

Abstract
Background
Orchids (Cymbidium spp.) exhibit significant variations in floral morphology, pollinator relations, and ecological habitats. Due to their exceptional economic and ornamental value, Cymbidium spp. have been commercially cultivated for centuries. SSR markers are extensively used genetic tools for biology identification and population genetics analysis.
Result
In this study, nine polymorphic EST-SSR loci were isolated from Cymbidium goeringii using RNA-Seq technology. All nine SSR loci showed transferability in seven other congeneric species, including 51 cultivars. The novel SSR markers detected inter-species gene flow among the Cymbidium species and intra-species sub-division of C. goeringii and C. ensifolium, as revealed by neighborhood-joining and Structure clustering analyses.
Conclusion
In this study, we developed nine microsatellites using RNA-Seq technology. These SSR markers aided in detecting potential gene flow among Cymbidium species and identified the intra-species sub-division of C. goeringii and C. ensifolium.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12870-023-04499-y.
Keywords: SSR-Marker, RNA-Seq, Cymbidium goeringii, Inter-species sub-division, Hybridization
Background
Orchidaceae is one of the most abundant species angiosperm families, constitutes approximately 10% of flowering plant species, and displays unique flower morphologies [1–3]. Orchids account for a large share of the global floriculture trade both as cut flowers and as potted plants and were estimated to comprise around 10% of the international fresh-cut flower trade [4, 5]. Orchids are fast-growing potted flowering plants in many countries in terms of sales [6]. Hybridization between species happens in nature and during culturing [7, 8]. The genus Cymbidium comprises 44 species that are widely distributed in East Asia [9–11]. Cymbidium spp. (Orchidaceae) are popular potted flowers which were considered to have great value in ornamental and economic and have been cultivated for several centuries [12]. Despite the great value, the richness of orchid species decreased dramatically, and a lot of orchid species have become rare or endangered in the world [5, 13]. Because of a long history of cultivation and nature hybridization, the genetic variation of Cymbidium spp. is high diversity and complex [14]. Consequently, the taxonomic classification of Cymbidium becomes very difficult [15]. Although several approaches have been attempted to understand genetic diversity [16–19], the genetic resources for the characterization of Cymbidium are still insufficient. Some microsatellite markers that developed for the genus Cymbidium are not well-tested in cross-species [16, 17, 19]. Additionally, the genetic relationship among many of the major lineages of Cymbidium species remains unclear and the genetic relationship between species is not clear [9, 20]. It is necessary to develop reliable markers to evaluate the genetic diversity and phylogenetic relationship of Cymbidium for effective conservation and utilization.
Microsatellites or simple sequence repeats (SSRs) are a subcategory of tandem repeats consisting of 1–6 nucleotides in length (motifs) found in genomes of all prokaryotes and eukaryotes [21, 22]. Microsatellites have been utilized liberally over previous years since they are profoundly informative with a high mutation rate per generation per locus (10−7 to 10−3) [21] and relatively selective neutrality [23, 24] As high polymorphism, abundance, co-dominance, selective neutrality and transferability across species, microsatellite markers have been widely used in species and cultivars identification [25]. The availability of high-throughput sequencing technologies (RNA-Seq) has enabled researchers to identify a substantial number of microsatellites at less cost and effort compared to traditional SSR development processes [26].
In this study, nine novel microsatellite markers were developed and characterized based on RNA-Seq data. Combined with four SSR markers from published literature, thirteen SSR markers were used to figure out: (1) how prevalent these SSR markers are in cross-species amplification; (2) is there sub-division population structure intra-species; (3) is there genetic hybridization inter-species in the genus Cymbidium.
Results
Sequencing and de novo assembly of transcriptome
In total, 11.07 Gb of clean data was obtained using the Illumina NovaSeq platform. RNA-Seq yielded 22,739,372 clean paired-end reads at least 150 bp in length, and 72,556 Unigenes were gained from the clean reads performed by de novo assembly with Trinity. The average length of Unigenes is 835 bp. The N50 of the Unigenes was 1,483 bp.
Unigenes annotation
The assembled Unigenes of C. goeringii were annotated against eight public databases (Table 1, Fig. 1A). A total of 49,636 Unigenes (42.13%) were successfully annotated against at least one database. In summary, 29.86% of Unigenes from C. goeringii were matched with Elaeis guineensis, and 23.56% matched Phoenix dactylifera (Fig. 1B). In total, 10,724 Unigenes (11.26%) were annotated and clustered into three main GO categories and 50 sub-categories (Fig. 2A). Based on the KOG database, 36,364 Unigenes were annotated and 19.37% of Unigenes were annotated into the ‘General function’ cluster (Fig. 2B). Based on the KEGG database, a total of 33,377 Unigenes were annotated (Fig. 2C).
Table 1.
Unigenes annotation of C. goeringii against eight databases
| Annotation database | Number of Unigenes | Percentage (%) |
|---|---|---|
| Annotated in NR | 44,934 | 47.19% |
| Annotated in NT | 23,672 | 24.86% |
| Annotated in KEGG | 33,377 | 35.05% |
| Annotated in SwissProt | 29,641 | 31.13% |
| Annotated in Interpro | 39,072 | 41.03% |
| Annotated in GO | 10,724 | 11.26% |
| Annotated in KOG | 36,354 | 38.18% |
| Annotated in Intersection | 39,072 | 41.03% |
| Annotated in at least one database | 49,636 | 52.13% |
| Total Unigenes | 95,224 | 100% |
Fig. 1.

Cymbidium goeringii Unigenes annotation against eight databases (A) and against different species (B)
Fig. 2.

