

Abstract
Infectious diseases (such as sepsis, influenza, and malaria), caused by various pathogenic bacteria and viruses, are widespread across the world. Early and rapid detection of disease-related pathogens is necessary to reduce their spread in the world and prevent their potential global pandemics. The clustered regularly interspaced short palindromic repeats (CRISPR) technology, as the next-generation molecular diagnosis technique, holds immense promise in the detection of infectious diseases because of its remarkable advantages, including supreme flexibility, sensitivity, and specificity. While numerous CRISPR-based biosensors have been developed for application in environmental monitoring, food safety, and point-of-care diagnosis, there remains a critical need to summarize and explore their potential in human health. This review aims to address this gap by focusing on the latest advancements in CRISPR-based biosensors for infectious disease detection. We provide an overview of the current status, pre-amplification methods, the unique feature of each CRISPR system, and the design of CRISPR-based biosensing strategies to detect disease-associated nucleic acids. Last but not least, the review analyzes the current challenges and provides future perspectives, which will contribute to developing more effective CRISPR-based biosensors for human health.
Keywords: CRISPR, Cas nuclease, Biosensor, Nucleic acids, Infectious disease
1. Introduction
Infectious diseases are constantly emerging and reemerging, posing a significant threat to human health and global economics. These diseases occur when pathogenic bacteria or viruses invade human bodies, causing various health symptoms and requiring medical attention for diagnosis and treatment.[1] It is estimated that infectious diseases caused 50,000 deaths each day and a total of over 17 million each year.[2] The continuing challenges posed by sepsis highlights this fact. Sepsis is one of the common infectious diseases, usually caused by bacterial or viral infections, accounting for approximately 20% of annual global deaths.[3] The pathogens responsible for infectious disease can be found all around the world, including in water and environmental samples, food matrices, as well as clinical and hospital settings.[4–6] Over the past two decades, antibiotics have been widely used to treat bacterial infections. However, the intensive use of antibiotics has resulted in these bacteria becoming resistant to various antibiotics.[7, 8] According to the World Health Organization (WHO), the existing antibiotics will lose their effectiveness to treat bacterial diseases in the next 1–2 decades.[9] In the United States, the rapid use of antibiotics causes more than 35,000 deaths each year, and it is estimated to rise to 10 million cases by 2050.[10, 11] Thus, there is an urgent need to develop novel strategies to sensitively detect infectious diseases at early stages, reducing subsequent outbreaks.
Due to the low infectious dose and biodiversity, it remains a substantial challenge to detect infectious diseases. Conventional culture-based colony counting remains the most reliable and accurate method to detect viable bacterial pathogens.[12] However, in some cases, viable bacteria and viruses are non-culturable, leading to false negative results. In addition, the culture-based colony counting process has a significant drawback as it is time-consuming. This is because it involves multiple steps, such as cell enrichment, colony isolation, and bacteria identification. As a result, there can be a time-lapse of several days from sample to results. Alternatively, quantitative polymerase chain reaction (qPCR) can detect a single bacterium or virus by amplifying its nucleic acids to hundreds and thousands of copies.[13] Because of its higher sensitivity than culture-based colony counting, real-time qPCR has been widely used to detect infectious diseases. With the requirement of bulky equipment, real-time qPCR for infectious disease detection is limited to non-laboratory settings. The immunoassay-based detections of infectious diseases rely on the specific recognition of antigen-antibody binding.[14] The most widely used recognition element is antibodies that can recognize the receptors on the surface of infectious diseases.[15, 16] The two typical examples of immunoassay-based detection methods are enzyme-linked immunosorbent assay (ELISA) and lateral flow assay (LFA).[17] Because infectious diseases may have multiple sub-strains that have different surface proteins expressed at different levels, the antibodies might fail to detect the infectious diseases.[18] Therefore, there is still a plenty of room to develop rapid, sensitive, and specific technologies to detect infectious diseases.
CRISPR (clustered regularly interspaced short palindromic repeats) technology is a promising tool to meet the needs of detecting infectious diseases in a rapid, sensitive, and specific manner. CRISPR was discovered in bacterial and archaeal cells as an adaptive defense system against phage infections or mobile genetic elements.[19] In 2012, the CRISPR/Cas9 system was first demonstrated to have the ability to cleave DNA in vitro, marking a significant milestone in the field of genetic research. This breakthrough discovery quickly led to widespread application for genome editing and gene therapies.[20] In 2016, another breakthrough occurred when the CRISPR/Cas9 system was reported to have the ability to detect nucleic acids. This development opened up new possibilities for CRISPR systems to detect infectious diseases by specifically targeting their nucleic acids.[21] CRISPR systems are classified into two major broad classes: class 1 and class 2. The class 1 systems use multi-subunit complexes that contain several different Cas nucleases, while the class 2 system contains only a single nuclease.[22] More specifically, the class 1 CRISPR/Cas system relies on multiple nucleases working together for the cleavage of nucleic acids, and each subunit nuclease offers a different function. For example, Cas6 processes mature crRNA from precursor RNA, and Cas8 recognizes the protospacer-adjacent motif (PAM) sequences.[23] In class 2 CRISPR/Cas systems, there is only a single Cas nuclease, including type II (Cas9), type V (Cas12, Cas14), and type VI (Cas13). Due to the simplicity of the assembled CRISPR/Cas nuclease complex, class 2 CRISPR/Cas systems have been widely studied and used to detect infectious diseases. It is worth mentioning that the US Food and Drug Administration (US FDA) has authorized the emergency usage of the Sherlock™ CRISPR SARS-CoV-2 kit to detect SARS-CoV-2 in May 2020, which is the first CRISPR-based detection product. Compared with other rapid detection methods, the CRISPR/Cas-based assay provides unique programmability and greater accuracy, enabling the efficient identification of specific gene sequences from pathogens, thereby facilitating its application in human health application.[24]
To highlight the recent advances of CRISPR-based biosensors for human health application, this review will cover the development of CRISPR-based biosensors to detect infectious diseases. In addition, the selection and design of effective CRISPR systems will be highlighted, including Cas9, Cas12, Cas13, and Cas14. We will also summarize the technologies of nucleic acid amplification to detect infectious diseases at low concentrations, and the mechanisms of signal transductions to visualize the detection results. Furthermore, the review will provide insights into future directions for developing CRISPR-based biosensors to address emerging infectious diseases. By combining recent advancements with future prospects, the review aims to significantly contribute to the advancement of diagnostic technologies and their impact on human health.
2. Development of CRISPR-based Biosensors
Advances in CRISPR technologies have made it feasible to detect infectious diseases, enabling their applications in various fields such as environmental monitoring, food safety, and clinical diagnostics. To ensure high detection sensitivity and specificity, it is important to design an effective CRISPR-based biosensor. As shown in Figure 1, the detection of infectious diseases using CRISPR-based biosensors includes target analyte identification, nucleic acid amplification, CRISPR system selection, and signal transduction. Since most infectious diseases are caused by either bacterial and/or viral pathogens, the first step is to identify the appropriate pathogens. However, complex sample matrices (such as food, blood, and feces) and trace amounts of target analytes pose challenges to the target identification process. To address this issue, the amplification is typically combined with a CRISPR-based biosensor to enhance the detection sensitivity and selectivity, including 1) DNA-to-DNA, 2) RNA-to-DNA, 3) DNA-to-RNA, and 4) RNA-to-DNA-to-RNA. The conversion between DNA and RNA can be achieved by in vitro transcription (IVT) and reverse transcription (RT). Theoretically, nucleic acid amplification technologies can amplify one DNA/RNA copy to one million copies within 30 min, ensuring detection sensitivity as low as one bacterial/viral pathogen.[13]
Figure 1.

Schematic illustration of CRISPR-based biosensors for infectious disease detection in various settings. The process of infectious disease detection using CRISPR-based biosensors involves four steps. 1) the target analyte (either bacterial and/or viral pathogens) need to be identified. 2) nucleic acid amplification techniques are employed to increase the amount of pathogen-associated nucleic acids. 3) an appropriate CRISPR system is selected to specifically recognize the amplified nucleic acid product. 4) a signal transduction strategy is utilized to transduce the specific recognition process into a measurable signal, allowing for the detection of infectious disease.
