

ABSTRACT
There are few treatments that slow neurodegeneration in Alzheimer’s disease (AD), and while therapeutic antibodies are being investigated in clinical trials for AD treatment, their access to the central nervous system is restricted by the blood–brain barrier. This study investigates a bispecific modular fusion protein composed of gantenerumab, a fully human monoclonal anti- amyloid-beta (Aβ) antibody under investigation for AD treatment, with a human transferrin receptor 1-directed Brainshuttle™ module (trontinemab; RG6102, INN trontinemab). In vitro, trontinemab showed a similar binding affinity to fibrillar Aβ40 and Aβ plaques in human AD brain sections to gantenerumab. A single intravenous administration of trontinemab (10 mg/kg) or gantenerumab (20 mg/kg) to non-human primates (NHPs, Macaca fascicularis), was well tolerated in both groups. Immunohistochemistry indicated increased trontinemab uptake into the brain endothelial cell layer and parenchyma, and more homogeneous distribution, compared with gantenerumab. Brain and plasma pharmacokinetic (PK) parameters for trontinemab were estimated by nonlinear mixed-effects modeling with correction for tissue residual blood, indicating a 4–18-fold increase in brain exposure. A previously developed clinical PK/pharmacodynamic model of gantenerumab was adapted to include a brain compartment as a driver of plaque removal and linked to the allometrically scaled above model from NHP. The new brain exposure-based model was used to predict trontinemab dosing regimens for effective amyloid reduction. Simulations from these models were used to inform dosing of trontinemab in the first-in-human clinical trial.
KEYWORDS: Alzheimer’s disease, gantenerumab, Brainshuttle™, monoclonal antibody, blood–brain barrier, pharmacokinetics
Introduction
Currently, the availability of disease-modifying pharmacologic treatments that slow neurodegeneration in Alzheimer’s disease (AD) is extremely limited.1,2 The recent US Food and Drug Administration (FDA) approvals of aducanumab and lecanemab3,4 mark a new era in the therapy of AD, but the observed clinical benefits with these new treatments are considered modest.5,6 Whilst small-molecule therapeutics can readily access the brain tissue after oral administration, small-molecule treatments for AD to date have only addressed cognitive, functional, and behavioral symptoms of the disease, without modifying the disease course.7,8 Therapeutic proteins offer access to fundamentally different modes of action; for example, by engaging the brain’s immune system in fighting the disease. Multiple therapeutic antibodies are currently under clinical investigation, which have mechanisms of action (MoAs) involving targeted amyloid-beta (Aβ) binding, recruitment of microglia via the antibody’s wild-type Fc domain binding to Fc gamma receptors, and Fc-mediated antibody – Aβ complex clearance.9,10 However, access to the central nervous system (CNS) for such therapeutic antibodies is restricted by the blood–brain barrier (BBB), a specialized feature of brain endothelial cells in conjunction with pericytes, astrocytes, and neuronal processes, and this likely contributes to the modest clinical benefits observed with current modalities.11–13 Only a small fraction of systemically circulating therapeutic antibodies can enter the brain to reach target sites in the CNS,14,15 and achieving adequate CNS exposure of antibody therapeutics is one of the largest hurdles to overcome for the successful development of antibody-based, disease-modifying therapies in AD and other CNS disorders.
Antibodies that can bind to endothelial transmembrane proteins expressed at the BBB have previously been investigated as ways to facilitate transport of molecules into the CNS.16–19 Transmembrane proteins used to transport antibodies across the BBB to date include the transferrin receptor 1 (TfR1),16,20 the insulin receptor (IR),17,21 TMEM30A,18 Glut-1, Bsg, CD98hc, and others.19 For TfR1 and IR, brain exposure data in non-human primates (NHPs) have been published.17,20,21 For the other receptors, only rodent data are available.18,19 However, none of these transmembrane proteins are unique to the BBB, and binding with subsequent internalization in other tissues constitutes an additional systemic elimination pathway referred to as target-mediated drug disposition (TMDD). TMDD can impair availability of the antibody for brain trafficking, as a function of binding affinity to the membrane receptor.22,23 In addition, binding to a receptor outside of the target tissue can have implications for safety, independent of the MoA against the pharmacologic target. For example, antibodies against mouse TfR1 have been shown to cause severe clinical signs in treated mice if these antibodies were able to bind and activate effector cell Fc-gamma receptors or complement receptors.24 Establishing a safe therapeutic window for such treatments that takes into account the therapeutic target, as well as the transmembrane receptor and its wider distribution within the body, is therefore a key consideration for advancing brain-targeted antibody constructs into clinical development. This, in turn, requires a robust working hypothesis for efficacious clinical dosing regimens.
We have previously reported a TfR1-based technology developed for enhanced delivery of biotherapeutics to the brain using a proprietary “2 + 1” format.25 The “2 + 1” format combines bivalent target binding of the “cargo” antibody to enable high-affinity target engagement, with monovalent binding to TfR1 to maintain normal transcytosis.25 By coupling the TfR1-binding module to the Fc C-terminus of the cargo antibody, the antigen-binding fragment (Fab) arms of the cargo antibody extend away from the TfR1-expressing cell surface, thereby preventing Fc gamma-receptor oligomerization on the effector cell due to steric hindrance.26 The resulting suppression of Fc effector function has been shown to improve the safety profile of the molecule in vivo, whereby the 2 + 1 format molecule was not associated with the systemic adverse effects seen with constructs lacking one or more Fab arms.26 However, productive Fc gamma receptor engagement has been demonstrated when the cargo antibody binds to its target, including amyloid plaques.26 Using the 2 + 1 format, the TfR1-directed Brainshuttle™ coupled to an Aβ antibody has been shown to maintain the antibody’s capacity to activate Fc gamma receptor-enhanced phagocytosis of microglia and thus clearance of amyloid plaques from the brains in AD-model mice.25,26
This TfR1-based technology was applied to develop a new human therapeutic antibody construct for AD, trontinemab, based on gantenerumab complementary-determining regions (CDRs) and a novel human/cynomolgus monkey cross-reactive TfR1-binding shuttle module. Gantenerumab is a fully human, anti-Aβ immunoglobulin G1 (IgG1) that binds with high affinity to aggregated forms of Aβ.10 It is thought to remove Aβ via microglia-mediated phagocytosis and has shown robust Aβ plaque removal in clinical trials (NCT01224106, NCT02051608).10–27–30 The recently released results of the GRADUATE Phase 3 studies evaluating the efficacy and safety of gantenerumab (NCT03444870; NCT03443973) showed a slowing of clinical decline, but the studies did not meet their primary endpoints of slowing clinical decline in people with early Alzheimer’s disease.31,32
Here, we describe the characterization and preclinical proof-of-concept studies of the receptor-mediated transcytosis, across the BBB, of the clinical candidate trontinemab in NHPs (cynomolgus macaque, Macaca fascicularis). Since the NHP does not allow a pharmacokinetic (PK)/pharmacodynamic (PD) or target-engagement assessment due to the absence of Aβ plaques, the data of quantitative brain concentration over time from this study were used to build a translational PK/PD model, back-translating the clinically established plaque removal profile of gantenerumab. The model was expanded into a model-informed drug development (MIDD) framework supporting the advancement of trontinemab into clinical development.33 The approach used in this study allowed for dose justification for the first-in-human, single-ascending dose, Phase 1 clinical trial (NCT04023994), and further helped to define a platform for model-based dose selection in trials at a later stage (NCT04639050).
Results
Development of human TfR1-directed trontinemab
The trontinemab molecule has been designed as a 2 + 1 bispecific monoclonal antibody, binding bivalently to Aβ plaques and monovalently to human TfR1 (Figure 1a–c). This molecule bound to fibrillar Aβ (Figure 1b) with a similar affinity and to Aβ plaques on human AD brain sections (Figure 1d) with a pattern comparable to that of gantenerumab, suggesting that the gantenerumab paratope maintained its binding affinity and selectivity to various species of Aβ aggregation in trontinemab. The TfR1-binding, cross-Fab moiety of trontinemab specifically bound to a preformed complex of the recombinant extracellular domain of the human TfR1 and holo-transferrin in a monovalent binding mode (Figure 1c), without disrupting the endogenous ligand – TfR1 interaction. The measured affinity of the 2 + 1 bispecific monoclonal antibody to cynomolgus TfR1 showed less than a two-fold difference compared with the affinity to human TfR1. Gantenerumab and trontinemab activated effector cells equally well, as demonstrated by induced cytokine secretion from pre-activated monocytes in the presence of Aβ (Figure 1e). A monovalent molecular shuttle with identical architecture had previously been shown to efficiently remove plaques in a mouse model of AD, at dose levels at which conventional mAbs had not shown noteworthy plaque clearance.25
Figure 1.
