

Abstract
Background & Aims:
Gain-of-function (GOF) mutations of CTNNB1 and loss-of-function (LOF) mutations of AXIN1 are recurrent genetic alterations in hepatocellular carcinoma (HCC). We aim to investigate the functional contribution of Hippo/YAP/TAZ in GOF CTNNB1 or LOF AXIN1 mutant HCCs.
Approach & Results:
The requirement of YAP/TAZ in c-Met/β-Catenin and c-Met/sgAxin1 driven HCC was analyzed using conditional Yap, Taz, and Yap;Taz knockout (KO) mice. Mechanisms of AXIN1 in regulating YAP/TAZ were investigated using AXIN1 mutated HCC cells. Hepatocyte-specific inducible TTR-CreERT2 KO system was applied to evaluate the role of Yap;Taz during tumor progression. Cabozantinib and G007-LK combinational treatment were tested in vitro and in vivo. Nuclear YAP/TAZ was strongly induced in c-Met/sgAxin1 mouse HCC cells. Activation of Hippo via overexpression of Lats2 or concomitant deletion of Yap and Taz significantly inhibited c-Met/sgAxin1 driven HCC development, whereas the same approaches had mild effects in c-Met/β-Catenin HCCs. YAP is the major Hippo effector in c-Met/β-Catenin HCCs, and both YAP and TAZ are required for c-Met/sgAxin1 dependent hepatocarcinogenesis. Mechanistically, AXIN1 binds to YAP/TAZ in human HCC cells and regulates YAP/TAZ stability. Genetic deletion of YAP/TAZ suppresses already formed c-Met/sgAxin1 liver tumors, supporting the requirement of YAP/TAZ during tumor progression. Importantly, tankyrase inhibitor G007-LK, which targets Hippo and Wnt pathways, synergizes with cabozantinib, a c-MET inhibitor, leading to tumor regression in the c-Met/sgAxin1 HCC model.
Conclusions:
Our studies demonstrate that YAP/TAZ are major signaling molecules downstream of LOF AXIN1 mutant HCCs, and targeting YAP/TAZ as an effective treatment against AXIN1 mutant human HCCs.
Keywords: Hepatocellular carcinoma, Wnt/β-Catenin, Hippo/YAP/TAZ, Tankyrase inhibitor
Introduction
Hepatocellular carcinoma (HCC), the most common form of primary liver cancer, continues to increase in incidence and mortality over decades(1). Treatment options for advanced staged HCC have expanded during the past several years. Immunotherapies and target therapies, such as atezolizumab plus bevacizumab, have become the first-line therapies for advanced HCC. Additional multi-tyrosine kinase inhibitors, such as sorafenib or lenvatinib, can also be applied to these patients(2). However, most of the HCC patients progress under immunotherapies or target therapies. Therefore, there is an urgent need to understand the mechanisms underlying HCC development and design novel therapeutics against this deadly malignancy.
The Wnt/β-Catenin pathway is a major cascade regulating liver development, homeostasis, regeneration, and tumorigenesis(3). AXIN1 loss of function (LOF) mutation and CTNNB1 gain of function (GOF) mutation are two important molecular events involved in HCC development(4). According to the TCGA dataset, CTNNB1 GOF mutations occur in ~30% of HCC cases, while AXIN1 LOF mutations are identified in ~8% of HCC cases, supporting the critical role of the Wnt/β-Catenin signaling during hepatocarcinogenesis(5). Importantly, CTNNB1 GOF mutations and AXIN1 LOF mutations are mutually exclusive (p<0.001; Supplementary Figure 1), suggesting that both mutants may promote HCC pathogenesis via aberrant activation of the Wnt/β-Catenin pathway(6, 7). Nevertheless, multiple studies also propose that CTNNB1 GOF mutations and AXIN1 LOF mutations may not be completely equivalent during hepatocarcinogenesis. For instance, it has been shown that canonical β-Catenin target genes, such as GS, AXIN2, TBX3, etc., are commonly upregulated in CTNNB1 GOF mutant HCCs, but not AXIN1 LOF mutant HCCs(8, 9). Another noticeable difference is the interaction with the Hippo cascade, an evolutionally conserved tumor suppressor pathway that has been implicated in liver tumor development(10), with YAP and TAZ as the downstream co-activators(11). Specifically, a previous study showed that YAP and β-Catenin nuclear concurrent localization is frequently observed in human hepatoblastoma (HB), but rarely detected in HCC(12). However, one has to be cautious as nuclear localization of β-Catenin may represent the activation of Wnt/β-Catenin cascade at very high levels. Importantly, Abitbol S et al. analyzed the gene signatures enriched in human HCCs with LOF AXIN1 mutations, and the results suggested the enrichment of activated YAP signatures in these HCC specimens(8). Thus, these studies indicate that the Hippo/YAP/TAZ cascade might be differentially regulated during AXIN1 LOF mutation- or CTNNB1 GOF mutation-driven hepatocarcinogenesis.
Activation of c-MET is an important oncogenic signaling event occurring in over 40% of human HCC samples. Also, simultaneous activation of c-MET and CTNNB1 GOF mutations could be identified in ~10% of human HCC samples(13), whereas c-MET activations with AXIN1 LOF mutations co-occur in ~4% of human HCCs(4). In our recent studies, we established two murine HCC models via hydrodynamic transfection and sleeping beauty mediated somatic integration: one with activated mutant forms of β-Catenin(ΔN90-β-Catenin) and c-Met (c-Met/β-Catenin)(13), and another one with CRISPR-based gene deletion of AXIN1 (sgAxin1) and c-Met (c-Met/sgAxin1)(4). Molecular analyses suggest that these two models recapitulate human HCCs with CTNNB1 GOF mutations and AXIN1 LOF mutations, providing the tool to genetically dissect the signaling pathways downstream of CTNNB1 GOF mutants and AXIN1 LOF mutants.
In the current study, using c-Met/β-Catenin and c-Met/sgAxin1 murine HCC models, we investigated whether the Hippo/YAP/TAZ axis is differentially regulated by distinct oncogenic stimuli affecting CTNNB1 and AXIN1 genes.
Materials and Methods
Constructs and reagents
The plasmids used in this study, including pT3-EF1a (Vector), pT3-EF1a-c-Met (human), pT3-EF1α-ΔN90-β-Catenin (human), pT3-EF1a-Lats2 (mouse), pX330-sgAxin1 (mouse), pX330-sgAXIN1 (human), pT3-EF1α-AXIN1 (human), pT3-EF1α-EGFP, pCMV, pCMV-Cre, pCMV-Sleeping Beauty transposase (SB), and pT3-TTRpro-CreERT2 were previously described(4, 14, 15). The characteristics of the plasmids were shown in Supplementary Table 1. Plasmids were purified using the GenElute HP Endotoxin-Free Plasmid Maxiprep Kit (Cat# NA0410, Sigma-Aldrich, St. Louis, MO, USA).
Mouse treatment and hydrodynamic injection
Wild-type FVB/N mice were obtained from Charles River (Wilmington, MA, USA). Yapflox/flox and Tazflox/flox mice were kindly provided by Dr. Eric Olson from the University of Texas Southwestern Medical Center (Dallas, TX), which were backcrossed to FVB/N genetic background for more than 5 generations respectively before using for the following experiments. Yapflox/flox;Tazflox/flox mice were generated by crossing the Yapflox/flox and Tazflox/flox mice followed by genotyping for validation. Mice used for hydrodynamic injection were 5.5- to 6-week-old. Hydrodynamic injections were performed as described(16). In brief, the plasmid-normal saline mixed solution was injected into the mice in 5~7 seconds, with the total volume equal to 0.1ml/g body weight. The dosages of each plasmid in the various tumor models are shown in Supplementary Table 2. Mice were monitored by abdominal palpation and euthanized when they developed a high burden of liver tumors, i.e., large abdominal masses. Mice were housed, fed, and monitored following protocols approved by the Committee for Animal Research at the University of California, San Francisco.
Statistical analysis
The Prism 6.0 software (GraphPad, San Diego, CA) was applied to analyze the data, which are presented as Means ± SD. Comparisons were performed with the two-tailed unpaired t-test or analysis of variance (ANOVA) when necessary. Kaplan-Meier survival data were evaluated using a log-rank (Mantel-Cox) test. p-value < 0.05 was considered statistically significant.
Please refer to the Supplementary data for detailed materials and methods.
Results
Differential activation of YAP/TAZ in c-Met/β-Catenin and c-Met/sgAxin1 mouse HCCs
As the first step to investigate the possible role(s) of the Hippo cascade in CTNNB1 GOF and AXIN1 LOF mutant HCCs, we analyzed the expression of the genes in this pathway in c-Met/β-Catenin(13) and c-Met/sgAxin1(4) mouse HCC samples. We found that LATS2 expression as well as LATS kinase activities (revealed by the levels of p-Aurora-BThr232)(17, 18) was downregulated in all mouse HCCs analyzed compared with normal livers. In contrast, YAP/TAZ was upregulated in the tumors compared with normal livers (Figure 1A). These data suggest the inactivation of the Hippo pathway in both mouse HCC models. Intriguingly, a side-by-side comparison between these two models demonstrated that total and nuclear YAP/TAZ expression was much higher in c-Met/sgAxin1 mouse HCCs (Figures 1B and 1C). CTGF and CYR61 are transcriptional targets of YAP/TAZ. Therefore, their mRNA levels could be used to measure the activation status of YAP/TAZ signaling. Consistently, upregulation of YAP/TAZ downstream target genes, Ctgf and Cyr61, was significantly more prominent in c-Met/sgAxin1 liver tumor samples (Figure 1D).