Cymbidium goeringii gene annotation based on GO, KOG, and KEGG databases. A Gene Ontology (GO) annotation graph of C. goeringii. B EuKaryotic Ortholog Groups (KOG) annotation graph of C. goeringii. C Kyoto Encyclopedia of Gene and Genomes (KEGG) annotation graph of C. goeringii
Frequency and distribution of SSRs in the transcriptome
Using the MISA software, a total of 95,224 Unigenes were scanned and 15,244 SSR loci were detected (Table 2). The SSR locus discovered from transcriptome data includes six types: mono-, di-, tri-, tetra-, penta-, and hexanucleotide repeat motifs. The content among different types varies greatly. The Di-nucleotide repeat motif ranked the most abundant type (accounting for 44.19%) and the penta-nucleotides was the least abundant type (accounting for 1.11%) (Fig. 3). The counts of four types of Di-nucleotide and ten types of Tri-nucleotide were presented at Fig. 3.
Table 2.
Prediction of SSRs out of the transcript datasets of C. goeringii
| Item | Number |
|---|---|
| Total number of sequences examined | 95224 |
| The total size of examined sequences (bp) | 79602818 |
| Total number of identified SSRs | 15244 |
| Number of SSR-containing sequences | 12745 |
| Number of sequences containing more than one SSR | 2051 |
| Number of SSRs present in compound formation | 962 |
| Mono nucleotide | 4783 |
| Di nucleotide | 6737 |
| Tri nucleotide | 3145 |
| Tetra nucleotide | 179 |
| Penta nucleotide | 161 |
| Hexa nucleotide | 239 |
Fig. 3.

Microsatellite loci distribution in the transcriptome data of C. goeringii
Genetic polymorphisms of 13 SSR loci
In total, nine loci were selected from the transcriptome data of C. goeringii. The sequences of the nine loci were submitted to NCBI (https://www.ncbi.nlm.nih.gov/nuccore/OP480183-OP480192) (Table 3). Combined with the four loci from published literature [17, 27], 167 alleles were detected from C. goeringii population (Table 3). The observed heterozygosities varied dramatically cross 13 loci, ranged from 0.09 to 1. The expected heterozygosities of most loci were lager than 0.75, only locus H93 present the observed heterozygosities of 0.54. Null allele frequency ranged from 0 to 0.8 across 13 SSR markers (Table 4). Linkage disequilibrium was not detected between any pair of loci.
Table 3.
Characteristics of 9 microsatellite loci isolated from Cymbidium goeringii
| Locus | Motif |
Primer sequence (5’- 3’) Forward/ reverse |
Ta(℃) | Product Size range (bp) | GenBank accession no |
|---|---|---|---|---|---|
| LH3 | (TTA)6 | TTTCTTGCTGAGCCTTTTATGTC/CCACTCCTTTTCTTCATCATTTG | 54 | 118–151 | OP480182 |
| LH4 | (AG)7 | AAAAATAGGCATAGTCGTCCGTC/TCTTATTTTATCCGGGGAGAGC | 54 | 132–168 | OP480183 |
| LH6 | (CT)26 | ACTCGACTCGACTCACTTCAAAA/AAGTTAAATAACCCCACCAGCAC | 54 | 121–149 | OP480184 |
| LH14 | (AG)6 | AGCTTGATGAAATTGCTGAAAAG/GGCAAAGGATCTGTATTCTTCCT | 54 | 107–109 | OP480186 |
| LH33 | (CT)6 | CTATCTGCAGTGTTTCTCAAGCA/CGCAATACCTCGATACCAAATAC | 54 | 159–189 | OP480188 |
| LH43 | (CT)15 | TTAAATTCAAAGTTTCACTCGCC/AACTCCCCAGTAGCTTTCAGTTT | 54 | 129–167 | OP480189 |
| LH45 | (GCA)5 | CTTTCTTCGCGCATATGTACTTT/ATCAAAGCTCACCATTTGTTCAT | 54 | 123–162 | OP480190 |
| LH88 | (TA)8 | AACTACAGCTTCATATTTGGCGA/GCTATGATTCCCCTTTTTCAATC | 54 | 112–142 | OP480191 |
| LH93 | (TCC)5 | TTCCACAATAGTTCCCCTGTCTA/AGGAAACAGAGGAAGAGGAAGAA | 54 | 84–96 | OP480192 |
Table 4.
Genetic diversity 13 SSR markers based on C. goeringii
| Locus | Product size(bp) | Na | Ne | fNA | I | PIC | Ho | He | F |
|---|---|---|---|---|---|---|---|---|---|
| L2 | 178–298 | 13 | 8.4 | 0.32 | 2.30 | 0.70 | 0.38 | 0.88 | 0.57 |
| L3 | 336–392 | 16 | 9.3 | 0.38 | 2.48 | 0.93 | 0.22 | 0.89 | 0.76 |
| X1 | 177–241 | 11 | 5.1 | 0.09 | 2.04 | 0.84 | 0.67 | 0.80 | 0.17 |
| X2 | 130–190 | 6 | 4.2 | 0.39 | 1.56 | 0.91 | 0.09 | 0.76 | 0.88 |
| LH3 | 118–160 | 8 | 4.8 | 0.15 | 1.79 | 0.86 | 0.50 | 0.79 | 0.37 |
| LH4 | 107–207 | 22 | 14.6 | 0.06 | 2.88 | 0.92 | 0.83 | 0.93 | 0.11 |
| LH6 | 110–160 | 21 | 15.8 | 0.00 | 2.89 | 0.94 | 1.00 | 0.94 | -0.07 |
| LH14 | 106–122 | 5 | 2.6 | 0.16 | 1.17 | 0.82 | 0.54 | 0.62 | 0.13 |
| LH33 | 158–212 | 15 | 9.4 | 0.22 | 2.44 | 0.86 | 0.57 | 0.89 | 0.37 |
| LH43 | 124–192 | 22 | 16.3 | 0.02 | 2.94 | 0.94 | 0.86 | 0.94 | 0.09 |
| LH45 | 123–163 | 10 | 5.4 | 0.19 | 1.91 | 0.91 | 0.54 | 0.81 | 0.33 |
| LH88 | 101–163 | 13 | 6.5 | 0.30 | 2.14 | 0.90 | 0.29 | 0.85 | 0.66 |
| LH93 | 63–93 | 5 | 2.2 | 0.80 | 1.08 | 0.85 | 0.04 | 0.54 | 0.92 |
Na number of alleles, Ne Effective number of alleles, fNA null-allele frequency, I Shannon's information index, PIC Polymorphism information content, F Fixed index, HO observed heterozygosity, HE expected heterozygosity
Cross-species analysis
The 9 polymorphic SSR loci isolated from C. goeringii (Table 3) and 4 loci from published literature (Table S1) were tested for cross-species amplification with 72 individuals from eight Cymbidium species. all these loci could be successfully amplified across eight Cymbidium species (Table 5). The genetic diversity for the eight Cymbidium species were listed in Table 5. The gene flow between species were presented in Fig. 4. Strong gene flow was detected between C. goeringii and C. ensifolium (Nm = 5.19). C. goeringii and C. faberi also present strong inter-species gene flow (Nm = 3.51). C. tortisepalum presents weak gene flow with other species (Fig. 4).