Based on the product of nucleic acid amplification, appropriate CRISPR/Cas systems should be selected to detect either DNA or RNA. There are two important components in the CRISPR system: a Cas endonuclease and a programmed guide RNA (gRNA). The Cas endonuclease acts like scissors to cut nucleic acids, and the gRNA can navigate the Cas endonuclease to cut the target nucleic acids specifically. For Cas9 and Cas14 systems, gRNA is fused with trans-activating crRNA (tracrRNA) and CRISPR RNA (crRNA), in which tracrRNA can bind to nucleases and crRNA is programmable for specifically binding to a target DNA. The tracrRNA-crRNA can be integrated to be a single guide RNA, called sgRNA. On the contrary, crRNA in the Cas12 and Cas13 systems serve both functions of binding to nucleases and targeting nucleic acids. The cleavage of target DNA by Cas9 and Cas12 nucleases begins with identifying the specific protospacer adjacent motif (PAM) which is subject to the specific Cas nucleases.[25, 26] Cas13 nuclease can recognize and cleave RNA without the requirement of PAM.[27] Like Cas12 nucleases, Cas14 can only cleave single-stranded DNA (ssDNA) and doesn’t require PAM.[28]
All these CRISPR systems are capable of recognizing and cleaving nucleic acids that are complementary to the crRNA, which is named cis-cleavage activity (specific). After the cis-cleavage activity, Cas12, Cas13, and Cas14 can offer additional indiscriminate ssDNA or ssRNA cleavage activity (termed as trans-cleavage activity, non-specific).[29–32] Due to the additional feature of trans-cleavage activity, Cas12, Cas13, and Cas14 offer advantages over Cas9 in detecting infectious diseases. For the CRISPR-based biosensors, the crRNA is highly target-specific, which serves as a recognition element to identify desired nucleic acids from infectious diseases. The specific recognition events can be amplified and transduced by Cas nucleases, which can be used to develop various signal transduction methods, including colorimetry, fluorescence, electrochemistry, and others.[33] The detection performance of CRISPR-based biosensors can be evaluated by the response time, detection limit, dynamic range, and specificity.[34]
3. Nucleic Acid Amplification
CRISPR-based biosensors provide highly specific target recognition. However, they frequently depend on the amplification of nucleic acids to detect infectious diseases when the target analytes are present in low concentrations in the sample. Amplification is crucial to enhance the detection sensitivity of CRISPR-based biosensors, allowing them to accurately detect and identify even trace amounts of the target, such as those found in the early stages of an infection. Various amplification techniques are available and can be combined with CRISPR-based biosensors to detect infectiouss diseases, including polymerase chain reaction (PCR), recombinase polymerase amplification (RPA), loop-mediated isothermal amplification (LAMP), rolling circle amplification (RCA), strand displacement amplification (SDA), and many more. [35–38] Each of these techniques has unique advantages and limitations, making it crucial to carefully consider the specific downstream CRISPR application when selecting an appropriate nucleic acid amplification method.
3.1. Polymerase Chain Reaction (PCR)
Polymerase chain reaction (PCR) is the gold standard and most widely used method for nucleic acid amplification. In PCR, a series of heating and cooling procedures are repeated to specifically amplify targeted double-stranded DNA (dsDNA). As shown in Figure 2a, the reaction mixture is initially heated to denature the target dsDNA, effectively separating its two strands. Subsequently, the reaction is cooled to create the optimal conditions for the primers to anneal, binding to each of the original DNA strands. After that, the temperature slightly increased to facilitate the extension of the new DNA strands from the primers. Typically, the “denaturing-annealing-extension” process is cycled around 30–35 times. In the past few years, CRISPR systems have been combined with PCR and reverse-transcription PCR (RT-PCR), generating highly sensitive and specific biosensors to detect infectious diseases.[39] For example, PCR-based CRISPR biosensors have demonstrated the detection of COVID-19,[39] H1N1 virus,[40] pathogenic bacteria,[41] antimicrobial resistance genes,[42] and many more analytes through the use of various CRISPR systems and readout signals. However, the requirement of thermal cycling for PCR-based amplification severely limits on-site amplification due to the reliance on expensive equipment and trained personnel. Therefore, there is a significant need to develop isothermal amplification for CRISPR-based biosensors.
Figure 2.

Nucleic acid amplification methods. a. PCR undergoes denaturalization, annealing, and extension via thermal cyclers. b. LAMP uses at least four primers (two inner primers: FIP, BIP; two outer primers: F3, B3) to generate extensive dumbbell-structures of amplified DNA. d. RCA amplifies the target DNA using a circular DNA template. c. RPA utilizes recombinases and single-stranded DNA binding proteins (SSBs) for isothermal amplification. e. EXPAR utilizes a strand displacement mechanism to exponentially amplify target sequences through repeated cycles of denaturation, annealing, extension, and nicking.
3.2. Loop-mediated Isothermal Amplification (LAMP)
Loop-mediated isothermal amplification (LAMP) is a powerful nucleic acid amplification technique that can be used to rapidly amplify target DNA or RNA. LAMP is performed under isothermal conditions, often providing a simpler and more readily available alternative to PCR, especially for CRISPR-based sensors designed to be used on-site.[43] LAMP utilizes a set of four or six primers and Bst polymerase to form dumbbell-loop structures that facilitate subsequent cycling amplification, generating extensively large amplified products (Figure 2b). Reactions are typically carried out between 60–70°C for 30 minutes with kits commercially available from several vendors. Fluorescent signal readouts are often combined with LAMP reactions for real-time LAMP (RT-LAMP) detection of infectious diseases. In addition, LAMP has been combined with CRISPR for the specific detection of Influenza A and B virus,[44] SARS-CoV-2,[45] and pathogenic bacteria.[46] While LAMP provides several advantages to PCR, drawbacks include complicated primer design and a high risk of false-positive signals derived from non-specific amplification and aerosol contamination.[47]
3.3. Recombinase Polymerase Amplification (RPA)
Recombinase polymerase amplification (RPA) is another commercially available isothermal amplification method and can be combined with CRISPR-based biosensors to detect infectious diseases. Significantly, RPA amplification avoids thermal template denaturation, achieving exponential amplification at the low-temperature range of 37–42 °C. In this strategy, a recombinase binds to a primer to form a complex capable of recognizing and displacing the template nucleic acids (Figure 2c). A single-stranded DNA binding protein (SSB) then binds and stabilizes the displaced strand. The entire exponential amplification can be completed within 30 min. Additionally, due to the similar reaction temperatures of the RPA and CRISPR nucleases, amplification and CRISPR-based detection can be performed simultaneously, drastically simplifying potential detection mechanisms.[48] RPA-based CRISPR systems have successfully detected SARS-CoV-2[49] and various pathogenic bacteria, including Staphylococcus aureus,[50] Trichomonas vaginalis,[51] Salmonella Typhimurium,[52] and Brucella spp..[53] Limitations of RPA amplification include the lack of specialized primer design software, limited availability of commercial kits, and the use of additional reagents, including recombinase enzymes and SSBs, which increase the cost and complexity of the reaction.
3.4. Rolling Circle Amplification (RCA)
Some other amplification methods provide unique versatility in certain situations. Among them, rolling circle amplification (RCA) has demonstrated highly sensitive and specific amplification of circular DNA. As shown in Figure 2d, the basic procedure of RCA is presented, including ligase binding, primer extension, and strand displacement amplification. The target nucleic acid pairs with a linear probe (padlock) through ligation, forming a circular template. Subsequently, polymerase extends the original target sequence as it moves around the template, creating a long linear nucleic acid strain complementary to the template nucleic acid.[54] Uniquely, this process can produce long single-stranded DNA/RNA molecules, making it highly compatible with the integration of Cas14 systems.[55] Despite the successful developments of RCA-based CRISPR biosensors for detecting several infectious diseases,[56, 57] it still encounters challenges in pricy padlock probes and non-specific amplification.
3.5. Others
Other isothermal amplification methods have been combined with CRISPR systems to detect infectious diseases, including exponential amplification reaction (EXPAR),[58] strand displacement amplification (SDA),[59] nicking-enzyme amplification (NEA),[60] recombinase-aided amplification (RAA),[61] and hybridization chain reaction (HCR).[62] For example, EXPAR employs a combination of nicking and strand displacement for extremely sensitive isothermal amplification of target DNA. As shown in Figure 2e, the EXPAR template is designed to have two repeat regions (X’) that perfectly match the target sequence (X). These repeat regions are separated by a cleavage site recognized by a nicking enzyme, which enables the template to be cleaved during the amplification process. EXPAR is initiated when target DNA pairs with the template sequence, resulting in the formation of a double-stranded duplex. Subsequently, DNA polymerase extends the duplex, generating an extended double-stranded DNA with a nicking site. In the presence of a nicking enzyme, the target strand is cleaved and then released through strand displacement mediated by DNA polymerase. After that, the displaced target strand is complementary to another template, initiating another round of amplification. EXPAR is typically carried out between 55–65 °C, even if the nicking enzyme works well at lower temperatures.[58] EXPAR-CRISPR sensors have been utilized with a variety of Cas enzymes for the detection of DNA mutations,[63] creatine kinase MB,[64] and microRNAs.[65] While EXPAR is extremely rapid and robust, the method requires an extremely complicated primer design and often results in non-specific interactions, leading to a high background amplification. Therefore, EXPAR is not a widely used amplification technique.
4. CRISPR/Cas Systems for Sensing Nucleic Acids
Benefiting from high programmability and specificity, CRISPR/Cas systems have become a powerful tool for infectious disease detection. In combination with nucleic acid amplification technologies mentioned above, CRISPR/Cas systems are designed to achieve a highly sensitive and specific detection of disease-related nucleic acids. However, the intrinsic differences of the CRISPR Cas nucleases vary in the detection methods, particularly for target types and trans-cleavage activities. Even when using the same CRISPR Cas nuclease, the underlying strategies of these detections may be entirely different. The following sub-sections will introduce various CRISPR/Cas systems and their underlying detection strategies for sensing disease-related nucleic acids.