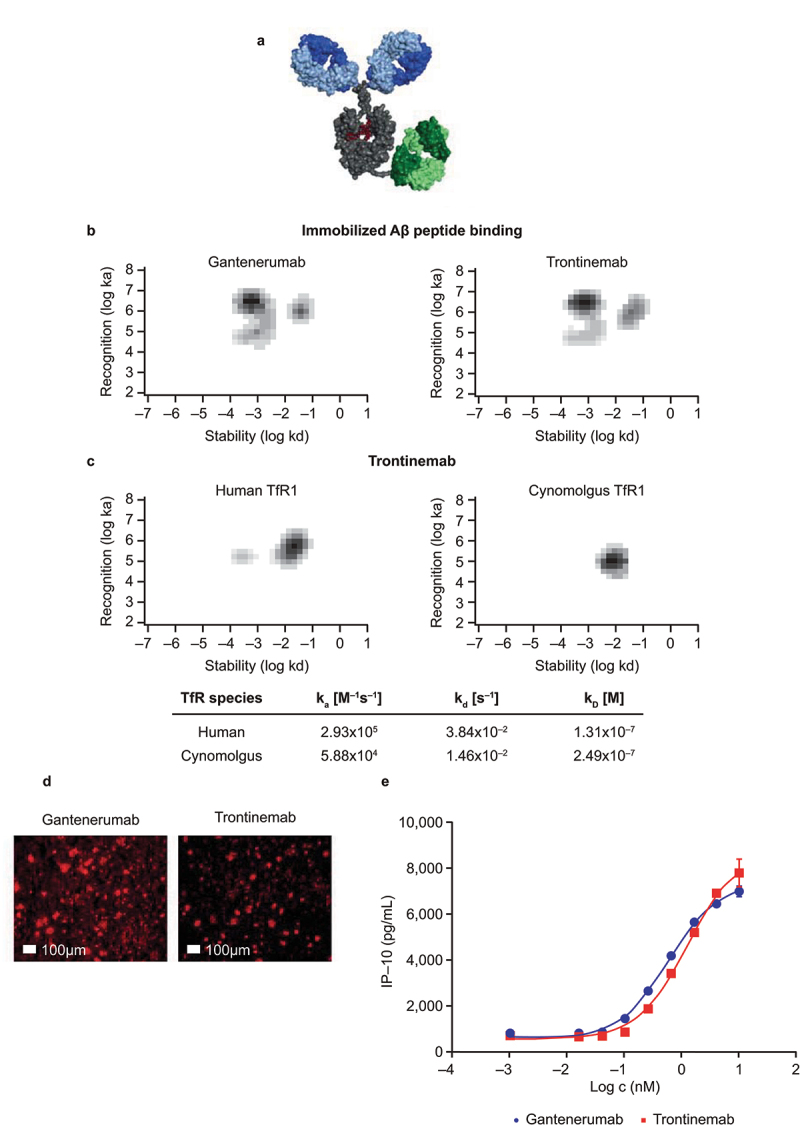
Characterization of trontinemab plaque binding. (a) image of trontinemab’s molecular format. Green = anti-TfR1 cross-Fab; gray = Fc component of IgG1; blue = Fab arms of IgG1; red = glycan. (b) rate constant maps showing the binding kinetics of gantenerumab (left) and trontinemab (right) binding to immobilized Aβ peptide. Darker areas represent higher binding of the mAbs to the immobilized Aβ peptide. Biacore SPR assay interaction maps were generated utilizing the Ridgeview Tracedrawer software for Figure 1b,c. (c) rate constant maps showing the kinetics of trontinemab binding to human TfR1 (left) and cynomolgus TfR1 (right) at 25°C, Biacore SPR assay. Darker areas represent higher binding of the trontinemab to the TfR1 species. (d) gantenerumab (left) and trontinemab (right) binding to human AD brain sections. Red represents the presence of the respective mAb. Staining of the mAbs was conducted by incubation with a secondary antibody (goat anti-human IgG(H+L)/AlexaFluor555, MolecularProbes, A21433) followed by nuclei staining (DAPI, 1 μg/mL in PBS), with intermediary wash buffer rinsing. (e) plot showing the induction of induced protein 10 (IP-10) release from activated U937 monocytes by gantenerumab (red squares) and trontinemab (blue circles). Aβ, amyloid-beta; DAPI, 4′,6-diamidino-2-phenylindole; ELISA, enzyme-linked immunosorbent assay; IgG, immunoglobulin G; ka, on-rate; kd, off-rate; KD, equilibrium dissociation constant; PBS, phosphate buffer saline; mAb, monoclonal antibody; TfR1, transferrin receptor 1.
NHP safety endpoints
Trontinemab and gantenerumab were administered to two groups of NHPs via a single intravenous (IV) bolus. Clinical assessments, clinical pathology, anatomic pathology, and immunofluorescence assays were performed. A 10 mg/kg IV administration of trontinemab or 20 mg/kg of gantenerumab was well tolerated. Transient post-dose, test item-related changes in red blood cell parameters were limited to animals given a single dose of 10 mg/kg of trontinemab and consisted of minimally decreased absolute reticulocyte counts in a few animals. No significant cytokine release was observed within the observation period and there was no evidence of acute infusion reactions.
PK disposition
Test compound concentrations were determined in plasma, cerebrospinal fluid (CSF), and tissue homogenates by enzyme-linked immunosorbent assay (ELISA), detecting the gantenerumab CDRs in both native and trontinemab molecules. All trontinemab-treated animals tested positive for anti-drug antibodies (ADAs) at the 336-h time point. A drop in plasma concentration coincided with these ADA findings at this time point.
Robust estimates of plasma PK parameters for trontinemab and gantenerumab in the NHPs were obtained using a two-compartment model (Supplementary Figure S1) constructed based on observed data. This model includes an element to capture the drop of concentrations seen at the 336-h time point for trontinemab. Table 1 provides a high-level summary of estimated PK parameters in NHPs and allometrically predicted parameters of PK in humans estimated for trontinemab, in addition to those derived from modeling of clinical gantenerumab data.34 Figure 2 shows the observed plasma concentrations overlaid on PK model prediction distributions. Parameters for trontinemab were typical for monoclonal antibodies, except for clearance, which was higher than the typical range in cynomolgus monkeys (0.21–0.5 mL/h/kg).35 In particular, with 1.01 mL/h/kg, trontinemab had a roughly two-fold higher clearance than gantenerumab (0.537 mL/h/kg) in this study.
Table 1.
Estimated PK parameters for trontinemab and gantenerumab in NHPs.
| Name | Cynomolgus monkey |
Human |
|||
|---|---|---|---|---|---|
| Units | Trontinemab | Gantenerumab | Trontinemab (projected) | Gantenerumaba | |
| Vcen | mL/kg | 45.4 | 50.8 | 45.4 | 50.3 |
| CL | mL/h/kg | 1.01 | 0.537 | 0.680 | 0.200 |
| Vper | mL/kg | 63.2 | 97.1 | 63.2 | 91.1 |
| Q | mL/h/kg | 0.746 | 2.86 | 0.502 | 0.517 |
Projected PK parameters for trontinemab in humans obtained by allometric scaling from NHPs and estimated human PK parameters for gantenerumab.
aOriginal parameters were transformed to different units (mL instead of L, h instead of day) and normalized by body weight (assuming 70 kg).
CL, clearance; NHP, non-human primate; Q, intercompartmental exchange clearance; PK, pharmacokinetic; Vcen, central volume; Vper, peripheral volume.
Figure 2.
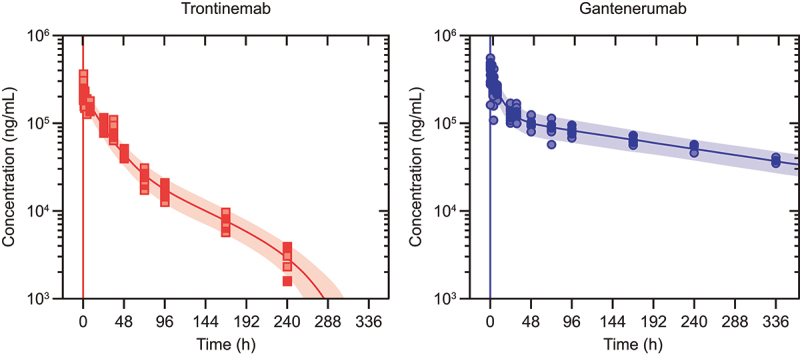
Observed plasma concentrations overlaid on pharmacokinetic model prediction distributions. Observed plasma concentrations (dots) after single IV administration of trontinemab (left, 10 mg/kg) and gantenerumab (right, 20 mg/kg) overlaid on pharmacokinetic model prediction distributions shown as median (lines) and 90% prediction intervals (shaded areas). Plasma concentrations were sampled at 0.083, 0.25, 0.5, 1, 2, 4, 8, 24, 31, 48, 72, 96, 168, and 240 h post-dose. For trontinemab, accelerated clearance is observed at late time points, presumably caused by ADAs and with negligible impact on total exposure (four observations for the 336-hour time point with median concentration of 69 ng/mL lie outside of the plot range). ADA, anti-drug antibodies; h, hour; IV, intravenous.
Brain uptake and modeling
Robust estimates of brain uptake parameters for trontinemab and gantenerumab were obtained for cortex, hippocampus, striatum, cerebellum, and CSF using a linear one-compartment model, which was driven by the plasma PK model described above. Particular care was taken to separate the parenchymal (extravascular) concentration from the contributions of residual plasma in the tissue homogenate samples (Supplementary Figure S1). Conceptually, this is achieved by estimating the fraction of residual plasma from blood tracer data and using that fraction to correct the total tissue concentration of the test compound for the contribution from residual plasma (product of plasma concentration with the fraction of residual plasma). In practice, applying this procedure to individual tissue samples leads to inflated measurement errors and, in some instances, negative concentration values. A sounder approach consisted of fitting the data of the test compounds and the blood tracer simultaneously, in a combined statistical and PK model (Nonlinear Mixed Effects [NLME]) and estimating the fraction of residual plasma alongside with the kinetic parameters (Supplementary Table S1). Rather than fitting the model to derived extravascular data, an effective tissue concentration was reconstituted from the simulated extravascular and residual plasma contributions then compared with the measured tissue concentrations. Furthermore, this procedure accounts for measurement errors of the concentration of test compounds and the blood tracer while constraining them to strictly positive values. Figure 3 shows observed concentrations (corrected for plasma contribution) overlaid with model simulations. Typical values for the estimated fraction of residual plasma (fpla) in tissues were between ~0.03% in striatum and ~0.1% in cerebellum. In all brain tissues, fpla was associated with very high inter-individual variability. Estimated outflow half-lives (calculated from outflow rate [kout]) from brain tissues and CSF were in the range of 10–20 h, except for gantenerumab in CSF with about three-fold longer half-life. The value for trontinemab in CSF was in line with those found in brain tissues. The estimation of parameters for choroid plexus was subject to substantially higher uncertainty compared with other brain regions, and was not included in further analyses (full parameters reported in Supplementary Table S1). Additionally, histologic examination of the tissues collected and analyzed as “choroid plexus” showed high variability in its composition that further limits our confidence in the quantitative assessment.