Figure 1. Differential activation of YAP/TAZ in c-Met/β-Catenin and c-Met/sgAxin1 mouse HCCs.
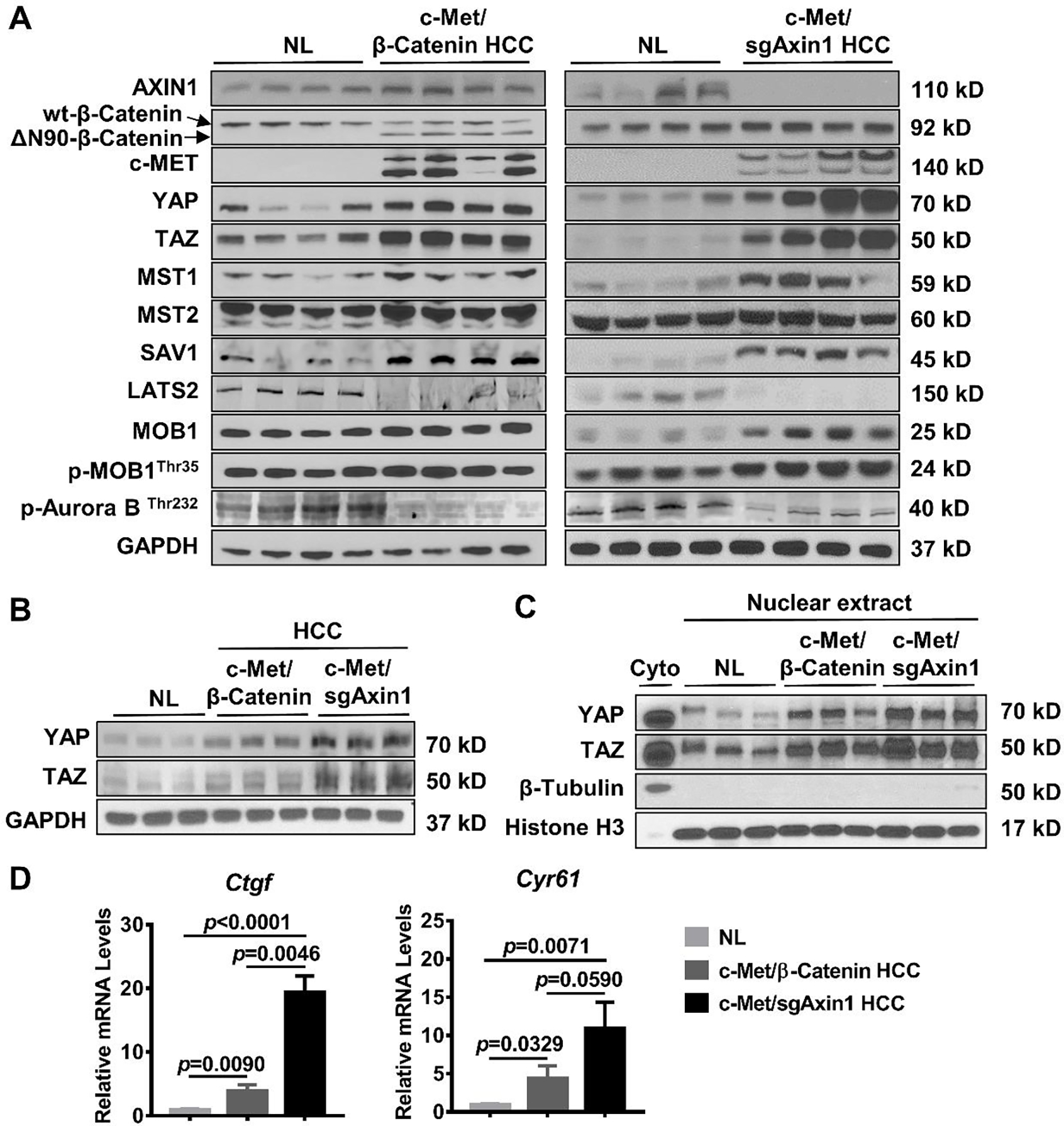
(A) Expression of AXIN1, β-Catenin, c-MET, YAP/TAZ, p-Aurora BThr232 and Hippo signaling kinases in c-Met/β-Catenin and c-Met/sgAxin1 mouse HCCs determined by Western blot analysis. GAPDH was used as a loading control. (B) Comparison of total YAP and TAZ expression in mouse normal liver (NL), c-Met/β-Catenin, and c-Met/sgAxin1 mouse HCC tissues. GAPDH was used as a loading control. (C) Comparison of nuclear YAP and TAZ expression in mouse normal liver (NL), c-Met/β-Catenin, and c-Met/sgAxin1 mouse HCC tissues. β-Tubulin and Histone H3 were used as the loading controls. (D) Comparison of Ctgf and Cyr61 mRNA expression in mouse normal liver (NL), c-Met/β-Catenin, and c-Met/sgAxin1 mouse HCC tissues.
Altogether, these data indicate a more robust activation of YAP/TAZ in the c-Met/sgAxin1 mouse HCC model than in c-Met/β-Catenin mice, thus recapitulating what has been described in human HCC.
Activation of the Hippo tumor suppressor strongly inhibits c-Met/sgAxin1 induced hepatocarcinogenesis in vivo
To investigate the function of the Hippo cascade during c-Met/sgAxin1- and c-Met/β-Catenin-driven hepatocarcinogenesis, we induced the activation of the Hippo onco-suppressor pathway in the two murine HCC models by overexpressing the Lats2 kinase(19). Both Lats1 and Last2 are core kinases that activate the Hippo signaling, and the two kinases are functionally redundant(19). Therefore, overexpression of either Lats1 or Last2 could readily activate the Hippo signaling. We co-injected the Lats2 plasmid with c-Met and sgAxin1 plasmids into the mouse liver (c-Met/sgAxin1/Lats2). Additional mice were injected with c-Met/sgAxin1 together with pT3-EF1α empty vector as control (c-Met/sgAxin1/pT3) (Figure 2A). Notably, Lats2 overexpression severely impaired the development of c-Met/sgAxin1 mouse HCCs (Figure 2B). This was accompanied by decreased liver weight and liver-to-body weight ratio in c-Met/sgAxin1/Lats2 mice (Figure 2C). Histopathological investigation revealed significant inhibition of cell proliferation as indicated by Ki67 (+) cell percentages (Figures 2D and 2E). Deletion of AXIN1 and overexpression of c-MET were confirmed by Western blotting in all c-Met/sgAxin1 tumors, while downregulation of YAP/TAZ occurred in c-Met/sgAxin1/Lats2 liver tumor tissues (Figure 2F). It is noticed that protein levels of c-MET were lower in the Lats2 overexpressed tumors, which was likely due to the fact the analyzed tumor nodules were very small as our studies found no evidence that Lats2 regulated c-MET protein levels in HCC cells. Nuclear levels of YAP and TAZ were significantly repressed upon Lats2 overexpression (Figure 2G). Consistently, the expression of Ctgf and Cyr61 was also downregulated (Figure 2H). Therefore, activation of Hippo via overexpressing Last2 profoundly inhibited Axin1 LOF mutant induced HCC in vivo.
Figure 2. Lats2 overexpression blocks c-Met/sgAxin1 mouse HCC development.