4.1. CRISPR/Cas9 System
CRISPR/Cas9 is the first CRISPR system to be used for infectious disease detection.[21] The core of its detection mechanism is the cis-cleavage activity of the Cas9 nuclease to cleave the target DNA, which is closely related to its two structural domains, namely HNH and RuvC. After recognizing the PAM from the target DNA, the Cas9 nuclease bounds the target with HNH and RuvC domains and triggers the cleavage activity on the target sequence. Besides, the endonuclease-deactivated Cas9 (dCas9) is a mutant form of Cas9 nuclease. Through silencing two mutations in the HNH and RuvC domain, the cleavage activity of Cas9 is removed while maintaining the binding activity toward the target DNA. In recent years, several CRISPR-based biosensors have been developed using Cas9 and dCas9.
Identification of single nucleotide mismatch.
Since the CRISPR/Cas9 system can recognize and perform blunt end cuts on target dsDNA, the CRISPR/Cas9-based biosensors usually use cleaved DNA as the template for further amplification. As shown in Figure 3a, in the presence of PAM-presenting oligonucleotide (PAMmer), the target ssDNA was recognized and cleaved by the CRISPR/Cas9 system, releasing the cleaved DNA for cyclic amplification with the assistance of nicking endonuclease.[66] As a result, numerous DNA amplicons were generated and conjugated to dsDNA binding dye (SYBR Green), and the signal was monitored by real-time fluorescence. This method achieved a detection limit of 0.82 amol within 1 hour (Figure 3b–c), providing superior specificity in the identification of single-base nucleotide mismatch. Furthermore, this method has been successfully extended to the detection of DNA methylation as well as L. monocytogenes total RNA, demonstrating its versatility in various application scenarios.
Figure 3.

CRISPR/Cas9-based sensing strategies. a. Principle of CIRSPR-Cas9 mediated isothermal amplification method for the identification of single nucleotide mismatch (PAMmer: PAM-presenting oligonucleotide). b. Real-time fluorescence intensity curve responding to different DNA concentrations. c. Calibration curve of point of inflection (POI) toward DNA concentrations. (Reproduced with permission from ref. [66]., Copyright 2018, American Chemical Society) d. Design of CRISPR/dCas9-based biosensor for the detection of Mycobacterium tuberculosis e. Detection sensitivity of CRISPR/dCas9 system targeting the Mycobacterium tuberculosis 16s rRNA gene. (Reproduced with permission from ref. [67], Copyright 2017, American Chemical Society)
Deactivated Cas9 (dCas9).
Due to the removal of its endonuclease activity, dCas9 can recognize and bind to target nucleic acids without causing any cleavage. The dCas9-based biosensors rely on synthetic biology to bring split enzymes at the terminal of dCas9 together. For example, a recent study investigated a paired dCas9 reporter system to re-functionalize split firefly luciferase for the detection of infectious diseases (Figure 3d).[67] The luciferase was divided into two parts and separately linked with dCas9. These recombinant dCas9 nucleases were programmed by sgRNA complementary to different segments of target DNA at a short distance. In the presence of the target nucleic acid, two recombinant dCas9-sgRNA complexes recognized and bound the target DNA sequence, integrating two parts of luciferase and triggering the bioluminescent reaction. With pre-amplification by PCR, this detection strategy allowed paired dCas9 to detect Mycobacterium tuberculosis with a detection limit of one genomic copy (Figure 3e).
4.2. CRISPR/Cas12 System
Although several studies have utilized CRISPR/Cas9 and CRISPR/dCas9 for the detection of infectious diseases, the complex design and tedious procedure hinder the transduction of the CRISPR/Cas9 system alone from target identification to readable outputs. Taking the gift of trans-cleavage toward nonspecific oligonucleotides by the RuvC domain, the Cas12 nucleases are able to provide direct signal outputs upon target identification, which greatly simplifies the detection procedure and extends the potential applications for infectious disease diagnosis.
Photoactivable CRISPR/Cas12 system.
In the conventional CRISPR/Cas12 system, Cas12 nucleases are triggered immediately in the presence of target DNA. To detect target DNA at low concentrations, nucleic acid amplifications are combined with the CRISPR/Cas12 system. But the conventional one-pot method suffered from poor compatibility because the activated CRISPR/Cas12 system could prevent nucleic acid amplification. To overcome this problem, a photoactivatable CRISPR/Cas12-based biosensor was developed for the detection of SARS-CoV-2 (Figure 4a).[68] The SARS-CoV-2 RNA was amplified by RT-RPA, which wasn’t affected by the CRISPR/Cas12 system because the crRNA was temporarily blocked by a protective oligo. The protective oligo is a nucleic acid strand containing photocleavable linker to change the crRNA structure. Once the RPA reaction was completed, the protective oligo was broken down by ultraviolet light at 365 nm, followed by recovering of crRNA and activating the CRISPR/Cas12 reaction. Compared with the conventional one-pot CRISPR assay, this method achieved more than two orders of magnitude higher sensitivity with a limit of detection of 10 copies (Figure 4b–c).
Figure 4.

CRISPR/Cas12-based sensing strategies. a. Schematic illustration of photoactivable CRISPR/Cas12 system to detect SARS-CoV-2. b. Comparison of the photoactivable one-pot system with the conventional one-pot system. c. Fluorescence signal response of photoactivable system toward different copies of SARS-CoV-2 N gene. (Reproduced with permission from ref. [68], Copyright 2022, National Academy of Sciences, USA) d. Principle of the AND logic gate to detect nucleic acids using split Cas12 enzyme with leucine zipper. e. The algorithm of 2-input AND gate. f. Gel electrophoresis results of split Cas12-mediated cleavage of dsDNA (960 bp). (Reproduced with permission from ref. [69], Copyright 2020, Elsevier)
Split Cas12 nuclease.
With the development of intelligent detection, the split Cas nuclease was explored to construct the molecular logic gate, providing an advanced approach for the precise control of biological reactions. As shown in Figure 4d, a two-input Boolean AND logic gate was developed through expressing a pair of split Cas12 nuclease with leucine zipper domains.[69] Without disrupting the native structure, the Cas12 protein was artificially split into two parts (N-terminal and C-terminal Cas12) as two inputs in the AND logic gate. A pair of leucine zippers were fused to the split nuclease for inducing the spontaneous assembly of the two parts. The operation mechanism of the molecular AND logic gate follows a binary operation (Figure 4e), where the presence and absence of inputs are defined as 1 and 0, respectively. For the output, the mature Cas12 enzyme with cleavage activity was indicated as 1, while 0 represents the incomplete Cas12 enzyme without cleavage activity. Only in the presence of both inputs, the Cas nuclease can be reassembled and interacted with crRNA, serving as a functional nuclease to cleave the target DNA. This split Cas12-based logic gate system was used to cleave the genomic CSCR4 locus of HEK293T cells (Figure 4f). Compared with the full-length Cas12 nuclease, the reassembled Cas12 nuclease (N+C) showed the comparable cleavage ability of target DNA fragments, offering higher flexibility and programmability.
4.3. CRISPR/Cas13 System
Cas13 is a type VI nuclease and is starkly different from the previously introduced nucleases because of two HEPN domains that act as cleavage sites to target RNA without the requirement of PAM. Besides, some of the Cas13 enzymes have specific dinucleotide cleavage motif preferences that facilitate the design of multiplexed detection. Due to its unique properties, Cas13 has been widely applied in infectious disease diagnosis.
Tandem CRISPR system.
To achieve high sensitivity without pre-amplification, a tandem CRISPR/Cas13-based biosensor was developed for the rapid detection of SARS-CoV-2 (Figure 5a).[70] By integrating three crRNAs targeting the N gene or E gene of SARS-CoV-2 genomic RNA, the Cas13 nucleases were synergistically activated, resulting in the cleavage of more fluorescent reporters and the improvement of detection sensitivity. Without any nucleic acid amplification, the tandem CRISPR system reached a detection limit of 31 copies/μL (Figure 5b) and correctly identified the nasal swab samples from positive patients (Figure 5c). Moreover, the utilization of multiple crRNAs toward different regions of the virus can safeguard against the potential false-negative results due to viral mutations.
Figure 5.

CRISPR/Cas13-based detection strategies. a. Schematic of tandem CRISPR/Cas13 system to detect SARS-CoV-2. b. The detection sensitivity of tandem CRISPR system to different concentrations of SARS-CoV-2 RNA. c. The detection performance of tandem CRISPR system on patient nasopharyngeal swabs. (Reproduced with permission from ref. [70], Copyright 2021, Elsevier) d. Design of CRISPR/Cas13-based multiplexed detection of pathogenic bacteria using dinucleotide preferences of Cas13 nucleases. e-f. Multiplexed detection of S. aureus thermonuclease and P. aeruginosa acyltransferase with LwaCas13a and PsmCas13b, respectively. (Reproduced with permission from ref. [71], Copyright 2018, American Association for the Advancement of Science)
Multiplexed detection.
