

Age- and differentiation-dependent apoptotic priming plays a pivotal role in driving radiotherapy-induced neurocognitive impairment and can be targeted for neuroprotection in pediatric patients.
Abstract
Although external beam radiotherapy (xRT) is commonly used to treat central nervous system (CNS) tumors in patients of all ages, young children treated with xRT frequently experience life-altering and dose-limiting neurocognitive impairment (NI) while adults do not. The lack of understanding of mechanisms responsible for these differences has impeded the development of neuroprotective treatments. Using a newly developed mouse model of xRT-induced NI, we found that neurocognitive function is impaired by ionizing radiation in a dose- and age-dependent manner, with the youngest animals being most affected. Histologic analysis revealed xRT-driven neuronal degeneration and cell death in neurogenic brain regions in young animals but not adults. BH3 profiling showed that neural stem and progenitor cells, neurons, and astrocytes in young mice are highly primed for apoptosis, rendering them hypersensitive to genotoxic damage. Analysis of single-cell RNA sequencing data revealed that neural cell vulnerability stems from heightened expression of proapoptotic genes including BAX, which is associated with developmental and mitogenic signaling by MYC. xRT induced apoptosis in primed neural cells by triggering a p53- and PUMA-initiated, proapoptotic feedback loop requiring cleavage of BID and culminating in BAX oligomerization and caspase activation. Notably, loss of BAX protected against apoptosis induced by proapoptotic signaling in vitro and prevented xRT-induced apoptosis in neural cells in vivo as well as neurocognitive sequelae. On the basis of these findings, preventing xRT-induced apoptosis specifically in immature neural cells by blocking BAX, BIM, or BID via direct or upstream mechanisms is expected to ameliorate NI in pediatric patients with CNS tumor.
Significance:
Age- and differentiation-dependent apoptotic priming plays a pivotal role in driving radiotherapy-induced neurocognitive impairment and can be targeted for neuroprotection in pediatric patients.
Introduction
External beam radiotherapy (xRT) is used to treat a wide range of malignancies that arise or metastasize to the brain in patients across the age spectrum, yet the doses that can be safely administered are limited by the potential injury to healthy brain tissue (1–4). Although technological improvements in therapeutic delivery of xRT attempt to limit the radiation exposure of healthy brain tissue, the doses administered to the normal cells adjacent to tumors cause DNA damage at levels sufficient to induce cell death and dysfunction (3, 5). In addition, high-dose xRT is intentionally delivered to the entire central nervous system (CNS) to control tumor spread or prevent seeding in patients with common tumors that have metastatic potential, such as medulloblastomas. In fact, most children diagnosed with this disease will undergo surgical resection and be conventionally treated with 23.4 Gy of craniospinal irradiation (IR) with an additional 32.4 Gy delivered to the posterior fossa where these tumors arise, even if there is no evidence of CNS dissemination after resection (3, 6). Interestingly, the long-term effects of radiotherapy are highly age dependent: young children exhibit impairments in learning, memory, executive processing, visual acuity, and fine motor coordination after xRT at vastly higher rates and with more severity than adults treated with similar doses (3). This impairment is associated with poor quality of life and poor economic status in childhood brain tumor survivors (7). Furthermore, this neurotoxicity is dose limiting and prevents the use of what could otherwise be potentially curative doses of xRT in children with brain tumors (3, 5). In fact, due to the high risk of severe neurotoxicity, xRT is no longer used in patients with medulloblastoma younger than 3 years of age despite its previously demonstrated effectiveness in tumor control (8). However, doses of xRT in excess of 50 Gy continue to be used in pediatric patients diagnosed with intracranial ependymoma as young as 1 year of age (9) even though it is associated with neurotoxic effects including hearing loss, hormone deficiency, cognitive impairment, and attentional deficits (9, 10). Clinical trials evaluating lower doses of xRT or region-sparing treatment strategies to reduce neurotoxicity have been able to reduce neurocognitive impairment (NI), yet this comes at the cost of decreased cure rates, increased tumor recurrence, and reduced survival (11).
The cognitive dysfunction resulting from cranial xRT in pediatric patients often manifests as deficits in executive processing, attention and hippocampal-dependent learning and memory (L/M; refs. 12, 13). These deficits are compounded by heightened anxiety and depression-like symptoms. Importantly, the severity of impairment depends upon the dose of radiation delivered and age of the patient. The direct causes of cognitive impairment after xRT are not fully clarified, in part due to the lack of rigorous mouse models defining the age-dependent effects of xRT. Varied mechanisms have been reported to be involved in xRT-induced NI including decreased hippocampal neurogenesis, altered neuronal function, neuroinflammation, disrupted white matter development, and impaired vascular and glial clonogenesis (14–16). While cellular necrosis has been shown to be the prominent cause of cell death in the adult brain in response to other stimuli, recent evidence suggests apoptosis is the key cell death pathway involved in neuronal cell death in young brain tissue (17, 18). Even at low doses, xRT reduces the number of proliferating cells in the dentate gyrus (DG) of rodents by increasing apoptotic cell death (19, 20), highlighting a potential vulnerability of progenitor cells to ionizing radiation in mice and humans.
The commitment to apoptotic cell death is controlled by the BCL2 family of proteins, which consists of prosurvival and proapoptotic proteins as well as the pore-forming proteins BAX and BAK that trigger mitochondrial release of cytochrome c and activation of caspases (21, 22). BAX has been previously shown to be the key proapoptotic effector in cells within the developing brain (18, 23). Collectively, these observations support the hypothesis that blocking apoptosis via BAX downregulation or inhibition may reduce neuronal cell death in various neuropathologic settings. Indeed, Bax−/− mice (24) exhibit reduced neuronal cell death in response to ethanol toxicity (25), brain injuries (26, 27), global ischemia (28), and ionizing radiation (18). However, previous studies have not identified the key cell types affected at different developmental time points nor explored the extent to which blockade of BAX-mediated apoptosis in the developing brain can improve neurobehavioral outcomes in response to brain injuries. Herein, we report on the development of a novel mouse model of pediatric xRT neurotoxicity that clearly defines the age-dependent, short- and long-term neurobehavioral effects of ionizing radiation and the potential for NI prevention by blocking BAX-mediated apoptosis.
Materials and Methods
Animal care and use
Mouse experiments were conducted in accordance with protocol IS00001059, which was approved by the Harvard T.H. Chan School of Public Health Institutional Animal Care and Use Committee (IACUC). C57BL/6J [wild-type (WT)], B6.129×1-Baxtm1Sjk/J (Bax−/−; Jackson Laboratories; Strain #002994), B6.129×1-Bidtm1Sjk/J (Bid−/−; Jackson Laboratories Strain #008887), and B6.129S2-Trp53tm1Tyj/J (Trp53−/−; Jackson Laboratories Strain #002101) mice were used for experiments evaluating neuroprotection. Heterozygous male and female mice were bred to produce WT, heterozygous, and knockout (KO) litter-matched animals for analysis. Genotypes were confirmed with PCR of DNA samples from tail snips (protocols from Jackson Laboratories). Mice were housed in colonies of 4–5 animals, with sham and irradiated (IR) animals comingled in the same cage, in a 12-hour light cycle (lights on from 7 am to 7 pm), with constant temperature and humidity (22°C and 40%). Food and water were available ad libitum. Animals’ weight was monitored weekly. Mice were 4–6 weeks old at the beginning of experiments and allowed 2 weeks of acclimation before experiments began. Behavioral testing was conducted between 9 am and 3 pm. Bcl2l11 (BIM)−/− mice were obtained from Jackson Laboratories (Strain #004525). All mice were of the species Mus musculus, maintained on the C57/BLK6 background. Experiments with Bcl2l11+/+ and Bcl2l11−/− mice were conducted in accordance with protocols #19-098 and 21-011, which were approved by the University of North Carolina at Chapel Hill's IACUC.
Brain irradiation
Brain-targeted irradiation was performed using a CT-guided small animal radiation research platform (SARRP, Xstrahl) a precision radiation device capable of delivering radiation beams down to 0.5 mm in diameter under CT guidance (29). It consists of an X-ray tube attached to a gantry that rotates over a range of 120° and generates 220 kVp. Radiation dose was calibrated according to Task Group 61 of the American Association of Physicist in Medicine (30). A 10-mm focal collimator and filtration of 0.25 mm Cu + 1 mm Al was used. The target was localized, and exposure times were calculated using the Muriplan treatment planning software. After localization on CT, the animal was moved to the appropriate treatment location by the robotic positioner, which has 4 degrees of freedom (X, Y, Z, θ). Through a combination of gantry and robot stage angles, beams were delivered to only central part of the head avoiding eyes, mouth, and neck. For irradiation of Bcl2l11+/+, ±, and −/− mice, animals were anesthetized using isoflurane and irradiated with one dose of 2 Gy using an X-RAD 320 irradiator. For irradiation of BAX+/+ and Bax−/− mice used for immunofluorescence of hippocampus, amygdala, and perirhinal cortex, mice were subjected to whole-body irradiation using a Cs-137 source (Mark 1 Irradiator, Shepherd & Associates) irradiator.
IHC, immunofluorescence, and histology
Brains were collected, rinsed with Hank's Balanced Salt Solution and placed into 4% formaldehyde solution (Electron Microscopy Sciences) for 24 hours at 4°C followed by 70% ethanol until processed. Sections were treated with antigen retrieval for 15 minutes, washed three times with PBS, and further treated with 0.3% Triton X-100 for 30 minutes followed by wash. Tissue was then blocked in normal horse serum for 30 to 60 minutes and incubated with primary antibodies against cleaved caspase-3 (Cell Signaling Technology #9664, RRID:AB_2070042), BAX (Cell Signaling Technology #2772, RRID:AB_10695870), BAK (Cell Signaling Technology #12105, RRID:AB_2716685), GFAP (Thermo Fisher Scientific 13-0300, RRID:AB_2532994), or MAP2 (Thermo Fisher Scientific #PA1-10005, RRID:AB_1076848) overnight at 4°C. Following day, slides are washed three times 5 minutes each in 0.1% PBS-Triton X100. Primary antibodies were detected with horseradish peroxidase–conjugated anti-rabbit and anti-mouse secondary antibodies (GE Amersham). In addition, sections were stained with hematoxylin and eosin stain. All slides were examined and scanned using microscope (Incell 6000) for further qualitative and semiquantitative analyses.
Immunofluorescent staining of brain tissues was performed on 4% paraformaldehyde (PFA)-fixed 5-μm-thick paraffin sections. Mouse brain samples were fixed in 4% PFA overnight, and further processed in a tissue processor (Thermo Fisher Scientific). Sections (5 μm) were cut from paraffin blocks using a CUT 6062 microtome (SLEE Medical GmbH). The sections were first warmed to 60°C in a vacuum incubator (Isotemp Vacuum Oven, Thermo Fisher Scientific) then washed immediately twice in xylene, gradually redehydrated in ethanol (100%, 95%, 70%, water), and then processed for antigen retrieval using sodium citrate buffer (pH 6.0) in a steamer for 30 minutes. Samples were allowed to cool down for 30 minutes on ice, rinsed in PBS followed by a wash with distilled water. Tissue autofluorescence was removed using a bleaching solution containing 3% hydrogen peroxide. Samples were permeabilized with 0.3% Triton X-100 for 30 minutes, followed by incubation with blocking buffer [5% normal goat serum (NGS)] for 1 hour at room temperature, then incubated overnight at 4°C with primary antibodies (diluted in 0.5% BSA per manufacturer's recommendation). The following day, samples were washed three times with PBS and incubated with appropriate fluorescent-labeled secondary antibodies (1:1,000 dilution in 0.5% BSA, Life Technologies) and the nuclear marker DAPI (BioLegend). Slides were mounted using anti-fade mounting media (Thermo Fisher Scientific) prior to imaging. Antibodies used for immunostaining are cleaved caspase-3 (Cell Signaling Technology #9664, RRID:AB_2070042), and MAP2 (Thermo Fisher Scientific #PA1-10005, RRID:AB_1076848). In addition, sections were stained with hematoxylin and eosin stain to observe histology.
For immunofluorescence of hippocampus, amygdala and perirhinal cortex in Bax+/+ and Bax−/− animals, postnatal day 4 (P4) animals were irradiated with 8 Gy and brain tissue was collected and fixed 4 hours later. 4% PFA-fixed 5-μm-thick paraffin sections were washed in Histo-Clear II followed by rehydration with a decreasing ethanol gradient. Antigen retrieval and autofluorescence photobleaching was performed, followed by permeabilization with PBS-Triton X 0.5%, blocking in 5% NGS (Vector Laboratories S-1000-20), and primary antibody incubation at 1:200 dilution in 5% NGS. Antibodies: cleaved caspase-3 (Cell Signaling Technology #9661, RRID:AB_2341188), SOX2 (Cell Signaling Technology #4900, RRID:AB_10560516), and NG2 (Abcam ab273443, RRID:AB_2940856). Secondary antibody incubation at 1:200 dilution (Thermo Fisher Scientific A-11008, RRID:AB_143165) and DAPI staining (Abcam ab228549) was performed the next day, with cover slipping using vector labs antifade mounting media (Vector Laboratories H-1000-10). Slides were imaged on a Zeiss Axio Observer and quantified using CellProfiler (31).