Table 5.
Genetic diversity of eight Cymbidium species
| Species | Sample size | Nps | Na | Ne | I | Ho | He |
|---|---|---|---|---|---|---|---|
| C. longibracteatum | 2 | 2.38 | 2.25 | 0.75 | 0.54 | 0.45 | -0.19 |
| C. goeringii | 21 | 11.77 | 7.47 | 2.04 | 0.52 | 0.81 | 0.38 |
| C. serratum | 2 | 2.23 | 2.18 | 0.69 | 0.23 | 0.44 | 0.57 |
| C. kanran | 3 | 3.92 | 3.42 | 1.22 | 0.58 | 0.65 | 0.18 |
| C. faberi | 16 | 8.00 | 5.25 | 1.57 | 0.35 | 0.69 | 0.55 |
| C. ensifolium | 17 | 10.08 | 6.11 | 1.90 | 0.44 | 0.78 | 0.46 |
| C. tortisepalum | 10 | 7.23 | 5.16 | 1.59 | 0.45 | 0.68 | 0.31 |
| C. sinense | 1 | 1.54 | 1.54 | 0.37 | 0.54 | 0.27 | -1.00 |
Nps Number of provenance samples, Na number of alleles, Ne Effective number of alleles, I Shannon's information index, HO: observed heterozygosity, HE expected heterozygosity
Fig. 4.
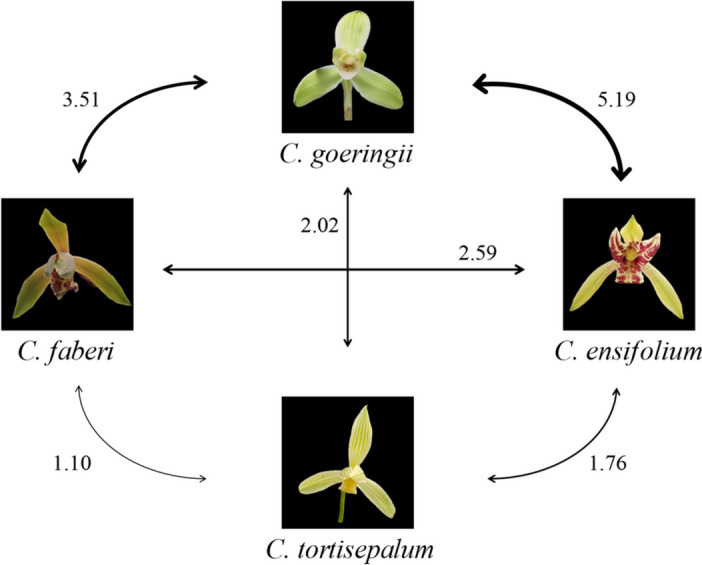
Gene flow among C. faberi, C. ensifolium, C. tortisepalum, and C. goeringii. The number noted on the line between species indicated the number of migrants (Nm) between species
Principal coordinate (PCoA) analysis of four Cymbidium species
In Principal coordinate (PCoA) analysis, the first two principal component accounts for 24.66% (Fig. 5). Most C. tortisepalum (LPL) individuals clustered together and most C. faberi (HUL) individuals clustered together. Two C. longibracteatum (CJ) individuals clustered together. But C. goeringii (CL) and C. ensifolium (JL) individuals are scattered (Fig. 5).
Fig. 5.

PCoA analysis of the 72 individuals from 8 species of Cymbidium. HUI: C. faberi; JL:C. ensifolium; ML: C. sinense; HAL: C. kanran; CJ: C. longibracteatum; LPL: C. tortisepalum; DBL: C. serratum; CL: C. goeringii
The Neighbor-Joining phylogenetic analysis
Based on the distance calculation method of Shared Allele, the Neighbor-Joining phylogenetic analysis presented the phylogenetic relationship of the 72 Cymbidium individuals (Fig. 6). Most of the individuals belong to the same species clustered together in the Neighbor-Joining tree. Such as all the C. tortisepalum individuals cluster into the LPL clade; ten C. faberi individuals clustered in the HUL clade and most C. goeringii and C. ensifolium individuals clustered in CL clades and JL clades separately (Fig. 6).
Fig. 6.

Neighbor-joining phylogenetic tree of 8 Cymbidium species. 72 individuals were included in the phylogenetic analysis. A phylogenetic tree. B partial pictures of the cultivars
But intra-species sub-division existed in both C. ensifolium (JL) and C. goeringii (CL), and each species contains three sub-clades (Fig. 6). The Neighbor-Joining tree also revealed some gene flow between Cymbidium species. For example, one C. goeringii (CL1) individual was mixed into C. ensifolium sub-clade (JL clade I); one C. sinense (ML99) individual was mixed into the C. tortisepalum (LPL) clade, and one C. ensifolium individual (JL 21) was mixed into C. ensifolium clade (HUL clade) Multiple mismatches exist between C. ensifolium and C. goeringii. One C. ensifolium individual (HUL 16) was mixed into C. goeringii sub-clade (CL clade II). There were multiple C. ensifolium and C. goeringii individuals clustered together and constituted mixed clades, such as CL-HUL mixed clade in Fig. 6.