In contrast to the indiscriminate trans-cleavage activity of Cas12, the Cas13 nucleases have unique dinucleotide preferences for trans-cleavage. Recent studies found that the trans-cleavage activities of LwaCas13a, CcaCas13b, and PsmCas13b could all be independently monitored with dinucleotide reporters of AA, AU, and UA, respectively (Figure 5d). Three dinucleotide reporters were modified with different fluorophores (FAM, TEX, and Cy5) to achieve three-channel signal output. In the presence of targets, the active Cas13 nucleases cleaved the preferred dinucleotide reporters to generate fluorescent signals in different channels.[71] Taking advantage of these dinucleotide preferences of Cas13 nucleases, a multiplexed detection assay with two fluorescent channels was explored for the simultaneous detection of Pseudomonas aeruginosa acyltransferase gene and the Staphylococcus aureus thermonuclease gene (Figure 5e–f). Impressively, the detection limit of this assay reached as low as 2 aM, demonstrating the high sensitivity of Cas13-based multiplexed detection assay and its capability for precise and reliable detection of infectious disease.
4.4. CRISPR/Cas14 System
As a compact type V nuclease, Cas14 shares a similar RuvC domain with Cas12 nuclease. The main feature of Cas14 is that it recognizes ssDNA without PAM, which serves as a double-edged sword in the development of the detection strategies for infectious diseases. Since it is independent of the PAM sequence, the design of target sequences is much more flexible than other Cas nucleases, allowing the detection of single-nucleotide polymorphisms (SNP) to be more feasible. However, the genetic code of most organisms in nature is dsDNA or RNA, which limits the applications of the CRISPR/Cas14-based detection systems.
PAM independent detection.
The PAM-independent characteristic provides the Cas14 enzyme with the ability for highly reliable SNP recognition. As shown in Figure 6a, a Cas14-mediated ssDNA targeting CRISPR system (Cas14-DETECTR) was designed to detect the human HERC2 gene, which contained a single-nucleotide difference between brown-eyed and blue-eyed individuals.[70] To generate the ssDNA for the Cas14 nuclease, the target gene was amplified by a phosphorothioate (PT)-modified primer, and then the T7 exonuclease was used to degrade the non-PT modified DNA strand (Figure 6a–b). When designing the crRNA toward the blue-eyed sequence, the Cas14-DETECTR showed superior SNP recognition ability with almost no background signal interference, while the Cas12-DETECTR failed to distinguish between two ssDNA sequences (Figure 6c). These results demonstrated that the Cas14-DETECTR expanded the toolbox for PAM-independent detection of SNP.
Figure 6.

CRISPR/Cas14-based detection strategies. a. Principle of CRISPR/Cas14-DETECTR for the detection of SNP. b. The effect of different concentration T7 exonuclease in CRISPR/Cas14-DETECTR. c. Comparison of the sensitivity of Cas14a-DETECTR and Cas12a-DETECTR for the detection of SNP in HERC2 gene. (Reproduced with permission from ref. [70]., Copyright 2018, American Association for the Advancement of Science) d. Design of aptamer-assisted CRISPR/Cas14 system for the detection of bacteria. e. The detection sensitivity of S. aureus using the aptamer-assisted CRISPR/Cas14 system. f. S.aureus. detection in clinical samples using the aptamer-assisted CRISPR/Cas14 system (Reproduced with permission from ref. [72], Copyright 2022, Elsevier)
Aptamer-assisted CRISPR detection.
They are excellent recognition elements for biosensing application due to high flexibility and specificity, offering alternative options for the design of CRISPR/Cas14-based biosensors[72] As shown in Figure 6d, aptamer-assisted Cas14 biosensors can be applied to detect live Staphylococcus aureus (S. aureus).[73] The aptamer and blocker hybridized to form a duplex strand DNA. In the presence of S. aureus, the aptamer binds specifically to the active protein molecules on the surface of live bacterial cell, releasing the blocker to trigger the trans-cleavage activity of the CRISPR/Cas14a system. With the assistance of the aptamer and blocker, this strategy simplified the detection process without the need of generating bacterial ssDNA through a series of tedious amplification and enzymatic digestion. By correlating the fluorescence intensity with bacterial concentration, it can distinguish live S.aureus with a detection limit of 400 CFU/mL (Figure 6e), and 100% accuracy in the clinic samples (Figure 6f).
5. Signal Transduction Mechanisms
Signal transduction is an important step to enable the output of sufficient signals for the detection of infectious diseases. So far, various signal transducers have been developed for CRISPR-based biosensors, including colorimetry, fluorescence, and electrochemistry. The following section discusses the most popular signal transduction mechanisms, each categorized based on their unique mechanisms.
5.1. Colorimetry
Due to their simplicity and visual readability, colorimetric biosensors have received wide attention for the point-of-care testing of infectious diseases. As optical sensors, colorimetric biosensors mainly rely on the color change caused by the target analytes. In recent years, the integration of colorimetric biosensors with smartphones has facilitated the quantification of color changes in the field. Taking the outstanding advantages of colorimetric assays, CRISPR-based colorimetric biosensors have been developed to detect infectious diseases.
Gold Nanoparticles (AuNPs).
Due to the localized surface plasma resonance (LSPR) effect, AuNPs have been widely used as ideal probes for colorimetric biosensors. By changing the distances between AuNPs, the solution can display a range of colors from red to blue.[34] Based on this distinct optical property, AuNPs have been applied as the signal transducer for CRISPR-based colorimetric biosensors. As shown in Figure 7a, ssDNA-modified AuNPs remained stabilized via the strong electrostatic interactions between AuNPs.[74] The addition of ssDNA crosslinkers can shorten the distance between AuNPs, changing the solution color from red to purple. In the presence of the Salmonella-associated target gene (invA), the CRISPR/Cas12 system was activated to degrade ssDNA crosslinkers into short oligo pieces. The degraded ssDNA crosslinkers failed to aggregate well-dispersed AuNPs, remaining the red color for the solution. The dispersed and crosslinker-induced aggregated AuNPs are characterized using transmission electron microscopy (TEM) (Figure 7b). Using this strategy, the detection limit of Salmonella was as low as 10 CFU/mL, which can be observed using naked eyes and measured using a spectrophotometer (Figure 7c–d). Similar CRISPR-based colorimetric biosensors have been applied to detect various infectious diseases.[75, 76] In addition to AuNPs, other plasmonic nanomaterials (e.g., silver nanoparticles, silver nanoprisms, gold nanorods, and gold nanostars) could also be combined with various CRISPR systems to detect infectious diseases.
Figure 7.

CRISPR-based colorimetric biosensors to detect infectious diseases. a. Principle of AuNPs-based colorimetric assay to detect Salmonella. b. TEM images of dispersed and aggregated AuNPs. c-d. Photographs and UV-vis spectra of AuNPs to detect different concentrations of Salmonella. (Reproduced with permission from ref. [74], Copyright 2021, American Chemical Society) e. Schematic illustration of LFA-based detection of SARS-CoV-2. f. Detection limit of a serial dilution of Ebola virus (EBOV) synthetic DNA, in which C and T indicate control and test line, respectively. g. Strip reader APP to analyze diagnostic results. The raw image data are sent to a backend server that runs the signal detection algorithm and returns the binary and semi-quantitative predictions of each strip. (Reproduced with permission from ref. [77], Copyright 2020, Springer Nature)
Lateral Flow Assay (LFA).
Over the past two decades, LAF has become one of the most widely used devices for the detection of infectious diseases. Instead of using antibodies as recognition elements, DNA hybridization combined with CRISPR systems has been developed in various infectious disease detection assays. As shown in Figure 7e, the control and test lines were modified with streptavidin and anti-rabbit antibodies, respectively. In the absence of Ebola virus (EBOV) analytes, the undamaged biotin-ssRNAFAM probes could bind the functional AuNPs (AuNPs conjugated with anti-FAM antibody) to streptavidin on the control line, resulting in a red band. In contrast, once the biotin-ssRNA-FAM probes were cleaved by the activated CRISPR/Cas13 system, the functional AuNPs would be captured by the anti-rabbit antibodies on the test line, indicating the presence of target analytes. Combined with RT-RPA and transcription of EBOV RNA, this assay allowed to detect EBOV down to 10 viral copies per uL (Figure 7f).[77] In addition, a smartphone app (HandLens) has been developed to interpret the colorimetric results and provided semi-quantification of the target viral concentration. As shown in Figure 7g, the smartphone APP can capture and analyze an image of one or more lateral flow strips for the semi-quantification of viral concentrations. This assay detected real samples with 93% accuracy, 91% sensitivity, and 100% specificity from a total of 21 strips.
5.2. Fluorescence
Due to their high sensitivity, fluorescent biosensors have been widely developed for the detection of various analytes. The concepts of most fluorescent biosensors are based on either fluorescence quenching or recovery/enhancement that is induced by the target analytes. To date, many fluorescent biosensors have been introduced to CRISPR systems for the detection of infectious diseases. Organic fluorophores (also known as fluorescent dye, a size under 1 nm) and inorganic fluorescent nanomaterials (a size ranges from 1 nm to 100 nm) are two of the most commonly used signaling transduction elements in CRISPR-based fluorescent biosensors.
Fluorophores.
