostwick DE, et al. Oxidative damage of DJ‐1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem 2006;281:10816–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lev N, Barhum Y, Lotan I, Steiner I, Offen D. DJ‐1 knockout augments disease severity and shortens survival in a mouse model of ALS. PLoS ONE 2015;10:e0117190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Seyfarth K, Poschmann G, Rozman J, Fromme T, Rink N, Hofmann A, et al. The development of diet‐induced obesity and associated metabolic impairments in Dj‐1 deficient mice. J Nutr Biochem 2015;26:75–81. [DOI] [PubMed] [Google Scholar]
- 17. Pham TT, Giesert F, Rothig A, Floss T, Kallnik M, Weindl K, et al. DJ‐1‐deficient mice show less TH‐positive neurons in the ventral tegmental area and exhibit non‐motoric behavioural impairments. Genes Brain Behav 2010;9:305–317. [DOI] [PubMed] [Google Scholar]
- 18. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng Y, Cui Z, Lu X, Gong H, Liu X, Wang H, et al. Transcriptomics dissection of calorie restriction and exercise training in brown adipose tissue and skeletal muscle. Nutrients 2023;15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tieland M, Trouwborst I, Clark BC. Skeletal muscle performance and ageing. J Cachexia Sarcopenia Muscle 2018;9:3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Raue U, Trappe TA, Estrem ST, Qian HR, Helvering LM, Smith RC, et al. Transcriptome signature of resistance exercise adaptations: mixed muscle and fiber type specific profiles in young and old adults. J Appl Physiol 1985;2012:1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McDonagh B, Sakellariou GK, Smith NT, Brownridge P, Jackson MJ. Differential cysteine labeling and global label‐free proteomics reveals an altered metabolic state in skeletal muscle aging. J Proteome Res 2014;13:5008–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petrany MJ, Swoboda CO, Sun C, Chetal K, Chen X, Weirauch MT, et al. Single‐nucleus RNA‐seq identifies transcriptional heterogeneity in multinucleated skeletal myofibers. Nat Commun 2020;11:6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen YW, Gregory C, Ye F, Harafuji N, Lott D, Lai SH, et al. Molecular signatures of differential responses to exercise trainings during rehabilitation. Biomed Genet Genom 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Wessel T, de Haan A, van der Laarse WJ, Jaspers RT. The muscle fiber type‐fiber size paradox: hypertrophy or oxidative metabolism? Eur J Appl Physiol 2010;110:665–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol 2008;7:97–109. [DOI] [PubMed] [Google Scholar]
- 27. Allen DL, Unterman TG. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol 2007;292:C188–C199. [DOI] [PubMed] [Google Scholar]
- 28. Quinn LS, Anderson BG, Drivdahl RH, Alvarez B, Argiles JM. Overexpression of interleukin‐15 induces skeletal muscle hypertrophy in vitro: implications for treatment of muscle wasting disorders. Exp Cell Res 2002;280:55–63. [DOI] [PubMed] [Google Scholar]
- 29. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aleyasin H, Rousseaux MW, Marcogliese PC, Hewitt SJ, Irrcher I, Joselin AP, et al. DJ‐1 protects the nigrostriatal axis from the neurotoxin MPTP by modulation of the AKT pathway. Proc Natl Acad Sci U S A 2010;107:3186–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kitamura Y, Watanabe S, Taguchi M, Takagi K, Kawata T, Takahashi‐Niki K, et al. Neuroprotective effect of a new DJ‐1‐binding compound against neurodegeneration in Parkinson's disease and stroke model rats. Mol Neurodegener 2011;6:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takahashi‐Niki K, Inafune A, Michitani N, Hatakeyama Y, Suzuki K, Sasaki M, et al. DJ‐1‐dependent protective activity of DJ‐1‐binding compound no. 23 against neuronal cell death in MPTP‐treated mouse model of Parkinson's disease. J Pharmacol Sci 2015;127:305–310. [DOI] [PubMed] [Google Scholar]
- 33. Moscovitz O, Ben‐Nissan G, Fainer I, Pollack D, Mizrachi L, Sharon M. The Parkinson's‐associated protein DJ‐1 regulates the 20S proteasome. Nat Commun 2015;6:6609. [DOI] [PubMed] [Google Scholar]