The population structure analysis
In the population structure analysis, the magnitude of Delta K as a function of K suggested the existence of 4 clusters for Cymbidium. when K = 4, the value Delta K was the largest (Fig. 7). We present the structure result when K = 4 (Fig. 8). The 4 clusters were presented using 4 colors: yellow, green, red, and blue, and the percentage of each color presented the proportion of each cluster individually. The yellow cluster takes more than 90% of most C. tortisepalum (LPL) individuals. C. tortisepalum was mainly constituted by a yellow cluster. Only one C. sinense individual (ML 99) was included in this work, and the was constituted mainly by yellow which is very similar to the constitution of C. tortisepalum. C. faberi was mainly constituted by green clusters. The green cluster was also contained in C. goeringii and C. ensifolium. The color constitution of C. goeringii and C. ensifolium were very complex. Both C. goeringii and C. ensifolium contain four clusters in Structure analysis (Fig. 8).
Fig. 7.

the magnitude of DeltaK (B) as a function of K suggested the existence of 4 clusters for Cymbidium. Results are from 10 replicates for each of 1 ≤ K ≤ 9
Fig. 8.

STRUCTURE genetic clusters of 72 individuals (up) and 8 species (down) of genus Cymbidium. Green, yellow, blue, and red represent the assignment probability for the four major clades
Discussion
The novel developed microsatellite markers by transcriptome sequencing for Cymbidium
In this study, nine Cymbidium SSR markers were developed using the RNA-seq technique. The availability of high-throughput sequencing technologies has recently assisted researchers, providing excellent opportunities for life sciences [28]. Generating transcriptome data through RNA sequencing has been successfully reported for SSR marker development in non-model plants with no reference genome as de novo sequencing [29–32]. Compared with the SSRs developed from genomic sequences, the SSR markers isolated from transcripts (ETS-SSR) displayed high transferability among related species and high genetic differentiation, low error rates, and low null allele frequencies but relatively low polymorphisms [33]. In this study, the transcriptome data provide abundant resources of the SSR sites, which would be useful in studies on the genetic diversity, and population genetics of C. goeringii and congeneric other species.
In this study, the newly developed microsatellite markers are highly transferable in the genus Cymbidium. The nine SSR loci could be successfully amplified across eight Cymbidium species (Table 5). Microsatellite was one of the most widely used neutral molecular markers [21–24]. Because of the high level of polymorphism, high abundance, co-dominance, selective, neutrality, and transferability across species, microsatellite markers have been utilized for a variety of applications in plant studies, including species/cultivars identification, paternity testing, genes mapping, construction of linkage maps, markers assisted selections and back-crosses, population genetics, gene flow, phylogenetics, and conservation genetics [25, 34, 35]. The nine microsatellite markers could be successfully amplified in eight Cymbidium species, and proforma highly polymorphic. Up to 22 alleles were detected in two loci (Table 4). As the urgent need for an identification method in orchid business marketing, our newly developed microsatellite markers will be useful in Cymbidium species and cultivars discrimination and identification both in orchid business and research.
The intra-species sub-division
Phylogenetic analysis was frequently used in resolving the genetic variation and structure of Orchidaceae species [36–40]. Using the novel developed SSR markers, the population genetic analysis in the genus Cymbidium revealed intra-species divergence and inter-species hybridization. The phylogenetic analysis presented the intra-species divergence in both C. ensifolium (JL clade I, JL clade II and JL clade III) and C. goeringii (CL clade I, CL clade II and CL clade III) species (Fig. 6). In the PCoA analysis, unlike C. tortisepalum and C. faberi, in which individuals clustered together, the C. goeringii and C. ensifolium individuals are scattered (Fig. 5). The intra-species divergence was also presented in the STRUCTURE analysis (Fig. 8). The cultivators (individuals) of C. goeringii and C. ensifolium presented complex constitutions. In natural populations, C. ensifolium and C. goeringii present low-level genetic diversity between populations [41, 42]. In this work, the genetic structure is more significant than the natural population. That may be the consequence of artificial breeding accelerated genetic divergence. In this work, all the C. ensifolium and C. goeringii individuals are cultivators. Genetic diversity analysis discovered more genetic divergence in cultivators. Using RAPD markers, two distinct groups were revealed among cultivators of C. goeringii [20]. Based on 38 C. ensifolium cultivars, high genetic diversity was discovered using RAPD analysis [43]. This indicated that higher genetic diversity exists in the cultivator than in the natural population of C. ensifolium and C. goeringii.
Inter-species gene flow among Cymbidium species
Neutral molecular markers were frequently used in detecting inter-species hybridization and gene flow [24, 34, 44]. In this work, based on SSR markers, using PCoA and phylogenetic analysis high-frequency gene flow was detected among C. goeringii, C. ensifolium, and C. faberi. High number of migrants (Nm) was detected among C. goeringii, C. ensifolium, and C. faberi (Fig. 4). In the PCoA analysis, two individuals were clustered into C. goeringii clade and one C. goeringii individual was clustered into C. faberi clade in (Fig. 5). Multiple C. ensifolium individuals were clustered into C. goeringii clade or C. goeringii clade in PCoA analysis (Fig. 5). In phylogenetic analysis, multiple C. goeringii and C. faberi individuals clustered together and formed mixed clades in NJ analysis (Fig. 6). One C. goeringii (CL 1) individual was clustered into C. ensifolium (JL clade I), one C. ensifolium (JL 21) individual was clustered into C. faberi (HUL clade) (Fig. 6). All these evidences indicated gene flow between C. goeringii and C. faberi.