The typical fluorescence probes for CRISPR-based biosensors are short ssDNA or ssRNA labeled with fluorophores and quenchers on each end, named ssDNA/RNA-FQ probes. Due to the short sequence of the ssDNA/ssRNA, the fluorophores are close to the quenchers, resulting in fluorophore quenching. After the trans-cleavage activity from CRISPR system toward ssDNA/RNA-FQ probes, fluorophores and quenchers are separated, resulting in fluorescence recovery. Common fluorophores include FAM, TEX, and Cy3. Instead of ssDNA/ssRNA fluorescence probes, a few studies are focusing on the development of dsDNA fluorescence probes for CRISPR systems.[78–80] As shown in Figure 8a, dsDNA reporters have been developed to expand the reporter toolbox for CRISPR/Cas12-based biosensors.[80] In this design, the fluorophore (FAM) and quenchers (Iowa Black FRET) were modified at opposite ends of each strand, respectively. In the presence of an activated Cas12 nuclease, the dsDNA reporter was degraded by the trans-cleavage activity, leading to an increase in fluorescence signal. Compared to the traditional linear ssDNA reporters, the dsDNA reporters showed a more stable fluorescence background that did not fluctuate significantly over time (Figure 8b). Under the optimized condition, the Cas12-based biosensor with dsDNA reporter was used to detect tobacco curly shoot virus (TCSV) DNA and hepatitis B virus (HepBV) DNA. This CIRSPR-based biosensing method has a significant signal response toward 50 pM targets, proving that dsDNA can broaden the fluorescence reporter library for CRISPR/Cas systems (Figure 8c).
Figure 8.

CRISPR-based fluorescent biosensors to detect infectious diseases. a. Design of ssDNA and dsDNA reporters for CRISPR trans-cleavage activity. b. Comparison of fluorescence background of ssDNA and dsDNA reporter probes. c. CRISPR-based fluorescence detection of tobacco curly shoot virus (TCSV) and hepatitis B virus (HepBV). (Reproduced with permission from ref. [80], Copyright 2022, American Chemical Society) d. Schematic illustration of QD-HP reporter for the detection of nucleic acids (DNA or RNA). Prior to the trans-cleavage of CRISPR systems, the Qdot is quenched by the surrounding Cy2 dye through FRET. Upon cleavage, the dyes are released and the Qdot fluorescence is recovered. e-f. Plots of the normalized PL ratio toward the target DNA and RNA concentrations, respectively. (Reproduced from ref. [82], Copyright 2020, American Chemical Society)
Fluorescent Nanomaterial.
Given the advantages of easy synthesis, photostability, and low fluorescent background, fluorescent nanomaterials have been used as ideal probes for fluorescence biosensors, including copper nanoclusters (CuNCs), upconversion nanoparticles (UCNPs), quantum dots (QDs), and carbon dots (CDs).[81] Among these nanomaterials, QDs offer excellent optical and electronic performances, including stable fluorescence intensity, high quantum yields, and a narrow emission spectrum. As shown in Figure 8d, a QD-nucleic acid hairpin (QD-HP) probe was developed to detect nucleic acids.[82] A peptide-peptide nucleic acid (peptide-PNA) was designed to hybrid with fluorescence-labeled DNA or RNA hairpins, and the resulting unit can self-assemble on the surface of ZnS-coated QDs. Based on the Förster resonance energy transfer (FRET), QDs are severed as an ideal donor to a fluorescence dye (Cy3). In the presence of an activated CRISPR system, the hairpin nucleic acids can be cleaved and released, enabling ratiometric detection of target DNA or RNA. In this strategy, the developed probes were used to detect DNA and RNA with a detection limit of 40 pM and 100 pM, respectively (Figure 8e–f).
5.3. Electrochemistry
Because of their easy operation, rapid signal output, and sensitive response, electrochemical biosensors are another powerful tool to detect infectious diseases. In most cases, the change of electrical signals (potential or current) is due to the changes in electron transfer on the electrode surface, which can be correlated to the analyte concentration. To date, several CRISPR systems (including Cas9, Cas12, and Cas13) have been used to develop electrochemical biosensors for the detection of infectious diseases.
Electrochemistry.
In CRISPR-based electrochemical biosensors, the electrode surfaces are usually modified with oligonucleotides labeled with redox labels, such as methylene blue (MB). The distance between the redox label and the electrode surface leads to changes in the electron transfer efficiency, which can be measured by electrochemical performance. As shown in Figure 9a, the electrodes were modified with MB-labeled hairpin DNA (MB-hpDNA).[83] Due to the specific design, the electrochemical active compounds (MB) were close to the electrodes, shortening the electron tunneling distance between MB and the electrode surface. With activated CRISPR/Cas12, the hpDNA was degraded and the MBs were detached from the electrode surfaces, resulting in a decrease of electrical response. Under the optimal conditions, the electrochemical biosensor was employed to detect human papillomavirus (HPV)-related DNA. As shown in Figure 9b, the differential pulse voltammetry (DPV) curves showed a gradual decrease with increased concentration of the target DNA. This strategy allowed for the detection of unamplified target DNA at concentrations as low as 30 pM within 60 min, and the dynamic range was from 50 pM to 100 nM (Figure 9c).
Figure 9.

CRISPR-based electrochemical biosensors to detect infectious diseases. a. Schematic illustration of CRISPR-based electrochemical biosensors to detect nucleic acids (MB: methylene blue). b. Typical differential pulse voltammetry (DPV) curve responding to different DNA concentrations. c. The calibrated ΔI % towards target DNA concentration. (Reproduced with permission from ref. [83], Copyright 2020, American Chemical Society) d. Schematic illustration of graphene-field effect transistor (g-FET) to detect nucleic acids. e. Representative of g-FET before and after the trans-cleavage of CRISPR system. The source-drain current (Ids) were monitored while keeping a constant source-drain voltage (Vds = 100 mV). (Reproduced with permission from ref. [85], Copyright 2022, Wiley-VCH GmbH)
Field Effect Transistor (FET).
FET has emerged as a promising technology for the detection of infectious diseases. A typical FET contains three electrodes (including gate, source, and drain electrodes), in which the gate electrode provides an electric field to control the current flow from the source electrode to the drain electrode.[84] Graphene as an exceptional semiconductor material has been widely used in FET. As shown in Figure 9d, an ultrasensitive graphene-field effect transistor (g-FET) for nucleic acids detection has been reported using CRISPR/Cas13 system.[85] Negatively charged reporters (polyU20) were modified on the surface of graphene. In the presence of the activated CRISPR/Cas13 system, the polyU20 probes were cleaved into small pieces, decreasing the number of electrons carried in the graphene channel. Consequently, a right shift of VCNP was observed, which was correlated with the target RNA concentrations (Figure 9e). In this detection strategy, clinic samples were detected, including 5 negative and 9 positive patient samples. Using a threshold of two standard deviations above the mean obtained from negative clinic samples, the developed CRISPR/Cas13 g-FET was able to distinguish between all 5 negative and 9 positive samples with 100% accuracy.
5.4. Other Signal Transduction Mechanisms
In addition to the signal transduction mechanisms described above, other novel strategies have been combined with CRISPR systems to detect infectious diseases, including chemiluminescence, surfaceenhanced Raman spectroscopy (SERS), nanopore, etc.
Surface-enhanced Raman Spectroscopy (SERS).
SERS is a Raman signal based on the enhanced localized surface plasmon resonances of noble metals (e.g., silver and gold).[86] Because of its extremely high sensitivity and rich molecular fingerprints, SERS has been used to detect a single molecule. Similarly, the combination of SERS and CRISPR/Cas systems is a promising strategy to sensitively detect infectious diseases. As shown in Figure 10a, the SERS substrate was coated with graphene oxide and triangle gold (Au) nanoflowers (GO-TANF), which can offer extremely strong electromagnetic and chemical enhancement.[87] The ssDNA-immobilized Raman probe-functionalized Au nanoparticles (RAuNPs) on the GO-TANF can provide strong SERS intensity. As a proof-of-concept, the developed RAuNP-modified GO-TANF has been demonstrated to detect the hepatitis B virus (HBV). For the detection of viral DNAs, the activated CRIPSR-Cas12 can cleave the ssDNA to detach RAuNPs from the SERS substrate, weakening the SERS intensity. Without the nucleic acid amplification, the spectra of SERS signals at different viral concertation were collected and the intensities at the Raman shift of 1656 cm−1 were plotted towards viral concentrations (Figure 10b–c). The detection limit of viral DNA was 1 aM with a dynamic range from 1 aM to 100 pM. In addition, the authors further developed a nanoarray for the multiplexed detection of three viruses, including HBV, human papillomavirus 16 (HPV-16), and HPV-18.
Figure 10.