Imaging and analysis
Imaging was carried out using a 20x/0.75 NA objective lens on a GE Healthcare IN Cell Analyzer 6000—Cytiva. Images overlapping by 20% were acquired for an entire section for every channel and stitched using ASHLAR7777, and a multi-channel composite image generated using Fiji (32). For quantification of immunofluorescence images, cells were segmented into individual objects based on the DAPI channel and cells stained positive for primary antibody were quantified based on Fiji measurements of mean intensity.
BH3 profiling
For BH3 profiling of WT animals, brain tissue samples from mice of different ages (P0-P6) were dissociated into a single-cell suspension using the collagenase/dispase for 30 minutes at 37°C. Samples were placed on ice and triturated with a cut 1 mL pipette, and were left to settle for 2–5 minutes before the cloudy cell suspension was transferred to new tubes and centrifuged at 200 × g for 5 minutes at 4°C. Supernatant was discarded and the pellet was resuspended in 100 μL FACS Stain Buffer (2% FBS in PBS) with 1 μL anti-CD45− AlexaFluor488 or anti-CD45− AlexaFluorPE (BioLegend). Cells were stained on ice for 25 minutes away from light, then centrifuged at 200 × g for 5 minutes and subjected to flow cytometry–based BH3 profiling as described previously (33). Briefly, cells were treated with activator or sensitizer BH3 peptides (New England Peptide) for 60 minutes at 28°C in MEB (10 mmol/L HEPES pH 7.5, 150 mmol/L mannitol, 50 mmol/L KCl, 0.02 mmol/L ethylene glycol tetraacetic acid, 0.02 mmol/L ethylenediaminetetraacetic acid, 0.1% BSA, 5 mmol/L succinate) with 0.001% digitonin. Peptide sequences are as follows: BIM (Ac-MRPEIWIAQELRRIGDEFNA-NH2), BID (Ac-EDIIRNIARHLAQVGDSMDRY-NH2), PUMA (Ac-EQWAREIGAQLRRMADDLNA-NH2), BAD (Ac-LWAAQRYGRELRRMSDEFEGSFKGL-NH2), HRK (Ac-WSSAAQLTAARLKALGDELHQ-NH2), and MS1 (Ac-RPEIWMTQGLRRLGDEINAYYAR-NH2). After peptide exposure, cells were fixed in 2% PFA for 15 minutes, which was then neutralized by addition of N2 buffer (1.7 mol/L Tris base, 1.25 mol/L glycine, pH 9.1). Cells were stained overnight with DAPI (1:1,000, Abcam), anti-PSA-NCAM-PE (1:500, Miltenyi Biotec 130-117-394, RRID:AB_2727931), GFAP-PE or GFAP-AlexaFluor488 (1:1,000 BioLegend 644701, RRID:AB_2109791), anti-CD133-PE (1:1,000 Millipore MAB4310, RRID:AB_2170527), and anti-Cytochrome c-AlexaFluor647 (1:2,000, clone 6H2.B4, BioLegend 612310, RRID:AB_2565241) in a saponin-based buffer (final concentration 0.1% saponin, 1% BSA) and then analyzed by flow cytometry. Cytochrome c release in response to BIM treatment was measured on an Attune NxT flow cytometer (Thermo Fisher Scientific). Data for marker positive cells were collected from the DAPI+/CD45− population.
For BH3 profiling of brain tissue from Bax−/− mice and Bax+/+ littermate controls, brain tissue samples were dissociated following an updated protocol, using a GentleMACS Octo Dissociator with Heaters (Miltenyi Biotec) and the corresponding Adult Brain Dissociation Kit. Tissue dissociation was carried out according to the manufacturer's protocol. Briefly, dissected brain tissue was cut into eight sagittal slices and added to an OctoMACS C tube with dissociation enzymes. Mechanical and enzymatic digestion was carried out on the GentleMACS Octo Dissociator using the protocol 37C_ABDK_01. Samples were strained through a 70 μm MACS Smart Strainer, then mixed with Debris Removal Solution to remove myelin and other debris by density centrifugation. Finally, cells were suspended in red blood cell Removal solution to lyse blood cells and yield a pure population of neural cells. These cells underwent staining for specific cell type markers, followed by BH3 profiling as described.
Primary neural cell cultures
Primary cortical and hippocampal cells taken from neonatal (P0) mouse brain tissue were cultured in a neurobasal medium (Thermo Fisher Scientific) with required growth factors to support neurons, astrocytes, microglia, and oligodendrocytes. B-27, GlutaMAX and glucose (Thermo Fisher Scientific) were added to growth media for cortical and hippocampal neurons in neurobasal media. Astrocytes were cultured and seeded in DMEM/F12 media with FBS. After 4–5 days, neurons and glia were collected using trypsin and processed for downstream analysis.
Immunoblotting
Immunoblotting was performed as described previously (23). Briefly, protein lysates were obtained by cell lysis in RIPA buffer (150 mmol/L sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate, 50 mmol/L Tris, ph 8.0). Protein loading was measured by Protein Assay Dye Reagent (Bio-Rad). Protein samples were electrophoretically separated on NuPAGE 4%–12% Bis-Tris polyacrylamide gels (Invitrogen). Antibodies used include: BAX (Cell Signaling Technology #2772, RRID:AB_10695870); BAK NT (Millipore 06-536, RRID:AB_310159); BIM (Cell Signaling Technology 2933, RRID:AB_1030947); BID (Cell Signaling Technology 2002, RRID:AB_10692485), cleaved BID (Millipore PC645, RRID:AB_2258975), cleaved caspase-3 (Cell Signaling Technology 9665, RRID:AB_2069872), Puma (Cell Signaling Technology #D30C10), p53 (Cell Signaling Technology 12450, RRID:AB_2797920), p21 (Cell Signaling Technology catalog no. 2947, RRID:AB_823586), Actin (Cell Signaling Technology 4967, RRID:AB_330288), GAPDH (Cell Signaling Technology #5174, RRID:AB_10622025).
Flow cytometric host cell reactivation analysis of DNA repair capacity
Reporter plasmids were prepared and the flow cytometric host cell reactivation (FM-HCR) assay was performed as described previously (34). HeLa WT, BAX KO (sgBAX), BAK1 KO (sgBAK1), and BAX KO BAK1 KO (sgBAX sgBAK1) were seeded in complete medium at a density of 105 cells per well in 12-well culture plates (Thermo Fisher Scientific). After 24 hours, medium was refreshed and transfection with reporter plasmids (1.5 μg plasmid DNA cocktail per well) was carried out using Lipofectamine 3000 following the manufacturer's protocol (Thermo Fisher Scientific, #L3000015). Each cell line was transfected with (i) an undamaged plasmid cocktail, (ii) damaged plasmid cocktail #1, and (iii) damaged cocktail #2 as described previously (34). After 24 hours incubation at 37°C, 5% CO2, transfected cells were trypsinized, suspended in culture medium, and analyzed using an Attune NxT flow cytometer (Thermo Fisher Scientific). Gating and compensation were set using single color controls. Reporter expression was normalized to transfection efficiency and repair capacity was calculated as described previously (34). Triplicate measurements were carried out by performing FM-HCR on 3 different days. HeLa cells were obtained from an authentic source (ATCC catalog no. CCL-2, RRID:CVCL_0030) and authenticated via short tandem repeat (STR) analysis every 6 months. Cells were confirmed to be free of Mycoplasma with the MycoAlert Plus Mycoplasma Detection Kit (Lonza # LT07-710) together with the MycoAlert Assay Control set (Lonza # LT07-518) upon receipt. Cells were tested regularly for Mycoplasma and used within 5 passages. Kruskal–Wallis test was performed with Dunn adjustment for multiple comparisons was computed using Prism 8.1 to assess statistical significance (P ≤ 0.05).
Flow cytometric analysis of cell viability
Radiation sensitivity of mouse embryonic fibroblasts (MEF) was assessed by irradiating cells in a X-RAD 320 (Precision X-Ray) followed by staining with Annexin V (R&D Systems; #4830-250-K) and propidium iodide (Thermo Fisher Scientific) after 48 hours. Knockdown of Bid was performed using ON-TARGETplus Mouse BID siRNA (Horizon Discovery L-058612-00-0010) transfected with Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific 13778075) according to manufacturer's protocol. Cells were analyzed on an Attune NxT flow cytometer. MEFs were obtained from a collaborating laboratory and authenticated via STR analysis. Cells were confirmed to be free of Mycoplasma as described above and used within 5 passages.
Caspase activation in tissues
Caspase activation was measured in irradiated tissues as described previously (18). Briefly, tissues were collected at indicated time points, mechanically dissociated and Dounce homogenized. Protein concentrations were measured using the Bradford assay (Thermo Fisher Scientific) and 5 μg of protein in 15 μL of PBS was loaded into triplicate wells in a 384-well plate. A total of 15 μL of Caspase-Glo 3/7 reagent (Promega; #G8090) was added to each well and luminescence was measured after 30 minutes on a Tecan microplate reader (Tecan Group).
Neurobehavioral analyses
Baseline and post-IR behavioral testing were performed with all mice together, in the same order each week. All animals are handled after IR everyday till day of testing for 5 minutes/mice. All subjects in the home cage are placed in the testing room for at least 1 hour before testing to minimize effects of stress on behavior during testing. During this 1 hour, behavioral equipment is cleaned and prepared for testing and weights are measured once per week. Apparatus was cleaned with 70% ethanol between each animal. Experiments were blinded as to the genotype of the animals or treatments administered whenever possible.
Open field test
Animals are placed into the center of a clear Plexiglas (40 × 40 × 30 cm) open field arena. Total distance (locomotor activity), center distance (the distance traveled in the center of the arena), rearing (standing on the hindlimbs without touching wall), grooming (licking and/or scratching the fur, licking the genitalia and tail), pausing (immobility/inactivity), stretching (stretching the whole body forward while keeping the hindlimbs in place), and fecal boli were recorded. Three trials after IR over the time course (30 days, 3 months, 6 months) are recorded for each animal. Statistical analysis was performed by one-way or two-way ANOVA.
Rotarod
Motor coordination was measured by the accelerating rotarod test. The rotarod apparatus (Stoelting Co. Ltd.) consists of a rod that is capable of rotating at different speeds. During test trials, the rod revolves with a steady rate of acceleration from 4 to 40 rpm over a 5-minute period. Animals were placed on the rod with constant low-speed rotation (4 rpm). After the mouse was accustomed to walking on the rod, the test was started. The number of falls and time spent on rotating rod was automatically measured.
Elevated plus maze and light-dark chamber
Both tasks are based on the innate aversion of animals to brightly illuminated open areas and on their tendency to exhibit spontaneous exploratory/locomotor behavior in response to novel environments, light and stress. The test is considered to be a measure of generalized anxiety because agents used to alleviate generalized anxiety disorder symptoms modify defensive behaviors evoked by the model. The elevated plus maze (EPM) apparatus consisted of a white wooden laminate structure with two open arms and two opposite closed arms (50 × 50 × 10 cm) that had 20 cm high walls. The open and closed arms were connected by a central square (10 × 10 cm). Mice were placed in the center of the maze, facing one closed arm, and allowed to explore for 5 minutes. Total distance traveled, number of entrances, and time spent in each arm was recorded. Light-dark (LD) apparatus has two connected compartments, one is lit (aversive area) and the other is darkened (safe area), with an opening between the two compartments. Mice were placed in the light area and they begin moving along the periphery of the compartment until they locate the opening to the dark compartment. Prior to the trial mice are kept in dark room for 1 hour. The time animal spends moving, rearing, and transitioning from the dark to light area is recorded over a 5-minute period. In addition, the number of crossings from the lit to the darkened side and vice versa and the level of locomotor activity in the entire chamber (darkened and lit sides combined) is measured. The first latency to enter dark compartment and time it takes to return to lit side are indices of anxiety. Transitions are index of activity exploration due to habituation over time.
Tail suspension and forced swim test
The tests are based on the same principle: measurement of the duration of immobility when rodents are exposed to an inescapable situation. In tail suspension task, mice are suspended by their tails with tape (1 cm from tip of tail), in such a position that it cannot escape or hold on to nearby surfaces. During this test, the mice were allowed to hang for 6 minutes and the duration of immobility was recorded using a stopwatch. Mice were considered immobile only when hanging passively and completely motionless. In forced swim test (FST), mice were placed individually in glass cylinders (40 × 20 cm) filled with water (22°C–25°C) to a depth of 30 cm; at this depth the mouse cannot stand on the cylinder bottom. The water was changed before the next animal was placed into the water tank. Passive/immobile behavior, floating, active/mobile behaviors, escape-oriented behaviors, and self-directed behaviors were scored.
Novel object recognition test and Y-maze
Tests are based on the spontaneous tendency of rodents to spend more time exploring a novel object/arm than a familiar one. The choice to explore the novel object reflects the use of learning and recognition memory. This is a non-force driving and spontaneous memory test, unlike most of the learning memory tasks that require trigger or stress stimulus. Mice are habituated to the test chamber by allowing them to freely explore the empty chamber for 5 to 10 minutes on the first day. A total of 24 hours following habituation, mice are placed in the test chamber for 2 minutes for rehabituation. The mice are removed, and two identical objects are placed in the test chamber. Then the mice are allowed to explore the objects (forepaw and/or nose contact) and time spent exploring and number of times is measured. On the third day of trial, mice are returned to the box with one novel object and one familiar that are counterbalanced to the preferred side from the training day (the less preferred side gets the novel object to eliminate any bias effect). The time spent exploring either object is measured. The discriminative index is calculated on the basis of:
 |
The discrimination in preferentially exploring the novel object over the familiar object provides an index of recognition memory. The Y-maze apparatus consisted of three arms at a 120° angle to one another with a triangular central zone. Each mouse was placed at the end of a randomly chosen arm and allowed to freely explore two arms for 5 minutes with the third arm blocked off. The blocked arm is opened in third trial and the time spent in the novel or familiar arm was measured.