Figure 3.
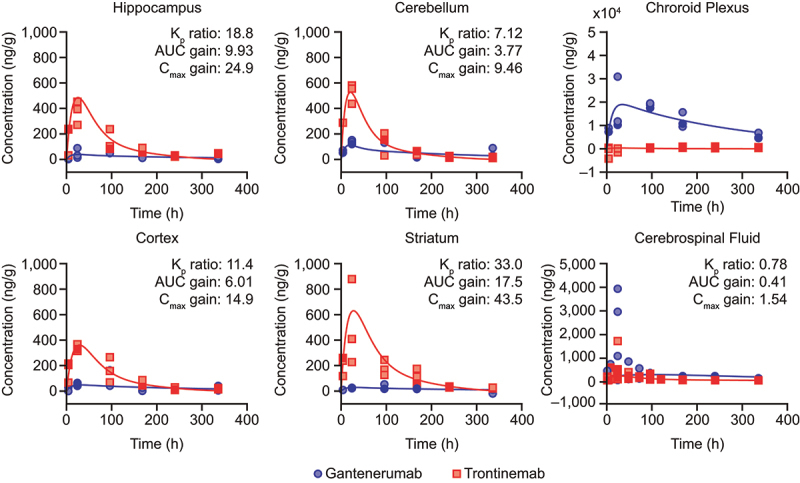
Observed brain concentration of gantenerumab (blue circles) and trontinemab (red squares) overlaid with model simulations (lines) in NHPs. Plots showing concentration of the mAb in the hippocampus, cerebellum, choroid plexus, cortex, striatum, and cerebrospinal fluid up to 336 h post-dose. For graphical representation, observed concentrations were corrected for the contribution of residual plasma using the naïve approach, leading to some concentrations being displayed with negative values. Kp ratio, AUC gain, and Cmax gain were not assessed for the choroid plexus. AUC, area under the curve; Cmax, maximum concentration; Kp, brain distribution coefficient; h, hour; NHP, non-human primate.
Various descriptors for quantifying the brain uptake of trontinemab versus gantenerumab were considered, as summarized in Figure 3. The brain distribution coefficient (Kp) compares the area under the curve (AUC) of brain extravascular concentrations with plasma AUC and was directly estimated from the model. For trontinemab, estimated Kp values of ~0.5% were found across different brain tissues and were between 7-fold (cerebellum) and 33-fold (striatum) higher than for gantenerumab. In contrast, trontinemab uptake into CSF seemed slightly inferior to gantenerumab (Figure 3). Importantly, the improvement of the brain uptake as quantified by Kp provides evidence that the Brainshuttle™ concept successfully overcomes the BBB in vivo. To estimate the net gain in brain exposure for a given dose, the faster systemic clearance of trontinemab has to be considered. The resulting differences in systemic exposure are accounted for by the net AUC gain of brain uptake, which is the AUC ratio for trontinemab versus gantenerumab for a given dose (labeled “AUC gain” in Figure 3). AUC gain in brain tissues was found to range between 3.77 (cerebellum) and 17.5 (striatum). In CSF, a much lower AUC gain of 0.41 was found, but with somewhat lower confidence in the estimation related to the high variability in the gantenerumab data. The maximum concentration ratio between trontinemab and gantenerumab for a given dose ranged from 9.46 (cerebellum) to 43.5 (striatum). Interestingly, the differences in AUC gain ratios between the different brain regions (excluding CSF) can mainly be attributed to regional variations in gantenerumab exposure (three-fold higher exposure in the cerebellum than in the striatum), while trontinemab regional exposures are more homogeneous (1.5-fold higher exposure in the striatum than in the cerebellum). An interesting observation is that the gain with the Brainshuttle™ approach is most notable in the deeper structures of the brain.
Imaging of brain tissue distribution by immunofluorescence
The distribution of both gantenerumab and trontinemab in the brain of cynomolgus monkeys at different time points following a single IV dose was assessed by two-plex immunofluorescence immunohistochemistry in the cortex, hippocampus, striatum, cerebellum, and choroid plexus. trontinemab showed vascular co-localization at the 4-h time point, as well as vascular and parenchymal (signal in brain tissue beyond the vascular basement membrane) localization at 24 h. Weak vascular and parenchymal trontinemab localization was detected 96 h post-dose, the latest time point assessed for trontinemab. This immunofluorescence assay did not detect extravascular gantenerumab signal in the brain parenchyma in any sample after a single dose of 20 mg/kg in the absence of target. The only noteworthy fluorescent signals were observed in the vasculature of poorly perfused areas and in the choroid plexus. These results demonstrate the increased uptake of trontinemab into the brain vasculature and parenchyma, compared with gantenerumab. To further characterize the parenchymal staining of trontinemab, an additional triplex immunofluorescence assay was performed on selected tissue sections. In this assay, trontinemab co-localized with vasculature (collagen IV) and microglia (IBA1) (Figure 4).
Figure 4.
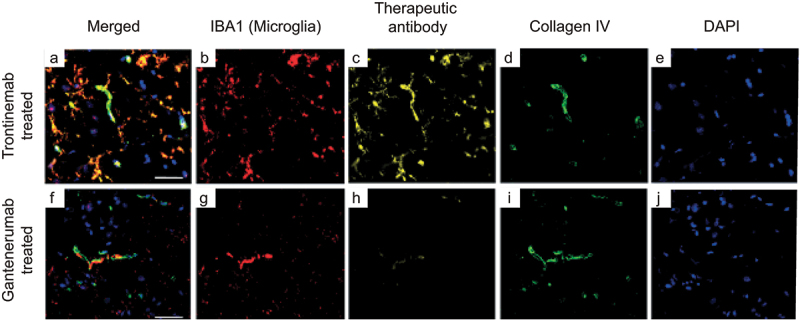
Triplex immunofluorescence of cynomolgus cortex brain sections 24 hours after a single IV injection of trontinemab (top) or gantenerumab (bottom). Immunofluorescent images showing the localization of microglia (IBA1, red), therapeutic antibody (anti-idiotypic) (yellow), vascular basement membrane (collagen IV, green) and nuclei (DAPI, blue) staining in brain sections 24 hours after trontinemab or gantenerumab dosing. At 24 hours post-dose, gantenerumab is limited to vasculature (co-localization with collagen IV), whereas trontinemab is present in the parenchyma (IBA1 co-localization) in addition to vasculature. Immunostaining was performed using the Ventana Discovery Ultra automated stainer. DAPI, 4′,6-diamidino-2-phenylindole; IBA1, ionized calcium-binding adaptor protein.
Translational PK/PD
Figure 5 shows simulated human patient profiles of amyloid load (positron emission tomography standardized uptake value ratio [PET SUVR]) following treatment with gantenerumab or trontinemab. The new brain exposure-based PK/PD model yielded almost identical results to the original model for gantenerumab, with small differences only apparent at short timescales, but with identical long-term trends. The amyloid reduction obtained with an equivalent of 600 mg gantenerumab every 4 weeks was predicted to be matched by 210 mg of trontinemab every 4 weeks, with effects accumulating to a 20% reduction over 1 year.
Figure 5.
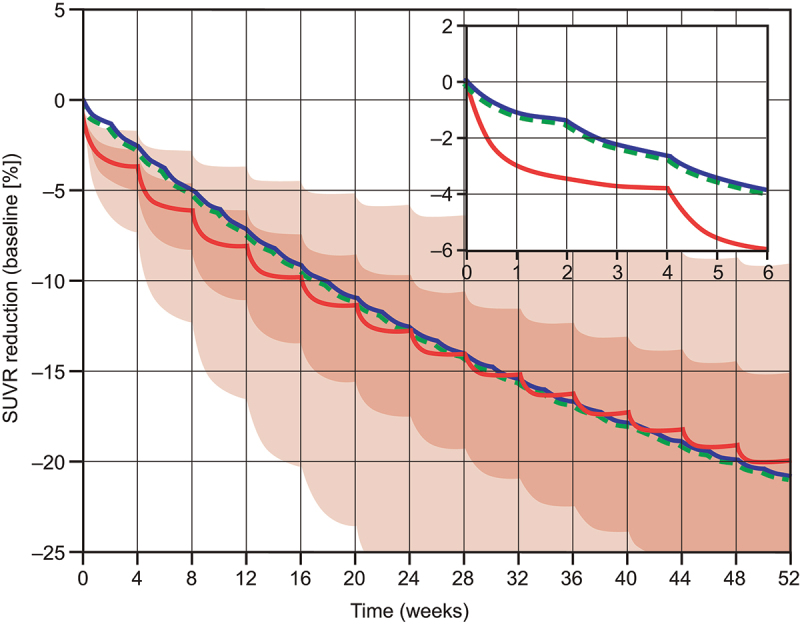
Model-based comparison of amyloid load reduction (PET SUVR) in the cortex following dosing with a cumulative dose of 600 mg gantenerumab every 4 weeks (300 mg IV of gantenerumab every 2 weeks, blue line) or with 210 mg of trontinemab every 4 weeks (red line and shaded confidence bands). Plots showing simulated SUVR reduction over time with an inset plot showing the dynamics for the first 6 weeks. For gantenerumab, simulations with the original plasma-exposure-driven model (dashed green line) overlay closely with the transformed brain-exposure-driven model (blue line). For the selected dosing regimen, similar amyloid reduction was predicted for trontinemab and gantenerumab over 1 year. Translational uncertainty (shown as 90% [lighter red] and 50% [darker red] confidence bands shaded in red) is illustrated assuming a 90% probability that the true typical values of the plasma clearance and the brain distribution coefficient are within two-fold of the predicted values based on log-normal distributions. Inset shows the dynamics for the first 6 weeks. IV, intravenous; PET, positron emission tomography; SUVR, standardized uptake value ratio.