- 34. Dolgacheva LP, Berezhnov AV, Fedotova EI, Zinchenko VP, Abramov AY. Role of DJ‐1 in the mechanism of pathogenesis of Parkinson's disease. J Bioenerg Biomembr 2019;51:175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choi MS, Nakamura T, Cho SJ, Han X, Holland EA, Qu J, et al. Transnitrosylation from DJ‐1 to PTEN attenuates neuronal cell death in parkinson's disease models. J Neurosci 2014;34:15123–15131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008;23:160–170. [DOI] [PubMed] [Google Scholar]
- 37. McCarthy JJ, Esser KA. Counterpoint: satellite cell addition is not obligatory for skeletal muscle hypertrophy. J Appl Physiol 1985;2007:1100–1102, discussion 2‐3. [DOI] [PubMed] [Google Scholar]
- 38. Miano JM, Ramanan N, Georger MA, de Mesy Bentley KL, Emerson RL, Balza RO Jr, et al. Restricted inactivation of serum response factor to the cardiovascular system. Proc Natl Acad Sci U S A 2004;101:17132–17137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Niu Z, Iyer D, Conway SJ, Martin JF, Ivey K, Srivastava D, et al. Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc Natl Acad Sci U S A 2008;105:17824–17829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Balza RO Jr, Misra RP. Role of the serum response factor in regulating contractile apparatus gene expression and sarcomeric integrity in cardiomyocytes. J Biol Chem 2006;281:6498–6510. [DOI] [PubMed] [Google Scholar]
- 41. Wang H, Hertlein E, Bakkar N, Sun H, Acharyya S, Wang J, et al. NF‐κB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol Cell Biol 2007;27:4374–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. DJ‐1 is increased in skeletal muscle during rehabilitation in human. (A) The information of muscles sample donors. (B) Dj‐1 Genome‐wide profiling expression data from immobilized human muscles during the 0‐day, 4‐week, 6‐week and 4‐month rehabilitation (n = 16–19). (C) The expression of Dj‐1 in mature myotubes of mice with 5‐month, 24‐month and 30‐month‐old. FBG: Fasting blood glucose, **p < 0.01, ***p < 0.001, a two‐tailed Student t test was used for statistical analysis.
Figure S2. Generation and verification of skeletal muscle specific DJ‐1 knockout mice. (A) Schematic of Flox targeting‐construct for the Dj‐1 locus. (B‐E) Western blot analysis of DJ‐1 in different muscle depots and heart of MDKO and Flox mice. (F) Muscles weight of male mice at the age of 18 weeks (n = 8). QUA, quadriceps (a). SOL, soleus (b). TA, tibialis anterior (c). EDL, extensor digitorum longus (d). Data represented the mean±s.e.m.. *p < 0.05, **p < 0.01, a two‐tailed Student t test was used for statistical analysis.
Figure S3. DJ‐1 knockout in skeletal muscle decreases muscle mass and impairs exercise performance in female mice. (A) Body weight of female mice fed on chow diet at 18‐week‐old (n = 8). (B‐C) Body composition of female mice at the age of 18 weeks (n = 8). Fat mass (B). Lean mass (C). (D) Muscles weight of female mice at the age of 20 weeks (n = 4). QUA (a). GAS (b). SOL (c). TA (d). EDL (e). (E) Exercise performance tests. Grip strength of female mice at the age of 13 weeks (n = 7–8) (a). Maximum load capacity of female mice in a resistance test at the age of 14 weeks (n = 7–8) (b). Maximum hanging time with four or fore limbs of mice at the age of 15 weeks (n = 7) (c‐d). Rotarod performance test of female mice at the age of 16 weeks (n = 8) (e). Total time, maximum speed and total distance of female mice in exhaustion test at the age of 17 weeks (n = 7) (f). Data represented the mean ± s.e.m.. *p < 0.05, ***p < 0.001, ****p < 0.0001, a two‐tailed Student t test was used for statistical analysis.
Figure S4. DJ‐1 knockout in skeletal muscle decreases muscle mass and impairs exercise performance in aged male mice. (A) Body weight of aged male mice fed on chow diet at 18‐month‐old (n = 5). (B) Lean mass of aged male mice at the age of 18 months (n = 5). (C) Grip strength of aged male mice at the age of 18 months (n = 5). (D) Maximum load capacity of aged male mice in a resistance test at the age of 18 months (n = 5). (E) Rotarod performance test of aged male at the age of 18 months (n = 5). Data represented the mean ± s.e.m.. *p < 0.05, ***p < 0.001, ****p < 0.0001, a two‐tailed Student t test was used for statistical analysis.