Gene flow between Cymbidium species was not discovered for the first time. In one molecular genetic analysis work on the genus Cymbidium, one C. faberi cultivator ‘Ruyisu’ was clustered into C. goeringii group in STRUCTURE analysis based on SSR markers [17]. Sympatric distribution may cause inters-species hybridization in Orchid. The natural distribution of C. goeringii and C. faberi overlaps frequently, and both distributes in southwest and southeast China [45]. Sympatric distributed interspecific hybridization was discovered in another genus of Orchid. Natural hybridization were detected and proved between sympatric distributed Geodorum eulophioides and G. densiflorum [46].
Artificial cross-breeding may be another reason for the inter-species gene flow in Cymbidium. Orchids have been cultivated for centuries, artificial cross-breeding in Cymbidium is quite frequency [47], and hybridization between species happens multiple times during culturing [7, 8, 12]. In this work, three cultivators of C. goeringii and C. faberi were clustered into CL-HUL mixed clade (Fig. 6), and indicated complex genetic background of these three cultivators (Fig. 6).
Conclusions
The newly developed microsatellite markers of Cymbidium goeringii with RNA-seq data were highly polymorphic, and successfully amplified across 8 Cymbidium species. Based on the SSR markers, intra-species sub-division was detected in C. goeringii and C. ensifolium; inter-species gene flow was detected among C. goeringii, C. ensifolium, and C. faberi. These SSR makers will be useful in the genus Cymbidium's cultivar and species identification and population genetic cultivar.
Methods
Materials
Fresh leave of Cymbidium goeringii ‘da fu gui’ was collected for RNA extraction and transcriptome sequencing. C. goeringii ‘da fu gui’ was a popular orchid cultivator and classic representative of spring orchids with lotus petal flowers. C. goeringii ‘da fu gui’ was collected from natural forests in 1909. The transcriptome sample used in this experiment was initially brought from the seedling and plant company at Shaoxing, Zhejiang Province, China, and then cultured at the Orchid greenhouse of Zhejiang A&F University by Dr. Hui-Juan Ning.
In total, 72 individuals from 8 Cymbidium species were collected for the SSR marker screening experiment. Including 21 C. goeringii individuals,16 C. faberi individuals, 17 C. ensifolium individuals, 10 C. tortisepalum individuals, 2 C. longibracteatum individuals, 2 C. serratum individuals, 3 C. kanran individuals, and 1 C. sinense individual (Table 5). All of these Cymbidium specimens were collected from southeast and southwest China (Table S2) and identified by Dr. Hui-Juan Ning (the author of this work) and preserved at the Orchid greenhouse of Zhejiang A&F University. The detail of the collection location, the cultivars’ name, and the morphology of all the samples were listed in supplementary table 1. The specimens used in this work were purchased from plant companies and these 8 species have not been listed in national key protected plants. We collected the samples without any required permissions. Our sample collection work and experimental research complied with local legislation and national and international guidelines. All the plant materials were cultured at the Orchid greenhouse of Zhejiang A&F University (ZAFU) or persevered deposited at the herbarium of ZAFU. The voucher no. of each specimen was listed in Table S2.
DNA extraction, RNA extraction, cDNA library construction and sequencing
The total RNA of one C. goeringii individual was extracted using a modified CTAB RNA extraction method for further transcriptome sequencing [48]. The genomic DNA of all the specimens was extracted using a modified DNA extraction method to detect polymorphisms of isolated microsatellite loci [49]. The quality and quantity of the exacted DNA and RNA was assessed using 1.5% agarose gel electrophoresis and NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). RNA-Seq library was constructed using Illumina TruSeq RNA Sample Preparation Kit (Illumina, San Diego, California, USA). The C. goeringii RNA was sequenced with RNA-Seq on the Illumina NovaSeq platform at BGI Tech (Shenzhen, China) generating 6.8 Gb reads.
Transcriptome assembly and Unigenes annotation
The raw data yielded from RNA-Seq was conducted through a quality assessment and credibility analysis using Trimmomatic [50]. Low-quality sequences were removed in the sequencing process. Trinity was used for conducting the de novo assembly [51, 52]. The transcripts were assembled and the main transcript was selected from the local area as Unigenes [53].
Unigenes sequences were compared against NCBI nr (National Center for Biotechnology Information non-redundant protein sequences), NT (Nucleotide Sequence Database) KOG (EuKaryotic Orthologous Groups of proteins), SwissProt (Swiss-Prot Sequence Database), KEGG (the Kyoto Encyclopedia of Genes and Genomes), Intersection, and Interpro databases to associate Unigenes with annotated proteins and functional information [54–56]. Gene ontology analyses were conducted using Blast2GO [57]. WEGO [58] was used to characterize GO annotations and statistics, and to describe the molecular functions of genes, cell components, and biological processes involved.
Microsatellites identification based on transcriptome data
The microsatellite tool (MISA-web) [59] was conducted to detect microsatellite loci with the following criteria [29]: mono-nucleotide repeat motifs with at least 12 repeats, di-nucleotide repeat motifs with at least six repeats and repeats of all other motif lengths extend at least five repeats.
Based on the Unigenes, SSR primers were designed using Primer Premier v5.0 software [60]. After primer designing, 120 pairs of primers were randomly selected with the condition of having targeted product sizes between 100 and 300 bp [29, 61]. Di-, tri-, tetra-, penta-, and hexanucleotide repeat loci have at least 9, 6, 5, 4, and 3 repeats, respectively.
PCR amplification and genotyping
Twenty-one C. goeringii individuals were amplified to survey the polymorphism of the SSR loci. PCR amplification was performed under an appropriate annealing temperature (Table 2).The primers were attached FAM or HEX fluorescent (Applied Biosystems, New York, USA). Fragment sizes were determined on an ABI 3100 Genetic Analyzer (Applied Biosystems). ROX 500 (Applied Biosystems) was used as the internal lane size standard.