Barnes maze
The Barnes maze is a spatial learning apparatus in which the animals search for an escape box (35). The maze is a dark circular platform (120 cm diameter), elevated 90 cm from the floor, containing 20 holes (10 cm in diameter) disposed circularly at the edge of the platform. One of the holes (target hole) is connected to an escape box (10 × 10 × 15 cm). There were visual cues on the walls, located 50 cm distant from the apparatus. During the behavioral sessions, the lights were turned on (420 lux) to increase escape motivation. In the habituation session, the animals were allowed to freely explore the apparatus during 150 seconds or until the entrance in escape box. Afterward, the animals were submitted to a set of four daily training sessions, each one with two trials. The intertrial interval was the duration of cleaning the apparatus (with a 5% alcohol solution). The first training session was performed immediately after the habituation. All trials lasted 300 seconds or until the animals reached the escape box. However, if the mice did not reach the target hole, the experimenter gently guided the animal toward it at the end of the trial. After reaching the escape box, animals remained inside for 60 seconds. The escape box is always located in the same place during the training set, but not during the previous habituation session. Retrieval of spatial learning was evaluated in the probe session, which was conducted 3 days after the last training day. The procedure was similar to the training trials, but the escape box was removed. At the beginning of each session, the animals were placed in an opaque container at the center of the maze. The container was then pulled up, and the animal was released to explore the maze. Between animals, the maze was cleaned and rotated to avoid odor cues
Composite behavioral scores
For each experiment, there are one or more contrasts of interest (e.g., sham vs. IR). For each contrast, we defined a composite test statistic for each category [anxiety and exploration (A/E), motor function (Mot), L/M, despair and depressive-like behavior (D/D)] by combining the measurements in that category as follows. For the jth measure, we computed a t-statistic (assuming unequal variances) comparing the case and control groups, excluding any missing values. Specifically, if
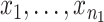 and
and
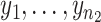 are the non-missing values of measure j for the control and case groups, respectively, then we computed
are the non-missing values of measure j for the control and case groups, respectively, then we computed
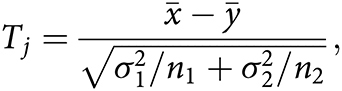 |
where
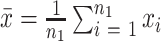 and
and
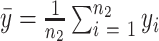 are the sample means, and
are the sample means, and
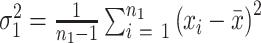 and
and
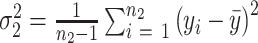 are the sample variances. Deceased subjects are treated as having missing values, this is a conservative approach to dealing with the missing values since nearly deceased subjects would be expected to have very poor performance.
are the sample variances. Deceased subjects are treated as having missing values, this is a conservative approach to dealing with the missing values since nearly deceased subjects would be expected to have very poor performance.
Then, for each category (A/E, Mot, L/M, D/D), these t-statistics are combined by taking a weighted average—for instance, for the A/E category,
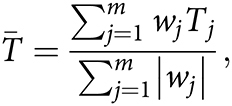 |
where the weights wj are −1, 0, or +1 depending on whether a higher value of measure j indicates better performance (+1), worse performance (−1), or is not included (0) in the A/E category. Occasionally, this results in undefined values because the case or control groups are too small due to having too many missing values. To deal with this issue, we set wj equal to 0 if n1 ≤ 1 or n2 ≤ 1 for measure j. For each contrast, this yields a test statistic
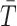 for each category (A/E, Mot, L/M, D/D).
for each category (A/E, Mot, L/M, D/D).
Calculation of P values for composite behavioral scores
We use random permutations to approximate the distribution of the test statistics
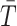 under the null hypothesis of no difference between case and control groups. Specifically, for a given category (A/E, Mot, L/M, D/D), we consider the subset of case and control subjects that have at least one non-missing value among the measurements in that category. Then we randomly permute the case–control group assignments for this subset of subjects and compute the resulting value of the test statistics
under the null hypothesis of no difference between case and control groups. Specifically, for a given category (A/E, Mot, L/M, D/D), we consider the subset of case and control subjects that have at least one non-missing value among the measurements in that category. Then we randomly permute the case–control group assignments for this subset of subjects and compute the resulting value of the test statistics
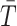 ; this yields a single simulated value from the approximate null distribution. We approximate the null distribution by computing
; this yields a single simulated value from the approximate null distribution. We approximate the null distribution by computing
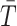 for R = 100, 000 such permutations. We then compute an initial estimate of the P value as the proportion of these
for R = 100, 000 such permutations. We then compute an initial estimate of the P value as the proportion of these
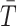 samples that are greater than the observed value of
samples that are greater than the observed value of
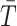 :
:
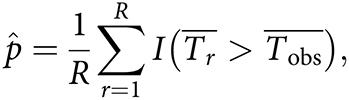 |
where I(·) denotes the indicator function, which equals 1 if the event is true and equals 0 otherwise. We use one-sided hypothesis tests because we define each contrast such that the alternative hypothesis is that the “control” group exhibits better performance than the “case” group.
When the P value is very small (≤1/R), this initial
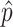 estimate will often be equal to zero because none of the
estimate will often be equal to zero because none of the
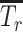 values will exceed
values will exceed
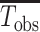 . To improve the approximation in this case, we use a normal approximation to the null distribution and compute the resulting P value. That is, if
. To improve the approximation in this case, we use a normal approximation to the null distribution and compute the resulting P value. That is, if
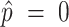 according to the initial estimate above, then we redefine
according to the initial estimate above, then we redefine
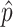 as follows:
as follows:
where Z is a normal random variable with mean
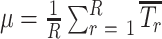 and variance
and variance
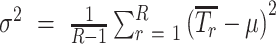 .
.
To adjust for multiple comparisons, we use the Bonferroni correction for M = 89 tests, which is the total number of tests performed across all parts using the composite score analyses. In the figures, we transform the estimated P values to −log10(P) for visualization purposes. The black dotted lines indicate the Bonferroni-corrected significance threshold when controlling the family-wise type I error rate at 0.05, specifically, the black dotted lines are at −log10(P) = −log10 (0.05/M) ≈ 3.25.
Single-cell RNA sequencing data analysis
We used previously published single-cell RNA sequencing (RNA-seq) data to examine expression of apoptosis-associated genes in the developing hypothalamus (36). Data were downloaded from Gene Expression Omnibus (GEO) accession number GSE132355 and analyzed using the Seurat package in R (37). The FindMarkers function in Seurat was used with default parameters to identify genes differentially expressed by Bax-positive hypothalamic neurons compared with hypothalamic neurons with no Bax expression. Enrichment of gene sets from the Molecular Signatures Database (38, 39) among the genes differentially expressed by Bax-positive hypothalamic neurons was done using the enrichr package (40). Enrichment plots for significant pathways were generated using enrichplot.
Statistical analysis
Mantel–Cox test, Kruskal–Wallis test, one-way and two-way ANOVA followed by Holm-Sidak post hoc multiple comparisons test, and t tests were performed using the GraphPad Prism software (GraphPad Software). Significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 throughout.
Data availability
The raw data generated in this study are available upon request from the corresponding author. Single-cell RNA-seq data analyzed in this study were obtained from GEO at GSE132355.
Results
High-dose 8 Gy xRT induces cognitive deficits and delay in learning memory in neonates
Existing mouse models of xRT-induced NI utilize juvenile or adult mice and are unsuitable for translational or mechanistic studies of xRT effects in pediatric patients with CNS tumor across the age spectrum and especially in young children, who are more likely to experience the most severe effects. To address this, we sought to develop a mouse model that allows for rigorous comparison of xRT-mediated neurocognitive effects from birth to adulthood (Fig. 1A). Neonatal mice (P3-P5) were initially treated with cranial xRT at a moderate/high dose (8 Gy) that was previously shown to induce mild but reproducible NI in juvenile and adult mice (18). Surprisingly, neonates irradiated at this dose exhibited an extremely high rate of mortality (Fig. 1B) and weight loss (Fig. 1C) compared with litter-matched shams, already demonstrating the heightened sensitivity of young animals to this treatment.
Figure 1.

Moderate/high-dose (8 Gy) brain irradiation induces cognitive deficits and delay in L/M in surviving neonates. A, Overall design of studies evaluating effects of age and apoptosis inhibition on xRT-induced NI and neural cell death. B, Overall survival of mice treated with 8 Gy CT-guided, whole-brain irradiation. C, xRT-induced changes in body weight. D and E, A/E as well as Mot was assessed using the open field test at 30 (D) and 60 (E) days after treatment. F, Rearing behavior (A/E, Mot) assessed at indicated number of days after treatment. G and H, A/E and Mot was assessed by counting crossings (G) between light and dark chamber and time spent in light chamber (H) at indicated number of days after treatment. I and J, Mot was assessed by rotarod test at indicated number of days after treatment by counting number of falls (I) and latency to first fall (J). K, Y maze was used to assess L/M at indicated number of days after treatment by timing the duration of animal exploration of novel arm of maze. L, NORT was used to assess L/M by testing the ability of the animal to discriminate between novel and familiar objects at indicated number of days after treatment. M, D/D was assessed using the tail suspension test by timing the duration of animal immobility (freezing) during a 5-minute test period. N, Neurobehavioral composite scoring indicates the significance of differences in indicated behavior types at indicated time after xRT. O, Histology of brain sections of indicated regions 60 days after 8 Gy xRT. Scale bars, 100 μm. P and Q, Overall survival (P) and weight (Q) of mice irradiated at P3-P6 with indicated dose of CT-guided brain xRT. Unless otherwise specified, each point on charts represents results from one animal in that treatment group, averaged over the number of tests performed as described in Materials and Methods. Unless otherwise specified, comparison between groups was conducted by one-way or two-way ANOVA followed by Holm-Sidak post hoc multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
We performed a battery of neurobehavioral tests on surviving mice to assess A/E, Mot, L/M as well as D/D to compare with previously reported results (Fig. 1A). These tests were chosen based on their widespread use for assessment of these factors, which are commonly altered in pediatric tumor survivors treated with xRT compared with age-matched or sibling controls (41, 42). For example, symptoms of anxiety and anxiety disorders are significantly more common in patients with cancer than the general population and this is particularly true of CNS tumor survivors treated with xRT (43). To assess levels of A/E following irradiation in our mouse models, open field test and LD transition test were used as they are based on the ingrained exploratory behavior of rodents along with their natural aversion for open spaces, which normally trigger fear and anxiety (44, 45). Animals tend to avoid the open areas, especially when they are brightly lit, favoring darker, more enclosed spaces and the activity/avoidance behaviors are indicators of physiologic stress levels. In the open field test (Fig. 1D), we noted that 8 Gy irradiated animals had prolonged periods of immobility and reduced exploration of the inner zone as compared with sham. The magnitude of these differences was reduced in animals surviving to 60 days after xRT but still highly significant (Fig. 1E). We also noted a near-total lack of rearing behavior in irradiated animals at 30 as well as 60 days after treatment (Fig. 1F), suggestive of higher anxiety and motor deficits. Irradiated mice also had fewer crossings between the light and dark areas and spent less time in the light chamber within the LD transition test (Fig. 1G and H).
The rotarod task was chosen to examine the effects of xRT on balance, coordination, and motor control (46, 47). As expected, sham mice maintained their balance on the accelerating rod and habituated quickly (Fig. 1I). However, 8 Gy xRT mice were unable to maintain balance and grip and fell as soon as the rod accelerated beyond 5 rpm. After two to three unsuccessful trials, animals were tested at a constant speed of 5 rpm and exhibited 90% shorter latency to fall and struggled to maintain balance, grip, and motor coordination (Fig. 1J). No significant improvement in Mot was observed after 60 days (Fig. 1I and J).
To assess the effect of xRT on hippocampus-dependent L/M, we performed the Y-maze and novel object recognition test (NORT). 8 Gy xRT mice exhibited impairment in learning and spatial memory by failing to explore the novel arm of the Y maze (Fig. 1K), although decreased overall exploratory behavior was noted. Irradiated mice also performed poorly and showed little to no ability to discern between familiar or novel objects, as evidenced by the reduced discrimination between these (Fig. 1L). Finally, we evaluated depressive-like behavior using the tail suspension test, which revealed that irradiated animals exhibited longer freeze duration (Fig. 1M), which is a measure of D/D.
Because we used several complementary assessments of multiple behavioral outcomes, we developed a cross-test composite scoring system that would enable more rigorous assessment of outcomes across varying doses, ages at treatment, and recovery times (see Materials and Methods). When applied to this initial dataset, the composite scoring system confirmed that the major neurobehavioral impairments were highly significant and the A/E and Mot impairments were somewhat improved with longer recovery times (Fig. 1N). Notably, the L/M impairments showed the least improvement with recovery time.
Molecular and histologic effects of xRT on the developing brain
We next examined brain tissue from 8 Gy xRT-treated animals via histologic analysis to better understand how ionizing radiation affects developing neural tissue. We observed significant infarcts in sections from 8 Gy xRT animals that were not present in litter-matched sham animals (Fig. 1O). xRT mouse brain tissue contained degenerated neurons as evidenced by chromatolysis and deep staining of neurons in neurogenic regions (Fig. 1O). This was suggestive of neural stem or progenitor cell death in the hippocampus, subgranular regions, and the striatum. Overall, these histologic findings were consistent with xRT causing neural cell loss, which is especially evident in neurogenic niches in the developing brain.