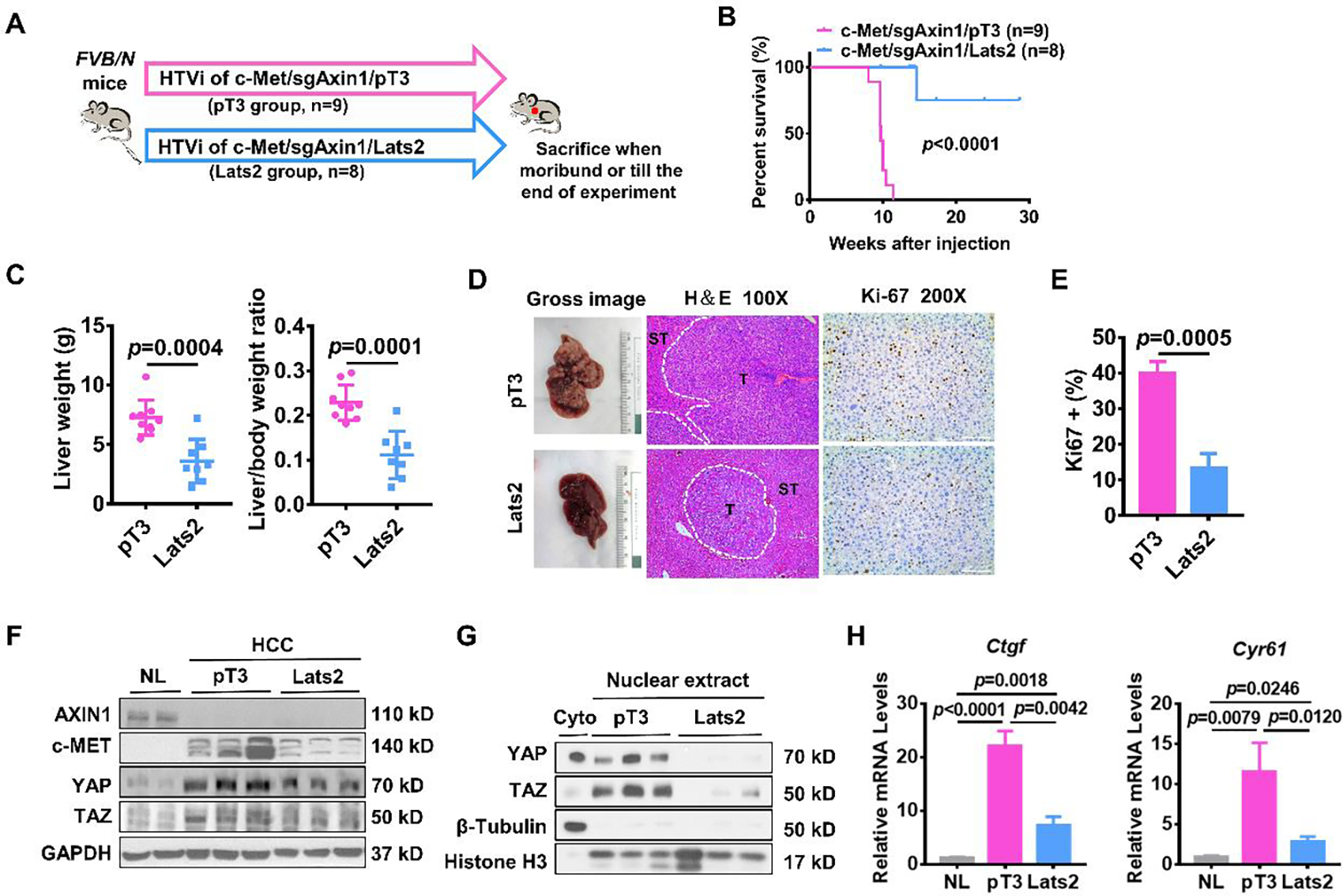
(A) Study design. FVB/N mice were injected with c-Met/sgAxin1/pT3 (n=9) and c-Met/sgAxin1/Lats2 (n=8) plasmids using hydrodynamic tail vein injection (HTVi). (B) Survival curve of mice in both groups. Kaplan-Meier comparison was performed, p<0.0001. (C) Comparisons of liver weight and liver weight to body weight ratio in both groups. (D) Representative images of macroscopic pictures, H&E, and Ki-67 staining of the tumors in both groups. Scale bars: 200μm for 100X, 100μm for 200X. (E) Quantification of Ki67 positive percentage in both groups. (F) Levels of AXIN1, c-MET, YAP, and TAZ determined by Western blot analysis. GAPDH was used as a loading control. (G) Expression of nuclear YAP and TAZ determined by Western blot analysis. β-Tubulin and Histone H3 levels were used as a loading control. (H) Comparisons of Ctgf and Cyr61 mRNA expression in mouse normal liver (NL), c-Met/sgAxin1/pT3, and c-Met/sgAxin1/Lats2 mouse HCC tissues.
Next, we co-injected Lats2 with c-Met/β-Catenin (c-Met/β-Catenin/Lats2) into mice. Mice injected with c-Met/β-Catenin/pT3 plasmids were used as control (Figure 3A). Overexpression of Lats2 slightly delayed c-Met/β-Catenin mouse HCC development. Mice in the control group were moribund due to high tumor burden at ~8 weeks after injection, while mice injected with c-Met/β-Catenin/Lats2 became moribund ~10–12 weeks post-injection (Figure 3B). Similar eventual liver weight and liver weight to body weight ratio between both groups were noted (Figure 3C). Inhibition of cell proliferation was observed in c-Met/β-Catenin/Lats2 tumors (Figures 3D and 3E). The expression of β-Catenin and c-MET in all mouse HCCs was validated by Western blotting. Simultaneously, overexpression of Lats2 triggered the downregulation of total and nuclear YAP/TAZ as the YAP/TAZ target gene Cyr61, but not Ctgf (Figures 3F–3H).
Figure 3. Lats2 overexpression slightly delays c-Met/β-Catenin mouse HCC development.
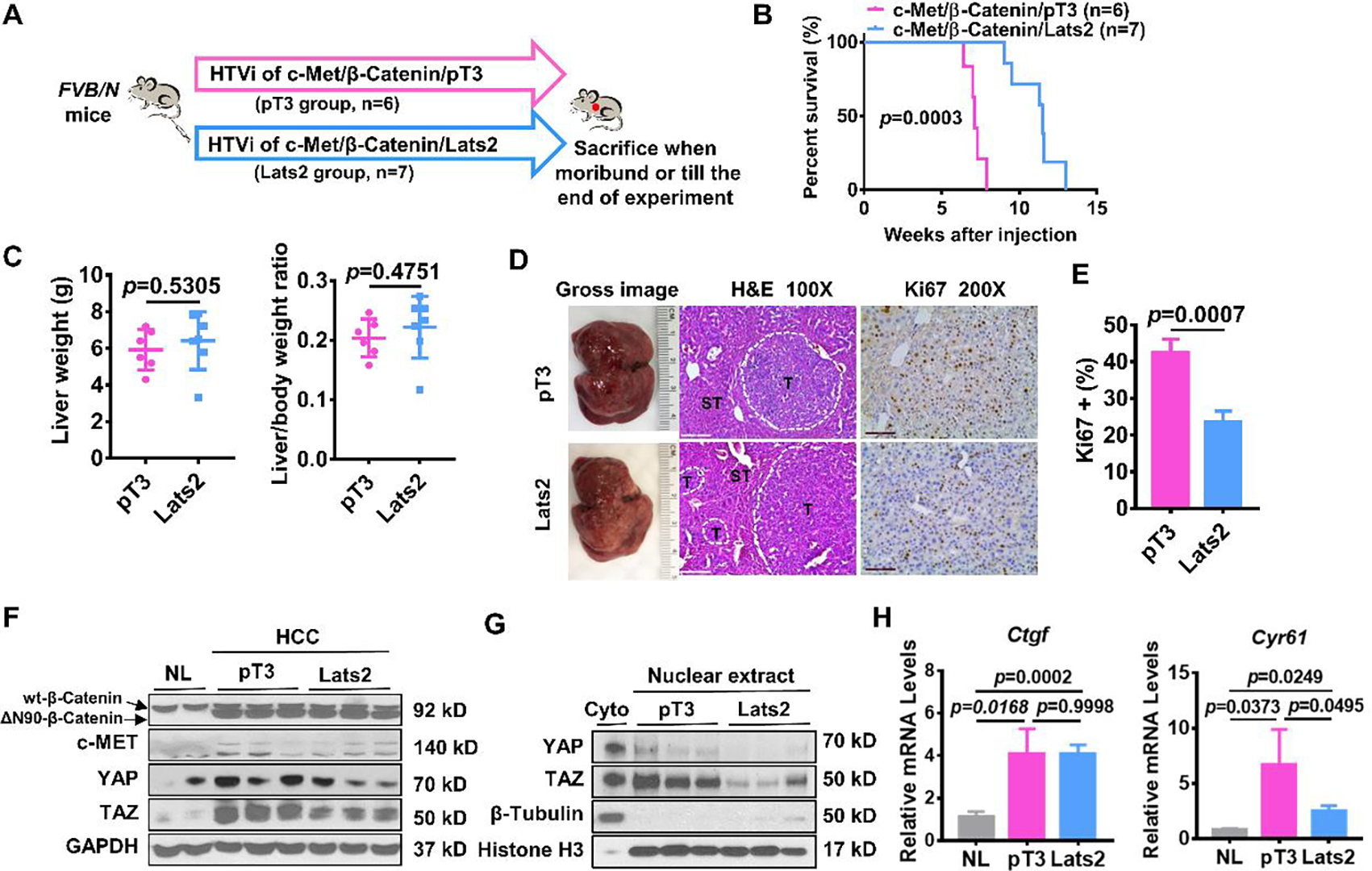
(A) Study design. FVB/N mice were injected with c-Met/β-Catenin/pT3 (n=6) and c-Met/β-Catenin/Lats2 (n=7) plasmids using hydrodynamic tail vein injection (HTVi). (B) Survival curve of mice in both groups. Kaplan-Meier comparison was performed, p=0.0003. (C) Comparisons of liver weight and liver weight to body weight ratio in both groups. (D) Representative images of macroscopic pictures, H&E, and Ki-67 staining of the tumor in both groups. Scale bars: 200μm for 100X, 100μm for 200X. (E) Quantification of Ki67 positive percentage in both groups. (F) Levels of β-Catenin, c-MET, YAP, and TAZ determined by Western blot analysis. GAPDH was used as a loading control. (G) Expression of nuclear YAP and TAZ determined by Western blot analysis. β-Tubulin and Histone H3 were used as a loading control. (H) Comparisons of Ctgf and Cyr61 mRNA expression in mouse normal liver (NL), c-Met/β-Catenin/pT3, and c-Met/β-Catenin/Lats2 mouse HCC tissues.
The role of the Hippo pathway in the regulation of HCC tumor growth was also analyzed in CTNNB1mut SNU398 cells, AXIN1null SNU449 cells, and AXIN1mut PLC/PRF/5 cells. Overexpression of LATS2 significantly suppressed cell growth in AXIN1 aberrant HCC cell lines as assessed by colony formation assays, whereas the growth inhibitory effects of LATS2 were relatively moderate in CTNNB1 mutated SNU398 cells (Supplementary Figure 2).