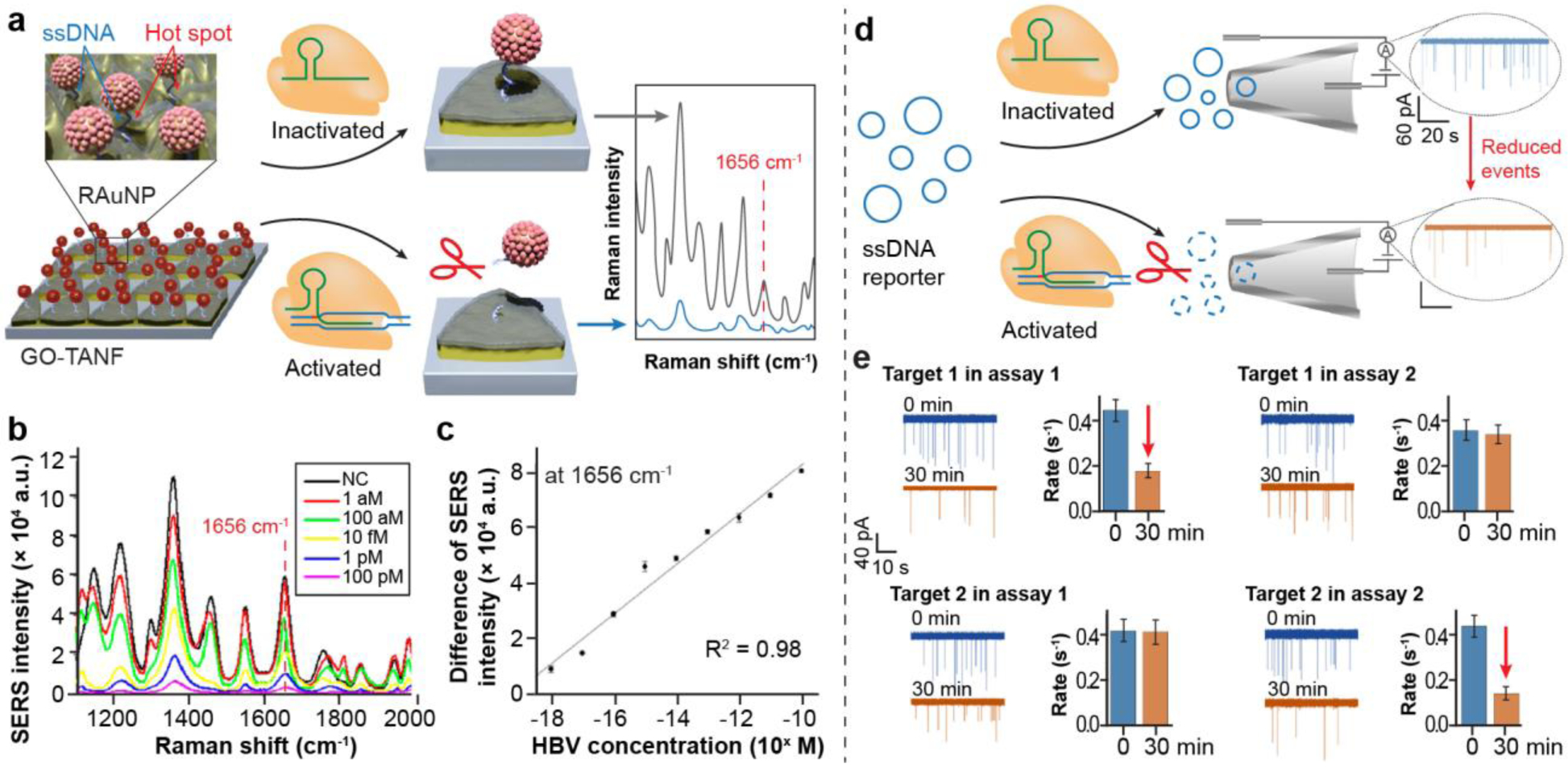
Other CRISPR-based novel biosensors to detect infectious diseases. a. Schematic illustration of the SERS-based strategy to detect viral DNA. RAuNP: Raman probe-modified AuNP. GO-TANF: graphene oxide-triangle Au nanoflower. b. Raman spectra of the RAuNP-modified GO-TANF array with varying concentrations of hepatitis B virus (HBV) DNA. c. the linear relationship of the decrease of SERS intensities towards the concentrations of HBV DNA. (Reproduced with permission from ref. [87] Copyright 2021, American Chemical Society) d. Schematic illustration of solid nanopore sensors to detect HIV. With the trans-cleavage of circular ssDNA reporter, fewer events are recorded. e. Detection specificity of HIV using CRISPR-based nanopore sensors. Only specific combinations (Assay 1 - Target 1 and Assay 2 - Target 2) lead to the degradation of circular ssDNA reporters. (Reproduced with permission from ref. [88], Copyright 2020, American Chemical Society)
Nanopore sensors.
Rapid advances in nanopore sensors have led to significant improvements in DNA sequencing. In a typical nanopore sensor, charged DNAs are electrophoretically driven through a nanoscale orifice to block the passage of ions, resulting in a dip in the current.[88] Each tip in the current indicates a reading event through the nanopore. As shown in Figure 10d, an abundance of the nanopore reading events was observed for circular ssDNA reporters without the trans-cleavage from CRISPR/Cas12 system. However, the degradation of ssDNA reporters by the activated CRISPR/Cas12 system caused a significant reduction of reading events. It is found that the CRISPR/Cas12-assisted nanopore method can detect over 10nM of target DNA within 1 h. To characterize the detection specificity, the authors designed two sets of HIV-1 DNA targets and assays, in which each assay was specific to its target (Assay 1 - Target 1 and Assay 2 - Target 2). As shown in Figure 10e, reading events were significantly reduced for target 1 in assay 1 and target 2 in assay 2, indicating the CRISPR-based nanopore sensors have a high detection specificity.
6. Conclusion and Prospective
The detection of infectious diseases has been greatly extended by the discovery of CRISPR systems, and greatly benefited by the cleavage mechanisms possessed by different CRISPR Cas nucleases. The integration of nucleic acid amplification methods with the CRISPR system is crucial for the early detection and prevention of infectious diseases, as it enables the amplification of DNA and RNA from even trace amounts. This capability is essential for accurate and sensitive detection in various scenarios, such as environmental monitoring, food safety, and clinical diagnostics. Furthermore, different signal transduction models, such as colorimetry, fluorescence, or electrochemistry, can be employed to provide the desired detection sensitivity, dynamic range, and compatibility with the intended detection platform or device. This adaptability allows the CRISPR-based biosensors to have diverse applications, from rapid in-field detection to high-throughput diagnostics. Even though substantial progress has been achieved in the development of CRISPR-based biosensors, there are still several potential directions that can be identified for further advancement (Figure 11).
Figure 11.

Future prospective in CRISPR-based biosensors.
Exploring potential properties of CRISPR Cas nucleases.
The target recognition mechanism of the CRISPR-based detection mainly depends on the properties of the Cas nuclease. The emergence of artificial intelligence, particularly tools like AlphaFold, has facilitated the prediction and analysis of protein structures.[89] By integrating this technology with protein engineering techniques, it becomes possible to modify Cas nucleases and explore their potential properties. These modifications can improve the properties of Cas nuclease, such as high-temperature resistance, PAM-site versatility, and high recognition accuracy. Therefore, the combination of artificial intelligence tools and protein engineering may open new avenues for improving and customizing the functionalities of Cas nucleases, leading to sensitive, specific, and multiplexed detection of infectious diseases.
Establishing a standardized sample pretreatment process.
While several CRISPR-based biosensors have already been developed for the detection of infectious diseases, a major challenge is that there is no standardized procedure for sample pretreatment. Different pretreatment methods are required for infectious disease detection from environmental, food, and clinical samples. The lack of standardization makes it difficult to objectively compare the performance and reliability of different detection methods. Therefore, establishing a standardized sample pretreatment procedure is essential for accurate and consistent comparisons between different CRISPR-based biosensors and to facilitate the development of rapid detection.
Detecting non-nucleic acid targets.
Currently, most CRISPR-based biosensor studies are centered around detecting nucleic acid targets. However, to expand the versatility of these biosensors, some researchers have focused on detecting non-nucleic acid targets, such as organic small molecules and proteins.[90–92] A noteworthy challenge in developing activable CRISPR-based biosensors for non-nucleic acid targets is devising effective methods to translate the target recognition process into the collateral cleavage activity of Cas enzymes. In this context, aptamers[90, 91] and bacterial allosteric transcription factors (aTFs)[92] emerge as potential solutions for enabling the detection of non-nucleic acid targets using the CRISPR system. By leveraging the unique properties of aptamers and aTFs, researchers can design innovative approaches to facilitate non-nucleic acid target detection and expand the application of CRISPR-based biosensors.
Developing amplification-free strategies.
The incorporation of nucleic acid amplification methods can significantly improve the sensitivity of CRISPR-based biosensors, but they also pose the risk of non-specific amplification and potential cross-contamination. Although several CRISPR-based biosensors have been developed for the amplification-free detection of infectious diseases, it is important to note that many of these methods are still in the laboratory validation stage.[93, 94] This is mainly because the limited number of targets in the real sample makes it difficult to achieve sensitive and reliable detection using amplification-free CRISPR-based biosensors. Addressing this limitation is crucial for advancing the practical application of amplification-free CRISPR-based biosensors in real-world scenarios.
Improving detection at a high throughput manner.
With the ongoing mutation of influenza viruses, the importance of high-throughput detection is becoming increasingly prominent. However, the vast amount of data generated from high-throughput assays presents a challenge in terms of data analysis. In this context, machine learning offers a promising solution for effective analysis of such data. The use of machine learning algorithms can improve the accuracy of results, allowing discrimination between different mutant viruses.[95] This application of machine learning in the analysis of high-throughput assay data has the potential to improve our understanding of influenza virus mutations and contribute to more effective detection and surveillance strategies.