Across these tests, our results demonstrated that whole-brain 8 Gy xRT caused extensive neural damage and degeneration and strongly impaired neurocognitive function. These effects appear shortly after irradiation and are largely irreversible, matching the clinical experiences evident with cranial xRT in young patients with CNS tumor. Importantly, these results contrasted sharply with the previously reported milder effects of this dose in juvenile or adult animals.
Neurotoxic effects of cranial xRT are dose dependent
We next sought to establish the degree to which the dose of xRT administered affected outcomes. We found that animals irradiated at 4 Gy exhibited a significant improvement in overall survival (Fig. 1P) and developmental weight gain (Fig. 1Q) compared with 8 Gy. Even lower doses of 1.8 and 1 Gy did not cause any mortality but did lead to a slight reduction in weight gain. Although there was no decrease in overall survival with 1 Gy treatment, we nonetheless detected NIs in mice irradiated with this low dose at a young age. Specifically, we detected impairments in anxiety, exploration, Mot, learning, and memory as well as D/D (Supplementary Fig. S1A–S1J). Neonates irradiated with even 1 Gy xRT did worse than sham-treated animals in NORT, indicative of impaired L/M (Supplementary Fig. S1G). Finally, we detected extended freeze times in 1 Gy irradiated mice in the tail suspension and FSTs, demonstrating potential increased levels of despair and depressive behaviors (Supplementary Fig. S1H and S1I). Although these effects were milder as compared with 8 Gy treated mice, they were consistently detectable and demonstrated that the developing brain is extremely sensitive to even mild doses of xRT and that the extent of NI is dose dependent.
Neurotoxic effects of cranial xRT are age dependent
Clarifying how xRT affects the brain during different phases of brain development is critical for understanding therapy-induced toxicities broadly and evaluating potential neuroprotective agents. We next investigated the effect of the moderate dose of xRT (4 Gy) when applied during key time points in brain development to identify vulnerable periods and to develop a rigorous mouse model of the age-dependent effects of xRT on neurocognition. The 4 Gy dose was chosen for neurobehavioral studies to maintain a high clinical relevance while also maintaining sufficient animal survival for long-term follow-up. To assess the differences in radiation sensitivity across key ages, we compared the effects of 4 Gy xRT on young (P3-6), adolescent (P10), juvenile (P20), and adult (P45+) mice. While the youngest irradiated mice exhibited nearly 50% mortality and a significant decrease in body weight at 60 days after irradiation, the adolescent, juvenile, and adult irradiated animals showed improved survival along with only mild declines in body weight (8%–12%; Fig. 2A and B). Consistent with these data, we detected a decrease in brain weight in animals treated at P3-P6 but not any other age group (Fig. 2C).
Figure 2.

Effects of brain xRT are age dependent. A, Overall survival of animals treated with 4 Gy CT-guided, whole-brain xRT at indicated ages. B and C, Body (B) and brain (C) weight of mice treated with 4 Gy xRT at indicated ages. D, Anxiety, exploration, and motor activity was assessed in animals treated at indicated ages using open field test 30 days after treatment. E, Rearing behavior assessed at 30 days after treatment. F, Anxiety was assessed by comparing time spent in light chamber in animals treated at indicated ages. G, Motor activity was assessed by using the rotarod test in animals treated at indicated age at indicated number of days after treatment. H, Y maze was used to assess L/M in animals treated at indicated ages by comparing time spent in novel arm. I, NORT was used to assess L/M by testing the ability of animals to discriminate between novel and familiar objects. J and K, Depression and despair-like behavior was assessed using the tail suspension and FSTs by timing the duration of animal immobility (freezing) during a 5-minute test period. L, Neurobehavioral composite scoring indicates the significance of differences in neurobehavioral impairments in animals irradiated at indicated ages. M, Histology of brain sections of indicated regions 60 days after 4 Gy xRT in animals treated at post hoc multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
Effects of 4 Gy xRT on neurobehavior in young, adolescent, juvenile, and adult mice
We next tested the effects of xRT on neurobehavioral outcomes across lifespan. After xRT at different ages, animals recovered for 30 days and then completed our comprehensive battery of neurocognitive tests. This dose of ionizing radiation caused the most deleterious long-term NI in the youngest animals (P3-6) followed by adolescent (P10) and juvenile (P20) animals as evidenced by their reduced exploration and rearing along with increased immobility and anxiety (Fig. 2D–F) in the open field and LD box tests. The effects on adult (P45) animals were milder and did not reach significance despite similar group sizes. Furthermore, we observed clear age-dependent effects of xRT on motor coordination with the youngest animals struggling to maintain their balance (Fig. 2G). Cognition was also affected in a similar manner—the ability of mice to learn to discriminate between novel and familiar areas (Fig. 2H) and objects (Fig. 2I) was more negatively impacted by IR treatment at young compared with older ages. Finally, we evaluated D/D using the tail suspension (Fig. 2J) and FSTs (Fig. 2K), both of which revealed that the youngest irradiated animals exhibited the largest increases in freeze duration. Our composite scoring system clearly demonstrated the age-dependent effects of xRT-induced NI with P20 and older animals not showing significant levels of NI in response to 4 Gy (Fig. 2L). Interestingly, we noted reduced intensity NI in P10 animals as compared with P3-6 in every category except Mot, suggesting that the brain regions responsible for optimal Mot have a prolonged sensitivity to xRT as compared with other regions.
Histologically, we observed that 4 Gy xRT caused loss of neuronal and glial cells in the cortex and hippocampus along with reduced cell density and granule cell dispersion in the neonatal brain (Fig. 2M). The adolescent, juvenile and adult irradiated animals did not show pronounced loss of granular or glial cells and only slight decreases in cell density were observed in adolescent and juvenile but not adult hippocampi and subgranular zones (SGZ).
Collectively, these results demonstrated that xRT of young brain tissue caused generalized anxiety, depressive-like behavior, and impaired learning, memory and Mot. These data are broadly in agreement with clinical experiences, in which younger children receiving higher doses of xRT present with more severe symptoms than adults or children receiving lower doses (48, 49).
Mechanisms driving xRT-induced NI
Because of the well-established link between ionizing radiation and the induction of apoptotic cell death via p53, we next sought to determine whether age-dependent changes in apoptosis sensitivity in neural cells may underlie the differences in xRT-induced NI. We have previously reported that apoptotic priming (proximity to the apoptotic threshold) is a critical determinant of whether a cell will commit to apoptosis when proapoptotic signals are produced by cell damage or stress (18, 50–53). Apoptotic priming is determined by the expression levels and interactions of the BCL2 family proteins that control mitochondrial apoptosis and can be measured using BH3 profiling. In our previous studies, BH3 profiling showed that young brain tissue, which is undergoing extensive remodeling and synaptic pruning postnatally, is highly primed for apoptosis and is consequently highly sensitive to ionizing radiation (18). Importantly, apoptotic priming is reduced during the first weeks of murine life and the tissue becomes apoptosis refractory by adulthood, supporting resistance to apoptotic stimuli including IR. However, although our previous studies showed that apoptosis is dynamically regulated during brain development and maturation (from embryo to adult), they did not directly compare priming among the various neural cell types and it is therefore unclear which cells in the brain are most vulnerable to stress-induced cell death and at which ages. To address this question, we utilized BH3 profiling on freshly collected neural tissue from mice across ages to compare mitochondrial apoptotic priming in relevant cell types and how it may change during the transition into adulthood. BH3 profiling showed that apoptotic priming in immature and migrating cortical neurons (PSA-NCAM+) decreases sharply during early postnatal life (Fig. 3A and B), as evidenced by the decrease in sensitivity to the proapoptotic BIM BH3 peptide at several doses and the combined area under the curve (log AUC). Astrocytes (GFAP+) followed a similar trend in priming but were more primed than neurons at every timepoint examined. Notably, neural progenitor cells (CD133+) were not only more primed at P0 but also remained primed during the first 5 weeks of life, suggesting that this population of cells is most vulnerable to proapoptotic stimuli of those we evaluated and maintains that vulnerability for a longer developmental duration.
Figure 3.

Mechanism of xRT-induced NI. A, BH3 profiling of neural stem cells (CD133+), astrocytes (GFAP+), and neurons (PSA-NCAM+) isolated from mice of indicated ages. B, Comparison of priming in indicated cell types isolated from brain tissue from P5 animal. C, BH3 profiling of astrocytes and neurons cultured from neural stem cells collected at indicated ages. D, Expression of mitochondrial apoptosis-associated genes in indicated cell types from mid embryogenesis to adulthood. E, Gene set enrichment analysis on BAX-expressing hypothalamic neurons. F and G, Enrichment plots for Myc target genes and genes associated with oxidative phosphorylation in BAX-expressing neurons. H, Immunoblotting for BCL2 family proteins in brain tissue isolated from animals of indicated ages. Relative molecular weight markers are shown on left for all immunoblots. I, BH3 profiling of in vitro cultured astrocytes and neurons treated with 8 Gy ionizing radiation for the indicated time period. J, Immunoblotting for BCL2 family proteins in neonatal (P1) or adult (P124) brain tissue after 8 Gy IR treatment in vivo for indicated time period. K, Immunoblotting for markers of apoptosis in brain tissue collected from animals irradiated at indicated age for indicated time period. L, Caspase-3/7 glow assay measuring caspase-3/7 activity in brain tissue collected from animals of indicated genotype 5 hours after 8 Gy xRT treatment. Unless otherwise specified, comparison between groups was conducted by one-way or two-way ANOVA followed by Holm-Sidak post hoc multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
To further understand the baseline level of priming and changes associated with differentiation, neurons and astrocytes were differentiated in vitro from neonatal, infant, and adolescent brain stem cells (Fig. 3C). After 2 days of differentiation, we found that astrocytes isolated from P7 animals were significantly more primed than neurons, mimicking the data seen from freshly isolated cells (Fig. 3A and B). In vitro differentiation of astrocytes and neurons again demonstrated an age-dependent decrease in priming (Fig. 3C).
We reasoned that the decreases in apoptotic priming across the major cell types we examined would be driven by changes in expression of BCL2 family proteins that control the mitochondrial apoptosis pathway. To uncover these potential changes, we used previously published single-cell RNA-seq data to examine expression of apoptosis-associated genes in the developing hypothalamus (36). Expression of apoptosis genes was relatively consistent in NPCs during mid to late embryogenesis with only minor variation in expression of Bax and Bcl2 observed (Fig. 3D). However, hypothalamic neurons exhibited temporal changes in expression of several key apoptosis genes including proapoptotic Bax and Bad, prosurvival Bcl2l1 and the inhibitor of IAP proteins Xiap. Particularly notable was the regulation of the pore-forming Bax, which was highly expressed in neurons in late embryonic development and strongly downregulated by adulthood (P45). Interestingly, almost all apoptosis genes, proapoptotic and prosurvival as well as those associated with initiation as well as execution of apoptosis, ceased to be expressed in adult neurons, suggesting that the apoptosis pathway is largely abandoned in these cells. Astrocytes also downregulated Bax postnatally but retained higher expression of this gene as well as Bcl2 in adulthood—these results were consistent with the somewhat higher priming evident in astrocytes as compared with neurons (Fig. 3A–C). Contrasting with neurons and astrocytes, oligodendrocytes increased expression of proapoptotic genes including Bax, Bad, and Bak1 postnatally with their highest expression being evident at P14 followed by a slight downregulation by P45. Although our BH3 profiling analysis did not measure apoptotic priming in oligodendrocytes, these data suggest that they maintain a higher level of apoptotic competency than other major cell types in the adult brain and consequently maintain higher sensitivity to damage or stress.
The particularly dynamic expression of Bax in neurons suggested that it was being regulated by upstream factors associated with developmental processes. To further explore this, we performed gene set enrichment analysis of genes differentially expressed by Bax-positive neurons and found that Myc target genes were significantly enriched as well as genes associated with oxidative phosphorylation, DNA repair, reactive oxygen species, and mTORC1 signaling (Fig. 3E–G). These gene sets are associated with growth, proliferation, protein translation, and highly active mitochondrial respiration, suggesting that these processes either increase apoptotic stress or that apoptotic sensitivity is specifically maintained in cells that are actively proliferating or capable of doing so, presumably to facilitate deletion in case of dysfunction or superfluity.
We next tested whether these changes would be evident at the protein level by performing immunoblotting for apoptosis-regulating proteins on mouse brain tissue from key developmental time points. We confirmed that BCL2 family proteins are dynamically regulated during postnatal brain development and detected decreases in proapoptotic BIM and the pore-forming BAX (Fig. 3H). We also noted decreases in expression of prosurvival proteins BCL2 and MCL1 while long-term expression of BCL-XL was maintained. The most notable changes in apoptosis-regulating protein expression occurred early in life, prior to P21, and were also accompanied by decreases in expression of apoptosis execution proteins caspase-3 and APAF-1. These changes confirm there is an active rewiring of the apoptosis pathway during postnatal brain maturation and broad suppression of apoptosis in the mammalian brain by adulthood via loss of BAX expression and other apoptosis-associated proteins.