SSR markers data analysis and cross-species analysis
GenALEX [62] was used to calculate the number of alleles (Na), the effective number of alleles (Ne), Shannon's information index (I), PIC Polymorphism information content (PIC), and the Fixed index (F) of each locus based on the data of C. goeringii. The likelihood ratio test was employed to estimate linkage disequilibrium using Genepop [63] and P-values were adjusted using the Bonferroni correction. The null-allele frequency was analyzed using Genepop [63].
To validate the transferability of the polymorphic loci isolated from C. goeringii, cross-species amplifications were tested for the 72 individuals from eight Cymbidium species using the same procedures as above, except that the annealing temperature was re-optimized for each locus. The number of provenance samples (Nps), number of alleles (Na), effective number of alleles (Ne), Shannon's information index (I), observed heterozygosity (Ho), and expected heterozygosity (He) was calculated for each species using GenALEX [62]. The pairwise species estimates of the number of migrants (Nm) were calculated among C. goeringii, C. ensifolium, C. tortisepalum, and C. faberi using GenALEX [62].
Cluster analysis of eight Cymbidium species
GenALEX [62] was used to calculate the Pairwise Population Matrix of Nei Genetic Identity between populations, followed by PcoA analysis using the Omic share website tool (https://www.omicshare.com/tools/).
Powermarker software [64] was used to calculate the genetic distance based on the Shared Allele algorithm, and then a phylogenetic tree was constructed based on the Neighbor-Joining method, and the final results were visualized with MEGA version X [65].
The population structure analysis was performed using Structure v2.3.4 [66], the parameters length of the burn-in period was set to 100,000 and the number of MCMC Reps after burn-in was set to 500,000, the optimal K value was calculated using the harvest online website (https://taylor0.biology.ucla.edu/struct_harvest/), then repeated sampling analysis was performed with CLUMPP [67], visualization was performed with distruct software [66].
Supplementary Information
Additional file 1: Supplementary Table S1. Characteristics of 4 microsatellite loci isolated from published literature. Supplementary Table S2. Sample collection of 72 individuals from 8 Cymbidium species.
Acknowledgements
We thank Jia-Yi Lou for editing the pictures.
Authors’ contributions
HJN collected all the plant samples and wrote the first draft of the manuscript; HJN and FFG conducted the molecular experiments; FFG, ENT, and LYY did the analysis. HJN and LYY design the experiment. ENT and LYY edited the manuscript. All authors participated in writing and reviewing the manuscript.
Funding
This study was supported by the Zhejiang Public Welfare Technology Application Research Project (LGN20C160004); Research Development Fund of Zhejiang A&F University (2019FR028); Open Fund of Zhejiang Provincial Key Laboratory of Germplasm Innovation and Utilization for Garden Plants (2020E10013-K202104). "Fourteenth Five-Year Plan" major scientific and technological project of new agricultural varieties breeding-new flower varieties breeding (2021C02071-5). Open Fund of MOE Key Laboratory of Biodiversity Science and Ecology Engineering (K202301). Open Fund of Hubei Key Laboratory of Economic Forest Germplasm Improvement and Resources Comprehensive Utilization (202141704).
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the NCBI (https://www.ncbi.nlm.nih.gov/nuccore/OP480183-OP480192).
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hui-Juan Ning and Fang-Fang Gui contributed equally to this work.
Contributor Information
En-Wei Tian, Email: tianenwei@126.com.
Li-Yuan Yang, Email: yangliyuan67@126.com.
References
- 1.Givnish TJ, Spalink D, Ames M, Lyon SP, Hunter SJ, Zuluaga A, Doucette A, Caro GG, McDaniel J, Clements MA, et al. Orchid historical biogeography, diversification, Antarctica and the paradox of orchid dispersal. J Biogeogr. 2016;43(10):1905–1916. [Google Scholar]
- 2.Givnish TJ, Spalink D, Ames M, Lyon SP, Hunter SJ, Zuluaga A, Iles WJD, Clements MA, Arroyo MTK, Leebens-Mack J, et al. Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc R Soc B: Biol Sci. 282:20151553. [DOI] [PMC free article] [PubMed]
- 3.Roberts DL, Dixon KW. Orchids. Curr Biol. 2008;18(8):R325–R329. doi: 10.1016/j.cub.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 4.Hinsley A, Lee TE, Harrison JR, Roberts DL. Estimating the extent and structure of trade in horticultural orchids via social media. Conserv Biol. 2016;30(5):1038–1047. doi: 10.1111/cobi.12721. [DOI] [PubMed] [Google Scholar]
- 5.Yang X-x, Gu W-Q, Zhang S-J, Xing M, Qu X-L, Luo L. Study on endangered degree and priority conservation sequence of orchids in the lower reaches of Yarlung Zangbo River. Bull Botan Res. 2023;43(02):169–178.
- 6.Palma MA, Chen Y-J, Hall C, Bessler D, Leatham D. Consumer preferences for potted orchids in the Hawaiian market. HortTechnol Hortte. 2010;20(1):239–244. [Google Scholar]
- 7.Sujii PS, Cozzolino S, Pinheiro F. Hybridization and geographic distribution shapes the spatial genetic structure of two co-occurring orchid species. Heredity. 2019;123(4):458–469. doi: 10.1038/s41437-019-0254-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson SD. Natural hybridization in the orchid flora of South Africa: Comparisons among genera and floristic regions. S Afr J Bot. 2018;118:290–298. [Google Scholar]
- 9.Obara-Okeyo P, Kako S. Genetic diversity and identification of cymbidium cultivars as measured by random amplified polymorphic DNA (RAPD) markers. Euphytica. 1998;99(2):95–101. [Google Scholar]
- 10.Puy DD, Cribb P. The genus Cymbidium. 1988.