Discussion
We have successfully engineered and produced a bispecific modular fusion protein of an anti-Aβ antibody with a human TfR1-directed Brainshuttle™ module. Formatting gantenerumab into its Brainshuttle™ version did not impair the Fc functionality, a critical property for the MoA of anti-Aβ antibodies, in vitro or in vivo.25 Trontinemab’s proprietary 2 + 1 format is considered crucial for this MoA, allowing monovalent binding to human TfR1 in systemic circulation whilst preserving the bivalent binding of Aβ plaques in the brain. Preclinical data show that a mouse surrogate of trontinemab clears Aβ plaques in the brain by Fc receptor-mediated phagocytosis, engaging microglia cells, demonstrating that Fc-receptor functionality is not compromised by C-terminal attachment of the shuttle module.25,26 At the same time, the 2 + 1 format provides steric hindrance to the wild-type Fc region as a means to overcome the safety challenges previously reported for other TfR1-targeting, wild-type IgG constructs.24 The cross-reactivity of the TfR1 binder used for this study was confirmed between humans and the NHP model, due to a similar range of binding affinities in both species. This enables meaningful and translatable assessment of safety and brain disposition in NHPs without the need for a human TfR-transgenic mouse model, prion-protein mouse models, or the use of surrogate compounds, each of which come with uncertain translatability to humans.
In the NHP model, trontinemab was cleared from plasma faster than a typical IgG, likely due to a TfR1-mediated clearance (CL) component. The development of ADAs in all animals does not allow for a clear distinction between ADA and TfR1-mediated CL in this study. However, the moderate affinity of the TfR1 binding suggests that the accelerated elimination observed in this study is likely due to ADA-mediated CL. The effects of ADAs on PK in humans are assessed in clinical trials. In line with the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) S6, immunogenicity in NHP is not considered to be predictive for humans.36 In the case of trontinemab only, ex vivo whole-slide fluorescence imaging data from this study demonstrated trontinemab co-localization with brain endothelial cells, with subsequent exposure to brain parenchyma, and its presence in microglial cells, consistent with TfR1-mediated transcytosis across the BBB and wild-type Fc-mediated interaction, respectively. These interactions occurred without the noteworthy clinical signs reported with other wild-type Fc TfR1 bispecific antibodies by Couch et al.24 at the trontinemab dose tested in the study (10 mg/kg). Effects on cytokine release and other safety parameters are further assessed in dedicated toxicology studies.
Terminal plasma, CSF, and tissue samples contained an inert and effector function silent human IgG (PGLALA Fc)37 as a blood tracer in addition to the test compound. The quantification of the tracer allowed for a specific correction for residual blood in tissue samples through the NLME model. This is of particular importance due to the large difference in concentrations between plasma and brain tissue, which leads to the risk of even small amounts of residual blood substantially contributing to the determined tissue homogenate concentrations. The test compound and the applied blood tracer could be quantified independently using bioanalytical ELISAs based on idiotype-specific only (test compound) or idiotype- and PGLALA-specific combined (tracer) capture and detection reagents. Since the same anti-idiotypic (anti-ID) antibody was used in the bioanalytical ELISA and immunofluorescent staining, both evaluations led to coherent results.
The dose-normalized maximal brain concentrations measured for trontinemab in our study are similar to those published by Kariolis et al.38 for a TfR1-targeted anti-BACE1 antibody, despite an approximately 10-fold lower affinity for the cynomolgus TfR1. This supports the hypothesis of a relatively broad range of shuttle module affinities enabling productive brain uptake.23
In contrast to the homogeneous tissue distribution of trontinemab throughout the cortex, hippocampus, striatum, and cerebellum, the choroid plexus stood out as the only tissue in which gantenerumab could be clearly detected by immunofluorescence under the conditions of our study (Supplementary Figure S2). Although high inter-individual variability prevented reliable quantitative assessments in the choroid plexus, gantenerumab concentrations appeared to exceed those of trontinemab. This supports the hypothesis that the choroid plexus plays a key role in providing access for IgGs to the brain as proposed by other groups,39 whilst the Brainshuttle™ construct enters the CNS via receptor-mediated transcytosis across the BBB. In line with this hypothesis, gantenerumab exposure varied considerably across different brain regions, while the distribution of trontinemab exposure was more homogeneous. This could potentially offer therapeutic advantages that cannot be estimated from the brain distribution coefficient alone, especially in brain regions distant from the cerebroventricular system. Amyloid PET data from a Phase 1b trial of aducanumab in patients with AD suggest that the extent of Aβ plaque removal may differ across different brain regions, and that certain brain regions like the medial temporal cortex may be less responsive to conventional monoclonal antibody treatment.40 While the observed differences may, in part, be explained by regional differences in Aβ plaque load at baseline, it is tempting to speculate that a more homogenous penetration of the brain tissue by trontinemab could lead to an improved clearance of amyloid deposits even in brain regions that are less responsive to conventional monoclonal antibodies targeting Aβ.
Consistent with the above observations and data from other groups, the enhancement in access to brain tissue observed with trontinemab was not reflected in increased CSF concentrations compared to gantenerumab in NHP. This is in principle in line with the hypothesis that if transferal across the BBB is the primary driving factor behind transport into the CNS, then exposure in the brain should be greater than exposure in the CSF. Equally, if blood – CSF transfer is predominant, then brain-exposure levels will be lower than CSF exposure levels to reflect this.38 Furthermore, brain homogenization during sample preparation can reduce the brain serum ratio artificially by diluting the concentration in the brain.41 This adds further complexity to the use of CSF concentrations to estimate CNS exposure levels, which has been shown to overestimate brain exposure for non-targeted antibodies in the case of blood – CSF transfer into the CNS,38 but could underestimate brain exposure for therapeutic proteins that cross the BBB. Higher brain exposure after receptor-mediated transcytosis may therefore not always lead to higher exposure in CSF.
CSF exposure in human was assessed in healthy volunteers during a single ascending dose study of trontinemab. The results of this study aid our understanding of how the NHP findings translate to human and will enable further refinement of the modeling and simulation framework presented here.
The same process that enables the transcytosis and enhanced BBB transport of the Brainshuttle™ is inevitably linked to TMDD and, therefore, to reduced plasma exposure. Antibodies with moderate affinities toward the transcytosis receptor were chosen to maintain plasma exposure of the engineered protein, thereby maintaining a favorable balance between increased brain distribution and reduced systemic exposure compared with a conventional IgG.9–11–15–42,43 In addition, the targeted endothelial cell receptors typically have vital functions in the homeostasis of their endogenous physiologic substrate in the brain and other tissues that could potentially be disrupted by brain-targeting antibodies. As demonstrated here, the anti-TfR1 Fab component of trontinemab did not interfere with the natural interaction between transferrin and TfR1.
In addition to the qualitative imaging data showing TfR1-mediated transcytosis across the BBB in vivo in NHPs, we presented a robust estimation of brain uptake profiles and Kp using a combination of brain perfusion and correction for residual plasma in tissue homogenates. The comparative exposure assessment of gantenerumab and trontinemab described herein enabled a robust comparison of brain AUC, maximum concentration (Cmax), and Kp, and provides a foundation for a translational MIDD framework.33,44
This framework was built to project pharmacodynamically active dosing regimens for trontinemab in humans by reverse translation of the gantenerumab clinical plaque removal data. For this, an existing PK/PD model of gantenerumab plaque removal was linked to the projected human exposure of trontinemab. This linkage was achieved by a new model of brain uptake in NHPs of trontinemab and gantenerumab, translating it to humans by allometric scaling of the PK portion.42 This underlines the critical role of the NHP study and the importance of a well-integrated study design, bioanalytical strategy, and modeling methodology. The latter was powered by an NLME approach in which the concentrations of trontinemab, gantenerumab, and the vascular tracer were modeled simultaneously, thus combining the kinetic analysis with an adequate statistical framework.45 The amyloid reduction obtained with 600 mg of IV gantenerumab every 4 weeks is predicted to be matched by 210 mg of IV trontinemab every 4 weeks, with effects accumulating to a 20% SUVR reduction over 1 year. Hence, a substantial relative and absolute increase in brain exposure may be achieved, which could result in improvements in target engagement and potential efficacy benefits for patients. Lower dosing, on the other hand, may be associated with potential safety benefits, reduced manufacturing burden, and increase flexibility in administration, including convenient subcutaneous dosing regimens using devices.