Figure S5. DJ‐1 ablation decreases CSA of soleus, but has no effects in mitochondrial DNA number or OXPHOS genes expression. (A) Physiological consequences of GAS in male mice fed on chow diet at the age of 18 weeks. Representative H&E staining of SOL (scale bars, 20 μm) (a). CSA distribution and median in SOL of male mice (n = 4–6) (b‐c). (B) Physiological consequences of SOL immunofluorescence staining in male mice. Representative immunofluorescence analysis of muscle fibre type in SOL (a). The different myosin heavy chain isoforms were stained in blue (MyHC‐I) or green (MyHC‐IIa) (scale bars, 20 μm). The ratio of muscle fibre type (n = 4–6) (b). CSA distribution in SOL (n = 4–6) (c). (C‐D) Representative electron micrographs of cross or longitudinal section in SOL of male mice (scale bars, 1 μm). Cross section (C), Longitudinal section (D). Blue arrows point to the healthy mitochondria in Flox mice and the impaired mitochondria in MDKO mice. (E). Mitochondria function in GAS of male mice. Mitochondrial DNA number (n = 4) (a). Expression of mitochondria complex related genes and proteins measured by qPCR (b) and western blots (c). Immunoblot analysis of PGC1α protein in GAS muscle of male KO and Flox mice (d). Data represented the mean±s.e.m.. *p < 0.05, a two‐tailed Student t test was used for statistical analysis.
Figure S6. DJ‐1 deletion shows progressive atrophy in SOL of immobilization models. (A) The weight of tissues of male mice at the age of 10 weeks in immobilization models (n = 6–7). BAT (a). iWAT (b). eWAT (c). Heart (d). Liver (e). (B). Physiological consequences of GAS H&E staining in immobilization models. Representative H&E staining of SOL (scale bars, 10 μm) (a). CSA distribution (b) and median (c) of SOL in immobilization models. (C) The ratio of muscle fibre type of GAS in immobilization models. (D) Physiological consequences of SOL immunofluorescence staining in immobilization models. Representative immunofluorescence analysis of muscle fibre type of SOL in immobilization models (a). The different myosin heavy chain isoforms were stained in blue (MyHC‐I) and green (MyHC‐IIa) (scale bars, 50 μm). The ratio of muscle fibre type (b) and CSA distribution (c) of SOL in immobilization models (n = 6–8). Data represented the mean±s.e.m.. ***p < 0.001, a two‐tailed Student t test was used for statistical analysis. (E) ROS was assessed by DHE staining in GAS (scale bars, 20 μm). Representative DHE staining images (a), DHE intensity analysis by Image J (b). (F) Mitochondria function was estimated in immobilized mice. Mitochondrial DNA number in GAS of immobilized mice (n = 3) (a). Expression of mitochondria complex related genes measured by western blots (b) and qPCR (c).
Figure S7. Myogenesis genes and autophagy related genes are not altered in DJ‐1 disrupted myotubes, but atrogenes are increased in GAS muscles. (A) Expression of myogenesis genes and proteins. Expression of muscle satellite cell related genes of GAS in immobilization models (n = 3–5) (a), Immunoblot analysis of MyoD and MyoG protein level in C2C12 myotubes (b). (B) Immunoblot analysis of phosphorylation and total mTOR of GAS in immobilization models. (C) Expression of GAS FoxO regulated genes, atrogenes, proteins and some transcript factors from mice on immobilization model (n = 5). Genes expression by RNA‐SEQ analysis (a). Atrogenes expression determined by qPCR of GAS (n = 3) (b). Immunoblot analysis of MSTN, MuRF1 and Atrogin‐1 protein level of GAS in immobilization models (n = 3) (c). Proteasome related genes (d), FoxOs regulated genes (e), other pathways related genes(f). (D) Ubiquitination expression level in GAS of immobilized mice (c). (E‐a) Expression of GAS atrogenes from mice on chow diet (n = 3–5). (E‐b) Immunoblot analysis of MuRF1 protein level in GAS of mice on chow diet. (F) Expression of GAS autophagy pathway proteins and genes in Immobilization model and autophagy flux model mice (n = 3–5). P62 and LC3 proteins level in GAS of immobilized mice and autophagy flux mice (a), Expression of P62 and LC3 proteins in autophagy flux model mice (b). (c) P62 and Lc3 genes level in GAS of immobilized mice. Data represented the mean±s.e.m.. *p < 0.05, a two‐tailed Student t test was used for statistical analysis. Ctsl, Cathepsin l; Gab, Gabarapl1; 4EPB, Eif4ebp1; Ncl, nucleolin.
Figure S8. FoxO1 pathway is responsible for DJ‐1‐ablation induced muscle atrophy. (A) Western blots analysis of phosphorylation and total FoxO1 protein level in GAS of mice fed on chow diet. (B) Immunoblot analysis of phosphorylation and total FoxO1 in C2C12 myotubes. (C) Protein level of FoxO1 in cytoplasmic fractions and nuclear fractions of GAS from mice fed on chow diet. (D) The effects of DJ‐1 knockdown or overexpression in 293 T cells. (D‐E) The distribution of fibre CSA in comp23‐treated mice. comp23, Compound23.
Table S1. Primer sequences.
Table S2. Antibody list.