These changes in priming and BCL2 family proteins suggested that cells in the young brain would more readily undergo apoptotic cell death in response to damage or stress. We next sought to confirm that DNA damage induced by ionizing radiation treatment produces proapoptotic signaling. We therefore BH3 profiled cells 8 to 72 hours after a single treatment with 8 Gy IR. Notably, we detected increased priming in both astrocytes and neurons (Fig. 3I), confirming that IR-induced DNA damage produces apoptotic signaling in neural cells. Further measurements over a 72-hour time course showed that apoptotic priming peaks shortly after irradiation (evident within 8 hours) and are subsequently reduced over the course of 72 hours. Interestingly, although priming in astrocytes returned to baseline by 48 hours after irradiation, it remained elevated above baseline levels for the full 72-hour time course in neurons.
To investigate the mechanisms driving differential responses to IR treatment at different developmental ages, we measured changes in apoptosis-related proteins in response to IR treatment. We first confirmed that IR induces apoptotic cell death in the young brain by detecting cleaved caspase-3 at 4 to 8 hours after xRT (Fig. 3J and K) and by measuring caspase-3 and -7 activity directly (Fig. 3L). Importantly, this effect was dependent on p53 as IR failed to induce activation of caspases-3/7 in neonatal brain tissue from Trp53−/− mice (Fig. 3L) with heterozygous mice exhibiting an intermediate level of caspase activation. Consistent with our previous report (18), 8 Gy IR did not induce apoptosis in adult brain tissue as measured by caspase-3/7 activation and apoptosis was reduced in an age-dependent manner (Fig. 3J and K). We previously reported that expression of BAX in the neonatal brain was transcriptionally driven by c-Myc (18), which was consistent with differences we detected in c-Myc expression between the young and adult mice (Fig. 3J). Unexpectedly, we also saw that c-Myc was upregulated shortly after irradiation in the adult brain, which is potentially responsible for the slight increase in BAX expression evident in irradiated adult tissue. We also detected an increase in the cleavage of PARP, a substrate for active caspase-3, in irradiated neonatal but not adult tissue. Interestingly, PARP was expressed predominantly as a cleaved fragment in the adult brain regardless of treatment status. Overall, these changes are consistent with the degree of apoptosis induced by xRT at various ages being linked to the extent of resulting NI.
Effects of BAX loss on xRT-induced molecular, biochemical, and histologic effects in the immature brain
On the basis of our single-cell RNA-seq and immunoblotting results, we hypothesized that BAX loss would protect the neonatal brain from xRT-induced apoptosis. To test this, we irradiated P1 Bax+/+ and Bax−/− animals and examined molecular responses. We found that p21 and PUMA are both upregulated shortly after IR treatment in both genotypes, confirming similar dynamics of p53 activation (Fig. 4A). We also detected high levels of cleaved caspase-3 in Bax+/+ but not Bax−/− animals, confirming prevention of apoptosis. Interestingly, we observed major differences in expression of proapoptotic tBID, which we and others have shown is necessary for maximal induction of apoptosis in response to genotoxic damage (54, 55). We found that full-length BID is rapidly cleaved (forming active, truncated, proapoptotic tBID) in response to 8 Gy IR in neonatal Bax+/+ brain tissue as indicated by the loss of full-length BID (Fig. 4A) and also the detectable accumulation of tBID at 4 hours after IR. BID can be cleaved by several caspases including active caspase-8 downstream of death receptor activation as well as active effector caspase-3 that can be activated by caspase-9 downstream of MOMP or active caspase-8. Cleaved BID participates in a feed-forward loop (56) that helps to ensure that initialization of apoptosis results in full commitment to cell death via this pathway—this is important as several groups have shown that partial cytochrome c release (referred to as “minority MOMP”) does not necessarily commit a cell fully to apoptosis (57–59). Notably, we found that BID cleavage was not detected in Bax−/− brain tissue after irradiation, suggesting that BID cleavage is driven by the mitochondrial amplification loop involving caspase-9 and caspase-3/7. To determine the extent to which BID is required for apoptosis execution in immature brain tissue, we measured caspase activation in irradiated brain tissue from Bid−/− animals and found that loss of BID eliminated radiation-induced apoptosis (Fig. 4B). The involvement of BID in radiation-induced apoptosis was further confirmed by detecting reduced radiation-induced apoptosis in Bid−/− MEFs compared with WT (Fig. 4C and D) and when transiently transfecting cells with siRNA for Bid compared with nontargeting control (Supplementary Fig. S1K).
Figure 4.

BAX ablation protects from xRT-induced apoptosis in neural stem and progenitor cells. A, Immunoblotting for BCL2 family proteins in Bax+/+ or Bax−/− P1 animals at indicated time period after 8 Gy xRT. B, Caspase-3/7 glow assay measuring caspase-3/7 activity in Bid+/+ and Bid−/− animals in cortex and hippocampus regions of brain 5 hours after 8 Gy xRT. C, Representative flow cytometry plots of viability analysis of MEFs of indicated genotype 48 hours after irradiation. D, Summary of viability analysis performed in MEFs of indicated genotype 48 hours after irradiation with indicated doses. E, BIM contributes toward the radiation sensitivity of CGNPs, as Bim−/− mice have decreased numbers of apoptotic cC3+ cells following radiation treatment (2 Gy, 3 hours after treatment) when compared with controls. F, Representative images of whole-brain sections immunostained for indicated markers (DAPI, GFAP, cleaved caspase-3). Scale bars, 200 μm. G, Quantification of cells positive for cleaved caspase-3 and GFAP in indicated brain regions. H, Representative images of whole-brain sections immunostained for indicated markers. Scale bars, 200 μm. I, Quantification of cells positive for cleaved caspase-3 and MAP2 in indicated brain regions. Values presented are means + SD from two to three separate experiments. All groups were compared with two-way ANOVA with Holm-Sidak adjustment. J, BH3 profiling of P2 mouse brain tissue from Bax+/+ and Bax−/− animals with stains for nucleated cells (DAPI+), neural stem cells (CD133+), and astrocytes (GFAP+). Unless otherwise specified, comparison between groups was conducted by one-way or two-way ANOVA followed by Holm-Sidak post hoc multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
Members of the BCL2 family of proteins generally have additive effects on the promotion or suppression of apoptosis. On the basis of this, we hypothesized that loss of proapoptotic BIM (encoded by Bcl2l11) would also contribute to radiation resistance. Indeed, we detected a reduction in radiation sensitivity in cerebellar granule neural precursor cells in Bcl2l11 knockout mice (Fig. 4E), suggesting that reducing apoptotic priming by lowering expression of proapoptotic protein expression or by increasing prosurvival protein expression would yield a similar survival benefit.
Taken together, these data suggest that xRT initially activates p53 in young neural cells, leading to the upregulation of p53 targets including proapoptotic PUMA. PUMA may then either activate BAX via a direct interaction or indirectly by inhibiting prosurvival proteins to free BIM. BAX is only activated in highly primed immature neural cells and causes initial, submaximal release of cytochrome c. Because of the low expression of XIAP in the brain (18) and the highly primed state of these immature cells, caspase-9 and then caspase-3 are activated, albeit at submaximal levels that are insufficient to fully commit cells to apoptosis. However, caspase-3 then cleaves available BID to activate the feed-forward loop to ensure apoptosis completion (see Discussion).
Preventing neurotoxicity by blocking BAX-mediated apoptosis
Apoptosis plays an important role in postnatal brain development and maturation by supporting neuronal pruning (60). Multiple reports have shown that BAX is the dominant pore-forming effector protein involved in this process and that BAX loss can reduce pruning (61) and, importantly, also reduce radiation-induced apoptosis (18). However, no studies have addressed how apoptosis blockade modulates cell fates in response to xRT or affects long-term survival and neurocognitive function after cranial xRT in pediatric mouse models. These are critically important for establishing the potential therapeutic utility of apoptosis suppression for prevention of radiation-induced impairment in pediatric patients with CNS tumor or other high-dose exposure settings. To address this, we compared xRT-induced changes in cell fates and neurocognitive performance in Bax+/+ versus Bax−/− mice.
We first measured the extent to which BAX loss can directly prevent the death of neural cells in response to IR at a young age by immunostaining brain tissue from sham- or xRT-treated neonatal Bax+/+ and Bax−/− mice for cleaved caspase-3 and markers for cells of interest. As expected, we detected cleaved caspase-3, indicative of caspase activation, in GFAP+ astrocytes across multiple regions of the xRT-treated brain including highly neurogenic niches such as the DG and Cornu Ammonis 3 (CA3; Fig. 4F and G). Importantly, we found that BAX loss prevents apoptosis in astrocytes across those same regions, reducing cleaved caspase-3 signals to background levels across nearly all regions (Fig. 4G). We repeated this analysis with the MAP2 marker to measure levels of apoptosis induced in neurons. We again noted extensive cleaved caspase-3 positivity in irradiated Bax+/+ brain tissue as compared with sham (Fig. 4H and I), which was almost completely abrogated in Bax−/− animals, including in neurogenic niches. Finally, BH3 profiling of neural stem cells (CD133+) and astrocytes (GFAP+) from P2 animals showed that loss of Bax strongly reduces, but does not completely eliminate, cytochrome c release in response to proapoptotic BH3 peptides (Fig. 4J). These results indicate that although BAX is the dominant pore-forming protein in neural cells within the early postnatal brain, BAK also does play a role in initiating apoptosis. This is consistent with our earlier findings showing that BAX loss was more protective against ionizing radiation-induced apoptosis than loss of BAK but that combined loss of both BAX and BAK was necessary to fully block neural cell death (18).
We next sought to test how BAX loss impacted long-term survival, brain integrity and neurocognition after xRT. We first found that while irradiated Bax+/+ animals had significantly worse survival compared with sham controls, Bax−/− mice did not exhibit a significant decrease in survival (Fig. 5A and B). Consistent with our immunofluorescence findings, histologic analysis of irradiated Bax+/+ and Bax−/− brain tissue revealed differences in the severity of damage induced by xRT compared with sham controls. Specifically, irradiated Bax+/+ mice exhibited signs of neuronal cell death, compromised hippocampal integrity and loss of the granule cell layer (Fig. 5C and D). This was particularly evident in the DG and SGZs—the regions where adult neurogenesis is typically most active. In contrast, Bax−/− animals exhibited reduced neural cell loss in these regions as a result of apoptosis prevention.
Figure 5.

Knockout of BAX reduces xRT-induced mortality and neurodegeneration. A, Overall survival of mice of indicated genotypes irradiated at P3-P6. B, Weight measurements of mice of indicated genotypes irradiated at P3-P6. C, Histology of indicated brain regions from Bax+/+ versus Bax−/− animals 180 days after irradiation or sham treatment. Arrows, areas of reduced or retained cellularity. Scale bars, 100 μm. D, DG areas of mouse brains 8 to 9 months after indicated treatment. E and F, Representative images of brain sections from hippocampus immunostained with DAPI and antibodies for cleaved caspase-3 as well as SOX2 (E) and NG2 (F). Scale bars, 25 μm. G, Quantification of cells positive for cleaved caspase-3 and SOX2 or NG2 in indicated brain regions in Bax+/+ versus Bax−/− animals after irradiation or sham treatment. Unless otherwise specified, comparison between groups was conducted by one-way or two-way ANOVA followed by Holm-Sidak post hoc multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
To elucidate the types of cells preferentially lost in response to xRT, we stained irradiated brain sections from the hippocampus (Fig. 5E and F), amygdala (Supplementary Fig. S2A and S2B) and perirhinal cortex (Supplementary Fig. S2C and S2D) for cleaved caspase-3 as well as SOX2 (expressed preferentially in neural stem cells; ref. 62) or NG2 (expressed preferentially in glial progenitor cells; ref. 63) from P4 animals. We focused on these brain regions due to their links with the neurobehavioral outcomes we were assessing including the hippocampus, which has a major role in L/M (41, 64), the amygdala, which controls emotion and fear (65, 66), and the perirhinal cortex, which supports object recognition (67). We found that both SOX2- and NG2-expressing cells underwent apoptosis in response to xRT within the hippocampus, with SOX2+ cells dying at higher rates (∼50% positive for cleaved caspase-3) than those that were NG2+ (∼17%; Fig. 5G). Interestingly, we did not detect significant levels of apoptosis within either of these cell populations within the amygdala. In the perirhinal cortex, we again detected cells undergoing apoptosis in response to xRT, with SOX2+ cells exhibiting slightly higher rates of apoptosis compared with NG2+ cells. All neural cell types were protected from xRT-induced apoptosis in Bax−/− animals, consistent with the dominant role this protein has in maintaining apoptotic priming (Fig. 4J) and mediating neural cell apoptosis (18).