The first-in-human, single-ascending dose, Phase 1 clinical trial (NCT04023994) assessing the safety, tolerability, and PK of trontinemab in healthy participants was completed recently with preliminary results released, and a Phase 1b/2a clinical trial in participants with prodromal or mild-to-moderate AD is currently ongoing (NCT04639050). The modular nature of the 2 + 1 Brainshuttle™ format of trontinemab allows for the application of the presented TfR1 shuttling technology to other therapeutic antibodies and further therapeutic modalities.45,46
Materials and methods
Molecule development (in vitro)
Gantenerumab human IgG1 (RO4909832)10 was produced from a clonal stable Chinese hamster ovary (CHO) cell line, with the genes encoding antibody heavy and light chains integrated in the CHO genome. After cultivation for 14 days, the supernatant was purified by a multistep column chromatography process. Trontinemab (RO7126209) was produced from a stable CHO pool with the genes encoding antibody heavy and light chains integrated in the CHO genome. After cultivation for 14 days, the supernatant was also purified by a multistep column chromatography process. DP47 PGLALA human IgG1 tracer (batch RO7126696) was produced by cultivation of a stable CHO cell line for 14 days. The supernatant was purified by MabSelectSure ProteinA affinity chromatography. The quality of all materials was verified by size-exclusion chromatography, caliper analysis, mass spectrometry, analytical ion-exchange chromatography, and affinity analysis by Biacore and fluorescence-activated cell sorting.
Plaque binding by immunohistochemistry
Immunofluorescence staining was done on a fresh, frozen human brain section (Braak VI) manually. After rehydration, slides were treated with ice-cold acetone for 3 min followed by rinsing with buffer solution and application of a hydrophobic barrier. Slides were rinsed with wash buffer (phosphate-buffered saline [PBS] + 0.01% Tween 20 [PBS-T], pH 7.4) and blocked for 20 min (PBS +1% bovine serum albumin + 1 ovalbumin + 1% normal goat serum). Primary antibody (gantenerumab, human trontinemab, or blank control) staining was conducted for 1 h at room temperature (RT) followed by wash buffer rinsing. Samples were incubated with a secondary antibody (Goat anti-Human IgG(H+L)/AlexaFluor555, MolecularProbes, A21433) for 1 h at RT followed by wash buffer rinsing. Nuclei staining was done for 3 min (4′,6-diamidino-2-phenylindole [DAPI], 1 μg/mL in PBS), followed by two rinses for 2 min each (wash buffer), and then the cover glass was mounted (ProLong Gold, Molecular Probes No. P36934/22×22x0.17 mm cover glasses No. 1.5 H, Marienfeld, #0107052).
Images were recorded on a Leica TCS SP8 AOBS confocal laser-scanning microscope. One optical layer was recorded at an excitation wavelength of 488 nm using an HC PL Fluotar 10×/0.30 objective at a pinhole setting of 70.7 μm. Instrument settings were kept constant for all images to allow a relational qualitative comparison. Specifically, laser power, gain, and offset were adjusted to allow for signal intensity monitoring within the dynamic range. For each antibody concentration, gray matter regions were recorded at comparable positions from consecutive sections to minimize potential variability arising from anatomical differences in plaque load.
SPR TfR binding assay
A CM5 biosensor chip was coated with about 14,000 resonance units (RU) of an anti-histidine antibody on a Biacore 8K instrument, using standard ethyl (dimethylaminopropyl) carbodiimide/N-hydroxy succinimide (EDC/NHS) chemistry. In a following measurement run, multiple cycles were recorded. All cycles included: a capturing step (60 s, 5 µl/min) resulting in a capturing level of about 15 RU, a preincubated 5 nM TfR-extra cellular domain:tissue factor complex, an analyte injection (association: 120 s, dissociation 300 s, 50 µl/min) with variable sample concentrations (1,800 nM −22.22 nM, three-fold dilutions) and two identical regeneration steps (50 s, 30 µl/min), followed by a final wait command (30 s). Resulting raw data were evaluated using the interaction map feature of the Ridgeview Tracedrawer software.
SPR Aß binding assay
Amyloid beta peptide 1–42 was coupled to a Biacore CM5 chip, using standard EDC/NHS amine coupling chemistry. Immobilized peptide was conditioned by three consecutive regeneration steps (two 30 s long injections of 15 mM sodium hydroxide). Then, concentration series of gantenerumab and trontinemab were injected successively, followed by a regeneration step after each individual injection. The obtained interaction curves were then evaluated using the interaction mapping technology provided by the Ridgeview Tracedrawer software.
Monocyte activation
Ninety-six well cell culture plates were coated with Aβ42 peptide (Bachem; 20 μg/mL in PBS) overnight, then incubated with anti-Aβ antibody solutions for 1 h at 37°C. After washing the plates, 105 U-937 human monocytes that had been pre-activated with 400 U/mL interferon- γ overnight were added per well. Plates were incubated for 24 h at 37°C and 5% carbon dioxide. The next day, supernatants were transferred to ELISA plates for determination of interleukin 8 (IL-8) and interferon γ-IP-10 kDa concentrations according to the manufacturer’s protocols (R&D Systems), as previously described.26
In vivo study in NHPs
The cynomolgus monkey is the only known TfR1 binder cross-reactive preclinical model species for this Brainshuttle™. All in-life procedures were conducted in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-certified facility after review and approval of the test procedure by local ethics committees. Purpose-bred Mauritian female cynomolgus monkeys, aged 25–47 months in the body weight range of 2.4–3.4 kg, were used in this study (15 per group). A sentinel group of four animals received a single IV bolus injection of 10 mg/kg of trontinemab prior to two additional groups of 15 animals each receiving a single IV bolus injection of either 10 mg/kg of trontinemab or 20 mg/kg of gantenerumab. The trontinemab dose was selected to be close to the maximum dose anticipated for clinical trials. Given the low brain distribution of regular antibodies, a two-fold higher dose of gantenerumab was chosen to increase the time interval during which concentrations remain quantifiable with the bioanalytical method and detectable by immunofluorescence. Blood was collected from the arm or leg veins of each animal until sacrifice, up to 336 h post-dose for groups one and two and up to 240 h post-dose for group three. Additionally, up to four CSF samples were collected from each animal under anesthesia via lumbar puncture (sampling schedule provided in Supplementary Table S2). Animals were sacrificed at 4, 24, 96, 168, 210 (trontinemab only), and 336 h post-dose for pathology assessment and collection of brain tissues.
Five minutes prior to termination, an IV bolus of 1 mg/kg of a non-binding IgG (DP47) was administered as a blood tracer followed by 200 IU/kg of heparin sodium. Immediately after administering a pentobarbitone overdose, animals were perfused transcardially with 0.001% sodium nitrite in 0.9% saline. One brain hemisphere was prepared for microscopic evaluation. From the contralateral hemisphere, two pieces of approximately 0.5 g (exactly weighed) were taken from the cortex, hippocampus, cerebellum, striatum, and choroid plexus and flash frozen at −70°C or below for the determination of test compound concentrations in tissue homogenates. Tissue samples of cortex, striatum, cerebellum, and choroid plexus for microscopic evaluation were frozen in optimal cutting temperature medium for evaluation by immunofluorescence microscopy.
Two-plex immunofluorescence immunohistochemistry
Samples of cortex, striatum, and cerebellum from all animals at 4-, 24-, and 96-h time points, as well as selected sections from other time points and regions, were sectioned onto Superfrost Plus slides. Staining was performed using the Ventana Discovery Ultra automated tissue stainer (Roche Tissue Diagnostics, Tucson AZ, USA). Anti-ID antibodies directed against the CDRs of gantenerumab (Roche Diagnostics GmbH, Penzberg, Germany) were used to detect gantenerumab and trontinemab. Collagen IV (ab19808, abcam, UK), a rabbit polyclonal antibody, was used to detect vascular basement membrane. Secondary antibodies were conjugated to fluorophores: polyclonal donkey anti-mouse IgG Alexa Fluor 555 conjugated (Invitrogen A31570), and donkey anti-rabbit IgG Alexa Fluor 647 conjugated. DAPI was used as a nuclear stain (Roche Tissue Diagnostics, Tucson, AZ, USA) and mounted using Brightmount (ab103746, abcam, UK) mounting media. Slides were digitized using a Pannoramic 250 (3D Histech, Budapest, Hungary) whole-slide scanner and Caseviewer software at 40× magnification. Digital slides were viewed using the 3D Histech Caseviewer Software.
NHP three-plex immunofluorescence immunohistochemistry methodology
For selected samples, cynomolgus frozen brain tissues were sectioned at 8 µm, tissue sections were fixed with 90% ice-cold ethanol for 10 min and washed in PBS before staining. Staining was performed using the Ventana Discovery Ultra automated stainer (Roche Tissue Diagnostics, Tucson AZ USA). Tyramide signal amplification multiplexing (R6G #253–6002, FAM #253–6000, Cy5 #253–4928, Roche Tissue Diagnostics, Tucson, AZ, USA) was applied to a combination of primary antibodies: anti-gantenerumab/IBA1/collagen IV. IBA1 (019–19741, Wako) was used to detect microglia. Primary antibodies were detected using horseradish peroxidase (HRP)-conjugated secondary antibodies (Roche Tissue Diagnostics, Tucson, AZ, USA): Omnimap anti mouse (760–4310), Omnimap anti rabbit (760–4311), Omnimap anti goat (760–4647), and Ultramap anti mouse (760–4313). Tissue was counter stained with DAPI (Roche Tissue Diagnostics, Tucson, AZ, USA) and mounted using Brightmount (ab103746, abcam, UK) mounting media. Slides were digitized using an AxioScan Z1 (Zeiss, Germany) whole-slide scanner at 40× magnification and visualized using Zeiss Zen software version 2.3.