- 11.Dressler R, Dressler RL, DRESSLER RL, Dressler R, Dressler RL, Dressler R, Dressler R, Dressler BL, Dressler G. The orchids: natural history and classification. Cambridge: Harvard University Press; 1981.
- 12.Chen SC, Tang T. A general review of the orchid flora of China. In: Orchid biology: reviews and perspectives. Edited by Arditti J, vol. II. New York: Cornell University Press; 1982. p. 39–87.
- 13.Phillips RD, Reiter N, Peakall R. Orchid conservation: from theory to practice. Ann Bot. 2020;126(3):345–362. doi: 10.1093/aob/mcaa093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hew CS. Ancient Chinese orchid cultivation: a fresh look at an age-old practice. Sci Hortic. 2001;87(1):1–10. [Google Scholar]
- 15.Garay LA, Sweet HR. Natural and artificial hybrid generic names of orchids. In: Withner CL, editor. The orchids scientific studies. New York: Wiley; 1974. pp. 1887–1973. [Google Scholar]
- 16.Moe KT, Hong W-J, Kwon S-W, Park Y-J. Development of cDNA-derived SSR markers and their efficiency in diversity assessment of Cymbidium accessions. Electron J Biotechnol. 2012;15(2):1–23. [Google Scholar]
- 17.Li X, Jin F, Jin L, Jackson A, Huang C, Li K, Shu X. Development of Cymbidium ensifolium genic-SSR markers and their utility in genetic diversity and population structure analysis in cymbidiums. BMC Genet. 2014;15(1):124. doi: 10.1186/s12863-014-0124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyun YS, Kim J, Chung KW. Development of polymorphic microsatellite markers for Cymbidium goeringii (Orchidaceae) Am J Bot. 2012;99(5):e193–e198. doi: 10.3732/ajb.1100505. [DOI] [PubMed] [Google Scholar]
- 19.Moe KT, Zhao W, Song H-S, Kim Y-H, Chung J-W, Cho Y-I, Park PH, Park H-S, Chae S-C, Park Y-J. Development of SSR markers to study diversity in the genus Cymbidium. Biochem Syst Ecol. 2010;38(4):585–594. [Google Scholar]
- 20.Sun C, Zhang M, Ye X, Liang C, Xia K. Studies on relationship between species cultivars of Cymbidium using RAPD. In: Acta Horticulturae Sinica. 2005. p. 1121–1124.
- 21.Selkoe KA, Toonen RJ. Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett. 2006;9(5):615–629. doi: 10.1111/j.1461-0248.2006.00889.x. [DOI] [PubMed] [Google Scholar]
- 22.Oliveira EJ, Pádua J, Zucchi MI, Vencovsky R, Vieira M. Origin, evolution and genome distribution of microsatellites. Genet Mol Biol. 2006;29(2):294–307. [Google Scholar]
- 23.Zane L, Bargelloni L, Patarnello T. Strategies for microsatellite isolation: a review. Mol Ecol. 2002;11(1):1–16. doi: 10.1046/j.0962-1083.2001.01418.x. [DOI] [PubMed] [Google Scholar]
- 24.Pokhriyal B, Thorat K, Limaye D, Joshi Y, Kadam VRD. Microsatellite markers – a novel tool in molecular genetics. Int J Res Pharm Chem. 2012;2:397–412.
- 25.Guichoux E, Lagache L, Wagner S, Chaumeil P, LÉger P, Lepais O, Lepoittevin C, Malausa T, Revardel E, Salin F, et al. Current trends in microsatellite genotyping. Mol Ecol Resourc. 2011;11(4):591–611. [DOI] [PubMed]
- 26.Taheri S, Lee Abdullah T, Yusop MR, Hanafi MM, Sahebi M, Azizi P, Shamshiri RR. Mining and development of novel SSR markers using Next Generation Sequencing (NGS) data in plants. Molecules. 2018;23(2):399. [DOI] [PMC free article] [PubMed]
- 27.Huang J-L, Zeng C-X, Li H-T, Yang J-B. Isolation and characterization of 15 microsatellite markers from the spring orchid (Cymbidium goeringii) (Orchidaceae) Am J Bot. 2011;98(4):e76–e77. doi: 10.3732/ajb.1000446. [DOI] [PubMed] [Google Scholar]
- 28.Egan AN, Schlueter J, Spooner DM. Applications of next-generation sequencing in plant biology. Am J Bot. 2012;99(2):175–185. doi: 10.3732/ajb.1200020. [DOI] [PubMed] [Google Scholar]
- 29.Liu Q, Lu Z, He W, Li F, Chen W, Li C, Chao Z, Tian E. Development and characterization of 16 novel microsatellite markers by Transcriptome sequencing for Angelica dahurica and test for cross-species amplification. BMC Plant Biol. 2020;20(1):152. doi: 10.1186/s12870-020-02374-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park S, Son S, Shin M, Fujii N, Hoshino T, Park S. Transcriptome-wide mining, characterization, and development of microsatellite markers in Lychnis kiusiana (Caryophyllaceae) BMC Plant Biol. 2019;19(1):14. doi: 10.1186/s12870-018-1621-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng X, Pan C, Diao Y, You Y, Yang C, Hu Z. Development of microsatellite markers by transcriptome sequencing in two species of Amorphophallus (Araceae) BMC Genomics. 2013;14(1):490–490. doi: 10.1186/1471-2164-14-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frenkel O, Portillo I, Brewer MT, Péros JP, Cadle-Davidson L, Milgroom MG. Development of microsatellite markers from the transcriptome of Erysiphe necator for analysing population structure in North America and Europe. Plant Pathol. 2012;61(1):106–119. [Google Scholar]
- 33.Postolache D, Leonarduzzi C, Piotti A, Spanu I, Roig A, Fady B, Roschanski A, Liepelt S, Vendramin GG. Transcriptome versus genomic microsatellite markers: highly informative multiplexes for genotyping Abies alba Mill. and Congeneric Species. Plant Mol Biol Report. 2014;32(3):750–760.