Behaviorally, irradiated Bax−/− mice exhibited improved ambulation and decreased immobility compared with irradiated Bax+/+ litter mates (Fig. 6A). As seen previously, the Bax+/+ animals did experience some improvement in exploration after 3 months of recovery, with reduced pausing behavior (Fig. 6A–C). In contrast, irradiated Bax−/− mice spent similar amounts of time exploring inner and outer zones and reduced immobile time at even 30 days after xRT and continued to exhibit improvement throughout the experimental period, suggesting that prevention of apoptosis had short-term as well as long-term beneficial effects. LD box testing showed similar results: irradiated Bax−/− animals exhibited reduced impairment at 30 days after treatment and recovered to sham-treated levels by 3 months while irradiated Bax+/+ animals did not (Fig. 6D). Rotarod testing of animal ambulation demonstrated that irradiated Bax+/+ mice had an impaired gait and grip that improved slightly by 6 months while irradiated Bax−/− mice showed no xRT-induced impairment across all time periods tested (Fig. 6E), although rotarod performance was noted to be reduced in sham-treated Bax−/− mice compared with Bax+/+. Evaluation of L/M via NORT also demonstrated the protective effects of BAX loss: irradiated Bax−/− mice exhibited a shorter period of L/M impairment that was less also less intense than litter-matched Bax+/+ animals (Fig. 6F). Additional long-term learning, memory, and synaptic plasticity tests demonstrated that irradiated Bax−/− mice had improved object novelty recognition (Fig. 6G) and improved task learning (Fig. 6H) than irradiated Bax+/+ litter mates. Tail suspension and FSTs again showed that xRT increases D/D (Fig. 6I and J) that was detected even after 6 months of recovery in Bax+/+ animals. Interestingly, Bax−/− animals also exhibited these depression-like behaviors after irradiation but demonstrated improved recovery compared with Bax+/+ mice, especially in the tail suspension test. Parallel studies performed with a reduced dose of irradiation (1.8 Gy xRT) demonstrated similar neuroprotection in Bax knockout animals (Supplementary Fig. S3A–S3H). When evaluating responses between sexes, we detected comparable levels of xRT-induced NI in male and female mice, which was similarly ameliorated by Bax loss (Supplementary Fig. S4A–S4C). Finally, using a fluorescence-based multiplex FM-HCR assay that measures the ability of cells to repair damaged plasmid reporters (34), we confirmed that BAX loss does not affect DNA repair capacity (Supplementary Fig. S5A–S5C), suggesting that blocking apoptosis by targeting this protein would not preclude DNA repair after xRT.
Figure 6.

Prevention of radiation-induced NI by targeting apoptosis. A–C, A/E was assessed using open field test of Bax+/+ versus Bax−/− mice irradiated at P3-P6 and tested at 30 (A), 90 (B), or 180 (C) days. D, LD chamber test performed at indicated number of days after xRT. E, Mot was assessed using the rotarod test performed at indicated number of days after xRT. F, L/M was assessed using NORT performed at indicated number of days after xRT. G, L/M was assessed using the object-location memory test performed at 180 days after xRT. H, L/M was assessed using the Barnes maze test performed at 180 days after xRT. I and J, Despair and depression-like behavior was assessed using tail suspension (I) and FSTs (J) performed to assess despair and depression in animals at indicated number of days after xRT. K–M, Neurobehavioral composite scoring indicates the significance of differences in neurobehavioral impairments in animals irradiated at P3-P6 and tested at indicated period after xRT. Unless otherwise specified, comparison between groups was conducted by one-way or two-way ANOVA followed by Holm-Sidak post hoc multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
Our composite scoring system demonstrated that Bax−/− mice were strongly protected from xRT-induced NI with most categories of neural function not significantly impaired by 4 Gy except D/D (Fig. 6K and L). Interestingly, L/M scoring showed that although L/M were both impacted early after xRT in both genotypes, the Bax−/− animals recovered more quickly and had significantly improved L/M by 90 days (Fig. 6M). Taken together, our results indicate that xRT produced neurobehavioral changes in Bax−/− mice that were reduced in intensity and resolved more quickly compared with irradiated Bax+/+ controls.
Discussion
Currently, there are no therapies that effectively treat long-term xRT-induced NI in childhood brain tumor survivors. This is largely due to our poor understanding of the major drivers of this neurotoxicity (68). In particular, the lack of clarity on the molecular mechanisms and key cell types involved has been a major impediment to development of optimized treatment strategies, as has the lack of a rigorous and reproducible mouse model of age-dependent effects. Our findings elucidate the molecular regulators of IR response in the developing brain and uncover the prominent role of apoptosis induction in driving acute cell loss as well as consequent long-term neurobehavioral deficits. Furthermore, our novel mouse modeling, including comprehensive assessment of the dose and age dependence of xRT-induced effects, provides a platform for future studies evaluating the neuroprotective effects of novel agents and treatment strategies as well more rigorous examination of NI induced by other injuries and agents across the age spectrum. It is important to note that medulloblastoma is typically treated with daily fractions of 1.8 Gy (11), which is lower than some of the higher doses (4 and 8 Gy) used here. However, as mentioned above, the total craniospinal irradiation dose administered to these patients is typically 23.4 Gy with a 54–55.8 Gy boost to the posterior fossa, which are far higher than those used herein. Also, although it would be ideal to replicate the 1.8 Gy fractionation schedule in our mouse model, administering repeated doses of xRT to anesthetized neonatal animals was not possible.
While the neurotoxic effects of ionizing radiation depend on the total dose, age at time of treatment, fractionation schedule, and volume treated, there is evidence to suggest that differential radiation sensitivity exists within various CNS subcompartments and cell types at critical developmental time points. Previous work has shown that the neonatal brain is exquisitely sensitive to apoptotic signaling and that this sensitivity decreases throughout childhood, eventually reaching an apoptosis refractory state by adulthood (18). Herein, we specifically identify CD133+ neural progenitor cells within the DG and CA3 regions as being highly primed for apoptosis in young brain tissue and, consequently, hypersensitive to clinically relevant doses of xRT. The loss of these cells causes deficits in learning, memory, anxiety, and exploration that we identified across our battery of tests and mirror the radiation-induced cognitive impairment observed clinically in timing and severity. The neurobehavioral deficits we observed after xRT demonstrate substantial disruption of L/M processes stemming from increased apoptosis in neurogenic niches—this impairs the hippocampal neurogenesis and synaptic plasticity necessary for higher cognitive function. These impairments were not present in adult animals, which is consistent with the apoptosis refractory nature of the adult brain and the near complete absence of radiation-induced apoptosis in our studies.
Although we anticipated some protective effects of apoptosis prevention via BAX loss for long-term NI after xRT, the breadth and scale of protection was beyond our expectations. The dramatic mitigating effects and recovery observed in Bax−/− animals are likely facilitated by the survival of neural progenitor pools, which likely maintain higher levels of hippocampal neurogenesis than WT animals and the positive influence of immature and hyperexcitable granule cell neurons upon the cortex-hippocampal circuitry. The persistence of these mitigating effects in Bax−/− animals even after 6–9 months after xRT demonstrates that apoptosis blockade is likely to be effective at preventing NI in the short and long term. Note that although the apoptosis inhibition afforded by Bax deletion is strong, it is not complete given that neural cells can also express the apoptosis effector BAK, albeit typically at much lower levels. We expect that complete inhibition of apoptosis via elimination of both effectors would produce even higher levels of neuroprotection and further improved outcomes. Clearly, the propensity to die via apoptosis can be a powerful determinant of cellular fate after damage or stress induction and protecting cells from apoptosis can be highly protective against cancer therapy–induced toxicity in pediatric models, as has been previously reported for doxorubicin-induced cardiotoxicity (18). Intriguingly, genome-wide association studies have uncovered certain gene polymorphisms associated with improved neurocognitive outcomes for pediatric patients treated with xRT (69, 70)—these polymorphisms may potentially modulate the apoptotic susceptibility of neural cells to exert their protective effects. Finally, we believe our models and findings are applicable to other neurotoxic agents that are particularly damaging to children including anesthesia (71) and metals (72).