Overall, our data suggest that Hippo signaling activation inhibits c-Met/sgAxin1 mouse HCC development while having relatively mild effects on c-Met/β-Catenin mouse dependent hepatocarcinogenesis.
Concomitant depletion of Yap and Taz significantly delays c-Met/sgAxin1 mouse HCC development
To further dissect the involvement of the Hippo pathway downstream co-effectors, YAP and TAZ, in regulating c-Met/sgAxin1 and c-Met/β-Catenin mouse HCCs, we applied Yapflox/flox and Tazflox/flox conditional knockout (KO) mice. We co-injected oncogenes with pCMV/Cre plasmid into the conditional KO mice, which allowed the co-expression of the oncogenes in Yap and/or Taz KO hepatocytes. Additional conditional KO mice were co-injected with oncogenes with pCMV empty vector as the control. Deletion of Yap did not affect c-Met/sgAxin1 mouse HCC development (Supplementary Figure 3). In contrast, c-Met/β-Catenin induced hepatocarcinogenesis was slightly retarded after Yap depletion (Supplementary Figure 4). Deleting Taz did not affect the development of c-Met/sgAxin1 liver tumors (Supplementary Figure 5) or c-Met/β-Catenin mouse HCCs (Supplementary Figure 6).
Because YAP and TAZ are likely redundant in regulating tumorigenesis, we tested Yap;Taz double KO (DKO) mice. Strikingly, depletion of Yap and Taz strongly delayed the development of c-Met/sgAxin1 tumors (Figures 4A and 4B), phenocopying Lats2 overexpression (Figures 2A and 2B). Specifically, all c-Met/sgAxin1/pCMV injected Yapflox/flox;Tazflox/flox mice developed a high tumor burden and needed to be euthanized by 15~20 weeks post-injection. In contrast, only small tumor nodules formed in c-Met/sgAxin1/Cre injected Yapflox/flox;Tazflox/flox mice at 20 weeks post-injection (Figure 4B). In addition, liver weight and liver weight to body weight ratio were significantly decreased in Yap;Taz DKO mice (Figure 4C). Expression of c-MET and loss of AXIN1 and ablation of YAP/TAZ were confirmed by Western blotting (Figure 4D), and deletion of YAP was also validated by IHC (Supplementary Figure 7A). Cell proliferation was significantly inhibited in Yap;Taz DKO liver tumor samples (Figures 4E and 4F).
Figure 4. Depletion of Yap/Taz inhibits c-Met/sgAxin1 while slightly delaying c-Met/β-Catenin mouse HCC development.
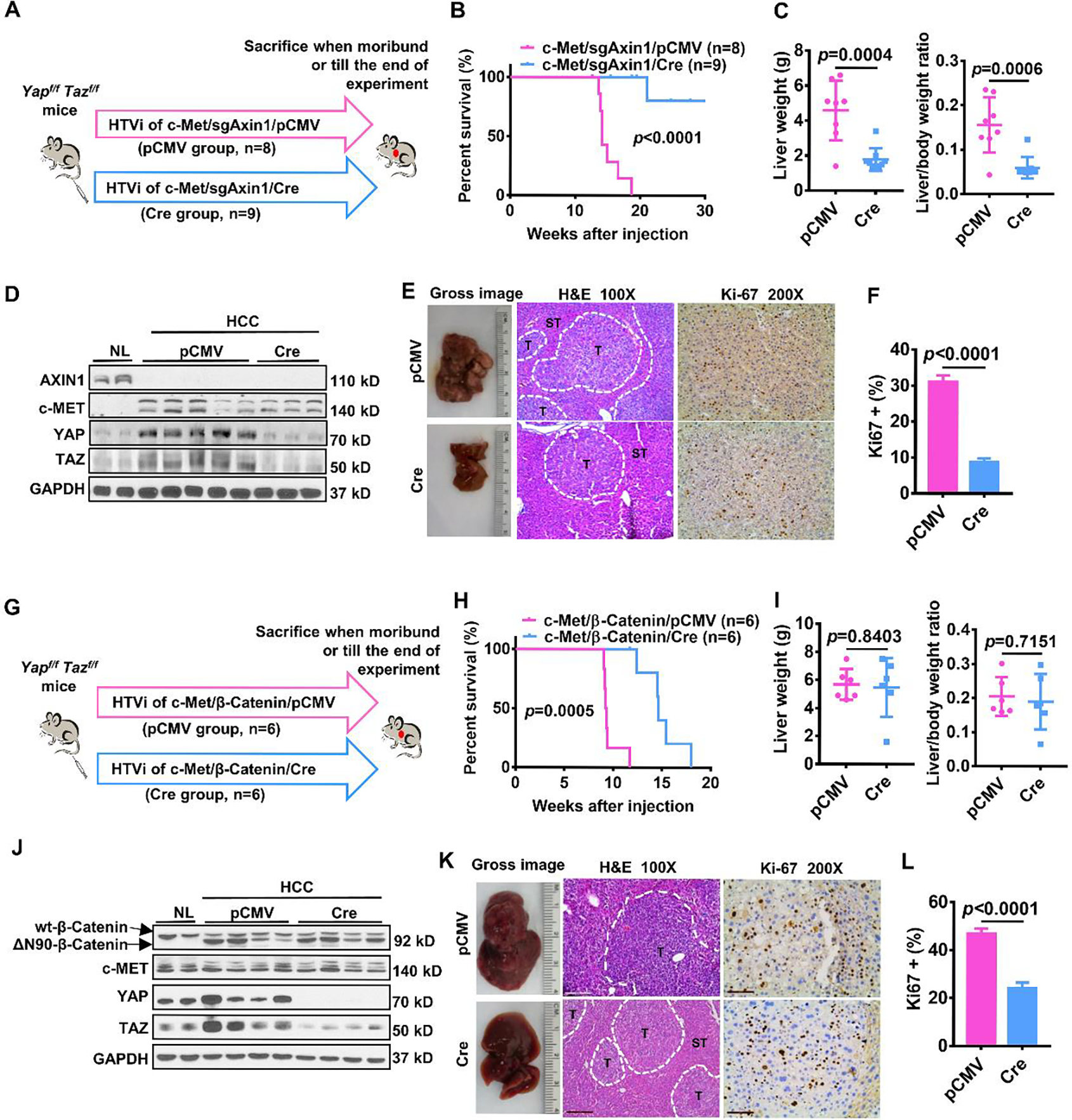
(A) Study design. Yapflox/floxTazflox/flox mice were injected with c-Met/sgAxin1/pCMV (n=8) and c-Met/sgAxin1/Cre (n=9) plasmids using hydrodynamic tail vein injection (HTVi), respectively. (B) Survival curve of mice in both groups. Kaplan-Meier comparison was performed, p<0.0001. (C) Comparisons of liver weight and liver weight to body weight ratio in both groups. (D) Levels of AXIN1, c-MET, YAP, and TAZ determined by Western blot analysis. GAPDH was used as a loading control. (E) Representative images of macroscopic pictures, H&E, and Ki-67 staining of the tumor in both groups. Scale bars: 200μm for 100X, 100μm for 200X. (F) Quantification of Ki67 positive percentage in both groups. (G) Study design. Yapflox/floxTazflox/flox mice were injected with c-Met/ β-Catenin/pCMV (n=6) and c-Met/β-Catenin/Cre (n=6) plasmids using hydrodynamic tail vein injection (HTVi), respectively. (H) Survival curve of mice in both groups. Kaplan-Meier comparison was performed, p=0.0005. (I) Comparisons of liver weight and liver weight to body weight ratio in both groups. (J) Expression of β-Catenin, c-MET, YAP, and TAZ determined by Western blot analysis. GAPDH was used as a loading control. (K) Representative images of macroscopic pictures, H&E, and Ki-67 staining of the tumor in both groups. Scale bars: 200μm for 100X, 100μm for 200X. (L) Quantification of Ki67 positive percentage in both groups.
In c-Met/β-Catenin-driven HCC, depletion of Yap/Taz mildly retarded c-Met/β-Catenin tumor formation (Figures 4G and 4H), similar to that observed in Yap knockout mice (Supplementary Figures 4A and 4B) as well as when Lats2 was overexpressed (Figures 3A and 3B). There was no significant difference in liver weight and liver weight to body weight ratio between pCMV and Cre injected mice (Figure 4I). The expression of c-MET, β-Catenin as well as ablation of YAP/TAZ were validated by Western blotting (Figure 4J). IHC did not detect immunoreactivity for YAP in Cre injected liver tumor tissues (Supplementary Figure 7B). Consistent with what we observed in c-Met/β-Catenin/Lats2 tumors, concomitant deletion of Yap and Taz led to significantly decreased tumor cell proliferation (Figures 4K and 4L).
In summary, our study demonstrates that the Hippo/YAP/TAZ cascade has a pivotal role in c-Met/sgAxin1 driven HCC, whereas it mildly contributes to c-Met/β-Catenin induced hepatocarcinogenesis. The results are consistent with the observation in human data supporting the differentially regulated Hippo/YAP/TAZ cascade in GOF CTNNB1 and LOF AXIN1 mutant HCCs.