- 34.Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK. Microsatellite markers: an overview of the recent progress in plants. Euphytica. 2011;177(3):309–334. [Google Scholar]
- 35.Barbará T, Palma-Silva C, Paggi GM, Bered F, Fay MF, Lexer C. Cross-species transfer of nuclear microsatellite markers: potential and limitations. Mol Ecol. 2010;16(18):3759–3767. doi: 10.1111/j.1365-294X.2007.03439.x. [DOI] [PubMed] [Google Scholar]
- 36.Chung MY, López-Pujol J, Maki M, Moon MO, Hyun JO, Chung MG. Genetic variation and structure within 3 endangered Calanthe species (Orchidaceae) from Korea: inference of population-establishment history and implications for conservation. J Hered. 2013;104(2):248–262. doi: 10.1093/jhered/ess088. [DOI] [PubMed] [Google Scholar]
- 37.Inda L, Pimentel M, Chase M. Chalcone synthase variation and phylogenetic relationships in Dactylorhiza (Orchidaceae) Bot J Linn Soc. 2010;163(2):155–165. [Google Scholar]
- 38.Deng H, Zhang G-Q, Lin M, Wang Y, Liu Z-J. Mining from transcriptomes: 315 single-copy orthologous genes concatenated for the phylogenetic analyses of Orchidaceae. Ecol Evol. 2015;5(17):3800–3807. doi: 10.1002/ece3.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jing X-J, Hu F-R. Research progress on molecular biology of Orchidaceae. Mol Plant Breed. 2018;16(17):5835–5848. [Google Scholar]
- 40.Kang J, Lu J, Qiu S, Chen Z, Liu J, Wang H. Dendrobium SSR markers play a good role in genetic diversity and phylogenetic analysis of Orchidaceae species. Sci Hortic. 2015;183:160–166. [Google Scholar]
- 41.Chung MY, Nason JD, López-Pujol J, Yamashiro T, Yang B-Y, Luo Y-B, Chung MG. Genetic consequences of fragmentation on populations of the terrestrial orchid Cymbidium goeringii. Biol Cons. 2014;170:222–231. [Google Scholar]
- 42.Liu C-H. Genetic diversity of Cymbidium ensifolium (Linn.) Sw from Jiangxi Province revealed by inter simple sequence repeats. Nanchang: Nanchang University; 2012.
- 43.Hu W, Huang RZ, Pan XH, Jin-Feng LI, Duan AS. RAPD analysis of thirty-eight Cymbidium ensifolium Cultivars. Acta Horticult Sin. 2008;35(2):289–294. [Google Scholar]
- 44.Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJ, Bierne N, Boughman J, Brelsford A, Buerkle CA, Buggs R. Hybridization and speciation. J Evol Biol. 2013;26(2):229–246. doi: 10.1111/j.1420-9101.2012.02599.x. [DOI] [PubMed] [Google Scholar]
- 45.Flora Committee of China CAoS . Flora of China: Orchidaceae. Beijing: Science Press; 1999. [Google Scholar]
- 46.Zhu X, Tang J, Jiang H, Yang Y, Chen Z, Zou R, Xu A, Luo Y, Deng Z, Wei X, et al. Genomic evidence reveals high genetic diversity in a narrowly distributed species and natural hybridization risk with a widespread species in the genus Geodorum. BMC Plant Biol. 2023;23(1):317. [DOI] [PMC free article] [PubMed]
- 47.Gen-Fa Z. Progress on international cross breeding of Cymbidium. Guangdong Agricult Ence. 2005;4(6):25–27. [Google Scholar]
- 48.Chang S, Puryear J, Cairney J. A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Report. 1993;11(2):113–116. [Google Scholar]
- 49.Yang J-B, Li D-Z, Li H-T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol Ecol Resour. 2014;14(5):1024–1031. doi: 10.1111/1755-0998.12251. [DOI] [PubMed] [Google Scholar]
- 50.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Trinity: reconstructing a full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44(D1):D279–285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007;36(suppl_1):D480–D484. [DOI] [PMC free article] [PubMed]
- 56.Mao X-Z, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 57.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 58.Ye J, Zhang Y, Cui H, Liu J, Wu Y, Cheng Y, Xu H, Huang X, Li S, Zhou A, et al. WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018;46(W1):W71–w75. [DOI] [PMC free article] [PubMed]
- 59.Beier S, Thiel T, Münch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33(16):2583–2585. doi: 10.1093/bioinformatics/btx198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh VK, Mangalam AK, Dwivedi S, Naik S. Primer premier: Program for design of degenerate primers from a protein sequence. Biotechniques. 1998;24(2):318–319. doi: 10.2144/98242pf02. [DOI] [PubMed] [Google Scholar]
- 61.Xu R, Wang Z, Su Y, Wang T. Characterization and development of microsatellite markers in Pseudotaxus chienii (Taxaceae) based on transcriptome sequencing. Front Genet. 2020;11:574304. [DOI] [PMC free article] [PubMed]
- 62.Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics. 2012;28(19):2537–2539. [DOI] [PMC free article] [PubMed]
- 63.Rousset F. GENEPOP ' 007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour. 2008;8(1):103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 64.Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21(9):2128–2129. doi: 10.1093/bioinformatics/bti282. [DOI] [PubMed] [Google Scholar]
- 65.Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2015;33(7):1870–1874. [DOI] [PMC free article] [PubMed]
- 66.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23(14):1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Table S1. Characteristics of 4 microsatellite loci isolated from published literature. Supplementary Table S2. Sample collection of 72 individuals from 8 Cymbidium species.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available in the NCBI (https://www.ncbi.nlm.nih.gov/nuccore/OP480183-OP480192).