On the basis of our results and current model for xRT-mediated apoptosis in the immature brain (Supplementary Fig. S6A and S6B), the transient pharmacologic inhibition or reduction of PUMA, tBID, BIM, or BAX in neural progenitor cells in patients undergoing brain xRT would ameliorate NI and improve outcomes. However, the inhibition of these proapoptotic proteins in patients with CNS or other tumors would be expected to also impede tumor responses to therapy due to the vital role of apoptosis in tumor regression after xRT (73). Thus, treatment strategies that specifically block mitochondrial apoptosis in normal neural cells but not tumor cells are necessary. Our results showing clear age- and differentiation-dependent decreases in apoptotic priming suggest that altering differentiation state via pharmacologic or other means would be accompanied by decreased BAX expression and reduced xRT-induced apoptosis in normal cells to improve cognitive outcomes. However, the 1.8 Gy daily fractions delivered over a period of several weeks complicate this strategy because a neuroprotective agent would need to be active during this entire protracted timespan. On the other hand, because xRT for pediatric brain tumors is administered in these daily fractions, comparing temporal apoptotic signaling dynamics in irradiated cells may uncover differences that can be leveraged to optimize patient outcomes. Indeed, the differences we detected in apoptotic priming after xRT in astrocytes and neurons indicate that the apoptotic sensitization induced by ionizing radiation remains present for varying durations in different CNS cell types—this finding may aid the development of treatment regimens that can take advantage of temporal differences in apoptotic signaling induced by xRT in healthy neural cells versus CNS tumors.
Supplementary Material
Supplementary Figure S6
{kind=link}
Supplementary Figure S5
{kind=link}
Supplementary Figure S4
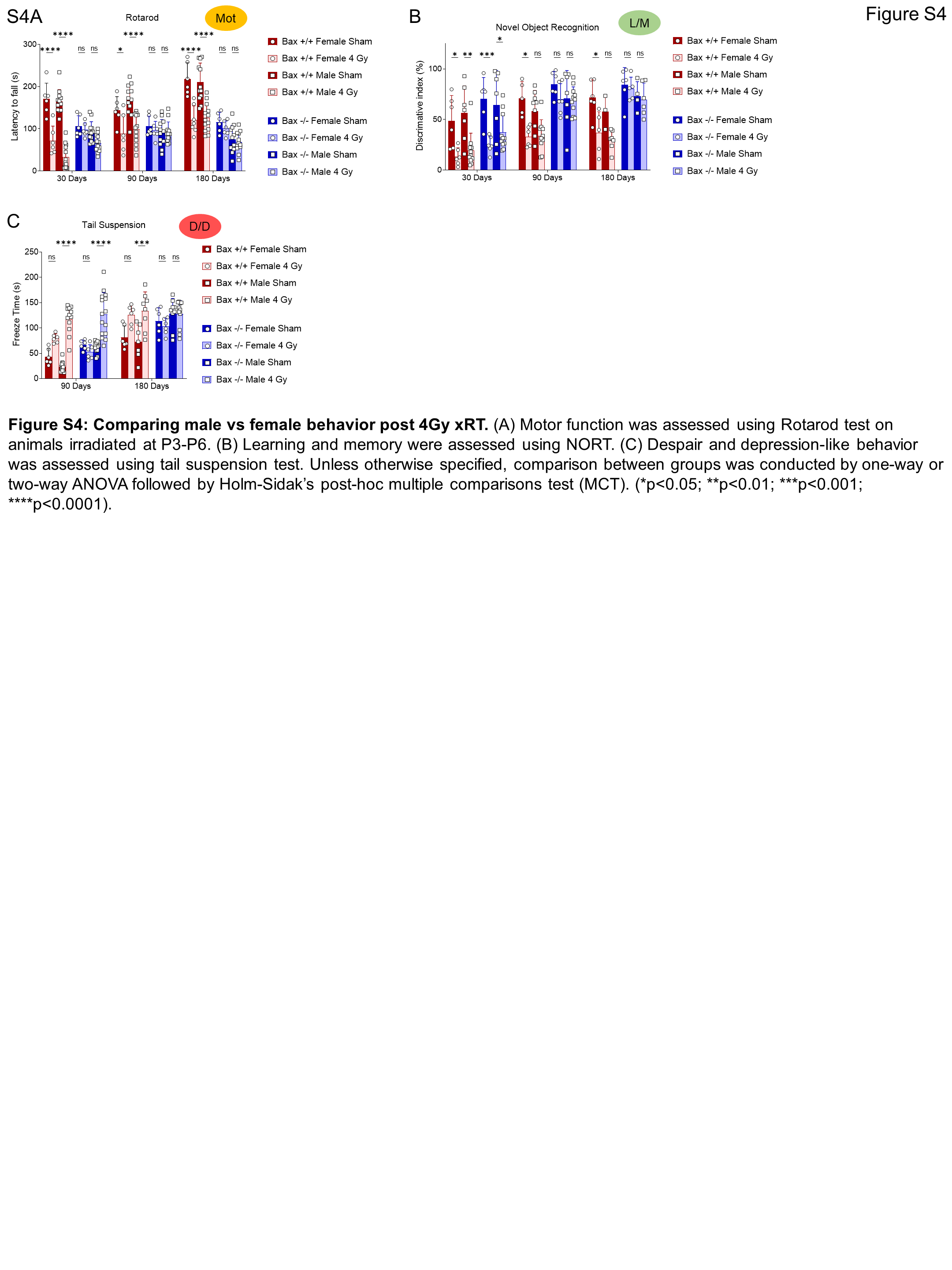{kind=link}
Supplementary Figure S3
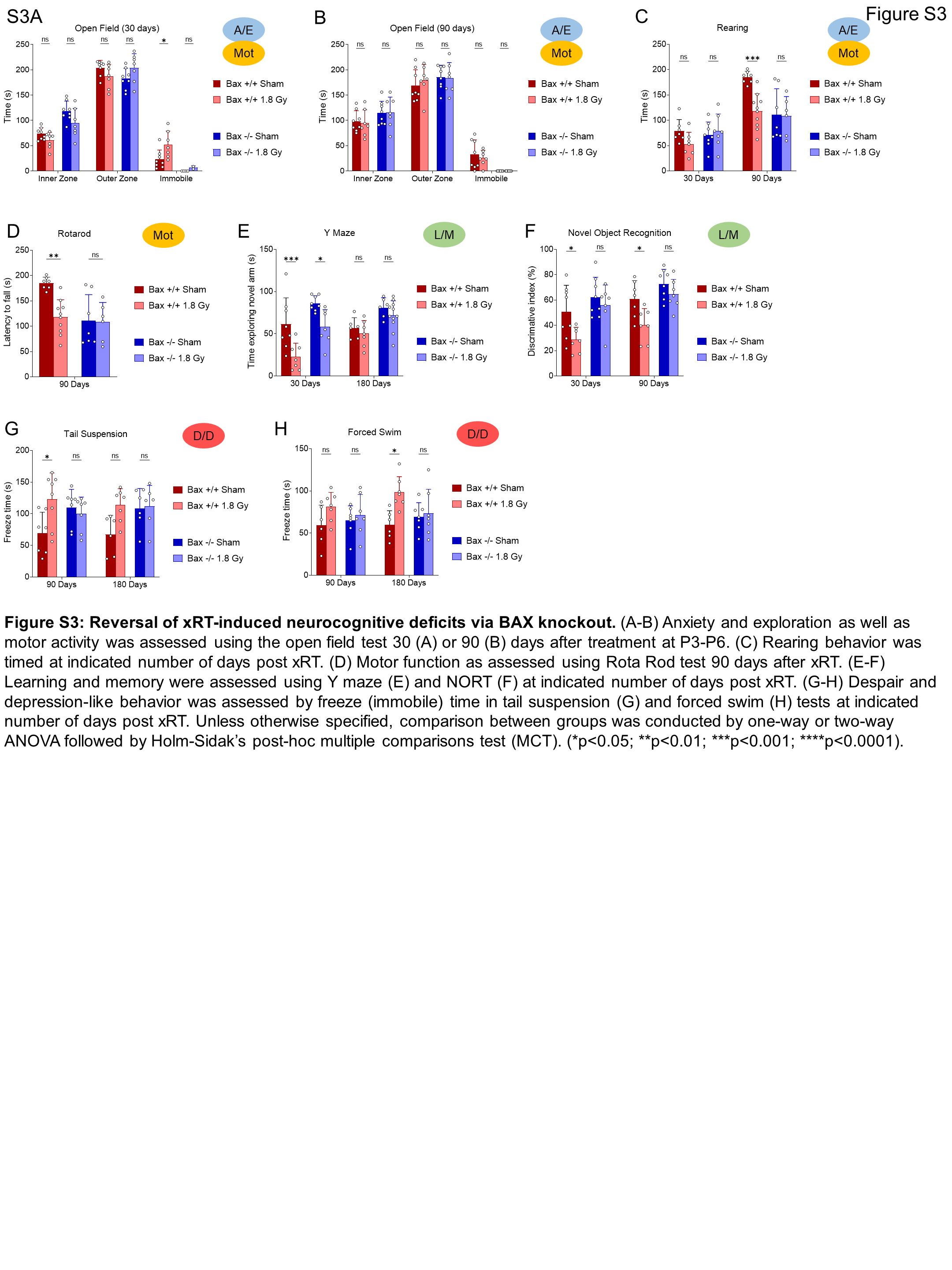{kind=link}
Supplementary Figure S2
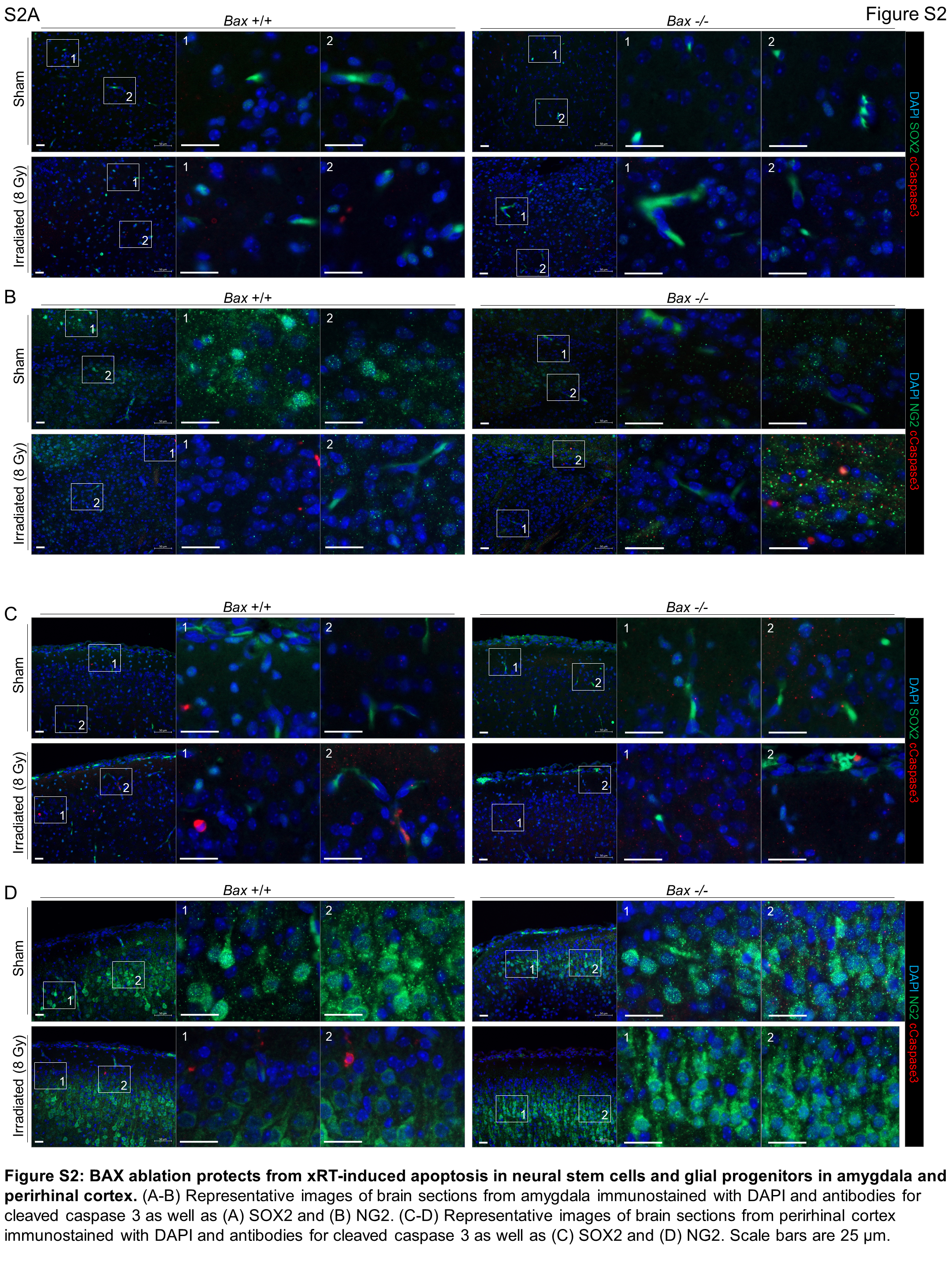{kind=link}
Supplementary Figure S1
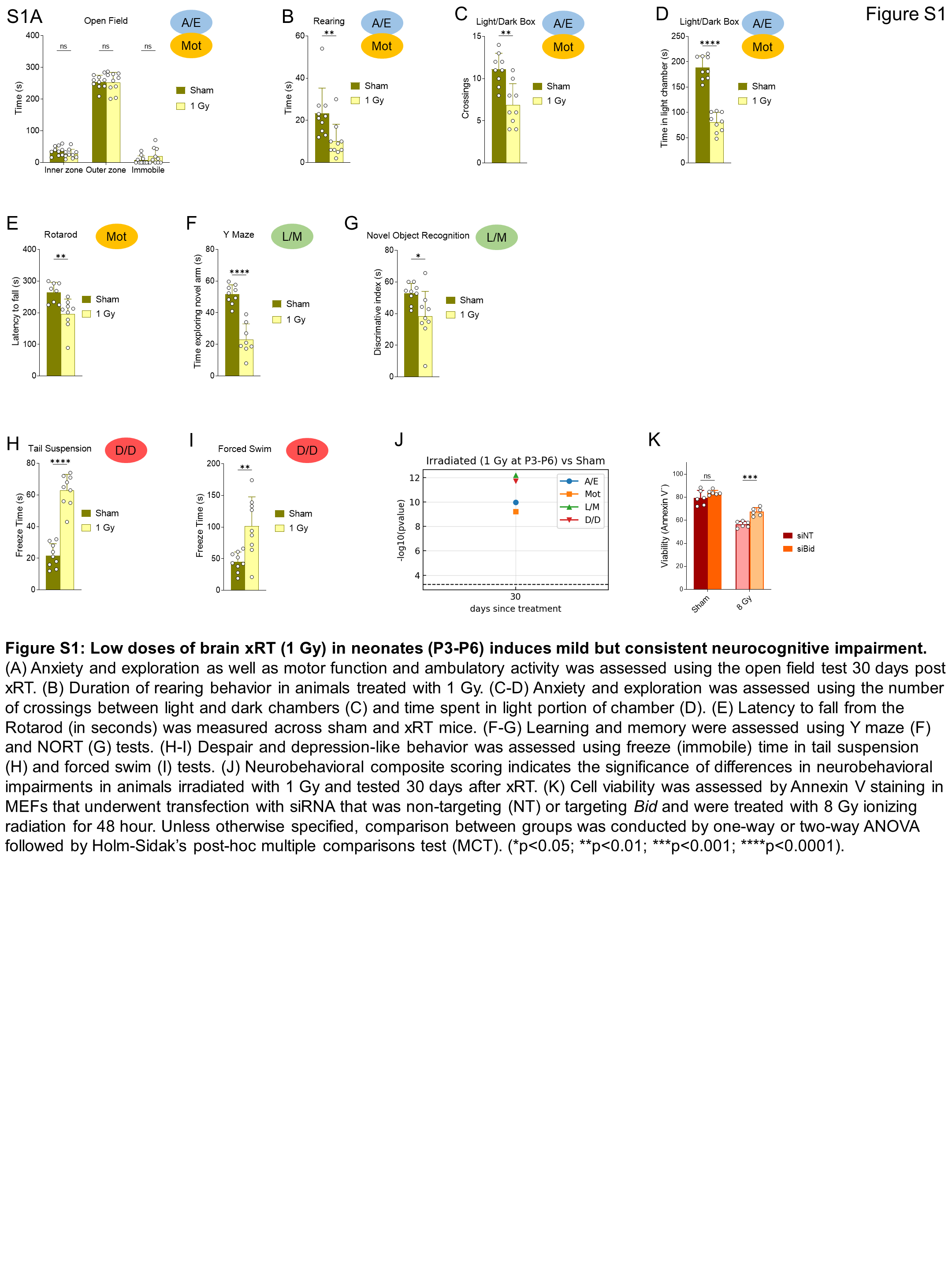{kind=link}
Acknowledgments
The authors thank the members of our labs for their thoughtful suggestions on this work. This work was supported by funding from: Alex's Lemonade Stand Foundation for Childhood Cancers (K.A. Sarosiek), Andrew McDonough B+ Foundation (K.A. Sarosiek), St. Baldrick's Foundation (K.A. Sarosiek), Making Headway Foundation (K.A. Sarosiek), Charles H. Hood Foundation Child Health Research Award (K.A. Sarosiek), NIH grant R00CA188679 (K.A. Sarosiek), NIH grant R37CA248565 (K.A. Sarosiek), NIH grant R01DK125263 (K.A. Sarosiek), NIH grant F31CA246811 (S. Yu), NIH grant F32AG077861 (Z. Inde), Blavatnik Institute at Harvard (K.A. Sarosiek), HSPH Dean's Fund for Scientific Advancement (K.A. Sarosiek), HSPH National Institute for Environmental Health Sciences (NIEHS) Center Grant, and NIH grant P30ES000002 (K.A. Sarosiek, J. Spetz).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
M. Osman reports grants from NIH during the conduct of the study. N. Almanzar reports grants from NIH during the conduct of the study. Z. Nagel reports grants from NIH during the conduct of the study and grants from NIH and American Cancer Society outside the submitted work. M.C. Hocking reports grants from NIH and Department of Defense outside the submitted work. T.I. Yock reports grants from Rising Tide Foundation, NCI Contracts, and nonfinancial support from MIM Software outside the submitted work. K.A. Sarosiek reports grants from NIH, NCI, National Institute of Diabetes and Digestive and Kidney Diseases, Alex's Lemonade Stand Foundation for Childhood Cancers, Andrew McDonough B+ Foundation, St. Baldrick's Foundation, Making Headway Foundation, Blavatnik Institute at Harvard, and Charles H. Hood Foundation Child Health Research Award during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
R. Singh: Investigation, visualization, methodology, writing–original draft, writing–review and editing. S. Yu: Investigation, visualization, methodology. M. Osman: Investigation, methodology. Z. Inde: Investigation, visualization, methodology. C. Fraser: Investigation, visualization, methodology. A.H. Cleveland: Investigation. N. Almanzar: Investigation, visualization, methodology. C.B. Lim: Investigation, visualization, methodology. G.N. Joshi: Investigation, visualization, methodology. J. Spetz: Investigation, visualization, methodology. X. Qin: Investigation, methodology. S.M. Toprani: Investigation, visualization, methodology. Z. Nagel: Methodology. M.C. Hocking: Conceptualization. R.A. Cormack: Methodology. T.I. Yock: Conceptualization. J.W. Miller: Formal analysis, methodology. Z.-M. Yuan: Methodology. T. Gershon: Conceptualization, investigation, methodology. K.A. Sarosiek: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Greenberger BA, Pulsifer MB, Ebb DH, Macdonald SM, Jones RM, Butler WE, et al. Clinical outcomes and late endocrine, neurocognitive, and visual profiles of proton radiation for pediatric low-grade gliomas. Int J Radiat Oncol Biol Phys 2014;89:1060–8. [DOI] [PubMed] [Google Scholar]
- 2. Jain N, Krull KR, Brouwers P, Chintagumpala MM, Woo SY. Neuropsychological outcome following intensity-modulated radiation therapy for pediatric medulloblastoma. Pediatr Blood Cancer 2008;51:275–9. [DOI] [PubMed] [Google Scholar]
- 3. Rieken S, Mohr A, Habermehl D, Welzel T, Lindel K, Witt O, et al. Outcome and prognostic factors of radiation therapy for medulloblastoma. Int J Radiat Oncol Biol Phys 2011;81:e7–13. [DOI] [PubMed] [Google Scholar]
- 4. Dropcho EJ. Neurotoxicity of radiation therapy. Neurol Clin 2010;28:217–34. [DOI] [PubMed] [Google Scholar]
- 5. De Braganca KC, Packer RJ. Treatment options for medulloblastoma and CNS primitive neuroectodermal tumor (PNET). Curr Treat Options Neurol 2013;15:593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Merchant TE, Pollack IF, Loeffler JS. Brain tumors across the age spectrum: biology, therapy, and late effects. Semin Radiat Oncol 2010;20:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stavinoha PL, Askins MA, Powell SK, Smiley NP, Robert RS. Neurocognitive and psychosocial outcomes in pediatric brain tumor survivors. Bioengineering 2018;5:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Merchant TE, Conklin HM, Wu S, Lustig RH, Xiong X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol 2009;27:3691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Merchant TE, Bendel AE, Sabin ND, Burger PC, Shaw DW, Chang E, et al. Conformal radiation therapy for pediatric ependymoma, chemotherapy for incompletely resected ependymoma, and observation for completely resected, supratentorial ependymoma. J Clin Oncol 2019;37:974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Indelicato DJ, Ioakeim-Ioannidou M, Bradley JA, Mailhot-Vega RB, Morris CG, Tarbell NJ, et al. Proton therapy for pediatric ependymoma: mature results from a bicentric study. Int J Radiat Oncol Biol Phys 2021;110:815–20. [DOI] [PubMed] [Google Scholar]
- 11. Michalski JM, Janss AJ, Vezina LG, Smith KS, Billups CA, Burger PC, et al. Children's oncology group phase III trial of reduced-dose and reduced-volume radiotherapy with chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 2021;39:2685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alvarez JA, Scully RE, Miller TL, Armstrong FD, Constine LS, Friedman DL, et al. Long-term effects of treatments for childhood cancers. Curr Opin Pediatr 2007;19:23–31. [DOI] [PubMed] [Google Scholar]
- 13. Dietrich J, Monje M, Wefel J, Meyers C. Clinical patterns and biological correlates of cognitive dysfunction associated with cancer therapy. Oncologist 2008;13:1285–95. [DOI] [PubMed] [Google Scholar]
- 14. Greene-Schloesser D, Moore E, Robbins ME. Molecular pathways: radiation-induced cognitive impairment. Clin Cancer Res 2013;19:2294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rola R, Raber J, Rizk A, Otsuka S, Vandenberg SR, Morhardt DR, et al. Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Exp Neurol 2004;188:316–30. [DOI] [PubMed] [Google Scholar]
- 16. Schindler MK, Forbes ME, Robbins ME, Riddle DR. Aging-dependent changes in the radiation response of the adult rat brain. Int J Radiat Oncol Biol Phys 2008;70:826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morrison RS, Kinoshita Y, Johnson MD, Guo W, Garden GA. P53-dependent cell death signaling in neurons. Neurochem Res 2003;28:15–27. [DOI] [PubMed] [Google Scholar]
- 18. Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, et al. Developmental regulation of mitochondrial apoptosis by c-Myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell 2017;31:142–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Madsen TM, Kristjansen PEG, Bolwig TG, Wörtwein G. Arrested neuronal proliferation and impaired hippocampal function following fractionated brain irradiation in the adult rat. Neuroscience 2003;119:635–42. [DOI] [PubMed] [Google Scholar]
- 20. Monje ML, Vogel H, Masek M, Ligon KL, Fisher PG, Palmer TD. Impaired human hippocampal neurogenesis after treatment for central nervous system malignancies. Ann Neurol 2007;62:515–20. [DOI] [PubMed] [Google Scholar]