AXIN1 regulates YAP/TAZ stability in human HCC cell lines
As our findings support the critical function of the Hippo/YAP/TAZ cascade in LOF AXIN1 mutant hepatocarcinogenesis, we investigated the responsible molecular mechanisms. We first examined whether AXIN1 might regulate YAP and TAZ mRNA expression. We analyzed YAP and WWTR1, which encodes TAZ, mRNA levels related to AXIN1 mutation status and AXIN1 mRNA expression in the TCGA LIHC dataset (Supplementary Figure 8A). As the control, low AXIN1 mRNA expression was detected in LOF AXIN1 mutant HCCs (Supplementary Figure 8B). In contrast, neither AXIN1 mutation status nor AXIN1 mRNA levels correlated with YAP or WWTR1 mRNA expression in human HCCs (Supplementary Figures 8B and 8C). In the murine HCC models, no consistent changes in Yap and Wwtr1 mRNA levels were found (Supplementary Figures 8D and 8E). The result suggests that AXIN1 is more likely to regulate YAP/TAZ at the post-transcriptional level.
To investigate the molecular mechanisms involved, we applied a panel of human HCC cells with different genetic backgrounds of AXIN1(20–22) and CTNNB1 mutation(22, 23) status (Figure 5A and Supplementary Table 7). Of note, all the AXIN1 mutated cells carry loss of function mutations, either resulting in releasing of β-Catenin from the β-Catenin destruction complex or deletion the critical phosphorylation (activating) site of AXIN1(21). Interestingly, among the AXIN1 LOF mutated human HCC cell lines, robust nuclear YAP accumulation was detected by immunofluorescence (Supplementary Figure 9A), supporting the activation of YAP in these cell lines. Consistently, the levels of YAP/TAZ target genes (CTGF and CYR61) from the Cancer Cell Line Encyclopedia (CCLE) cell line gene expression profiles dataset(24) were all higher than the average in the five AXIN1 mutant or null HCC cell lines (Supplementary Figure 9B), which independently verified that YAP/TAZ pathway was activated in AXIN1 mutant/null HCC cell lines.
Figure 5. AXIN1 regulates the stability of YAP/TAZ in human HCC cell lines.
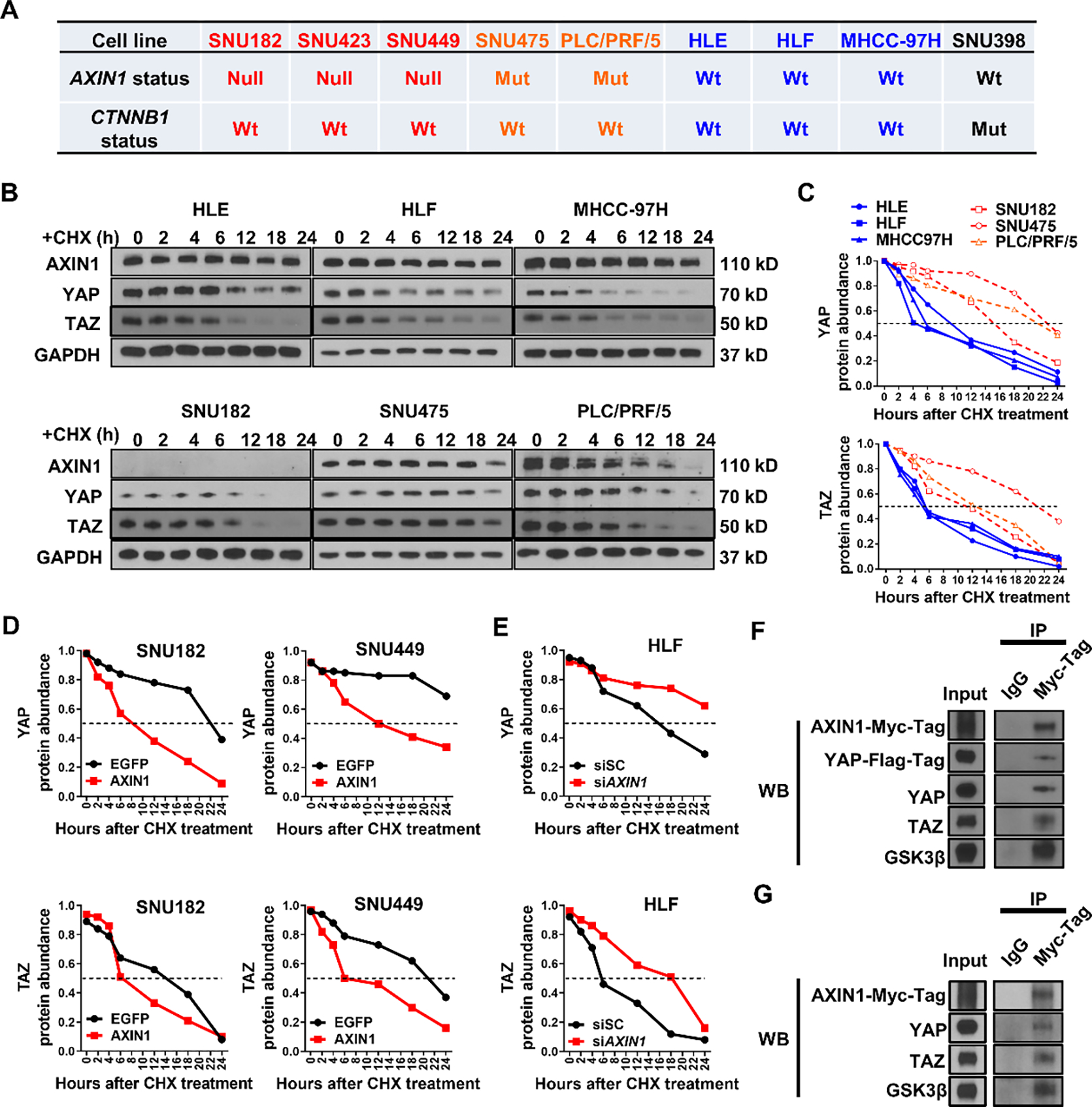
(A) AXIN1 and CTNNB1 mutation status in human HCC cell lines. Null, extremely low or undetectable of AXIN protein expression; Mut, mutation; Wt, wild-type. (B) Western blot analysis showing AXIN1, YAP, and TAZ levels in human HCC cell lines 0, 2, 4, 6, 12, 18, and 24 hours of CHX administration. GAPDH was used as a loading control. (C) Quantification of YAP and TAZ protein abundance in human HCC cells 0, 2, 4, 6, 12, 18, and 24 hours of CHX administration. (D) Quantification of YAP and TAZ protein abundance in EGFP or AXIN1 transfected SUN182 and SNU449 cells 0, 2, 4, 6, 12, 18, and 24 hours of CHX administration. (E) Quantification of YAP and TAZ protein abundance in scrambled siRNA (siSC) or siAXIN1 transfected HLF cells 0, 2, 4, 6, 12, 18, and 24 hours of CHX administration. (F, G) Co-immunoprecipitation assay showing AXIN1-YAP/TAZ interaction. GSK3β was used as a positive control. B-G are representative results of three independent experiments.
The scaffold protein AXIN1 was generally regarded to function through protein-protein interaction(25). In addition, studies have shown that AXIN1 might interact with YAP and modulate its stability(26). Therefore, we analyzed the protein stability of YAP, TAZ, and AXIN1 in the panel of HCC cell lines using cycloheximide (CHX) chase assay. Protein abundance of endogenous YAP, TAZ, and AXIN1 were measured by Western blotting 0, 2, 4, 6, 12, 18, and 24 hours after CHX administration. We found that YAP and TAZ protein levels significantly decreased at ~6 hours after CHX administration in 3 AXIN1wt human HCC cell lines, while degradation of YAP and TAZ was considerably slower in 3 AXIN1 LOF mutated human HCC cell lines (Figure 5B). Further quantification of YAP and TAZ protein levels suggested that both had longer half-time in AXIN1 LOF mutated HCC cells (Figure 5C).
Subsequently, AXIN1null (extremely low or undetectable of AXIN protein expression) human HCC cell lines, including SNU182 and SNU449 cells, were transfected with Myc-AXIN1 or EGFP. The protein abundance of endogenous AXIN1, YAP, TAZ, and exogenous AXIN1 (Myc-tag) was determined by CHX chase assays and Western blotting. Strikingly, AXIN1 overexpression significantly shortened the protein half-life of YAP and TAZ (Figure 5D, Supplementary Figures 10A and 10B). In addition, AXIN1 overexpression also led to decreased nuclear levels of YAP and TAZ (Supplementary Figure 11). Consistently, mRNA levels of CTGF and CYR61 were also diminished upon AXIN1 transfection (Supplementary Figures 12A and 12B). Next, we silenced AXIN1 in the AXIN1wt HLF cells. We found that this led to the increased stability of YAP and TAZ proteins and triggered the upregulation of CTGF and CYR61 (Figure 5E and Supplementary Figures 10C and 12C). These findings indicate that AXIN1 regulates YAP and TAZ protein stability by promoting YAP/TAZ degradation.
Next, we tested the hypothesis that AXIN1 triggers YAP/TAZ degradation through AXIN1-YAP/TAZ interaction. Thus, we transfected AXIN1null SNU182 cell line with AXIN1 (with Myc-Tag) and YAP (with Flag-Tag), followed by Co-Immunoprecipitation (Co-IP) with an anti-Myc-Tag antibody. As a result, we found that AXIN1 could Co-IP with YAP and TAZ (Figure 5F). Furthermore, when SNU182 cells were transfected with AXIN1-Myc-Tag followed by Co-IP studies, AXIN1 could Co-IP with endogenous YAP and TAZ (Figure 5G).
In summary, AXIN1 binds to YAP/TAZ proteins, leading to their destabilization and limiting the intracellular concentration of YAP/TAZ in human HCC cell lines.