Integrating CRISPR-based biosensors onto portable devices.
To overcome the limitations of sophisticated instrumentation for detection, the CRISPR-based methods can be integrated into portable devices, such as smartphones, portable electrochemical stations, and handheld Raman spectrometers.[96] Most of these portable devices are equipped with their own batteries, which facilitate data collection and analysis directly in the field. Furthermore, the real-time uploading of data to the cloud through network connections enables infectious disease monitoring and facilitates the establishment of risk prediction models. Taking advantage of portable devices, the accessibility and applicability of CRISPR-based biosensors can be expanded, allowing them to be deployed in resource-limited settings and remote areas where sophisticated laboratory equipment is limited.
Highlights:
The design principle of CRISPR-based biosensors are systematically introduced.
The sensing strategies for each Cas enzymes are summarized.
The signal transduction mechanisms of CRISPR-based biosensors are elaborated.
Challenges and future perspectives for infectious disease detection are discussed.
Acknowledgements
This work was supported by NIH NIGMS (R35GM147069) and USDA NIFA (2021-67017-33439).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References:
- [1].Fauci AS, Cell 124 (2006) 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Prentice T, World Health Organization (1996).
- [3].W.H. Organization, WHO Calls for Global Action on Sepsis—Cause of 1 in 5 deaths Worldwide. 2020.
- [4].Whitesides GM, Nature 442 (2006) 368. [DOI] [PubMed] [Google Scholar]
- [5].Chang D, Zakaria S, Esmaeili Samani S, Chang Y, Filipe CD, Soleymani L, Brennan JD, Liu M, Li Y, Accounts of Chemical Research 54 (2021) 3540. [DOI] [PubMed] [Google Scholar]
- [6].Chen J, Andler SM, Goddard JM, Nugen SR, Rotello VM, Chemical Society Reviews 46 (2017) 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ben Y, Fu C, Hu M, Liu L, Wong MH, Zheng C, Environmental research 169 (2019) 483. [DOI] [PubMed] [Google Scholar]
- [8].Mancuso G, Midiri A, Gerace E, Biondo C, Pathogens 10 (2021) 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ray PC, Khan SA, Singh AK, Senapati D, Fan Z, Chemical Society Reviews 41 (2012) 3193. [DOI] [PubMed] [Google Scholar]
- [10].Uddin TM, Chakraborty AJ, Khusro A, Zidan BRM, Mitra S, Emran TB, Dhama K, Ripon MKH, Gajdács M, Sahibzada MUK, Journal of infection and public health 14 (2021) 1750. [DOI] [PubMed] [Google Scholar]
- [11].van Duin D, Paterson DL, Infectious Disease Clinics 34 (2020) 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Amiri M, Bezaatpour A, Jafari H, Boukherroub R, Szunerits S, ACS sensors 3 (2018) 1069. [DOI] [PubMed] [Google Scholar]
- [13].Batt CA, Science 316 (2007) 1579. [DOI] [PubMed] [Google Scholar]
- [14].Iqbal SS, Mayo MW, Bruno JG, Bronk BV, Batt CA, Chambers JP, Biosensors and Bioelectronics 15 (2000) 549. [DOI] [PubMed] [Google Scholar]
- [15].Lin Y-H, Chen S-H, Chuang Y-C, Lu Y-C, Shen TY, Chang CA, Lin C-S, Biosensors and Bioelectronics 23 (2008) 1832. [DOI] [PubMed] [Google Scholar]
- [16].Koubova V, Brynda E, Karasova L, Škvor J, Homola J, Dostalek J, Tobiška P, Rošický J, Sensors and Actuators B: Chemical 74 (2001) 100. [Google Scholar]
- [17].Velusamy V, Arshak K, Korostynska O, Oliwa K, Adley C, Biotechnology advances 28 (2010) 232. [DOI] [PubMed] [Google Scholar]
- [18].Dos Santos WG, Biomedicine & Pharmacotherapy 136 (2021) 111272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Marraffini LA, Sontheimer EJ, Nature Reviews Genetics 11 (2010) 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E, science 337 (2012) 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pardee K, Green AA, Takahashi MK, Braff D, Lambert G, Lee JW, Ferrante T, Ma D, Donghia N, Fan M, Cell 165 (2016) 1255. [DOI] [PubMed] [Google Scholar]
- [22].Dai Y, Wu Y, Liu G, Gooding JJ, Angewandte Chemie International Edition 59 (2020) 20754. [DOI] [PubMed] [Google Scholar]
- [23].Morisaka H, Yoshimi K, Okuzaki Y, Gee P, Kunihiro Y, Sonpho E, Xu H, Sasakawa N, Naito Y, Nakada S, Nature communications 10 (2019) 5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fapohunda FO, Qiao S, Pan Y, Wang H, Liu Y, Chen Q, Lü P, Microbiological Research 259 (2022) 127000. [DOI] [PubMed] [Google Scholar]
- [25].Anders C, Niewoehner O, Duerst A, Jinek M, Nature 513 (2014) 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yan F, Wang W, Zhang J, CRISPR-Cas12 and Cas13: the lesser known siblings of CRISPR-Cas9, Cell biology and toxicology. 35 (2019) 489. [DOI] [PubMed] [Google Scholar]
- [27].Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DB, Kellner MJ, Regev A, Nature 550 (2017) 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Harrington LB, Burstein D, Chen JS, Paez-Espino D, Ma E, Witte IP, Cofsky JC, Kyrpides NC, Banfield JF, Doudna JA, Science 362 (2018) 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen JS, Ma EB, Harrington LB, Da Costa M, Tian XR, Palefsky JM, Doudna JA, Science 360 (2018) 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Myhrvold C, Freije CA, Gootenberg JS, Abudayyeh OO, Metsky HC, Durbin AF, Kellner MJ, Tan AL, Paul LM, Parham LA, Science 360 (2018) 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, Zhang F, Science 360 (2018) 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Harrington LB, Burstein D, Chen JS, Paez-Espino D, Ma E, Witte IP, Cofsky JC, Kyrpides NC, Banfield JF, Doudna JA, Science 362 (2018) 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Guo Y, Guo L, Su Y, Xiong Y, Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology 15 (2022) e1851. [DOI] [PubMed] [Google Scholar]
- [34].Saha K, Agasti SS, Kim C, Li X, Rotello VM, Chemical Reviews 112 (2012) 2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhou W, Hu L, Ying L, Zhao Z, Chu PK, Yu X-F, Nature communications 9 (2018) 5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tian B, Minero GAS, Fock J, Dufva M, Hansen MF, Nucleic Acids Research 48 (2020) e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang M, Wang H, Wang H, Wang F, Li Z, Analytical Chemistry 93 (2021) 7942. [DOI] [PubMed] [Google Scholar]
- [38].You J, Park H, Lee H, Jang K, Park J, Na S, Biosensors and Bioelectronics 224 (2023) 115078. [DOI] [PubMed] [Google Scholar]
- [39].Gupta N, Augustine S, Narayan T, O’Riordan A, Das A, Kumar D, Luong JHT, Malhotra BD, Biosensors 11 (2021) 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jiao C, Sharma S, Dugar G, Peeck NL, Bischler T, Wimmer F, Yu Y, Barquist L, Schoen C, Kurzai O, Sharma CM, Beisel CL, Science 372 (2021) 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ma L, Peng L, Yin LJ, Liu GZ, Man SL, Acs Sensors 6 (2021) 2920. [DOI] [PubMed] [Google Scholar]
- [42].Kasputis T, Hilaire SS, Xia K, Chen J, ACS Food Science & Technology (2022).