- 21. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol 2019;20:175–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010;11:621–32. [DOI] [PubMed] [Google Scholar]
- 23. Krajewska M, Mai JK, Zapata JM, Ashwell KWS, Schendel SL, Reed JC, et al. Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ 2002;9:145–57. [DOI] [PubMed] [Google Scholar]
- 24. Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 1995;270:96–9. [DOI] [PubMed] [Google Scholar]
- 25. Young C, Klocke BJ, Tenkova T, Choi J, Labruyere J, Qin YQ, et al. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ 2003;10:1148–55. [DOI] [PubMed] [Google Scholar]
- 26. Shi L, Molina DP, Robbins ME, Wheeler KT, Brunso-Bechtold JK. Hippocampal neuron number is unchanged 1 year after fractionated whole-brain irradiation at middle age. Int J Radiat Oncol Biol Phys 2008;71:526–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tehranian R, Rose ME, Vagni V, Pickrell AM, Griffith RP, Liu H, et al. Disruption of Bax protein prevents neuronal cell death but produces cognitive impairment in mice following traumatic brain injury. J Neurotrauma 2008;25:755–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hetz C, Vitte P-A, Bombrun A, Rostovtseva TK, Montessuit S, Hiver A, et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem 2005;280:42960–70. [DOI] [PubMed] [Google Scholar]
- 29. Ghita M, McMahon SJ, Thompson HF, McGarry CK, King R, Osman SO, et al. Small field dosimetry for the small animal radiotherapy research platform (SARRP). Radiat Oncol 2017;12:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma C-M. AAPM TG-61 report on kilovoltage X-ray dosimetry: formalisms and applications. Proceedings of the 22nd Annual International Conference of the IEEE Engineering in Medicine and Biology Society (Cat No. 00CH37143). Chicago, IL2000. p. 2308–12 vol.3. [Google Scholar]
- 31. McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, et al. CellProfiler 3.0: next-generation image processing for biology. PLoS Biol 2018;16:e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fraser C, Ryan J, Sarosiek K. BH3 profiling: a functional assay to measure apoptotic priming and dependencies. Methods Mol Biol 2019;1877:61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagel ZD, Margulies CM, Chaim IA, McRee SK, Mazzucato P, Ahmad A, et al. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc Natl Acad Sci U S A 2014;111:E1823–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pitts MW. Barnes maze procedure for spatial learning and memory in mice. Bio Protoc 2018;8:e2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim DW, Washington PW, Wang ZQ, Lin SH, Sun C, Ismail BT, et al. The cellular and molecular landscape of hypothalamic patterning and differentiation from embryonic to late postnatal development. Nat Commun 2020;11:4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinfrmatics 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shi L, Adams MM, Long A, Carter CC, Bennett C, Sonntag WE, et al. Spatial learning and memory deficits after whole-brain irradiation are associated with changes in NMDA receptor subunits in the hippocampus. Radiat Res 2006;166:892–9. [DOI] [PubMed] [Google Scholar]
- 42. Raghubar KP, Mahone EM, Yeates KO, Cecil KM, Makola M, Ris MD. Working memory and attention in pediatric brain tumor patients treated with and without radiation therapy. Child Neuropsychology 2017;23:642–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bates GE, Mostel JL, Hesdorffer M. Cancer-related anxiety. JAMA Oncol 2017;3:1007. [DOI] [PubMed] [Google Scholar]
- 44. Kulesskaya N, Voikar V. Assessment of mouse anxiety-like behavior in the light-dark box and open-field arena: role of equipment and procedure. Physiol Behav 2014;133:30–8. [DOI] [PubMed] [Google Scholar]
- 45. Serchov T, van Calker D, Biber K. Light/dark transition test to assess anxiety-like behavior in mice. Bio Protoc 2016;6:e1957. [Google Scholar]
- 46. Carter RJ, Morton J, Dunnett SB. Motor coordination and balance in rodents. Curr Protoc Neurosci 2001Aug;Chapter 8:Unit 8.12. doi: 10.1002/0471142301.ns0812s15. [DOI] [PubMed] [Google Scholar]
- 47. Crawley JN. Behavioral phenotyping strategies for mutant mice. Neuron 2008;57:809–18. [DOI] [PubMed] [Google Scholar]
- 48. Krasin MJ, Constine LS, Friedman DL, Marks LB. Radiation-related treatment effects across the age spectrum: differences and similarities or what the old and young can learn from each other. Semin Radiat Oncol 2010;20:21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Packer RJ, Zhou T, Holmes E, Vezina G, Gajjar A. Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of children's oncology group trial A9961. Neuro Oncol 2013;15:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011;334:1129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Inde Z, Croker BA, Yapp C, Joshi GN, Spetz J, Fraser C, et al. Age-dependent regulation of SARS-CoV-2 cell entry genes and cell death programs correlates with COVID-19 severity. Sci Adv 2021;7:eabf8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Spetz JKE, Florido MHC, Fraser CS, Qin X, Choiniere J, Yu SJ, et al. Heightened apoptotic priming of vascular cells across tissues and life span predisposes them to cancer therapy-induced toxicities. Sci Adv 2022;8:eabn6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fraser CS, Spetz JKE, Qin X, Presser A, Choiniere J, Li C, et al. Exploiting endogenous and therapy-induced apoptotic vulnerabilities in immunoglobulin light chain amyloidosis with BH3 mimetics. Nat Commun 2022;13:5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maas C, de Vries E, Tait SWG, Borst J. Bid can mediate a pro-apoptotic response to etoposide and ionizing radiation without cleavage in its unstructured loop and in the absence of p53. Oncogene 2011;30:3636–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, et al. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell 2013;51:751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shelton SN, Shawgo ME, Robertson JD. Cleavage of Bid by executioner caspases mediates feed forward amplification of mitochondrial outer membrane permeabilization during genotoxic stress-induced apoptosis in Jurkat cells. J Biol Chem 2009;284:11247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell 2015;57:860–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ichim G, Tait SWG. A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer 2016;16:539–48. [DOI] [PubMed] [Google Scholar]
- 59. Liu X, He Y, Li F, Huang Q, Kato TAA, Hall RPP, et al. Caspase-3 promotes genetic instability and carcinogenesis. Mol Cell 2015;58:284–96.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kole AJ, Annis RP, Deshmukh M. Mature neurons: equipped for survival. Cell Death Dis 2013;4:e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cusack CL, Swahari V, Hampton Henley W, Michael Ramsey J, Deshmukh M. Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat Commun 2013;4:1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang S, Cui W. Sox2, a key factor in the regulation of pluripotency and neural differentiation. World J Stem Cells 2014;6:305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci 2003;24:476–88. [DOI] [PubMed] [Google Scholar]
- 64. Opitz B. Memory function and the hippocampus. Front Neurol Neurosci 2014;34:51–9. [DOI] [PubMed] [Google Scholar]
- 65. Riccio A, Li Y, Moon J, Kim K-S, Smith KS, Rudolph U, et al. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell 2009;137:761–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fadok JP, Markovic M, Tovote P, Lüthi A. New perspectives on central amygdala function. Curr Opin Neurobiol 2018;49:141–7. [DOI] [PubMed] [Google Scholar]
- 67. Murray EA, Richmond BJ. Role of perirhinal cortex in object perception, memory, and associations. Curr Opin Neurobiol 2001;11:188–93. [DOI] [PubMed] [Google Scholar]
- 68. Hocking MC, Walsh KS, Hardy KK, Conklin HM. Addressing neurocognitive late effects in pediatric cancer survivors: current approaches and future opportunities. J Clin Oncol 2021;39:1824–32. [DOI] [PubMed] [Google Scholar]
- 69. Howarth RA, Adamson AM, Ashford JM, Merchant TE, Ogg RJ, Schulenberg SE, et al. Investigating the relationship between COMT polymorphisms and working memory performance among childhood brain tumor survivors. Pediatr Blood Cancer 2014;61:40–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wefel JS, Noll KR, Scheurer ME. Neurocognitive functioning and genetic variation in patients with primary brain tumours. Lancet Oncol 2016;17:e97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Andropoulos DB. Effect of anesthesia on the developing brain: infant and fetus. Fetal Diagn Ther 2018;43:1–11. [DOI] [PubMed] [Google Scholar]
- 72. Grandjean P, Landrigan P. Developmental neurotoxicity of industrial chemicals. Lancet 2006;368:2167–78. [DOI] [PubMed] [Google Scholar]
- 73. Crowther AJ, Ocasio JK, Fang F, Meidinger J, Wu J, Deal AM, et al. Radiation sensitivity in a preclinical mouse model of medulloblastoma relies on the function of the intrinsic apoptotic pathway. Cancer Res 2016;76:3211–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S6
Supplementary Figure S5
Supplementary Figure S4
Supplementary Figure S3
Supplementary Figure S2
Supplementary Figure S1
Data Availability Statement
The raw data generated in this study are available upon request from the corresponding author. Single-cell RNA-seq data analyzed in this study were obtained from GEO at GSE132355.