YAP/TAZ inhibition leads to c-Met/sgAxin1 mouse HCC regression
Having demonstrated the critical role of YAP/TAZ in LOF AXIN1 mutant HCC formation, we explored whether YAP and TAZ are required for c-Met/sgAxin1 mouse HCC progression as these studies would provide direct evidence to support targeting YAP/TAZ for HCC treatment. To this end, we introduced pT3-TTR-CreERT2 together with c-Met/sgAxin1 plasmids into Yapflox/flox;Tazflox/flox mice. Upon administration of Tamoxifen, Cre recombinase shall be activated and expressed in tumor cells. FVB/N mice (as control) and Yapflox/flox;Tazflox/flox mice were injected with c-Met/sgAxin1/TTR-CreERT2. A group of mice was sacrificed at 13 weeks post injection, and all mice had moderate liver tumor burdens and were destinated as a pre-treatment control. Additional tumor-bearing mice were treated with Tamoxifen at this time point (Figure 6A). FVB/N control mice have to be euthanized due to the high tumor burden by ~16 weeks post-injection. Surprisingly, Tamoxifen treated Yapflox/flox;Tazflox/flox mice survived until the end of observation (Figure 6B). The liver weights were significantly reduced in the Yapflox/flox;Tazflox/flox mice compared to the FVB/N group and pre-treatment group (Figure 6C), indicating that genetic deletion of YAP and TAZ led to the regression c-Met/sgAxin1 HCC. At the histological level, the cell proliferation was significantly inhibited in the Yap;Taz DKO tumors (Figures 6D and 6E). Thus, YAP and TAZ are required for c-Met/sgAxin1 HCC progression, supporting YAP/TAZ as effective targets for the treatment of HCC with LOF AXIN1 mutations.
Figure 6. Genetic depletion of Yap;Taz leads to regression of c-Met/sgAXIN1 tumor.
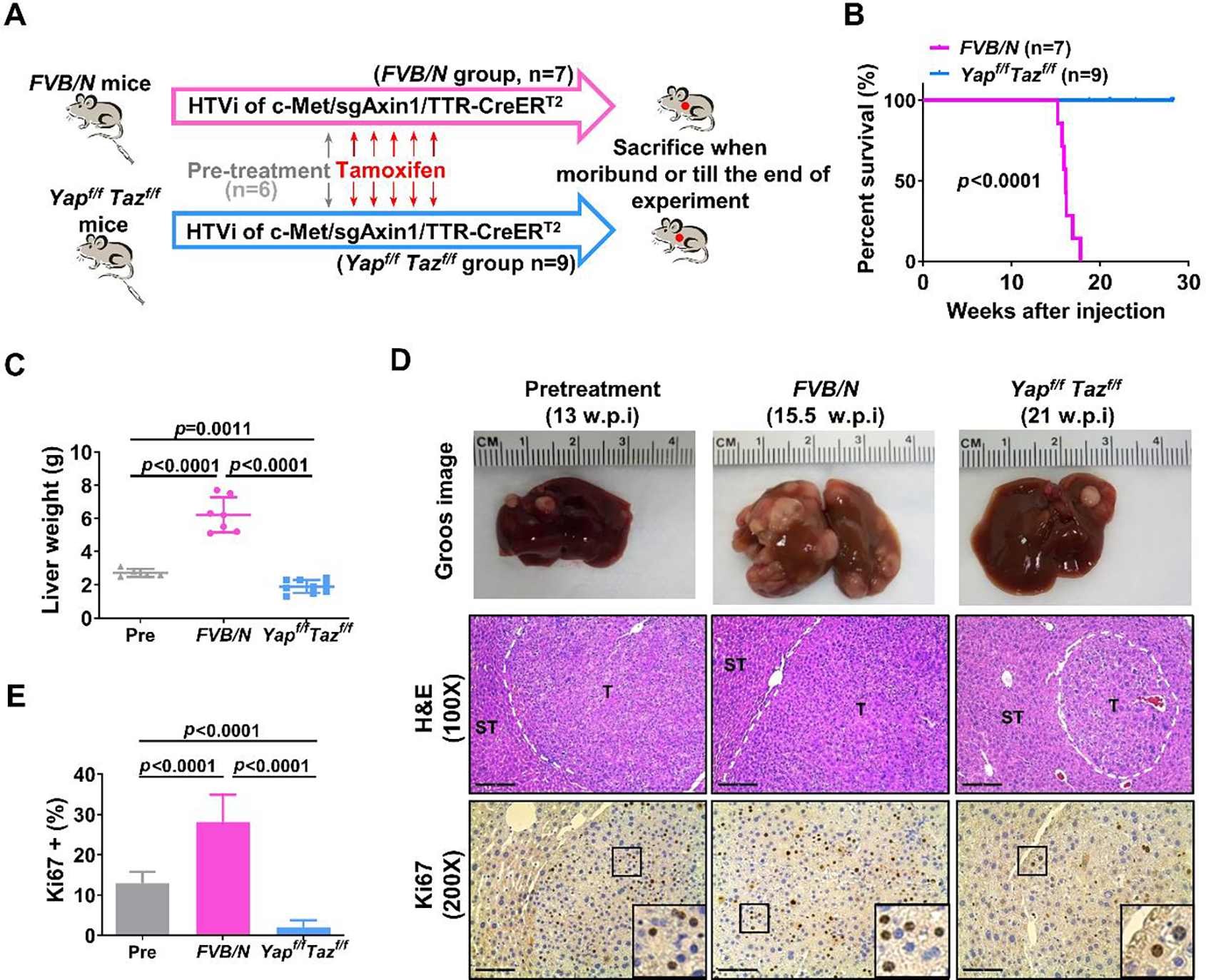
(A) Study design. FVB/N and Yap;Tazflox/flox mice were hydrodynamically injected (HTVi) with c-Met/sgAxin1/TTR-CreERT2 constructs. Mice (n=6) were sacrificed at 13 weeks post-injection (w.p.i) as a pre-treatment group, while the remaining mice were injected with Tamoxifen. All mice were sacrificed when moribund or till the end of observation. (B) Survival curves of FVB/N (n=7) and Yap;Tazflox/flox (n=9) mice treated with Tamoxifen. p-value was calculated by Log-rank (Mantel-Cox) test. (C) Comparison of liver weight in the three groups. (D) Representative images of macroscopic pictures, H&E, and Ki-67 staining of the tumors in the three groups. Scale bars: 200μm for 100X, 100μm for 200X. (E) Quantification of Ki67 positive percentage in the three groups.
Targeting YAP/TAZ cascade for c-Met/sgAxin1 HCC treatment
The results suggest that c-Met/sgAxin1 dependent hepatocarcinogenesis depends on YAP/TAZ for its development and progression. Thus, targeting this pathway may be effective for the treatment of HCC with LOF AXIN1 mutations. Previously, we have shown that G007-LK, a tankyrase inhibitor, suppressed HCC cell growth by modulating the Hippo cascade(27), and cabozantinib inhibited c-MET induced HCC development via suppressing c-MET/p-ERK cascade(14). Therefore, we hypothesized that G007-LK might be effective for the treatment of c-Met/sgAxin1 HCC, either alone or in combination with cabozantinib. Firstly, SNU449 (Axin1null) and SNU475 (Axin1Mut) cells, both expressing p-MET(14), were treated with G007-LK and cabozantinib, either alone or combined. Compared to single treatments, concomitant administration of G007-LK and cabozantinib induced decreased cell viability in HCC cells. The combination index was calculated, and all combination index values were less than 1 (Supplementary Figure 13), supporting that the two drugs synergized to inhibit HCC cell growth in vitro.
Subsequently, we tested the therapeutic potential of G007-LK, either alone or in combination with cabozantinib, in the c-Met/sgAxin1 model. Preliminary dosing studies suggested that treatment with cabozantinib (60mg/kg/day) and G007-LK (40mg/kg/day) were well-tolerated in mice, and were selected for the in vivo studies. Mice were injected with c-Met/sgAxin1 plasmids. Subsequently, these c-Met/sgAxin1 tumor-bearing mice were randomly separated into 5 cohorts. The first group was harvested at 8.5 weeks post hydrodynamic injection as the pre-treatment group. All mice exhibited moderate HCC liver tumor burden at this point, with an average liver weight of ~3g to mimic the early-stage HCC tumors in the clinical scenario. The remaining mice were treated with vehicle, cabozantinib, G007-LK, or cabozantinib/G007-LK for 3 weeks (Figure 7A). All mice developed large tumors in the vehicle treatment cohort. G007-LK-treated mice exhibited a slower but still progressive tumor growth, indicated by the lower tumor burden than the vehicle cohort but higher tumor burden than the pre-treatment cohort. Consistent with our previous study, cabozantinib was effective against mouse HCCs with activated c-MET(14). No differences in liver weight between the pre-treatment and cabozantinib therapy groups were detected, suggesting that cabozantinib induced stable disease in c-Met/sgAxin1 mice. Strikingly, the cabozantinib/G007-LK combination therapy exhibited a strong anti-neoplastic effect with the lowest liver weight, lower than that of pre-treatment mice (Figure 7B), indicating that the combination therapies caused tumor regression. The suppression of YAP/TAZ signaling by G007-LK was further confirmed by qPCR, showing downregulation of Ctgf and Cyr61 in the G007-LK treated tumors compared to the vehicle-treated tumors (Figure 7C). Of note, protein levels of YAP and TAZ decreased upon G007-LK/Cabozantinib treatment (Figure 7D). At the histological level, compared to the vehicle-treated group, cell proliferation (Ki67) was inhibited, and cell apoptosis (Cleaved Caspase 3) was promoted. In addition, the cabozantinib-treated and the combinational treatment group showed profound inhibition of microvascular density, as measured by anti-CD34 immunohistochemistry (Figures 7E–H).