- [43].Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T, Nucleic Acids Research 28 (2000) e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Park BJ, Park MS, Lee JM, Song YJ, Biosensors 11 (2021) 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rezaei M, Bazaz SR, Rad DM, Shimoni O, Jin DY, Rawlinson W, Warkiani ME, Biosensors 11 (2021) 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang Y, Li JQ, Li SJ, Zhu X, Wang XX, Huang JF, Yang XG, Tai J, Microchimica Acta 188 (2021) 1.33386503 [Google Scholar]
- [47].Ho NRY, Lim GS, Sundah NR, Lim D, Loh TP, Shao H, Nature Communications 9 (2018) 3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vatankhah M, Azizi A, Langeroudi AS, Azimi SA, Khorsand I, Kerachian MA, Motaei J, Critical Reviews in Clinical Laboratory Sciences 58 (2021) 225. [DOI] [PubMed] [Google Scholar]
- [49].Aman R, Marsic T, Rao GS, Mahas A, Ali Z, Alsanea M, Al-Qahtani A, Alhamlan F, Mahfouz M, Frontiers in Bioengineering and Biotechnology 9 (2022) 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang HY, Wu Q, Yan C, Xu JG, Qin XS, Wang J, Chen W, Yao L, Huang L, Qin PZ, Sensors and Actuators B-Chemical 369 (2022) 132293. [Google Scholar]
- [51].Li S, Wang XC, Yu YH, Cao SG, Liu J, Zhao PP, Li JH, Zhang XC, Li X, Zhang N, Sun M, Cao LL, Gong PT, Parasites & Vectors 15 (2022) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].He YW, Jia F, Sun YX, Fang WH, Li YB, Chen JH, Fu YC, Sensors and Actuators B-Chemical 369 (2022) 132301. [Google Scholar]
- [53].Xu JH, Ma JF, Li YW, Kang L, Yuan B, Li SQ, Chao J, Wang LH, Wang JL, Su S, Yuan Y, Sensors and Actuators B-Chemical 364 (2022) 131864. [Google Scholar]
- [54].Zhou T, Huang MQ, Lin JQ, Huang R, Xing D, Analytical Chemistry 93 (2021) 2038. [DOI] [PubMed] [Google Scholar]
- [55].Yang H, Chen J, Yang S, Zhang T, Xia X, Zhang K, Deng S, He G, Gao H, He Q, Analytical Chemistry 93 (2021) 12602. [DOI] [PubMed] [Google Scholar]
- [56].Zhu Z, Guo Y, Wang C, Yang Z, Li R, Zeng Z, Li H, Zhang D, Yang L, Biosensors and Bioelectronics 228 (2023) 115179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sun X, Wang Y, Zhang L, Liu S, Zhang M, Wang J, Ning B, Peng Y, He J, Hu Y, Analytical chemistry 92 (2020) 3032. [DOI] [PubMed] [Google Scholar]
- [58].Reid MS, Le XC, Zhang H, Angewandte Chemie International Edition 57 (2018) 11856. [DOI] [PubMed] [Google Scholar]
- [59].Shi C, Liu Q, Ma C, Zhong W, Analytical Chemistry 86 (2014) 336. [DOI] [PubMed] [Google Scholar]
- [60].Wang WH, Liu JH, Wu LA, Ko CN, Wang XL, Lin CK, Liu JQ, Ling LS, Wang J, Analytica Chimica Acta 1195 (2022) 339479. [DOI] [PubMed] [Google Scholar]
- [61].Huang D, Qian JJ, Shi ZW, Zhao JR, Fang MJ, Xu ZN, Acs Synthetic Biology 9 (2020) 3114. [DOI] [PubMed] [Google Scholar]
- [62].Ke XX, Ou YJ, Lin Y, Hu T, Biosensors & Bioelectronics 212 (2022) 114428. [DOI] [PubMed] [Google Scholar]
- [63].Song J, Kim S, Kim HY, Hur KH, Kim Y, Park HG, Nanoscale 13 (2021) 7193. [DOI] [PubMed] [Google Scholar]
- [64].Chen M, Zhang J, Peng Y, Bai J, Li S, Han D, Ren S, Qin K, Zhou H, Han T, Wang Y, Gao Z, Biosensors and Bioelectronics 218 (2022) 114792. [DOI] [PubMed] [Google Scholar]
- [65].Yang Y, Yang J, Gong F, Zuo P, Tan Z, Li J, Xie C, Ji X, Li W, He Z, Sensors and Actuators B: Chemical 367 (2022) 132158. [Google Scholar]
- [66].Huang M, Zhou X, Wang H, Xing D, Analytical chemistry 90 (2018) 2193. [DOI] [PubMed] [Google Scholar]
- [67].Zhang Y, Qian L, Wei W, Wang Y, Wang B, Lin P, Liu W, Xu L, Li X, Liu D, ACS Synthetic Biology 6 (2017) 211. [DOI] [PubMed] [Google Scholar]
- [68].Hu M, Qiu Z, Bi Z, Tian T, Jiang Y, Zhou X, Proceedings of the National Academy of Sciences 119 (2022) e2202034119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kempton HR, Goudy LE, Love KS, Qi LS, Molecular cell 78 (2020) 184. [DOI] [PubMed] [Google Scholar]
- [70].Fozouni P, Son S, de León Derby MD, Knott GJ, Gray CN, D’Ambrosio MV, Zhao C, Switz NA, Kumar GR, Stephens SI, Cell 184 (2021) 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, Zhang F, Science 360 (2018) 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Tombelli S, Minunni M, Mascini M, Biosensors and Bioelectronics 20 (2005) 2424. [DOI] [PubMed] [Google Scholar]
- [73].Wei Y, Tao Z, Wan L, Zong C, Wu J, Tan X, Wang B, Guo Z, Zhang L, Yuan H, Biosensors and Bioelectronics 211 (2022) 114282. [DOI] [PubMed] [Google Scholar]
- [74].Ma L, Peng L, Yin L, Liu G, Man S, ACS Sensors 6 (2021) 2920. [DOI] [PubMed] [Google Scholar]
- [75].Jiang Y, Hu M, Liu A-A, Lin Y, Liu L, Yu B, Zhou X, Pang D-W, ACS sensors 6 (2021) 1086. [DOI] [PubMed] [Google Scholar]
- [76].ChrisáLe X, Chemical Communications 57 (2021) 6871. [DOI] [PubMed] [Google Scholar]
- [77].Barnes KG, Lachenauer AE, Nitido A, Siddiqui S, Gross R, Beitzel B, Siddle KJ, Freije CA, Dighero-Kemp B, Mehta SB, Carter A, Uwanibe J, Ajogbasile F, Olumade T, Odia I, Sandi JD, Momoh M, Metsky HC, Boehm CK, Lin AE, Kemball M, Park DJ, Branco L, Boisen M, Sullivan B, Amare MF, Tiamiyu AB, Parker ZF, Iroezindu M, Grant DS, Modjarrad K, Myhrvold C, Garry RF, Palacios G, Hensley LE, Schaffner SF, Happi CT, Colubri A, Sabeti PC, Nature Communications 11 (2020) 4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Smith CW, Nandu N, Kachwala MJ, Chen Y-S, Uyar TB, Yigit MV, Biochemistry 59 (2020) 1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Liu S, Xie T, Huang Z, Pei X, Li S, He Y, Tong Y, Liu G, Sensors and Actuators B: Chemical 373 (2022) 132746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Smith CW, Kachwala MJ, Nandu N, Yigit MV, ACS synthetic biology 10 (2021) 1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wang G, Wang Y, Chen L, Choo J, Biosensors and Bioelectronics 25 (2010) 1859. [DOI] [PubMed] [Google Scholar]
- [82].Green CM, Spangler J, Susumu K, Stenger DA, Medintz IL, Díaz SA, ACS Nano 16 (2022) 20693. [DOI] [PubMed] [Google Scholar]
- [83].Zhang D, Yan Y, Que H, Yang T, Cheng X, Ding S, Zhang X, Cheng W, ACS Sensors 5 (2020) 557. [DOI] [PubMed] [Google Scholar]
- [84].Weng Z, You Z, Yang J, Mohammad N, Lin M, Wei Q, Gao X, Zhang Y, Angewandte Chemie International Edition 62 (2023): e202214987. [DOI] [PubMed] [Google Scholar]
- [85].Li H, Yang J, Wu G, Weng Z, Song Y, Zhang Y, Vanegas JA, Avery L, Gao Z, Sun H, Chen Y, Dieckhaus KD, Gao X, Zhang Y, Angewandte Chemie International Edition 61 (2022) e202203826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Chen J, Li Y, Huang K, Wang P, He L, Carter KR, Nugen SR, ACS Applied Materials & Interfaces 7 (2015) 22106. [DOI] [PubMed] [Google Scholar]
- [87].Choi J-H, Shin M, Yang L, Conley B, Yoon J, Lee S-N, Lee K-B, Choi J-W, ACS Nano 15 (2021) 13475. [DOI] [PubMed] [Google Scholar]
- [88].Nouri R, Jiang Y, Lian XL, Guan W, ACS Sensors 5 (2020) 1273. [DOI] [PubMed] [Google Scholar]
- [89].Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, Nucleic acids research 50 (2022) 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Xiong Y, Zhang J, Yang Z, Mou Q, Ma Y, Xiong Y, Lu Y, Journal of the American Chemical Society 142 (2019) 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Samanta D, Ebrahimi SB, Ramani N, Mirkin CA, Journal of the American Chemical Society 144 (2022) 16310. [DOI] [PubMed] [Google Scholar]
- [92].Liang M, Li Z, Wang W, Liu J, Liu L, Zhu G, Karthik L, Wang M, Wang K-F, Wang Z, Nature communications 10 (2019) 3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Bruch R, Baaske J, Chatelle C, Meirich M, Madlener S, Weber W, Dincer C, Urban GA, Advanced materials 31 (2019) 1905311. [DOI] [PubMed] [Google Scholar]
- [94].Li H, Xie Y, Chen F, Bai H, Xiu L, Zhou X, Guo X, Hu Q, Yin K, Chemical Society Reviews 52 (2023) 361. [DOI] [PubMed] [Google Scholar]
- [95].Ackerman CM, Myhrvold C, Thakku SG, Freije CA, Metsky HC, Yang DK, Ye SH, Boehm CK, Kosoko-Thoroddsen T-SF, Kehe J, Nature 582 (2020) 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Qiu M, Zhang J, Pang L, Zhang Y, Zhao Q, Jiang Y, Yang X, Man C, Trends in Food Science & Technology 129 (2022) 364. [Google Scholar]