Preparation of cynomolgus brain tissue homogenates for ELISA
Frozen cynomolgus brain tissue samples of 300 mg were thawed at RT for 2 h. 800 µL of lysis buffer (one tablet of complete protease inhibitor cocktail [Roche Diagnostics GmbH] dissolved in 50 mL Tissue Extraction Reagent I [Invitrogen]) was added to the thawed brain tissue. Next, the sample was homogenized in a MagNA Lyser instrument (Roche Diagnostics) for 20 s at 6,500 rpm. The tissue homogenate was then centrifuged for 10 min at 12,000 rpm using a centrifuge 5,430 (Eppendorf). Finally, the supernatant was transferred to a 1.5 mL vial for further analysis or stored at −80°C.
Quantification of antibody and blood tracer concentrations in cynomolgus monkey brain homogenate, plasma, and CSF
To quantify trontinemab or gantenerumab, samples from cynomolgus monkey (brain, plasma, or CSF), calibration standards, and quality control samples were pre-incubated with a mixture of biotinylated (Bi) and digoxigenylated (Dig) anti-ID antibodies directed against the CDRs of gantenerumab (Roche Diagnostics GmbH) for 30 min at RT. The mixture contained 1% (v/v) brain homogenate, 10% (v/v) plasma, or 10% (v/v) CSF cynomolgus sample, as well as 100 ng/mL anti-ID-Bi, and 100 ng/mL anti-ID-Dig. Formed immunocomplexes were transferred to a streptavidin-coated microtiter plate (MTP) and incubated for 1 h at RT. The supernatant was aspirated, and unbound substances were removed by subsequent washing steps in PBS-T. Samples were then incubated with 50 mU/mL of HRP-conjugated anti-digoxigenin Fab fragments (Roche Diagnostics GmbH) for 1 h at RT, washed, and then incubated at RT in 2,2´-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) substrate solution (Roche Diagnostics GmbH). The color intensity was photometrically monitored at 405 nm (490 nm reference wavelength). Concentrations were determined from the standard curve with a four-parameter non-linear regression Wiemer – Rodbard curve fitting function with weighting. The lower limit of quantification (LLOQ) was 0.78 ng/mL for trontinemab and gantenerumab in plasma or CSF, and 7 ng/mL for trontinemab and gantenerumab in brain homogenate samples. Dependent on the homogenized brain tissue weights, lower limits of quantification can be calculated in ng/g for each brain tissue compartment specifically.
To quantify DP47 tracer in cynomolgus monkey samples (brain, plasma, or CSF), 500 ng/mL biotinylated capture antibody directed against the idiotype of DP47 tracer (Roche Diagnostics GmbH) was transferred to a streptavidin-coated MTP and incubated for 1 h at RT. After another PBS washing step, calibration standards, quality controls, and samples, diluted to 1% (v/v), were added and incubated for 1 h at RT. The supernatant was aspirated, and unbound substances were removed by subsequent washing steps in PBS. 125 ng/mL digoxigenylated blood tracer PGLALA Fc-specific detection antibody (Roche Diagnostics GmbH) was added for 1 h at RT. After an additional PBS washing step, samples were then incubated with 50 mU/mL HRP-conjugated anti-digoxigenin Fab fragments for 1 h at RT, washed, and incubated at RT in ABTS substrate solution. The color intensity and concentrations were determined as above. The lower limit of quantitation (LLOQ) was 7.4 ng/mL in plasma, CSF, and brain homogenate samples. Dependent on the homogenized brain tissue weights, lower limits of quantification can be calculated in ng/g for each brain tissue compartment specifically.
ADA assay
For the qualitative detection of antibodies directed against the test compounds, a sandwich ELISA was used. A biotinylated capture antibody directed at the human Fc antibody part, mAb<Hu-IgG>M-R10Z8E9 (Roche Diagnostics), was bound to streptavidin-coated MTP at a concentration of 0.5 μg/mL and incubated for 1 h at RT. After washing the plate, samples and standards were diluted to 1% cynomolgus monkey plasma in LowCross buffer (Candor Bioscience GmbH) spiked with 1 µg/mL trontinemab or gantenerumab. 100 μL was added to each well and incubated for 1 h at RT. After washing, 0.5 μg/mL of digoxigenylated anti-cynomolgus IgG detection antibody (Roche Diagnostics GmbH) was added to the cavities of the MTP and incubated for 1 h. After washing, 25 mU/mL of polyclonal anti-digoxigenin-HRP conjugate was added and incubated for 1 h. The HRP of the antibody–enzyme conjugate catalyzed the color reaction after addition of the substrate solution ABTS to the MTP. Absorption was measured by an ELISA reader at a 405 nm wavelength (reference wavelength: 490 nm). Twice the absorption signal of pre-dose samples of every individual cynomolgus monkey animal was set as the cut point to assess post-dose samples for ADAs. A sample was defined as ADA positive if the signal of a post-dose sample of an individual was higher than the corresponding cut point of the same individual.
Safety monitoring
In-life parameters for safety assessment included clinical observations, hematology, coagulation, clinical chemistry, soluble transferrin receptor, and cytokines (interferon γ, tumor necrosis factor-alpha, monocyte chemoattractant protein-1, IL-6, and IL-8), as well as post-dose observations of body weight, body temperature, and feces. Following sacrifice, macroscopic examination and organ weights were assessed, and microscopic evaluation of a full tissue list was performed.
PK modeling
Plasma PK for trontinemab and gantenerumab, as well as the single concentration point of antibody used as blood tracer, were analyzed in a first step, independent of any further analyses of concentrations in brain compartments. A linear two-compartment model was used to describe the plasma concentrations (Cpla) of trontinemab and gantenerumab, with parameters central volume (Vcen), clearance (CL), peripheral volume (Vper), and intercompartmental exchange clearance (Q). For trontinemab, an additional clearance term was included to account for the accelerated clearance, likely due to immunogenicity. NLME modeling was used together with the assumption that the typical parameter values (fixed effects) differed between the test compounds, but that the inter-individual variabilities (random effects) and the residual errors were equal. The serum concentration of the blood tracer was modeled as a constant (Dpla), for which it was assumed that inter-individual variability is consistent with the PK model for trontinemab and gantenerumab. Parameters were estimated in Monolix version 4.3.3 (Lixoft SAS, Antony, France).47 Model quality was checked and compared based on the identifiability of parameters, objective function, and diagnostic plots.
Allometric scaling, closely following the methods reviewed in Wang et al.,48 was used for scaling of the PK of trontinemab to humans: volumes (Vcen, Vper) were scaled with body weight. CL parameters Q were scaled with the body weight ratio to the power 0.85. Body weights used were 5 kg for monkeys and 70 kg for humans. Note that the human projection of PK does not include the projection of immunogenicity and its effect on exposure.
Brain uptake modeling
The conceptual model for the brain uptake and the correction for residual plasma are shown in Supplementary Figure S1. Brain and CSF uptake data were modeled separately for each brain tissue and CSF. The structural model for the extravascular brain concentration (Cext) is a linear one-compartment model parameterized by the Kp and the kout:
| (1) |
This model used the individual Cpla of trontinemab and gantenerumab as input, for which the individual (post hoc) PK parameters were used as regressors. The total brain concentration (Cbrn) of test compound was the sum of the vascular contribution (with fpla) and the brain extravascular contribution:
| (2) |
On average, , and thus the contribution of residual plasma for the estimation of Kp is negligible only when fpla << Kp. With a plasma content of ~2.5% in a living brain,47,49 perfusion would be required to remove >>99% from the brain (fpla <<0.025%) to enable unbiased estimate of a Kp of e.g., 0.025%. Blood tracer data were therefore used to correct for the contamination by residual plasma by estimating fpla and thereby improving the accuracy of the estimation. It was assumed that the tracer antibody was constrained to the vascular space and hence the concentration of blood tracer in the brain (Dbrn) was related to the concentration in serum by:
| (3) |
Conceptually, the brain uptake model can be fitted to empirically calculated extravascular concentrations , obtained from the Cbrn and from fpla using Equations (2) and (3):
| (4) |
However, this naive approach can lead to negative values of in the case of low tissue uptake, which is a problem in particular for gantenerumab with its lower brain uptake and longer plasma half-life. Furthermore, this way of calculating mixes up the statistical distributions of the various variables, making a sound assessment difficult.
Instead, the approach taken here is to fit Cbrn and Dbrn simultaneously and estimating fpla alongside the parameters of the brain uptake Kp and kout. Again, NLME modeling was used. For the brain tissues, the test compound was assumed to affect only the typical values (fixed effect) of Kp, while kout and fpla were shared between the two compounds. For CSF, the test compound was allowed to affect also the kout, while fpla was assumed to be negligible. In brain tissues, inter-individual variability was considered for the in all tissues. As only single observations were available for each animal in brain tissues, the width of distribution of residual errors (proportional error model) was fixed, and a value of 0.3 was chosen. In CSF, where observations at multiple time points were available, inter-individual variability was considered for all parameters and the width of distribution of residual errors was freely estimated. Note that values below the LLOQ were dealt with as ‘censored’ and that the limit was sample specific. Parameter estimation was done using Monolix version 4.3.3.47 Model quality was checked and compared based on the identifiability of parameters, objective function, and diagnostic plots. For the translation of the uptake to brain and CSF, it was assumed that model parameters Kp and kout were equal between monkey and human.