Figure 7. G007-LK synergizes with cabozantinib to inhibit c-Met/sgAxin1 tumor development.
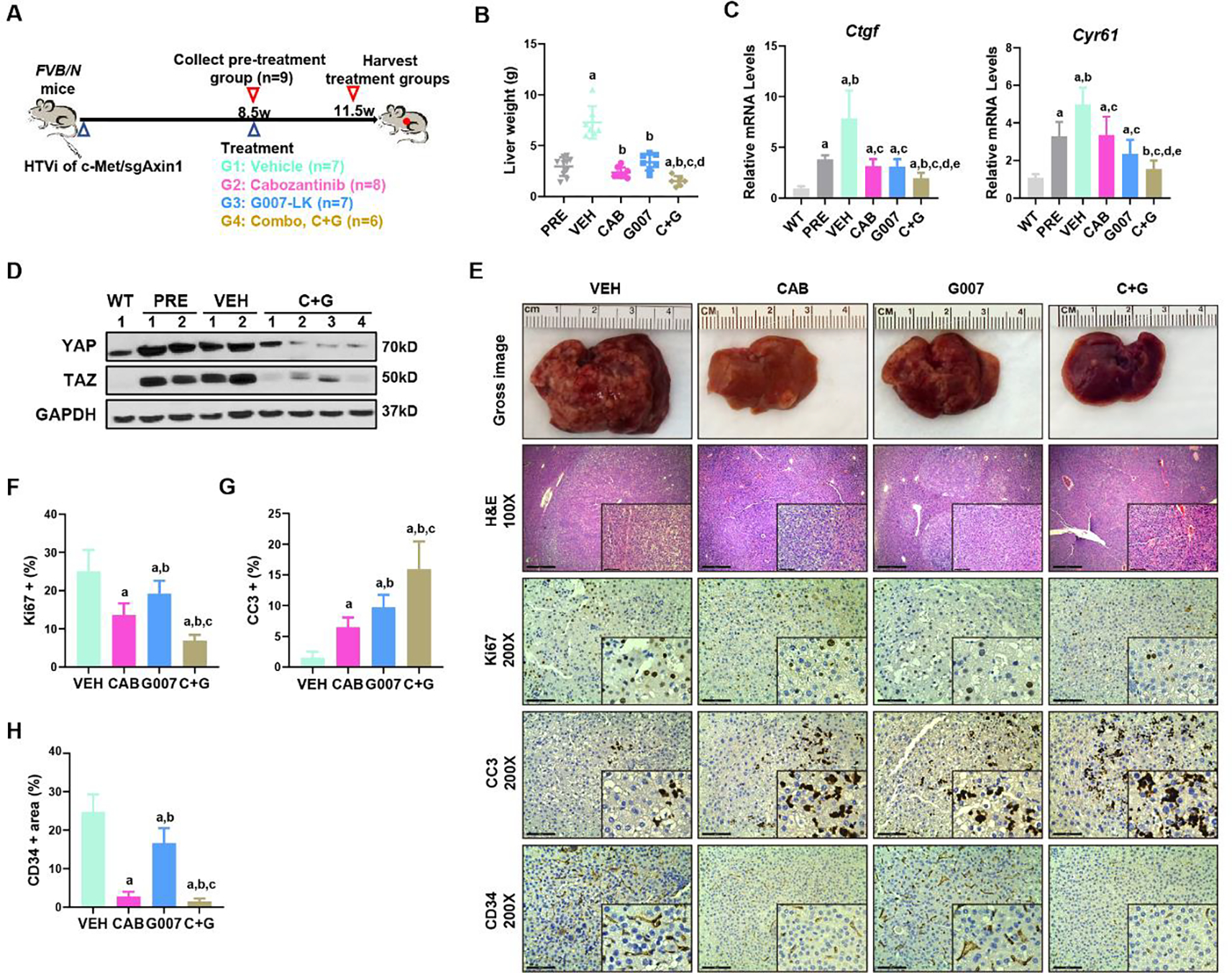
(A) Study design. FVB/N mice were hydrodynamically injected (HTVi) with c-Met/sgAxin1/SB constructs. At 8.5 weeks after injection, one group of mice (n=9) were sacrificed, and liver tissues were harvested as pre-treatment. Other mice were randomly assigned into the vehicle (VEH, n=7), Cabozantinib (CAB, n=8), G007-LK (G007, n=7), or Cabozantinib and G007-LK combinational (C+G, n=6) treated groups. Mice were treated for three weeks and subsequently were sacrificed. (B) Comparisons of the liver weight in the five groups. Mean ± SD; One-way ANOVA test. p<0.05, (a) vs PRE; (b) vs.VEH; (c) vs. CAB; (d) vs. G007. (C) qPCR results of Ctgf and Cyr61 in the normal liver (WT), pre-treatment (PRE), vehicle (VEH), and drug-treated liver tumors. Mean ± SD; One-way ANOVA test. p<0.05, (a) vs WT; (b) vs.PRE; (c) vs. VEH; (d) vs. CAB; (e) vs. G007. (D) Expression of YAP and TAZ determined by Western blot analysis. GAPDH was used as a loading control. (E) Representative images of macroscopic pictures, H&E, Ki-67, Cleaved caspase-3 (CC3), and CD34 staining of the tumors in the four groups. Scale bars: 200μm for 100X, 100μm for 200X. (F-H) Quantifications of Ki67 (F), CC3 (G), and CD34 (H) positive percentage in the four groups. Mean ± SD; One-way ANOVA test. p<0.05, (a) vs VEH; (b) vs. CAB; (c) vs. G007.
Overall, the data indicate that combined YAP/TAZ and cabozantinib targeting might be an effective therapy for human HCC with LOF AXIN1 mutation.
Discussion
Recent comprehensive studies have uncovered the genetic landscape of human HCC, suggesting that this tumor type is a highly heterogeneous disease and underlining the importance of a precision medicine approach for HCC treatment(5). Activation of the Wnt/β-Catenin cascade plays a significant role during hepatocarcinogenesis. Mutations in CTNNB1, the gene encoding for β-Catenin, interfere with its degradation leading to its oncogenic activation, and are implicated in 20–35% of HCCs. In addition, approximately 8% of HCCs have LOF mutations in AXIN1, which encodes for the scaffolding protein AXIN1, essential for β-Catenin degradation(4). While both GOF CTNNB1 mutations and LOF AXIN1 mutations are thought to function via activating the Wnt/β-Catenin cascade, recent studies suggest that these genetic events may lead to distinct cellular signaling and phenotypes(8). Based on human HCC gene profiling, it has been suggested that most HCCs could be classified into a “proliferating group” or “non-proliferating group”. While HCCs with CTNNB1 mutations belong to the “non-proliferating group”; HCCs with LOF AXIN1 mutations are classified into “proliferating group”(28). A detailed analysis of genes enriched in LOF AXIN1 mutant HCC suggests the activation of the YAP/TAZ pathway. However, the functional contribution of YAP/TAZ in HCCs with GOF CTNNB1 mutations and LOF AXIN1 mutations has not been investigated to date.
In the current study, we applied the c-Met/β-Catenin and c-Met/sgAxin1 murine HCC models to investigate the functional role of the Hippo/YAP/TAZ cascade in GOF CTNNB1 and LOF AXIN1 mutations driven HCC. The fact that both the murine models have c-MET as the common second “oncogenic hit” allowed us to accurately compare the downstream signals elicited by activated β-Catenin or loss of Axin1 in vivo. We found strong activation of YAP/TAZ in c-Met/sgAxin1 mouse HCC, recapitulating what has been described in LOF AXIN1 mutant human HCC. Next, via activation of the Hippo kinase Lats2, or deletion of Yap and Taz, we demonstrated that YAP/TAZ are critically required for c-Met/sgAxin1 induced HCC but have relatively mild effects on c-Met/β-Catenin driven hepatocarcinogenesis. Therefore, our investigation provides powerful mechanistic insights into how GOF CTNNB1 and LOF AXIN1 mutations might function via common and distinct pathways to promote HCC development (Supplementary Figure 14). Specifically, both genetic aberrations lead to the activation of β-Catenin and its downstream oncogenic effectors, including c-Myc and Cyclin D1. In addition, GOF CTNNB1 mutation induces canonical liver-specific genes, such as GS. This molecular event, in turn, leads to the activation of mTORC1(8, 9). Together with activated c-Met signaling, it eventually leads to HCC formation. On the other hand, AXIN1 interacts with YAP/TAZ, leading to its degradation and inactivation. LOF AXIN1 mutation results in increased protein stability and, subsequently, the activation of YAP/TAZ, which is required for its cooperation with c-MET to promote hepatocarcinogenesis. In addition, it has been shown that YAP/TAZ could bind to AXIN1 at its LRP6 binding domain, which locates between the 496 to 811 amino acids (exon 5–10) of AXIN1(26). Therefore, it is likely that AXIN1 mutation in the HCC cell lines, including SNU475 and PLC/PRF/5, could prohibit the interaction between AXIN1 and YAP/TAZ, resulting in the activation status of YAP/TAZ.