Translational PK/PD
The impact of the increased brain uptake of trontinemab, compared with gantenerumab, on the dynamics of the amyloid reduction in the brain was investigated by coupling the human projection of PK and brain uptake to the PD model for amyloid reduction in individuals with AD as assessed by SUVR from PET imaging as published previously.34 In short, this model describes the relationship between exposure and PET SUVR reduction by a power model combined with an effect compartment to account for the delay between exposure and PET response. In order to account for differences in brain uptake, this model is transformed to a related model such that it is driven by the Cext rather than by the Cpla. The differential equation for the concentration in the effect compartment (Ceff) is then:
Here, Kp* makes the connection to the clinical results with gantenerumab34 and is chosen such that the same average Ceff is obtained as in the original model. Since the cortex dominates the signal of PET SUVR in these studies, the Kp for gantenerumab in cortex will be used. Given the disparity of time scales related to the effect compartment (half-life 398 days) and the brain uptake (<1 day), it can be shown that solutions to this equation are good approximations to the original equation for gantenerumab. The dynamics of the reduction of amyloid (Apet) is then:
Here, is the PET SUVR baseline value (“Base” in the original paper). The reference concentration Cref and the power (“POW” in the original notation) are the parameters driving the effect. For gantenerumab, Cref is related to the original parameters as:
Based on the design of the molecule, on its affinity to amyloid and its potency24 (see also cytokine release data from Figure 1e), equal molar concentrations of gantenerumab and trontinemab are assumed to lead to the same effects. Therefore, introducing the molecular weights MWgant and MWbrsh (for gantenerumab and trontinemab), the reference concentration for trontinemab, Cref,brsh is expressed as:
In order to illustrate the translational uncertainty, simulations were repeated 1,000 times while randomly varying the CL and Kp around their predicted values using log-normal distributions and setting the width of the distribution such that 90% of the values fall within two-fold of the respective predicted values.
Abbreviations
- AAALAC
Association for Assessment and Accreditation of Laboratory Animal Care
- Aβ
amyloid-beta
- AD
Alzheimer’s disease
- ADAs
anti-drug antibodies
- AUC
area under the curve
- BBB
blood – brain barrier
- CDRs
complementary determining regions
- CHO
Chinese hamster ovary
- CL
clearance
- Cmax
maximum concentration
- CNS
central nervous system
- CSF
cerebrospinal fluid
- DAPI
4′,6-diamidino-2-phenylindole
- EDC/NHS
ethyl (dimethylaminopropyl) carbodiimide/N-hydroxy succinimide
- ELISA
enzyme-linked immunosorbent assay
- Fab
fragment antigen-binding
- FDA
US Food and Drug Administration
- fpla
fraction of residual plasma
- h
hour
- HRP
horseradish peroxidase
- IBA1
ionized calcium-binding adaptor protein
- ICH
International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use
- IgG
immunoglobulin G
- IgG1
immunoglobulin G1
- IR
insulin receptor
- IV
intravenous
- ka
on-rate
- kd
off-rate
- KD
equilibrium dissociation constant
- kout
outflow rate
- Kp
brain distribution coefficient
- LLOQ
lower limit of quantification
- MoAs
mechanisms of action
- MIDD
model-informed drug development
- MPT
microtiter plate
- NHP
non-human primate
- NLME
Nonlinear Mixed Effects
- PD
pharmacodynamic
- PK
pharmacokinetic
- PBS
phosphate buffer saline
- PBS-T
phosphate buffer saline + 0.01% Tween 20
- PET
positron emission tomography
- PET SUVR
positron emission tomography standardized uptake value ratio
- Q
intercompartmental exchange clearance
- RT
room temperature
- SUVR
standardized uptake value ratio
- TfR1
transferrin receptor 1
- TMDD
target-mediated drug disposition
- Vcen
central volume
- Vper
peripheral volume
Supplementary Material
Acknowledgments
The authors would like to acknowledge Marie-Helene Gouy, Nadege Foiselle, Roland Staack, Michael Winter, and Luisa Bell (Pharmaceutical Sciences, F. Hoffmann-La Roche Ltd), as well as for their support in study execution and data management. The authors thank Luka Kulic and Kerstin Hahn for valuable discussions and his critical review of the manuscript.
Funding Statement
This work was funded by F. Hoffmann-La Roche Ltd, Basel, Switzerland. Medical writing for the development of this draft was provided by Chris Ackroyd, MBiochem, of Health Interactions, funded by F. Hoffmann-La Roche Ltd, Basel, Switzerland.
Disclosure statement
HPG, VS, POF, KB, CH, SR, SS, RN, NJ were employees and shareholders of F. Hoffmann-La Roche Ltd at the time the work was completed.
MS, SIJ, PR, TS, UG, MH, AZ, JN were employees of Roche Diagnostics GmbH and shareholders of F. Hoffmann-La Roche Ltd at the time the work was completed.
Author contributions
Conceptualization: KB, NJ, CH, VS
Methodology: HPG, RN
Investigation: VS, HPG, SS, MS, NJ
Resources: JN, SIJ, TS, PR, UG, MH, AZ
Formal Analysis: VS, MS, NJ, HPG
Writing (original draft): VS, HPG, JN, NJ
Writing (review, edit, and approval): All authors
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2261509
References
- 1.Wang S, Mims PN, Roman RJ, Fan F.. Is beta-amyloid accumulation a cause or consequence of Alzheimer’s disease? J Alzheimers Parkinsonism Dement. 2016;1:007. [PMC free article] [PubMed] [Google Scholar]
- 2.Alzheimer’s Association . Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 2021;17(3):327–14. doi: 10.1002/alz.12328. [DOI] [PubMed] [Google Scholar]
- 3.US Food and Drug Administration (FDA) . FDA’s decision to approve new treatment for Alzheimer’s disease [press release]; 2021. June 7 [accessed 2023 June 28]. https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-alzheimers-disease.
- 4.US Food and Drug Administration (FDA) . FDA grants accelerated approval for Alzheimer’s disease treatment [press release]; 2023. January 6 [accessed 2023 June 28]. https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment.
- 5.Aging NNIo . NIA statement on report of lecanemab reducing cognitive decline in Alzheimer’s clinical trial; 2023. [accessed 2023 March 16]. https://www.nia.nih.gov/news/nia-statement-report-lecanemab-reducing-cognitive-decline-alzheimers-clinical-trial
- 6.van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388(1):9–21. doi: 10.1056/NEJMoa2212948. [DOI] [PubMed] [Google Scholar]
- 7.Grossberg GT, Tong G, Burke AD, Tariot PN, Fink A.. Present algorithms and future treatments for Alzheimer’s disease. J Alzheimers Dis. 2019;67(4):1157–71. doi: 10.3233/JAD-180903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yiannopoulou KG, Papageorgiou SG. Current and future treatments in Alzheimer disease: an update. J Cent Nerv Syst Dis. 2020;12:1179573520907397. doi: 10.1177/1179573520907397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537(7618):50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 10.Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, Messer J, Oroszlan K, Rauchenberger R, Richter WF, et al. Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J Alzheimers Dis. 2012;28(1):49–69. doi: 10.3233/JAD-2011-110977. [DOI] [PubMed] [Google Scholar]
- 11.Saunders NR, Dreifuss JJ, Dziegielewska KM, Johansson PA, Habgood MD, Møllgård K, Bauer HC. The rights and wrongs of blood-brain barrier permeability studies: a walk through 100 years of history. Front Neurosci. 2014;8:404. doi: 10.3389/fnins.2014.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonkowski D, Katyshev V, Balabanov RD, Borisov A, Dore-Duffy P. The CNS microvascular pericyte: pericyte-astrocyte crosstalk in the regulation of tissue survival. Fluids Barriers CNS. 2011;8(1):8. doi: 10.1186/2045-8118-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beshir SA, Aadithsoorya AM, Parveen A, Goh SSL, Hussain N, Menon VB, Abate G. Aducanumab therapy to treat Alzheimer’s disease: a narrative review. Int J Alzheimers Dis. 2022;2022:1–10. doi: 10.1155/2022/9343514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardridge WM. Drug transport across the blood–brain barrier. J Cereb Blood Flow Metab. 2012;32(11):1959–72. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freskgård PO, Urich E. Antibody therapies in CNS diseases. Neuropharmacol. 2017;120:38–55. doi: 10.1016/j.neuropharm.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Johnsen KB, Burkhart A, Thomsen LB, Andresen TL, Moos T. Targeting the transferrin receptor for brain drug delivery. Prog Neurobiol. 2019;181:101665. doi: 10.1016/j.pneurobio.2019.101665. [DOI] [PubMed] [Google Scholar]
- 17.Pardridge WM, Kang YS, Buciak JL, Yang J. Human insulin receptor monoclonal antibody undergoes high affinity binding to human brain capillaries in vitro and rapid transcytosis through the blood–brain barrier in vivo in the primate. Pharm Res. 1995;12(6):807–16. doi: 10.1023/A:1016244500596. [DOI] [PubMed] [Google Scholar]
- 18.Haqqani AS, Delaney CE, Brunette E, Baumann E, Farrington GK, Sisk W, Eldredge J, Ding W, Tremblay TL, Stanimirovic DB. Endosomal trafficking regulates receptor-mediated transcytosis of antibodies across the blood brain barrier. J Cereb Blood Flow Metab. 2018;38(4):727–40. doi: 10.1177/0271678X17740031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zuchero YJ, Chen X, Bien-Ly N, Bumbaca D, Tong RK, Gao X, Zhang S, Hoyte K, Luk W, Huntley MA, et al. Discovery of novel blood–brain barrier targets to enhance brain uptake of therapeutic antibodies. Neuron. 2016;89(1):70–82. doi: 10.1016/j.neuron.2015.11.024. [DOI] [PubMed] [Google Scholar]
- 20.Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C, Bien-Ly N, Hersom M, Maloney JA, Meilandt WJ, et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med. 2014;6(261):261ra154. doi: 10.1126/scitranslmed.3009835. [DOI] [PubMed] [Google Scholar]
- 21.Boado RJ, Pardridge WM. Brain and organ uptake in the rhesus monkey in vivo of recombinant iduronidase compared to an insulin receptor antibody–iduronidase fusion protein. Mol Pharmaceut. 2017;14(4):1271–77. doi: 10.1021/acs.molpharmaceut.6b01166. [DOI] [PubMed] [Google Scholar]
- 22.Gadkar K, Yadav D, Zuchero J, Couch J, Kanodia J, Kenrick M, Atwal J, Dennis M, Prabhu S, Watts R, et al. Mathematical PKPD and safety model of bispecific TfR/BACE1 antibodies for the optimization of antibody uptake in brain. Eur J Pharm Biopharm. 2016;101:53–61. doi: 10.1016/j.ejpb.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 23.Kanodia JS, Gadkar K, Bumbaca D, Zhang Y, Tong RK, Luk W, Hoyte K, Lu Y, Wildsmith KR, Couch JA, et al. Prospective design of anti‐transferrin receptor bispecific antibodies for optimal delivery into the human brain. CPT Pharmacometrics Syst Pharmacol. 2016;5(5):283–91. doi: 10.1002/psp4.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Couch JA, Yu YJ, Zhang Y, Tarrant JM, Fuji RN, Meilandt WJ, Solanoy H, Tong RK, Hoyte K, Luk W, et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med. 2013;5(183):183ra157. doi: 10.1126/scitranslmed.3005338. [DOI] [PubMed] [Google Scholar]
- 25.Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P, Rueger P, Stracke JO, Lau W, Tissot AC, et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron. 2014;81(1):49–60. doi: 10.1016/j.neuron.2013.10.061. [DOI] [PubMed] [Google Scholar]
- 26.Weber F, Bohrmann B, Niewoehner J, Fischer JAA, Rueger P, Tiefenthaler G, Moelleken J, Bujotzek A, Brady K, Singer T, et al. Brain shuttle antibody for Alzheimer’s disease with attenuated peripheral effector function due to an inverted binding mode. Cell Rep. 2018;22(1):149–62. doi: 10.1016/j.celrep.2017.12.019. [DOI] [PubMed] [Google Scholar]
- 27.Klein G, Delmar P, Voyle N, Rehal S, Hofmann C, Abi-Saab D, Andjelkovic M, Ristic S, Wang G, Bateman R, et al. Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alz Res Therapy. 2019;11(1):101. doi: 10.1186/s13195-019-0559-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Novakovic D, Feligioni M, Scaccianoce S, Caruso A, Piccinin S, Schepisi C, Errico F, Mercuri NB, Nicoletti F, Nisticò R. Profile of gantenerumab and its potential in the treatment of Alzheimer’s disease. Drug Des Devel Ther. 2013;7:1359–64. doi: 10.2147/DDDT.S53401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, Klunk WE, Ashford E, Yoo K, Xu ZX, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198–207. doi: 10.1001/archneurol.2011.1538. [DOI] [PubMed] [Google Scholar]
- 30.Klein G, Delmar P, Kerchner G, Hofmann C, Abi-Saab D, Davis A, Voyle N, Baudler M, Fontoura P, Doody R. Thirty-six-month amyloid positron emission tomography results show continued reduction in amyloid burden with subcutaneous gantenerumab. J Prev Alzheimers Dis. 2021;8:1–4. doi: 10.14283/jpad.2020.68. [DOI] [PubMed] [Google Scholar]
- 31.Bateman R, Smith J, Donohue MC, Delmar P, Abbas R, Salloway S, Wojtowicz J, Blennow K, Bittner T, Black SE, et al. Topline results of phase III GRADUATE I & II pivotal trials with subcutaneous gantenerumab. J Prev Alzheimers Dis. 2022;9:S10. [Google Scholar]
- 32.Ltd. FH-LR . [Ad Hoc Announcement Pursuant To Art. 53 LR] Roche Provides Update On Phase III GRADUATE Programme Evaluating Gantenerumab In Early Alzheimer’s Disease. 2022. [accessed 2023 March 16]. https://www.roche.com/media/releases/med-cor-2022-11-14.
- 33.Madabushi R, Wang Y, Zineh I. A holistic and integrative approach for advancing model-informed drug development. CPT Pharmacom & Syst Pharma. 2019;8(1):9–11. doi: 10.1002/psp4.12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Retout S, Gieschke R, Serafin D, Weber C, Frey N, Hofmann C. Disease modeling and model-based meta-analyses to define a new direction for a phase III program of gantenerumab in Alzheimer’s disease. Clin Pharma And Therapeutics. 2022;111(4):857–66. doi: 10.1002/cpt.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: What have we learned? mAbs. 2011;3(1):61–66. doi: 10.4161/mabs.3.1.13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.ICH S6 (R1) Preclinical safety evaluation of biotechnology-derived pharmaceuticals . International Council For Harmonisation Of Technical Requirements For Pharmaceuticals For Human Use (ICH); 1997. [accessed 2023 14/08/2023]. https://www.ich.org/.
- 37.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29(10):457–66. doi: 10.1093/protein/gzw040. [DOI] [PubMed] [Google Scholar]
- 38.Kariolis MS, Wells RC, Getz JA, Kwan W, Mahon CS, Tong R, Kim DJ, Srivastava A, Bedard C, Henne KR, et al. Brain delivery of therapeutic proteins using an Fc fragment blood–brain barrier transport vehicle in mice and monkeys. Sci Transl Med. 2020;12(545):12. doi: 10.1126/scitranslmed.aay1359. [DOI] [PubMed] [Google Scholar]
- 39.Pardridge WM. CSF, blood–brain barrier, and brain drug delivery. Expert Opin Drug Deliv. 2016;13(7):963–75. doi: 10.1517/17425247.2016.1171315. [DOI] [PubMed] [Google Scholar]
- 40.Chiao P, Bedell BJ, Avants B, Zijdenbos AP, Grand’maison M, O’Neill P, O’Gorman J, Chen T, Koeppe R. Impact of reference and target region selection on amyloid PET SUV ratios in the phase 1b PRIME study of aducanumab. J Nucl Med. 2019;60(1):100–06. doi: 10.2967/jnumed.118.209130. [DOI] [PubMed] [Google Scholar]
- 41.Chang H, Morrow K, Bonacquisti E, Zhang W, Shah D. Antibody pharmacokinetics in rat brain determined using microdialysis. MAbs. 2018;10(6):843–53. doi: 10.1080/19420862.2018.1473910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Villaseñor R, Schilling M, Sundaresan J, Lutz Y, Collin L. Sorting tubules regulate blood–brain barrier transcytosis. Cell Rep. 2017;21(11):3256–70. doi: 10.1016/j.celrep.2017.11.055. [DOI] [PubMed] [Google Scholar]
- 43.Villaseñor R, Ozmen L, Messaddeq N, Grüninger F, Loetscher H, Keller A, Betsholtz C, Freskgård PO, Collin L. Trafficking of endogenous immunoglobulins by endothelial cells at the blood–brain barrier. Science Reports. 2016;6(1):25658. doi: 10.1038/srep25658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madabushi R, Benjamin JM, Grewal R, Pacanowski MA, Strauss DG, Wang Y, Zhu H, Zineh I. The US Food and drug administration’s model-informed drug development paired meeting pilot program: early experience and impact. Clin Pharma And Therapeutics. 2019;106(1):74–78. doi: 10.1002/cpt.1457. [DOI] [PubMed] [Google Scholar]
- 45.Ruderisch N, Schlatter D, Kuglstatter A, Guba W, Huber S, Cusulin C, Benz J, Rufer AC, Hoernschemeyer J, Schweitzer C, et al. Potent and selective BACE-1 peptide inhibitors lower brain Aβ levels mediated by brain shuttle transport. EBioMedicine. 2017;24:76–92. doi: 10.1016/j.ebiom.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campos CR, Kemble AM, Niewoehner J, Freskgård PO, Urich E, Ginsberg SD. Brain shuttle neprilysin reduces central amyloid-β levels. PloS One. 2020;15(3):e0229850. doi: 10.1371/journal.pone.0229850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Monolix . Ver. 4.3.3 [software]. Antony (France): Lixoft; 2014. September. [Google Scholar]
- 48.Wang J, Iyer S, Fielder PJ, Davis JD, Deng R. Projecting human pharmacokinetics of monoclonal antibodies from nonclinical data: comparative evaluation of prediction approaches in early drug development. Biopharm & Drug Disp. 2016;37(2):51–65. doi: 10.1002/bdd.1952. [DOI] [PubMed] [Google Scholar]
- 49.Valentin J. Basic anatomical and physiological data for use in radiological protection: reference values: ICRP publication 89. Ann ICRP. 2002;32(3–4):1–277. doi: 10.1016/S0146-6453(03)00002-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.