Previously, Bisso et al. reported that YAP/TAZ is required for activated β-Catenin to accelerate MYC-promoted hepatocarcinogenesis using MYC;β-CatEx3 transgenic mice (29). Here, we showed that Yap;Taz DKO also mildly delayed c-Met/β-Catenin tumor development. All these data support that YAP/TAZ have a role, although relatively moderate, in GOF CTNNB1 mutation driven hepatocarcinogenesis. It is important to note as key oncogenic molecules, YAP and TAZ activities are regulated at multiple levels. Indeed, we noted the lower expression of LATS2 in mouse HCC samples, suggesting the inaction of Hippo kinases as a key mechanism leading to YAP/TAZ activation in these mouse HCC models. Loss of AXIN1 function leads to a further increased YAP/TAZ stability. All these mechanisms function together to promote a high level of YAP/TAZ activation in AXIN1 mutant/null HCC cells. While YAP and TAZ are paralogs, recent studies suggest that the two molecules may have distinct and/or redundant roles in liver tumor development, depending on the driver oncogenes(30, 31). In our current studies, we demonstrated that in mouse HCCs induced by c-Met/sgAxin1 or c-Met/β-Catenin, ablation of Yap or Taz alone did not prevent HCC formation, suggesting that under these oncogenic drivers, YAP and TAZ are functionally redundant; and targeting both YAP and TAZ is required to inhibit HCC progression.
Our study has clear translational implications, especially in precision medicine-based therapeutics against HCC. While activated β-Catenin has been implicated in LOF AXIN1 mutant HCCs, targeting β-Catenin has proven to be complicated(32, 33). Our investigation demonstrates that inactivation of Hippo and activation of YAP/TAZ have pivotal roles in LOF AXIN1 mutation, but not GOF CTNNB1 mutation-induced HCC, suggesting the targeting YAP/TAZ or reactivating the Hippo cascade for the treatment of HCC with LOF AXIN1 mutations. Furthermore, using the inducible Cre system, we proved that simultaneous deletion of Yap;Taz resulted in the regression of already formed c-Met/sgAxin1 tumors. Also, the combinational administration of the tankyrase inhibitor G007-LK and the tyrosine kinase inhibitor cabozantinib induced significant survival benefits, providing a novel therapeutic strategy against AXIN1 mutant HCCs. By the end of the 3-week-therapy, the combinational treatment had already resulted in significant tumor reduction. Therefore, we anticipate that longer treatment might be able to exhibit more effectiveness. Therefore, the current study supports further investigation of combined cabozantinib and tankyrase inhibitors for HCC treatment in clinical trials. Importantly, screening patients with LOF AXIN1 mutations, such as via next-generation sequencing, could assist in identifying patients who are most likely to benefit from the combination treatment.
Supplementary Material
Financial support:
This study is supported by NIH grants R03CA208311, R01CA250227, and R01CA239251 to Xin Chen, P30DK026743 for UCSF Liver Center; grants from China Scholarship Council (grant number 201806165021) and Natural Science Foundation of Hubei province (grant number 2017CFB258) to Binyong Liang, grants from National Natural Science Foundation (grant number 82002967) to Haichuan Wang.
List of abbreviations:
- AXIN1
Axis inhibition protein 1
- AXIN2
Axis inhibition protein 2
- CCLE
Cancer Cell Line Encyclopedia
- CHX
Cycloheximide
- c-Met
c-mesenchymal-epithelial transition factor
- Co-IP
Co-Immunoprecipitation
- CTNNB1
β-Catenin (cadherin-associated protein) beta 1
- CTGF
connective tissue growth factor
- CYR61
Cysteine-rich angiogenic inducer 61
- DKO
Double knockout
- GOF
Gain of function
- GS
Glutamine synthetase
- HB
Hepatoblastoma
- HCC
Hepatocellular carcinoma
- H&E
Hematoxylin-Eosin staining
- HTVi
Hydrodynamic tail vein injection
- IHC
Immunohistochemistry
- KO
Knockout
- LATS 2
Large tumor suppressor kinase 2
- qRT-PCR
quantitative reverse transcription PCR
- SB
Sleeping beauty
- TAZ
PDZ-binding motif
- TBX3
T-box transcription factor 3
- TCGA
The Cancer Genome Atlas
- wt
wild-type
- WWTR1
WW domain-containing transcription regulator 1
- YAP
Yes-associated protein
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest.
References
- 1.Vogel A, Cervantes A, Chau I, et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv238–iv255. [DOI] [PubMed] [Google Scholar]
- 2.Gordan JD, Kennedy EB, Abou-Alfa GK, et al. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J Clin Oncol. 2020;38(36):4317–4345. [DOI] [PubMed] [Google Scholar]
- 3.Russell JO, Monga SP. Wnt/beta-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu Rev Pathol. 2018;13(351–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiao Y, Wang J, Karagoz E, et al. Axis inhibition protein 1 (Axin1) Deletion-Induced Hepatocarcinogenesis Requires Intact beta-Catenin but Not Notch Cascade in Mice. Hepatology. 2019;70(6):2003–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169(7):1327–1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abitbol S, Dahmani R, Coulouarn C, et al. AXIN deficiency in human and mouse hepatocytes induces hepatocellular carcinoma in the absence of beta-catenin activation. J Hepatol. 2018;68(6):1203–1213. [DOI] [PubMed] [Google Scholar]
- 9.Zucman-Rossi J, Benhamouche S, Godard C, et al. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene. 2007;26(5):774–780. [DOI] [PubMed] [Google Scholar]
- 10.Yimlamai D, Fowl BH, Camargo FD. Emerging evidence on the role of the Hippo/YAP pathway in liver physiology and cancer. J Hepatol. 2015;63(6):1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S, Zhou D. Role of the transcriptional coactivators YAP/TAZ in liver cancer. Curr Opin Cell Biol. 2019;61(64–71. [DOI] [PubMed] [Google Scholar]
- 12.Tao J, Calvisi DF, Ranganathan S, et al. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology. 2014;147(3):690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tao J, Xu E, Zhao Y, et al. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant beta-catenin. Hepatology. 2016;64(5):1587–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shang R, Song X, Wang P, et al. Cabozantinib-based combination therapy for the treatment of hepatocellular carcinoma. Gut. 2021;70(9):1746–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song X, Xu H, Wang P, et al. Focal adhesion kinase (FAK) promotes cholangiocarcinoma development and progression via YAP activation. J Hepatol. 2021;75(4):888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol. 2014;184(4):912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yabuta N, Mukai S, Okada N, Aylon Y, Nojima H. The tumor suppressor Lats2 is pivotal in Aurora A and Aurora B signaling during mitosis. Cell Cycle. 2011;10(16):2724–2736. [DOI] [PubMed] [Google Scholar]
- 18.Yabuta N, Yoshida K, Mukai S, et al. Large tumor suppressors 1 and 2 regulate Aurora-B through phosphorylation of INCENP to ensure completion of cytokinesis. Heliyon. 2016;2(7):e00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furth N, Aylon Y. The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death Differ. 2017;24(9):1488–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pineau P, Marchio A, Nagamori S, et al. Homozygous deletion scanning in hepatobiliary tumor cell lines reveals alternative pathways for liver carcinogenesis. Hepatology. 2003;37(4):852–861. [DOI] [PubMed] [Google Scholar]
- 21.Satoh S, Daigo Y, Furukawa Y, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24(3):245–250. [DOI] [PubMed] [Google Scholar]
- 22.Caruso S, Calatayud AL, Pilet J, et al. Analysis of Liver Cancer Cell Lines Identifies Agents With Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology. 2019;157(3):760–776. [DOI] [PubMed] [Google Scholar]
- 23.Qiu Z, Li H, Zhang Z, et al. A Pharmacogenomic Landscape in Human Liver Cancers. Cancer Cell. 2019;36(2):179–193 e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harnos J, Rynes J, Viskova P, et al. Analysis of binding interfaces of the human scaffold protein AXIN1 by peptide microarrays. J Biol Chem. 2018;293(42):16337–16347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Azzolin L, Panciera T, Soligo S, et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell. 2014;158(1):157–170. [DOI] [PubMed] [Google Scholar]
- 27.Jia J, Qiao Y, Pilo MG, et al. Tankyrase inhibitors suppress hepatocellular carcinoma cell growth via modulating the Hippo cascade. PLoS One. 2017;12(9):e0184068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. [DOI] [PubMed] [Google Scholar]
- 29.Bisso A, Filipuzzi M, Gamarra Figueroa GP, et al. Cooperation Between MYC and beta-Catenin in Liver Tumorigenesis Requires Yap/Taz. Hepatology. 2020;72(4):1430–1443. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Wang J, Zhang S, et al. Distinct and Overlapping Roles of Hippo Effectors YAP and TAZ During Human and Mouse Hepatocarcinogenesis. Cell Mol Gastroenterol Hepatol. 2021;11(4):1095–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Zhang S, Zhang Y, et al. TAZ is indispensable for c-MYC-induced hepatocarcinogenesis. J Hepatol. 2022;76(1):123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vilchez V, Turcios L, Marti F, Gedaly R. Targeting Wnt/beta-catenin pathway in hepatocellular carcinoma treatment. World J Gastroenterol. 2016;22(2):823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perugorria MJ, Olaizola P, Labiano I, et al. Wnt-beta-catenin signalling in liver development, health and disease. Nat Rev Gastroenterol Hepatol. 2019;16(2):121–136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.